 Li Qi
Li Qi Mengke Shi
Mengke Shi Fang-Chao Zhu
Fang-Chao Zhu Chun-Ang Lian
Chun-Ang Lian Li-Sheng He
Li-Sheng He- 1Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences, Sanya, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Key Laboratory of Tropical Marine Ecosystem and Bioresource, Fourth Institute of Oceanography, Ministry of Natural Resources, Beihai, China
The genus Rimicaris is the dominant organism living in hydrothermal vents. However, little research has been done on the functions of their intestinal flora. Here, we investigated the potential functions of Deferribacterota, which is dominant in the intestine of Rimicaris kairei from the Central Indian Ridge. In total, six metagenome-assembled genomes (MAGs) of Deferribacterota were obtained using the metagenomic approach. The six Deferribacterota MAGs (Def-MAGs) were clustered into a new branch in the phylogenetic tree. The six Def-MAGs were further classified into three species, including one new order and two new genera, based on the results of phylogenetic analysis, relative evolutionary divergence (RED), average nucleotide identity (ANI), average amino acid identity (AAI) and DNA–DNA hybridization (DDH) values. The results of the energy metabolism study showed that these bacteria can use a variety of carbon sources, such as glycogen, sucrose, salicin, arbutin, glucose, cellobiose, and maltose. These bacteria have a type II secretion system and effector proteins that can transport some intracellular toxins to the extracellular compartment and a type V CRISPR–Cas system that can defend against various invasions. In addition, cofactors such as biotin, riboflavin, flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD) synthesized by R. kairei gut Deferribacterota may also assist their host in surviving under extreme conditions. Taken together, the potential function of Deferribacterota in the hydrothermal R. kairei gut suggests its long-term coevolution with the host.
1. Introduction
The Rimicaris shrimp is dominant in many hydrothermal vents. There are at least ten species in this genus, including R. chacei, R. paulexa, R. parva, R. susannae, R. exoculata, R. falkorae, R. hybisae, R. kairei, R. vandoverae, and R. variabilis. They mainly live in the hydrothermal fields with some difference in distribution among these species. For examples, R. exoculata is mainly living at the hydrothermal vent of the Mid-Atlantic Ridge (MAR), R. kairei is mainly resident at the Central Indian Ridge (Watabe and Hashimoto, 2002), and R. chacei also lives in the Atlantic Ridge, but most at the periphery of R. exoculata aggregates (Apremont et al., 2018). Within the Rimicaris genus, R. exoculata, R. kairei, and R. hybisae have an enlarged branchial chamber with a highly dense population of epibiotic bacteria (Zbinden and Cambon-Bonavita, 2020). R. exoculata is reported to house a density of epibiotic bacteria in cephalothoracic chamber and modified mouthparts (Gebruk et al., 2000). Due to that, a body of studies has focused on the epibionts and their benefits to Rimicaris species. The main epibiotic communities in R. exoculata are Gamma-, Alpha-, Beta-, Delta-, Zetaproteobacteria, Campylobacteria, and Bacteroidetes (Zbinden et al., 2008; Petersen et al., 2010; Hügler et al., 2011; Guri et al., 2012; Jan et al., 2014; Jiang et al., 2020). A few studies have also reported the gut microbial composition of R. exoculata. Three major groups, Deferribacteres, Mollicutes, and Campylobacteria, were found in the midgut, along with small amounts of Gammaproteobacteria (Zbinden and Cambon-Bonavita, 2003; Durand et al., 2010; Cowart et al., 2017). Additionally, the three main lineages were still present after 72 h of starvation, so they have designated resident epibionts rather than transient microflora (Durand et al., 2010). Besides, Apremont et al. showed that Deferribacteres, Mollicutes, and Epsilon- and Gammaproteobacteria were the main microbes existed in the digestive tract of R. chacei (Apremont et al., 2018). The difference between the intestinal microbial composition of R. exoculata and R. chacei may be due to the different living environments. In our previous studies, we showed that the intestinal microbiota of R. kairei, which was from the Central Indian Ridge, was dominant by Deferribacterota, Campylobacter, Bacteroidetes, Firmicutes, and Proteobacteria, although there is a significant difference in the composition between different developing stages (Qi et al., 2021). To date, some studies have been reported on the intestinal microbial community of Rimicaris species, but there are few studies on their functions, even though the functions of the gill symbiotic microbiota have been extensively investigated. Therefore, we will further investigate the functions of the gut microbiota and present the relationship with their host Rimicaris species.
Deferribacteres, a new phylum recorded in 2001 (Garrity et al., 2001), was emended in 2009 to represent only the family Deferribacteriaceae (Jumas-Bilak et al., 2009). In 2011, the phylogeny of the phylum was repositioned (Kunisawa, 2011), and the new name Deferribacterota was given in 2018 (Whitman et al., 2018). At present, there are few reports on the genomic function of Deferribacterota. Members of the phylum Deferribacterota are organized into a single order and six families. A deep lineage is the Deferribacteraceae family, whose genus Deferribacter includes four species, D. thermophilus, D. desulfuricans, D. abyssii, and D. autotrophicus. D. thermophilus was isolated from a high-temperature around 60°C, seawater-flooded oil reservoir in the North Sea (Greene et al., 1997), while the other three species were all isolated from deep-sea hydrothermal vents (Miroshnichenko et al., 2003; Takaki et al., 2010; Slobodkin et al., 2019). D. desulfuricans was obtained from the Suiyo Seamount hydrothermal chimney (Takaki et al., 2010), D. abyssi was isolated from the Rainbow hydrothermal vent field of the Mid-Atlantic Ridge (Miroshnichenko et al., 2003), and D. autotrophicus was isolated from Ashadze hydrothermal chimney on the Mid-Atlantic Ridge at a depth of 4,100 m (Slobodkin et al., 2019). All recognized Deferribacter species are strictly anaerobic and thermophilic organisms, which can oxidize various complex organic compounds and organic acids in the presence of diverse electron acceptors. D. desulfurican can use formate, acetate, and pyruvate as substrates; D. abyssii is capable of using molecular hydrogen, acetate, succinate, pyruvate, and proteinaceous compounds as electron donors and elemental sulfur, nitrate, or Fe (III) as electron acceptors; D.autotrophicu used molecular hydrogen, acetate, lactate, succinate, pyruvate and complex proteinaceous compounds as electron donors, and Fe (III), Mn (IV), nitrate or elemental sulfur as electron acceptors; D. thermophilus obtained energy from the reduction of manganese (IV), iron(III), and nitrate in the presence of yeast extract, peptone, casamino acid, tryptone, hydrogen, malate, acetate, citrate, pyruvate, lactate, succinate, and valerate. D. desulfurican is a heterotrophic bacterium, while all of the other three species are chemolithoautotrophic bacteria. In addition, Flexistipes sinusarabici, which was most closely related to Deferribacter and isolated from the Atlantis Deep brines of the Red Sea, is tolerant to high temperature, high salt concentration and heavy metals, and strictly anaerobic. This organism prefers complex growth substrates such as yeast extract, meat extract, peptone, and trypsin, while formate, lactate, citrate, malate, carbohydrate, amino acid, and alcohol do not support cell growth (Lapidus et al., 2011). Up to now, only one species of Mucispirillum schaedleri in the phylum of Deferribacterota has been isolated from the intestine. M. schaedleri inhabits the intestinal mucus layer of rodents and other animals in abundance and are considered pathogenic. M. schaedleri harbors a complete Embden-Meyerhof-Parnas (EMP) pathway and a nonoxidative pentose phosphate pathway as well as a complete tricarboxylic acid cycle. M. schaedleri can also alter gene expression in mucosal tissues, suggesting an intimate interaction with the host (Loy et al., 2017).
Different from the reported Deferribacterota in the hydrothermal zone, the six Deferribacterota from R. kairei guts are heterotrophic according to the MAG analysis. Def-MAGs do not have genes related to carbon, nitrogen, and sulfur utilization. Instead, they have a complete glycolysis pathway and genes for transporting and degrading polysaccharides. Although slight differences among the six Def-MAGs, the main functions of carbohydrate metabolism, polysaccharide degradation, vitamin synthesis and so on were similar. This study further elucidated the diversity of Deferribacterota and their host interaction relationships, supplementing the understanding of existing Deferribacterota.
2. Materials and methods
2.1. Sample collection
The shrimp were collected from two sites in the Central Indian Ridge by the manned submersible Deep-Sea Yongshi during R/V Tansuoyihao research cruise TS10 (February 2019). Sampling sites were located in Edmond (69.59667°E, 23.87782°S) and Kairei (70.04010°E, 25.32048°S) at depths of 3,281 m and 2,421 m, respectively (Supplementary Figure S1). All shrimp were obtained using the suction sampler. Once onboard, individuals were immediately frozen at −80°C or stored in 75% ethanol at −20°C.
2.2. DNA extraction and metagenome sequencing
The samples were identified as R. kairei in our previous article (Qi et al., 2021). The intestinal anatomy of R. kairei was performed in a sterile environment, and the total DNA of the gut was extracted using a PowerSoil DNA isolation kit (Qiagen, Germany) following the manufacturer’s procedures. The quality and quantity of genomic DNA were checked by gel electrophoresis. The DNA concentration was determined by using a Qubit dsDNA HS assay kit with a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA). A total of 100 ng DNA was used for library preparation. High-throughput sequencing was performed with the Novaseq 6000 platform to produce 2 × 150 bp paired-end reads (Illumina).
2.3. Metagenomic assembly and genome binning
Metagenomic DNA sequencing produced a total of 154,387,934 reads, with a total length of 45 Gbp. Trimmomatic v0.36 was used for trimming with parameters (LEADING: 3; The TRAILING: 3; HEADCROP: 4; SLIDINGWIDOW: 4:15; MINLEN: 80) (Bolger et al., 2014), and FastQC v0.11.9 was used to evaluate the quality of data before and after filtering (Brown et al., 2017). The qualified reads were further assembled into contigs by SPAdes-3.11 (Bankevich et al., 2012) software with a k-mer range of 21 to 127. MetaWRAP v1.2.1 (Uritskiy et al., 2018) was used for genome binning and subsequent refinement with parameters (three different algorithms MaxBin2, metaBAT2, and CONCOCT for metagenomic binning; contig length: >2000 bp; completeness: >50%; and contamination: <10%). The MAGs (metagenome assembled genomes) were checked by CheckM v1.1.3 (Parks et al., 2015) to filter those with low completeness (<50%) and high contamination (>10%). Taxonomic annotation of the MAGs was performed using GTDB-tk v1.4.0 software (Chaumeil et al., 2019). The whole-genome average nucleotide identity (ANI) between genomes was calculated by fastANI v1.33 software (Jain et al., 2018). Average Amino acid Identity (AAI) was calculated by AAI calculator online tool (Rodríguez-R and Konstantinidis, 2014). DNA–DNA hybridization (DDH) was calculated by Genome-to-Genome Distance Calculator 3.0 online tool (Meier-Kolthoff et al., 2021). GTDB-tk software was used to calculate the relative evolutionary divergence (RED) values when a query genome could not be classified based on the ANI values. Then, the MAGs belonging to Deferribacterota bacteria were retrieved for downstream analyses.
2.4. Phylogenetic analyses
There were 79 genomes in total for phylogenetic analyses, including 57 genomes from Tenericutes, Firmicutes, Acidobacteria, Chrysiogenetes, and Proteobacteria, and the others were from the phylum Deferribacterota. Firstly, 43 ribosome proteins were obtained by CheckM analysis from all of these genomes and then aligned by MAFFT v7.487 (Katoh et al., 2002) with the default settings, and poorly aligned regions were removed by trimAI v1.4 (Capella-Gutiérrez et al., 2009). The maximum likelihood (ML) phylogenomic tree was constructed using the concatenated aligned protein sequences with the IQ-TREE tool (Nguyen et al., 2015) and the best-fit substitution model (LG + R10 model) for 1,000 replicates. In addition, all phylogenetic trees were visualized using the interactive Tree of Life (iTOL) online tool (Letunic and Bork, 2019).
2.5. Genome annotation
Reference genomes included D. autotrophicus, D. desulfuricans SSM1, F. sinusarabici DSM4947, and M. schaedleri ASF457, of which D. autotrophicus, D. desulfuricans SSM1 and F. sinusarabici DSM4947 were free-living bacteria from hydrothermal environments, and M. schaedleri ASF457 was from the mouse intestine. The studied MAGs and reference genomes were annotated using Prokka v1.14.6, and the parameters were set to metagenome and kingdom bacteria (Seemann, 2014). This annotation pipeline relied on several external prediction tools, including Prodigal for coding sequences, Aragon for transfer RNA genes, RNAmmer for ribosomal RNA genes, and Infernal for noncoding RNAs. Kofamscan v1.3.0 (Aramaki et al., 2020) was used for functional annotations of the predicted genes, and the amino acid sequences of each genome were inputted and then compared with the KEGG database using the mapper model. KEGG-Decoder was performed to determine the completeness of various metabolic pathways by Kofamscan results (Graham et al., 2018). GO annotation was analysed using eggNOG-mapper v2.1.3 software (Huerta-Cepas et al., 2017), and the parameters were set as follows: -seed_ortholog_evalue, 0.00001; −m, hmmer; and -d, bact. Carbohydrate enzyme annotation was performed by searching the CAZyme database using dbCAN2 software, which used default settings (Zhang et al., 2018). Annotation of the deduced proteins was also performed using BLASTP against the NCBI Nr, KEGG (Kanehisa et al., 2016), Pfam (El-Gebali et al., 2019) and COG (Tatusov et al., 2000; Galperin et al., 2021) databases, with a maximum e-value cut-off of 1e-05. The CRISPR region was identified using CRISPRCasTyper v1.6.1 (Russel et al., 2020) and the CRISPRCasFinder online tool (Grissa et al., 2007).
Data availability
The MAGs obtained from the samples of Gut of Rimicaris kairei in this study have been submitted to the NCBI database under BioProject ID PRJNA931729.
3. Results
3.1. Metagenome assembly and characteristics
Deferribacterota was the dominant phylum in both microbial communities of the juvenile and adult R. kairei intestinal tracts in our previous study (Qi et al., 2021). To further investigate the potential functions of the intestinal Deferribacterota, metagenomes were assembled and analysed. A total of 45 Gbps of raw data were obtained by high-throughput sequencing, and 42.4 Gbps of clean data were retained. After de novo assembly based on three different algorithms (MaxBin2, metaBAT2, and CONCOCT), genomic bins with each longer than 2 kbp, completeness >50%, and contamination <10% were selected. There were 18 MAGs obtained in total, including Firmicutes, Bacteroidetes, Spirochaetota, Campylobacterota and Deferribacterota, based on the analysis by GTDB-tk (Supplementary Table S1). The six Def-MAGs (Deferribacterota MAGs, defined as Def_J1, Def_J3, Def_J5, Def_J6, Def_A4, and Def_A7, respectively) with four related reference genomes, including D. autotrophicus, D. desulfuricans SSM1, F. sinusarabici DSM4947, and M. schaedleri ASF457, are shown in Table 1. Among the six Def-MAGs, the completeness of Def_J6 is close to 90%, and its genome size is 2.5 Mb, similar to the four reference MAGs with a genome size of 2.2–2.5 Mb. The completeness of other Def-MAGs ranged from 55.33 to 88.76%, with genome sizes ranging from 0.9 to 1.8 Mb. Notably, the average G + C content of the six Def-MAGs ranged from 46.4 to 50.7%, much higher than that of the reference genomes, in which the G + C content ranged from 30 to 38%. We also listed the number of tRNAs and rRNAs and the number and percentage of genes annotated by each genome in the KEGG and COG databases (Table 1).
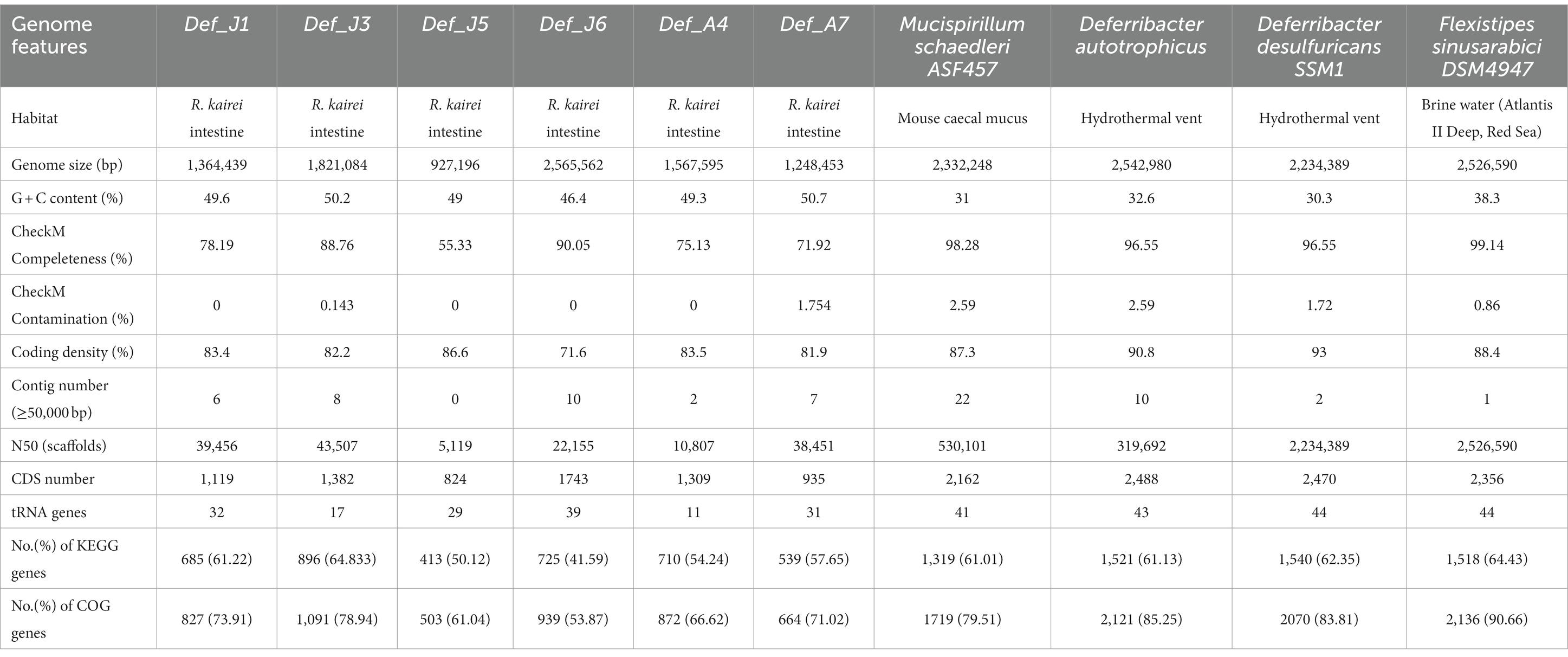
Table 1. Genomic features of gut Deferribacterota in R. kairei and reference genomes.
3.2. Phylogenetic analysis of Deferribacterota
To further investigate the taxonomic status of the six species from the R. kairei intestine, phylogenetic analysis was performed using 43 conserved proteins with five phyla adjacent to the phylum Deferribacterota: Tenericutes, Firmicutes, Acidobacteria, Chrysiogenetes, and Proteobacteria. In addition to the six studied species from Deferribacterota, 16 other genome sequences in the phylum Deferribacterota, including 8 identified and 8 unidentified species, were selected from the RefSeq database, and a total of 79 genomes were constructed for phylogenetic analyses (Figure 1). The results showed that the six Def-MAGs were clustered into a single branch and separated from other strains in the phylum Deferribacterota. We also tested the average nucleotide identity (ANI) value (Table 2). The results, consistent with the phylogenetic tree, were that the six Def-MAGs clustered together (Supplementary Figure S2). The Average Amino acid Identity (AAI) and DNA–DNA hybridization (DDH) values are similar to the ANI values, and the six Def-MAGs are classified into three species (Table 2). According to the classification analysis by GTDB-tk software, Def_J3, Def_J5, Def_J6, and Def_A7 are classified as the Deferribacteres class but could not be further classified into any known order. The relative evolutionary divergence (RED) values are 0.467, 0.460, 0.467, and 0.455 for Def_J3, Def_J5, Def_J6, and Def_A7, respectively. Def_J1 and Def_A4 are classified as the Mucispirillaceae family but could not be further classified into any known genus. The RED values were 0.747 and 0.749 for Def_J1 and Def_A4, respectively (Table 2). Therefore, Def_J3, Def_J5, Def_J6, and Def_A7 could be in new orders, while Def_J1 and Def_A4 could be in new genera. Together with the phylogenetic results and ANI values, the six Def-MAGs were classified into three species, with Def_J1 representing a new genus, Def_J3, Def_J6, and Def_A7 representing a new order, and Def_J5 and Def_A4 representing another new genus. Here, the classification of Def_J5 is slightly confused, which is probably due to its genome with only 50% integrity and needs to be further clarified by obtaining a longer length.
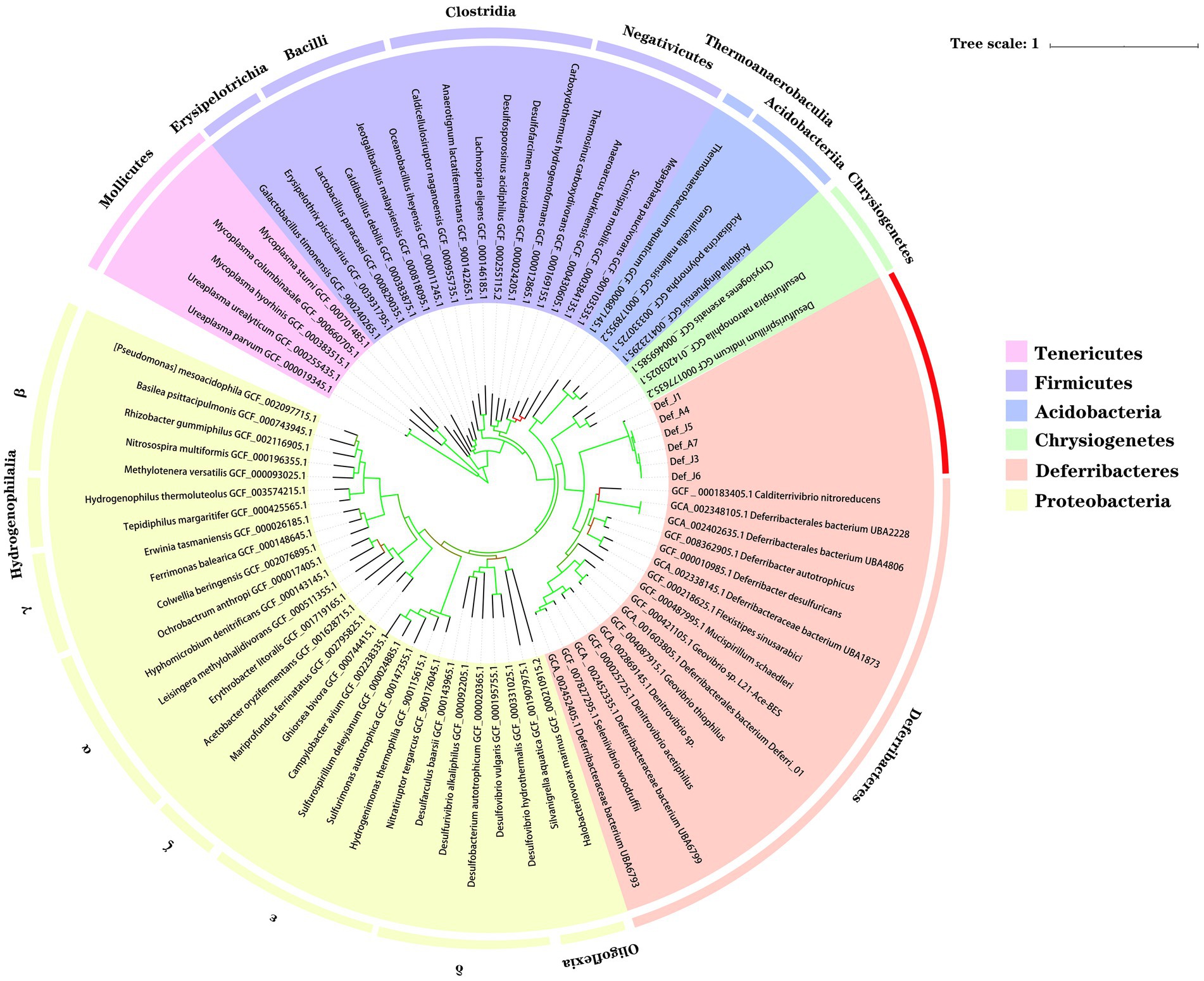
Figure 1. Phylogenetic position of gut Deferribacterota in R. kairei. Phylogenomic analysis was conducted on 43 concatenated conserved proteins to reconstruct a maximum-likelihood tree with 1,000 replications. Bootstrap values are colour-coded in the branches, with a colour range of red to green indicating values from 47 to 100. Different background colours represent different phyla. The ones marked in the outer circle are the class names, and the red ones are the samples of this study.
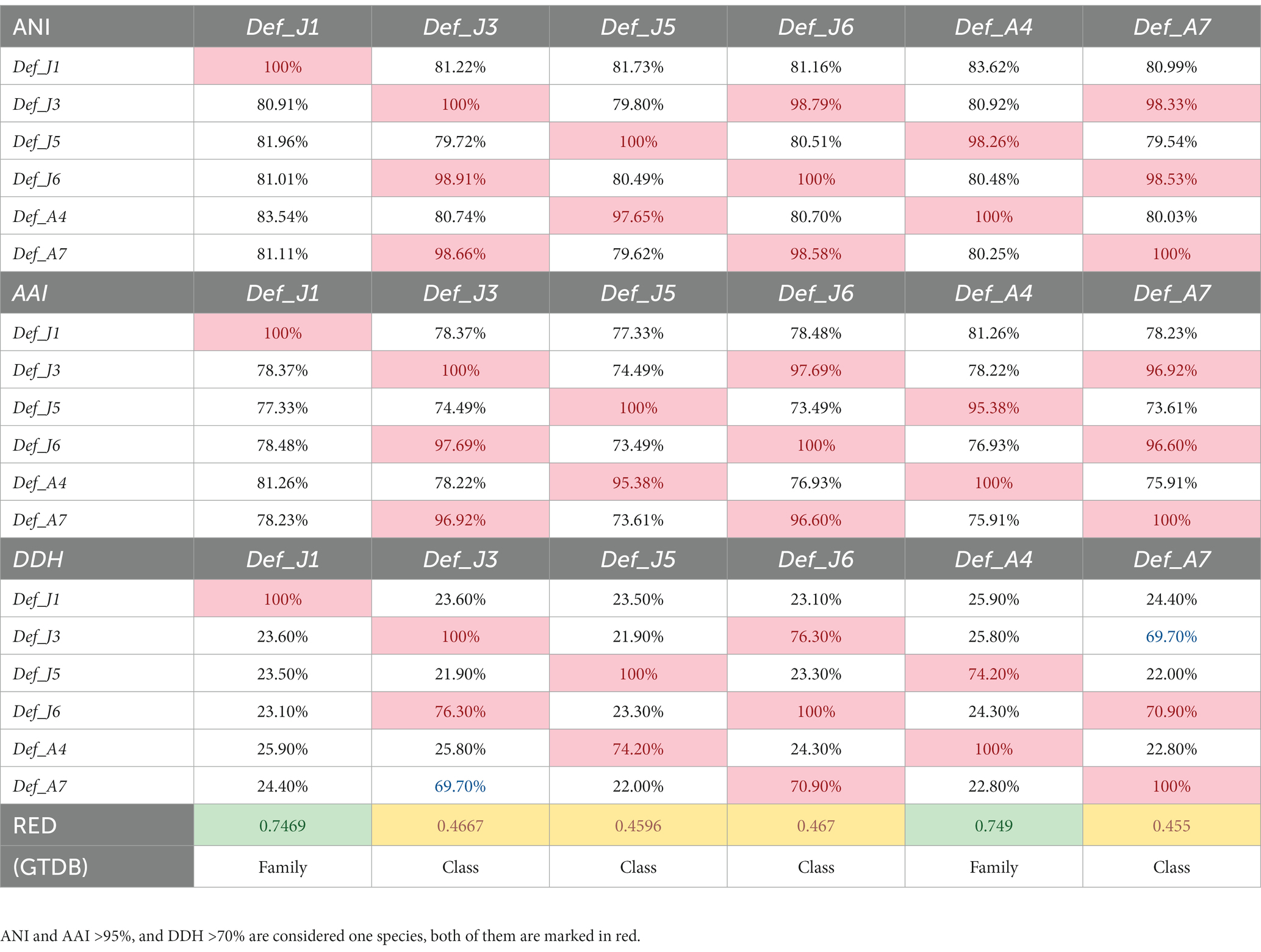
Table 2. Average nucleotide identity, average amino acid identity, DNA–DNA hybridization and relative evolutionary branching values of gut Deferribacterota in R. kairei.
3.3. Metabolic features
3.3.1. Energy metabolism
Six Def-MAGs and four reference MAGs were used to comparatively analyse the metabolic pathways by KEGG decoding. Based on the heatmap, both groups have a similar integrity of metabolism for the glycolysis and gluconeogenesis pathways, but the gluconeogenesis pathway was almost absent in the genome of M. schaedleri ASF457 (Figure 2). Genes related to the tricarboxylic acid cycle were severely absent in Def-MAG compared to the reference MAGs (Figure 2). Taking Def_J6-MAG, with the highest completeness, as an example, only malate dehydrogenase (EC1.1.1.37), fumarate hydratase (EC4.2.1.2), and succinate dehydrogenase (ubiquinone) flavoprotein subunit (EC1.3.5.1) were found, and most of the enzymes, including key enzymes such as citrate synthase (EC2.3.3.1) and ATP citrate lyase (EC2.3.3.8), were absent. In Def-MAG and M. schaedleri ASF457, starch or glycogen has 33–66% integrity for the synthesis pathway and 100% integrity for the degradation pathway. However, both pathways were absent in the three environmental reference genomes (Supplementary Table S2). We found that all six Def-MAGs have the key enzyme glycogen phosphorylase (EC2.4.1.1.1), which is involved in glycogen degradation, while 1,4-α-glucan branching enzyme (EC2.4.1.18), which is essential for increasing the solubility of glycogen molecules and reducing the osmotic pressure within cells, is only present in Def_J1, Def_J3, Def_J6 and Def_A4 (Figure 3). Notably, the carbon degradation of the six Def-MAGs was mainly via β-N-acetylhexosaminidase and β-glucosidase, while the three environmental reference genomes were mainly via D-galacturonate epimerase, and the carbon degradation process was not present in the M. schaedleri ASF457 MAG from the mouse intestine (Figure 2). β-N-acetylhexosaminidase and β-glucosidase are key enzymes for the degradation of chitin and cellulose, respectively, while D-galacturonate epimerase is able to convert UDP-d-glucuronide into D-galacturonide, a monosaccharide that is one of the activating precursors necessary for the synthesis of pectinas, indicating that the six Def-MAGs from the R. kairei intestine are able to degrade cellulose and chitin, while the reference genomes have the potential to degrade pectin. Def-MAGs contain many phosphotransferases, which transport extracellular glucose (EC22.7.1.199), salicin (EC2.7.1.-), arbutin (EC2.7.1--), cellobiose (EC2.7.1.205), maltose (EC2.7.12.08), and other carbon sources into cells (Figure 3). The large number of PTSs identified in the Def-MAG genomes suggests that Def-MAG may utilize multiple carbon sources. In addition, we found that Def-MAG has carbamate kinase (EC2.7.2.2) and ornithine carbamoyltransferase (EC2.1.3.3) (Figure 3), both of which convert NH3 to citrulline and over the process reversibly. Although no nitric oxide synthase (EC1.14.14.17) was found for the conversion from citrulline to arginine, arginine deiminase (EC3.5.3.6), which catabolizes arginine to citrulline, is present and can irreversibly hydrolyse L-arginine to L-citrulline and ammonia, suggesting that Def-MAGs could provide energy by arginine metabolism generating ATP.
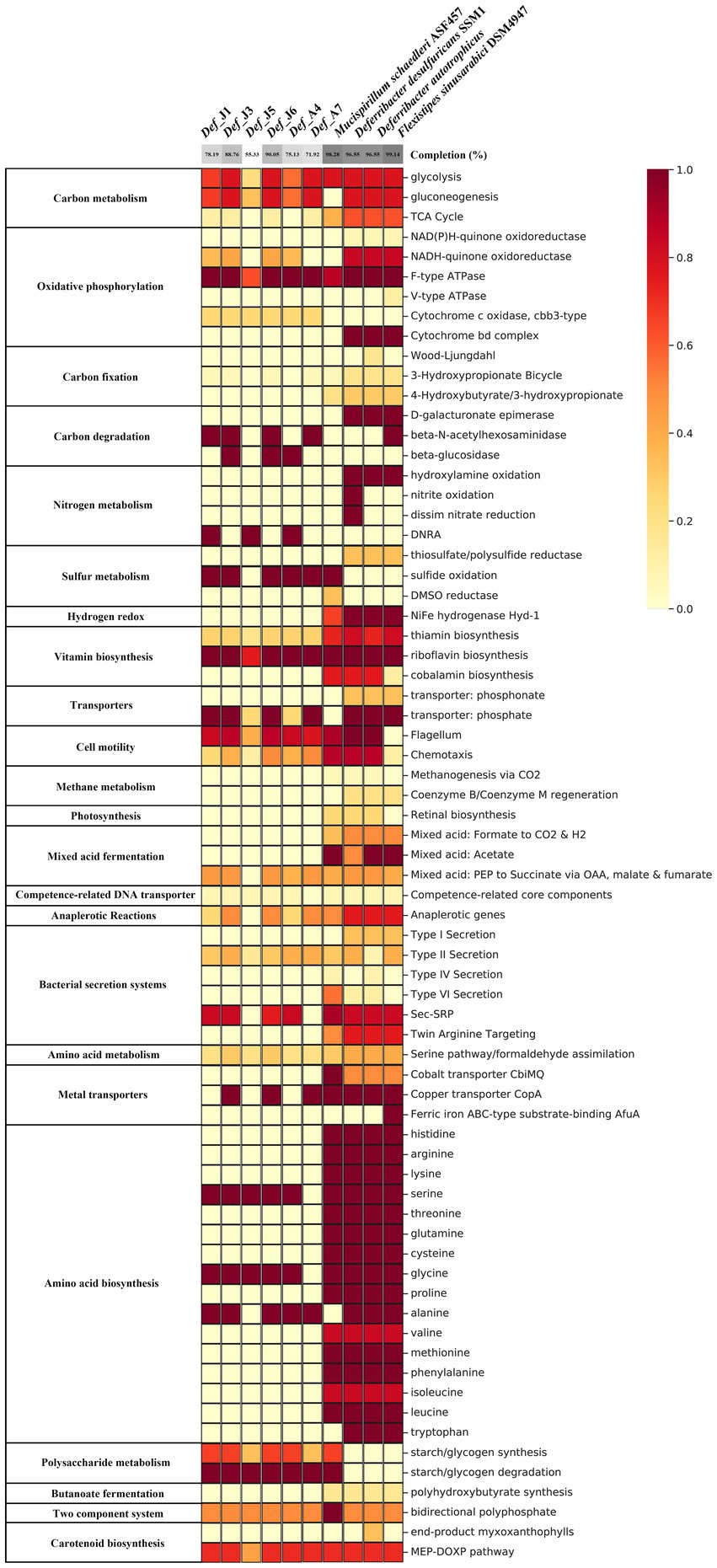
Figure 2. Heatmap based on KEGG annotation. The heatmap represents the integrity of the metabolic pathways of MAGs based on the presence or absence of genes identified by the KEGG decoder. The absence or presence of genes identified by the KEGG decoder. The grey colour at the top indicates the degree of genome integrity.
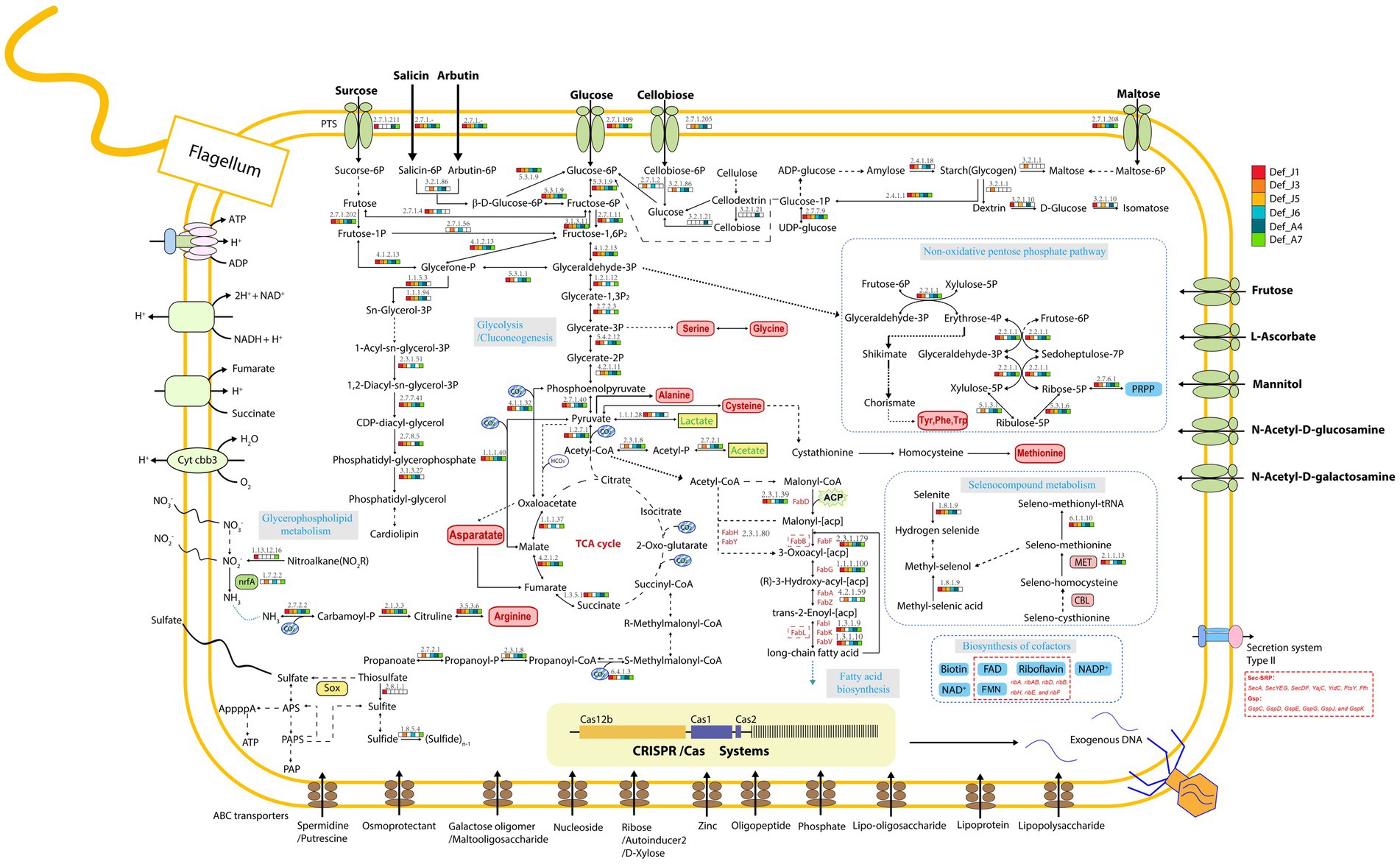
Figure 3. Diagram of the major predicted metabolic pathways. Metabolites are shown in black, amino acids in red, cofactors in blue, and the enzymes predicted for each MAG in each process are shown in different small blocks of colour in the diagram, with white indicating that the enzyme is not present. PTS, ABC transporters, CRISPR–Cas systems, etc., are indicated in the diagram.
3.3.2. Carbohydrate enzymes
In the six Def-MAGs and four reference genomes, a total of 33 carbohydrate-active enzymes were identified, which were classified into 5 types: AA (auxiliary activities), CBM (carbohydrate-binding modules), CE (carbohydrate esterases), GH (glycoside hydrolases) and GT (glycosyltransferases). Among them, a total of 21 carbohydrate enzymes were in six De-MAGs, nine of which were specific in the six Def-MAGs, including one CBM, CBM67; one CE, CE9; and 7 GHs, GH1, GH133, GH19, GH3, GH4, GH42, and GH78 (Figure 4). A total of 18 carbohydrate enzymes were identified in the genome of M. schaedleri, among which CBM13 and GH57 were specific (Figure 4). A total of 20 carbohydrate enzymes were identified in the genomes of the three environmental Deferribacterota isolates, five of which were specific to these genomes, namely, AA4, CE4, GH109, GH114, and GH130 (Figure 4). In addition, the six Def-MAGs were predominantly rich in glycoside hydrolases compared to the mouse intestine, where the main type of carbohydrase was glycosyltransferase. Compared with the environmental Deferribacterota genomes, the six Def-MAGs have specific GT35 and GT5 glycosyltransferases (Figure 4). These two glycosyltransferases can phosphorylate or synthesize glycogen, suggesting that Deferribacterota in the gut may provide a carbon source for the host.
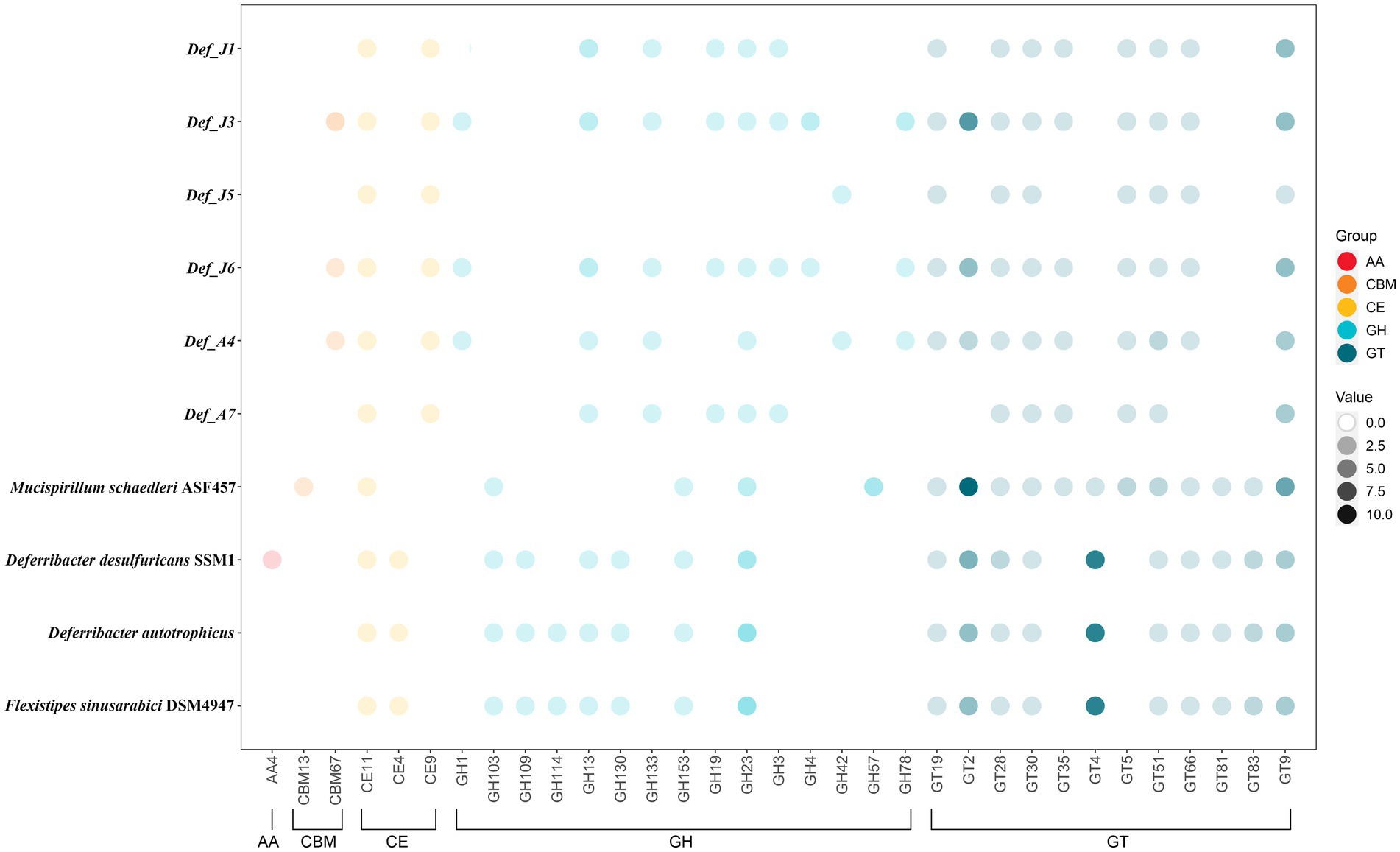
Figure 4. Relative abundance of carbohydrate-active enzyme genes. The different coloured circles represent the different types of enzymes, auxiliary activity (AA), carbohydrate-binding module (CBM), carbohydrate esterase (CE), glycoside hydrolase (GH), and glycosyltransferase (GT). The darkness of the circles indicates the number of each carbohydrate-active enzyme gene in different MAGs. Gene distribution, classification, and functions are reported in Supplementary Table S3.
3.3.3. Amino acids, vitamins, and cofactors
Compared to the three environmental reference genomes, the six Def-MAGs lost most amino acid synthesis genes except serine, glycine, and alanine; however, for the genome of M. schaedleri ASF457, the Deferribacterota from the mouse intestine, only the alanine and tryptophan synthesis pathways were absent (Figure 2). This result suggests the interdependent relationship between Def-MAGs and their host R. kairei. Except for Def_J5, the integrity of the pathways involved in riboflavin synthesis was 100% in the remaining five Def-MAGs and four reference genomes (Supplementary Table S2), and the flavin-like compound biosynthesis-related genes ribA, ribAB, ribD, ribB, ribH, ribE, and ribF were identified in the six Def-MAG genomic metabolic pathway reconstructions (Figure 3). In addition, Def-MAGs also have biotin, flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD) and other cofactor synthesis pathways (Figure 3).
3.3.4. Secretome and immune protection
The secretory system is essential for prokaryotic cell growth and other physiological processes (Green and Mecsas, 2016). Compared with the reference genomes, the six Def-MAGs contained only a type II secretion system (T2SS), but the reference genomes also have type I, type IV, and type VI secretion systems (Figure 2). T2SS effectors are transferred from the cytoplasm to the outer membrane or into the extracellular environment in two steps. First, Protein translocation across the inner membrane via the general secretion (Sec) pathway or twin arginine translocation (Tat) pathway (Pugsley et al., 1991; Voulhoux et al., 2001). Second, protein folded in the periplasm crosses the outer membrane by transport of T2SS (Johnson et al., 2006; Douzi et al., 2011; Nivaskumar and Francetic, 2014). Key proteins of the Sec pathway and the signal recognition particle (SRP) pathway were blasted and found in six Def-MAGs, including SecA, SecYEG, SecDF, YajC, YidC, FtsY, and Ffh, but the Secret monitor (SecM) protein was absent. In Def-MAGs, six of different genral secretory pathway (Gsp) protein of T2SS were also found, namely, GspC, GspD, GspE, GspG, GspJ, and GspK (Figure 3). These results indicate that the protein secretion of Deferribacterota in the R. kairei intestine mainly transfers the protein to the periplasmic space through the Sec pathway, and then the protein folds, while the Tat pathway existing in reference genomes transfers the folded protein to the periplasmic space.
The CRISPR-cas system was investigated in six Def-MAGs, and which in Def_J1, Def_J3, Def_J6, and Def_A7 consisted of three cas genes, cas1, cas2, and cas12b, and 70–190 spacers (Figure 5). These Def-MAGs were dominated by the type V CRISPR-cas system, belonging to the class II CRISPR-cas system due to the cas 12b gene, the key enzyme for type V CRISPR-cas. In contrast, in the reference genomes, only D. desulfuricans SSM1 and F. sinusarabici DSM4947 have a complete CRISPR–Cas system and belong to types I and III of class I, respectively. Furthermore, by blasting the spacer sequences of the Def-MAGs in the CRISPRCasdb database, most of them are unknown spacer sequences. A total of 17 spacer sequences were matched with identities of 92–100%, and most of them were pathogenic bacteria (Supplementary Table S4). These results suggest that the CRISPR system of hydrothermal R. kairei intestinal Deferribacterota genomes is likely to provide immune protection to the host against invasion by other pathogenic bacteria.
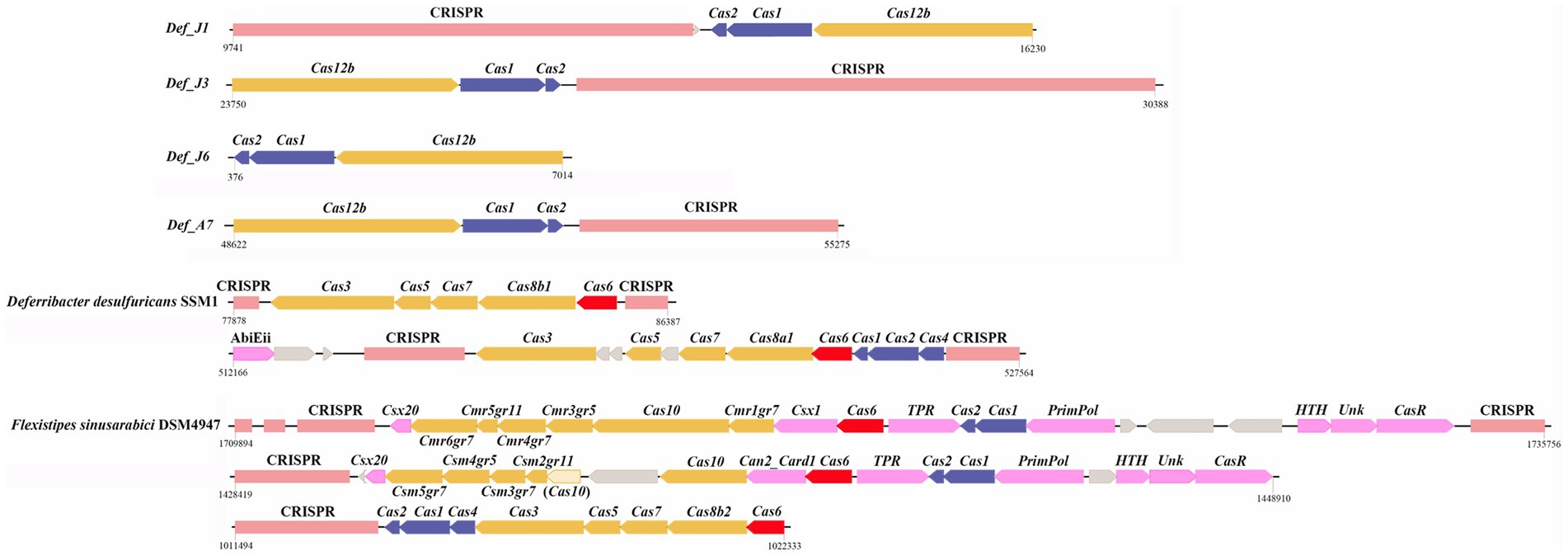
Figure 5. Structures of CRISPR–Cas systems. Structures of CRISPR-Cas systems in Deferribacterota and reference genomes. The CRISPR sequence is in pink. The interference module is in yellow. The adaptation module is in blue. Cas6 is in red. Accessory genes are in purple. Genes with alignment scores below the thresholds are lighter and with parentheses around names. Unknown genes are in gray. The number represents the starting and ending positions on the contig. The arrow indicates the direction of the gene. The spacer BLAST results for spacers in the database of CRISPR/Casdb database are shown in Supplementary Table S4.
4. Discussion
In this study, functional annotation analysis was comparably performed between the six Def-MAGs and three free-living bacteria in the phylum Deferribacterota from the environment and one Deferribacterota from the intestinal tract of mice. Based on the features of the six Def-MAGs, Deferribacterota from the intestinal tract of R. kairei can utilize various carbon sources, including glycogen, sucrose, salicin, arbutin, glucose, cellobiose, and maltose.
Glycogen in bacteria plays a significant role in carbon and energy storage. It presently has been found in species such as archaea, bacteria, and heterotrophic eukaryotes (Ball et al., 2015). Glycogen is a highly soluble homogeneous polysaccharide and contains hundreds of thousands of glucose units. Glycogen can accumulate in bacteria when the carbon content exceeds that of another nutrient and limits growth (Wilson et al., 2010). During disadvantageous periods, glycogen is decomposed as a carbon and energy reserve to support the long-term survival of bacteria (Wang and Wise, 2011). Glycogen plays a central role in the widespread connectivity of various cellular pathways and can be involved in bacterial energy metabolism, environmental durability, dormancy, and virulence (McMeechan et al., 2005; Jones et al., 2008; Chandra et al., 2011; Sawers, 2016; Gupta et al., 2017; Klotz and Forchhammer, 2017). The classical pathway for glycogen synthesis is the GlgC-GlgA pathway, which generates activated glucose nucleotide diphosphate from glucose 1-phosphate via nucleotide diphosphate glucose pyrophosphorylase (GlgC), which then polymerizes by glycogen synthase (GlgA) to produce linear glucans. Finally, a non-reducing-end oligoglucan transfer mediated by branching enzymes (GlgB or GBE) to the 6-position of residues within a chain generates side branches that convert it to glycoge (Preiss, 2006). GBE (EC 2.4.1.18) is a determinant of glycogen structure and highly conserved (Zmasek and Godzik, 2014). Bacterial GBE belongs to the GH13 family of glucose hydrolases (α-amylase family; Feng et al., 2015). In prokaryotes, glycogen has been considered to be degraded by highly conserved glycogen phosphorylases (GlgP) and debranching enzymes (GlgX; Wilson et al., 2010; Wang and Wise, 2011). GlgP can remove non-reducing terminal glycosyl residues until four glycosyl residues remain at the branching point, then GlgX acts on the short chain by truncating the α-1,6-glycosidic bond (Dauvillée et al., 2005; Alonso-Casajús et al., 2006). The glucose 1-phosphate generated by glycogen degradation readily enters the primary generation. We found GlgP in six of the Def-MAGs, and GBE was in Def_J1, Def_J3, Def_J6, and Def_A4, indicating that Def-MAGs can participate in glycogen degradation and synthesis.
Based on the carbohydrate enzyme annotation, we found that there were nine specific enzymes in the six Def-MAGs of R. kairei, namely, CBM67, CE9, GH1, GH133, GH19, GH3, GH4, GH42, and GH78, compared with the four reference genomes of Deferribacterota. Of these, CBM67 is mainly able to bind L-rhamnose. CE9 esterases catalyse the deacetylation of N-acetylglucosamine-6-phosphate to glucosamine-6-phosphate. This reaction has been demonstrated to be important for bacterial amino sugar metabolism and peptidoglycan cell wall recycling (Park, 2001; Ahangar et al., 2018). GH1 mainly includes β-glucosidases and β-galactosidases but also includes 6-phosphate-β-glucosidase and 6-phosphate-β-galactosidases, β-mannosidase, β-D-fucosidase, and β-glucuronidase (Michalska et al., 2013). Def-MAGs mainly have β-glucosidases and β-galactosidases. GH133 is mainly amylo-α 1,6-glucosidase (Stam et al., 2006). GH19 is an endo-acting enzyme that hydrolyses glycosidic bonds within chitin, a partially deacetylated chitin, with high degrees of acetylation, even though it lacks a CBM (Kawase et al., 2006). GH3 is widely distributed in bacteria, fungi, and plants and has a variety of functions, including cellulose biomass degradation, plant and bacterial cell wall remodelling, energy metabolism, and pathogen defence. It has been reported that GH3 can hydrolyse cellulose disaccharides and hydrolyse the nonreducing end β-1,4 bond of cellulose dextrins via β-glucosidase (Karkehabadi et al., 2018). GH4 differs from the other glycoside hydrolases in the family with different substrate specificities from each other. This family contains α-glucosidases, α-galactosidases, α-glucosidase, 6-phospho-α-glucosidases and 6-phospho-β-glucosidases. Similar to GH1, some enzymes prefer phosphorylated substrates to nonphosphorylated substrates (Henrissat and Bairoch, 1996; Henrissat and Davies, 1997). GH42 is active against lactose (Yuan et al., 2008). The only activity identified for GH78 was the hydrolysis of α-L-rhamnosides (Mutter et al., 1994). These results indicate that the intestinal Def-MAGs can hydrolyse a variety of polysaccharides and can hydrolyse and utilize residues such as cellulose and chitin that are ingested by the hydrothermal shrimp R. kairei as an energy source.
In this study, we found that the six Def-MAGs have a type II secretory system for transferring endotoxin and exotoxin to the extracellular system. Many gram-negative bacteria secrete toxic factors and effectors for molecular communication with their hosts through the bacterial secretory system. There are six types of protein-secreting systems in gram-negative bacteri (Costa et al., 2015). Among them, the type II secretion system is mainly centred on the general secretory pathway (GSP) gene cluster. The other reference genomes mainly contain the bacterial type VI secretory system (T6SS), which is widely found in gram-negative bacteria and is an important “weapon” of bacterial competition. Its structure is similar to that of a phage’s caudal tube, which is inverted on the inside of the bacterial cell membrane (Cianfanelli et al., 2016). T6SS is commonly produced in multiple processes associated with bacterial virulence and had shown to attack bacterial competitors or defeat host defense mechanisms to survive in competition or colonized host ecological niches (Russell et al., 2011; Basler et al., 2013). The presence of a type VI secretion system and a putative effector protein in the mouse gut of M. schaedleri has been reported to alter gene expression in mucosal tissue, suggesting a close interaction with the host and a possible role in inflammation (Loy et al., 2017). The type II secretory system is widely found in animal and plant pathogens and can secrete various proteins for exocytosis, which is very common in gram-negative bacteria. We also identified a type V CRISPR–Cas immune protection system in the intestinal Def-MAGs of the hydrothermal shrimp R. kairei, which is capable of providing immune protection to the host against multiple pathogen invasions. In conclusion, intestinal Def-MAGs of the hydrothermal shrimp R. kairei may enhance its viability and competitiveness in the host intestinal environment through interaction with the host and provide immune protection to the host.
Only serine, glycine, and alanine synthesis pathways are present in Def-MAGs, but other amino acid synthesis pathways are missing, suggesting that these bacteria may acquire some amino acids from the host. However, synthetic pathways of riboflavin, biotin, FMN, and FAD are found in Def-MAGs, and these cofactors can provide nutritional help for the host under extreme environments. Although it has been reported in terrestrial animals that the Deferribacterota bacterium may be a pathogen causing some diseases, we found that the intestinal Deferribacterota of the hydrothermal shrimp R. kairei may provide a variety of nutritional and immune protection to the host. Perhaps due to long-term coevolution, the intestinal Deferribacterota bacterium of the hydrothermal shrimp R. kairei has formed a mutualistic relationship with the host.
5. Conclusion and outlook
There is an interdependent relationship between the Deferribacterota bacterium and its host R. kairei in terms of material and energy, suggesting that Deferribacterota is a symbiont in the gut of R. kairei. The predominant occupation of Deferribacterota in the intestine of R. kairei plays an important role in survival. The study of the functions of Deferribacterota not only further explains the survival strategy and mechanism of blind shrimp in extreme environments but also deepens the understanding of the viability and living conditions of microorganisms.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
QL and L-SH conceived and designed the experiments. QL performed the experiments. QL, MS, F-CZ, and C-AL analysed the data. QL and L-SH wrote the manuscript with input from all other authors. L-SH directed and supervised all of the research. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the general projects of National Natural Science Foundation of China (42176125); the Scientific research and manufacture projects of Sanya City, Grant (NO. 2020KS01); Hainan Provincial Natural Science Foundation of China, grant (322CXTD531); the major scientific and technological projects of Hainan Province (ZDKJ2019011); and the major scientific and technological projects of Hainan Province (ZDKJ2021028).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1179935/full#supplementary-material
References
Ahangar, M. S., Furze, C. M., Guy, C. S., Cooper, C., Maskew, K. S., Graham, B., et al. (2018). Structural and functional determination of homologs of the Mycobacterium tuberculosis N-acetylglucosamine-6-phosphate deacetylase (NagA). J. Biol. Chem. 293, 9770–9783. doi: 10.1074/jbc.RA118.002597
Alonso-Casajús, N., Dauvillée, D., Viale, A. M., Muñoz, F. J., Baroja-Fernández, E., Morán-Zorzano, M. T., et al. (2006). Glycogen phosphorylase, the product of the glgP gene, catalyzes glycogen breakdown by removing glucose units from the nonreducing ends in Escherichia coli. J. Bacteriol. 188, 5266–5272. doi: 10.1128/jb.01566-05
Apremont, V., Cambon-Bonavita, M. A., Cueff-Gauchard, V., François, D., Pradillon, F., Corbari, L., et al. (2018). Gill chamber and gut microbial communities of the hydrothermal shrimp Rimicaris chacei Williams and Rona 1986: a possible symbiosis. PLoS One 13:e0206084. doi: 10.1371/journal.pone.0206084
Aramaki, T., Blanc-Mathieu, R., Endo, H., Ohkubo, K., Kanehisa, M., Goto, S., et al. (2020). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252. doi: 10.1093/bioinformatics/btz859
Ball, S., Colleoni, C., and Arias, M. C. (2015). “The transition from glycogen to starch metabolism in Cyanobacteria and eukaryotes” in Starch: Metabolism and Structure. ed. Y. Nakamura (Tokyo: Springer Japan), 93–158.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Basler, M., Ho, B. T., and Mekalanos, J. J. (2013). Tit-for-tat: type VI secretion system counterattack during bacterial cell-cell interactions. Cells 152, 884–894. doi: 10.1016/j.cell.2013.01.042
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brown, J., Pirrung, M., and McCue, L. A. (2017). FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. doi: 10.1093/bioinformatics/btx373
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chandra, G., Chater, K. F., and Bornemann, S. (2011). Unexpected and widespread connections between bacterial glycogen and trehalose metabolism. Microbiology (Reading) 157, 1565–1572. doi: 10.1099/mic.0.044263-0
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Cianfanelli, F. R., Monlezun, L., and Coulthurst, S. J. (2016). Aim, load, fire: the type VI secretion system, a bacterial Nanoweapon. Trends Microbiol. 24, 51–62. doi: 10.1016/j.tim.2015.10.005
Costa, T. R. D., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., et al. (2015). Secretion systems in gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13, 343–359. doi: 10.1038/nrmicro3456
Cowart, D. A., Durand, L., Cambon-Bonavita, M. A., and Arnaud-Haond, S. (2017). Investigation of bacterial communities within the digestive organs of the hydrothermal vent shrimp Rimicaris exoculata provide insights into holobiont geographic clustering. PLoS One 12:e0172543. doi: 10.1371/journal.pone.0172543
Dauvillée, D., Kinderf, I. S., Li, Z., Kosar-Hashemi, B., Samuel, M. S., Rampling, L., et al. (2005). Role of the Escherichia coli glgX gene in glycogen metabolism. J. Bacteriol. 187, 1465–1473. doi: 10.1128/jb.187.4.1465-1473.2005
Douzi, B., Ball, G., Cambillau, C., Tegoni, M., and Voulhoux, R. (2011). Deciphering the Xcp Pseudomonas aeruginosa type II secretion machinery through multiple interactions with substrates. J. Biol. Chem. 286, 40792–40801. doi: 10.1074/jbc.M111.294843
Durand, L., Zbinden, M., Cueff-Gauchard, V., Duperron, S., Roussel, E. G., Shillito, B., et al. (2010). Microbial diversity associated with the hydrothermal shrimp Rimicaris exoculata gut and occurrence of a resident microbial community. FEMS Microbiol. Ecol. 71, 291–303. doi: 10.1111/j.1574-6941.2009.00806.x
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–d432. doi: 10.1093/nar/gky995
Feng, L., Fawaz, R., Hovde, S. L., Gilbert, L., Chiou, J., and Geiger, J. H. (2015). Crystal structures of Escherichia coli branching enzyme in complex with linear oligosaccharides. Biochemistry 54, 6207–6218. doi: 10.1021/acs.biochem.5b00228
Galperin, M. Y., Wolf, Y. I., Makarova, K. S., Vera Alvarez, R., Landsman, D., and Koonin, E. V. (2021). COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–d281. doi: 10.1093/nar/gkaa1018
Garrity, G. M., Holt, J. M., Huber, H., Stetter, K. O., Greene, A. C., Patel, B. K. C., et al. (2001). “Phylum BIX. Deferribacteres phy. nov,” in Bergey’s Manual® of Systematic Bacteriology: Volume One: The Archaea and the Deeply Branching and Phototrophic Bacteria. eds. D. R. Boone, R. W. Castenholz, and G. M. Garrity (New York, NY: Springer New York), 465–471.
Gebruk, A., Southward, E., Kennedy, H., and Southward, A. (2000). Food sources, behaviour, and distribution of hydrothermal vent shrimps at the mid-Atlantic ridge. J. Mar. Biol. Assoc. U K 80, 485–499. doi: 10.1017/S0025315400002186
Graham, E. D., Heidelberg, J. F., and Tully, B. J. (2018). Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 12, 1861–1866. doi: 10.1038/s41396-018-0091-3
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems: an overview. Microbiol Spectr. 4:VMBF-0012-2015. doi: 10.1128/microbiolspec.VMBF-0012-2015
Greene, T., Patel, B., and Sheehy, A. J. (1997). Deferribacter thermophilus gen nov, sp nov, a novel thermophilic manganese- and iron-reducing bacterium isolated from a petroleum reservoir. Int. J. Syst. Bacteriol. 47, 505–509. doi: 10.1099/00207713-47-2-505
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Gupta, A. K., Singh, A., and Singh, S. (2017). “Glycogen as key energy storehouse and possibly responsible for multidrug resistance in mycobacterium tuberculosis,” in Drug Resistance in Bacteria, Fungi, Malaria, and Cancer. eds. G. Arora, A. Sajid, and V. C. Kalia (Cham: Springer International Publishing), 263–285.
Guri, M., Durand, L., Cueff-Gauchard, V., Zbinden, M., Crassous, P., Shillito, B., et al. (2012). Acquisition of epibiotic bacteria along the life cycle of the hydrothermal shrimp Rimicaris exoculata. ISME J. 6, 597–609. doi: 10.1038/ismej.2011.133
Henrissat, B., and Bairoch, A. (1996). Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 316, 695–696. doi: 10.1042/bj3160695
Henrissat, B., and Davies, G. (1997). Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644. doi: 10.1016/s0959-440x(97)80072-3
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through Orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Hügler, M., Petersen, J., Dubilier, N., Imhoff, J., and Sievert, S. (2011). Pathways of carbon and energy metabolism of the Epibiotic community associated with the Deep-Sea hydrothermal vent shrimp Rimicaris exoculata. PLoS One 6:e16018. doi: 10.1371/journal.pone.0016018
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jan, C., Petersen, J. M., Werner, J., Teeling, H., Huang, S., Glöckner, F. O., et al. (2014). The gill chamber epibiosis of deep-sea shrimp Rimicaris exoculata: an in-depth metagenomic investigation and discovery of Zetaproteobacteria. Environ. Microbiol. 16, 2723–2738. doi: 10.1111/1462-2920.12406
Jiang, L., Liu, X., Dong, C., Huang, Z., Cambon-Bonavita, M. A., Alain, K., et al. (2020). “Candidatus Desulfobulbus rimicarensis,” an uncultivated Deltaproteobacterial Epibiont from the Deep-Sea hydrothermal vent shrimp Rimicaris exoculata. Appl. Environ. Microbiol. 86:e02549-19. doi: 10.1128/aem.02549-19
Johnson, T. L., Abendroth, J., Hol, W. G., and Sandkvist, M. (2006). Type II secretion: from structure to function. FEMS Microbiol. Lett. 255, 175–186. doi: 10.1111/j.1574-6968.2006.00102.x
Jones, S. A., Jorgensen, M., Chowdhury, F. Z., Rodgers, R., Hartline, J., Leatham, M. P., et al. (2008). Glycogen and maltose utilization by Escherichia coli O157:H7 in the mouse intestine. Infect. Immun. 76, 2531–2540. doi: 10.1128/iai.00096-08
Jumas-Bilak, E., Roudière, L., and Marchandin, H. (2009). Description of ‘Synergistetes’ phyl. Nov. and emended description of the phylum ‘Deferribacteres’ and of the family Syntrophomonadaceae, phylum ‘Firmicutes’. Int. J. Syst. Evol. Microbiol. 59, 1028–1035. doi: 10.1099/ijs.0.006718-0
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Karkehabadi, S., Hansson, H., Mikkelsen, N. E., Kim, S., Kaper, T., Sandgren, M., et al. (2018). Structural studies of a glycoside hydrolase family 3 β-glucosidase from the model fungus Neurospora crassa. Acta Crystallogr F Struct Biol Commun 74, 787–796. doi: 10.1107/s2053230x18015662
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kawase, T., Yokokawa, S., Saito, A., Fujii, T., Nikaidou, N., Miyashita, K., et al. (2006). Comparison of enzymatic and antifungal properties between family 18 and 19 chitinases from S. coelicolor A3(2). Biosci. Biotechnol. Biochem. 70, 988–998. doi: 10.1271/bbb.70.988
Klotz, A., and Forchhammer, K. (2017). Glycogen, a major player for bacterial survival and awakening from dormancy. Future Microbiol. 12, 101–104. doi: 10.2217/fmb-2016-0218
Kunisawa, T. (2011). Inference of the phylogenetic position of the phylum Deferribacteres from gene order comparison. Antonie Van Leeuwenhoek 99, 417–422. doi: 10.1007/s10482-010-9492-7
Lapidus, A., Chertkov, O., Nolan, M., Lucas, S., Hammon, N., Deshpande, S., et al. (2011). Genome sequence of the moderately thermophilic halophile Flexistipes sinusarabici strain (MAS10T). Stand. Genomic Sci. 5, 86–96. doi: 10.4056/sigs.2235024
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Loy, A., Pfann, C., Steinberger, M., Hanson, B., Herp, S., Brugiroux, S., et al. (2017). Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a Core member of the murine gut microbiota. mSystems 2:e00171-16. doi: 10.1128/mSystems.00171-16
McMeechan, A., Lovell, M. A., Cogan, T. A., Marston, K. L., Humphrey, T. J., and Barrow, P. A. (2005). Glycogen production by different Salmonella enterica serotypes: contribution of functional glgC to virulence, intestinal colonization and environmental survival. Microbiology (Reading) 151, 3969–3977. doi: 10.1099/mic.0.28292-0
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Göker, M. (2021). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Michalska, K., Tan, K., Li, H., Hatzos-Skintges, C., Bearden, J., Babnigg, G., et al. (2013). GH1-family 6-P-β-glucosidases from human microbiome lactic acid bacteria. Acta Crystallogr. D Biol. Crystallogr. 69, 451–463. doi: 10.1107/s0907444912049608
Miroshnichenko, M. L., Slobodkin, A. I., Kostrikina, N. A., L’Haridon, S., Nercessian, O., Spring, S., et al. (2003). Deferribacter abyssi sp. nov., an anaerobic thermophile from deep-sea hydrothermal vents of the mid-Atlantic ridge. Int. J. Syst. Evol. Microbiol. 53, 1637–1641. doi: 10.1099/ijs.0.02673-0
Mutter, M., Beldman, G., Schols, H. A., and Voragen, A. G. (1994). Rhamnogalacturonan alpha-L-rhamnopyranohydrolase. A novel enzyme specific for the terminal nonreducing rhamnosyl unit in rhamnogalacturonan regions of pectin. Plant Physiol. 106, 241–250. doi: 10.1104/pp.106.1.241
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nivaskumar, M., and Francetic, O. (2014). Type II secretion system: a magic beanstalk or a protein escalator. Biochim. Biophys. Acta 1843, 1568–1577. doi: 10.1016/j.bbamcr.2013.12.020
Park, J. T. (2001). Identification of a dedicated recycling pathway for anhydro-N-acetylmuramic acid and N-acetylglucosamine derived from Escherichia coli cell wall murein. J. Bacteriol. 183, 3842–3847. doi: 10.1128/jb.183.13.3842-3847.2001
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Petersen, J. M., Ramette, A., Lott, C., Cambon-Bonavita, M.-A., Zbinden, M., and Dubilier, N. (2010). Dual symbiosis of the vent shrimp Rimicaris exoculata with filamentous gamma- and epsilonproteobacteria at four mid-Atlantic ridge hydrothermal vent fields. Environ. Microbiol. 12, 2204–2218. doi: 10.1111/j.1462-2920.2009.02129.x
Preiss, J. (2006). “Bacterial glycogen inclusions: enzymology and regulation of synthesis,” in Inclusions in Prokaryotes. ed. J. M. Shively (Berlin, Heidelberg: Springer Berlin Heidelberg), 71–108.
Pugsley, A. P., Kornacker, M. G., and Poquet, I. (1991). The general protein-export pathway is directly required for extracellular pullulanase secretion in Escherichia coli K12. Mol. Microbiol. 5, 343–352. doi: 10.1111/j.1365-2958.1991.tb02115.x
Qi, L., Lian, C. A., Zhu, F. C., Shi, M., and He, L. S. (2021). Comparative analysis of intestinal microflora between two developmental stages of Rimicaris kairei, a hydrothermal shrimp from the central Indian ridge. Front. Microbiol. 12:802888. doi: 10.3389/fmicb.2021.802888
Rodríguez-R, L., and Konstantinidis, K. (2014). Bypassing cultivation to identify bacterial species: culture-independent genomic approaches identify credibly distinct clusters, avoid cultivation bias, and provide true insights into microbial species. Microbe Magazine 9, 111–118. doi: 10.1128/microbe.9.111.1
Russel, J., Pinilla-Redondo, R., Mayo-Muñoz, D., Shah, S. A., and Sørensen, S. J. (2020). CRISPRCasTyper: automated identification, annotation, and classification of CRISPR-Cas loci. Crispr J 3, 462–469. doi: 10.1089/crispr.2020.0059
Russell, A. B., Hood, R. D., Bui, N. K., LeRoux, M., Vollmer, W., and Mougous, J. D. (2011). Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347. doi: 10.1038/nature10244
Sawers, R. G. (2016). Dormancy: illuminating how a microbial sleeping beauty awakens. Curr. Biol. 26, R1139–r1141. doi: 10.1016/j.cub.2016.08.039
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Slobodkin, A., Slobodkina, G., Allioux, M., Alain, K., Jebbar, M., Shadrin, V., et al. (2019). Genomic insights into the carbon and energy metabolism of a thermophilic Deep-Sea bacterium Deferribacter autotrophicus revealed new metabolic traits in the phylum Deferribacteres. Genes (Basel) 10:849. doi: 10.3390/genes10110849
Stam, M. R., Danchin, E. G., Rancurel, C., Coutinho, P. M., and Henrissat, B. (2006). Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of alpha-amylase-related proteins. Protein Eng. Des. Sel. 19, 555–562. doi: 10.1093/protein/gzl044
Takaki, Y., Shimamura, S., Nakagawa, S., Fukuhara, Y., Horikawa, H., Ankai, A., et al. (2010). Bacterial lifestyle in a deep-sea hydrothermal vent chimney revealed by the genome sequence of the thermophilic bacterium Deferribacter desulfuricans SSM1. DNA Res. 17, 123–137. doi: 10.1093/dnares/dsq005
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Uritskiy, G. V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1
Voulhoux, R., Ball, G., Ize, B., Vasil, M. L., Lazdunski, A., Wu, L. F., et al. (2001). Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. EMBO J. 20, 6735–6741. doi: 10.1093/emboj/20.23.6735
Wang, L., and Wise, M. J. (2011). Glycogen with short average chain length enhances bacterial durability. Naturwissenschaften 98, 719–729. doi: 10.1007/s00114-011-0832-x
Watabe, H., and Hashimoto, J. (2002). A new species of the genus Rimicaris (Alvinocarididae: Caridea: Decapoda) from the active hydrothermal vent field, “Kairei field,” on the central Indian ridge, the Indian Ocean. Zool. Sci. 19, 1167–1174. doi: 10.2108/zsj.19.1167
Whitman, W. B., Oren, A., Chuvochina, M., da Costa, M. S., Garrity, G. M., Rainey, F. A., et al. (2018). Proposal of the suffix -ota to denote phyla. Addendum to ‘Proposal to include the rank of phylum in the international code of nomenclature of Prokaryotes’. Int. J. Syst. Evol. Microbiol. 68, 967–969. doi: 10.1099/ijsem.0.002593
Wilson, W. A., Roach, P. J., Montero, M., Baroja-Fernández, E., Muñoz, F. J., Eydallin, G., et al. (2010). Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev. 34, 952–985. doi: 10.1111/j.1574-6976.2010.00220.x
Yuan, T., Yang, P., Wang, Y., Meng, K., Luo, H., Zhang, W., et al. (2008). Heterologous expression of a gene encoding a thermostable beta-galactosidase from Alicyclobacillus acidocaldarius. Biotechnol. Lett. 30, 343–348. doi: 10.1007/s10529-007-9551-y
Zbinden, M., and Cambon-Bonavita, M.-A. (2003). Occurrence of Deferribacterales and Entomoplasmatales in the deep-sea Alvinocarid shrimp Rimicaris exoculata gut. FEMS Microbiol. Ecol. 46, 23–30. doi: 10.1016/S0168-6496(03)00176-4
Zbinden, M., and Cambon-Bonavita, M. A. (2020). Rimicaris exoculata: biology and ecology of a shrimp from deep-sea hydrothermal vents associated with ectosymbiotic bacteria. Mar. Ecol. Prog. Ser. 652, 187–222. doi: 10.3354/meps13467
Zbinden, M., Shillito, B., le Bris, N., de Villardi de Montlaur, C., Roussel, E., Guyot, F., et al. (2008). New insigths on the metabolic diversity among the epibiotic microbial communitiy of the hydrothermal shrimp Rimicaris exoculata. J. Exp. Mar. Biol. Ecol. 359, 131–140. doi: 10.1016/j.jembe.2008.03.009
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–w101. doi: 10.1093/nar/gky418
Keywords: Deferribacterota, gut microbiota, Rimicaris kairei, metagenomic, hydrothermal vent
Citation: Qi L, Shi M, Zhu F-C, Lian C-A and He L-S (2023) Genomic evidence for the first symbiotic Deferribacterota, a novel gut symbiont from the deep-sea hydrothermal vent shrimp Rimicaris kairei. Front. Microbiol. 14:1179935. doi: 10.3389/fmicb.2023.1179935
Edited by:
Zhiyong Li, Shanghai Jiao Tong University, ChinaCopyright © 2023 Qi, Shi, Zhu, Lian and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Sheng He, aGUtbGlzaGVuZ0BpZHNzZS5hYy5jbg==