 Feng He1,2,3
Feng He1,2,3 Xueying Jin1,2,3
Xueying Jin1,2,3 Chunfeng Wang1,2,3
Chunfeng Wang1,2,3 Jingtao Hu1,2,3Shuai Su1,2,3
Jingtao Hu1,2,3Shuai Su1,2,3 Lei Zhao4Tingting Geng1,2,3
Lei Zhao4Tingting Geng1,2,3 Yuan Zhao1,2,3
Yuan Zhao1,2,3 Li Pan1,2,3Nan Bao1,2,3Hui Sun1,2,3*
Li Pan1,2,3Nan Bao1,2,3Hui Sun1,2,3*- 1College of Animal Science and Technology, Jilin Agricultural University, Changchun, China
- 2Ministry of Education Laboratory of Animal Production and Quality Security, Jilin Agricultural University, Changchun, China
- 3Jilin Provincial Key Laboratory of Animal Nutrition and Feed Science, Jilin Agricultural University, Changchun, China
- 4College of Animal Science and Veterinary Medicine, Heilongjiang Bayi Agricultural University, Daqing, Heilongjiang, China
At present, most studies have shown that probiotics have a positive regulatory effect on the nutritional metabolism of the body, but the mechanism is still unclear. Here, 48 piglets were divided into four groups. The control group was not fed probiotics, the Lac group was fed L. Rhamnosus GG ATCC53103, the Rha group was fed L. Plantarum JL01, and the mix group was fed two types of probiotics. Nitrogen metabolism and mRNA levels of mTOR and S6K in skeletal muscle were observed in each group. Then, metagenome and non-targeted metabonomics were used to observe the changes of intestinal microorganisms and plasma metabolites in portal channels after probiotics feeding. Finally, we combined the results of omics analysis to reveal the mechanism of probiotics on nitrogen metabolism in weaned piglets. The results showed that L. Rhmnosus GG ATCC53103 and L. Plantarum JL01 increased nitrogen apparent digestibility, nitrogen deposition rate, and nitrogen utilization rate of weaned piglets (P < 0.05); the relative expression of mTOR and SK6 mRNA in skeletal muscle increased significantly (P < 0.05). When L. rhamnosus GG ATCC53103 and L. plantarum JL01 were combined, we found that Clostridium and Prevotella significantly increased in the jejunum (P < 0.05). The relative abundance of Lactobacillus, Ruminococcus, Streptococcus, and Prevotella in the ileum increased significantly (P < 0.05). Compared with the control group, L-Tryptophan, 3-Phosphonyloxypyruvate, cis-Aconitate, and Carbamoyl phosphate were significantly increased in the mixed group portal vein. The result of the combinatorial analysis showed that the significantly increased microorganisms could encode the enzyme genes for the synthesis of L-Tryptophan, 3-Phosphonooxypyruvate, cis-Aconitate, and Carbamoyl phosphate. In summary, our results demonstrated that L. Rhamnosus GG ATCC53103 and L. Plantarum JL01 could stimulate the expression of skeletal muscle protein synthesis genes of weaned piglets by modulating the structure of the gut microbiota and its metabolites, thereby improving nitrogen metabolism in weaned piglets.
1. Introduction
The gastrointestinal microbial community is diverse and vibrant and plays a crucial role in the health and nutrition of the host (Roager and Licht, 2018). In previous studies, nitrogen metabolism and utilization were generally considered the role of digestive enzymes in the digestive tract. However, gut microbes are rich in functional genes that encode amino acid metabolism and protein synthesis. Prevotella ruminicola, Butyrivibrio fibrisolvens, Mitsuokella multiacidas, and Streptococcus bovis can secrete highly active dipeptidyl peptidase and dipeptidase for digestion and absorption of proteins. Therefore, gut microbes play a vital role in the metabolism of amino acids (Krajmalnik-Brown et al., 2012; Zhao et al., 2019). It has been reported that small intestinal microbes can use various nutrients in the diet, endogenous secreted proteins, and circulating urea in the intestine to synthesize amino acids (Bergen and Wu, 2009). A study showed that microbes in the small intestine can extensively metabolize various amino acids, with a metabolic rate of more than 50% (Yang et al., 2014). The isotope labeling method can better confirm this conclusion. In a report on lysine, lysine was synthesized by intestinal microorganisms and utilized by the body (Metges, 2000). Although the contribution rate may be small, it is still not negligible in the case of limited intake (Matthews, 2020). Moreover, various amino acids synthesized by intestinal microorganisms may promote muscle metabolism as regulatory factors (Lin et al., 2017). Such as tryptophan stimulates the mTOR pathway in muscle cells, promoting gene expression involved in myofibril synthesis (Dukes et al., 2015).
It has been reported that intestinal microorganisms use proteins to produce a large number of microbial metabolites, some of which are intermediate products and others are final products (Bishu, 2016). Some of these bacterial metabolites are absorbed by intestinal epithelial cells and have a positive effect. Unabsorbed bacterial metabolites are transported to the portal vein and exert physiological effects on the liver and peripheral organs (Davila et al., 2013). For example, tryptophan is metabolized by microorganisms to produce indole and its derivatives (Roager and Licht, 2018), which may induce intestinal endocrine cells to secrete glucagon-like peptide 1, inhibit gastric secretion and peristalsis, and promote satiety (Jardon et al., 2022). The gut microbiota metabolizes and produces many of the vitamins the body needs, especially folic acid and vitamin B12 (LeBlanc et al., 2013), which may improve muscle anabolism (Kuo et al., 2007). Therefore, there is a close relationship between gut microbes and nitrogen metabolism.
Weaning stress disrupts the gut microbial balance and reduces mammalian absorptivity for amino acid and protein synthesis (Wang et al., 2019; Yi et al., 2019). Probiotics have been shown to balance gut microbes and affect the body's absorption of amino acids (Utzschneider et al., 2016; Geng et al., 2021). Furthermore, probiotics have beneficial effects on growth performance and nitrogen metabolism during weaning in mammals (Khanian et al., 2019; Wang and Kim, 2021). Our previous study showed that L. rhamnosus GG ATCC53103 and L. plantarum JL01 improved the growth performance and intestinal immune function of weaned pigs (Geng et al., 2021). However, the effects of L. rhamnosus GG ATCC53103 and L. plantarum JL01 on the nitrogen metabolism of weaned pigs were still unclear. Therefore, the present study explored the effects of feeding L. rhamnosus GG ATCC53103 and L. plantarum JL01 on nitrogen metabolism and gut microbes in weaned piglets and used metagenomics to analyze the role of dominant flora. Finally, metagenomics and metabolomics are linked to explore the mechanism of its impact on nitrogen metabolism.
2. Materials and methods
2.1. Ethics statement
All animal experimental procedures were approved by the Ethics Committee of Jilin Agricultural University (Changchun, Jilin, China).
2.2. Animals and experimental design
Forty-eight 35-day-old “Yorkshire-Landrace-Duroc” weaned piglets (average 11.59 kg) were randomly divided into four diet groups (n = 4 each, i = 3 each). Every three piglets are kept in a cage. After a 7-day acclimation period, a 28-day animal experiment was started. The probiotics were cultured in MRS Medium at 37°C for 18h. The control group was fed the basal diet of the National Research Council (2012) nutritional requirements, and each piglet was fed 10 mL of MRS without bacteria per day. The experimental group was fed the basal diet with MRS containing probiotics, and the dose of probiotics per pig per day was included 10 mL containing L. plantarum JL01 (1 × 109 CFU/mL Lac group), L. rhamnosus GG ATCC53103 (1 × 109 CFU/mL, Rha group) or both probiotics (0.5 × 109 CFU/mL L. plantarum JL01 and 0.5 × 109 CFU/mL L. rhamnosus GG ATCC53103, mix group). Probiotics or MRS were mixed into a small amount of the basic diet and then fed to the piglets separately with a spoon to ensure that all weaned piglets would eat the whole dose. All piglets can get free water and feed. The composition of the diet is shown in Table 1.
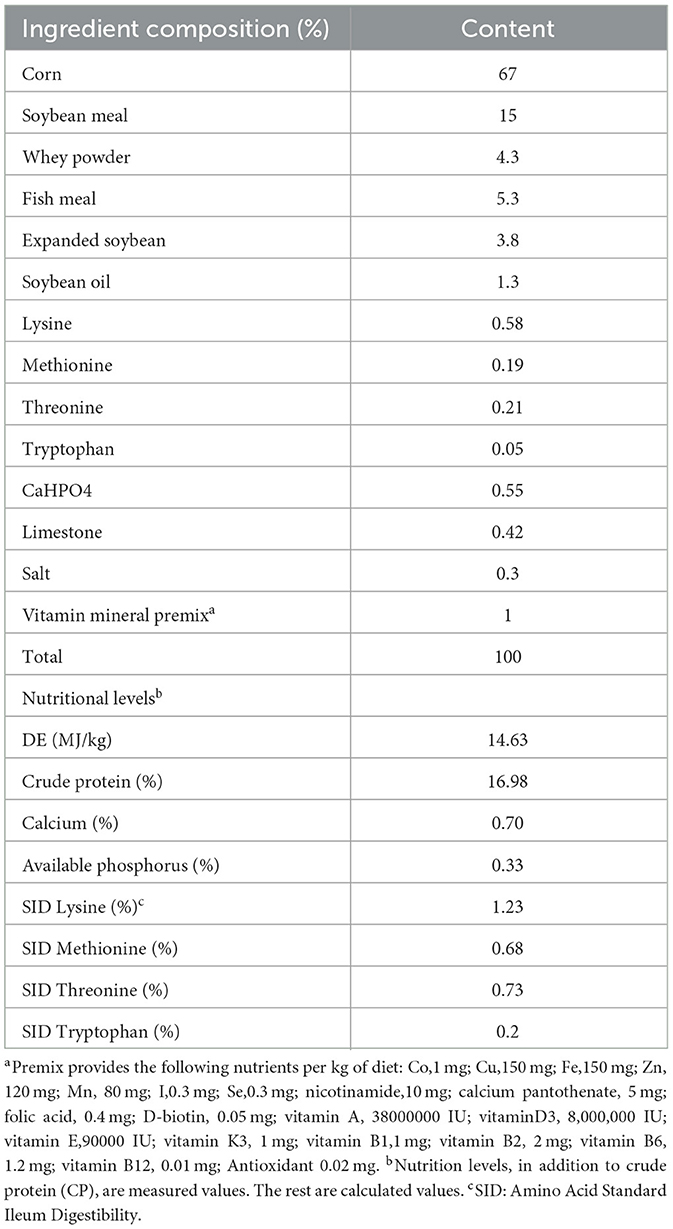
Table 1. Composition and nutritional level of the diets.
2.3. Sample collection
On the 23rd day of the rearing period. The metabolic experiments were conducted for 5 days. Using the method of collecting feces and urine, we randomly selected three piglets from each group for the nitrogen metabolism test, fecal weight and urine volume were measured using a separate collection device for feces and urine in metabolic cages, and feces and urine were collected at 8:00 and 16:00 every day (Feyera et al., 2019). Urine samples were added with 10% sulfuric acid for nitrogen fixation and stored at −80°C for testing. We mixed the fecal samples collected every day, kept them in zip lock bags, recorded the weight, took out 200 g of fecal samples, quickly added 10% sulfuric acid to fix nitrogen, baked them at 60 °C to constant weight, then pulverized, and stored at −80 °C refrigerator for testing. On the morning of the 28th day, three weaned piglets were randomly selected from each group to collect intestinal samples, and a 3% sodium barbital solution was used as anesthesia for weaned piglets. The jejunum and ileum were dissected, and the contents of the jejunum and ileum were carefully squeezed out, and then, chyme was collected in sterile cryogenic vials. Afterward, 10 mL of hepatic portal vein blood was collected and placed in a blood collection tube containing heparin sodium. After centrifugation, plasma layers were extracted and distributed into 1.5 mL cryopreservation tubes. Afterward, the skeletal muscle (gluteus maximus) of piglets was quickly collected with a sterile EP tube and quickly frozen in liquid nitrogen. The samples were stored in a −80°C refrigerator.
2.4. Nitrogen metabolism experiment
The nitrogen content in feed and feces was measured using Nitrogen determination apparatus (FOSS Kjeltec 8420 Kjeldahl +nitrogen determination apparatus, FOSS [Technology and Trade Co. Ltd., Beijing, China]). The formulas used were as follows: nitrogen emission (g/d) = fecal nitrogen (FN) (g/d) + urine nitrogen (UN) (g/d), nitrogen apparent digestibility (%) = [(nitrogen intake (NI) - FN)/NI] × 100%, nitrogen deposition rate (%) = [(NI-FN-UN)/NI] × 100%, and nitrogen utilization rate (%) = [(NI-FN-UN) / (NI-FN)] × 100%.
2.5. Real-time quantitative PCR
Total RNA from the muscle tissue was extracted using TRIzol. The steps for RNA-reverse transcription and RT-qPCR were the same as before (Ding et al., 2020). PrimeScript™ RT reagent kit and SYBR® Premix Ex T aq™ II were used for reverse transcription and RT-qPCR processes, respectively. RT-qPCR analysis was conducted using the Agilent Stratagene Mx3005P qPCR system (Agilent Technologies, Inc.). These RT-qPCR assays were run in triplicate for each sample, and the results were normalized to the expression of β-actin. Expression was analyzed using 2–ΔΔCt comparison method. Primer sequences are provided in Table 2.
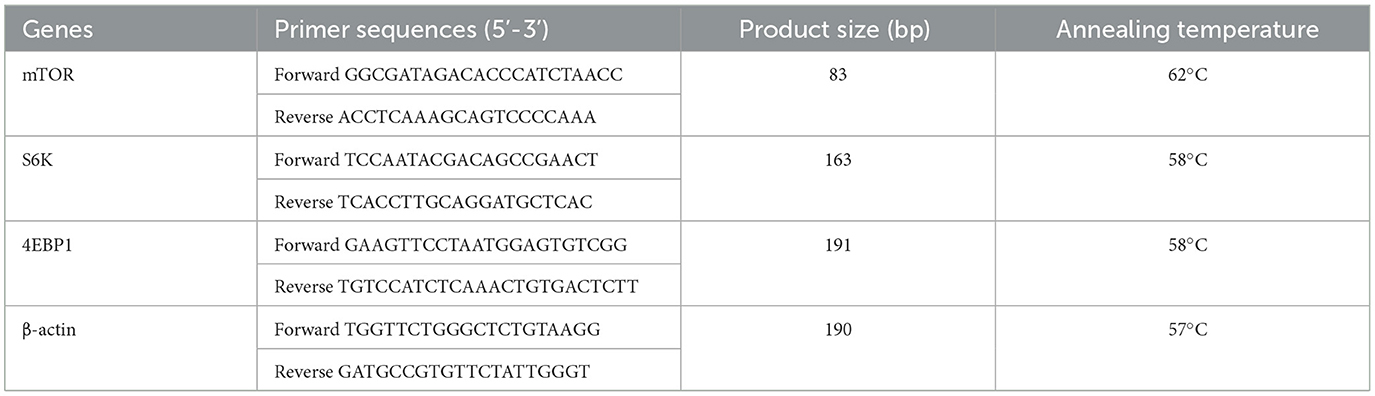
Table 2. Parameters of primer for real-time PCR.
2.6. DNA extraction and 16S rRNA sequencing
Gut chyme microbial DNA was extracted using the TIANamp Stool DNA kit (Tiangen, Beijing, China).
The sequences of 16S rDNA genes were analyzed using the Illumina HiSeq™ 2500 sequencing platform. The raw data were filtered using the FASTP software. All samples were clustered, and the effective sequence similarity ≥ 97% was clustered into operation classification units (OTUs). The OTU sparsity curve and rank abundance curve were plotted in QIIME. An inter-group Venn analysis was performed in R (version 3.4.1) to identify unique and common otus. We use QIIME to calculate Chao1, Simpson, etc. α Diversity Index.
2.7. Sample preparation for LC-MS analysis
Portal vein plasma samples weighing 50 mg were carefully measured and transferred into an EP tube. Subsequently, the EP tube was filled with the solvent (acetylene-methane-water, 2: 2: 1, including internal standards). After rotating for 30s at an average speed of 45 Hz, the sample was subjected to a 4-min ice water bath. Then, it was ultrasonicated for 5min. The average ultrasound is repeated three times and then stored at −20 °C for 1 h and for 15 min for 12, 000 RPM and 4 °C. The obtained Shangqing was transferred to the LC-MC bottle, analyzed in UHPLC-QE or orBITRAP MS, and mixed with the equivalent of all samples to clear the quality control (QC) sample.
2.8. Functional analysis
We used the T-test as a variable analysis to screen the different metabolites, and the VIP threshold was set to 1, and the differences between the two groups of the metabolites of a P-value of < 0.05 and a VIP of ≥ 1 were selected. The metabolites were enriched in the KEGG metabolic pathway for enrichment analysis. Compared with the entire background, the enrichment analysis determined the significant enrichment of metabolites or signal transition channels in the difference.
2.9. Sequence analysis
We used the Nextera XT DNA Library to prepare the library for the macro-base group and sequence the Illumina Hiseq platform. We also used Metaphlan (V.2.2) (Truong et al., 2015) and Humann2 to analyze macroscopic group data. Then, we use the Unigene to query the enzyme committee (EC) database through Diamond software. NCBI's RefSeq database was used to build a microbial index with significant differences in pigs annotated by Metaothello.
2.10. Statistical analysis
The results were expressed as the mean ± SD. Statistical analysis was performed using Prism software (GraphPad Software, San Diego, CA) to carry out one-way ANOVA followed by Bonferroni post-hoc analysis. Values of p < 0.05 were considered significant. Different microbial, genetic functions, metabolites, and metabolic channels were drawn through Adobe Illustrator software (USA).
3. Results
3.1. Nitrogen metabolism
The effects of L. plantarum JL01 and L. rhamnosus GG ATCC53103 on the nitrogen metabolism of weaned piglets are shown in Table 3. Compared to the Con group, the ingested nitrogen of the piglets in the Rha group, Lac group, and mix group was significantly increased (P < 0.05), fecal nitrogen, urinary nitrogen, and total nitrogen emissions showed a decreasing trend, the apparent nitrogen digestibility of Rha and Lac groups tended to increase; the apparent nitrogen digestibility of the mix group increased by 1.88% (P < 0.05), and the nitrogen deposition rate increased respectively increased by 4.17, 4.49, and 5.65% (P < 0.05), and the nitrogen utilization rate increased by 2.82, 3.23, and 3.70%, respectively (P < 0.05).
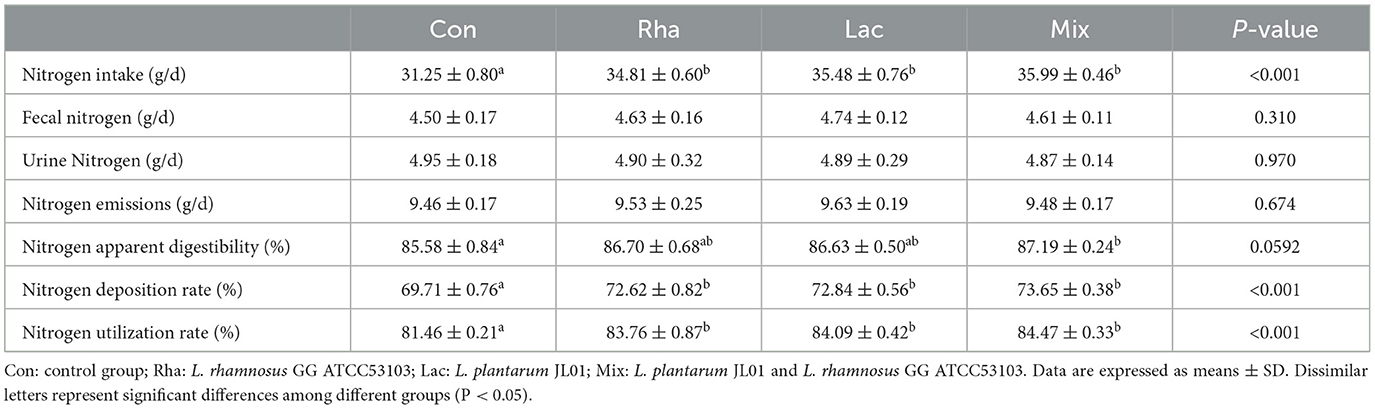
Table 3. Effects of L. plantarum JL01, L. rhamnosus GG ATCC53103 on nitrogen metabolism of weaned piglets.
3.2. Gene expression of skeletal muscle protein deposition
As shown in Figure 1, the relative expression of mTOR and S6K mRNA in the skeletal muscle of piglets in the Rha, Lac, and mix groups was significantly increased compared with the Con group (P < 0.05).
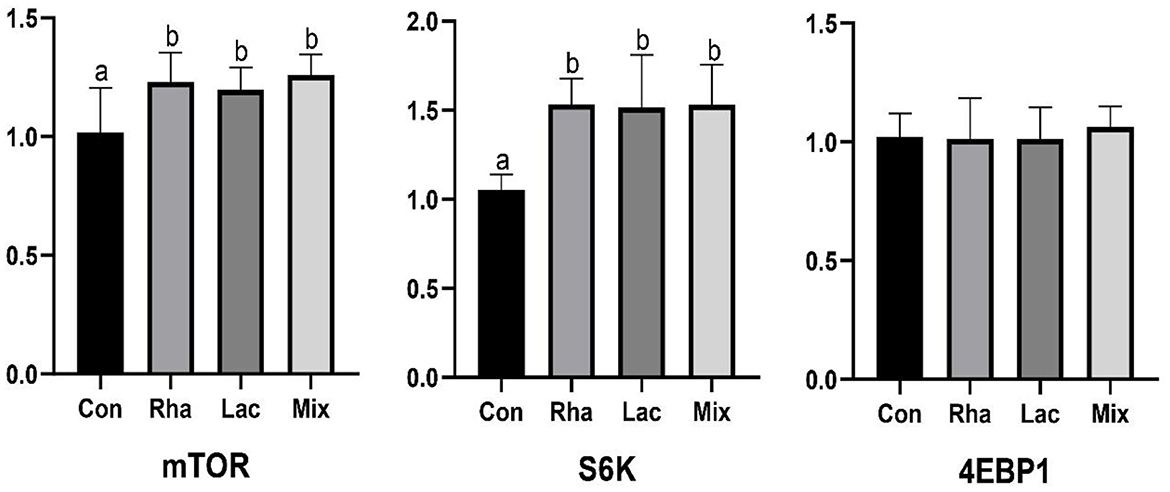
Figure 1. The relative expression of mTOR, S6K, 4EBP1 mRNA of the dorsal longest muscle of weaned piglets. Con: control group; Rha: L.rhamnosus GG ATCC53103; Lac: L. plantarum JL01; Mix: L. plantarum JL01 and L. rhamnosus GG ATCC53103. Data are expressed as means ± SD. Dissimilar letters represent significant differences among different groups (P < 0.05).
3.3. Data acquired from high-throughput sequencing and alpha-diversity measurements
A total of 2, 800, 880 sequences were obtained in this experiment, and the original sequences were filtered, and finally, 2, 591, 938 valid sequences were obtained, with an average of 128, 160 valid sequences per sample for subsequent analysis. Microbial diversity is the diversity in a particular habitat or ecosystem and uses two parameters, species richness, and evenness, to reflect the diversity and abundance of microorganisms (Grice et al., 2009). Chao1 and ACE indices were used to estimate species richness information of samples, and Simpson and Shannon were used to estimate species diversity information (Segata et al., 2011). The effects of L. plantarum JL01 and L. rhamnosus GG ATCC53103 on the microbial diversity of the small intestine of weaned piglets are shown in Figure 2. Compared with the Con group, the Shannon and Simpson indices of the Rha, Lac, and mix groups showed an increasing trend in the jejunum but were insignificant. In the ileum, the ACE, Chao1, Shannon, and Simpson indexes of piglets in the mix group were significantly increased compared with the Con group (P < 0.05), while the uniformity and abundance of other intestinal contents were not significant.
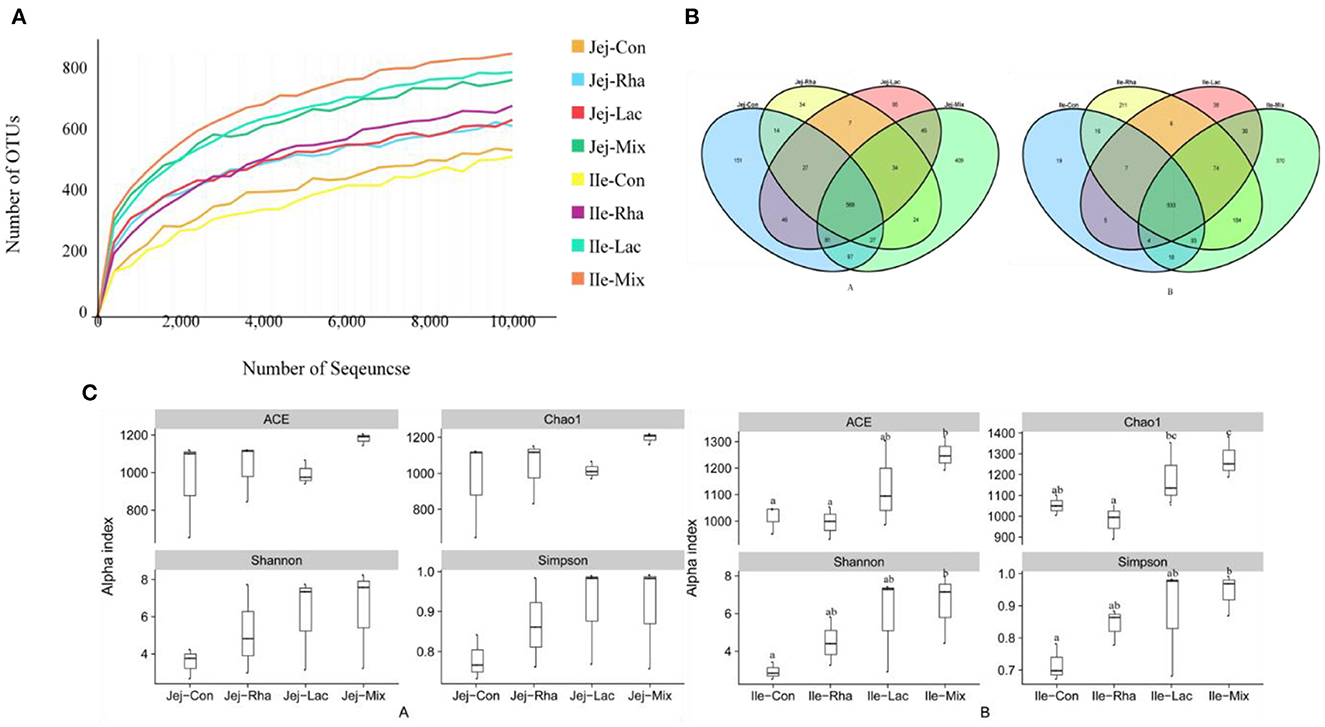
Figure 2. (A) Rarefaction curves of OTUs from microbiota in the jejunum and ileum of weaned piglets. (A) OTU comparative analysis of intestinal microbiota of weaned piglets (A) jejunum (B) ileum. (C) Box pattern of microbial Alpha diversity index of weaned piglets (A) jejunum (B) ileum; Con: control group; Rha: L. rhamnosus GG ATCC53103; Lac: L. plantarum JL01; Mix: L. plantarum JL01 and L. rhamnosus GG ATCC53103. Data are expressed as means ± SD. Dissimilar letters represent significant differences among different groups (P < 0.05).
3.4. Changes in gut microbiota
The microbial composition of the gut contents of weaned piglets was analyzed. Figure 3A shows the 10 most abundant phyla among all phyla. Firmicutes, Proteobacteria, and Bacteroidetes are the common dominant phyla in each treatment group, and their relative abundances together account for the bacterial abundances in each treatment group. They accounted for more than 93% of the total bacteria. As shown in Figure 3B, compared with the Con group, in the jejunum, the Firmicutes in the mix group were significantly decreased (P < 0.05). The relative abundance of Bacteroidetes significantly increased (P < 0.05), and the relative abundance of Proteus significantly decreased (P < 0.05) in the Lac and mix groups. Firmicutes/Bacteroidetes were significantly increased in piglets in the Lac and mix groups (P < 0.05). In the ileum, the relative abundance of Firmicutes in the Rha and mix groups was significantly lower than those in the Con group (P < 0.05). The relative abundance of Bacteroidetes increased significantly in Rha, Lac, and mix groups (P < 0.05). Firmicutes/Bacteroidetes were significantly increased in Rha, Lac, and mix groups (P < 0.05).
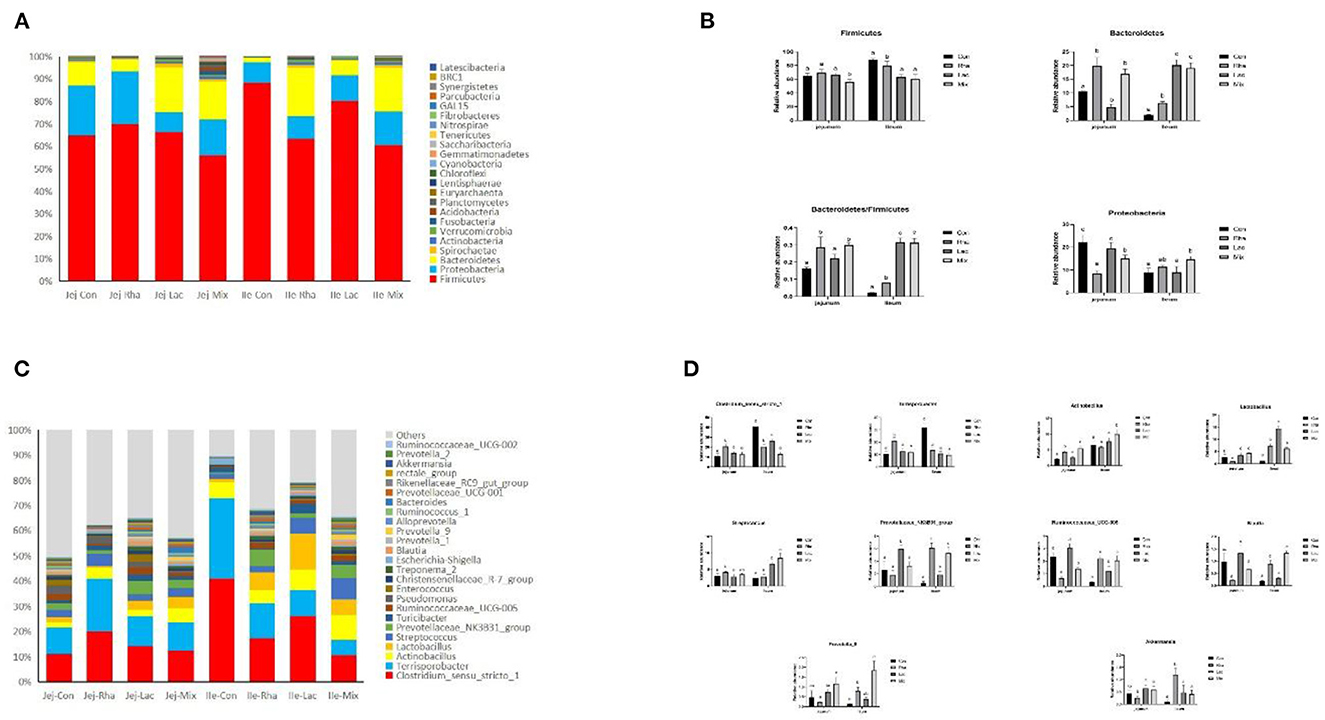
Figure 3. (A) The top 10 most abundant phyla are presented. (B) Bacterial composition differences in the small intestine at the phylum level. Con: control group; Rha: L. rhamnosus GG ATCC53103; Lac: L. plantarum JL01; Mix: L. plantarum JL01 and L. rhamnosus GG ATCC53103. Data are expressed as means ± SD. Dissimilar letters represent significant differences among different groups (p < 0.05). (C) Relative abundances of the top ten genera. (D) Bacterial composition differences in the small intestine at the genus level. Con: control group; Rha: L. rhamnosus GG ATCC53103; Lac: L. plantarum JL01; Mix: L. plantarum JL01 and L. rhamnosus GG ATCC53103. Data are expressed as means ± SD. Dissimilar letters represent significant differences among different groups (P < 0.05).
At the genus level, 181 genera were identified from all samples. The relative abundances of the top 25 genera are shown in Figure 3C. As shown in Figure 2D, compared with the control group, the relative abundance of Lactobacillus, Actinobacillus, and Prevotella_9 in the mix group increased significantly (P < 0.05), and the relative abundance of Ruminococcaceae_UCG-005 decreased significantly (P < 0.05). The relative abundances of Ruminococcaceae_UCG-005, Actinobacillus, Streptococcus, Blautia, and Prevotella_9 increased significantly (P < 0.05), while the relative abundances of Clostridium_sensu_stricto_1 and Terrisporobacter decreased significantly (P < 0.05). Rha group in the jejunum, including Clostridium_sensu_stricto_1, Terrisporobacter, and Actinobacillus, were found to be significantly different. The relative abundance of Lactobacillus, Ruminococcaceae_UCG-005, and Streptococcus decreased significantly (P < 0.05). Conversely, in the ileum, the relative abundance of the Rha group, Prevotellaceae_NK3B31_group, Lactobacillus, Ruminococcaceae_UCG-005, and Akkermansia, increased significantly (P < 0.05). Additionally, the relative abundance of Clostridium_sensu_stricto_1 and Terrisporobacter decreased significantly (P < 0.05). The relative abundance of the Lac group, including Prevotellaceae_NK3B31_group, Lactobacillus, Ruminococcaceae_UCG-005, Prevotella_9, showed a significant increase (P < 0.05). Similarly, in the ileum, the relative abundance of the Lac group, comprising Prevotellaceae_NK3B31_group, Lactobacillus, Ruminococcaceae_UCG-005, and Streptococcus, also increased significantly (P < 0.05). In contrast, the relative abundance of Clostridium_sensu_stricto_1 and Terrisporobacter exhibited a significant decrease (P < 0.05).
3.5. Small intestine differentiated microbial and functional gene network map
This experiment only focuses on the differentiated microorganisms in the small intestine and the functional genes encoding amino acid metabolism and protein synthesis. As shown in Figure 4, Clostridium_ sensu_ stricto_ 1 and ruminocaceae_ Ucg-005 have a gene encoding d-3-phosphoglycerate dehydrogenase; Actinobacillus, Clostridium, Lactobacillus, and Streptococcus have genes encoding carbamate kinase and Carbamoyl phosphate synthase; Akkermansia and Ruminococcus have genes encoding Carbamoyl phosphate synthase; Actinomycetes, Clostridium, Prevotellaceae_ NK3B31_ Group, Prevotella_ 9, Rumencocci and Streptococcus have genes encoding aconitate hydratase; Actinomycetes, Klebsiella, Streptococcus, Rumencocci, Prevotellaceae_ NK3B31_ Group, Prevotella_ 9 and Clostridium has a gene encoding tryptophan synthase.
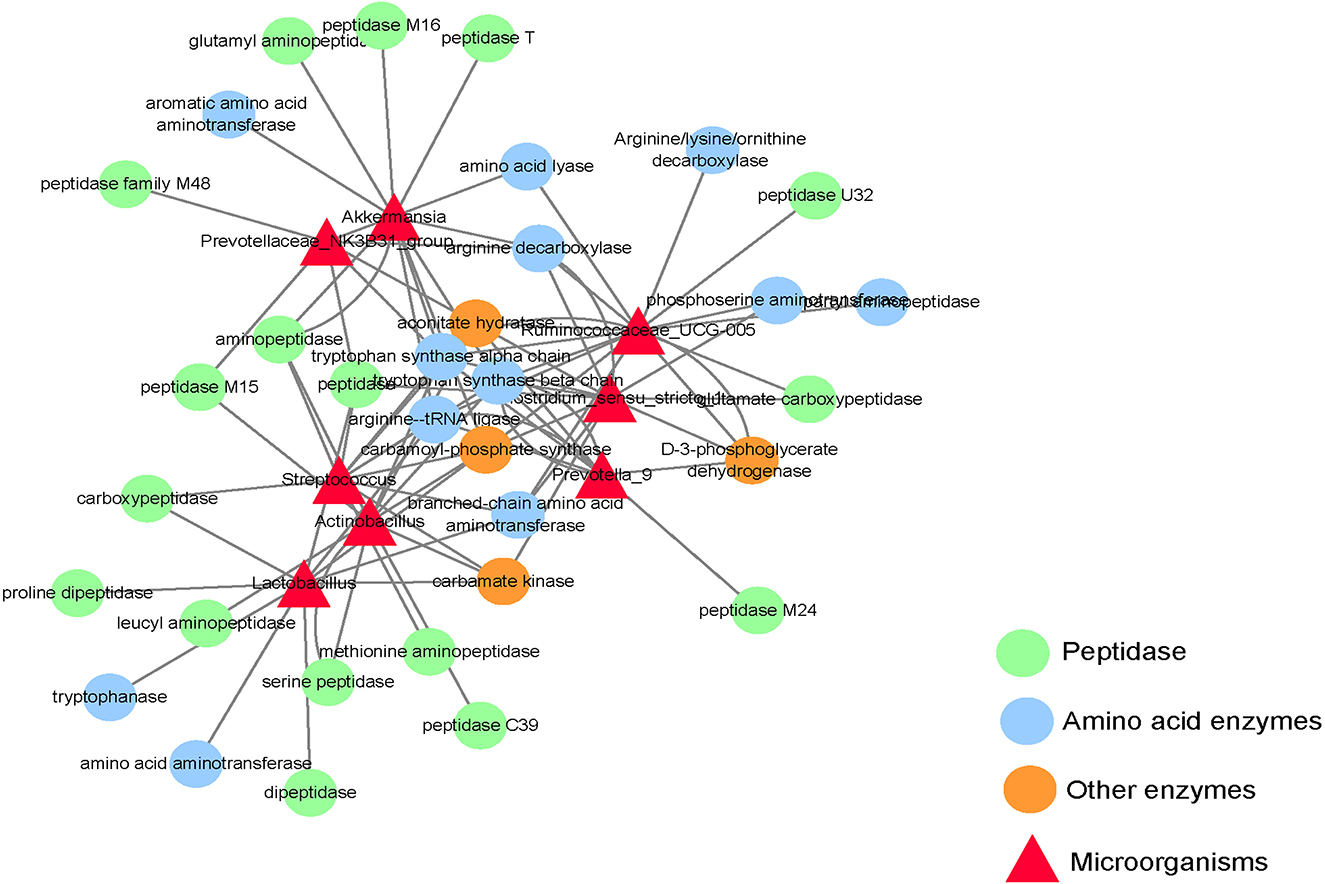
Figure 4. Network diagram of differential microbes and functional genes in the small intestine of the weaned piglets.
3.6. Metabolomic profiling of portal vein
In this test, no significant peaks were detected in the blank sample, indicating that there was no cross-contamination between samples in this test. Substance identification was conducted using a self-written R program package and a self-built secondary mass spectrometry database. The second-order mass spectrometry matching degree cutoff = 0.2. A total of 11, 875 metabolites were obtained from the detection results of this study, and 617 substances were identified by the secondary spectrum.
As shown in Figure 5A, each treatment group had apparent separation compared with the control group, indicating that feeding L. rhamnosus and L. plantarum had a significant effect on the plasma metabolites of the hepatic portal vein of weaned piglets. In this study, the differential metabolites related to nitrogen metabolism were selected for analysis. As shown in Table 4, in the portal vein metabolites, compared with Con, the differential metabolites produced by the Rha group were 3-Phosphonooxypyruvate (P < 0.05), the differential metabolites produced by the Lac group were L-Tryptophan (P < 0.05), and the differential metabolites produced by the mix group were L-Tryptophan, 3-Phosphonooxypyruvate, cis-Aconitate and Carbamoyl phosphate (P < 0.05). As shown in Figures 4A–D, after the metabolites were enriched, the first 20 paths with the lowest enrichment q value were selected to draw the map. This experiment only focused on the metabolic pathway of nitrogen nutrients. The results showed that the differential metabolite 3-phosphodeoxypyruvate enriched in the portal vein of weaned piglets produced by feeding the mixed bacteria of L. rhamnosus and L. plantarum was glycine, serine, and threonine metabolism pathway; the metabolic pathway enriched by the differential metabolite L-Tryptophan is the tryptophan metabolic pathway; the metabolic pathway enriched by the differential metabolite cis-Aconitate is the Citrate cycle (TCA cycle) pathway; The metabolic pathway for the differential metabolite Carbamoyl phosphate is the Arginine biosynthesis pathway.
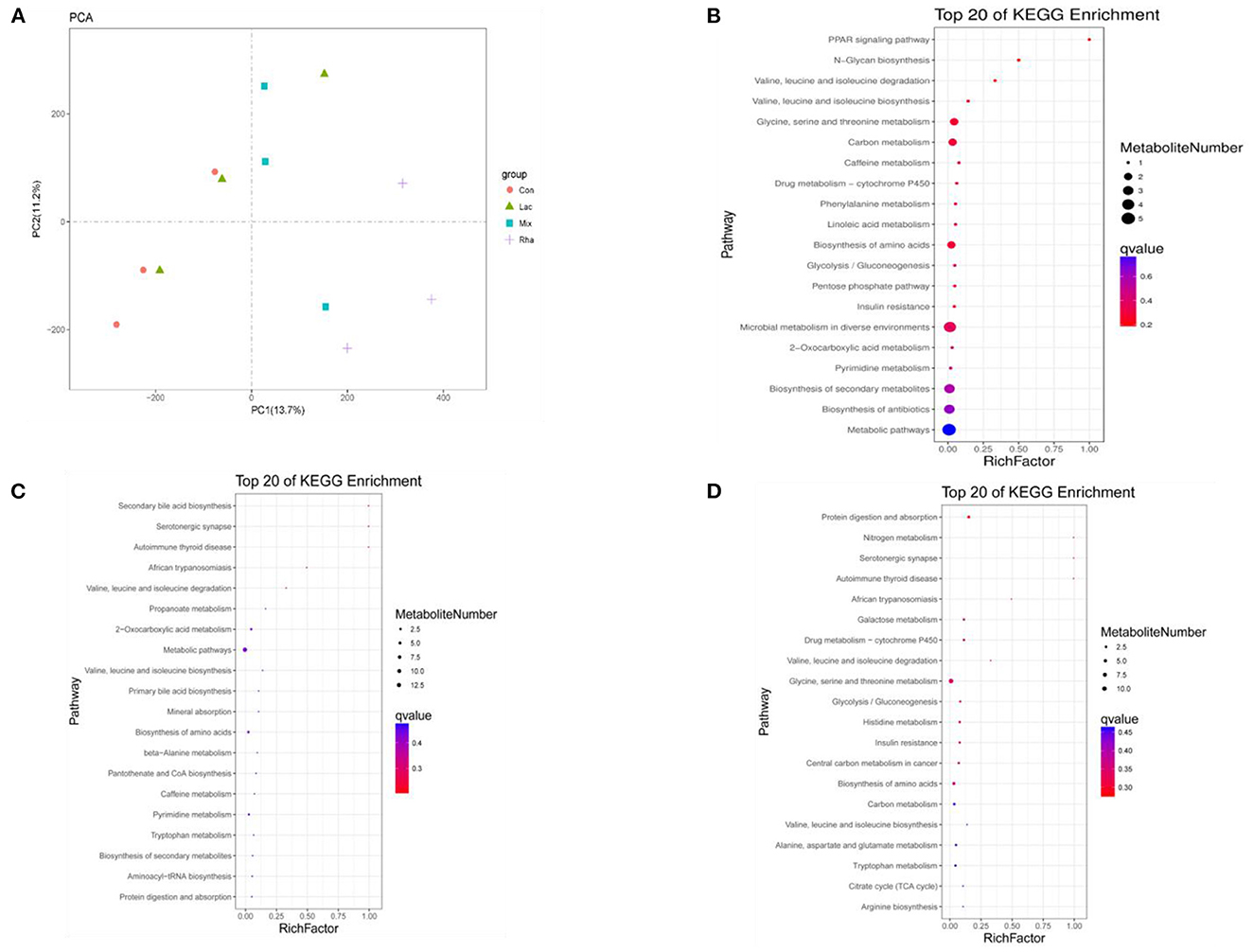
Figure 5. (A) PCA score map of metabolites in plasma of hepatic portal vein. Distribution of enriched KEGG pathway of differential metabolites in plasma of hepatic portal vein (B) Con vs. Rha (C) Con vs. Lac (D) Con vs. Mix.
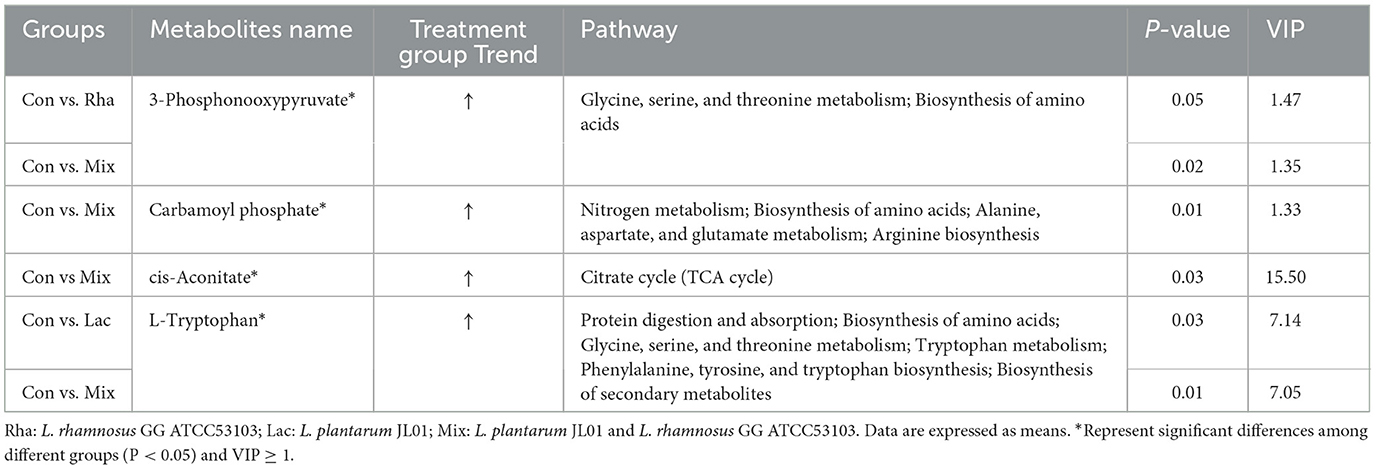
Table 4. Differential metabolites in plasma of hepatic portal vein.
3.7. Relationship between the intestinal microbiome and non-targeted metabolomics
To further study the relationship between intestinal microflora and metabolites, we visualized the correlation between jejunum and ileum microflora and metabolites and carried out Spearman correlation analysis on the relative abundance of different microbial species and metabolites. As shown in Figure 6A, in the jejunum, L-Tryptophan and cis-Aconitate were positively correlated with Streptococcus, Lactobacillus, and Actinobacillus and negatively correlated with Akkermansia. Carbamoyl phosphate was positively correlated with Akkermansia, Lactobacillus, and Actinobacillus and negatively correlated with Streptococcus. 3-Phosphonoxypyruvate was positively correlated with Lactobacillus and Actinobacillus and negatively correlated with Akkermansia and Streptococcus. As shown in Figure 6B, in the ileum, we found 3-Phosphonooxypyruvate, Carbamoyl phosphate, cis-Aconitate, L-Tryptophan and Actinobacillus, Blautia, Lactobacillus, Streptococcus, Ruminococcaceae_UCG-005, Prevotella_9, Prevotellaceae_NK3B31_group, and Akkermansia were positively correlated and negatively correlated with Clostridium_sensu_stricto_1.
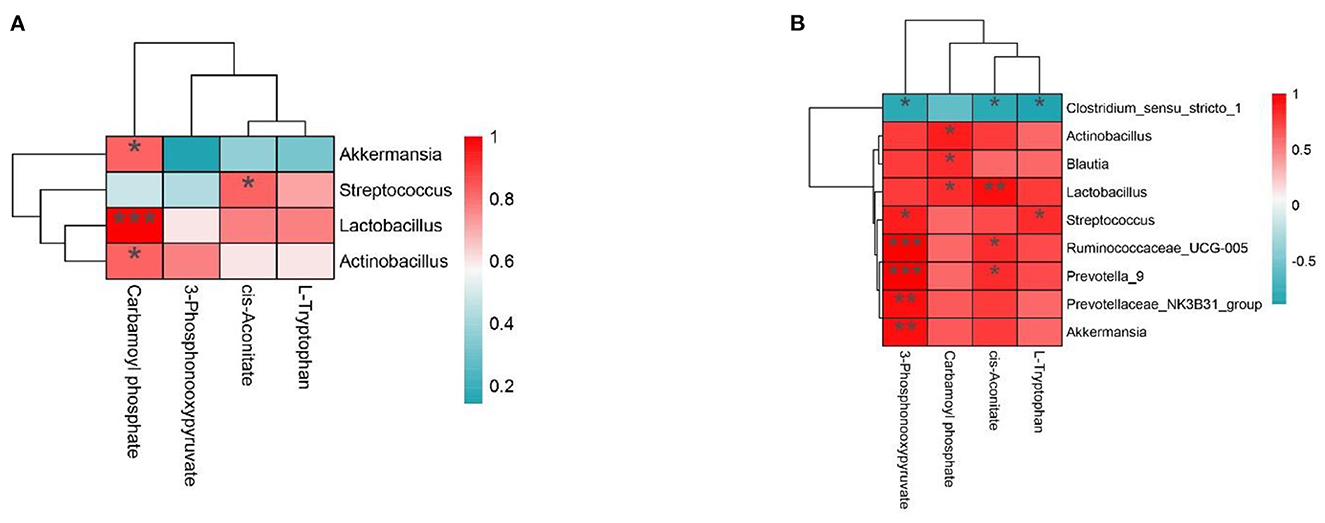
Figure 6. (A, B) The interaction between intestinal flora and portal vein metabolome in the Con group vs. Mix group.*P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences between the microbiota and metabolites. Blue represents positive correlations; red represents negative correlations.
4. Discussion
During pig production, approximately 46% of nitrogen was retained in the animal's body, and the remaining 54% was excreted mostly in feces and urine. Therefore, improving the efficiency of converting plant protein feed into animal protein could reduce pollution caused during pork production and possibly increase economic efficiency (Millet et al., 2018). The small intestine is the main site of absorption of dietary protein. Most of the nitrogenous nutrients in the diet are digested in the small intestine and converted into amino acids or small peptides; the unabsorbed part is directly excreted with the feces, and part of the absorbed part is used by the animal body to become deposited nitrogen, and the other part is passed through the ornithine cycle to generate urea nitrogen and excreted in the urine (Zhao et al., 2020). At the same time, gut microbes also play an important role. They use nitrogen sources in the gut to synthesize amino acids and participate in nitrogen metabolism (van der Wielen et al., 2017). Through the analysis of nitrogen metabolism, the preliminary situation of nitrogen utilization in the body is reflected. In the present study, we found that the combined feeding of L. plantarum JL01 and L. rhamnosus GG ATCC53103 had the best effect, increasing the apparent nitrogen digestibility by 1.31%, the nitrogen deposition rate by 5.65%, and the nitrogen utilization rate by 3.70% in weaned piglets and was consistent with previous reports (Zhu et al., 2017).
Intestinal microorganisms play an important role in the digestive system of animals (Sun et al., 2019). It has been reported that the higher the diversity of intestinal microorganisms, the higher the stability of the body's intestinal microecosystem, thus promoting the growth of animals and the absorption and utilization of nutrients (Cani and Delzenne, 2009; McCann et al., 2017). Previous studies have shown that dietary supplementation with probiotics improves the diversity of gut flora, which is consistent with our results (Sun et al., 2023). Moreover, a large number of amino acids in the small intestine are transformed, metabolized, and utilized by intestinal microorganisms, which not only maintains the composition and quantity of the microorganisms themselves but also produces metabolites that regulate the physiological functions of the host (Nicholson et al., 2012), and could also utilize various substrates to synthesize new microbial amino acids (Torrallardona et al., 2003; Metges and Petzke, 2005). In our study, we found that co-administration of the two probiotics significantly improved the abundance of Prevotella, Streptococcus, Actinomyces, and Klebsiella in the jejunum and ileum chyme significantly increased in piglets. At present, there are few articles on the relationship between small intestinal microorganisms and amino acids, so we detected the functional genes of small intestinal microorganisms through the macrogenome to predict the relationship between small intestinal microorganisms and amino acids. In our experiment, macrogenomic data showed that Prevotella, Streptococcus, Actinomyces, and Klebsiella secreted proteinases and alkaline peptidases that hydrolyze proteins and amino acids. This plays an important role in the digestion of mammalian proteins and the absorption of amino acids. Actinomyces, Klebsiella, Streptococcus, Ruminococcus, Prevotella, Lactobacillus, and Clostridium all have genes encoding various peptidases. Clostridium, Lactobacillus, Ruminococcus, and Streptococcus have genes encoding branched-chain amino acid aminotransferases. In addition, Actinomyces, Klebsiella, Streptococcus, Ruminococcus, Prevotella, Lactobacillus, and Clostridium have genes encoding arginase. Previous studies have demonstrated that Klebsiella may synthesize aspartic acid, glutamic acid, and tryptophan, Streptococcus may synthesize leucine, lysine, and serine, and Clostridium may synthesize serine, proline, and valine, which aligns with our results (Hooper et al., 2002; Dai et al., 2011). In summary, when the L. rhamnosus GG ATCC53103 and L. plantarum JL01 are combined, it can increase the number of beneficial bacteria in the pig's intestine and adjust the intestinal microorganisms.
The hepatic portal vein is a large vein that enters the liver from the hepatic portal after the mesenteric veins of the digestive tract converge. It is closely related to the metabolism of proteins, lipids, carbohydrates, vitamins, and hormones (Johnson et al., 2016). In this study, portal vein plasma analysis was performed in the Con, Rha, Lac, and mix groups. Compared with the Con group, four important metabolites, 3-Phosphonooxypyruvate, Carbamoyl phosphate, cis-Aconitate, and L-Tryptophan, were detected in the mix group. To detect the relationship between metabolites and intestinal microflora, we analyzed the functional genes of intestinal microflora through macrogenome to analyze the reasons for the increase in metabolites (Figure 7). Our data show that D-3-phosphoglycerate dehydrogenase was encoded by Clostridium, Prevotella, and Ruminococcus, which is an enzyme that converts 3-phosphoglycerate to phosphopyruvate. The genes of carbamate kinase and carbamoyl phosphate synthase are encoded by Actinomycetes, Clostridium, Lactobacillus, and Streptococcus, which can convert ammonia to carbamate. Aconitic acid hydratase is encoded by Actinomycetes, Clostridium, Prevotella, Ruminococcus, and Streptococcus, which can convert citric acid and isocitric acid into cis-Aconitic acid. L-tryptophan is encoded by Actinomycetes, Klebsiella, Streptococcus, Ruminococcus, Prevotella, and Clostridium, which have genes for tryptophan synthase. Compared with the Con group, these microorganisms were significantly increased in the mix group. In addition, our results showed that most microorganisms were positively correlated with metabolites. Through KEGG enrichment analysis, it was found that cis-Aconitic acid was enriched in the TCA cycle and was an important intermediate product of the TCA cycle. Its increase may promote the TCA cycle and lead to an increase in α-ketoglutarate (AKG). It is well known that AKG plays an important role in protein metabolism and transmembrane transport of amino acids (Wernerman and Hammarqvist, 1999; Zhao et al., 2020) found that adding AKG to a low-protein diet can improve the nitrogen metabolism efficiency of weaned piglets. In addition, the levels of S6K, mTOR, and 4EBP1 mRNA and phosphorylated protein in muscle were higher. In an in vitro study by Cai et al. (2020), it was shown that AKG promotes protein synthesis in C2C12 myotubes through the Akt/mTOR signaling pathway. Our data showed that the relative expression of mTOR and S6K mRNA in skeletal muscle increased significantly. Therefore, cis-Aconitic acid may promote protein synthesis in skeletal muscle by increasing the content of AKG, thereby improving the efficiency of nitrogen metabolism. Moreover, the significantly elevated metabolites 3-Phosphonooxypyruvate and Carbamyl phosphate in the portal vein may also promote protein synthesis by promoting the increase of AKG caused by the TCA cycle. 3-Phosphonooxypyruvate is an intermediate substance in the metabolic pathway of glycine, serine, and threonine and has the function of promoting the metabolism of glycine, serine, and threonine to produce metabolites such as acetyl CoA, propionic acid, and α-aminobutyric acid (Cogo et al., 2018). Acetyl-CoA produced by 3-Phosphonooxypyruvate metabolism may be involved in the TCA cycle by synthesizing citric acid. Carbamoyl phosphate could participate in the urea cycle to generate fumaric acid and participate in the TCA cycle. Tryptophan is a special amino acid that can participate in host protein synthesis, increase host feed intake, and ensure host growth performance (Sterndale et al., 2020). Previous studies have shown that dietary supplementation of moderate tryptophan significantly reduces serum urea and improves growth performance (Ruan et al., 2014; Rao et al., 2021), which is consistent with our results. In addition to producing acetyl-CoA to participate in the TCA cycle, tryptophan can also be used as a raw material for protein synthesis to directly promote protein synthesis.
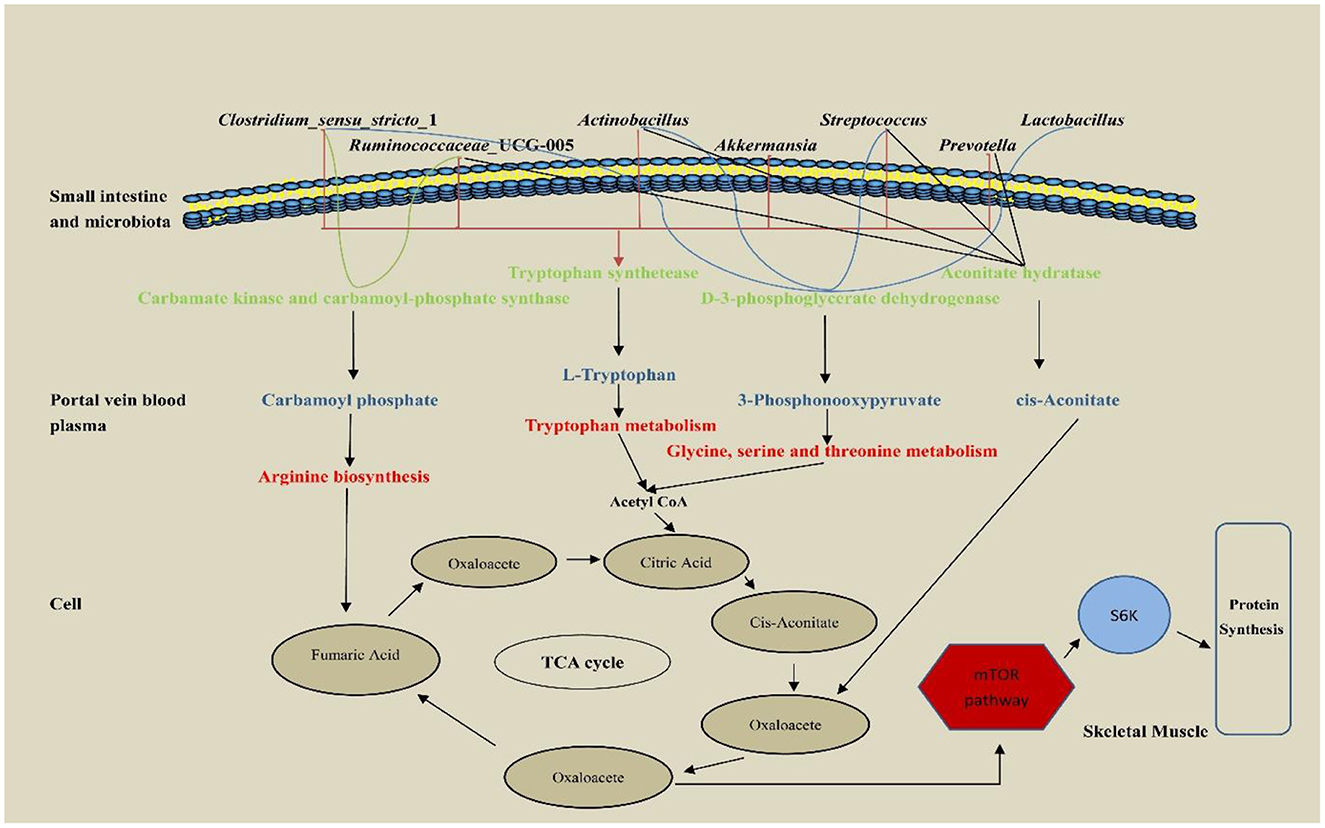
Figure 7. The pathway of hepatic portal vein differential metabolites regulatory protein synthesis of weaned piglets.
Interestingly, carbamoyl phosphate may also metabolize arginine to promote protein synthesis. A previous study has shown that arginine is the most abundant nitrogen carrier for protein synthesis in the body (Wu et al., 1999), and in mammals, arginine has the ability to stimulate the secretion of growth factors in the anterior pituitary gland (Flynn et al., 2002), so young animals are more sensitive to arginine requirements that are particularly high (Watanabe et al., 2009). In addition, dietary arginine supplementation can improve nitrogen use efficiency and enhance host protein synthesis (Guoyao et al., 2007), which is consistent with our results. Therefore, the increased nitrogen metabolism in the co-administration of L. rhamnosus GG ATCC53103 and L. plantarum JL01 in weaned piglets may be mediated by L-Tryptophan, 3-Phosphonooxypyruvate, cis-Aconitate and Carbamoyl phosphate. The increase of these metabolites may be related to the functional genes contained in the significantly increased flora.
5. Conclusion
In sum, oral administration of L. plantarum JL01 and L. rhamnosus GG ATCC53103 to weaned piglets increased apparent nitrogen digestion. We found that dietary supplementation of L. plantarum JL01 and L. rhamnosus GG ATCC53103 improved the gut microbial diversity of weaned piglets. With the help of functional genes encoded by dominant intestinal bacteria, the production of dominant metabolites L-tryptophan 3-Phosphonooxypyruvate, cis-Aconitate, and Carbamoyl phosphate is promoted, and these metabolites promote the TCA cycle in different ways, which may lead to the increase of AKG, and then play a positive regulatory role in skeletal muscle protein deposition. Moreover, L-tryptophan has a positive regulatory effect on nitrogen metabolism. This finding further highlights the importance of gut microbiota for nitrogen metabolism and demonstrates an effect of L. plantarum JL01 and L. rhamnosus GG ATCC53103 to promote nitrogen metabolism in weaned piglets.
Data availability statement
The data presented in the study are deposited in the figshare repository, accession number 10.6084/m9.figshare.23537733.
Ethics statement
The animal study was reviewed and approved by the Ethics Committee of Jilin Agricultural University.
Author contributions
FH, XJ, TG, SS, and HS designed. FH wrote this manuscript. CW, JH, LZ, YZ, NB, and LP were involved in modifying the manuscript. HS supervised the writing of this manuscript. All authors read and approved the final version of the manuscript.
Funding
This study was supported by the National Key Research and Development Plan of China (2017YFD0500506) and the Natural Science Foundation of Jilin Province (20210101019JC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bergen, W. G., and Wu, G. (2009). Intestinal nitrogen recycling and utilization in health and disease. J. Nutr. 139, 821–825. doi: 10.3945/jn.109.104497
Bishu, S. (2016). Sensing of nutrients and microbes in the gut. Curr. Opin. Gastroenterol. 32, 86–95. doi: 10.1097/MOG.0000000000000246
Cai, X., Zhu, C., Xu, Y., Jing, Y., Yuan, Y., Wang, L., et al. (2020). Retraction note: alpha-ketoglutarate promotes skeletal muscle hypertrophy and protein synthesis through akt/mtor signaling pathways. Sci Rep. 10, 18721. doi: 10.1038/s41598-020-72330-x
Cani, P. D., and Delzenne, N. M. (2009). The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Design. 15, 1546–1558. doi: 10.2174/138161209788168164
Cogo, C., Pérez-Giménez, J., Rajeswari, C. B., Luna, M. F., and Lodeiro, A. R. (2018). Induction by bradyrhizobium diazoefficiens of different pathways for growth in d-mannitol or l-arabinose leading to pronounced differences in co2 fixation, o2 consumption, and lateral-flagellum production. Front. Microbiol. 9, 1189. doi: 10.3389/fmicb.2018.01189
Dai, Z. L., Wu, G., and Zhu, W. Y. (2011). Amino acid metabolism in intestinal bacteria: links between gut ecology and host health. Front. Biosci. 16, 1768–1786. doi: 10.2741/3820
Davila, A. M., Blachier, F., Gotteland, M., Andriamihaja, M., Benetti, P. H., Sanz, Y., et al. (2013). Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host. Pharmacol. Res. 68, 95–107. doi: 10.1016/j.phrs.2012.11.005
Ding, H., Zhao, J., Liu, H., Wang, J., and Lu, W. (2020). Bmal1 knockdown promoted apoptosis and reduced testosterone secretion in tm3 leydig cell line. Gene. 747, 144672. doi: 10.1016/j.gene.2020.144672
Dukes, A., Davis, C., El, R. M., Upadhyay, S., Mork, S., Arounleut, P., et al. (2015). The aromatic amino acid tryptophan stimulates skeletal muscle igf1/p70s6k/mtor signaling in vivo and the expression of myogenic genes in vitro. Nutrition. 31, 1018–1024. doi: 10.1016/j.nut.2015.02.011
Feyera, T., Zhou, P., Nuntapaitoon, M., Sorensen, K. U., Krogh, U., Bruun, T. S., et al. (2019). Mammary metabolism and colostrogenesis in sows during late gestation and the colostral period. J. Anim. Sci. 97, 231–245. doi: 10.1093/jas/sky395
Flynn, N. E., Meininger, C. J., Haynes, T. E., and Wu, G. (2002). The metabolic basis of arginine nutrition and pharmacotherapy. Biomed. Pharmacother. 56, 427–438. doi: 10.1016/S0753-3322(02)00273-1
Geng, T., He, F., Su, S., Sun, K., Zhao, L., Zhao, Y., et al. (2021). Probiotics lactobacillus rhamnosus gg atcc53103 and lactobacillus plantarum jl01 induce cytokine alterations by the production of tcda, dha, and succinic and palmitic acids, and enhance immunity of weaned piglets. Res. Vet. Sci. 137, 56–67. doi: 10.1016/j.rvsc.2021.04.011
Grice, E. A., Kong, H. H., Conlan, S., Deming, C. B., Davis, J., Young, A. C., et al. (2009). Topographical and temporal diversity of the human skin microbiome. Science. 324, 1190–1192. doi: 10.1126/science.1171700
Guoyao, W., Fuller, W. B., Teresa, A. D., Laurie, A. J., Greg, A. J., Sung, W. K., et al. (2007). Important roles for the arginine family of amino acids in swine nutrition and production. Livest. Sci. 112, 3. doi: 10.1016/j.livsci.2007.07.003
Hooper, L. V., Midtvedt, T., and Gordon, J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307. doi: 10.1146/annurev.nutr.22.011602.092259
Jardon, K. M., Canfora, E. E., Goossens, G. H., and Blaak, E. E. (2022). Dietary macronutrients and the gut microbiome: a precision nutrition approach to improve cardiometabolic health. Gut. 71, 1214–1226. doi: 10.1136/gutjnl-2020-323715
Johnson, C. H., Ivanisevic, J., and Siuzdak, G. (2016). Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. 17, 25. doi: 10.1038/nrm.2016.25
Khanian, M., Karimi-Torshizi, M. A., and Allameh, A. (2019). Alleviation of aflatoxin-related oxidative damage to liver and improvement of growth performance in broiler chickens consumed lactobacillus plantarum 299v for entire growth period. Toxicon. 158, 57–62. doi: 10.1016/j.toxicon.2018.11.431
Krajmalnik-Brown, R., Ilhan, Z. E., Kang, D. W., and DiBaise, J. K. (2012). Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. 27, 201–214. doi: 10.1177/0884533611436116
Kuo, H. K., Liao, K. C., Leveille, S. G., Bean, J. F., Yen, C. J., Chen, J. H., et al. (2007). Relationship of homocysteine levels to quadriceps strength, gait speed, and late-life disability in older adults. J. Gerontol. Ser. A-Biol. Sci. Med. Sci. 62, 434–439. doi: 10.1093/gerona/62.4.434
LeBlanc, J. G., Milani, C., de Giori, G. S., Sesma, F., van Sinderen, D., and Ventura, M. (2013). Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr. Opin. Biotechnol. 24, 160–168. doi: 10.1016/j.copbio.2012.08.005
Lin, R., Liu, W., Piao, M., and Zhu, H. (2017). A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids. 49, 2083–2090. doi: 10.1007/s00726-017-2493-3
Matthews, D. E. (2020). Review of lysine metabolism with a focus on humans. J. Nutr. 150, 2548S−2555S. doi: 10.1093/jn/nxaa224
McCann, J. C., Elolimy, A. A., and Loor, J. J. (2017). Rumen microbiome, probiotics, and fermentation additives. Vet. Clin. N. Am.-Food Anim. Pract. 33, 539–553. doi: 10.1016/j.cvfa.2017.06.009
Metges, C. C. (2000). Contribution of microbial amino acids to amino acid homeostasis of the host. J. Nutr. 130, 1857S−1864S. doi: 10.1093/jn/130.7.1857S
Metges, C. C., and Petzke, K. J. (2005). Utilization of essential amino acids synthesized in the intestinal microbiota of monogastric mammals. Br. J. Nutr. 94, 621–622. doi: 10.1079/BJN20051509
Millet, S., Aluwe, M., Van den Broeke, A., Leen, F., Boever, D. E., De Campeneere, J., et al. (2018). Review: pork production with maximal nitrogen efficiency. Animal. 12, 1060–1067. doi: 10.1017/S1751731117002610
National Research Council. (2012). Nutrient Requirements of Swine: Eleventh Revised Edition. Washington, DC: The National Academies Press.
Nicholson, J. K., Holmes, E., Kinross, J., Burcelin, R., Gibson, G., Jia, W., et al. (2012). Host-gut microbiota metabolic interactions. Science. 336, 1262–1267. doi: 10.1126/science.1223813
Rao, Z., Li, J., Shi, B., Zeng, Y., Liu, Y., Sun, Z., et al. (2021). Dietary tryptophan levels impact growth performance and intestinal microbial ecology in weaned piglets via tryptophan metabolites and intestinal antimicrobial peptides. Animals. 11, 817. doi: 10.3390/ani11030817
Roager, H. M., and Licht, T. R. (2018). Microbial tryptophan catabolites in health and disease. Nat. Commun. 9, 3294. doi: 10.1038/s41467-018-05470-4
Ruan, Z., Yang, Y., Wen, Y., Zhou, Y., Fu, X., Ding, S., et al. (2014). Metabolomic analysis of amino acid and fat metabolism in rats with l-tryptophan supplementation. Amino Acids. 46, 2681–2691. doi: 10.1007/s00726-014-1823-y
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Sterndale, S. O., Miller, D. W., Mansfield, J. P., Kim, J. C., and Pluske, J. R. (2020). Increasing dietary tryptophan in conjunction with decreasing other large neutral amino acids increases weight gain and feed intake in weaner pigs regardless of experimental infection with enterotoxigenic escherichia coli. J. Anim. Sci. 98, 190. doi: 10.1093/jas/skaa190
Sun, W., Chen, W., Meng, K., Cai, L., Li, G., Li, X., et al. (2023). Dietary supplementation with probiotic bacillus licheniformis s6 improves intestinal integrity via modulating intestinal barrier function and microbial diversity in weaned piglets. Biology-Basel. 12, 238. doi: 10.3390/biology12020238
Sun, Y., Sun, Y., Shi, Z., Liu, Z., Zhao, C., Lu, T., et al. (2019). Gut microbiota of wild and captive alpine musk deer (moschus chrysogaster). Front. Microbiol. 10, 3156. doi: 10.3389/fmicb.2019.03156
Torrallardona, D., Harris, C. I., and Fuller, M. F. (2003). Lysine synthesized by the gastrointestinal microflora of pigs is absorbed, mostly in the small intestine. Am. J. Physiol.-Endocrinol. Metab. 284, E1177–E1180. doi: 10.1152/ajpendo.00465.2002
Truong, D. T., Franzosa, E. A., Tickle, T. L., Scholz, M., Weingart, G., and Pasolli, E., et al. (2015). Metaphlan2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 12, 902–903. doi: 10.1038/nmeth.3589
Utzschneider, K. M., Kratz, M., Damman, C. J., and Hullar, M. (2016). Mechanisms linking the gut microbiome and glucose metabolism. J. Clin. Endocrinol. Metab. 101, 1445–1454. doi: 10.1210/jc.2015-4251
van der Wielen, N., Moughan, P. J., and Mensink, M. (2017). Amino acid absorption in the large intestine of humans and porcine models. J. Nutr. 147, 1493–1498. doi: 10.3945/jn.117.248187
Wang, H., and Kim, I. H. (2021). Evaluation of dietary probiotic (lactobacillus plantarum bg0001) supplementation on the growth performance, nutrient digestibility, blood profile, fecal gas emission, and fecal microbiota in weaning pigs. Animals. 11, 1–14. doi: 10.3390/ani11082232
Wang, Q., Sun, Q., Qi, R., Wang, J., Qiu, X., Liu, Z., et al. (2019). Effects of lactobacillus plantarum on the intestinal morphology, intestinal barrier function and microbiota composition of suckling piglets. J. Anim. Physiol. Anim. Nutr. 103, 1908–1918. doi: 10.1111/jpn.13198
Watanabe, K., Arozal, W., Tanaka, H., Ma, M., Satoh, S., Veeraveedu, P. T., et al. (2009). Beneficial effect of food substitute containing l-arginine, omega-3 poly unsaturated fatty acid, and ribonucleic acid in preventing or improving metabolic syndrome: a study in 15 overweight patients and a study of fatty acid metabolism in animals. J. Clin. Biochem. Nutr. 44, 266–274. doi: 10.3164/jcbn.08-241
Wernerman, J., and Hammarqvist, F. (1999). Glutamine: a necessary nutrient for the intensive care patient. Int. J. Colorectal Dis. 14, 137–142. doi: 10.1007/s003840050199
Wu, G., Ott, T. L., Knabe, D. A., and Bazer, F. W. (1999). Amino acid composition of the fetal pig. The J. Nutr. 129, 1031 doi: 10.1093/jn/129.5.1031
Yang, Y. X., Dai, Z. L., and Zhu, W. Y. (2014). Important impacts of intestinal bacteria on utilization of dietary amino acids in pigs. Amino Acids. 46, 2489–2501. doi: 10.1007/s00726-014-1807-y
Yi, H., Yang, G., Xiong, Y., Wu, Q., Xiao, H., Wen, X., et al. (2019). Integrated metabolomic and proteomics profiling reveals the promotion of lactobacillus reuteri lr1 on amino acid metabolism in the gut-liver axis of weaned pigs. Food Funct. 10, 7387–7396. doi: 10.1039/C9FO01781J
Zhao, J., Zhang, X., Liu, H., Brown, M. A., and Qiao, S. (2019). Dietary protein and gut microbiota composition and function. Curr. Protein Pept. Sci. 20, 145–154. doi: 10.2174/1389203719666180514145437
Zhao, L., Guo, H., and Sun, H. (2020). Effects of low-protein diet supplementation with alpha-ketoglutarate on growth performance, nitrogen metabolism and mtor signalling pathway of skeletal muscle in piglets. J. Anim. Physiol. Anim. Nutr. 104, 300–309. doi: 10.1111/jpn.13230
Keywords: metabolites, nitrogen metabolism, weaned piglets, Lactobacillus rhamnosus GG ATCC53103, Lactobacillus plantarum JL01
Citation: He F, Jin X, Wang C, Hu J, Su S, Zhao L, Geng T, Zhao Y, Pan L, Bao N and Sun H (2023) Lactobacillus rhamnosus GG ATCC53103 and Lactobacillus plantarum JL01 improved nitrogen metabolism in weaned piglets by regulating the intestinal flora structure and portal vein metabolites. Front. Microbiol. 14:1200594. doi: 10.3389/fmicb.2023.1200594
Received: 05 April 2023; Accepted: 30 May 2023;
Published: 29 June 2023.
Edited by:
Hao Zhong, Zhejiang University of Technology, ChinaReviewed by:
Yu Pi, Feed Research Institute (CAAS), ChinaMiaomiao Bai, Institute of Subtropical Agriculture (CAS), China
Liping Gan, Henan University of Technology, China
Copyright © 2023 He, Jin, Wang, Hu, Su, Zhao, Geng, Zhao, Pan, Bao and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Sun, aHVpMTY4OEAxNjMuY29t