 Lingzhi Dong1,2
Lingzhi Dong1,2 Yaben Li1,2
Yaben Li1,2 Caixin Yang1,2Jian Gong1,2
Caixin Yang1,2Jian Gong1,2 Wentao Zhu3
Wentao Zhu3 Yuyuan Huang2Mimi Kong1,2Lijun Zhao1,2Feifei Wang1,2
Yuyuan Huang2Mimi Kong1,2Lijun Zhao1,2Feifei Wang1,2 Shan Lu1,2,4
Shan Lu1,2,4 Ji Pu1,2*
Ji Pu1,2* Jing Yang1,2,4*
Jing Yang1,2,4*- 1Department of Epidemiology, School of Public Health, Shanxi Medical University, Taiyuan, China
- 2State Key Laboratory of Infectious Disease Prevention and Control, Chinese Center for Disease Control and Prevention, National Institute for Communicable Disease Control and Prevention, Beijing, China
- 3Department of Infectious Diseases and Clinical Microbiology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China
- 4Research Units of Discovery of Unknown Bacteria and Function, Chinese Academy of Medical Sciences, Beijing, China
Introduction: Ticks and fleas, as blood-sucking arthropods, carry and transmit various zoonotic diseases. In the natural plague foci of China, monitoring of Yersinia pestis has been continuously conducted in Marmota himalayana and other host animals, whereas other pathogens carried by vectors are rarely concerned in the Qinghai-Tibet Plateau.
Methods: In this study, we investigated the microbiota of ticks and fleas sampling from M. himalayana in the Qinghai-Tibet Plateau, China by metataxonomics combined with metagenomic methods.
Results: By metataxonomic approach based on full-length 16S rDNA amplicon sequencing and operational phylogenetic unit (OPU) analyses, we described the microbiota community of ticks and fleas at the species level, annotated 1,250 OPUs in ticks, including 556 known species and 492 potentially new species, accounting for 48.50% and 41.71% of the total reads in ticks, respectively. A total of 689 OPUs were detected in fleas, consisting of 277 known species (40.62% of the total reads in fleas) and 294 potentially new species (56.88%). At the dominant species categories, we detected the Anaplasma phagocytophilum (OPU 421) and potentially pathogenic new species of Wolbachia, Ehrlichia, Rickettsia, and Bartonella. Using shotgun sequencing, we obtained 10 metagenomic assembled genomes (MAGs) from vector samples, including a known species (Providencia heimbachae DFT2), and six new species affliated to four known genera, i.e., Wolbachia, Mumia, Bartonella, and Anaplasma. By the phylogenetic analyses based on full-length 16S rRNA genes and core genes, we identified that ticks harbored pathogenic A. phagocytophilum. Moreover, these potentially pathogenic novel species were more closely related to Ehrlichia muris, Ehrlichia muris subsp. eauclairensis, Bartonella rochalimae, and Rickettsia limoniae, respectively. The OPU 422 Ehrlichia sp1 was most related to Ehrlichia muris and Ehrlichia muris subsp. eauclairensis. The OPU 230 Bartonella sp1 and Bartonella spp. (DTF8 and DTF9) was clustered with Bartonella rochalimae. The OPU 427 Rickettsia sp1 was clustered with Rickettsia limoniae.
Discussion: The findings of the study have advanced our understanding of the potential pathogen groups of vectors in marmot (Marmota himalayana) in the Qinghai-Tibet Plateau.
Introduction
Vector-borne diseases (VBDs) are infections that are primarily transmitted through an invertebrate, generally, insects. With the global climatic alteration and human activity amplification, the VBDs pose a significant burden on public health and economic development. It is estimated that 80% of the population in the world is at risk of two or more VBDs. They account for 17% of all infectious diseases and cause more than 700,000 deaths annually (Golding et al., 2015; WHO, 2017; Chala and Hamde, 2021). As the largest developing country in the world, China has made great progress in the prevention and treatment of infectious diseases. However, emerging infectious diseases are a new challenge for China (Wang et al., 2008).
Ticks are obligate blood-sucking arthropods that can parasitize vertebrates and transmit a wide variety of infectious microorganisms such as viruses, bacteria, spirochetes, protozoans, and parasites (Parola, 2004). As a major contributor to vector-borne diseases, a huge number of novel tick-borne disease agents have been reported such as borreliosis, ehrlichioses, anaplasmosis, and tick-borne rickettsial diseases. Among the 109 tick-borne pathogens identified in China (Zhao et al., 2021) between 1950 and 2018, 65 were newly identified. Plenty of human cases have been confirmed for tick-borne infections with Borrelia, Anaplasmataceae, Babesia, and spotted fever group Rickettsiae (Fang et al., 2015). Fleas are blood-sucking arthropods that parasitize mammals and birds. They are known for transmitting flea-borne Yersinia pestis causing plague (Zeppelini et al., 2016). In recent years, increasing numbers of flea-borne diseases, including murine typhus, epidemic typhus, and flea-borne spotted fever have been reported across the world (Bitam et al., 2010). Fleas have also been reported to transmit Bartonella, including Bartonella henselae, the agent cat-scratch disease (Chomel et al., 2006; Gutiérrez et al., 2015).
Marmots (Marmota himalayana) were identified as the predominant host of Yersinia pestis in the Qinghai-Tibet Plateau, China (Ben-Ari et al., 2012). In the natural plague foci of China, such as Qinghai and Xinjiang, marmots and other animal hosts have been continuously monitored under China's Plague Surveillance Program (Qin et al., 2022). Our previous studies have found the possibility of marmots to be hosts and reservoirs for pathogens including tick-born encephalitis virus (Dai et al., 2018) and delta-Coronaviruses (Zhu et al., 2021). In recent years, there have been some studies on vector-borne pathogens in this area, such as the research of spotted fever group Rickettsia infecting ticks, yaks, and Tibetan sheep (He et al., 2021b) and the molecular detection of Anaplasm, Babesia, and Theileria in yaks and Tibetan sheep (He et al., 2021a). However, the characterization of species-level microbiota of vectors, especially in fleas, is lacking in marmots. In the present study, we described the microbiota community of ticks and fleas sampling from marmots in Qinghai-Tibet Plateau, China. We aimed to discover the profile of tick-borne and flea-borne pathogens or potentially new pathogenic bacterial species, providing etiological information for vector infectious disease prevention and control.
Materials and methods
Samples collection
The ticks and fleas were collected from M. himalayana in Tongren, Huangnan Tibetan Autonomous Prefecture (Altitude: 3273 m; N: 35°49′, E: 102°28′), Qinghai Province, China, from July to September 2018. Marmots were collected for routine surveillance and screened for plague in Autumn 2018. All the marmots were captured and preliminarily detected Y. pestis F1 antibody by the colloidal gold method. Then, ticks and fleas parasitized on the surface of marmots were collected and stored on the principle of one marmot per frozen tube. All the vectors were transported to the local lab, and subsequently, ticks and fleas were identified as adults by professionals using a DM-500 binocular biological microscope, referring to the reference book of Cai et al. (1997). All the samples were transported to our laboratory in Beijing and stored at −80°C prior to DNA extraction. The sampling work was conducted by Qinghai Institute for Endemic Disease Prevention and Control for seasonal vector surveillance. The ethical practice was approved by the Ethical Committee of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, China (No. ICDC-2018004).
DNA extraction and full-length 16S rDNA amplification sequencing
A total of 25 ticks and 24 fleas were enrolled in our experiment, which were divided into eight pools for each sample ticks and fleas according to the engorgement level (Supplementary Table S1). Ticks and fleas were surface-sterilized in 10% bleach and then washed twice in 99% ethanol to remove bacteria from the external body. Sixteen vector samples and two blank control groups were homogenized (Liu et al., 2017; Adegoke et al., 2020) separately in the PBS solution by the TissueLyser II (Qiagen, Germany) at 24 Hz, 37°C for 2 min, three times. The TissueLyser II system was achieved through high-speed shaking with beads, which beat and grid samples, and also simultaneously homogenized samples to facilitate subsequent purification procedures. The total DNA was then extracted from the supernatant using the QIAamp DNA Mini Kit (Qiagen, Germany). The quality of DNA extraction was checked by NanoDrop 2000 (Thermo Fisher Scientific, USA). The almost full-length 16S rRNA gene was amplified using the universal primers 27F/1492R with 16 nt symmetric (reverse complement) barcodes tagged at the 5′ end, as described previously (Yang et al., 2020). The libraries of PCR products were generated, followed by sequencing on the PacBio Sequel platform at Tianjin Biochip Corporation, China.
The raw sequences generated were filtered and cleaned using the previous pipeline described by Meng et al. (2017) and Yang et al. (2020), i.e., the raw sequencing data were initially processed based on the PacBio SMRT Link (version 6.0.0) pipeline, QIIME (Bolyen et al., 2019) was used to split data and filter out adapters, primers, ambiguous bases, and chimeras were detected and removed using the UCHIME (Edgar et al., 2011) algorithm implemented in USEARCH (version 11.0.667; option: -uchime_ref -strand plus -nonchimeras) with the RDP Gold reference database.
Species-level taxonomy by operational phylogenetic unit analyses
High-quality sequences of the full-length 16S rRNA gene were clustered into operational taxonomic units (OTUs) with an identity threshold of 98.7% by the USEARCH pipeline. Then, all the representative sequences for each OTU were grouped into operational phylogenetic units (OPUs) using the tool Arb by the visual inspection of the final phylogenetic trees. The reference database used was LTP132 (which was the latest version at the time of this study). The detailed analysis process and classification criteria of OPU were shown in our previous studies (Meng et al., 2017; Bai et al., 2018; Yang et al., 2020). In brief, each OPU consisting of one or more OTUs can be equated to a species-level taxon. When an identity value of OTU representatives with a type strain was >98.7%, it was identified as known species. A putative new species within a specific genus was defined when the identity values of OTU representatives with the closest type strain were <98.7 and >94.5%. When OTUs represent a sequence whose membership position was not clear but was closely affiliated to a known genus, family, or higher taxa, it was listed as a potential new “higher taxa.” Taxa around the OTU sequences that were represented by a large number of uncultured organisms and were not attached to any of the internal taxa were referred to as “uncultured taxa” categories.
Shotgun metagenomic sequencing, assembly, and contig binning
The DNA material used for shotgun metagenomic sequencing was the same as that described in 16S rRNA-based amplicon sequencing section. DNA degradation, contamination, and concentration were monitored by agarose gel electrophoresis and Qubit® 2.0 Fluorometer. Then, DNA libraries were constructed, purified, analyzed, quantified, and sequenced on the Illumina PE150 platform (150-bp, paired-end) at the Novogene Co., Ltd. (Beijing, China).
The raw paired-end reads were quality-filtered to remove the adapter and low-quality sequences using the Readfq (V8, https://github.com/cjfields/readfq). Then, the reads mapped to the host genomic DNA by Bowtie2 (version 2.2.4, http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) with the following parameter settings: –end-to-end, –sensitive, -I 200, and -X 400 were filtered out. The sequence data were assembled individually by MEGAHIT (version 1.0.4, https://github.com/voutcn/megahit) using the parameter settings: –presets meta-large (–end-to-end, –sensitive, -I 200, -X 400). Next, the assembled contigs were binned with three different tools using the default settings in MetaWRAP (Uritskiy et al., 2018) (version 1.3.2) pipeline, and the completeness and contamination of the reassembled metagenomic assembled genomes (MAGs) were estimated by CheckM (Parks et al., 2015) (version 1.0.13). The reassembled MAGs were classified using the Genome Taxonomy Database Toolkit (GTDB-Tk version 1.3.0) based on the GTDB RefSeq_95 database. The taxonomic assignment was defined by a combination of the placement of the query genome in the GTDB reference tree, the RED of the query genome, and Average Nucleotide Identity (ANI) (Ciufo et al., 2018; Jain et al., 2018) to reference genomes.
Phylogenetic analyses of the potential vector-borne pathogens
The phylogenetic analyses of full-length 16S rRNA gene amplicon sequencing were performed with MEGA X (https://www.megasoftware.net/) using the maximum-likelihood (ML) algorithm. Multiple alignments were performed with the program CLUSTAL_W, and all cases were evaluated based on 1,000 bootstrap replicates. The phylogenetic analyses of reassembled MAGs were performed with FastTree (Price et al., 2009) by using the neighbor-joining (NJ) algorithm, which was generated based on core genes that were extracted from the reference whole genomes and visualized in Dendroscope 3.0.
Results
Species-level microbiota of ticks and fleas
A total of 16 vector pools were sequenced, with eight pools each for ticks and fleas (Supplementary Table S1). The PacBio sequel sequencing platform rendered a total of 135,040 and 98,090 raw reads for ticks and fleas, respectively. After quality filtering and chimera removal, a total of 110,760 (80.50%) and 70,502 (71.10%) high-quality reads were obtained from ticks and fleas, with an average of 13,845.00 ± 6,588.00 per tick pool and 8,812.75 ± 2,681.68 per flea pool. The average length was 1,437.00 ± 15.07 and 1,453.63 ± 7.02 nucletide (nt) per read for ticks and fleas, respectively (Supplementary Table S2). The near full-length 16S rRNA gene sequences were clustered into 4,789 and 2,170 OTUs in respective ticks and fleas at an identity threshold of 98.7% using the USEARCH pipeline. Subsequently, a total of 1,250 and 689 operational phylogenetic units (OPUs) were obtained for ticks and fleas, respectively, with an average of 303.25 ± 164.84 per tick pool and 153.36 ± 96.37 per flea pool (Supplementary Table S3). We further classified these OPUs into four categories: (i) known species (556 OPUs, relative abundance 48.50% vs. 277 OPUs, 40.62% in ticks and fleas, respectively); (ii) potentially new species (492 OPUs, 41.71% vs. 294 OPUs, 56.88%); (iii) higher taxa (133 OPUs, 6.19% vs. 83 OPUs, 2.02%); and (iv) uncultured bacteria (69 OPUs, 3.60% vs. 35 OPUs, 0.48%; Supplementary Table S3 and Supplementary Figure S1). It can be noted that the vast majority of the flora was identified at the species level (known species and potentially new species), indicating the relative abundance of dominant taxa reached 90.21% and 97.50% in ticks and fleas, respectively.
The potential vector-borne pathogens detected in ticks and fleas at the species level
In ticks, the 556 known species and 492 putative new species affiliated to 19 phyla, 49 classes, 96 orders, 177 families, and 431 genera, accounting for 90.21% of the total reads (Figure 1A and Supplementary Figure S1). The top 10 most abundant species in ticks represented 74.76% of the total reads, ranging from 27.25% to 97.38% in individual tick pools (Figure 1C). Of these 10 species, the Anaplasma phagocytophilum (OPU 421) and Ehrlichia sp1 (OPU 422) were presented, and their relative abundance were 5.11% and 3.37%, respectively, and which were indicated by red arrows and words in Figures 1A, C. However, Bartonella sp1 (OPU 230) had a relatively lower abundance of 0.01%. In flea samples, most of the reads (97.50%) were annotated to the species level, including 277 known species and 294 putative new species (Figure 1B and Supplementary Figure S1). The top 10 species accounted for 90.51% of the total reads, ranging from 84.51% to 96.07% in individual flea pools (Figure 1D). The potential novel species affiliated to vector-borne pathogenic genera including Rickettsia and Bartonella were founded in these top 10 species, and their relative abundance were 3.01% and 0.75%, respectively.
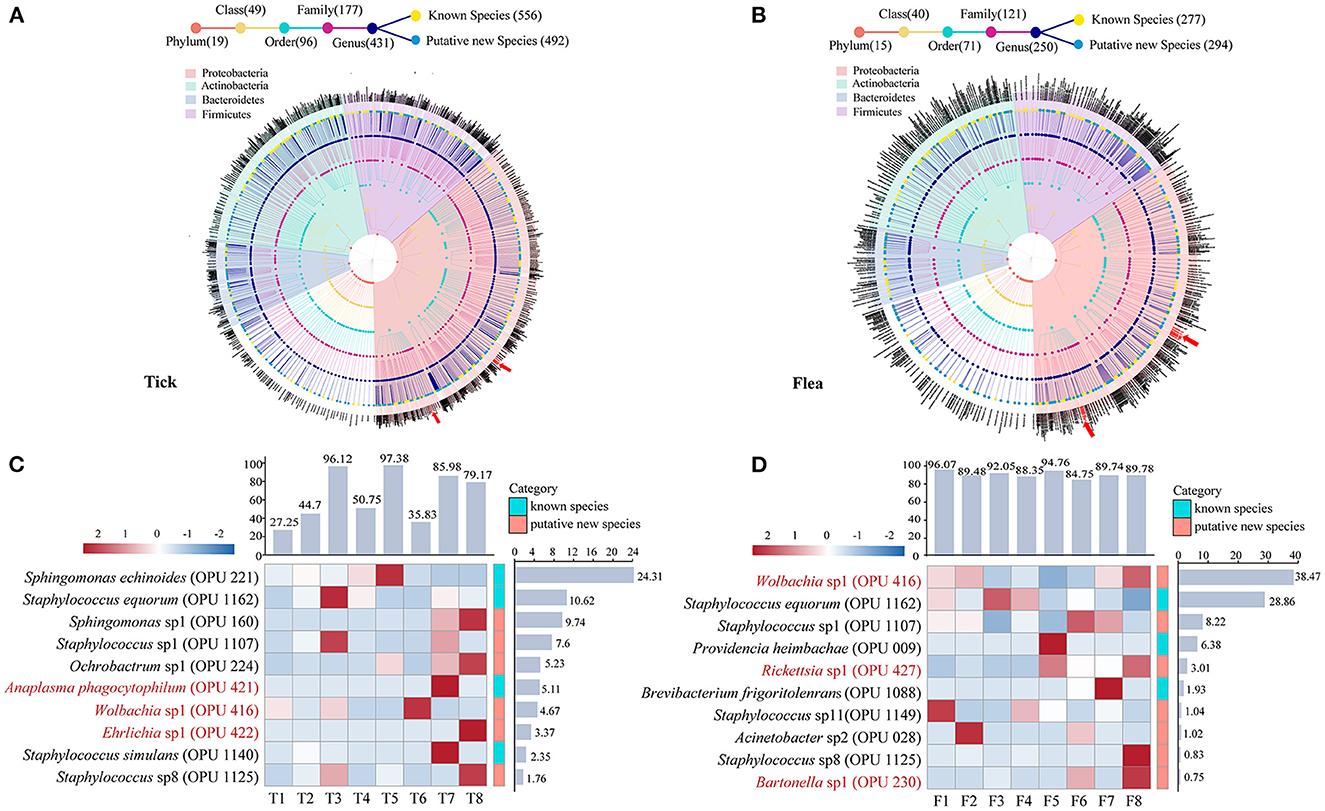
Figure 1. Taxonomic profile and dominant taxa in ticks and fleas at species level. Taxonomic trees of (A) 556 known species and 492 putative new species in ticks and (B) 277 known species and 294 putative new species in fleas. The heat map and relative abundance of the top ten abundant bacteria at the species level (C) in ticks and (D) in fleas. Each dot represents a taxonomic entity. From the inner to the outer circles, the taxonomic levels range from phylum to species. Dots of different colors indicate different taxonomic levels according to the color key shown. The total number at different hierarchical levels is displayed in brackets. The potential pathogens are indicated by red arrows and words in the figure. Different colored squares represent different phyla according to the color key shown.
In addition, Wolbachia were detected in the top 10 abundance at the species level in both ticks and fleas. The relative abundance of Wolbachia sp1 (OPU 416) were 4.67% in ticks and 38.47% in fleas.
Notably, the known vector-borne pathogen A. phagocytophilum and potentially novel species affiliated with vector-borne pathogenic genera, including Ehrlichia, Rickettsia, and Bartonella were detected in the dominant species categories of the microbiota community in ticks and fleas, accounting for 13.16% and 42.23% of the total reads in ticks and fleas, respectively (Figure 2). Hence, we intend to acquire the complete genomes of these potential pathogens for deep analysis.
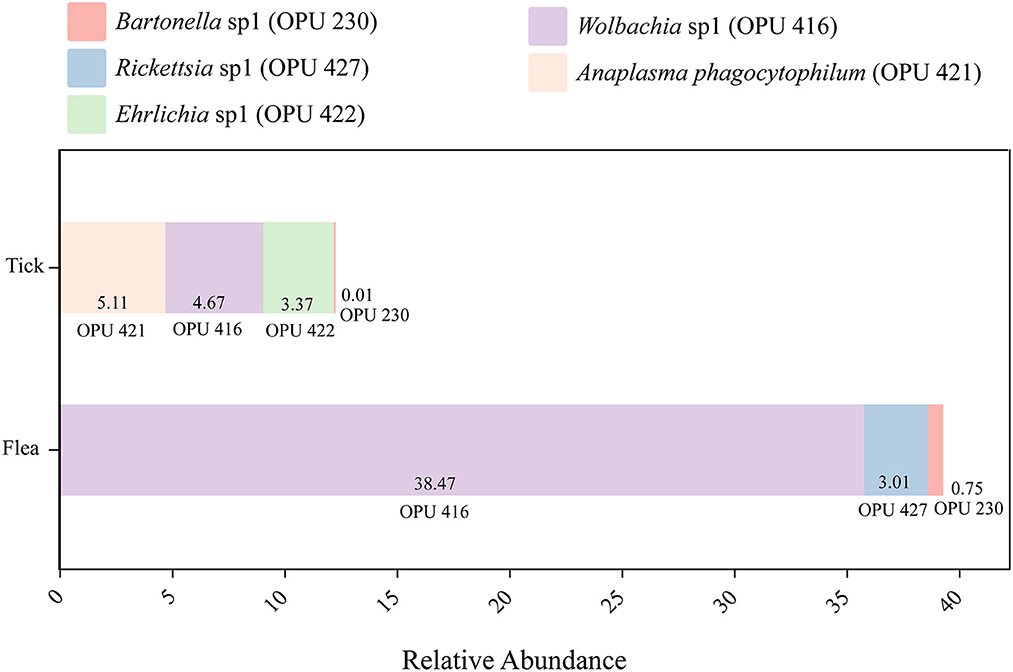
Figure 2. The relative abundance of the potential pathogens in ticks and fleas including Anaplasma phagocytophilum (OPU 421), Ehrlichia sp1 (OPU 422), Bartonella sp1 (OPU 230), Rickettsia sp1 (OPU 427), and Wolbachia sp1 (OPU 416).
The metagenomic assembled genomes of ticks and fleas
We used the same DNA samples as full-length 16S rDNA sequencing for shotgun metagenomic sequencing. An average of 7,040.24 ± 747.37 and 7,396.88 ± 627.33 Mb high-quality reads data (paired-end, 150-bp) per sample pools were generated in ticks and fleas, respectively. The de novo assembly produced a mean of 474,197 ± 48,301 and 287,426 ± 84,212 kb contigs per sample pool comprising respective ticks and fleas (Supplementary Table S4). Finally, we recovered nine MAGs (bins) from the assembled contigs in fleas and one MAG in ticks. Detailed descriptions of the 10 MAGs and their proposed nomenclatures were provided in Supplementary Table S5. A total of six out of 10 MAGs had >90% completeness and <5% contamination, while three MAGs showed >70% completeness and <10% contamination, and one MAG had <50% completeness and <15% contamination (Supplementary Table S5). Moreover, 10 MAGs (>1 Mbp) were subsequently identified using the Genome Taxonomy Database Toolkit (GTDB-Tk) combined with ANI value calculation. It was shown that one MAG (f5_bin2) belonged to known bacterial species (Providencia heimbachae DFT2; >95% ANI with a complete genome), six MAGs could be assigned to undescribed species affiliated to four known genera, including Wolbachia, Mumia, Bartonella, and Anaplasma, and the remaining three MAGs had low ANI values (<85%), demonstrating that they may belong to the family Rickettsiaceae (f5_bin1, f6_bin6) and order Acidimicrobiales (f6_bin5).
Phylogenetic analyses of the potential vector-borne pathogens
We identified the known vector-borne pathogen A. phagocytophilum and potentially new species affiliated with vector-borne pathogenic genera, including Ehrlichia, Rickettsia, and Bartonella, using full-length 16S rRNA gene sequencing. Further through metagenomic data, we recovered the MAGs of Bartonella and Anaplasma from fleas and ticks. To further confirm the taxonomic positions of these potential vector-borne pathogens, we constructed the phylogenetic trees based on the full-length 16S rRNA genes and core genes using the ML and NJ algorithms, respectively. The OPU 421 identified in ticks with 5,431 reads affiliated as an independent cluster with A. phagocytophilum (U02521, NR 044762, and AY537213) with an average 16S rRNA gene sequence similarity of 99.72%, possibly representing a known species A. phagocytophilum (Figure 3A). The phylogenomic tree based on 208 core genes showed that Anaplasma sp. DTF10 (tick_bin1) formed a branch with A. phagocytophilum (NC 021880; Figure 3B). The ANI value between Anaplasma sp. DTF10 and A. phagocytophilum NC 021880 was 89.3%, indicating that it could be assigned as a new species of Anaplasma. Phylogenetic analysis based on the 16S rRNA gene sequences revealed that OPU 422 (3,586 reads in ticks) was related to Ehrlichia muris (HM543745, NRO25962, NR121714, and U15527) and Ehrlichia muris subsp. eauclairensis (HM543745; Figure 5A) with a maximum 16S rRNA gene sequence similarity of 98.4%. Thus, OPU 422 could represent a new species belonging to the genus Ehrlichia.
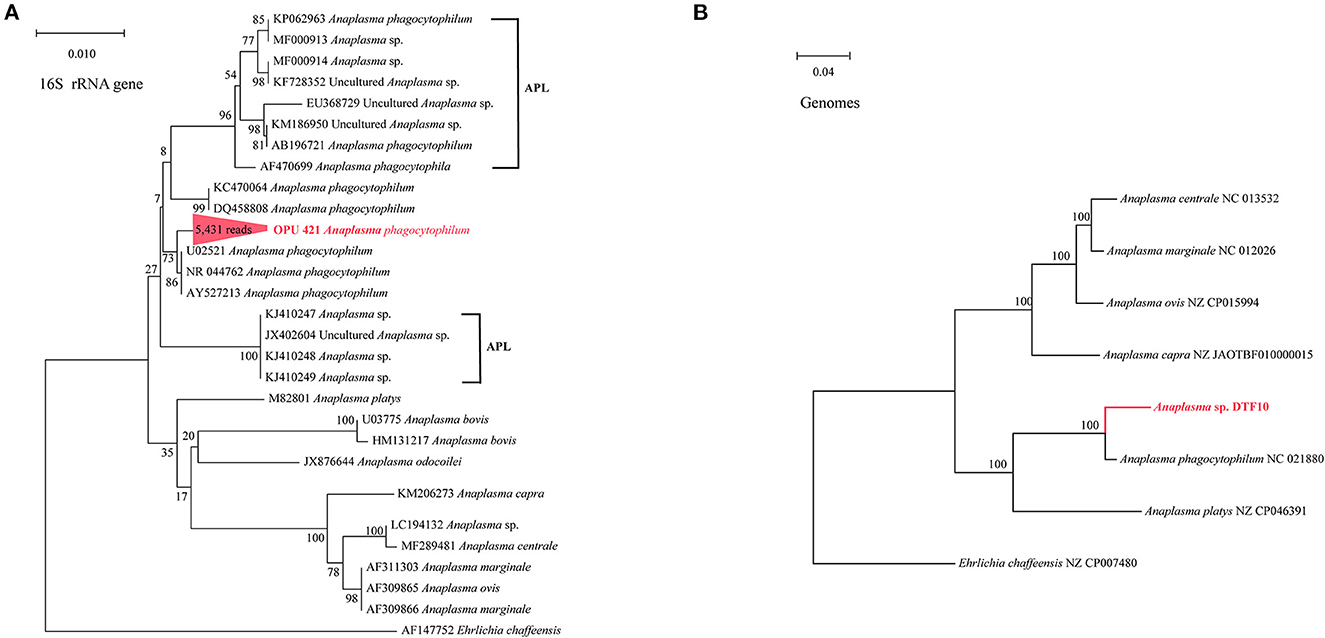
Figure 3. Phylogenetic trees based on full-length 16S rRNA gene sequences and genomic core genes revealed the positions of (A) the OPU 421 Anaplasma phagocytophilum (B) the Anaplasma sp. DTF10 (208 core genes). The phylogenetic trees were constructed using maximum-likelihood algorithms based on full-length 16S rRNA gene sequences and neighbor-joining (NJ) algorithms based on core genes. The APL represents genetically related A. phagocytophilum-like (APL) species. Trees showed the precise position of the OPUs and genomes of the identified species compared with members of the most closely related reference sequences. Red labels are the OPUs and genomes of the identified species, while known species are shown in black.
The OPU 230 (469 reads in fleas) were placed in a clade with Bartonella rochalimae (DQ683196) based on the phylogenetic analyses of the 16S rRNA gene (Figure 4A). The representative sequences of OPU 230 Bartonella sp1 showed an average identity of 98.40 ± 0.01% with B. rochalimae (DQ683196). A phylogenomic tree based on 333 core genes showed that the two MAGs (f6_bin8, f8_bin9) of Bartonella spp. (DTF8 and DTF9) formed a distinct clade and were clustered with B. rochalimae (NZ KL407337), which was consistent with the phylogenetic tree of 16S rRNA gene sequences (Figure 4B). The ANI values between Bartonella spp. (DTF8 and DTF9) and B. rochalimae (NZ KL407337) ranged from 91.79% to 94.52 %, indicating two uncultured new species within the genus Bartonella. The representative sequences of OPU 427 Rickettsia sp1 (1,897 reads in fleas) were shown to be clustered with Rickettsia limoniae (AF322442; Figure 5B), with an average identity of 97.23 ± 0.025 %, indicating that OPU 427 could be a new species affiliated to genus Rickettsia.
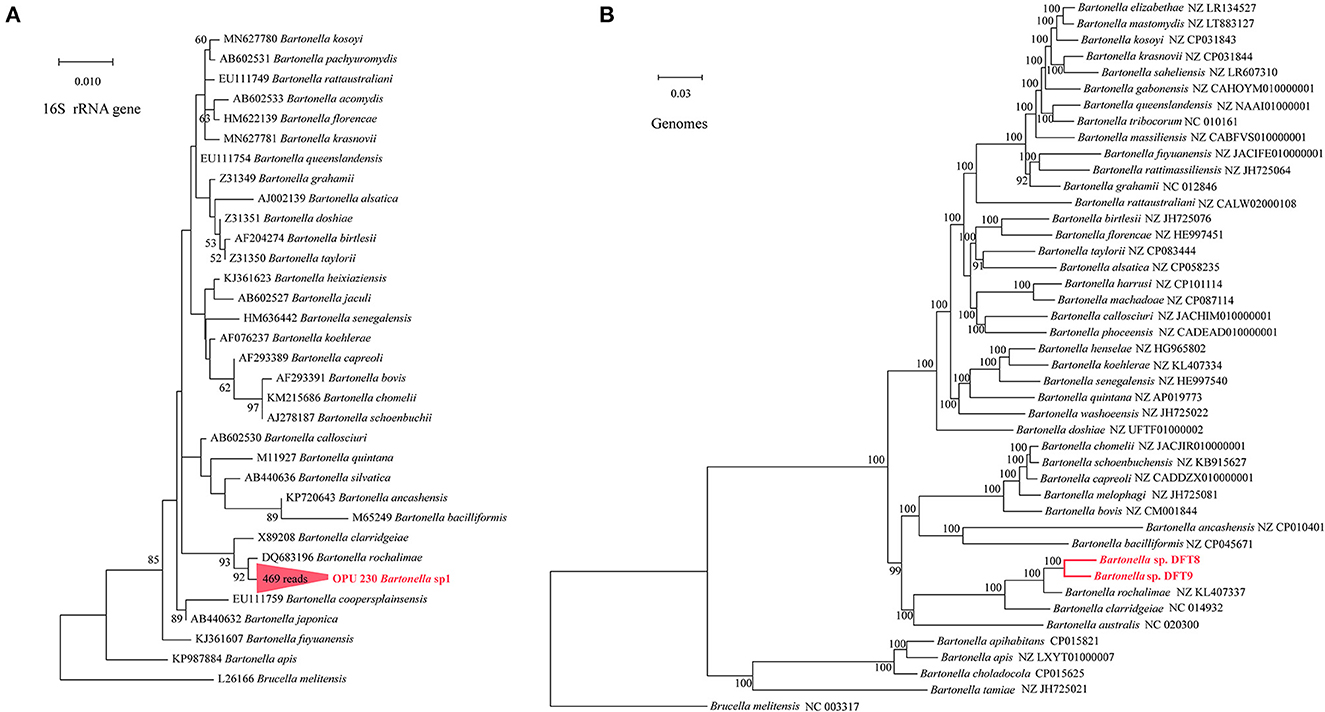
Figure 4. Phylogenetic trees based on full-length 16S rRNA gene sequences and genomic core genes revealed the positions of (A) the OPU 230 Bartonella sp1 and (B) the Bartonella sp. DTF8 and the Bartonella sp. DTF9 (333 core genes). The phylogenetic trees were constructed using the maximum-likelihood and neighbor-joining (NJ) algorithms based on full-length 16S rRNA gene sequences and core genes. Trees showed the precise position of the OPUs and genomes of the identified species compared with members of the most closely related reference sequences. Red labels are the OPUs and genomes of the identified species, while known species are shown in black.
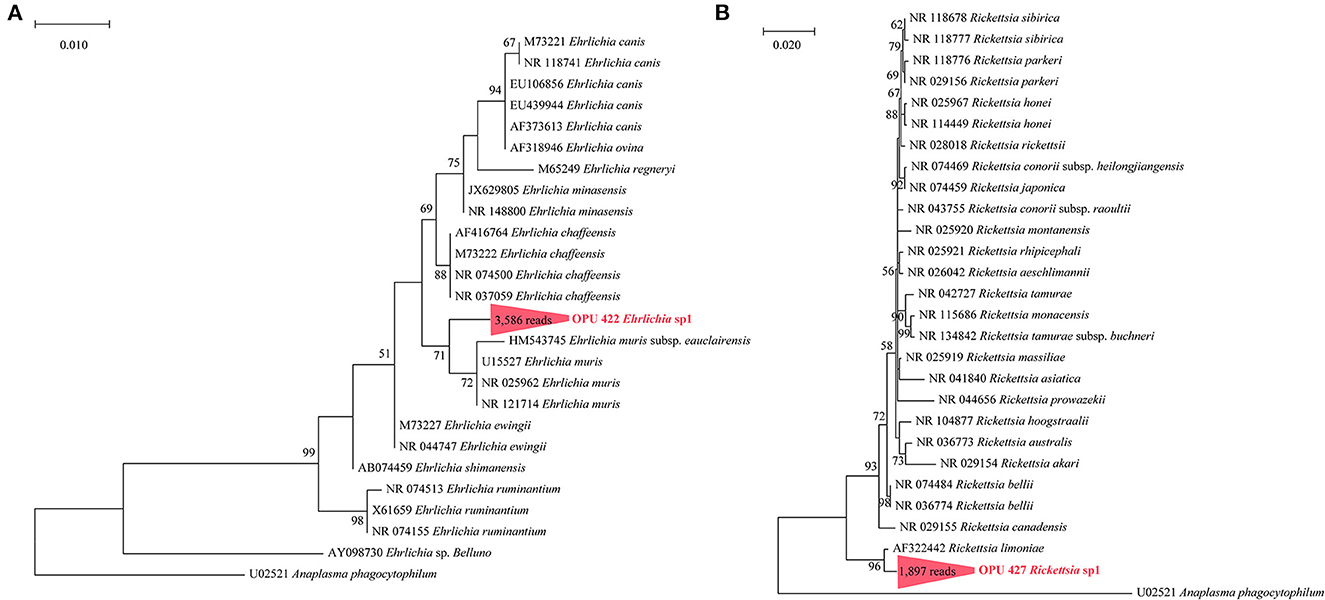
Figure 5. Phylogenetic trees based on full-length 16S rRNA gene sequences revealed the positions of (A) the OPU 422 Ehrlichia sp1 and (B) the OPU 427 Rickettsia sp1. The phylogenetic trees was constructed using a maximum-likelihood algorithm based on full-length 16S rRNA gene sequences. Trees showed the precise position of the OPUs of the identified species compared with members of the most closely related reference sequences. Red labels are the OPUs of the identified species, while known species are shown in black.
Discussion
In the present study, we have described the microbiota community in ticks and fleas collected from M. himalayana in Qinghai Tibet Plateau, China. By using the metataxonomic analyses combined with shotgun metagenomics, we obtained species-level microbiota profiles of ticks and fleas, with 90.21% and 97.50% of the total reads annotated at the species level (known species and potentially new species), respectively. The metataxonomic method based on the full-length 16S rRNA gene amplicon sequencing had been applied in the investigation of the microbiota in vultures, Tibet antelopes, and human gut microbiota (Meng et al., 2017; Bai et al., 2018; Yang et al., 2020) in our previous studies. We identified a total of 1,250 and 689 OPUs (species-level taxon) in ticks and fleas, respectively. The major bacterial at phylum-level microbiota of ticks and fleas in our study included Proteobacteria and Firmicutes, followed by Cyanobacteria and Actinobacteria, which was consistent with the previous studies of fleas (Supplementary Figure 2; Hawlena et al., 2013), ticks (Andreotti et al., 2011; Thapa et al., 2019), and other vectors (Minard et al., 2013; Yun et al., 2014).
Notably, at the species level, we detected the known vector-borne pathogen A. phagocytophilum (OPU 421) and several novel species affiliated with vector-borne pathogenic genera, Ehrlichia, Bartonella, and Rickettsia, in the present study and performed deep analysis combined with metagenomic assembly and binning. The A. phagocytophilum, known as the causative agent of anaplasmosis, is responsible for approximately 6,000 cases of human granulocytic anaplasmosis per year in the USA (Bakken and Dumler, 2015). Previous studies have reported that the A. phagocytophilum has been divided into pathogenic A. phagocytophilum and the closely related nonpathogenic A. phagocytophilum-like (APL) species (Ben Said et al., 2017; Seo et al., 2018; Yan et al., 2022). From the phylogenetic analyses based on 16S rRNA gene sequences, the OPU 421 identified ticks in this study clustered with pathogenic A. phagocytophilum (Figure 3A), indicating that the ticks in the Qinghai-Tibet Plateau harbored pathogenic A. phagocytophilum. Recently, Duan et al. (2022) in their study detected the A. phagocytophilum for the first time in M. himalayana in Tibet, China, confirming that M. himalayana is a reservoir for A. phagocytophilum. Most importantly, they have found that plague may be exacerbated in the presence of A. phagocytophilum and humans that are at risk of infection by exposure to such marmots through ticks and fleas, potentially leading to a complicated disease.
In addition, using the phylogenetic analyses based on full-length 16S rRNA genes and core genes, we found that these novel species were most close to E. muris, E. muris subsp. eauclairensis, B. rochalimae, and R. limoniae, respectively. E. muris was originally isolated in 1983 and was identified as the cause of the patients in Minnesota and Wisconsin in 2009 (Pritt et al., 2011). In addition, the E. muris subsp. eauclairensis was recognized as the etiological agent of human ehrlichiosis in Minnesota and Wisconsin (Lynn et al., 2019). Although transmission to human appears to be limited to Minnesota and Wisconsin, this species could be a potential hazard to humans in the studied area. Bartonella were the most frequently identified bacteria in the fleas collected from rodents in southern Indiana, USA (Hawlena et al., 2013). Bartonella rochalimae can cause intra-erythrocytic infections in mammals and can cause disease manifestations that include bacteremia, splenomegaly, fever, and myalgia in humans. This pathogen has been identified in Xenopsylla cheopis fleas collected from Rattus norvegicus rats in Los Angeles, California (Billeter et al., 2011). Rickettsia can cause a group of diseases such as spotted fever group rickettsioses. However, with more Rickettsia species identified, plenty of Rickettsia were classified into endosymbionts rather a pathogen. Rickettsia limoniae has been reported from the microbiome of non-agricultural insects including the cranefly Limonia chorea (Perlman et al., 2006). However, its pathogenicity is unclear to date. In conclusion, these undescribed novel species, whether they can lead to diseases, need further study and surveillance.
Another important observation from our study was the detection of the vertically transmitted Wolbachia bacteria among these ticks and fleas in the present study. Wolbachia are obligate endosymbionts of bacteria that have been identified in various arthropods, including ticks, fleas, and mosquitos (Plantard et al., 2012; Duan et al., 2020; Flatau et al., 2021). Previous studies have reported that Wolbachia are essential for the survival and reproduction of the host and are involved in molting, embryogenesis, growth, and survival of the host. It can profoundly influence the ecology and evolution of its host through a wide range of symbiotic interactions. Currently, Wolbachia are the staple means of controlling vector-borne diseases due to their unique ability to spread through insect populations and to affect vector competence through pathogen protection. However, our understanding of their biology is still in its infancy and needs further study (Landmann, 2019; Manoj et al., 2021).
There are some shortcomings in this study. Although we obtained several metagenomic assembled genomes from ticks and fleas, we did not isolate potentially pathogenic species successfully. By controlling the density of marmots, the plague was rarely reported in the Qinghai-Tibet Platea in recent years. Thus, vector sample collection becomes more difficult, because it is only possible to collect ticks and fleas after a live marmot has been captured. This leads to another limitation in this study, which is a small sample size of ticks and fleas that we collected.
Conclusion
By using the metataxonomic analysis based on full-length 16S rRNA gene amplicon sequencing, we comprehensively characterized the microbiota community in ticks and fleas collected from M. himalayana found in the Qinghai-Tibet Plateau at the species level for the first time. Notably, we identified the pathogenic A. phagocytophilum and potentially new vector-borne pathogens affiliated with genera Ehrlichia, Bartonella, and Rickettsia, and we performed deep analysis combined with metagenomic assembly and binning. The findings of the study have advanced our understanding of the potential pathogen groups of vectors in marmots in the Qinghai-Tibet Plateau, and we hope to provide etiological information for vector infectious disease prevention and control in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA898891.
Ethics statement
The animal study was reviewed and approved by Ethical Committee of National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, China (No. ICDC-2018004).
Author contributions
Conceptualization: LD and JY. Methodology: LD, JP, and JY. Data curation: JP and JY. Formal analysis: LD, JP, and CY. Visualization: LD, YL, CY, and JG. Writing—original draft: LD. Writing—review and editing: JY, JP, YL, WZ, YH, MK, LZ, and FW. All authors contributed to the article and approved the final version of the manuscript.
Funding
This work was supported by grants from the National Key R&D Program of China (2019YFC1200501) and Research Units of Discovery of Unknown Bacteria and Function (2018RU010).
Acknowledgments
We would like to appreciate Professor Hongjian Chen and his team for providing help on vector sampling and identification. We also thank Professor Yaming Sun and his team for the technical assistance with PacBio sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1188155/full#supplementary-material
References
Adegoke, A., Kumar, D., Bobo, C., Rashid, M. I., Durrani, A. Z., Sajid, M. S., et al. (2020). Tick-borne pathogens shape the native microbiome within tick vectors. Microorganisms 8, 1299. doi: 10.3390/microorganisms8091299
Andreotti, R., Pérez De León, A. A., Dowd, S. E., Guerrero, F. D., Bendele, K. G., Scoles, G. A., et al. (2011). Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol. 11, 6. doi: 10.1186/1471-2180-11-6
Bai, X., Lu, S., Yang, J., Jin, D., Pu, J. Díaz Moyá, S., et al. (2018). Precise fecal microbiome of the herbivorous tibetan antelope inhabiting high-altitude alpine plateau. Front. Microbiol. 9, 2321. doi: 10.3389/fmicb.2018.02321
Bakken, J. S., and Dumler, J. S. (2015). Human granulocytic anaplasmosis. Infect. Dis. Clin. North Am. 29, 341–355. doi: 10.1016/j.idc.2015.02.007
Ben Said, M., Belkahia, H., El Mabrouk, N., Saidani, M., Ben Hassen, M., Alberti, A., et al. (2017). Molecular typing and diagnosis of Anaplasma spp. closely related to Anaplasma phagocytophilum in ruminants from Tunisia. Ticks Tick Borne Dis. 8, 412–422. doi: 10.1016/j.ttbdis.2017.01.005
Ben-Ari, T., Neerinckx, S., Agier, L., Cazelles, B., Xu, L., Zhang, Z., et al. (2012). Identification of Chinese plague foci from long-term epidemiological data. Proc. Natl. Acad. Sci. USA. 109, 8196–8201. doi: 10.1073/pnas.1110585109
Billeter, S. A., Gundi, V. A., Rood, M. P., and Kosoy, M. Y. (2011). Molecular detection and identification of Bartonella species in Xenopsylla cheopis fleas (Siphonaptera: Pulicidae) collected from Rattus norvegicus rats in Los Angeles, California. Appl. Environ. Microbiol. 77, 7850–7852. doi: 10.1128/AEM.06012-11
Bitam, I., Dittmar, K., Parola, P., Whiting, M. F., and Raoult, D. (2010). Fleas and flea-borne diseases. Int. J. Infect. Dis. 14, e667–e676. doi: 10.1016/j.ijid.2009.11.011
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Cai, L., Zhan, X., Wu, W., and Li, C. (1997). The Annals of Fleas on the Tibetan Plateau. Xi'an: Shaanxi Science and Technology Press.
Chala, B., and Hamde, F. (2021). Emerging and re-emerging vector-borne infectious diseases and the challenges for control: a review. Front. Public Health 9, 715759. doi: 10.3389/fpubh.2021.715759
Chomel, B. B., Boulouis, H. J., Maruyama, S., and Breitschwerdt, E. B. (2006). Bartonella spp. in pets and effect on human health. Emerging Infect. Dis. 12, 389–394. doi: 10.3201/eid1203.050931
Ciufo, S., Kannan, S., Sharma, S., Badretdin, A., Clark, K., Turner, S., et al. (2018). Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 68, 2386–2392. doi: 10.1099/ijsem.0.002809
Dai, X., Shang, G., Lu, S., Yang, J., and XU, J. (2018). A new subtype of eastern tick-borne encephalitis virus discovered in Qinghai-Tibet Plateau, China. Emerg. Microbes Infect. 7, 74. doi: 10.1038/s41426-018-0081-6
Duan, R., Lv, D., Fan, R., Fu, G., Mu, H., Xi, J., et al. (2022). Anaplasma phagocytophilum in Marmota himalayana. BMC Genom. 23, 335. doi: 10.1186/s12864-022-08557-x
Duan, X. Z., Sun, J. T., Wang, L. T., Shu, X. H., Guo, Y., Keiichiro, M., et al. (2020). Recent infection by Wolbachia alters microbial communities in wild Laodelphax striatellus populations. Microbiome 8, 104. doi: 10.1186/s40168-020-00878-x
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fang, L. Q., Liu, K., Li, X. L., Liang, S., Yang, Y., Yao, H. W., et al. (2015). Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect. Dis. 15, 1467–1479. doi: 10.1016/S1473-3099(15)00177-2
Flatau, R., Segoli, M., and Hawlena, H. (2021). Wolbachia endosymbionts of fleas occur in all females but rarely in males and do not show evidence of obligatory relationships, fitness effects, or sex-distorting manipulations. Front. Microbiol. 12, 649248. doi: 10.3389/fmicb.2021.649248
Golding, N., Wilson, A. L., Moyes, C. L., Cano, J., Pigott, D. M., Velayudhan, R., et al. (2015). Integrating vector control across diseases. BMC Med. 13, 249. doi: 10.1186/s12916-015-0491-4
Gutiérrez, R., Krasnov, B., Morick, D., Gottlieb, Y., Khokhlova, I. S., Harrus, S., et al. (2015). Bartonella infection in rodents and their flea ectoparasites: an overview. Vector Borne Zoonotic Dis. 15, 27–39. doi: 10.1089/vbz.2014.1606
Hawlena, H., Rynkiewicz, E., Toh, E., Alfred, A., Durden, L. A., Hastriter, M. W., et al. (2013). The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. ISME J. 7, 221–223. doi: 10.1038/ismej.2012.71
He, Y., Chen, W., Ma, P., Wei, Y., Li, R., Chen, Z., et al. (2021a). Molecular detection of Anaplasma spp., Babesia spp. and Theileria spp. in yaks (Bos grunniens) and Tibetan sheep (Ovis aries) on the Qinghai-Tibetan Plateau, China. Parasit, Vectors 14, 613. doi: 10.1186/s13071-021-05109-2
He, Y. C., Li, J. X., Sun, Y. L., Kang, M., He, H. X., Guo, Y. H., et al. (2021b). Spotted fever group rickettsia infecting ticks (Acari: Ixodidae), Yak (Bos grunniens), and Tibetan Sheep (Ovis aries) in the Qinghai-Tibetan Plateau Area, China. Front. Vet. Sci. 8, 779387. doi: 10.3389/fvets.2021.779387
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114. doi: 10.1038/s41467-018-07641-9
Landmann, F. (2019). The wolbachia endosymbionts. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.BAI-0018-2019
Liu, X. Y., Gong, X. Y., Zheng, C., Song, Q. Y., Chen, T., Wang, J., et al. (2017). Molecular epidemiological survey of bacterial and parasitic pathogens in hard ticks from eastern China. Acta Trop. 167, 26–30. doi: 10.1016/j.actatropica.2016.12.010
Lynn, G. E., Burkhardt, N. Y., Felsheim, R. F., Nelson, C. M., Oliver, J. D., Kurtti, T. J., et al. (2019). Ehrlichia isolate from a minnesota tick: characterization and genetic transformation. Appl. Environ. Microbiol. 85, e00866-1. doi: 10.1128/AEM.00866-19
Manoj, R. R. S., Latrofa, M. S., Epis, S., and Otranto, D. (2021). Wolbachia: endosymbiont of onchocercid nematodes and their vectors. Parasit. Vectors 14, 245. doi: 10.1186/s13071-021-04742-1
Meng, X., Lu, S., Yang, J., Jin, D., Wang, X., Bai, X., et al. (2017). Metataxonomics reveal vultures as a reservoir for Clostridium perfringens. Emerg. Microbes Infect. 6, e9. doi: 10.1038/emi.2016.137
Minard, G., Mavingui, P., and Moro, C. V. (2013). Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vectors 6, 146. doi: 10.1186/1756-3305-6-146
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parola, P. (2004). Tick-borne rickettsial diseases: emerging risks in Europe. Comp. Immunol. Microbiol. Infect. Dis. 27, 297–304. doi: 10.1016/j.cimid.2004.03.006
Perlman, S. J., Hunter, M. S., and Zchori-Fein, E. (2006). The emerging diversity of Rickettsia. Proc. Biol. Sci. 273, 2097–2106. doi: 10.1098/rspb.2006.3541
Plantard, O., Bouju-Albert, A., Malard, M. A., Hermouet, A., Capron, G., Verheyden, H., et al. (2012). Detection of Wolbachia in the tick Ixodes ricinus is due to the presence of the hymenoptera endoparasitoid Ixodiphagus hookeri. PLoS ONE 7, e30692. doi: 10.1371/journal.pone.0030692
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Pritt, B. S., Sloan, L. M., Johnson, D. K., Munderloh, U. G., Paskewitz, S. M., Mcelroy, K. M., et al. (2011). Emergence of a new pathogenic Ehrlichia species, Wisconsin and Minnesota, 2009. N. Engl. J. Med. 365, 422–429. doi: 10.1056/NEJMoa1010493
Qin, S., Liang, J., Tang, D., Chen, Y., Duan, R., Lu, X., et al. (2022). Serological investigation of plague and brucellosis infection in Marmota himalayana plague foci in the Altun Mountains on the Qinghai-Tibet Plateau. Front. Public Health 10, 990218. doi: 10.3389/fpubh.2022.990218
Seo, M. G., Ouh, I. O., Kwon, O. D., and Kwak, D. (2018). Molecular detection of Anaplasma phagocytophilum-like Anaplasma spp. and pathogenic A. phagocytophilum in cattle from South Korea. Mol. Phylogenet. Evol. 126, 23–30. doi: 10.1016/j.ympev.2018.04.012
Thapa, S., Zhang, Y., and Allen, M. S. (2019). Bacterial microbiomes of Ixodes scapularis ticks collected from Massachusetts and Texas, USA. BMC Microbiol. 19, 138. doi: 10.1186/s12866-019-1514-7
Uritskiy, G. V., Diruggiero, J., and Taylor, J. (2018). MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158. doi: 10.1186/s40168-018-0541-1
Wang, L., Wang, Y., Jin, S., Wu, Z., Chin, D. P., Koplan, J. P., et al. (2008). Emergence and control of infectious diseases in China. Lancet 372, 1598–1605. doi: 10.1016/S0140-6736(08)61365-3
WHO (2017). Global Vector Control Response 2017–2030. Geneva: WHO. Availabel online at: https://www.who.int/publications/i/item/WHO-HTM-GVCR-2017.01 (accessed February 24, 2023).
Yan, Y., Lu, C., Gong, P., Pei, Z., Peng, Y., Jian, F., et al. (2022). Molecular detection and phylogeny of Anaplasma spp. closely related to Anaplasma phagocytophilum in small ruminants from China. Ticks Tick Borne Dis. 13, 101992. doi: 10.1016/j.ttbdis.2022.101992
Yang, J., Pu, J., Lu, S., Bai, X., Wu, Y., Jin, D., et al. (2020). Species-level analysis of human gut microbiota with metataxonomics. Front. Microbiol. 11, 2029. doi: 10.3389/fmicb.2020.02029
Yun, J. H., Roh, S. W., Whon, T. W., Jung, M. J., Kim, M. S., Park, D. S., et al. (2014). Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 80, 5254–5264. doi: 10.1128/AEM.01226-14
Zeppelini, C. G., de Almeida, A. M., and Cordeiro-Estrela, P. (2016). Zoonoses as ecological entities: a case review of plague. PLoS Negl. Trop. Dis. 10, e0004949. doi: 10.1371/journal.pntd.0004949
Zhao, G. P., Wang, Y. X., Fan, Z. W., Ji, Y., Liu, M. J., Zhang, W. H., et al. (2021). Mapping ticks and tick-borne pathogens in China. Nat. Commun. 12, 1075. doi: 10.1038/s41467-021-21375-1
Keywords: tick, flea, Marmota himalayana, Anaplasma phagocytophilum, Wolbachia, Bartonella
Citation: Dong L, Li Y, Yang C, Gong J, Zhu W, Huang Y, Kong M, Zhao L, Wang F, Lu S, Pu J and Yang J (2023) Species-level microbiota of ticks and fleas from Marmota himalayana in the Qinghai-Tibet Plateau. Front. Microbiol. 14:1188155. doi: 10.3389/fmicb.2023.1188155
Received: 17 March 2023; Accepted: 31 May 2023;
Published: 21 June 2023.
Edited by:
Joerg Graf, University of Connecticut, United StatesReviewed by:
David H. Walker, University of Texas Medical Branch at Galveston, United StatesOlivier Andre Sparagano, City University of Hong Kong, Hong Kong SAR, China
Copyright © 2023 Dong, Li, Yang, Gong, Zhu, Huang, Kong, Zhao, Wang, Lu, Pu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji Pu, cHVqaUBpY2RjLmNu; Jing Yang, eWFuZ2ppbmdAaWNkYy5jbg==