 Tao Tang1,2
Tao Tang1,2 Fanfan Wang1,2Guobin Fang3Ting Mao1
Fanfan Wang1,2Guobin Fang3Ting Mao1 Jie Guo1,2Hui Kuang3Guangzhong Sun3Xiaoliang Guo1
Jie Guo1,2Hui Kuang3Guangzhong Sun3Xiaoliang Guo1 Yuanyuan Duan2
Yuanyuan Duan2 Jingmao You1,2*
Jingmao You1,2*- 1Key Laboratory of Chinese Herbal Medicine Biology and Cultivation, Ministry of Agriculture and Rural Affairs, Institute of Chinese Herbal Medicine, Hubei Academy of Agricultural Science, Enshi, China
- 2Hubei Engineering Research Center of Good Agricultural Practices (GAP) Production for Chinese Herbal Medicines, Institute of Chinese Herbal Medicines, Hubei Academy of Agricultural Sciences, Enshi, China
- 3Hubei Provincial Plant Protection Station, Wuhan, China
Introduction: The ecological balance of the plant microbiome, as a barrier against pathogens, is very important for host health. Coptis chinensis is one of the important medicinal plants in China. In recent years, Illumina Miseq high-throughput sequencing technology was frequently used to analyze root rot pathogens and the effects of root rot on rhizosphere microorganisms of C. chinensis. But the effects of root rot infection on rhizosphere microecological balance of C. chinensis have received little attention.
Methods: In this study, Illumina Miseq high-throughput sequencing technology was applied to analyze the impact on microbial composition and diversity of C. chinensis by root rot.
Results: The results showed that root rot infection had significant impact on bacterial α-diversity in rhizome samples, but had no significant effect on that in leaf samples and rhizosphere soil samples, while root rot infection exhibited significant impact on the fungal α-diversity in leaf samples and rhizosphere soil samples, and no significant impact on that in rhizome samples. PCoA analysis showed that the root rot infection had a greater impact on the fungal community structure in the rhizosphere soil, rhizome, and leaf samples of C. chinensis than on the bacterial community structure. Root rot infection destroyed the microecological balance of the original microbiomes in the rhizosphere soil, rhizome, and leaf samples of C. chinensis, which may also be one of the reasons for the serious root rot of C. chinensis.
Discussion: In conclusion, our findings suggested that root rot infection with C. chinensis disrupts microecological balance of rhizosphere soil and endophytic microbiomes. The results of this study can provide theoretical basis for the prevention and control of C. chinensis root rot by microecological regulation.
1. Introduction
Coptis chinensis is a perennial herb belonging to family Ranunculaceae, and genus Coptis. Its rhizome is one of the most commonly used materials in traditional Chinese medicine (He et al., 2017). The using of C. chinensis in traditional Chinese medicine has a long history, which can be traced back to the earliest known work on Chinese medicine in my country, Divine Farmer’s Materia Medica (Shennong Bencao Jing) (Liu et al., 2021). C. chinensis rhizome has broad antibacterial, antiviral, and antioxidant effects, and it is often used to treat dysentery, jaundice, acute fever-induced sore throat, fever, and diarrhea, and other diseases (Tseng et al., 2020). The latest research showed that the active substances in Huanglian Jiedu Decoction (toxicity-relief soup) can treat new coronavirus pneumonia through multiple targets and multiple pathways (Huang et al., 2020). Part of the mountainous areas above 1,200 meters above sea level in Enshi Tujia and Miao Autonomous Prefecture are one of the main C. chinensis-producing areas with a total planting area of about 5,000 hm2 and a total annual output of about 4,000 t. However, with the continuous expansion of the planting scale and continuous cultivation for many years, the root rot of C. chinensis occurred in Lichuan city and other places, resulting in a large-scale production reduction or even no harvest, which seriously decreasing the yield and quality of C. chinensis and adversely affecting the planting enthusiasm of farmers. Fusarium avenaceum and Fusarium solani have been reported to be pathogen of C. chinensis root rot disease (Luo et al., 2014; Mei et al., 2021b). Diaporthe eres has also been reported to cause C. chinensis root rot (Mei et al., 2021a). Our previous study has found that Phoma aquilegiicola and Colletotrichum boninense can cause rhizome rot of C. chinensis. There are many types of pathogens causing C. chinensis root rot disease making prevention and control of C. chinensis root rot difficult.
In animal disease theory, the “disease triangle” is often used to describe the dynamic interactions between hosts, pathogens, and environmental factors including microorganisms (Scholthof, 2007). This theory also applies to plants. The plant microbiome plays an indispensable role in various biological processes of the host. As the same, the microbial community in environment can affect various biological processes of the host and the host microbiome. Recent studies have shown that biological and environmental factors (including microorganisms) have a strong influence on host disease dynamics (Hawkes et al., 2020). Gut microbes, just like the skin, are the first barrier of immunity and play an important role in host defense against pathogens (Bernardo-Cravo et al., 2020). With the maturity of Illumina Miseq high-throughput sequencing technology, researchers began to pay more attention to the effect of pathogen infection on plant microbiome. Relevant studies have shown that the microbial community structure in plant rhizosphere soil, stem, and phyllosphere will change after infection by pathogen. The analysis of the soil fungal community structure in verticillium wilt disease-infected cotton fields used the Illumina Miseq high-throughput sequencing technology has shown that the number and abundance of fungal OTUs in severely diseased cotton fields were higher than those in mildly-diseased or disease-free fields, while the fungal diversity was reduced in diseased cotton fields (Liu et al., 2019). Zhang et al. have reported that the number of Fusarium and Verticillium in the rhizosphere soil of diseased cotton was higher, while the number of beneficial microorganisms such as Trichoderma in the rhizosphere soil of healthy cotton was significantly higher than that of diseased cotton (Zhang et al., 2022). Liao et al. (2021) quickly identify potential pathogenic and antagonistic microorganisms related to root rot of C. chinensis through high-throughput sequencing technology. The results indicated that Fusarium, Volutella, Cladorrhinum, Cylindrocarpon, Cylindrocarpon and Exophiala were potential pathogenic agents of C. chinensis root rot, and Bacillus may be potential antagonists. However, most of the previous studies focused on the influence of root rot infection on rhizosphere microbiome structure of C. chinensis, but little attention was paid to the influence on ecological balance. The destruction of the ecological balance of host microorganisms by pathogens may be an important reason for the difficulty of disease control. Microecological regulation refers to the purpose of preventing and controlling plant diseases by regulating the microbial environment of plants so that the microbial environment was beneficial to the host and not conducive to pathogens (Berendsen et al., 2012). Bai et al. (2015) isolated more than 400 culturable endophytic bacteria from the roots and leaves of Arabidopsis thaliana, and reconstructed the microbiome in the roots and leaves of A. sterile shoots, creating a research model for reconstructing the microbiome. Li et al. (2021) analyzed the characteristics of the microbial community in the rhizosphere and root of Astragalus mongholicus with root rot, and constructed a synthetic community based on significantly enriched bacteria in the rhizosphere of A. mongholicus with root rot. The synthetic community can inhibit the growth of pathogen and activate plant-induced systemic resistance of A. mongholicus to achieve the purpose of reducing the incidence of root rot (Li et al., 2021). Using the theory of microecology and the method of microecological regulation to adjust the microbial community of plants to improve their disease resistance can be an important research idea for the prevention of C. chinensis root rot.
In this study, Illumina Miseq high-throughput sequencing technology was used to analyze the composition and diversity of rhizomes, leaf endophytic microorganisms, and rhizosphere soil microorganisms of healthy and diseased C. chinensis from two different locations. The objective of this study was as followed: (1) to clarify the differences of rhizosphere soil, rhizosphere and leaf microbial community structure in C. chinensis; (2) to clarify the effects of F. avenaceum and F. solani infection on rhizosphere soil, rhizosphere and leaf microbial community structure; (3) to provide theoretical support for using micro-ecological control methods or SynComs to prevent and control C. chinensis root rot.
2. Materials and methods
2.1. Sampling site and sampling method
All the sequencing samples were collected from the C. chinensis Planting Base (108° 35′ E, 30° 22′ N) in Jianzhuxi Town, Lichuan City, Enshi Prefecture at an altitude of 1,300 m in October, 2019. The samples were collected from two different sites and divided into 12 groups as shown in Table 1. The samples were taken from 5 different positions in the same field ridge and mixed uniformly into one sample. The rhizosphere soil around 5–10 cm below the ground part of the C. chinensis plant was collected by using the JC-802 root soil sampler. The rhizome samples obtained from the same 5 positions of the rhizosphere soil samples and leaf samples were mixed uniformly into one sample. The rhizome samples and the leaf samples were surface-sterilized with 75% ethanol immediately after sampling. Three samples in each group were collected from different positions, and the experiments were conducted in three biological repetitions. The samples were put into sterile 50 ml centrifuge tubes, transported to the laboratory at low temperature, and stored at 4°C for later use. The distance between the two sampling sites was more than 3 km. F. avenaceum and F. solani, isolated and identified in both locations, were the main pathogens of root rot on C. chinensis from Lichuan City.
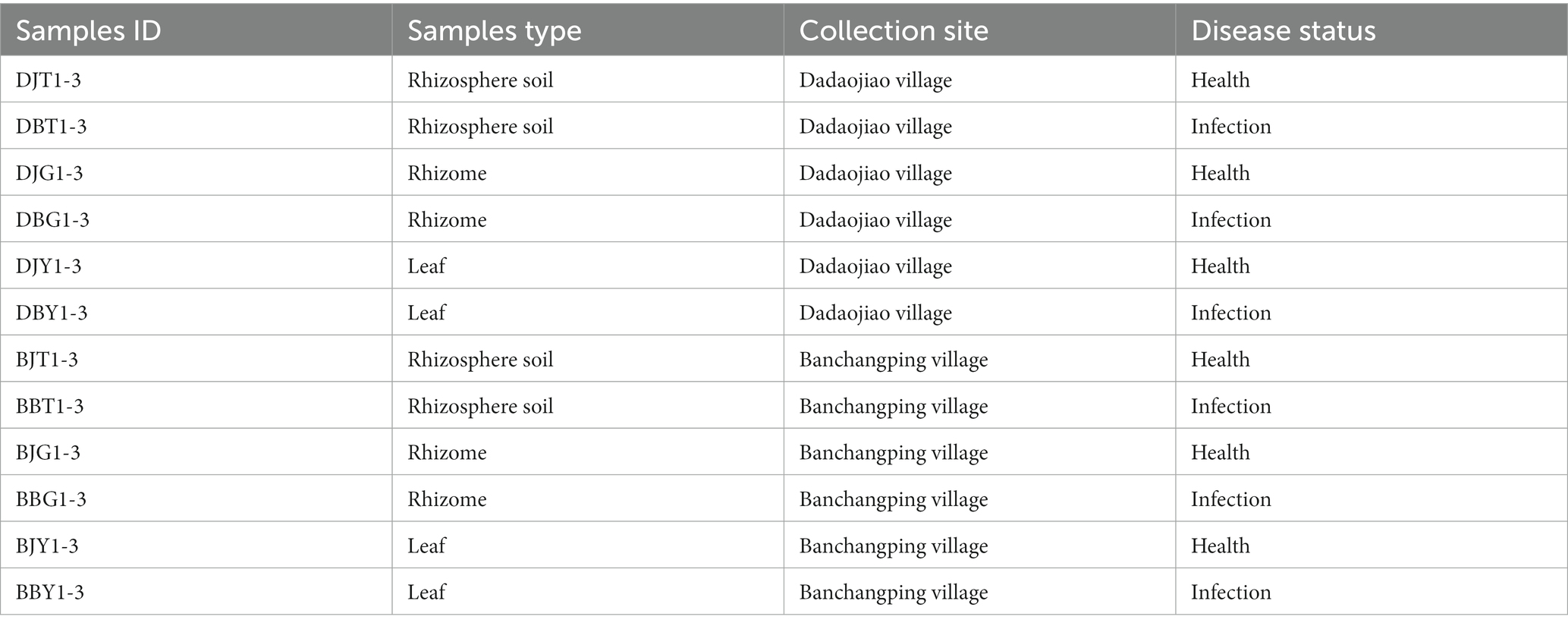
Table 1. The collection information of experimental samples.
2.2. Analysis of physicochemical properties of rhizosphere soil
The soil physicochemical indexes were determined as follows. The deionized distilled water (1:20 w/v) was used to make soil suspension, and Mettler-Toledo FE 20 was used to measure soil pH. Soil organic matter (TOM) was determined using the potassium dichromate external heating method [38]. Soil total nitrogen (TCN) was detected by Kjeldahl determination (Bremner, 1960). Soil total phosphorus (TCP) was determined by the molybdenum antimony blue colorimetry (Murphy and Riley, 1962). Soil total potassium (TCK) was determined by flame atom absorption method (Wu et al., 2017). Soil available nitrogen (AN) was determined by diffusion absorption method (Michrina et al., 1982). Soil available phosphorus (AP) was analyzed following the methods described by Li et al., (2020). Soil available potassium (AK) was determined by atomic absorption spectrometry (Tian et al., 2021). Soil physicochemical factors were determined by the Institute of Plant Protection and Soil and Fertilizer of Hubei Academy of Agricultural Sciences.
2.3. DNA extraction and PCR amplification
The total microbial DNA of rhizosphere soil samples was extracted with PowerSoil® DNA Isolation Kit (MOBIO, United States). The rhizomes and leaves were chopped and ground with liquid nitrogen, and the total DNA was extracted by PowerSoil® DNA Isolation Kit. The DNA concentration and purity were determined by Nanodrop ultraviolet spectrophotometer (Thermo Scientific, United States). The quality of DNA extract was detected by 1.2% agarose gel electrophoresis, and then the DNA extraction solution was stored at −80°C for later use. The V5-V7 region of fungal 18S rRNA was amplified by using universal primers SSU0817F (5’-TTAGCATGGAATAATRRAATAGGA-3′) and 1196R (5’-TCTGGACCTGGTGAGTTTCC-3′; Jiang et al., 2017). The V3-V4 region of 16S rRNA was PCR amplified by using universal primers 338\u00B0F (5’-ACTCCTACGGGAGGCAGCA-3′) and 806 R (5’-GGACTACHVGGGTWTCTAAT-3′; Li et al., 2020). Pfu high-fidelity DNA polymerase from TransGen Biotech was used for PCR amplification with the number of amplification cycles strictly controlled to make cycle number as small as possible and the amplification conditions for the same batch of samples consistent. The PCR products were purified and recovered with the AxyPrep DNA Gel extraction kit. Subsequently, each sample was quantified using BioTek Flx800 microplate reader with Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, P7589).
2.4. Library preparation and on-board sequencing
Sequencing libraries were prepared using the TruSeq Nano DNA LT Library Prep Kit from Illumina. After passing the test, the constructed library was sequenced by Shanghai Parsenor Biotechnology Co., Ltd. using the Illumina Miseq platform. Before computer sequencing, it is necessary to adopt Agilent High Sensitivity DNA Kit to conduct quality of constructed library inspection on Agilent Bioanalyzer. After that, Quant-iT PicoGreen dsDNA Assay Kit was used to quantify the library on the Promega QuantiFluor system, and the qualified concentration of the library should be above 2 nM. The qualified computer sequencing libraries were diluted in gradient, mixed in proportion to the required sequencing volume, and denatured into single strand by NaOH for computer sequencing. All raw data has been uploaded to GSA with the GSA number: PRJCA008804.1
2.5. Sequencing data processing
For the raw data obtained from sequencing, firstly, qiime cutadapt trim-paired command was executed to excise the primer fragments of the sequence. The unmatched primer sequences were discarded, and then the qiime DADA2 denoise-paired command was executed for quality control, denoising, splicing, and chimera clearance (Bolyen et al., 2019). The above steps were performed separately for each library. After all the libraries were denoised, the ASVs feature sequences and ASV tables were merged, and singletons ASVs were removed. The classify-sklearn algorithm in QIIME22 with default parameters was adopted to preform species annotation for the feature sequence of each ASV using a pre-trained Naive Bayes classifier (Bokulich et al., 2018). The qiime feature-table rarefy function in the QIIME2 software was employed to subsample all samples with subsampling depth set as 95% of the minimum sample sequence size. For bacteria 16 s rRNA genes, the Greengenes database (Release13.83) was selected by default. For fungal ITS sequence, the UNITE database (Release 8.04) was selected by default.
2.6. Statistical analysis
Statistical analysis was performed using SPSS software (versions 18.0) and R (version 4.0.2). Principal Component Analysis (PCoA) was performed on “the Bray–Curtis distances” using the R package VEGAN (version 2.5–7) package (Bray and Curtis, 1957). The PCoA plots were compared using the procrustes analyzes in the VEGAN. Venn plots were generated using the R package ‘VennDiagram’ (version 1.6.20) (Zaura et al., 2009). Differential core microorganisms in each sample were statistically analyzed using the R software package “DESeq2 version 1.28.1.” The correlation network between core microorganisms and the correlations between core microorganisms and soil physicochemical factors were analyzed by using Spearman algorithm in R package WGCNA (version 1.69) and the network diagram was drawn by Gephi 0.9.2 (Langfelder and Horvath, 2008). GraphPad Prism software (version 7.0) was used for statistical analysis and data plotting. Significant differences among microbial diversity were analyzed using t-test in GraphPad Prism software (version 7.0). The level of significance was set at *p < 0.05 and **p < 0.01.
3. Results
3.1. Soil physicochemical properties
As shown in Table 2, the pH of healthy C. chinensis rhizosphere soil in two different locations was significantly higher than that of root rot C. chinensis rhizosphere soil (p < 0.05). In addition, the rhizosphere soil organic matter, alkali-hydrolyzed nitrogen, available phosphorus, total phosphorus, and total nitrogen of healthy C. chinensis rhizosphere soil were significantly higher than those of root rot C. chinensis rhizosphere soil (p < 0.05). However, the contents of available potassium and total potassium were inconsistent in the two different locations. In the samples from Daojiao Village, the content of available potassium in root rot C. chinensis rhizosphere soil was significantly higher than that in the healthy C. chinensis rhizosphere soil (p < 0.05), whereas the content of total potassium was not significantly different. The contents of available potassium and total potassium in the root rot C. chinensis rhizosphere soil samples from Banchangping Village were significantly lower than those in the healthy C. chinensis rhizosphere soil samples (p < 0.05). This indicated that soil pH, organic matter, alkali-hydrolyzed nitrogen, available phosphorus, total phosphorus, and total nitrogen content may have a certain relationship with the occurrence of C. chinensis root rot.
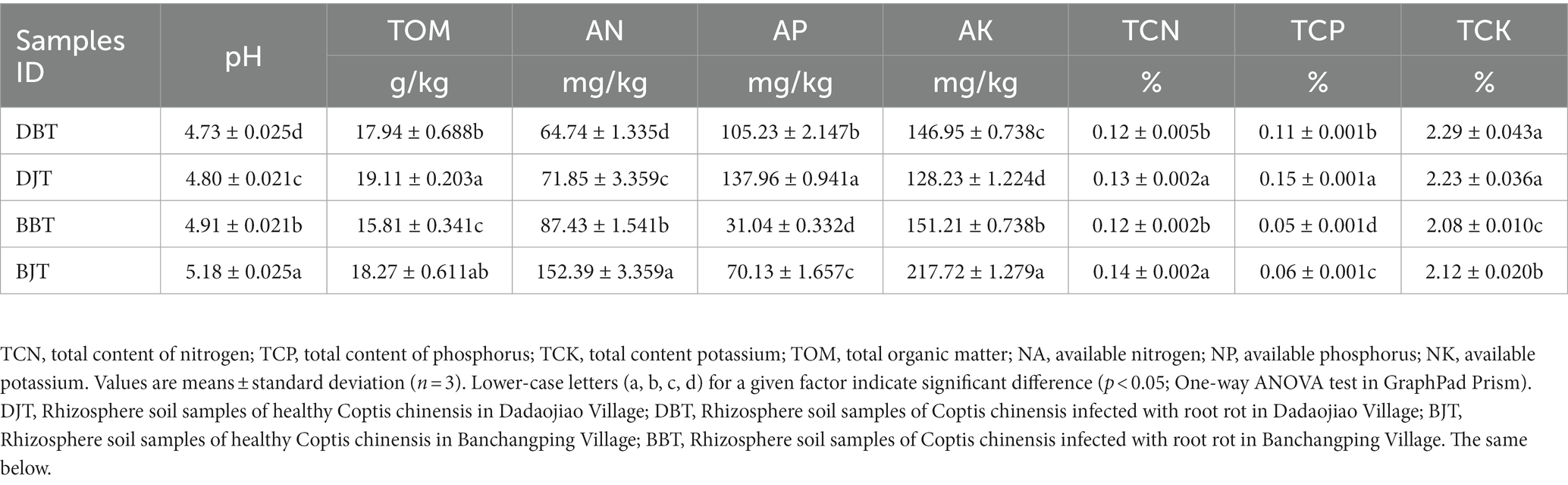
Table 2. Physiochemical characteristics of rhizosphere soil of healthy and infected Coptis chinensis.
3.2. Alpha diversity
The α-diversity indices Chao1, Shannon, Simpson, Observed-species, and Pielou-e were used to evaluate the diversity and richness of each sample. The Observed species index referred to the number of OTUs actually contained in the sample, while the chao1 index referred to the number of OTUs estimated in the sample, both of which reflected the number of OTUs (species) in the sample. Shanon index was used to describe the disorder and uncertainty in OTUs. Simpson index referred to the probability of taking two sequences randomly from the sample, which belong to different OTUs. Shannon and Simpson index not only reflect the number of species in the sample (richness) but also reflect the abundance distribution of each species in the sample (evenness). The group analysis of healthy C. chinensis and those infected with root rot disease was performed. As shown in Figure 1, the infection with root rot disease significantly increased the α-diversity index including Chao1, Shannon, Simpson and Observed-species in C. chinensis rhizome samples, but it had no significant effect on the α-diversity index in the rhizosphere soil samples and leaf samples of C. chinensis. As for fungal α-diversity, the infection with root rot disease significantly increased the α-diversity indices including Simpson, Shannon, Observed-species, and Pielou-e in the rhizosphere soil samples of C. chinensis, but significantly decreased Simpson and Shannon indices in C. chinensis leaf samples, and the infection had no significant effect on the α-diversity index in C. chinensis rhizome samples. The effects of root rot infection on the bacterial and fungal community structure in C. chinensis rhizosphere soil, rhizome, and leaf samples exhibited opposite trends. Namely, root rot infection had the greatest effect on the bacterial alpha diversity of rhizome samples, but no effect on leaf and rhizosphere soil samples. However, root rot infection had the greatest effect on the fungal alpha diversity of leaf samples and rhizosphere soil samples, but the least effect on the rhizome samples. In addition, in terms of the Chao1 and Observed-species indices of bacterial α-diversity, the bacterial abundance in the C. chinensis rhizosphere soil samples were significantly higher than that in the rhizome samples, which in turn was significantly higher than that in the leaf samples. However, the fungal abundances were not significantly different.
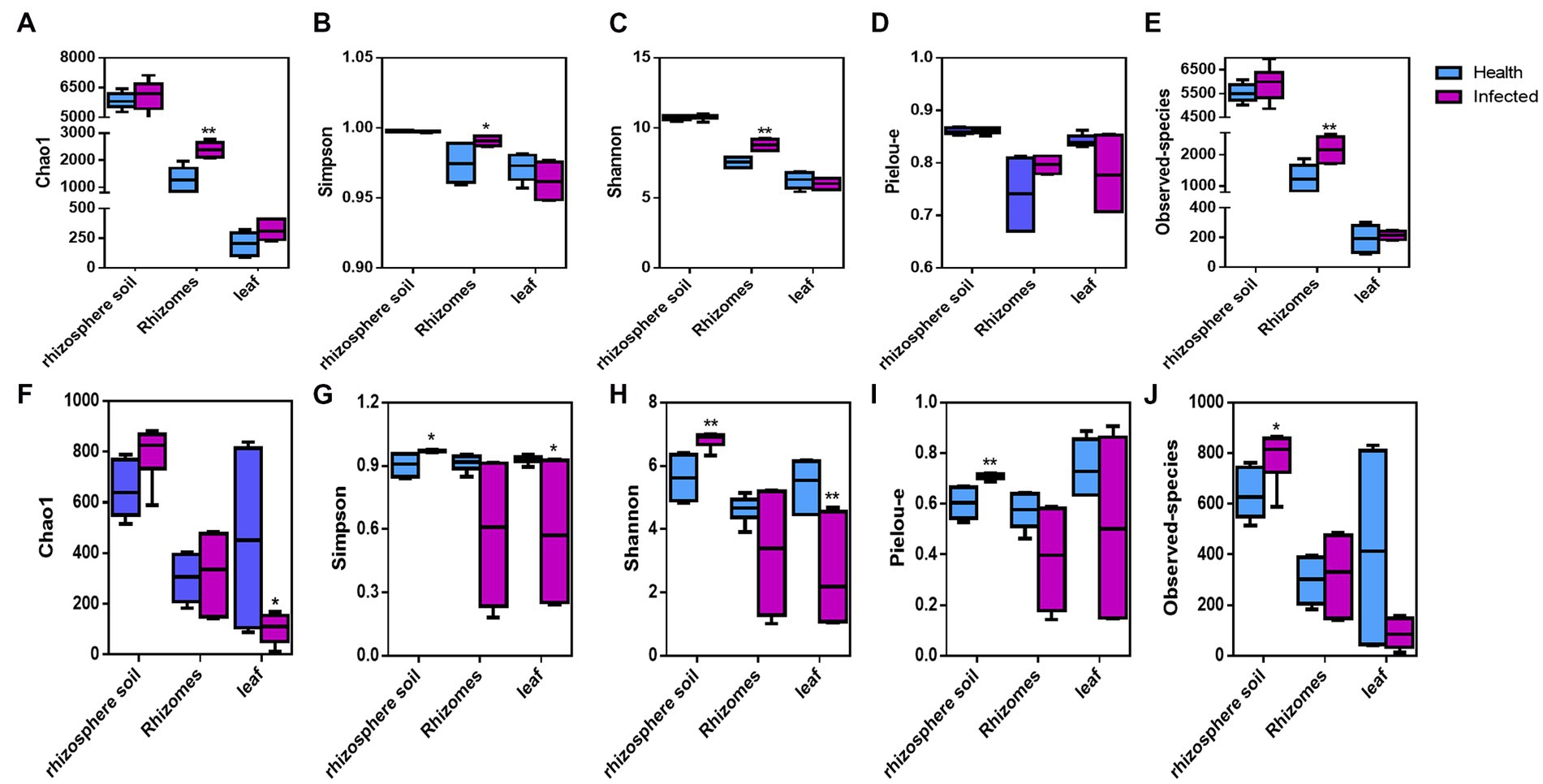
Figure 1. Alpha diversity indices of the bacteria and fungal community in rhizosphere soils, rhizome, and leaf of C. chinensis. (A) Chao1 index of the bacteria community in all samples; (B) Simpson diversity index of the bacteria community in all samples; (C) Shannon diversity index of the bacteria community in all samples; (D) Pielou evenness of the bacteria community in all samples; (E) Observed species of the bacteria community in all samples; (F) Chao1 index of the fungi community in all samples; (G) Simpson diversity index of the fungi community in all samples; (H) Shannon diversity index of the fungi community in all samples; (I) Pielou evenness of the fungi community in all samples; (J) Observed species of the fungi community in all samples. ‘*’ and ‘**’ represent significant differences between health and infected of rhizosphere soils, rhizome, and leaf of C. chinensis, respectively (Meltiple t-tests-one per row in GrahPad Prism).
3.3. β diversity
β diversity differences between healthy group and root rot infection group samples were analyzed using Bray-Curtis distance matrix. As shown in Figure 2, bacterial communities in healthy rhizome samples and those infected with root rot were clustered separately, with the first two axes explaining 45.89 and 35.77% of the total variance (Figure 2B). The bacterial communities of healthy C. chinensis and root rot-infected C. chinensis rhizosphere soil and leaf samples exhibited a certain intersection in the PCoA map (Figures 2A,C). In contrast, the fungal communities in the rhizosphere soil, rhizome, and leaf samples of healthy C. chinensis and root rot disease-infected C. chinensis were all clustered separately (Figures 2D–F). This result showed that root rot disease had a great impact on the fungal community structure of the rhizosphere soil, rhizome, and leaf samples of C. chinensis, and it had a great impact on the bacterial community of the rhizome samples of C. chinensis, but less impact on the bacterial community of rhizosphere soil and leaf samples. This result was consistent with that of alpha diversity.
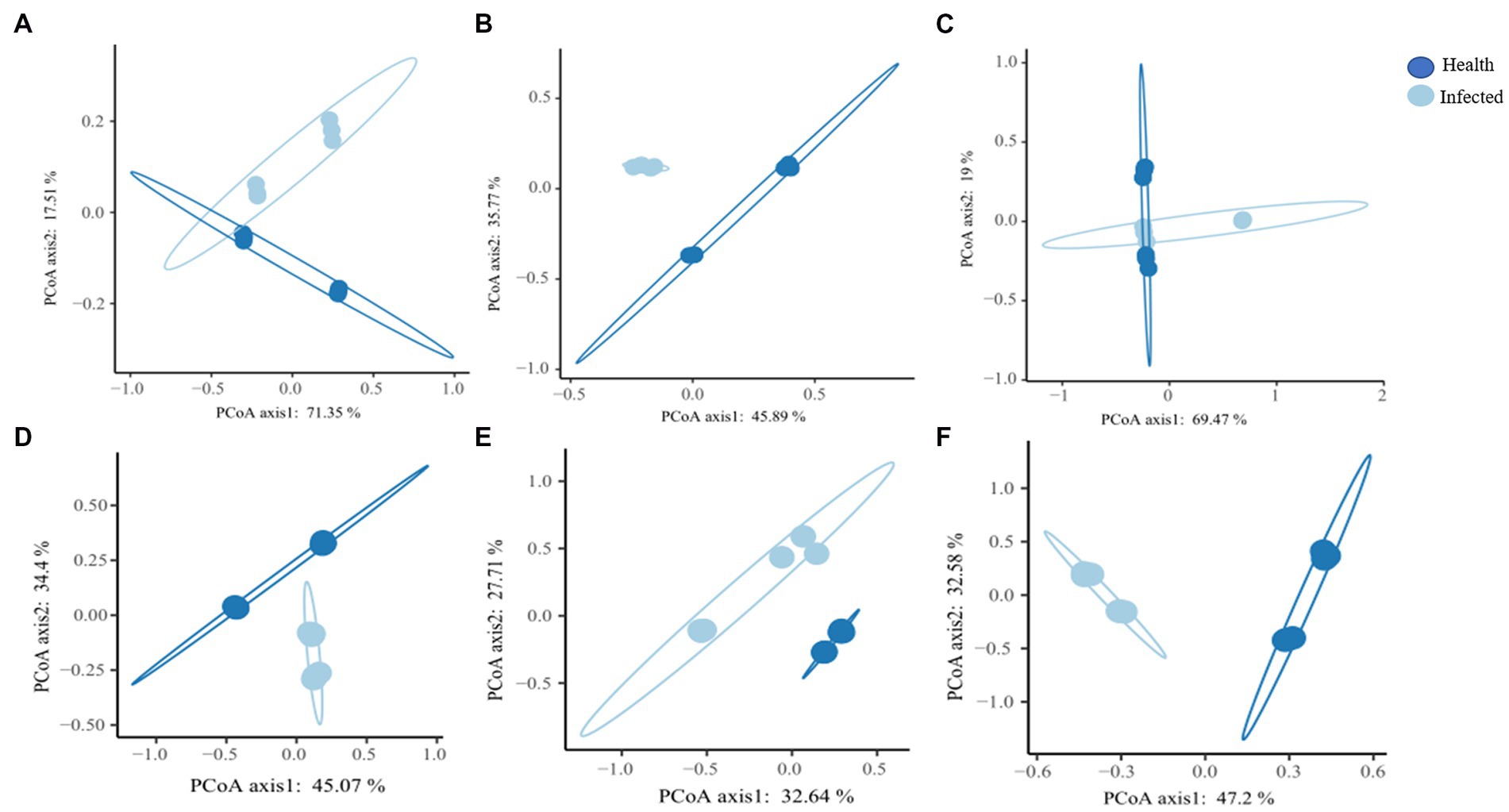
Figure 2. Principle coordinate analysis (PcoA) of bacteria and fungal community in rhizosphere soils, rhizome, and leaf of health and infected C. chinensis. (A) Principle coordinate analysis (PcoA) of bacteria community in rhizosphere soils of bacteria of health and infected C. chinensis; (B) Principle coordinate analysis (PcoA) of bacteria community in rhizome of health and infected C. chinensis; (C) Principle coordinate analysis (PcoA) of bacteria community in leaf of health and infected C. chinensis; (D) Principle coordinate analysis (PcoA) of fungal community in rhizosphere soils of bacteria of health and infected C. chinensis; (E) Principle coordinate analysis (PcoA) of fungal community in rhizome of health and infected C. chinensis; (F) Principle coordinate analysis (PcoA) of fungal community in leaf of health and infected C. chinensis.
3.4. Analysis of bacterial and fungal community structure
At the phylum level, the main bacterial communities in the rhizosphere soil, rhizome, and leaf samples of Coptis chinensis were Proteobacteria, Acidobacteria, Actinobacteria, Chloroflexi, and Bacteroidetes. The main fungal communities in the rhizosphere soil, rhizome, and leaf samples of C. chinensis were Ascomycota and Basidiomycota.
At the genus level, the fungal and bacterial community compositions from the different samples (rhizosphere soil, rhizome, and leaf) were quite different. The sequencing results showed that the dominant bacterial genera in the rhizosphere soil samples of C. chinensis were Holophaga and Bryobacter, but most others were not annotated at the genus level (Figure 3A). The dominant bacterial genera in the rhizome samples of C. chinensis were Burkholderia, Bradyrhizobia, Parafrigoribacteria, and Rhizobia. The infection with root rot disease significantly suppressed the relative abundance of Burkholderia. Bradyrhizoloium, Acidibacter, and Streptemyces were significantly enriched in the diseased rhizome samples (Figure 3B). The dominant bacterial genera in C. chinensis leaf samples were Methylobacteria and Sphingomonas (Figure 3C). Mortierella and a type of unannotated fungus were dominant fungal communities in the rhizophere soil samples of C. chinensis (Figure 3D). The dominant fungal genera of the healthy C. chinensis rhizome samples and the diseased C. chinensis rhizome samples were completely different. The dominant fungal community genera of the healthy C. chinensis rhizome samples were Ilyonectria, while that of the diseased C. chinensis rhizome samples was Paraboeremia (Figure 3E). The dominant bacteria in the fungal community of healthy C. chinensis leaf samples and diseased C. chinensis leaf samples were quite different. The dominant fungal genus in the fungal community in healthy C. chinensis samples from Daojiao Village was Pseudocercospora and that from Banchangping Village was Pyrenochaetopsis, while the dominant fungal genus in the infected C. chinensis leaf samples collected from Daojiao Village was Calophoma, and the dominant fungal community genera in the infected C. chinensis leaf samples collected from Banchangping Village were unidentified Agaricomycetes and unidentified_Capnodiales (Figure 3F). The influence of root rot infection was greater on the fungal community structure of various C. chinensis samples than on the bacterial community structure, implying that C. chinensis root rot was mainly caused by fungal pathogens.
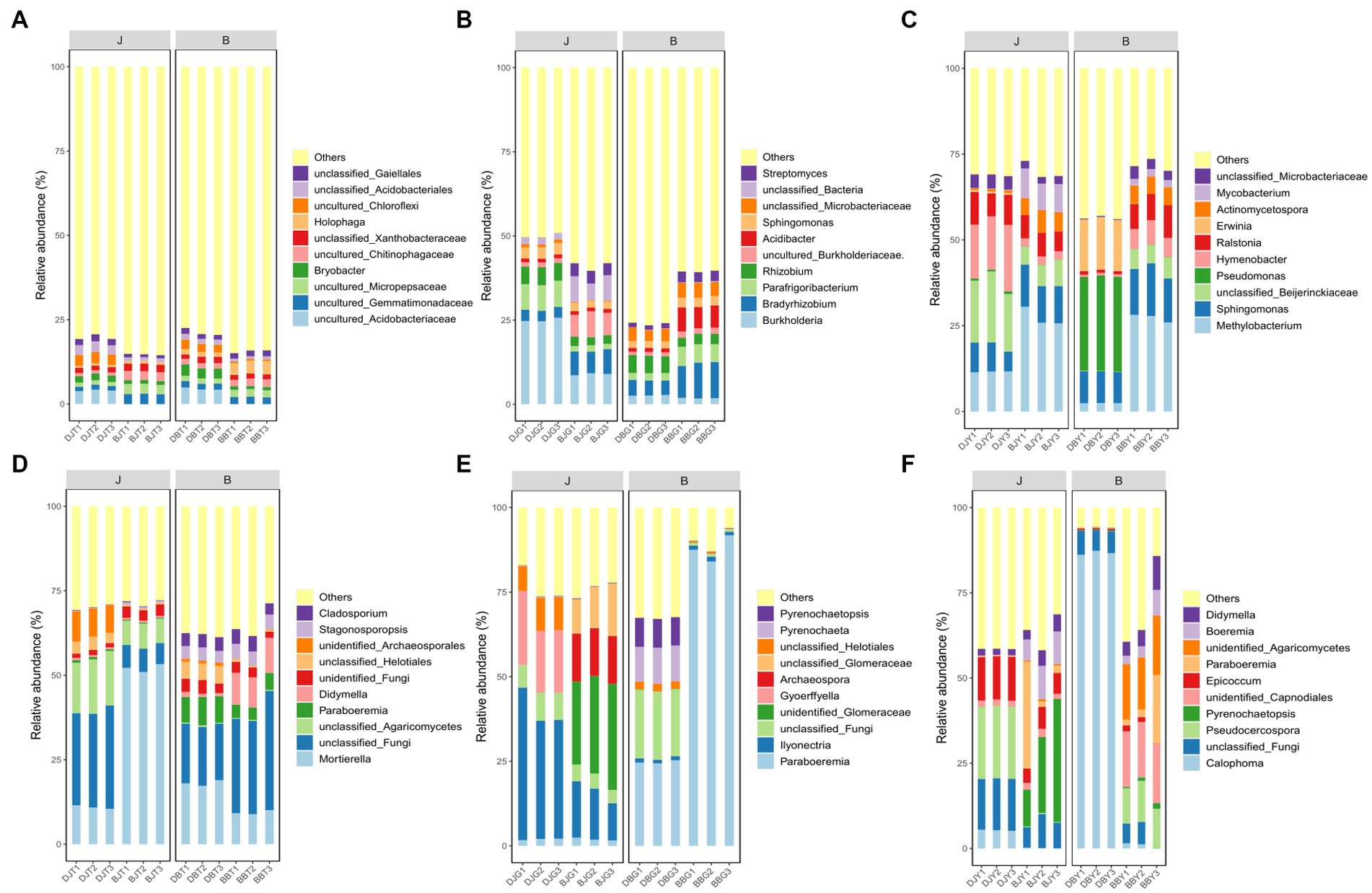
Figure 3. Bacterial and fungus composition in rhizosphere soils, rhizome, and leaf of health and infected C. chinensis at the phylum level. (A) Bacterial composition in rhizosphere soils of health and infected C. chinensis at the phylum level; (B) Bacterial composition in rhizome of health and infected C. chinensis at the phylum level; (C) Bacterial composition in leaf of health and infected C. chinensis at the phylum level; (D) Fungus composition in rhizosphere soils of health and infected C. chinensis at the phylum level; (E) Fungus composition in rhizome of health and infected C. chinensis at the phylum level; (F) Fungus composition in leaf of health and infected C. chinensis at the phylum level.
3.5. Overall association analysis of fungal and bacterial microbiomes
The overall correlation between fungal and bacterial microorganisms in healthy C. chinensis samples and diseased C. chinensis samples was analyzed by procrustes analysis. As shown in Figure 4, the overall structure between bacterial microbiome and fungal microbiome in healthy C. chinensis rhizosphere soils, rhizome, and leaf samples was significantly correlated (m122 values <0.45, correlation values >0.775, and significance values ≤0.001; Figure 4A). However, the overall structural correlation between bacterial microbiome and fungal microbiome in the rhizosphere soil, rhizome, and leaf samples of C. chinensis disappeared after root rot infection (Figure 4B). This result indicated that root rot infection of C. chinensis severely damaged the ecological balance of the original bacterial microbiome and fungal microbiome of healthy C. chinensis, and root rot infection significantly inhibited the interaction of endophytic fungi and bacteria in C. chinensis.
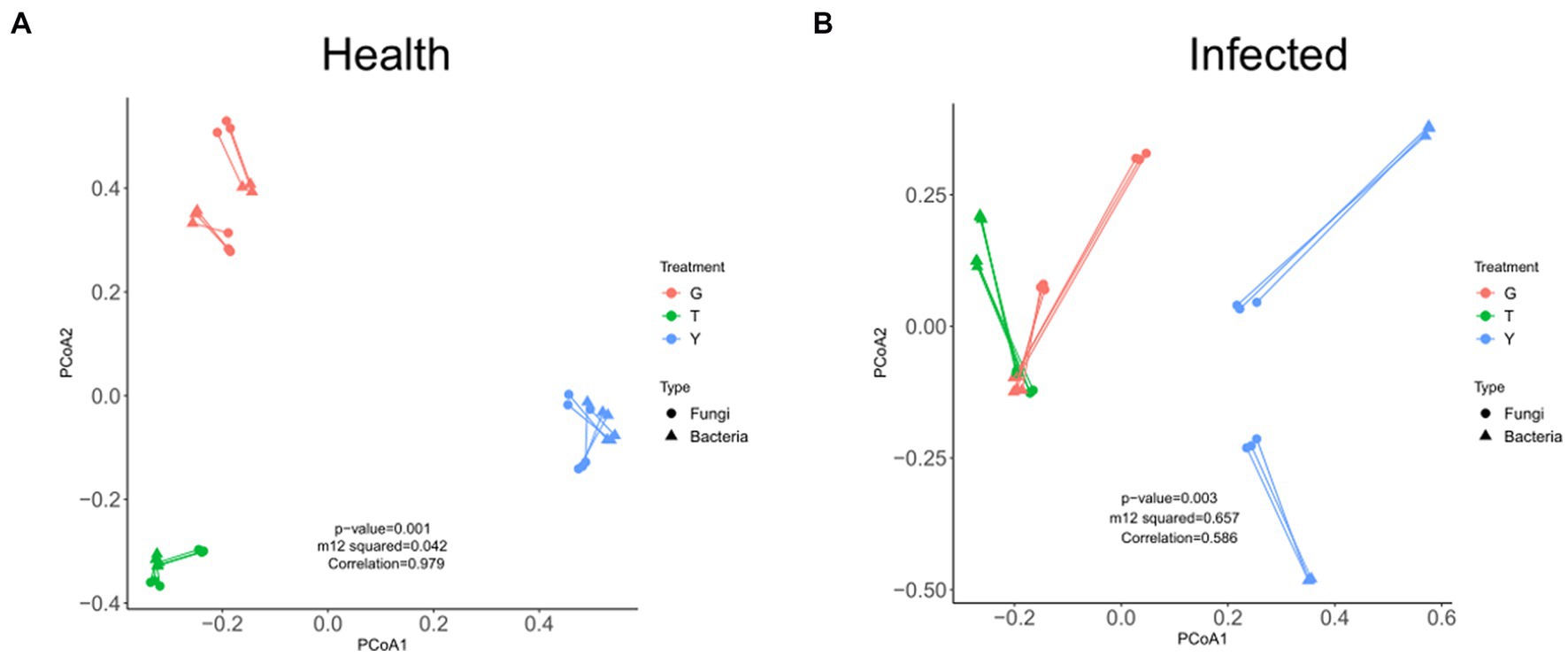
Figure 4. Procrustes analysis under fungal microbiome and bacterial microbiome of health and infected samples. (A) Procrustes analysis under fungal microbiome and bacterial microbiome of health samples; (B) Procrustes analysis under fungal microbiome and bacterial microbiome of infected samples. The color of each dot/triangle represents fungal microbiome/bacterial microbiome. Satisfying conditions were significant (m122 values of <0.45, correlation values of >0.775, and significance values of ≤0.001).
Core microbes refer to those whose relative abundance are greater than 0.05% and who are present in at least half of the samples. In this study, an interaction network analysis was performed of the core microbes of fungi and bacteria in healthy and diseased C. chinensis samples. Sixty core microbes were identified from healthy C. chinensis rhizosphere soil, rhizomes, and leaf samples, including 26 fungi and 34 bacteria (Supplementary Table S1). A total of 60 core microbes were identified from the infected C. chinensis samples, including 38 fungi and 22 bacteria (Supplementary Table S2). Based on the R package WGCNA, the Spearman algorithm was used to analyze the correlation network between the healthy and diseased core microbes. The results showed that the fungal microbes in the healthy C. chinensis samples were significantly separated from bacterial microbes, and the bacterial distribution was denser and the correlation was more complex (Supplementary Figure S1A). The independent effects between bacteria and fungi in the infected C. chinensis samples were disrupted, and the correlation of fungi was also significantly weakened, while bacterial correlation network became denser (Supplementary Figure S1A). This result corroborates with the results of Procrustes analysis.
3.6. Microbial difference analysis of healthy and diseased Coptis chinensis samples
In order to further determine the microbes whose abundance changes lead to the disturbance of soil and plant microbial homeostasis and are related to the occurrence of C. chinensis root rot, the DESeq2 package in R was used to analyze the relative abundance of microbes in the rhizosphere soil, rhizomes, and leaves of healthy and diseased C. chinensis sample. As shown in Figure 5, the differential bacterial communities of C. chinensis leaf samples were relatively less, indicating that the infection had the weakest effect on the bacterial community of C. chinensis leaf samples. The effects of infection on the fungal community in the rhizosphere soil, rhizome, and leaf samples of C. chinensis were relatively comparable. However, no common intersection in differential microbial communities was found among the three types of C. chinensis samples. Because the rhizosphere soil and roots in space contact more closely, we speculated that the differential microbial communities shared by rhizosphere soil and rhizome samples in healthy versus infected pairwise comparison played an important role in affecting microbial homeostasis. In total, there were 30 shared genera of differential bacterial communities (Supplementary Table S3), and 7 shared genera of differential fungal communities (Supplementary Table S4). The abundance of 22 shared genera of differential bacterial communities including Aridibacter, Sphingopyxis, Oligoflexus, Pseudorhodoferax, Polaromonas in the infected group was higher than that in the healthy group, which may play an important role in inhibiting the fungal pathogens of C. chinensis (Supplementary Table S5). Subsequent screening of antagonists should focus on these genera. In terms of fungi, Clonostachys, Plenodomus, and unclassified_Ceratobasidiaceae significantly enriched in the infected group, they may promote disease occurs or were the potential pathogen of C. chinensis root rot. While the abundance of Meliniomyces, Oidiodendron, Xenochalara and unidentified_Glomeraceae in healthy groups was significantly higher than that in infected groups, and they may play a certain role in inhibiting the root rot of C. chinensis (Supplementary Table S6).
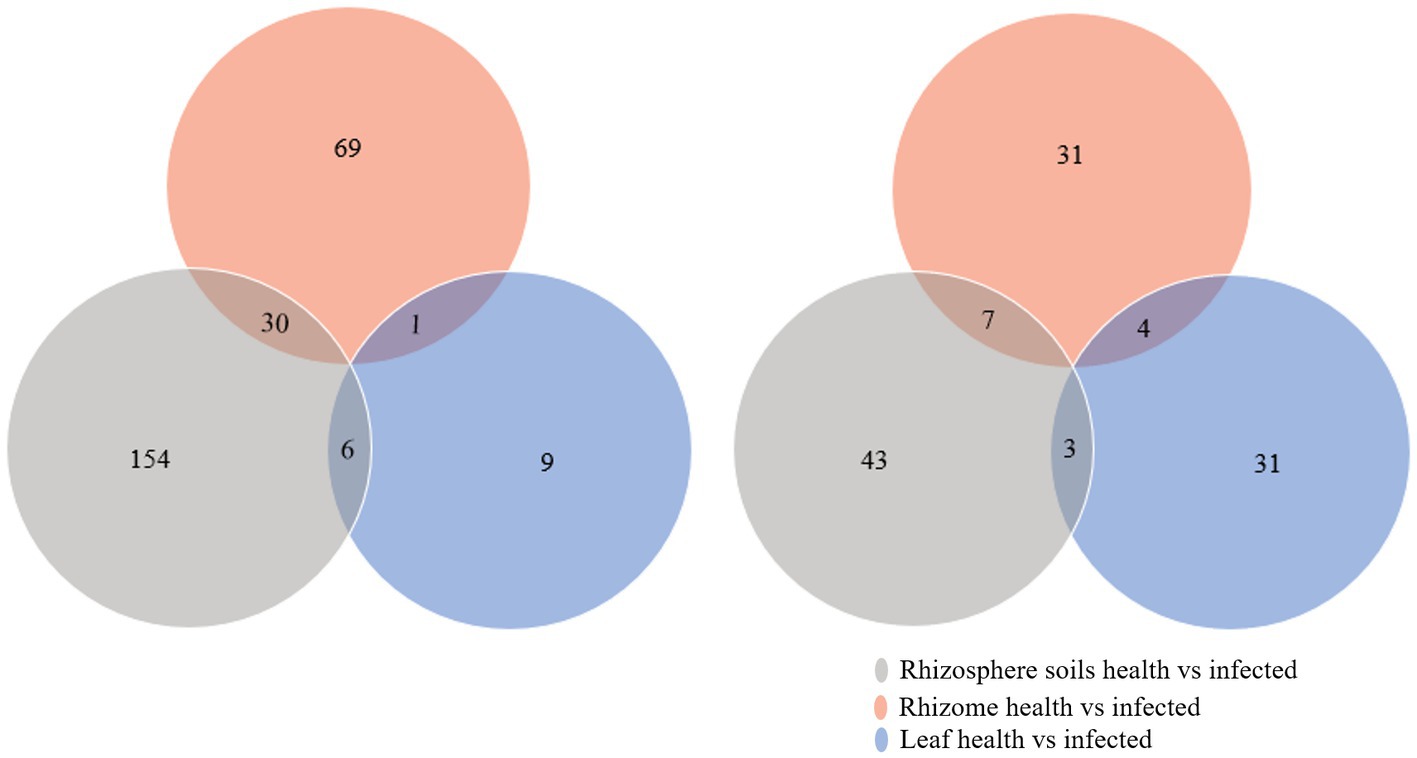
Figure 5. Venn diagram of the differential microbe among health and infected rhizosphere soils, rhizome, and leaf of C. chinensis. (A) Venn diagram of the differential bacteria among health and infected rhizosphere soils, rhizome, and leaf of C. chinensis; (B) Venn diagram of the differential fungi among health and infected rhizosphere soils, rhizome, and leaf of C. chinensis.
3.7. Correlation analysis between core microorganisms and soil physical and chemical factors
Since the rhizosphere soil is in close contact with the rhizome, the root is the main site for the interaction between plants and microorganisms (Lundberg et al., 2012), we performed the correlation analyzes between soil physicochemical factors and the differential microbes (30 bacteria and 7 fungi) shared by the rhizosphere soil samples and rhizome samples (in Figures 5A,B). First, based on Bary-curtics distance, the Mantel analysis was performed using Vegan package in R. The results showed that the core differential microbial structure was significantly correlated with the structure of soil factors (Significance = 0.021; Supplementary Table S7). In order to explore the specific one-to-one correlation between the core differential microbes and the physicochemical properties of the rhizosphere soil, the Spearman correlation analysis was conducted using WGCNA package in R. As shown in Figure 6, a significant correlation was observed between the soil factors and the differential microbes (healthy vs. infected) shared by rhizosphere soil samples and rhizome samples. It could also be seen from the Figure 6 that fungi and rhizosphere soil physicochemical factors were mostly positively correlated, while bacteria and rhizosphere soil physicochemical factors were mostly negatively correlated. Physicochemical properties such as pH, organic matter, total nitrogen, total phosphorus, available nitrogen, and available phosphorus were relatively strongly correlated with microbial communities, while total potassium and available potassium were weakly correlated with microbial communities (Figure 6).
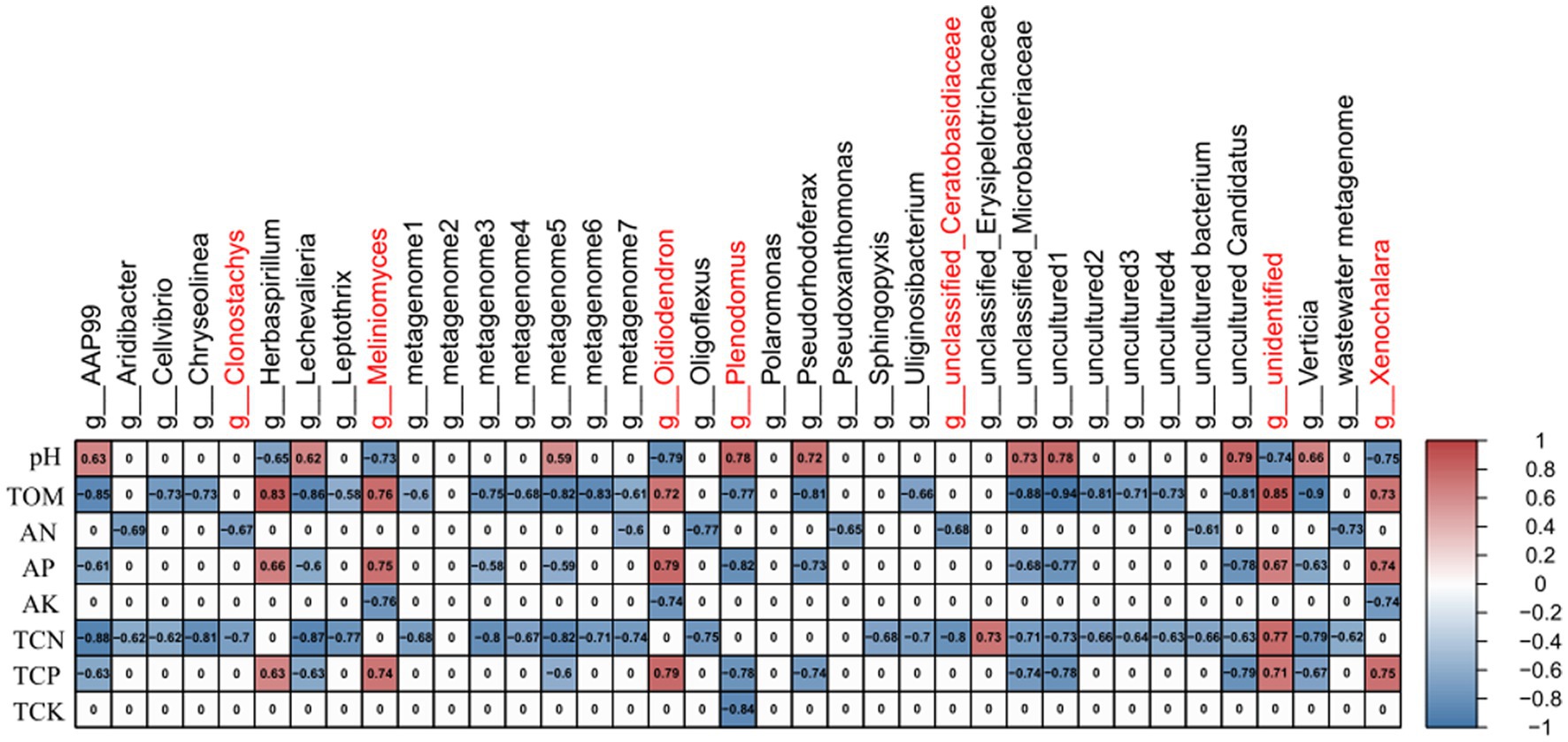
Figure 6. Correlation between soil physicochemical factors of rhizosphere soil and core microorganisms. Red font is marked as fungi; black font is marked as bacteria; red represents positive correlation; blue represents negative correlation in table.
4. Discussion
Soil microbial communities are widely considered as an integrated component of soil quality due to their important roles in many ecological processes (Fierer et al., 2012). Soil microbial community richness and diversity play a crucial role in soil quality, function, and productivity, and soil properties may be an important factor controlling microbial community structure (Lauber et al., 2008; Song et al., 2018). Spearman correlation analysis (based on the WGCNA package) showed that pH, organic matter, total nitrogen, total phosphorus, available nitrogen, and available phosphorus were significantly correlated with microbial communities. This result is consistent with previous study report that physicochemical properties such as soil fertility can significantly affect soil microbial community structure (Bai et al., 2018). The differential microbes in diseased versus healthy C. chinensis comparison were strongly correlated with total nitrogen, total phosphorus, available nitrogen, and available phosphorus, but weakly correlated with total potassium and available potassium, suggesting that excessive nitrogen and phosphate were applied in Coptis chinensis planting area, but relatively less potassium was applied. Fungi and rhizosphere soil physicochemical factors were mostly positively correlated, while bacteria were mostly negatively correlated with rhizosphere soil physicochemical factors, indicating that excessive fertilization might also be one of the important reasons for the serious fungal disease of C. chinensis.
In this study, we found that infection with root rot disease had a great impact on the diversity and abundance of microbial communities in the rhizosphere soil, rhizome, and leaf samples of C. chinensis. It is well known that many microorganisms can induce or promote plant defense against various pathogens (Maksimov et al., 2011; Pieterse et al., 2014). The reduction in microbial diversity and abundance has been recognized as a major threat to ecosystems and a major cause of soil function loss, which is also the main cause of continuous cropping obstacles (Singh et al., 2014; Maron et al., 2018). In this study, infection with root rot did not significantly reduce microbial diversity and abundance, but infection with root rot significantly altered the fungal and bacterial community structure in the rhizosphere soil, rhizome, and leaf samples of C. chinensis. In addition, root rot infection of C. chinensis severely disrupted the ecological balance of the original bacterial and fungal microbiomes of healthy C. chinensis, and root rot infection significantly inhibited the interaction of endophytic fungi and bacteria in C. chinensis Zhang et al. (2017) found that Huanglongbing infection led to a decrease in the relative abundance of the microbiomes enriched on the surface of citrus roots and the expression activity of specific functions, thus leading to impaired plant-host-microbiome interactions. The rhizosphere surface microbes and endophyte microbes in the cotton infected with Verticillium wilt were significantly altered, and the numbers of rhizosphere fungi and endophyte fungi were significantly increased in diseased plants (Zhang et al., 2022). Beneficial microbial communities are involved in plant-pathogen interactions, and they possess the energy and potential to effectively enhance plant growth and productivity. The dominant microbiotas in citrus rhizosphere microorganisms are Burkholderia and Bradyrhizobium, and previous experiments have shown that inoculation of Burkholderia strain isolated from the related microorganisms in healthy citrus roots can induce the expression of systemic resistance-related genes in the inoculated plants, thus functioning in resisting citrus Huanglongbing (Zhang et al., 2017). Our sequencing data also showed that Burkholderia and Bradyrhizobium were dominant in the rhizome samples of C. chinensis, and the relative abundance of Burkholderia in healthy rhizome samples was significantly higher than that in diseased rhizome samples. The pathogen of C. chinensis root rot disease destroyed the ecological balance of Burkholderia during the infection process, which indirectly indicated that Burkholderia might play a certain role in resisting C. chinensis root rot. Plant pathogens need to break through the barrier of microorganisms on the rhizosphere surface to invade plants, which provides a new perspective for plant protection. Namely, crops could be protected from diseases by building a healthy microbial system. Cucumber plants susceptible to Fusarium tend to enrich beneficial microorganisms such as Comamonadaceae and Xanthomonadaceae to control Fusarium wilt [37]. Previous studies have found that leaf fall of healthy adult cacao (Theobroma cacao) brings the intact microbiome to the ground, thereby protecting seedlings from disease (Christian et al., 2017). Li et al. (2021) have analyzed the characteristics of the microbial community in the rhizosphere and root of Astragalus membranaceus infected with root rot disease by sequencing, and have constructed an artificial microbial community based on the significantly enriched bacteria in the rhizosphere of A. membranaceus infected with root rot disease. This artificial microbial community can inhibit the growth of pathogens and trigger the induced systemic resistance (ISR) of the diseased A. membranaceus, thus reducing the incidence of A. membranaceus root rot disease (Li et al., 2021). In our sequencing data, Bradyrhizoloium sp., Acidibacter sp., and Streptemyces sp. were significantly enriched in the diseased rhizome samples. Although these bacterial communities were not the same as those enriched in A. membranaceus root rot samples, these bacterial communities also will be the focus of our follow-up research. Potato common scab disease affects the rhizosphere soil microbial community structure, which in turn is significantly associated with the expression of Scab toxin gene txtAB (Shi et al., 2019). All these findings indicate that plant roots can selectively accumulate beneficial microorganisms and directly inhibit pathogens to improve crop yield. In addition, the healthy microbial system of plants can also be artificially constructed to improve the induced systemic resistance of plants or to directly participate in the resistance to pathogens. The destruction of the microecological balance between the rhizosphere soil microbiome and the host endogenous microbiome may also be an important reason for the aggravation of disease damage and the difficulty of prevention and control.
This study focused on the influence of root rot infection on rhizosphere microbial community structure of C. chinensis, but the specific process of this influence was not analyzed. Was it the pathogen that affected the root exudates of C. chinensis, then affected the rhizosphere soil microbial community structure, and then leads to changes in the microbial community structure of rhizome and leaf of C. chinensis? Or does the pathogen directly produce some compound that stresses on the structure of the microbial community? These problems were urgent for us to try to solve. In addition, Aridibacter, Sphingopyxis, Oligoflexus, Pseudorhodoferax, Polaromonas Meliniomyces, Oidiodendron, and Xenochalara play an important role in inhibiting the root rot of C. chinensis. However, these microorganisms have not been isolated, identified and functionally verified. Later work will focus on the isolation and identification of rhizosphere soil microorganisms, the verification of bacteriostatic and growth-promoting functions, and the construction of synthetic bacteria for root rot prevention and control.
5. Conclusion
In this study, we confirmed that infection with root rot would destroy the ecological balance of the microbiomes in the rhizosphere soil, rhizome and leaf samples, and that some microbiotas were strongly correlated with soil physicochemical factors. Unfortunately, the sequencing data failed to reveal the main pathogen for C. chinensis root rot disease. It is speculated that C. chinensis root rot may be caused by the combined infection of multiple pathogens. Root rot infection destroyed the ecological balance of the original microbiomes in the rhizosphere soil, rhizome, and leaf samples of C. chinensis. Bradyrhizoloium sp., Acidibacter sp., and Streptemyces sp. which would be the focus of following microecological regulation research, were significantly enriched in the diseased rhizome samples. These results provide direction for the use of micro-ecological regulation methods to prevent and control the root rot of C. chinensis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
TT, FW, TM, JG, XG, and YD performed the experiments and analyzed the data. GF, HK, GS, and JY designed the experiments, supervised the study, evaluated the data, and wrote and revised the manuscript for publication. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the central government guides local science and technology development fund projects (2022BGE256), Hubei Technology Innovation Center for Agricultural Sciences-Key Research and Development Project of Science and Technology (2020–620–000-002-04).
Acknowledgments
The authors thank Liu Ping (College of Foreign Languages, Huazhong Agriculture University, Wuhan, China) for her critical reading of the manuscript. They also thank the Institute of Plant Protection and Soil and Fertilizer of Hubei Academy of Agricultural Sciences for soil physicochemical factors determining.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1180368/full#supplementary-material
Footnotes
1. ^ https://ngdc.cncb.ac.cn/gsa/
2. ^ https://docs.qiime2.org/2020.11/tutorials/
References
Bai, Y., Müller, D. B., Srinivas, G., Garrido-Oter, R., Potthoff, E., Rott, M., et al. (2015). Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369. doi: 10.1038/nature16192
Bai, L., Sun, H., Zhang, X., and Cai, B. (2018). Next-generation sequencing of root fungal communities in continuous cropping soybean. Chil. J. Agric. Res. 78, 528–538. doi: 10.4067/S0718-58392018000400528
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. H. M. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bernardo-Cravo, A. P., Schmeller, D. S., Chatzinotas, A., Vredenburg, V. T., and Loyau, A. (2020). Environmental factors and host microbiomes shape host–pathogen dynamics. Trends Parasitol. 36, 616–633. doi: 10.1016/j.pt.2020.04.010
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland Forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Bremner, J. M. (1960). Determination of nitrogen in soil by the Kjeldahl method. J. Agric. Sci. 55, 11–33. doi: 10.1017/S0021859600021572
Christian, N., Herre, E. A., Mejia, L. C., and Clay, K. (2017). Exposure to the leaf litter microbiome of healthy adults protects seedlings from pathogen damage. Proc. R. Soc. B 284:20170641. doi: 10.1098/rspb.2017.0641
Fierer, N., Lauber, C. L., Ramirez, K. S., Zaneveld, J., Bradford, M. A., and Knight, R. (2012). Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 6, 1007–1017. doi: 10.1038/ismej.2011.159
Hawkes, C. V., Bull, J. J., and Lau, J. A. (2020). Symbiosis and stress: how plant microbiomes affect host evolution. Philos. Trans. R. Soc. B 375:20190590. doi: 10.1098/rstb.2019.0590
He, Y., Xiao, H., Deng, C., Fan, G., Qin, S., and Peng, C. (2017). Complete chloroplast genome sequence of Coptis chinensis Franch. And its evolutionary history. Biomed. Res. Int. 2017, 1–7. doi: 10.1155/2017/8201836
Huang, L. L., Wang, J. A., Xu, R., and Liu, Z. Y. (2020). Study on mechanism of Huanglian Jiedu decoction in treating novel coronavirus pneumonia based on network pharmacology. J. Chin. Med. Mater. 3, 779–785. doi: 10.13863/j.issn1001-4454.2020.03.050
Jiang, J., Song, Z., Yang, X., Mao, Z., Nie, X., Guo, H., et al. (2017). Microbial community analysis of apple rhizosphere around Bohai gulf. Sci. Rep. 7:8918. doi: 10.1038/s41598-017-08398-9
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Lauber, C. L., Strickland, M. S., Bradford, M. A., and Fierer, N. (2008). The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 40, 2407–2415. doi: 10.1016/j.soilbio.2008.05.021
Li, Z., Bai, X., Jiao, S., Li, Y., Li, P., Yang, Y., et al. (2021). A simplified synthetic community rescues Astragalus mongholicus from root rot disease by activating plant-induced systemic resistance. Microbiome 9:217. doi: 10.1186/s40168-021-01169-9
Li, H. Z., Li, N., Wang, J. J., Li, H., Huang, X., Guo, H., et al. (2020). Dysbiosis of gut microbiome affecting small intestine morphology and immune balance: a rhesus macaque model. Zool. Res. 41, 20–31. doi: 10.24272/j.issn.2095-8137.2020.004
Li, H., Li, Y., Xu, Y., and Lu, X. (2020). Biochar phosphorus fertilizer effects on soil phosphorus availability. Chemosphere 244:125471. doi: 10.1016/j.chemosphere.2019.125471
Liao, H. L., Huang, L., Li, N., Ke, W. J., Xiang, Y. Q., and Ma, Y. T. (2021). Auxiliary rapid identification of pathogenic and antagonistic microorganisms associated with Coptis chinensis root rot by high-throughput sequencing. Sci. Rep. 11:11141. doi: 10.1038/s41598-021-90489-9
Liu, Y., Wang, B., Shu, S., Li, Z., Song, C., Liu, D., et al. (2021). Analysis of the Coptis chinensis genome reveals the diversification of protoberberine-type alkaloids. Nat. Commun. 12:3276. doi: 10.1038/s41467-021-23611-0
Liu, H. Y., Wang, W., Zhang, R. F., Raxida, A., and Yao, J. (2019). Fungal community structure of cotton-field soil under different incidences of cotton verticillium wilt. Sci. Agric. Sin. 52, 455–465. doi: 10.3864/j.issn.0578-1752.2019.03.006
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Luo, X.-M., Li, J.-L., Dong, J.-Y., Sui, A.-P., Sheng, M.-L., and Yang, X.-Y. (2014). First report of fusarium solani causing root rot on Coptis chinensis in southwestern China. Plant Dis. 98:1273. doi: 10.1094/PDIS-02-14-0164-PDN
Maksimov, I. V., Abizgildina, R. R., and Pusenkova, L. I. (2011). Plant growth promoting rhizobacteria as alternative to chemical crop protectors from pathogens (review). Appl. Biochem. Microbiol. 47, 333–345. doi: 10.1134/S0003683811040090
Maron, P.-A., Sarr, A., Kaisermann, A., Lévêque, J., Mathieu, O., Guigue, J., et al. (2018). High microbial diversity promotes soil ecosystem functioning. Appl. Environ. Microbiol. 84, e02738–e02717. doi: 10.1128/AEM.02738-17
Mei, P. Y., Song, X. H., Zhu, Z. Y., and Li, L. Y. (2021a). First report of Diaporthe eres causing root rot of Coptis chinensis. Plant Dis. 105:1854. doi: 10.1094/PDIS-12-20-2564-PDN
Mei, P. Y., Song, X. H., Zhu, Z. Y., and Li, L. Y. (2021b). First report of root rot caused by fusarium avenaceum on Coptis chinensis in Chongqing, China. Plant Dis. 105:496. doi: 10.1094/PDIS-05-20-1110-PDN
Michrina, B. P., Fox, R. H., and Piekielek, W. P. (1982). Chemical characterization of two extracts used in the determination of available soil nitrogen. Plant Soil 64, 331–341. doi: 10.1007/BF02372516
Murphy, J., and Riley, J. P. (1962). A modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta 27, 31–36. doi: 10.1016/S0003-2670(00)88444-5
Pieterse, C. M. J., Zamioudis, C., Berendsen, R. L., Weller, D. M., Van Wees, S. C. M., and Bakker, P. A. H. M. (2014). Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 52, 347–375. doi: 10.1146/annurev-phyto-082712-102340
Scholthof, K.-B. G. (2007). The disease triangle: pathogens, the environment and society. Nat. Rev. Microbiol. 5, 152–156. doi: 10.1038/nrmicro1596
Singh, B. K., Quince, C., Macdonald, C. A., Khachane, A., Thomas, N., Al-Soud, W. A., et al. (2014). Loss of microbial diversity in soils is coincident with reductions in some specialized functions: microbial diversity and ecosystem functions. Environ. Microbiol. 16, 2408–2420. doi: 10.1111/1462-2920.12353
Shi, W. C., Li, M. C., Wei, G. S., Tian, R. M., and Li, C. P. (2019). The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome. Microbiome. 7:14. doi: 10.1186/s40168-019-0629-2
Song, X., Pan, Y., Li, L., Wu, X., and Wang, Y. (2018). Composition and diversity of rhizosphere fungal community in Coptis chinensis Franch. Continuous cropping fields. PLoS One 13:e0193811. doi: 10.1371/journal.pone.0193811
Tian, H., Qiao, J., Zhu, Y., Jia, X., and Shao, M. (2021). Vertical distribution of soil available phosphorus and soil available potassium in the critical zone on the loess plateau, China. Sci. Rep. 11:3159. doi: 10.1038/s41598-021-82677-4
Tseng, C.-Y., Sun, M.-F., Li, T.-C., and Lin, C.-T. (2020). Effect of Coptis chinensis on biofilm formation and antibiotic susceptibility in Mycobacterium abscessus. Evid. Based Complement. Altern. 2020, 1–9. doi: 10.1155/2020/9754357
Wen, T., Yuan, J., He, X., Lin, Y., Huang, Q., and Shen, Q. (2020). Enrichment of beneficial cucumber rhizosphere microbes mediated by organic acid secretion. Hortic. Res. 7:154. doi: 10.1038/s41438-020-00380-3
Wu, Q., Liang, Y., Li, Y., Wang, X., Yang, L., and Wang, X. (2017). Factors acquisition and content estimation of farmland soil organic carbon based upon internet of things. Chin. Geogr. Sci. 27, 431–440. doi: 10.1007/s11769-017-0875-9
Zaura, E., Keijser, B. J., Huse, S. M., and Crielaard, W. (2009). Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 9:259. doi: 10.1186/1471-2180-9-259
Zhang, Y., Xu, J., Riera, N., Jin, T., Li, J., and Wang, N. (2017). Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 5:97. doi: 10.1186/s40168-017-0304-4
Keywords: Coptis chinensis , rhizosphere soil and endophytic microbiomes, microecological regulation, microecological balance, soil physicochemical factors
Citation: Tang T, Wang F, Fang G, Mao T, Guo J, Kuang H, Sun G, Guo X, Duan Y and You J (2023) Coptischinensis Franch root rot infection disrupts microecological balance of rhizosphere soil and endophytic microbiomes. Front. Microbiol. 14:1180368. doi: 10.3389/fmicb.2023.1180368
Edited by:
Zhang Chuanqing, Zhejiang Agriculture and Forestry University, ChinaReviewed by:
Sher Muhammad, Government College University, Faisalabad, PakistanWei Zhou, Zhejiang Chinese Medical University, China
Copyright © 2023 Tang, Wang, Fang, Mao, Guo, Kuang, Sun, Guo, Duan and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingmao You, amluZ21hb3lvdUAxMjYuY29t