 Qing Niu
Qing Niu Xiaoxu Wang2
Xiaoxu Wang2 Qizhong Huang
Qizhong Huang- 1Animal Husbandry and Veterinary Research Institute, Shanghai Academy of Agricultural Science, Shanghai, China
- 2Shanghai Animal Disease Control Center, Shanghai, China
- 3Shanghai Runzhuang Agricultural Technology Limited Liability Company, Shanghai, China
Introduction: Pullorum disease is one of the common bacterial infectious diseases caused by Salmonella pullorum (S. pullorum), which can result in a decrease in the reproductive performance of laying hens, thus causing considerable economic losses. However, studies about the characteristics of intestinal microbiota with pullorum and their potential association with reproductive performance in hens are still limited. This study was to identify the gut microbiota associated with S. pullorum in poultry.
Methods: A total of 30 hens with S. pullorum-negative (PN) and 30 hens with S. pullorum-positive (PP) were analyzed for hatching eggs laid in 2 weeks (HEL), fertilization eggs (FE), chick number (CN), and microbial structure.
Results: There were significant differences in HEL (p < 0.01), FE (p < 0.01), and CN (p < 0.01) between PP and PN. Histomorphological observations showed abnormal morphology of the ovaries and fallopian tubes and low integrity of epithelial tissue in the ileum and cecum in PP. 16S rRNA gene sequencing revealed that beneficial cecal microbes, such as Bacteroides, Desulfovibrio, and Megamonas, were positively correlated with reproductive performance and had lower abundance in PP (p = 0.001). Furthermore, diminished phosphotransferase system (PTS) and pentose phosphate pathway, butanoate metabolism and oxidative phosphorylation were also found in PP.
Discussion: Taken together, this study clarified the morphological characteristics of the reproductive tract and intestines of chickens infected with S. pullorum and preliminarily explored the potential association between cecal microbiota and reproductive performance in hens. Our data may provide a reference for revealing the intestinal microbial characteristics of hens in resisting pullorum and exploring novel approaches to infection control in future studies.
Introduction
Pullorum disease, caused by Salmonella pullorum (S. pullorum), is transmitted both vertically and horizontally in chickens (Berchieri et al., 2001; Li et al., 2018; Zhou et al., 2022). It is an acute systemic disease and is more common in young birds (Soria et al., 2012; Wang et al., 2020), and some of the infected adult birds are asymptomatic carriers that transmit the bacteria to the offspring and other chickens in the flock, some of which show the symptoms of diarrhea, decreased fertility and laying, reproductive tract abnormalities, inappetence, and weight loss (Shivaprasad, 2000; Wigley et al., 2001; Setta et al., 2012; Ding et al., 2021; Shen et al., 2022). Every year, Salmonella infection leads to serious economic losses in the poultry industry, especially in developing countries (Wigley et al., 2002; Barrow and Freitas Neto, 2011; Li et al., 2018, 2019). Previous studies suggested that in mainland China, PD was highly prevalent in the autumn, followed by the winter. Their findings also demonstrated that PD still posed a major threat to the poultry industry and that comprehensive and stringent strategies should be used to prevent and control this disease (Lv et al., 2022; Jiang et al., 2023). There is conclusive evidence that increased stocking density and larger farms result in an increased occurrence, persistence, and spread of Salmonella in laying hen flocks (Hazards et al., 2019). It causes decreased production performance and even the death of poultry, as well as being a threat to public health. Although eradication programs have been carried out as a prevention and control measure, the agglutination test results are erratic, including false-negative reactions and a lack of sensitivity, so Salmonella infection is still one of the most important problems worldwide (Barrow et al., 2012; Wang et al., 2020).
The use of antibiotics to prevent and treat bacterial diseases, such as Salmonella, leads to an increase in multiple drug-resistant bacteria worldwide (Zhou et al., 2020). Sulfonamides have been used in the treatment of pullorum disease, including sulfadiazine, sulfamerazine, sulfathiaole, sulfamethazine, and sulfaquinoxaline. However, most studies have indicated that no drug or combination of drugs has been found to be capable of eliminating infection from treated flocks (Shivaprasad, 2000). Globally, the animal industries are moving toward restricting and eventually a total ban on the usage of antibiotic growth promoters (Liao and Nyachoti, 2017). Meanwhile, the Chinese government is promoting a reduction in antibiotic use currently (Zhang et al., 2015). This trend prompts people to actively seek an ideal alternative to antibiotics.
The inclusion of alternative feed additives in lieu of antibiotics in animal diets is definitely required to support a profitable and sustainable poultry industry (Liao and Nyachoti, 2017). As is known, the gut microbiota can be manipulated by feed additives such as exclusion products, probiotics, prebiotics, organic acids, plant extracts, essential oils, and feed enzymes (Shin et al., 2008; De Lange et al., 2010; Le Bon et al., 2010; Heo et al., 2013). However, there is no report on preventing and controlling pullorum disease in the Chinese local chicken breeds. An animal's character can be affected by changes in the microbiota. As it is an opportunistic pathogen, the occurrence of pullorum disease is mainly caused by an imbalance of the intestinal microbiota (Shivaprasad, 2000). Probiotics are defined as a microbial feed supplement that beneficially affects the host animal by improving its intestinal balance (Fuller, 1989). They are a category of feed additives that can be used to replenish the gut microbial population while recuperating the host immune system (Liao and Nyachoti, 2017). Many recent studies have shown that humans and animals fed probiotics have altered intestinal microbiota, increased intestinal immunity, improved resistance to disease, reduced shedding of pathogens and disease symptoms, and improved health status (Zhao et al., 2013; Chiu et al., 2014; Upadhaya et al., 2015; Gao et al., 2017; Liao and Nyachoti, 2017; Li et al., 2017; Chen et al., 2020; Wang et al., 2021). The gut microbiota affecting the pullorum and its functions in chickens need further study.
Due to the large diversity of bacterial species, the gut microbiome contains circa 9 million unique protein-coding genes, but they have not been studied extensively (Rosenberg and Zilber-Rosenberg, 2018). The availability of high-throughput sequencing will shortly enable the sequencing of whole bacterial populations, enabling a more comprehensive view of bacterial evolution among related bacterial species (Barrow and Freitas Neto, 2011). This study aimed to analyze the intestinal microbial characteristics of hens resisting pullorum and their relationships with hatching eggs laid in 2 weeks (HEL), fertilization eggs (FE), and chick number (CN) in hens.
Materials and methods
Ethics statement
All procedures and the use of animals were carried out in accordance with the Guidelines for the Ethics and Animal Welfare Committee of the Shanghai Academy of Agricultural Sciences (No. SAASPZ0522051).
Animal experiment and sample collection
S. pullorum infections in chickens were diagnosed by S. pullorum and S. gallinarum polyvalent antigen rapid slide agglutination test reagents (Beijing Zhonghai Biotech Co., Ltd., China). On a clean glass slide, 50 μl of polyvalent antigen and 50 μl of venous blood were placed. The samples were deemed positive if 50% or more agglutination occurred in the mixture within 2 min, and samples without agglutination were considered negative. In this study, two chicken groups were obtained after three times of the slide agglutination tests, and the results of the S. pullorum infection texts were positive or negative all three times. A total of 60 New Pudong chickens [samples from 30 hens with S. pullorum-negative (PN) and 30 hens with S. pullorum-positive (PP)] were selected from the experimental farm of the Shanghai Academy of Agricultural Sciences, Shanghai, China. These 60 hens were artificially inseminated at the age of 47 weeks. Recording the number of eggs laid, fertilized, and hatched chicks per hen and the performance, including HEL, FE, and CN, were analyzed during chick hatching processes. All eggs received disinfection before hatching. All hens were in the same poultry house (PN hens were distributed in the east of the poultry house and PP hens were distributed in the west of the poultry house), selected according to a unified breed standard, and fed antibiotic-free corn-soybean diets (Supplementary Table S1). Antibiotics in the feed or for any therapeutic purposes were not provided for hens after the age of 1 week. These 60 hens were slaughtered at the age of 49 weeks. After slaughter, tissue samples of the ovaries, fallopian tubes, ileum, and cecum with the size of about 2 × 3 cm were cut with a sterile scalpel and quickly stored in 4% paraformaldehyde for separate morphological observation. Cecal content was individually collected in 2 ml centrifuge tubes for 16S rRNA gene sequencing. All samples were kept in an ice box for preservation and transportation and then stored at −80°C in the laboratory (Janssen and Kersten, 2015).
Morphological observation of the ovaries, fallopian tubes, ileum, and cecum
Tissues were routinely embedded in paraffin wax blocks, sectioned at 5 μm thickness, mounted on glass slides, and stained with hematoxylin & eosin (H&E). Morphological observations were conducted by a Nikon ECLIPSE 80i light microscope with a computer-assisted morphometric system (Nikon Corporation, Tokyo, Japan).
16S rRNA sequencing and bioinformatics analysis
The gut microbiota population in the hens with S. pullorum-negative (n = 30) and S. pullorum-positive (n = 30) was analyzed by 16S rRNA gene sequencing, respectively. Microbial community genomic DNA was extracted from cecum samples using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, United States) according to the manufacturer's instructions. The DNA extract was checked on 1% agarose gel, and the DNA concentration and purity were determined using NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, United States). The hypervariable V3-V4 region of the 16S rRNA gene with a length of ~468 bp was targeted for sequencing. PCR amplification was performed with gene-specific primers 338 F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806 R (5′-GGACTACHVGGGTWTCTAAT-3′) under the following conditions: initial denaturation at 95°C for 3 min, followed by 27 cycles of denaturing at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 45 s, single extension at 72°C for 10 min, and end at 4°C.
The PCR mixtures contain 5 × TransStartFastPfu buffer 4 μl, 2.5 mM dNTPs 2 μl, forward primer (5 μM) 0.8 μl, reverse primer (5 μM) 0.8 μl, TransStartFastPfu DNA Polymerase 0.4 μl, template DNA 10 ng, and finally ddH2O up to 20 μl. PCR reactions were performed in triplicate. The PCR product was extracted from a 2% agarose gel, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) according to the manufacturer's instructions, and quantified using the Quantus™ Fluorometer (Promega, United States). Purified amplicons were pooled in equimolar amounts and paired-end sequenced on an Illumina MiSeq PE300 platform (Illumina, San Diego, United States) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). All obtained raw sequence datasets have been deposited into the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) with the accession number PRJNA799073.
The raw 16S rRNA gene sequencing reads were demultiplexed, quality-filtered by the FastP version 0.20.0 (Chen et al., 2018) and merged by the FLASH version 1.2.7 (Magoc and Salzberg, 2011) with the following criteria: the 300 bp reads were truncated at any site receiving an average quality score of <20 over a 50 bp sliding window, and the truncated reads shorter than 50 bp were discarded; reads containing ambiguous characters were also discarded; only overlapping sequences longer than 10 bp were assembled according to their overlapped sequence. The maximum mismatch ratio of the overlap region is 0.2. Reads that could not be assembled were discarded; samples were distinguished according to the barcode and the primers, and the sequence direction was adjusted for exact barcode matching and a 2-nucleotide mismatch in primer matching. Operational taxonomic units (OTUs) with a 97% similarity cutoff (Schloss et al., 2009) were clustered using the UPARSE version 7.1, and chimeric sequences were identified and removed. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier version 2.2 (Wang et al., 2007) against the 16S rRNA database (Silva version 138) using a confidence threshold of 0.7.
Statistical analysis
Reproductive performance, including HEL, FE, and CN, was calculated using the SAS 9.4 software (Shen et al., 2014). Alpha diversity was calculated using the Mothur (Schloss et al., 2009). Venn diagrams and rank abundance distribution curves were performed using Mothur. Linear discriminate analysis effect size (LEfSe) was used to identify the bacteria enriched (Segata et al., 2011). The pair-wise phylogenetic distance was measured by weighted UniFrac (Lozupone et al., 2006) to compare community compositions across samples. Principal component analysis (PCA) was used to compress dimensionality into 2D principal coordinate analysis plots (Vazquez-Baeza et al., 2013), enabling visualization of sample relationships. PICRUSt was used to explore the functional composition of that bacterial community that the data might convey (Langille et al., 2013). The visualization of conventional results was achieved by Origin 2023. The co-occurrence network is implemented through the Gephi 0.10 software.
Results
Comparisons of reproductive performances in S. pullorum-negative hens and S. pullorum-positive hens
The reproductive performance showed that the average hatching eggs laid in 2 weeks (HEL), average fertilization eggs (FE), and average chick number (CN) of S. pullorum-negative hens were higher than that of S. pullorum-positive hens (p < 0.01, Figure 1). The average HEL of S. pullorum-negative hens was 12.5, which was 0.5 more than that of S. pullorum-positive hens. The average FE of S. pullorum-negative hens was 11.7, which was 8.4 times more than that of S. pullorum-positive hens. The average CN of S. pullorum-negative hens was 11.0, which was 9.8 times more than that of S. pullorum-positive hens.
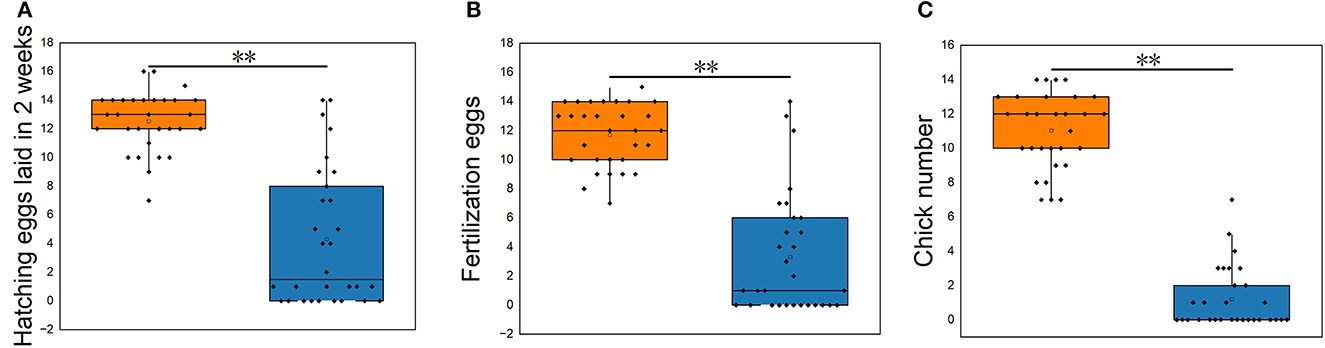
Figure 1. (A–C) Comparisons of reproductive performances in S. pullorum-negative hens and S. pullorum-positive hens. PN, S. pullorum-negative group; PP, S. pullorum-positive group.
Morphological observation of the reproductive tract and intestinal tract
Morphological observations showed that the presence of deformed follicles, follicular dysplasia, or even necrosis occurred in the S. pullorum-positive hens (Figures 2A, B). Moreover, in the S. pullorum-positive hens, epithelial cells of the oviductal mucosa consisted mostly of ciliated cells, with few secretory cells and an underdeveloped lamina propria (Figures 2C, D). Histomorphological observations of the ileum and cecum in PP revealed that the intestinal epithelial integrity was low and there was severe intestinal epithelial damage. Severe detachment of mucosal epithelial cells and exposure of the lamina propria in the intestinal lumen were mainly observed (Figures 2E, H).
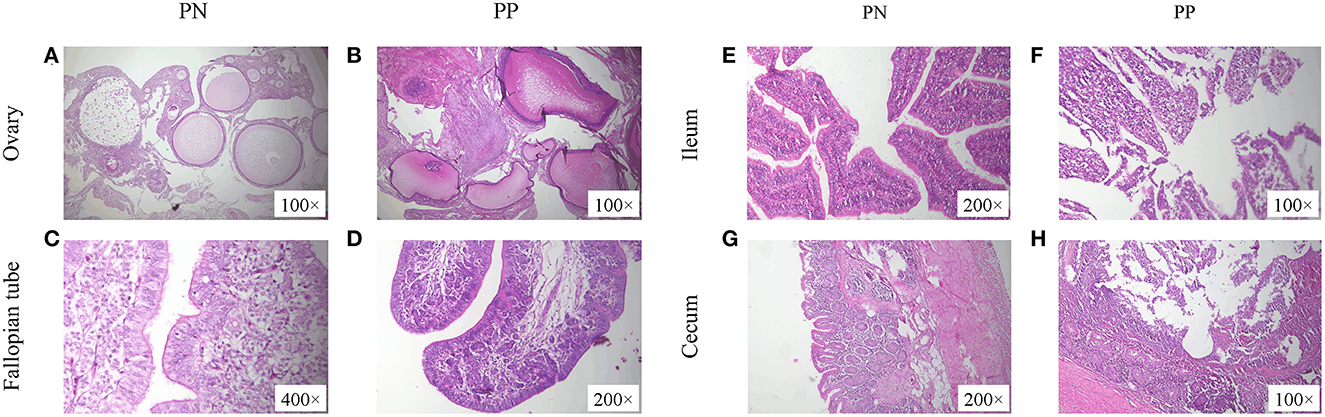
Figure 2. (A–H) Morphological observation of ovaries, fallopian tubes, ileum, and cecum of PN and PP. PN, S. pullorum-negative group; PP, S. pullorum-positive group.
Bacterial community structure and prediction functions of S. pullorum-negative hens and S. pullorum-positive hens
More than 4 million sequences were obtained from all samples, and there were 16,888 high-quality sequences per sample. The average sequence length was 417 bp. Microbial composition analysis showed that, at the phylum level, the two most dominant phyla were Firmicutes and Bacteroidetes, which comprised 72.6% of the total sequences in PN and 71.6% of the total sequences in PP, respectively (Figure 3). At the genus level, a total of 357 genera were identified from all samples, and the enrichment of these genera in the PN and PP had a large variation (Figure 3). The two most dominant genera were Bacteroides and Rikenellaceae_RC9_gut_group belonging to the phylum Bacteroidota and comprised 25.70% of the total sequences in PN and 24.42% of the total sequences in PP, respectively (Figure 3). A total of 1,880 OTUs were identified from all samples, and the VENN diagram showed the unique OTUs and genera of the two groups and the shared OTUs and genera (Supplementary Figure S1). In addition, principal component analysis (PCA) at the genus level revealed a significant separation between the samples of PN and PP, indicating a large difference in the cecum microbiota of these two groups (Figure 4). The diversity and richness of the cecum microbiota were significantly lower in the PP group than in the PN group (Figures 5A, E).
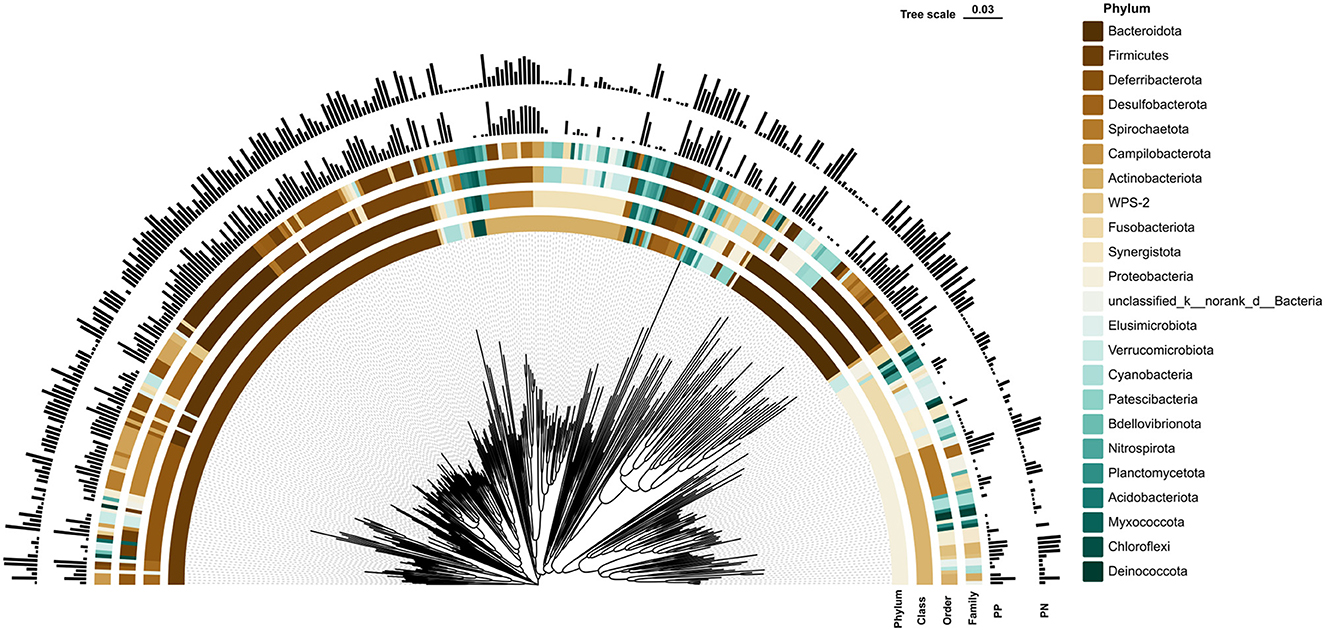
Figure 3. Phylogenetic tree of cecal microbiota and its microbial community composition in PN and PP. PN, S. pullorum-negative group; PP, S. pullorum-positive group.
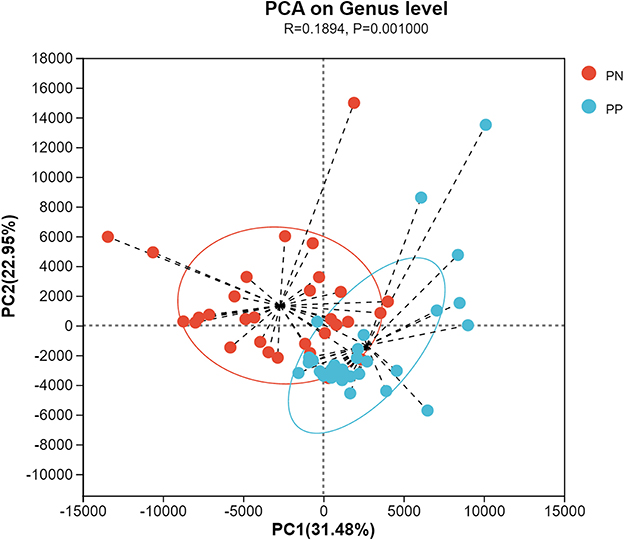
Figure 4. Principal component analysis (PCA) at genus level between PN and PP. PN, S. pullorum-negative group; PP, S. pullorum-positive group.
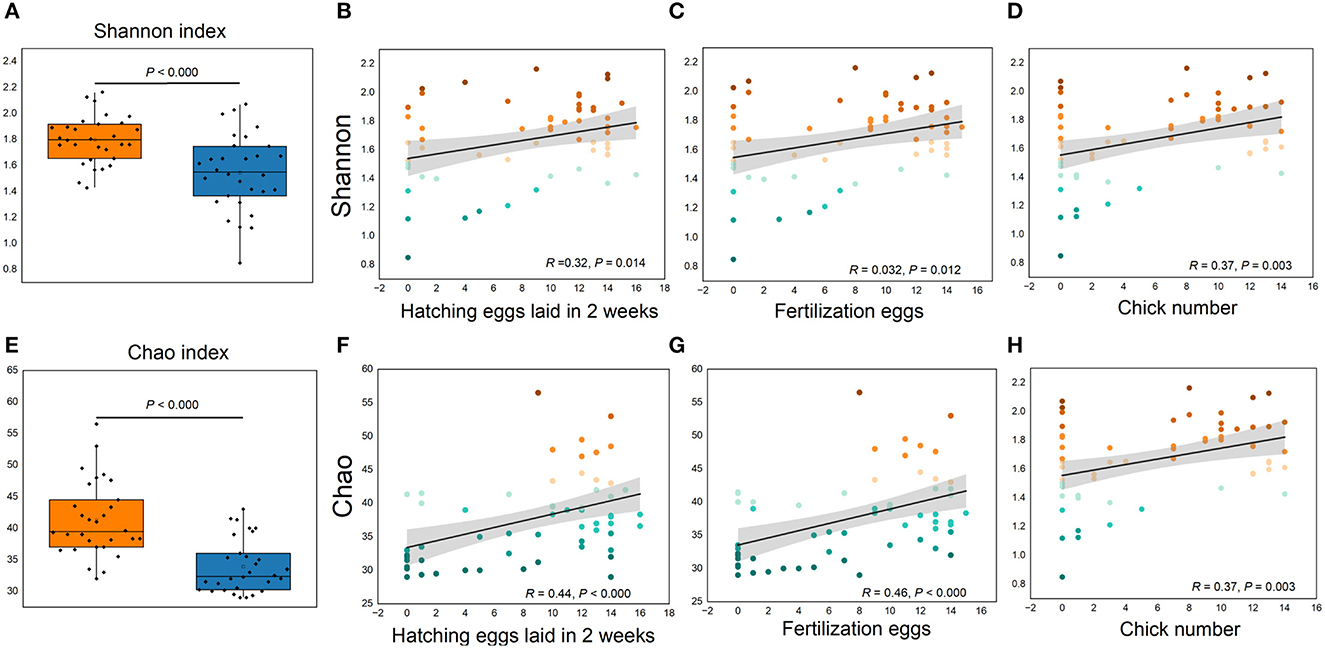
Figure 5. (A–H) Cecum microbial alpha diversity and its relationships with reproductive performance. (A, E), Shannon and Chao indexes, respectively. (B, D, F, H) Correlations between reproductive performance with Shannon and Chao indexes, respectively.
A Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) analysis was performed to investigate the functional properties of microbiota. Using the KEGG pathway annotation information, we found that among the major microbial functions (top 100), the PP cecum microbial functions were generally weaker than the PN group (Figure 6A), where the phosphotransferase system (PTS), pentose phosphate pathway, butanoate metabolism, and oxidative phosphorylation were significantly less abundant in PP than PN (Figures 6B–E).
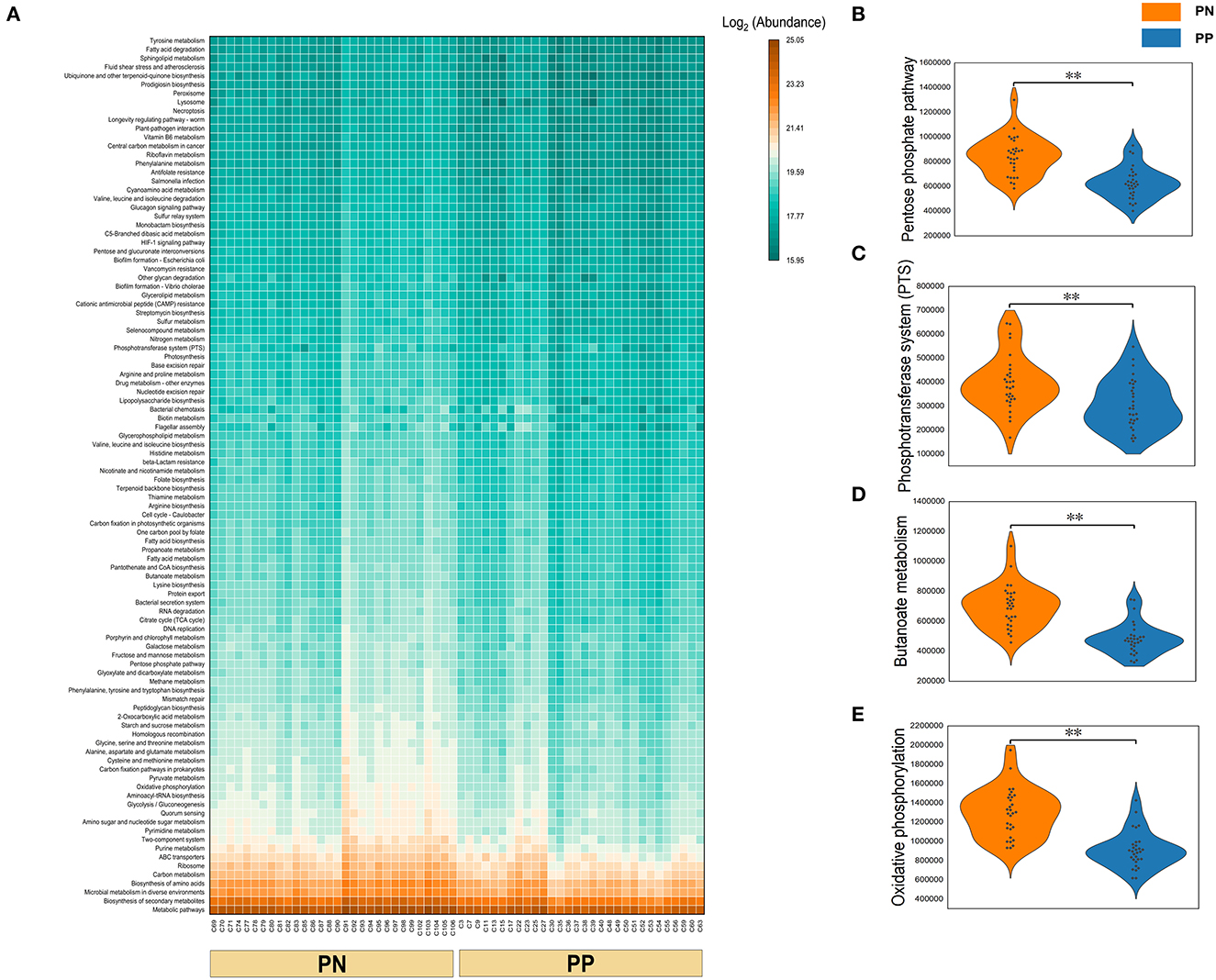
Figure 6. (A–E) Functional composition of microbiota and their differences between two groups. Microbial functional composition. (A) Heatmap of the functional composition (top 100) of microbiota. (B–E) Comparison of abundance of phosphotransferase system (PTS) and pentose phosphate pathway, butanoate metabolism, and oxidative phosphorylation.
Correlations of reproductive performances with the gut microbiota
It is noteworthy that the alpha diversity of the cecum microbiota was positively correlated with the reproductive performance (HEL, FE, and CN) of hens (p < 0.01, Figures 5B–D, F–H). To further investigate the microbial differences between the PP and PN groups, the Wilcoxon rank-sum test was used, and it was found that there were 68 significantly different genera between these two groups (p < 0.05, Supplementary Table S2). LEfSe was used to further determine the taxa that most likely explain the differences between PN and PP samples. A total of 65 genera were found to be potential biomarkers between PN and PP. There was a significant enrichment of 50 genera (including Bacteroides, Desulfovibrio, and Megamonas) in PN (Figure 7 A). Additionally, enrichment of 15 genera, including Salmonella, was found in PP (Figure 7A). Notably, we found significant positive correlations between Bacteroides, Desulfovibrio, and Megamonas, with reproductive performance (HEL, FE, and CN, Figures 7B–D).
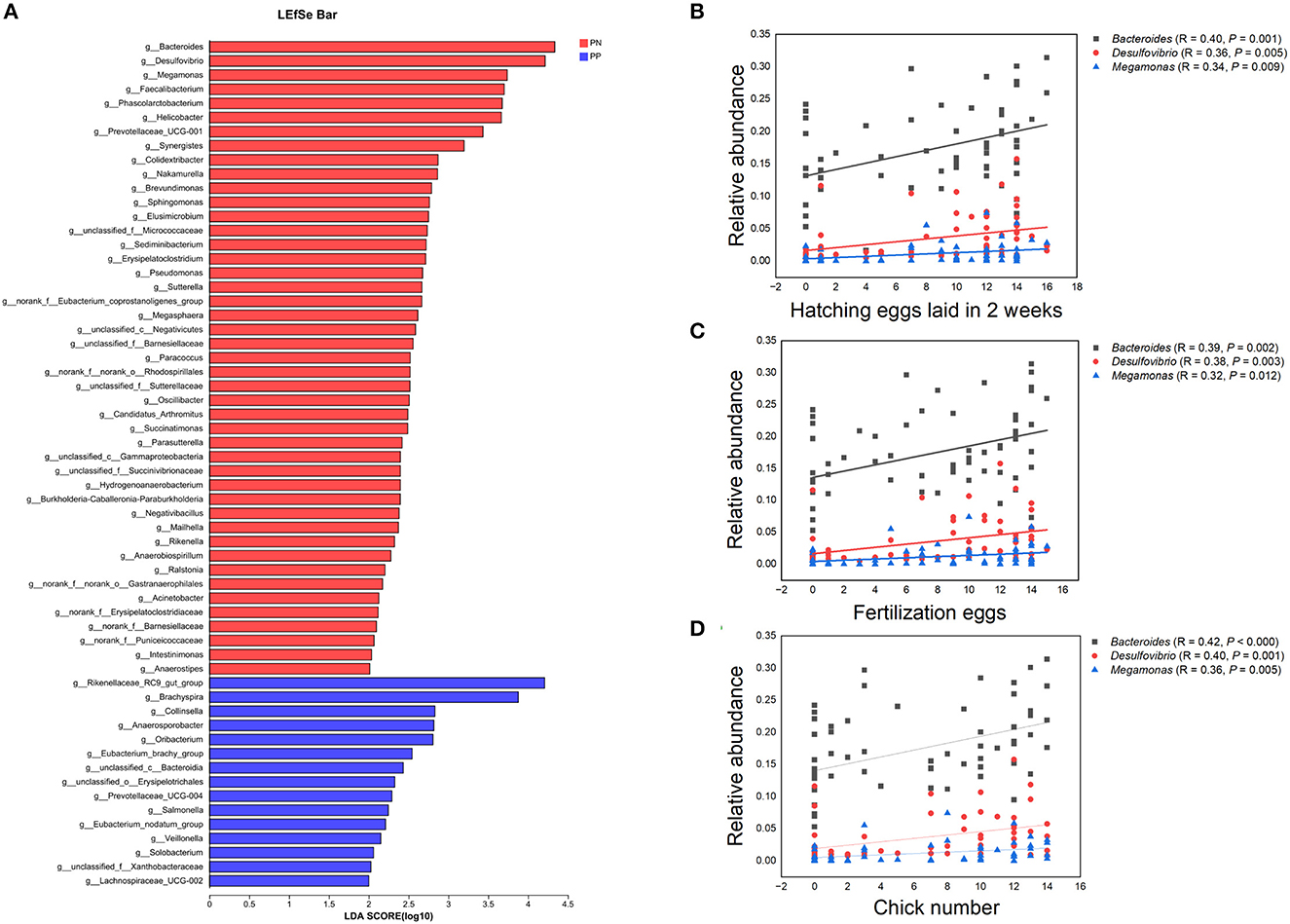
Figure 7. (A–D) Identification of differential microbes and the associations between microbial abundance and reproductive performance. (A) Linear discriminant analysis (LDA) effect size (LEfSe) analysis. (B–D) Correlation analysis microbial abundance and reproductive performance.PN, S. pullorum-negative group, PP, S. pullorum-positive group.
Analysis of the co-occurrence network of microorganisms between S. pullorum-negative hens and S. pullorum-positive hens
Analysis of the co-occurrence network of microorganisms at the genus level found that in the PP and PN groups, the genus Firmicutes is the core microbe in the co-occurrence network and has extensive connections with microbes from other phyla (Figure 8). In addition, compared with the control group, the association between members of the Firmicutes phylum in the PP group seems to be closer (Figure 8B).
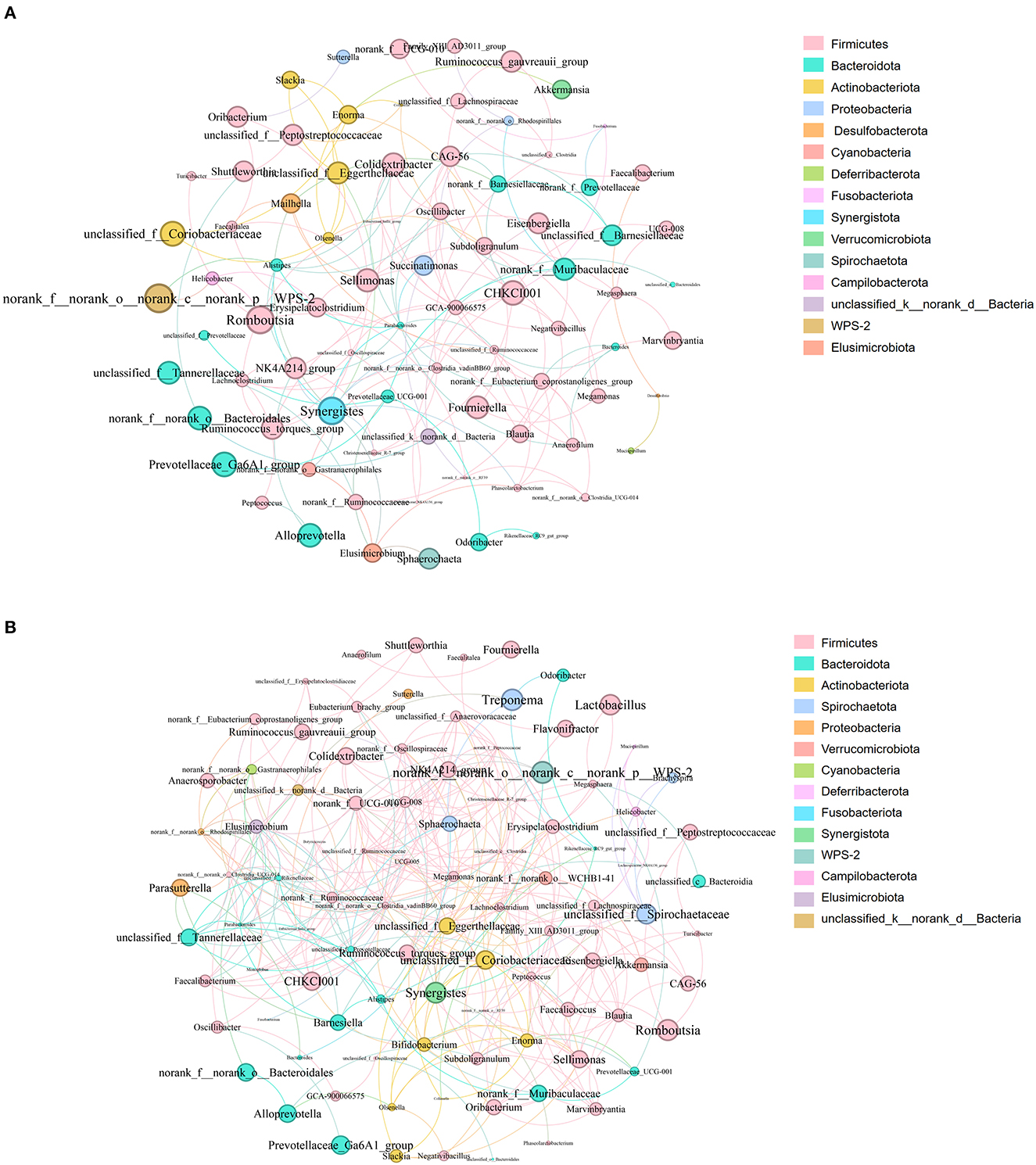
Figure 8. (A, B) Microbial co-occurrence network analysis at the genus level. (A) was PN, (B) was PP. Node size indicates relative abundance. Red edges indicate significant positive correlation (P < 0.05), and green edges indicate significant negative correlation (p < 0.05). PN, S. pullorum-negative group; PP, S. pullorum-positive group.
Discussion
Pullorum is a disease caused by S. pullorum through both horizontal and vertical transmission. It is known that a percentage of birds that survive clinical disease when they are infected as young chicks may show few signs of infection but may become carriers. Intestinal carriage of S. pullorum in the poultry host does not cause substantive gastrointestinal disease and is asymptomatic. Therefore, it is of great significance to identify gut microbiota related to pullorum disease in chickens. This will lead to the development of novel prevention and control strategies for S. pullorum infection in poultry production.
Among the different diseases occurring in poultry, those caused by the genus Salmonella are the most common, leading to serious economic losses to the poultry industry in terms of mortality, reduced growth, and loss of egg production (Shivaprasad, 2000; Wigley et al., 2001). One aim of the current study was to investigate and characterize the effect that S. pullorum infection had on reproductive performances. The results illustrated that S. pullorum infection in chickens reduced their reproductive performances and altered the gut microbial composition, resulting in variations in the microbial metabolic pathways and functions. Previous studies have reached the same conclusion (Ding et al., 2021). This study illustrated that pullorum disease reduced reproductive performance including HEL, FE, and CN. Pullorum disease is manifested by decreased egg production, fertility, and hatchability in hens. Shivaprasad HL showed that regressing ovarian follicles can be found in the ovaries of chickens and microscopic lesions in adults include fibrinosuppurative to pyogranulomatous inflammation of ovarian follicles characterized by necrosis. Caeca may contain necrotic caseous debris within the lumen and necrosis of the mucosa with the infiltration of heterophils into the lamina propria (Shivaprasad, 2000). The other aim of this study was to investigate the different bacterial communities between S. pullorum-negative hens and S. pullorum-positive hens and their microbial functions. The availability of high-throughput sequencing will shortly enable the sequencing of whole bacterial populations, giving us a more comprehensive view of bacterial evolution among related bacterial species (Barrow and Freitas Neto, 2011). Shen et al. studied the dissemination pattern of S. pullorum in different organs and at different time points. It showed that the cecum carried S. pullorum throughout the experiment duration, while the small intestine did not carry S. pullorum during the last few days of the experiment (Shen et al., 2022). Therefore, this study examined DNA sequence data and the bacterial community structure of S. pullorum-negative hens and S. pullorum-positive hens in the cecum. It obtained a large number of effective sequences. The two most dominant phyla were Firmicutes and Bacteroidota. The similar results were obtained in previous studies (Wei et al., 2013; Khan et al., 2020; Rychlik, 2020). The two most dominant genera were Bacteroides and Rikenellaceae_RC9_gut_group, which belong to the phylum Bacteroidota comprised 34.1% of the total sequences in PN and 16.2% of the total sequences in PP, respectively. Ding et al. investigated that the dominant phyla were Firmicutes (65.5% in group N and 62.1% in group P), Fusobacteria (16.3% in group N and 18.7% in group P), and Proteobacteria (9.37% in group N and 9.95% in group P); the preponderant genera were Lactobacillus, Fusobacterium, Peptoclostridium, and Gallibacterium (Ding et al., 2021). The reasons that lead to different results in similar studies are complicated. Many factors can cause different microbiome compositions (e.g., breeds, age, gender, nutritional level, and sample selection). It is interesting that although significant enrichment of Salmonella was found in PN, there was a low abundance of Salmonella in PP. The basis of host specificity in salmonellosis continues to elude scientific explanation (Barrow et al., 2012). The outcome of infection is the combined effect of the microbial gene set and the host's genetic background. After an intestinal infection, where are the sites of Salmonella serovar Pullorum persistence in convalescent birds? Wigley et al. showed that Salmonella serovar Pullorum evades the immune response by surviving intracellularly within macrophages, which would be required by Salmonella serovar Pullorum to persist in both the spleen and the reproductive tract (Wigley et al., 2001). Salmonella serovar Pullorum localizes in the reproductive tract of chickens and, as a consequence, may be transmitted vertically to chicks by transovarian transmission of the bacteria into developing hatching eggs (Wigley et al., 2001). The correlation analysis between reproductive performances (HEL, FEN, and CN) and gut microbiota was therefore significant.
After a systemic disease, there may be negative effects on immunity, such as the balance of gut microbes, which could be upset, and the ability of harmful bacteria to secrete toxins, which could be increased. In this study, the ileum and cecum in PP revealed low intestinal epithelial integrity, and this should cause negative effects in the absorption and digestion of feed nutrients. Intestinal integrity is essential to prevent animal bacterial diseases (Citi, 2018). A stable and healthier intestinal state can inhibit pathogenic bacteria and reduce the production of toxins to ensure better capacity for digestion and absorption in hens (Jackman et al., 2020; Papadopoulos et al., 2022; Cui et al., 2023). The gut microbiota is widely perceived as being closely related to gut health and the growth performance of the host (Wang et al., 2016). The results elaborated that the abundances of 29, 32, and 39 genera were separately positively correlated with HEL, FEN, and CN. Meanwhile, the abundances of 7, 6, and 2 genera were separately negatively correlated with HEL, FEN, and CN.
The 50 genera, which were significant enrichments for S. pullorum-negative hens, were used to make PICRUSt1 function prediction analyses in all samples. The most important functions and metabolic pathways of the above different potential biomarkers were amino acid transport and metabolism and amino acid metabolism, respectively. Among these 50 genera, previous studies showed that Bacteroides and Megamonas could produce short-chain fatty acids (SCFA) by fermenting carbohydrates to provide energy for the gut and promote the growth performance of animals (Hooper et al., 2002; Shimizu et al., 2019; Ni et al., 2021; Zhu et al., 2021). Desulfovibrio consumes free hydrogen for the reduction of sulfate, thus contributing to the removal of free hydrogen formed during anaerobic fermentation in the gut environment (Rychlik, 2020). Faecalibacterium as a probiotic candidate has shown promising results toward enhancing food safety and gut health (Khan et al., 2020). The major source energies of Suterrella and Parasutterella originate from protein, amino acid, and fatty acid metabolism (Line et al., 2010; Polansky et al., 2015). Rychlik showed that Megasphaera and Phascolarctobacterium were capable of butyrate production (Rychlik, 2020). Zhang indicated that high body weight chickens contained Sphingomonas more abundantly (p < 0.05) (Zhang et al., 2021). In-feed supplementations of probiotics strengthen the gut microbiota for improved host performance and colonization resistance to gut pathogens such as Salmonella and Campylobacter. The mechanisms of action of prebiotics and probiotics come through the production of organic acids, the activation of the host immune system, and the production of antimicrobial agents. Many probiotic preparations contain high numbers of lactobacilli that normally produce large quantities of volatile fatty acids such as formic acid. The incorporation of these into feed has been shown to inhibit gut colonization by zoonotic serovars of Salmonella (Barrow and Freitas Neto, 2011). Mixed probiotics effectively reduced the mortality of pullorosis in chicks, promoted growth performance, regulated the balance of the intestinal flora, improved immune function, resisted pullorosis disease, completely prevented chicks from pullorosis after infection, and reduced economic loss in the poultry industry (Chen et al., 2020). The probiotics reported above could be in culture, but most of these were poor or unclear, and most studies are empirical in nature.
Many studies have revealed the importance of probiotics in the context of infectious diseases, including pullorum. Zhou et al. studied the effect of a selected yeast fraction (Safmannan, SYF) on the prevention of pullorum disease in commercial breeder chickens and demonstrated that SYF supplementation could significantly decrease SP and SG infection rates and improve the body weight of birds challenged with S. pullorum (Zhou et al., 2020). Mon et al. examined the three-way interaction that occurred between host metabolites, resident gut microbiota, and Salmonella following inoculation of Salmonella enteritidis in 2-week-old layer chicks. It showed that there was differential regulation in many of the metabolites in association with Salmonella enteritidis colonization in chickens; perturbation in metabolic pathways related to arginine and proline metabolism as well as the TCA cycle was most prominently detected (Mon et al., 2020). Alrubaye et al. learned that the microbial metabolite deoxycholic acid shapes microbiota against Campylobacter jejuni chicken colonization and suggested that there was a bidirectional interaction between microbiota and microbial metabolites (Alrubaye et al., 2019). In this study, most of the genera we obtained by 16S rRNA sequencing were uncultured. The use of competitive exclusion gut flora preparations has the same protective effect as the normal flora in animal intestines. There is the possibility of a very intimate interaction between host bacteria and pathogens in the cecum (Barrow et al., 2012), and one area for future Salmonella control exploration is the development of probiotic organisms that have a rational basis for protection. Utilization of the information generated in this study should improve the efficacy of surveillance and biological interventions, both for intestinal carriage.
Conclusion
Pullorum disease reduced reproductive performance. Abnormal morphology of the ovaries and fallopian tubes and low integrity of epithelial tissue in the ileum and cecum were found in PP. Pullorum disease reduced the cecal microbial alpha diversity and relative abundance of Bacteroides, Desulfovibrio, and Megamonas, which were positively correlated with reproductive performance. Diminished phosphotransferase systems (PTS) and pentose phosphate pathways, butanoate metabolism, and oxidative phosphorylation were also found in PP. Taken together, this study clarified the morphological characteristics of the reproductive tract and intestines of hens infected with S. pullorum and preliminarily explored the potential association between cecal microbiota and reproductive performance in hens.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA799073.
Ethics statement
The animal study was reviewed and approved by the Ethics and Animal Welfare Committee of Shanghai Academy of Agricultural Sciences (No. SAASPZ0521009). Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
QN and QH conceived and designed the experiments. CG, ZZ, and KY raised the experimental animals. XW, CC, XQ, and QN participated in the sample collection. XQ and QN participated in the data analysis. QN wrote the article. QH, XW, CC, XQ, CG, KY, and ZZ assisted with experiments and provided advice on manuscript content. All authors read and approved the final manuscript.
Acknowledgments
We would like to thank the financial support provided by the Shanghai Agriculture Applied Technology Development Program, China (Grant No. X2022-02-08-00-12-F01184) and the China Agriculture Research System (Grant No. CARS-41-Z07) for funding this study.
Conflict of interest
CC was employed by Shanghai Runzhuang Agricultural Technology Limited Liability Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1216542/full#supplementary-material
Supplementary Figure S1. Venn diagrams of OTUs (A) and genera (B) between PN and PP, respectively. PN, S. pullorum-negative group; PP, S. pullorum-positive group.
Supplementary table S1. Composition of experimental diet.
Supplementary table S2. The microbes with significant differences between PN and PP.
References
Alrubaye, B., Abraha, M., Almansour, A., Bansal, M., Wang, H., Kwon, Y. M., et al. (2019). Microbial metabolite deoxycholic acid shapes microbiota against Campylobacter jejuni chicken colonization. PLoS ONE 14, e0214705. doi: 10.1371/journal.pone.0214705
Barrow, P. A., and Freitas Neto, O. C. (2011). Pullorum disease and fowl typhoid–new thoughts on old diseases: a review. Avian Pathol. 40, 1–13. doi: 10.1080/03079457.2010.542575
Barrow, P. A., Jones, M. A., Smith, A. L., and Wigley, P. (2012). The long view: Salmonella–The last forty years. Avian Pathol. 41, 413–420. doi: 10.1080/03079457.2012.718071
Berchieri, A. Jr., Murphy, C.K., Marston, K., and Barrow, P. A. (2001). Observations on the persistence and vertical transmission of Salmonella enterica serovars Pullorum and Gallinarum in chickens: effect of bacterial and host genetic background. Avian Pathol. 30, 221–231. doi: 10.1080/03079450120054631
Chen, C., Li, J., Zhang, H., Xie, Y., Xiong, L., Liu, H., et al. (2020). Effects of a probiotic on the growth performance, intestinal flora, and immune function of chicks infected with Salmonella pullorum. Poult. Sci. 99, 5316–5323. doi: 10.1016/j.psj.2020.07.017
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chiu, Y. H., Lin, S. L., Tsai, J. J., and Lin, M. Y. (2014). Probiotic actions on diseases: implications for therapeutic treatments. Food Funct. 5, 625–634. doi: 10.1039/c3fo60600g
Citi, S. (2018). Intestinal barriers protect against disease. Science. 359, 1097–1098. doi: 10.1126/science.aat0835
Cui, Z., Zhang, R., Dai, B., Fu, C., Zhao, G., Xu, Y., et al. (2023). Effects of glyceryl monolaurate on production performance, egg quality, oviduct cytokines and intestinal microflora of 66 weeks old laying hens. Animals 13, 215. doi: 10.3390/ani13020215
De Lange, Pluske, C. F. M., Gong, J. J., and Nyachoti, C. M. (2010). Strategic use of feed ingredients and feed additives to stimulate gut health and development in young pigs. Livest. Sci. 134, 124–134. doi: 10.1016/j.livsci.2010.06.117
Ding, J., Zhou, H., Luo, L., Xiao, L., Yang, K., Yang, L., et al. (2021). Heritable gut microbiome associated with salmonella enterica serovar pullorum infection in chickens. mSystems 6, 20. doi: 10.1128/mSystems.01192-20
Fuller, R. (1989). Probiotics in man and animals. J. Appl. Bacteriol. 66, 365–378. doi: 10.1111/j.1365-2672.1989.tb05105.x
Gao, P., Ma, C., Sun, Z., Wang, L., Huang, S., Su, X., et al. (2017). Feed-additive probiotics accelerate yet antibiotics delay intestinal microbiota maturation in broiler chicken. Microbiome 5, 91. doi: 10.1186/s40168-017-0315-1
Hazards, E. P. O. B., Koutsoumanis, K., Allende, A., Alvarez-Ordonez, A., Bolton, D., Bover-Cid, S., et al. (2019). Salmonella control in poultry flocks and its public health impact. EFSA J. 17, e05596. doi: 10.2903/j.efsa.2019.5596
Heo, J. M., Opapeju, F. O., Pluske, J. R., Kim, J. C., Hampson, D. J., Nyachoti, C. M., et al. (2013). Gastrointestinal health and function in weaned pigs: a review of feeding strategies to control post-weaning diarrhoea without using in-feed antimicrobial compounds. J. Anim. Physiol. Anim. Nutr. 97, 207–237. doi: 10.1111/j.1439-0396.2012.01284.x
Hooper, L. V., Midtvedt, T., and Gordon, J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307. doi: 10.1146/annurev.nutr.22.011602.092259
Jackman, J. A., Hakobyan, A., Zakaryan, H., and Elrod, C. C. (2020). Inhibition of African swine fever virus in liquid and feed by medium-chain fatty acids and glycerol monolaurate. J. Anim. Sci. Biotechnol. 11, 114. doi: 10.1186/s40104-020-00517-3
Janssen, A. W., and Kersten, S. (2015). The role of the gut microbiota in metabolic health. FASEB J. 29, 3111–3123. doi: 10.1096/fj.14-269514
Jiang, Z., Kang, X., Song, Y., Zhou, X., and Yue, M. (2023). Identification and evaluation of novel antigen candidates against salmonella pullorum infection using reverse vaccinology. Vaccines 11, 865. doi: 10.3390/vaccines11040865
Khan, S., Moore, R. J., Stanley, D., and Chousalkar, K. K. (2020). The gut microbiota of laying hens and its manipulation with prebiotics and probiotics to enhance gut health and food safety. Appl. Environ. Microbiol. 86, 20. doi: 10.1128/AEM.00600-20
Langille, M. G., Zaneveld, J., Caporaso, J. G., Mcdonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Le Bon, M., Davies, H. E., Glynn, C., Thompson, C., Madden, M., Wiseman, J., et al. (2010). Influence of probiotics on gut health in the weaned pig. Livest. Sci. 133, 179–181. doi: 10.1016/j.livsci.2010.06.058
Li, P., Niu, Q., Wei, Q., Zhang, Y., Ma, X., Kim, S. W., et al. (2017). Microbial shifts in the porcine distal gut in response to diets supplemented with Enterococcus Faecalis as alternatives to antibiotics. Sci. Rep. 7, 41395. doi: 10.1038/srep41395
Li, X., Nie, C., Liu, Y., Chen, Y., Lv, X., Wang, L., et al. (2019). A genome-wide association study explores the genetic determinism of host resistance to Salmonella pullorum infection in chickens. Genet. Sel. Evol. 51, 51. doi: 10.1186/s12711-019-0492-4
Li, X., Nie, C., Zhang, Z., Wang, Q., Shao, P., Zhao, Q., et al. (2018). Evaluation of genetic resistance to Salmonella Pullorum in three chicken lines. Poult. Sci. 97, 764–769. doi: 10.3382/ps/pex354
Liao, S. F., and Nyachoti, M. (2017). Using probiotics to improve swine gut health and nutrient utilization. Anim. Nutr. 3, 331–343. doi: 10.1016/j.aninu.2017.06.007
Line, J. E., Hiett, K. L., Guard-Bouldin, J., and Seal, B. S. (2010). Differential carbon source utilization by Campylobacter jejuni 11,168 in response to growth temperature variation. J. Microbiol. Methods 80, 198–202. doi: 10.1016/j.mimet.2009.12.011
Lozupone, C., Hamady, M., and Knight, R. (2006). UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform. 7, 371. doi: 10.1186/1471-2105-7-371
Lv, Q., Ran, X., Qiu, H., Zhao, S., Hu, Z., Wang, J., et al. (2022). Seroprevalence of pullorum disease in chicken across mainland China from 1982 to 2020: a systematic review and meta-analysis. Res. Vet. Sci. 152, 156–166. doi: 10.1016/j.rvsc.2022.08.003
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mon, K. K. Z., Zhu, Y., Chanthavixay, G., Kern, C., and Zhou, H. (2020). Integrative analysis of gut microbiome and metabolites revealed novel mechanisms of intestinal Salmonella carriage in chicken. Sci. Rep. 10, 4809. doi: 10.1038/s41598-020-60892-9
Ni, Z., Cheng, W., Ding, J., Yao, R., Zhang, D., Zhai, D., et al. (2021). Impact of Buzhong Yiqi prescription on the gut microbiota of patients with obesity manifesting polycystic ovarian syndrome. Evid. Based Complement. Alternat. Med. 2021, 6671367. doi: 10.1155/2021/6671367
Papadopoulos, G. A., Poutahidis, T., Chalvatzi, S., Kroustallas, F., Karavanis, E., Fortomaris, P., et al. (2022). Effects of a tributyrin and monolaurin blend compared to high ZnO levels on growth performance, faecal microbial counts, intestinal histomorphometry and immunohistochemistry in weaned piglets: a field study in two pig herds. Res. Vet. Sci. 144, 54–65. doi: 10.1016/j.rvsc.2022.01.011
Polansky, O., Sekelova, Z., Faldynova, M., Sebkova, A., Sisak, F., Rychlik, I., et al. (2015). Important metabolic pathways and biological processes expressed by chicken cecal microbiota. Appl. Environ. Microbiol. 82, 1569–1576. doi: 10.1128/AEM.03473-15
Rosenberg, E., and Zilber-Rosenberg, I. (2018). The hologenome concept of evolution after 10 years. Microbiome 6, 78. doi: 10.1186/s40168-018-0457-9
Rychlik, I. (2020). Composition and function of chicken gut microbiota. Animals 10, 103 doi: 10.3390/ani10010103
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Setta, A. M., Barrow, P. A., Kaiser, P., and Jones, M. A. (2012). Early immune dynamics following infection with Salmonella enterica serovars Enteritidis, Infantis, Pullorum and Gallinarum: cytokine and chemokine gene expression profile and cellular changes of chicken cecal tonsils. Comp. Immunol. Microbiol. Infect. Dis. 35, 397–410. doi: 10.1016/j.cimid.2012.03.004
Shen, X., Zhang, A., Gu, J., Zhao, R., Pan, X., Dai, Y., et al. (2022). Evaluating Salmonella pullorum dissemination and shedding patterns and antibody production in infected chickens. BMC Vet. Res. 18, 240. doi: 10.1186/s12917-022-03335-z
Shen, Z., Wang, D., Ruan, Y., Xue, C., Zhang, J., Li, R., et al. (2014). Deep 16S rRNA pyrosequencing reveals a bacterial community associated with Banana Fusarium Wilt disease suppression induced by bio-organic fertilizer application. PLoS ONE 9, e98420. doi: 10.1371/journal.pone.0098420
Shimizu, J., Kubota, T., Takada, E., Takai, K., Fujiwara, N., Arimitsu, N., et al. (2019). Relative abundance of Megamonas hypermegale and Butyrivibrio species decreased in the intestine and its possible association with the T cell aberration by metabolite alteration in patients with Behcet's disease (210 characters). Clin. Rheumatol. 38, 1437–1445. doi: 10.1007/s10067-018-04419-8
Shin, M. S., Han, S. K., Ji, A. R., Kim, K. S., and Lee, W. K. (2008). Isolation and characterization of bacteriocin-producing bacteria from the gastrointestinal tract of broiler chickens for probiotic use. J. Appl. Microbiol. 105, 2203–2212. doi: 10.1111/j.1365-2672.2008.03935.x
Shivaprasad, H. L. (2000). Fowl typhoid and pullorum disease. Rev. -. Off. Int. Epizoot. 19, 405–424. doi: 10.20506/rst.19.2.1222
Soria, M. C., Soria, M. A., and Bueno, D. J. (2012). Comparison of 2 culture methods and PCR assays for Salmonella detection in poultry feces. Poult. Sci. 91, 616–626. doi: 10.3382/ps.2011-01831
Upadhaya, S. D., Kim, S. C., Valientes, R. A., and Kim, I. H. (2015). The effect of bacillus-based feed additive on growth performance, nutrient digestibility, fecal gas emission, and pen cleanup characteristics of growing-finishing pigs. Asian-australas. J. Anim. Sci. 28, 999–1005. doi: 10.5713/ajas.15.0066
Vazquez-Baeza, Y., Pirrung, M., Gonzalez, A., and Knight, R. (2013). EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2, 16. doi: 10.1186/2047-217X-2-16
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang, W., Li, Z., Han, Q., Guo, Y., Zhang, B., D'inca, R., et al. (2016). Dietary live yeast and mannan-oligosaccharide supplementation attenuate intestinal inflammation and barrier dysfunction induced by Escherichia coli in broilers. Br. J. Nutr. 116, 1878–1888. doi: 10.1017/S0007114516004116
Wang, X., Wang, H., Li, T., Liu, F., Cheng, Y., Guo, X., et al. (2020). Characterization of Salmonella spp. isolated from chickens in Central China. BMC Vet. Res. 16, 299. doi: 10.1186/s12917-020-02513-1
Wang, Y., Li, J., Xie, Y., Zhang, H., Jin, J., Xiong, L., et al. (2021). Effects of a probiotic-fermented herbal blend on the growth performance, intestinal flora and immune function of chicks infected with Salmonella pullorum. Poult. Sci. 100, 101196. doi: 10.1016/j.psj.2021.101196
Wei, S., Morrison, M., and Yu, Z. (2013). Bacterial census of poultry intestinal microbiome. Poult. Sci. 92, 671–683. doi: 10.3382/ps.2012-02822
Wigley, P., Berchieri, A. Jr., Page, K.L., Smith, A.L., and Barrow, P.A. (2001). Salmonella enterica serovar Pullorum persists in splenic macrophages and in the reproductive tract during persistent, disease-free carriage in chickens. Infect. Immun. 69, 7873–7879. doi: 10.1128/IAI.69.12.7873-7879.2001
Wigley, P., Jones, M. A., and Barrow, P. A. (2002). Salmonella enterica serovar Pullorum requires the Salmonella pathogenicity island 2 type III secretion system for virulence and carriage in the chicken. Avian Pathol. 31, 501–506. doi: 10.1080/0307945021000005879
Zhang, Q. Q., Ying, G. G., Pan, C. G., Liu, Y. S., and Zhao, J. L. (2015). Comprehensive evaluation of antibiotics emission and fate in the river basins of China: source analysis, multimedia modeling, and linkage to bacterial resistance. Environ. Sci. Technol. 49, 6772–6782. doi: 10.1021/acs.est.5b00729
Zhang, X., Hu, Y., Ansari, A. R., Akhtar, M., Chen, Y., Cheng, R., et al. (2021). Caecal microbiota could effectively increase chicken growth performance by regulating fat metabolism. Microb. Biotechnol. 4, 841. doi: 10.1111/1751-7915.13841
Zhao, X., Guo, Y., Guo, S., and Tan, J. (2013). Effects of Clostridium butyricum and Enterococcus faecium on growth performance, lipid metabolism, and cecal microbiota of broiler chickens. Appl. Microbiol. Biotechnol. 97, 6477–6488. doi: 10.1007/s00253-013-4970-2
Zhou, C., Liang, J., Jiang, W., He, X., Liu, S., Wei, P., et al. (2020). The effect of a selected yeast fraction on the prevention of pullorum disease and fowl typhoid in commercial breeder chickens. Poult. Sci. 99, 101–110. doi: 10.3382/ps/pez567
Zhou, X., Kang, X., Zhou, K., and Yue, M. (2022). A global dataset for prevalence of Salmonella Gallinarum between 1945 and 2021. Sci Data 9, 495. doi: 10.1038/s41597-022-01605-x
Keywords: gut microbiota, poultry, pullorum, microbial structure, prediction functions
Citation: Niu Q, Wang X, Qi X, Cao C, Yang K, Gu C, Zhou Z and Huang Q (2023) Identification of the gut microbiota affecting Salmonella pullorum and their relationship with reproductive performance in hens. Front. Microbiol. 14:1216542. doi: 10.3389/fmicb.2023.1216542
Received: 04 May 2023; Accepted: 03 July 2023;
Published: 27 July 2023.
Edited by:
Yan Wang, South China Agricultural University, ChinaReviewed by:
Lin Xi, North Carolina State University, United StatesMin Yue, Zhejiang University, China
Copyright © 2023 Niu, Wang, Qi, Cao, Yang, Gu, Zhou and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qizhong Huang, c2hzbnlreHlAMTYzLmNvbQ==