 Jun-Peng Jiang
Jun-Peng Jiang Xuan Liu
Xuan Liu Yi-Fan Liao1
Yi-Fan Liao1 Jun Shan
Jun Shan Chang-Hong Liu
Chang-Hong Liu- 1State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, China
- 2State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing, China
Introduction: Aspergillussydowii is an important filamentous fungus that inhabits diverse environments. However, investigations on the biology and genetics of A. sydowii in subseafloor sediments remain limited.
Methods: Here, we performed de novo sequencing and assembly of the A. sydowii 29R-4-F02 genome, an isolate obtained from approximately 2.4 km deep, 20-million-year-old coal-bearing sediments beneath the seafloor by employing the Nanopore sequencing platform.
Results and Discussion: The generated genome was 37.19 Mb with GC content of 50.05%. The final assembly consisted of 11 contigs with N50 of 4.6 Mb, encoding 12,488 putative genes. Notably, the subseafloor strain 29R-4-F02 showed a higher number of carbohydrate-active enzymes (CAZymes) and distinct genes related to vesicular fusion and autophagy compared to the terrestrial strain CBS593.65. Furthermore, 257 positively selected genes, including those involved in DNA repair and CAZymes were identified in subseafloor strain 29R-4-F02. These findings suggest that A. sydowii possesses a unique genetic repertoire enabling its survival in the extreme subseafloor environments over tens of millions of years.
Introduction
Ocean Drilling Program revealed a new ecosystem composed of microorganisms in subseafloor sediments (D’Hondt et al., 2004; Schrenk et al., 2010; Lomstein et al., 2012), which vertically extends for ~2.5 km below the seafloor (Selbmann et al., 2013; Inagaki et al., 2015). This deep subseafloor biosphere harbors organisms from three major domains of life: Archaea (Orsi et al., 2013), Bacteria (Schippers et al., 2005), and Eukarya (Borgonie et al., 2015). Fungi, as the dominant component of eukaryotes, also widely exist in the subseafloor sediments (Rédou et al., 2015; Nagano et al., 2016; Liu et al., 2017). Most subseafloor fungi were affiliated with the phyla Ascomycota, Basidiomycota and Chytridiomycota (Nagano and Nagahama, 2012). Phylogenetic analysis indicated that subseafloor fungi were closely related to the isolates from terrestrial, freshwater and marine environments (Liu et al., 2017; Quemener et al., 2020). Various culture-dependent and culture-independent investigations show that fungi are active and well-adapted to subseafloor environments (Rédou et al., 2015; Liu et al., 2017). Some fungi, such as Aquamyces sp. and Orbilia sp., can live in a saprophytic mode and maintain their life in anaerobic habitats by degrading organic substances (Ortega-Arbulu et al., 2019). Our previous studies have shown that Schizophyllum commune isolated from ~2 km coal-bearing sediment below seafloor has a variety of anaerobic survival strategies, including improving the metabolic efficiency of ethanol and amino acids, increasing the number of mitochondria in cells, and forming autophagosomes, but it cannot complete sexual propagation (Zain Ul Arifeen et al., 2021a,b). Genome analysis of S. commune revealed significant expansion of FunK1 protein kinase, the NmrA family, and transposon, but significant reduction of nucleotide diversity, substitution rates, and homologous recombination, reflecting the unique genetic evolution of fungi to maintain their long-term life in subseafloor environments (Liu et al., 2022). However, due to the limited number of pure culture strains of subseafloor fungi, the genomes of only a few fungi have been sequenced, and there is still a lack of understanding of the genetic characteristics of the fungi in deep biosphere.
Aspergillus is one of the best-studied filamentous fungi, mainly because some species of this genus are widely used in the fields of medicine (e.g., A. fumigatus, A. terreus), food spoilage (e.g., A. flavus, A. parasiticus, and A. hancockii), and industry (e.g., A. niger, A. aculeatus, and A. oryzae; de Vries et al., 2017; Pitt et al., 2017). A. sydowii, in particular, is distributed throughout the world and can survive as saprophytes (Raper and Fennell, 1965; Geiser et al., 1998; Klich, 2002). Our previous study found that this fungus is also one of the dominant fungi in ~2 km sediments below the seafloor (Liu et al., 2017). Some strains of A. sydowii isolated from marine ecosystems can cause an epizootic infection of sea fan corals (Kim and Harvell, 2004; Hayashi et al., 2016), while others can produce a series of secondary metabolites with novel structure and bioactivities (Wang et al., 2019; Niu et al., 2020), and degrade the refractory substances such as xylan (Brandt et al., 2020), lignocellulose (Cong et al., 2017), and waste engine oil (Kumar et al., 2021). Given the wide distribution and important application value of A. sydowii, five strains of this species have been sequenced, assembled and annotated (de Vries et al., 2017; Brandt et al., 2021; Kumar et al., 2021; Jing et al., 2022), of which strain CBS593.65 had been fully studied. Of the sequenced strains, AS31, AS42, Fsh102, CBS593.65, and BOBA1 were isolated from terrestrial land and deep sea, respectively. However, the genome of any A. sydowii strains derived from subseafloor sediments is not yet available.
In order to acquire insight into the survival and environmental adaptation mechanism of subseafloor fungi, we performed the de novo whole genome assembly and comprehensive analysis of A. sydowii 29R-4-F02 buried in 2,405 m coal-bearing sediment below the seafloor. A large number of genes were identified to be involved in the secondary metabolism and carbohydrate metabolism. The carbohydrate-active enzymes (CAZymes) repertoire of A. sydowii 29R-4-F02 was identified and compared with that of other fungi. Moreover, genes or gene families were investigated through comparative genomes to reveal the special genetic evolution process of the strain.
Materials and methods
Fungal strain and culture conditions
A. sydowii 29R-4-F02 (CCTCC AF 2022080) strain was isolated from ~2.4 km coal-bearing sediments below the seafloor at drilling Site C0020 (41°10′35″N, 142°12′01″E) off the Shimokita Peninsula, Japan (Liu et al., 2017) and maintained aerobically on potato dextrose agar (PDA, 200 g/L potato, 20 g/L glucose, and 15 g/L agar) slants at 4°C. Details of the habitat and culture conditions of A. sydowii 29R-4-F02 have been described previously (Liu et al., 2017). For DNA isolation, mycelium of A. sydowii 29R-4-F02 was grown in 250 ml conical flask containing 100 ml PD (200 g/L potato and 20 g/L glucose) and incubated at 30°C and 200 rpm for 2–3 days.
Genome sequencing and assembly
DNA extraction was carried out by HP fungal DNA mini kit (Omega Bio-Tek, GA, USA) according to the manufacturer’s instructions. The DNA concentration (>50 ng/μl), quality and integrity were determined by using a Qubit Flurometer (Invitrogen, USA) and a NanoDrop Spectrophotometer (Thermo Scientific, USA). Genome sequencing was performed using the Nanopore sequencing platform at the Beijing Novogene Bioinformatics Technology Co., Ltd. After filtering out low quality reads (less than 500 bp) by Nanoplot v 1.41.0 software, a total of 441,200 bases reads was retained (De Coster et al., 2018). The clean reads were mapped with each other to find the overlap between sequences by Minimap2 v 2.26 software (Li, 2018), and then assembled by Miniasm software to constructed scaffolds (Li, 2016). Finally, the Racon v 1.5.0 software was used to constructed consensus sequence and correct the assembly results (Vaser et al., 2017). BUSCO v 5.4.7 was used to assess the completeness of gene regions using the dataset fungi_odb9 (Waterhouse et al., 2017). Quast v 5.1.0 software was further used to evaluate the assembly quality of A. sydowii 29R-4-F02 genome with the default parameters (Gurevich et al., 2013). Reference-based assembly of the A. sydowii 29R-4-F02 mitochondrial genome was conducted using the Rebaler v 0.2.0 software (Baeza and García-De León, 2022). We executed Rebaler using A. nidulans mitochondrial genome as reference genome. Also, we ran Rebaler with the option “circular = true” indicating that the reference genome was circular so that Rebaler “rotated” contigs between polishing rounds to ensure improved accuracy of the final assembled mitochondrial genome.
Genome annotation
After obtaining the whole-genome data of A. sydowii 29R-4-F02, we used two strategies to annotated genes: (i) Augustus v 2.7 was used for de novo prediction (Stanke et al., 2008); (ii) Homologous species was predicted in Genewise v 2.4.1 using the reference gene models of A. sydowii CBS593.65 (Birney et al., 2004). Gene models from these two approaches were combined using the EVidenceModeler v 2.1.0 and updated by PASA v2.4.1 (Haas et al., 2008). Dispersed repeat sequences (DRs) and tandem repeat sequences (TRs) were identified by RepeatMasker v open-4.0.5 (Saha et al., 2008) and TandemRepeat v 4.07b Finder (TRF), respectively (Benson, 1999). The tRNA and rRNA were predicted by tRNAscan-SE v 1.3.1 (Chan et al., 2021) and rRNAmmer v 1.2 (Lagesen et al., 2007). The sRNA, snRNA and miRNA were first performed Rfam database v 31.0 (Gardner et al., 2009) comparison annotation, and then use the cmsearch program (v 1.1rc4; default parameter) to determine the final sRNA, snRNA and miRNA.
Gene functions were annotated based on Gene Ontology (GO, v 20171011), Kyoto Encyclopedia of Genes and Genomes (KEGG, v 20181107), Eukaryotic Clusters of Orthologous Groups (KOG, v 201711), Non-redundant protein database (Nr, v 201703), PFAM databases (v 31.0), SwissProt (v 20180716), and Carbohydrate-Active enZYmes Database (v 20181107; Wang et al., 2021; Liu et al., 2022). The secreted proteins were predicted by the SingalP 4.1 and TMHMM 2.0c (Petersen et al., 2011) signal database. Meanwhile, fungal secondary metabolite pathways were predicted using the online tool AntiSMASH v 4.0.2 (Medema et al., 2011). The cytochrome P450 gene family were predicted using Cytochrome P450 Database (Fischer et al., 2007).
Phylogenetic and synteny analyses
A total of 21 fungal species (i.e., 22 strains) of Aspergillus with whole genome sequence, including A. sydowii (two strains 29R-4-F02 and CBS593.65), A. aculeatus, A. brasiliensis, A. carbonarius, A. clavatus, A. fischeri, A. flavus, A. fumigatus, A. glaucus, A. homomorphus, A. ibericus, A. luchuensis, A. nidulans, A. niger, A. oryzae, A. phoenicis, A. terreus, A. tubingensis, A. versicolor, A. wentii, and A. zonatus were used to perform gene family analysis. Except A. sydowii 29R-4-F02, protein sequences of these species were downloaded from NCBI and JGI databases, and analyzed by OrthoFinder with default parameters to find orthologous genes families (Emms and Kelly, 2015).
The single copy orthologous genes of the 22 analyzed strains were aligned using MAFFT and create a super-gene for each species (Katoh and Standley, 2013). After Gblocks alignment optimization (Castresana, 2000), the conserved blocks of super-genes were used for phylogenetic tree construction using RAxML (Stamatakis, 2014). The final phylogenetic tree was visualized and edited in iTOL9 (Letunic and Bork, 2016).
In addition, synteny analysis of strains 29R-4-F02 and CBS593.65 was performed using MUMmer sequence alignment package (Delcher et al., 2002). Conserved syntenic blocks between the two strains were identified using the MCScanX package with default parameters based on a minimum requirement of five collinear orthologous genes (Wang et al., 2012). Gene Ontology (GO) enrichment was analyzed by Cytoscape software.
Identification and classification of CAZymes families
For comparative analysis of the CAZymes, HmmScan in the HMMER v 3.1b2 package (Eddy, 1998) was used to search protein sequences of A. sydowii 29R-4-F02 against the family-specific HMM profiles of CAZymes from dbCAN database v 9 (Yin et al., 2012) to detect putative CAZymes. The identified CAZymes were then classified into glycoside hydrolases (GHs), glycosyltransferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), auxiliary activities (AAs), and carbohydrate-binding modules (CBMs) base on the CAZy database (Lombard et al., 2014).
Positively selected genes
The positive selected genes (PSGs) between A. sydowii 29R-4-F02 and A. sydowii CBS593.65 were detected using the KaKs_Calculator v 2.0 software with the default parameters (Zhang et al., 2006).
Results
Genome sequencing and assembly
The genome of A. sydowii 29R-4-F02 was sequenced using Nanopore sequencing platform. After filtering out low-quality reads, over 3.11 Gb reads (~83.62×) were assembled into 11 contigs with a contig N50 of 4.6 Mb, and the GC content of 50.05% (Figure 1; Table 1), resulting in a total assembly size of 37.19 Mb that was consistent with the estimated genome size of 29.00–39.00 Mb and GC content of 48.00–53.00% of different Aspergillus isolates (de Vries et al., 2017; Kumar et al., 2021). The integrity of the gene region was evaluated using BUSCO, which showed high quality of sequence assembly, with 92.8% of single copies intact and only 3.7% missing. Quast analysis showed that the genomic quality of strain 29R-4-F02 was superior to that of strains Fsh102 and BOBA1 (Supplementary Figure 1), and comparable to that of strain CBS593.65 (de Vries et al., 2017). 86.10% of genes of strain 29R-4-F02 were collinear with those of strain CBS593.65 (Supplementary Figure 2). In addition, we assembled the mitochondrial genome of strain 29R-4-F02, which has a length of 26,185 bp and contains 13 genes, including 7 NADH–ubiquinone oxidoreductase genes, 3 cytochrome c oxidase genes, 1 cytochrome b gene, 1 ribosomal protein gene, and 1 ATP synthase gene, with a GC content of 23.67%. The size and number of genes in the mitochondrial genome of strain 29R-4-F02 are similar to those of other Aspergillus species, with genome sizes ranging from 27,817 to 77,649 bp and gene numbers ranging from 14 to 21 (Zhao et al., 2012; Takahashi et al., 2021; Jing et al., 2022).
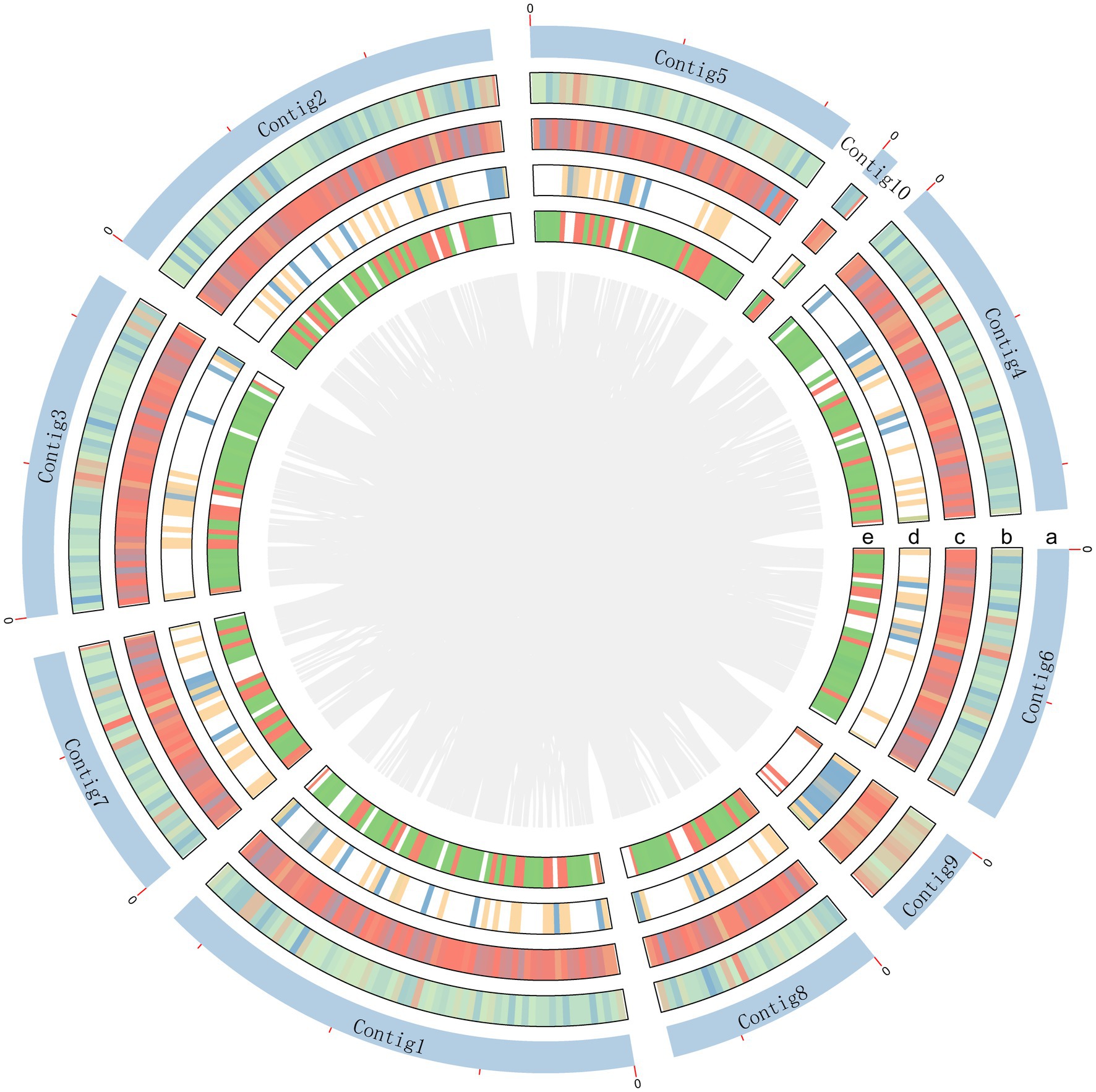
Figure 1. Schematic representation of genomic characteristics of A. sydowii 29R-4-F02. Circle a represents contigs, but excluding contig11 without annotation gene. Circle b and c are CDS on the positive and negative chains. Circle d and e are gene density on the positive and negative chains.
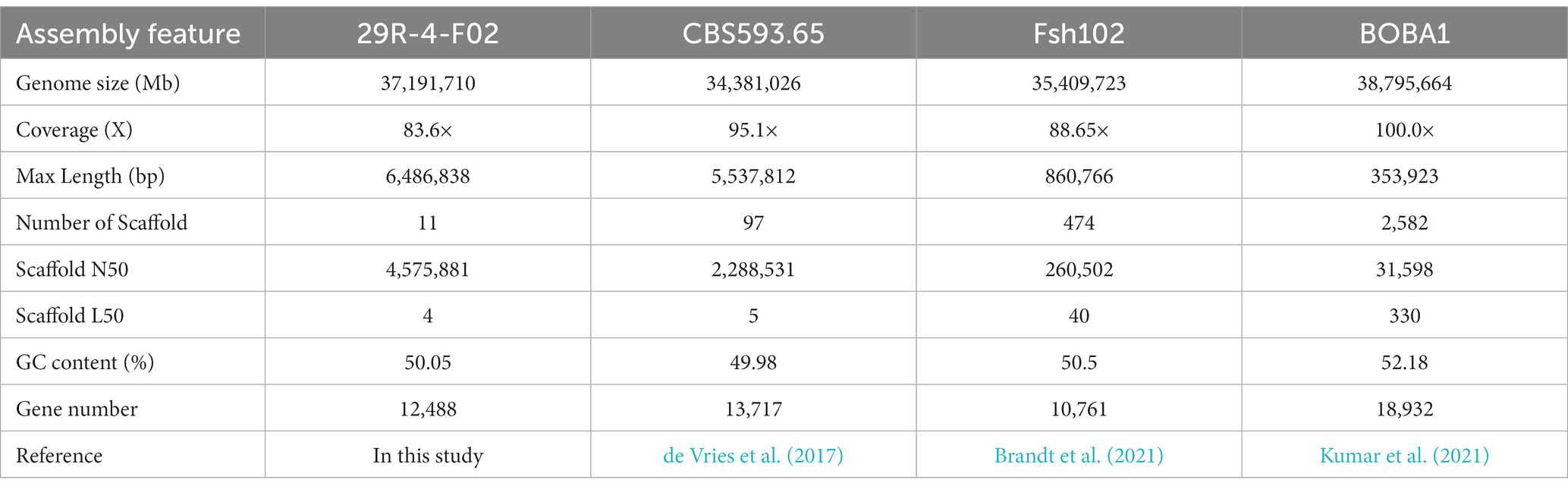
Table 1. Genome assembly and annotation summary of A. sydowii strains.
Genome and functional annotation
A total of 12,488 protein-coding genes were predicted with an average sequence length of 1,567 bp and a cumulative length of 19.6 Mb, accounting for 52.62% of the total genome sequence. The total length of repeat sequences was about 0.76 Mb, accounting for 2.06% of the genomic size. Among the repetitive elements, tandem and interspersed repeats account for 0.74 and 1.31%, respectively. Moreover, 160 tRNA genes, 35 sRNA genes, 15 snRNA genes, and 48 rRNA genes were predicted in the genome (Supplementary Table 1). Additionally, functional annotation of the predicted genes was performed based on the public database of Nr, SwissProt, KEGG, KOG, GO, and Pfam, protein databases (Supplementary Table 2).
Based on the GO database, 68.51% of protein-coding genes (8,555) were annotated into three categories: biological process, cellular components, and molecular function (Figure 2; Supplementary Table 2). For biological processes, genes involved in “metabolic process” (4,783) are the most abundant, followed by “cellular process” (4,637), “localization” (1,606), “establishment of localization” (1,570), “biological regulation” (1,150), and “regulation of biological process” (1,150). Among the genes involved in cellular components, 3,368, 3,368, 1,453, and 712 were associated with “cell,” “cell parts,” “organelle,” and “macromolecular complex,” respectively. Furthermore, for genes related to molecular function, 4,585, 4,487, 769, and 531 were involved in “binding,” “catalytic activity,” “transporter activity,” and “nucleic acid binding transcription factor activity,” respectively. These results revealed that the subseafloor A. sydowii 29R-4-F02 had abundant genes related to metabolic activities. Additionally, 2,237 protein-coding genes (17.91%) were annotated based on the KOG database (Figure 3; Supplementary Table 2). The majority of the genes were associated with “general function prediction only” (287), followed by “Energy production and conversion,” “post-translational modification, protein turnover, chaperones,” “Amino acid transport and metabolism,” and “Translation, ribosomal structure and biogenesis.” These findings indicated that A. sydowii 29R-4-F02 has a variety of protein metabolism and energy metabolism processes, which may help the fungus to better absorb and utilize nutrients from the subseafloor environment and maintain long-term survival. To further investigate the gene functions in A. sydowii 29R-4-F02, 10,893 (87.22%) protein-coding genes were assigned to their orthologs in the KEGG database. The KEGG function classification was shown in Figure 4 and Supplementary Table 2, including Cellular Processes (460), Environmental Information Processing (230), Genetic Information Processing (649), Human Diseases (578), Metabolism (1,756), and Organismal Systems (428). Consistent with the KOG annotation, metabolism and biosynthesis categories in KEGG were significantly annotated.
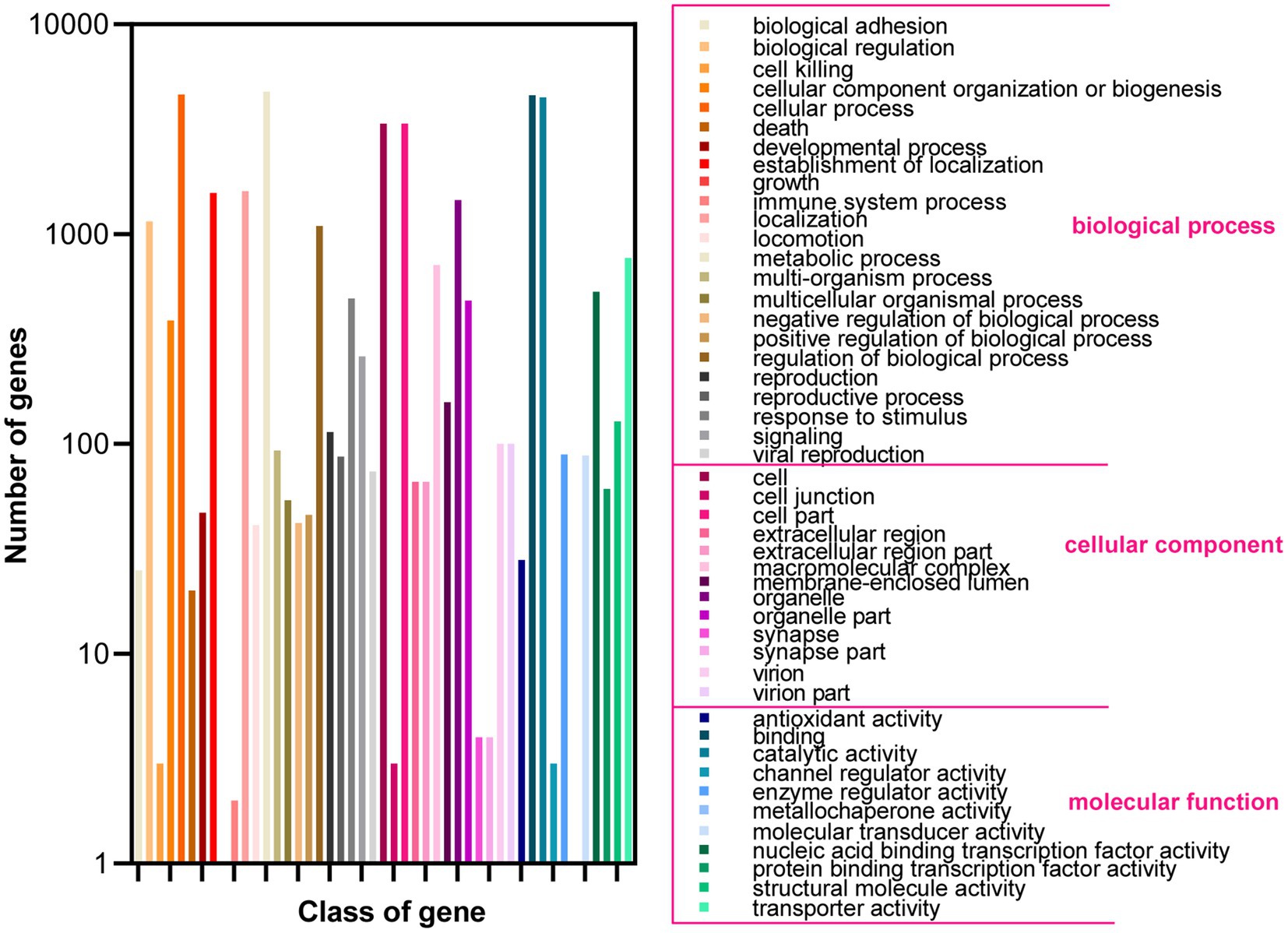
Figure 2. Functional annotation of A. sydowii 29R-4-F02 in Gene Ontology (GO) database.
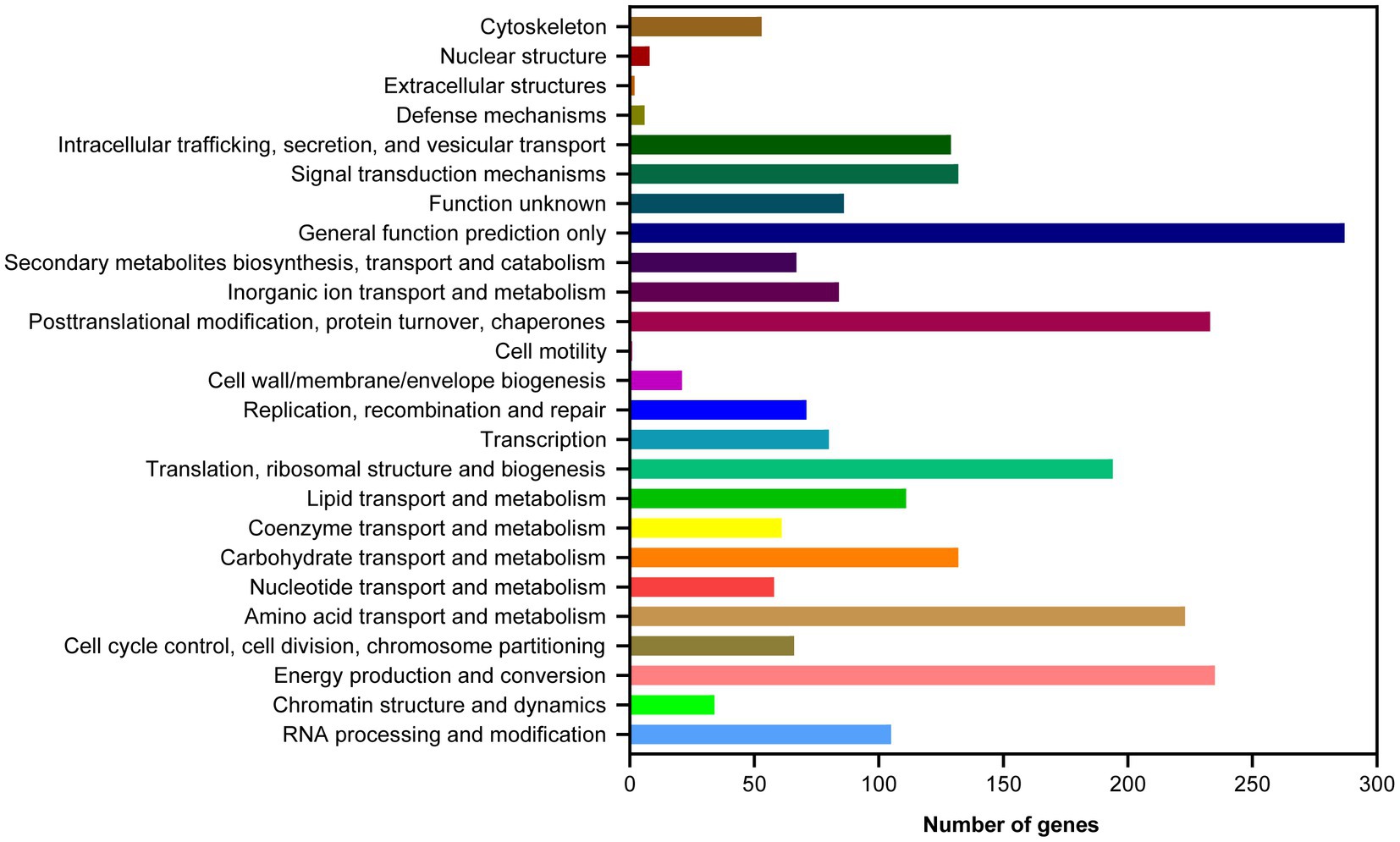
Figure 3. Clusters of Orthologous Groups of proteins (KOG) function classification of proteins in A. sydowii 29R-4-F02.
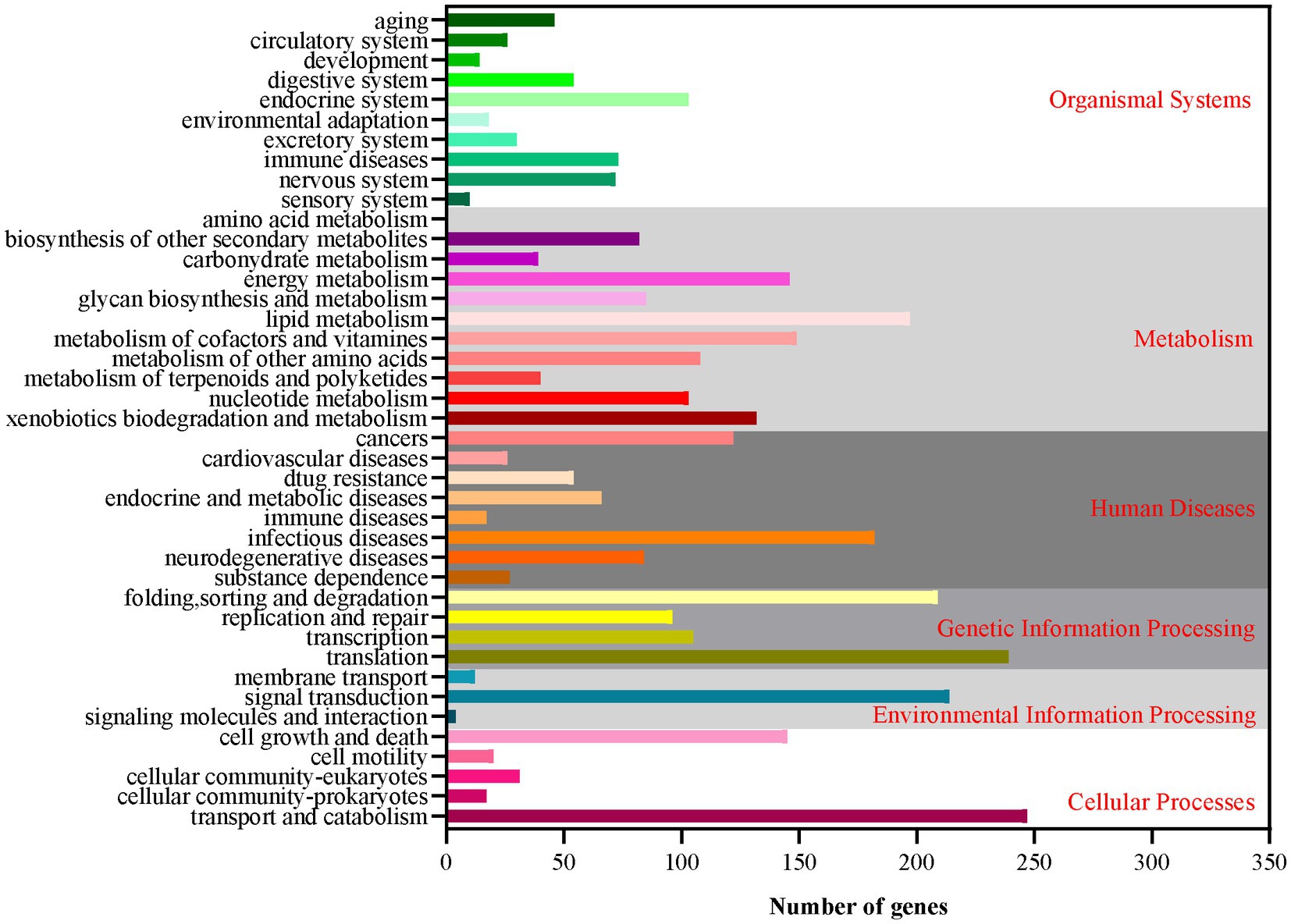
Figure 4. The Kyoto Encyclopedia of Genes and Genomes (KEGG) function annotation of A. sydowii 29R-4-F02.
A total of 52 gene clusters involved in the secondary metabolism, 231 cytochrome P450 genes, and 920 secreted proteins were identified in A. sydowii 29R-4-F02 genome (Figure 5; Supplementary Table 3). Among these P450 genes (Figure 5A), 13 were classified as P450, CYP52 class, 9 as E − class P450, CYP2D class, 9 as E − class P450, CYP3A class, 1 as E − class P450, CYP2A class, 93 as E − class P450, group I class, 27 as E − class P450, group IV class, 15 as Pisatin demethylase−like class, 1 as B − class P450 class, 7 as Cytochrome P450 class, and 56 as undetermined class. The E-class P450, group I class encoded by the most genes are involved in the oxidation–reduction reactions, while 56 undetermined class were predicted to participate in various secondary metabolic processes, such as xenobiotic biodegradation and carbohydrate metabolism. Similarly, among the secondary metabolite gene clusters (Figure 5B), 12 were classified as T1pks, 10 as Nrps, 8 as Terpenes, 6 as Indole, 4 as T1pks-Nrps, 2 as T1pks-Terpene, 1 as Indole-T1pks, and 9 others. These clusters contain whole or partial genes from known clusters such as Indole-T1pks and T1pks-Terpene cluster from Penicillium digitatum (CBS130527; Wang et al., 2021), T1pks and Terpene cluster from Calcarisporium sp. (KF525; Kumar et al., 2018), Nrps cluster from Aspergillus hancockii (FRR3425; Pitt et al., 2017), T1pks-Nrps cluster from Aspergillus westerdijkiae (CBS112803; Han et al., 2016), and T1pks-Nrps cluster from Pestalotiopsis sp. (KF079; Kumar et al., 2018), suggesting that A. sydowii 29R-4-F02 is capable of synthesizing these metabolites. These genes play a crucial role in fungi adapting to extreme environments and maintaining population competitive advantages (Choi et al., 2010; Cresnar and Petric, 2011; Keller, 2019; Yurchenko et al., 2021).
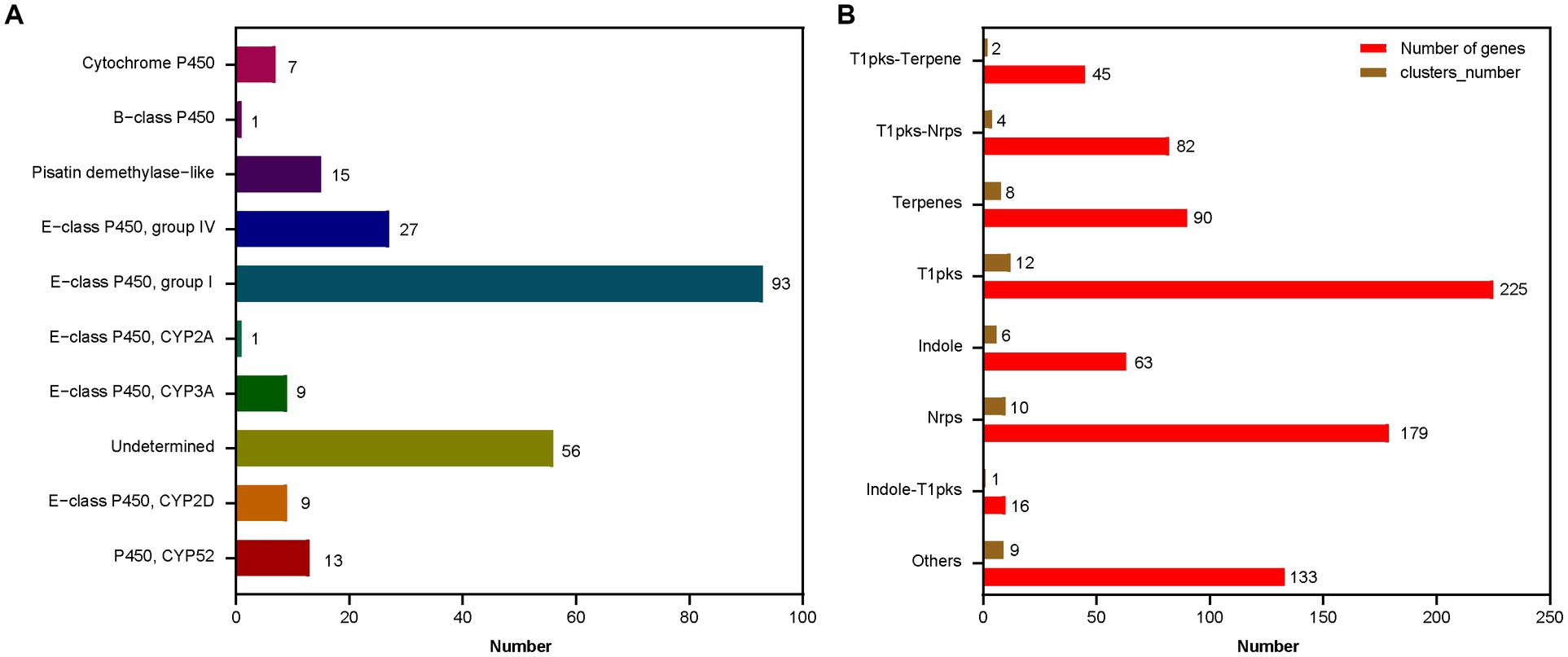
Figure 5. The function classification of the P450 genes (A) and the secondary metabolic gene clusters (B) in A. sydowii 29R-4-F02 genome. The horizontal axis represents the number of genes or gene clusters.
The CAZyme family
CAZymes play important roles in degrading cellulose, hemicellulose, pectin, and lignin polysaccharides and are responsible for the acquisition of nutrients for fungi (de Vries et al., 2017). In A. sydowii 29R-4-F02 genome, 670 CAZyme coding genes were identified, including 337 encoding genes for GHs, 78 encoding genes for AAs, 126 encoding genes for GTs, 39 encoding genes for CEs, 67 encoding genes for CBMs, and 23 encoding genes for PLs (Supplementary Table 4), which was much more than the genes in other species of Aspergillus (Table 2). Strain 29R-4-F02 also contains a large number of CAZyme coding genes related to the degradation of plant cell wall polysaccharides, including (hemi-) cellulases degrading enzymes (GH10, GH11, GH5, GH6, GH7, GH45, and GH115), pectin degrading enzymes (GH28, GH43, GH51, GH53, GH78, GH88, GH93, PL1, PL3, PL4, and PL9), xylan (GH10, GH11, GH62, GH67, CE1, and CE15), laccase (AA1), cellobiose dehydrogenase (AA3_1), vanillyl alcohol oxidase (AA7), lytic polysaccharide monooxygenases (AA9), and rhamnogalacturonan lyases (PL11) coding genes. Some CAZyme coding genes related to the degradation of non-plant polysaccharides (GH79 and GH88 families) were identified as well in the strain. These data indicate that A. sydowii 29R-4-F02 has a strong ability to degrade organic matter to facilitate its growth energy in lignite-containing subseafloor sediments.
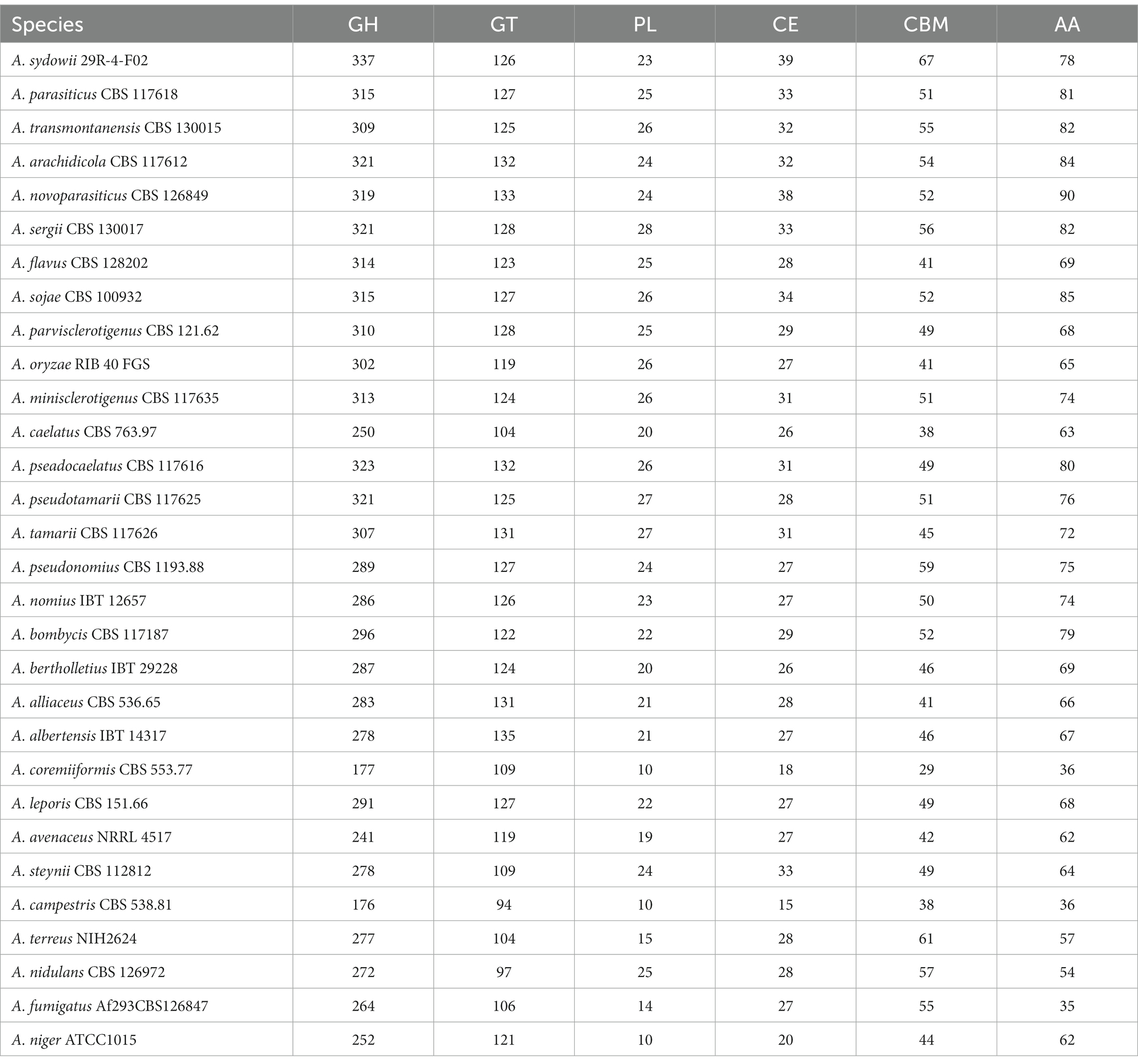
Table 2. Comparison of CAZyme encoding genes in different species of Aspergillus.
Gene family evolution and phylogenetic analysis
OrthoFinder analysis showed that 9,678 gene families were identified from 22 sequenced fungi. Among them, 3,559 gene families were shared by 22 fungi and 364 gene families were unique to A. sydowii 29R-4-F02 (Figure 6). These 364 gene families are composed of species-specific genes (8) and uncluster genes (356). Of these, the species-specific genes (A00461, A03330, A06713, and A12243) with functional annotation were found to be primarily associated with vesicle fusion and autophagy. 185 uncluster genes (Supplementary Figure 3), as determined through gene functional annotation, were found to be mainly involved in biological processes, cell components and molecular functions according to the results of GO enrichment analysis (Supplementary Figure 4; Supplementary Table 5). These data reveal that A. sydowii 29R-4-F02 has the ability to adapt to complex subseafloor environments.
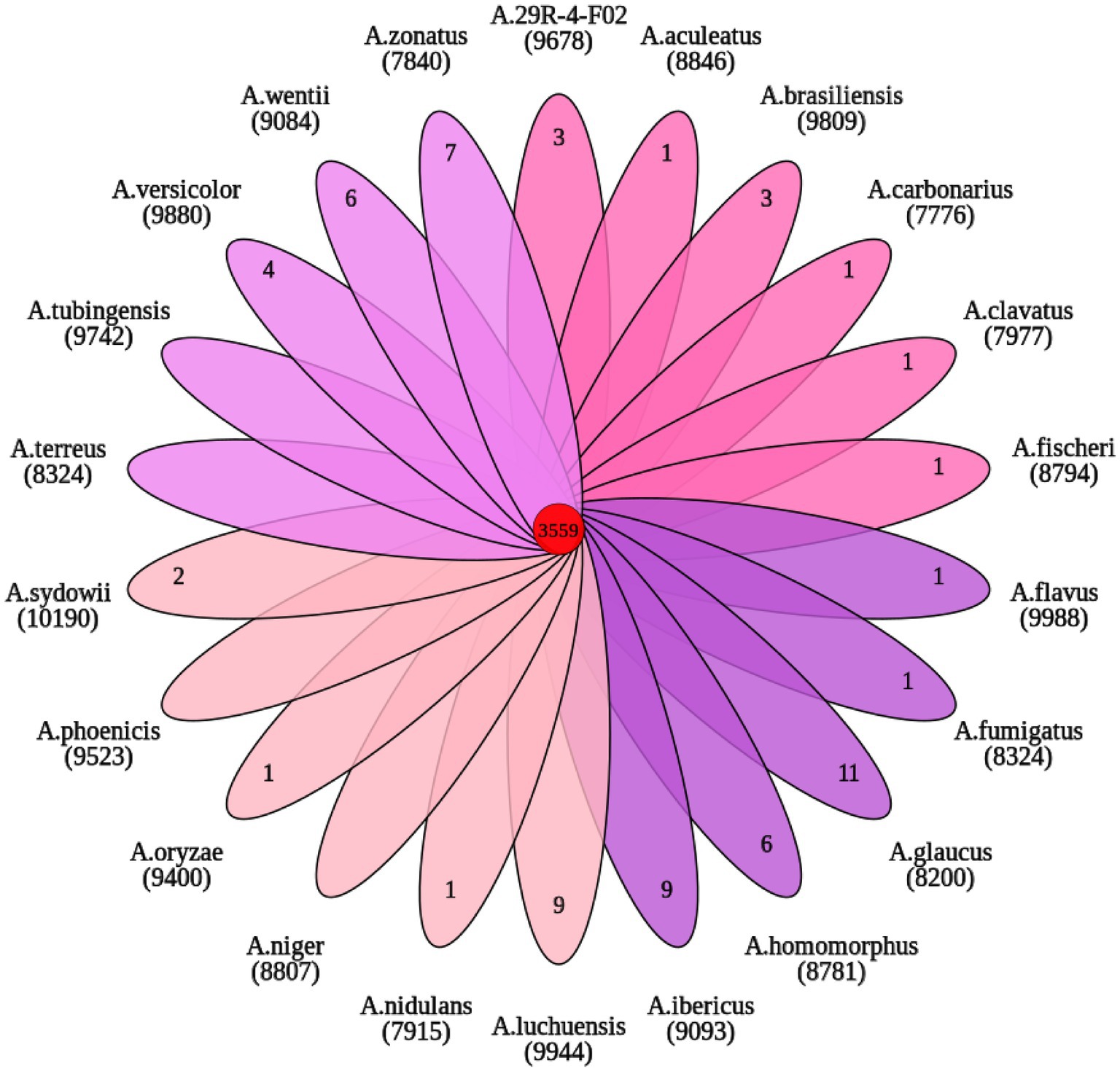
Figure 6. Venn diagram of orthologous gene families.
A phylogenetic tree was constructed based on 386 single copy orthologous genes identified from 22 fungi. The inference shows that A. sydowii 29R-4-F02 and A. sydowii CBS593.65 were clustered into the same clade (Figure 7). Compared with strain CBS593.65, strain 29R-4-F02 had 1,168 unique genes, including 12 genes belonging to 2 species-specific families (OG0000029 and OG0000322) and 1,156 uncluster genes (Supplementary Figures 5, 6; Supplementary Table 6). Among them, OG0000029 and OG0000322 were involved in ZEB2-regulated ABC transporter and glycopeptide α-N-acetylgalactosaminidase, respectively. ZEB2 regulated ABC transporters have been shown to play a crucial role in secreting virulence factors or compounds in pathogenic fungi (Lee et al., 2011; Zhang et al., 2023). Glycopeptide α-N-acetylgalactosaminidase belongs to glycoside hydrolase family (GH101) and has been primarily demonstrated to hydrolyze core 1-type O-glycans from glycoproteins (Fujita et al., 2005). Although this enzyme has been reported in bacteria, its presence in fungi has not been documented yet. In addition, 36 gene families involved in energy metabolism and environmental adaptation were significantly expanded in the genome of A. sydowii 29R-4-F02 (Supplementary Table 7). For example, OG0000024, OG0000315, OG0000220, and OG0000464 were associated with NADP-dependent alcohol dehydrogenase, short-chain dehydrogenase, heat shock protein, and nucleotide-excision repair, respectively. Given the nucleotide-excision repair mechanism has been proven to play an important role in genome conservation among subsurface microbes (Becraft et al., 2021; Liu et al., 2022), we believe that these extended genes may provide the genetic basis necessary for the long-term survival of A. sydowii 29R-4-F02 in extreme subseafloor sedimentary environments.
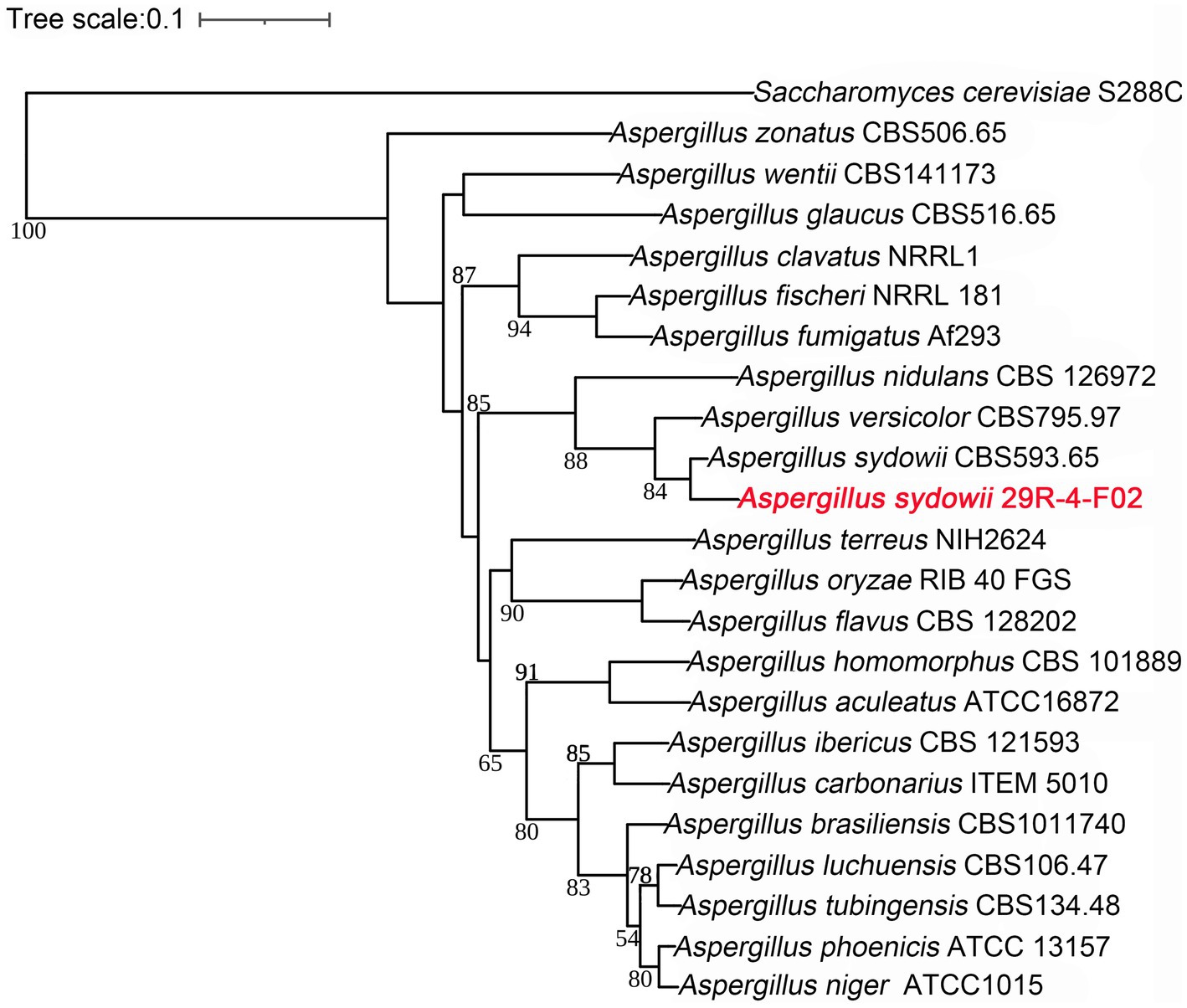
Figure 7. Phylogenetic tree of 21 Aspergillus genomes constructed based on single copy ortholog genes. The newly sequenced species in this study are represented in red font. Saccharomyces cerevisiae S288C as an outgroup species, the scale bar indicates evolutionary distance. Bootstrap values of <50 have been removed for clarity.
Positive selection improves the environmental adaptation of A. sydowii 29R-4-F02
Positive selection, acting on the evolution of functionally important gene families, is an important driving force for microorganisms in adaptation to the complex environment. To investigate those genes related to the adaptability of Aspergillus to the subseafloor environment, we conducted a positive selection analysis between A. sydowii 29R-4-F02 and A. sydowii CBS593.65 using KaKs_Calculator (Ka/Ks > 1 referred to positive selection; Wang et al., 2010). The results showed that 362 genes were under positive selection, including 257 annotated genes and 105 non-annotated genes. Among the annotation genes, 10 genes were involved in CAZymes family, and 5 genes were associated with DNA repair. These genes may help A. sydowii 29R-4-F02 to acquire nutrients and energy in lignite containing subseafloor sediments.
Discussion
A. sydowii was a filamentous fungus with a worldwide distribution and attracted our attention due to its abundantly metabolic profile (de Vries et al., 2017; Brandt et al., 2020; Kumar et al., 2021; Jing et al., 2022). In this study, a strain of A. sydowii 29R-4-F02 isolated from seafloor sediments was sequenced de novo using Nanopore long read sequencing technique. Collinearity and BUSCO analysis showed that strain 29R-4-F02 had high genome quality and integrity. Gene annotation indicated that the strain has rich metabolic activities, especially protein metabolism and energy metabolism, which provided an opportunity for us to explore the lifestyle and ecological adaptation mechanism of fungi in the subseafloor environment.
CAZymes are thought to play an important role in the utilization of organic matter such as cellulose, hemicellulose, pectin and lignin polysaccharides by fungi to obtain nutrients and energy for growth, and are also closely related to host preference and lifestyle adaptation of fungi (Couturier et al., 2012; Ohm et al., 2012; Sista Kameshwar and Qin, 2018). Compared with other species of Aspergillus, A. sydowii 29R-4-F02 had extremely rich CAZymes encoding genes, especially GHs, CEs, and CBMs encoding genes, suggesting that A. sydowii 29R-4-F02 may have stronger environmental adaptability than other species of Aspergillus. In addition, the genome of A. sydowii 29R-4-F02 also contains GH79 and GH88 gene families, indicating the ability of this fungus to utilize non-plant polysaccharides, such as animal and bacterial polysaccharides (de Vries et al., 2017; Yu et al., 2020). As more and more sedimentary fungi are obtained (Nagahama et al., 2011; Bengtson et al., 2014; Xu et al., 2014; Burgaud et al., 2015; Drake et al., 2017; Hassett et al., 2019), the analysis of the diversity and abundance of CAZymes encoding genes in these fungi is expected to provide a perspective on how fungi adapt to the extreme subseafloor environment where nutrients are scarce and difficult to utilize.
Based on orthology analysis, we found that A. sydowii 29R-4-F02 possesses 364 distinct gene families, among which, the OG0011277 gene family is annotated as a SNARE domain associated with vesicular fusion and autophagy in fungi. Vesicle fusion is the main form of intracellular transmembrane transport of macromolecules and particulate matter (Mohrmann et al., 2010), and autophagy is a strategy for cell renewal of cell components in response to stress or hunger (Glick et al., 2010). In addition, compared with the terrestrial strain A. sydowii CBS593.65, A. sydowii 29R-4-F02 had more unique genes, including ZEB2-regulated ABC transporters, glycopeptide α-N-acetylgalactoamidosidase, heat shock proteins and nucleotide excision repair genes. ZEB2-regulated ABC transporters are involved in the secretion of virulence factors or compounds of fungi (Coleman and Mylonakis, 2009; Lee et al., 2011), and glycopeptide α-N-acetylgalactosidase is involved in intercellular communication in higher eukaryotes (Fujita et al., 2005). Given that A. sydowii 29R-4-F02 was isolated from subseafloor sediment samples at in situ temperatures of 50–55°C, such high temperatures may cause damage to DNA, proteins and other biomolecules (Inagaki et al., 2015; Lever et al., 2015). Therefore, strain 29R-4-F02 possesses large number of nucleotides excision, DNA repair, and heat shock protein-related genes, which may be an evolutionary mechanism of heat protection in response to this environment.
Conclusion
The genome of A. sydowii 29R-4-F02 isolated from ~2.4 km of coal-bearing sediments below the seafloor was de vivo sequenced and assembled using a Nanopore long-read sequencing platform. Compared with other species or strains of Aspergillus, A. sydowii 29R-4-F02 has more abundant CAZymes encoding genes, which facilitate the fungus to obtain nutrients and energy in the subseafloor sediments. Gene expansion associated with vesicular fusion and autophagy, as well as positive selection of CAZymes and enzymes associated with DNA repair, may be one of the adaptive selection mechanisms that allowed fungi to survive 20 million years in extreme subseafloor environment. The results of this study will help us to understand the life style, evolution and survival mechanism of deep biosphere fungi.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.biosino.org/elmsg/index, LMSG_G000000742.1; https://github.com/liuxuan-425lab/A.sydowii-29R-4-F02.
Author contributions
J-PJ, XL, and Y-FL performed data analysis. J-PJ wrote the first draft of this manuscript. XL and Y-PZ extracted DNA for genome sequencing. J-PJ and JS cultivated strain of A. sydowii 29R-4-F02. C-HL conceived the study and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (nos. 41973073, 42273077, and 91951121) and the Science and Technology Innovation Program of Jiangsu Province (no. BK20220036).
Acknowledgments
The authors are grateful to all crews, drilling team members, lab technicians, and shipboard scientists on board the drilling vessel Chikyu for supporting core sampling and onboard measurements during IODP Expedition 337.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1216714/full#supplementary-material
References
Baeza, J. A., and García-De León, F. J. (2022). Are we there yet? Benchmarking low-coverage nanopore long-read sequencing for the assembling of mitochondrial genomes using the vulnerable silky shark Carcharhinus falciformis. BMC Genomics 23:320. doi: 10.1186/s12864-022-08482-z
Becraft, E. D., Vetter, M. C. Y. L., Bezuidt, O. K. I., Brown, J. M., Labont, J. M., Kauneckaite-Griguole, K., et al. (2021). Evolutionary stasis of a deep subsurface microbial lineage. ISME J. 15, 2830–2842. doi: 10.1038/s41396-021-00965-3
Bengtson, S., Ivarsson, M., Astolfo, A., Belivanova, V., Broman, C., Marone, F., et al. (2014). Deep-biosphere consortium of fungi and prokaryotes in Eocene subseafloor basalts. Geobiology 12, 489–496. doi: 10.1111/gbi.12100
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Borgonie, G., Linage-Alvarez, B., Ojo, A. O., Mundle, S. O., Freese, L. B., Van Rooyen, C., et al. (2015). Eukaryotic opportunists dominate the deep-subsurface biosphere in South Africa. Nat. Commun. 6:8952. doi: 10.1038/ncomms9952
Brandt, S. C., Brognaro, H., Ali, A., Ellinger, B., Maibach, K., Ruhl, M., et al. (2021). Insights into the genome and secretome of Fusarium metavorans DSM105788 by cultivation on agro-residual biomass and synthetic nutrient sources. Biotechnol. Biofuels 14:74. doi: 10.1186/s13068-021-01927-9
Brandt, S. C., Ellinger, B., van Nguyen, T., Harder, S., Schlüter, H., Hahnke, R. L., et al. (2020). Aspergillus sydowii: genome analysis and characterization of two heterologous expressed, non-redundant xylanases. Front. Microbiol. 11:2154. doi: 10.3389/fmicb.2020.573482
Burgaud, G., Hue, N. T. M., Arzur, D., Coton, M., Perrier-Cornet, J. M., Jebbar, M., et al. (2015). Effects of hydrostatic pressure on yeasts isolated from deep-sea hydrothermal vents. Res. Microbiol. 166, 700–709. doi: 10.1016/j.resmic.2015.07.005
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Choi, J., Park, J., Kim, D., Jung, K., Kang, S., and Lee, Y. H. (2010). Fungal secretome database: integrated platform for annotation of fungal secretomes. BMC Genomics 11:105. doi: 10.1186/1471-2164-11-105
Coleman, J. J., and Mylonakis, E. (2009). Efflux in fungi: la piece de resistance. PLoS Pathog. 5:e1000486. doi: 10.1371/journal.ppat.1000486
Cong, B. L., Wang, N. F., Liu, S. H., Liu, F., Yin, X. F., and Shen, J. H. (2017). Isolation, characterization and transcriptome analysis of a novel Antarctic Aspergillus sydowii strain MS-19 as a potential lignocellulosic enzyme source. BMC Microbiol. 17:129. doi: 10.1186/s12866-017-1028-0
Couturier, M., Navarro, D., Olive, C., Chevret, D., Haon, M., Favel, A., et al. (2012). Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genomics 13:57. doi: 10.1186/1471-2164-13-57
Cresnar, B., and Petric, S. (2011). Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta, Proteins Proteomics 1814, 29–35. doi: 10.1016/j.bbapap.2010.06.020
De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
de Vries, R. P., Riley, R., Wiebenga, A., Aguilar-Osorio, G., Amillis, S., Uchima, C. A., et al. (2017). Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 18:28. doi: 10.1186/s13059-017-1151-0
Delcher, A. L., Phillippy, A., Carlton, J., and Salzberg, S. L. (2002). Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30, 2478–2483. doi: 10.1093/nar/30.11.2478
D’Hondt, S., Jorgensen, B. B., Miller, D. J., Batzke, A., Blake, R., Cragg, B. A., et al. (2004). Distributions of microbial activities in deep subseafloor sediments. Science 306, 2216–2221. doi: 10.1126/science.1101155
Drake, H., Ivarsson, M., Bengtson, S., Heim, C., Siljestrom, S., Whitehouse, M. J., et al. (2017). Anaerobic consortia of fungi and sulfate reducing bacteria in deep granite fractures. Nat. Commun. 8:55. doi: 10.1038/s41467-017-00094-6
Eddy, S. R. (1998). Profile hidden markov models. Bioinformatics 14, 755–763. doi: 10.1093/bioinformatics/14.9.755
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Fischer, M., Knoll, M., Sirim, D., Wagner, F., Funke, S., and Pleiss, J. (2007). The Cytochrome P450 engineering database: a navigation and prediction tool for the cytochrome P450 protein family. Bioinformatics 23, 2015–2017. doi: 10.1093/bioinformatics/btm268
Fujita, K., Oura, F., Nagamine, N., Katayama, T., Hiratake, J., Sakata, K., et al. (2005). Identification and molecular cloning of a novel glycoside hydrolase family of core 1 type O-glycan-specific endo-alpha-N-acetylgalactosaminidase from Bifidobacterium longum. J. Biol. Chem. 280, 37415–37422. doi: 10.1074/jbc.M506874200
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140. doi: 10.1093/nar/gkn766
Geiser, D. M., Taylor, J. W., Ritchie, K. B., and Smith, G. W. (1998). Cause of sea fan death in the West Indies. Nature 394, 137–138. doi: 10.1038/28079
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Han, X. L., Chakrabortti, A., Zhu, J. D., Liang, Z. X., and Li, J. M. (2016). Sequencing and functional annotation of the whole genome of the filamentous fungus Aspergillus westerdijkiae. BMC Genomics 17:633. doi: 10.1186/s12864-016-2974-x
Hassett, B. T., Borrego, E. J., Vonnahme, T. R., Rämä, T., Kolomiets, M. V., and Gradinger, R. (2019). Arctic marine fungi: biomass, functional genes, and putative ecological roles. ISME J. 13, 1484–1496. doi: 10.1038/s41396-019-0368-1
Hayashi, A., Crombie, A., Lacey, E., Richardson, A. J., Vuong, D., Piggott, A. M., et al. (2016). Aspergillus sydowii marine fungal bloom in australian coastal waters, its metabolites and potential impact on symbiodinium dinoflagellates. Mar. Drugs 14:59. doi: 10.3390/md14030059
Inagaki, F., Hinrichs, K. U., Kubo, Y., Bowles, M. W., Heuer, V. B., Hong, W. L., et al. (2015). Exploring deep microbial life in coal-bearing sediment down to ~2.5 km below the ocean floor. Science 349, 420–424. doi: 10.1126/science.aaa6882
Jing, M. Y., Xu, X. H., Peng, J., Li, C., Zhang, H. C., Lian, C. L., et al. (2022). Comparative genomics of three Aspergillus strains reveals insights into endophytic lifestyle and endophyte-induced plant growth promotion. J. Fungi 8:690. doi: 10.3390/jof8070690
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keller, N. P. (2019). Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180. doi: 10.1038/s41579-018-0121-1
Kim, K., and Harvell, C. D. (2004). The rise and fall of a six-year coral-fungal epizootic. Am. Nat. 164, S52–S63. doi: 10.1086/424609
Klich, M. A. (2002). Identification of common Aspergillus species. Centraalbureau voor Schimmelcultures, Utrecht.
Kumar, A. G., Manisha, D., Sujitha, K., Peter, D. M., Kirubagaran, R., and Dharani, G. (2021). Genome sequence analysis of deep sea Aspergillus sydowii BOBA1 and effect of high pressure on biodegradation of spent engine oil. Sci. Rep. 11:9347. doi: 10.1038/s41598-021-88525-9
Kumar, A., Sorensen, J. L., Hansen, F. T., Arvas, M., Syed, M. F., Hassan, L., et al. (2018). Genome sequencing and analyses of two marine fungi from the north sea unraveled a plethora of novel biosynthetic gene clusters. Sci. Rep. 8:10187. doi: 10.1038/s41598-018-28473-z
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lee, S., Son, H., Lee, J., Lee, Y. R., and Lee, Y. W. (2011). A putative ABC transporter gene, ZRA1, is required for zearalenone production in Gibberella zeae. Curr. Genet. 57, 343–351. doi: 10.1007/s00294-011-0352-4
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Lever, M. A., Rogers, K. L., Lloyd, K. G., Overmann, J., Schink, B., Thauer, R. K., et al. (2015). Life under extreme energy limitation: a synthesis of laboratory- and field-based investigations. FEMS Microbiol. Rev. 39, 688–728. doi: 10.1093/femsre/fuv020
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. doi: 10.1093/bioinformatics/btw152
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Liu, X., Huang, X., Chu, C., Xu, H., Wang, L., Xue, Y. R., et al. (2022). Genome, genetic evolution, and environmental adaptation mechanisms of Schizophyllum commune in deep subseafloor coal-bearing sediments. iScience 25:104417. doi: 10.1016/j.isci.2022.104417
Liu, C. H., Huang, X., Xie, T. N., Duan, N., Xue, Y. R., Zhao, T. X., et al. (2017). Exploration of cultivable fungal communities in deep coal-bearing sediments from similar to 1.3 to 2.5 km below the ocean floor. Environ. Microbiol. 19, 803–818. doi: 10.1111/1462-2920.13653
Lombard, V., Ramulu, H. G., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Lomstein, B. A., Langerhuus, A. T., D'Hondt, S., Jorgensen, B. B., and Spivack, A. J. (2012). Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment. Nature 484, 101–104. doi: 10.1038/nature10905
Medema, M. H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. doi: 10.1093/nar/gkr466
Mohrmann, R., de Wit, H., Verhage, M., Neher, E., and Sorensen, J. B. (2010). Fast vesicle fusion in living cells requires at least three snare complexes. Science 330, 502–505. doi: 10.1126/science.1193134
Nagahama, T., Takahashi, E., Nagano, Y., Abdel-Wahab, M. A., and Miyazaki, M. (2011). Molecular evidence that deep-branching fungi are major fungal components in deep-sea methane cold-seep sediments. Environ. Microbiol. 13, 2359–2370. doi: 10.1111/j.1462-2920.2011.02507.x
Nagano, Y., Konishi, M., Nagahama, T., Kubota, T., Abe, F., and Hatada, Y. (2016). Retrieval of deeply buried culturable fungi in marine subsurface sediments, Suruga-Bay, Japan. Fungal Ecol. 20, 256–259. doi: 10.1016/j.funeco.2015.12.012
Nagano, Y., and Nagahama, T. (2012). Fungal diversity in deep-sea extreme environments. Fungal Ecol. 5, 463–471. doi: 10.1016/j.funeco.2012.01.004
Niu, S. W., Yang, L. H., Chen, T. T., Hong, B. H., Pei, S. X., Shao, Z. Z., et al. (2020). New monoterpenoids and polyketides from the deep-sea sediment-derived fungus Aspergillus sydowii MCCC 3A00324. Mar. Drugs 18:561. doi: 10.3390/md18110561
Ohm, R. A., Feau, N., Henrissat, B., Schoch, C. L., Horwitz, B. A., Barry, K. W., et al. (2012). Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 8:e1003037. doi: 10.1371/journal.ppat.1003037
Orsi, W. D., Edgcomb, V. P., Christman, G. D., and Biddle, J. F. (2013). Gene expression in the deep biosphere. Nature 499, 205–208. doi: 10.1038/nature12230
Ortega-Arbulu, A. S., Pichler, M., Vuillemin, A., and Orsi, W. D. (2019). Effects of organic matter and low oxygen on the mycobenthos in a coastal lagoon. Environ. Microbiol. 21, 374–388. doi: 10.1111/1462-2920.14469
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pitt, J. I., Lange, L., Lacey, A. E., Vuong, D., Midgley, D. J., Greenfield, P., et al. (2017). Aspergillus hancockii sp. nov., a biosynthetically talented fungus endemic to southeastern Australian soils. PLoS One 12:e0170254. doi: 10.1371/journal.pone.0170254
Quemener, M., Mara, P., Schubotz, F., Beaudoin, D., Li, W., Pachiadaki, M., et al. (2020). Meta-omics highlights the diversity, activity and adaptations of fungi in deep oceanic crust. Environ. Microbiol. 22, 3950–3967. doi: 10.1111/1462-2920.15181
Rédou, V., Navarri, M., Meslet-Cladière, L., Barbier, G., and Burgaud, G. (2015). Species richness and adaptation of marine fungi from deep-subseafloor sediments. Appl. Environ. Microbiol. 81, 3571–3583. doi: 10.1128/aem.04064-14
Saha, S., Bridges, S., Magbanua, Z. V., and Peterson, D. G. (2008). Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294. doi: 10.1093/nar/gkn064
Schippers, A., Neretin, L. N., Kallmeyer, J., Ferdelman, T. G., Cragg, B. A., Parkes, R. J., et al. (2005). Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 433, 861–864. doi: 10.1038/nature03302
Schrenk, M. O., Huber, J. A., and Edwards, K. J. (2010). Microbial provinces in the subseafloor. Annu. Rev. Mar. Sci. 2, 279–304. doi: 10.1146/annurev-marine-120308-081000
Selbmann, L., Egidi, E., Isola, D., Onofri, S., Zucconi, L., de Hoog, G. S., et al. (2013). Biodiversity, evolution and adaptation of fungi in extreme environments. Plant Biosyst. 147, 237–246. doi: 10.1080/11263504.2012.753134
Sista Kameshwar, A. K., and Qin, W. (2018). Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 9, 93–105. doi: 10.1080/21501203.2017.1419296
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Takahashi, H., Umemura, M., Ninomiya, A., Kusuya, Y., Shimizu, M., Urayama, S., et al. (2021). Interspecies genomic variation and transcriptional activeness of secondary metabolism-related genes in Aspergillus section Fumigati. Front. Fungal Biol. 2:656751. doi: 10.3389/ffunb.2021.656751
Vaser, R., Sovic, I., Nagarajan, N., and Sikic, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Wang, W. Y., Gao, M. L., Luo, Z. H., Liao, Y. Y., Zhang, B. B., Ke, W. Q., et al. (2019). Secondary metabolites isolated from the deep sea-derived fungus Aspergillus sydowii C1-S01-A7. Nat. Prod. Res. 33, 3077–3082. doi: 10.1080/14786419.2018.1519561
Wang, M. S., Ruan, R. X., and Li, H. Y. (2021). The completed genome sequence of the pathogenic ascomycete fungus Penicillium digitatum. Genomics 113, 439–446. doi: 10.1016/j.ygeno.2021.01.001
Wang, Y. P., Tang, H. B., DeBarry, J. D., Tan, X., Li, J. P., Wang, X. Y., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinform. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2017). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Xu, W., Pang, K. L., and Luo, Z. H. (2014). High fungal diversity and abundance recovered in the deep-sea sediments of the pacific ocean. Microb. Ecol. 68, 688–698. doi: 10.1007/s00248-014-0448-8
Yin, Y. B., Mao, X. Z., Yang, J. C., Chen, X., Mao, F. L., and Xu, Y. (2012). dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451. doi: 10.1093/nar/gks479
Yu, F., Song, J., Liang, J. F., Wang, S. K., and Lu, J. K. (2020). Whole genome sequencing and genome annotation of the wild edible mushroom, Russula griseocarnosa. Genomics 112, 603–614. doi: 10.1016/j.ygeno.2019.04.012
Yurchenko, A. N., Girich, E. V., and Yurchenko, E. A. (2021). Metabolites of marine sediment-derived fungi: actual trends of biological activity studies. Mar. Drugs 19:88. doi: 10.3390/md19020088
Zain Ul Arifeen, M., Chu, C., Yang, X., Liu, J., Huang, X., Ma, Y., et al. (2021a). The anaerobic survival mechanism of Schizophyllum commune 20R-7-F01, isolated from deep sediment 2 km below the seafloor. Environ. Microbiol. 23, 1174–1185. doi: 10.1111/1462-2920.15332
Zain Ul Arifeen, M., Ma, Z. J., Wu, S., Liu, J. Z., Xue, Y. R., and Liu, C. H. (2021b). Effect of oxygen concentrations and branched-chain amino acids on the growth and development of sub-seafloor fungus, Schizophyllum commune 20R-7-F01. Environ. Microbiol. 23, 6940–6952. doi: 10.1111/1462-2920.15738
Zhang, Y., He, K., Guo, X., Jiang, J., Qian, L., Xu, J., et al. (2023). Transcriptomic profiling of Fusarium pseudograminearum in response to Carbendazim, Pyraclostrobin, Tebuconazole, and Phenamacril. J. Fungi 9:334. doi: 10.3390/jof9030334
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, G. K., and Yu, J. (2006). KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinform. 4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
Keywords: Aspergillus sydowii , subseafloor sediments, fungi, CAZymes, adaptive evolution
Citation: Jiang J-P, Liu X, Liao Y-F, Shan J, Zhu Y-P and Liu C-H (2023) Genomic insights into Aspergillus sydowii 29R-4-F02: unraveling adaptive mechanisms in subseafloor coal-bearing sediment environments. Front. Microbiol. 14:1216714. doi: 10.3389/fmicb.2023.1216714
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Paul Greenfield, Commonwealth Scientific and Industrial Research Organisation (CSIRO), AustraliaStefano Ghignone, National Research Council (CNR), Italy
Copyright © 2023 Jiang, Liu, Liao, Shan, Zhu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chang-Hong Liu, Y2hsaXVAbmp1LmVkdS5jbg==