 Xiaoying Zhang1,2
Xiaoying Zhang1,2 Wenqiang Zhao
Wenqiang Zhao Yongping Kou
Yongping Kou Qing Liu
Qing Liu- 1CAS Key Laboratory of Mountain Ecological Restoration and Bioresource Utilization & Ecological Restoration and Biodiversity Conservation Key Laboratory of Sichuan Province, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Faculty of Agriculture, Forestry and Food Engineering, Yibin University, Yibin, China
Knowledge of variations in abundant and rare soil microbial communities and interactions during secondary forest succession is lacking. Soil samples were gathered from different secondary successional stages (grassland, shrubland, and secondary forest) to study the responses of abundant and rare bacterial and fungal communities, interactions and driving factors to secondary forest succession by Illumina sequencing of the 16S and ITS rRNA genes. The results showed that the α-diversities (Shannon index) of abundant bacteria and fungi revealed no significant changes during secondary forest succession, but increased significantly for rare bacteria. The abundant and rare bacterial and fungal β-diversities changed significantly during secondary forest succession. Network analysis showed no obvious changes in the topological properties (nodes, links, and average degree) of abundant microbial networks during secondary forest succession. In contrast, these properties of the rare microbial networks in the secondary forest were higher than those in the grassland and shrubland, indicating that rare microbial networks are more responsive to secondary forest succession than abundant microorganisms. Additionally, rare microbial networks revealed more microbial interactions and greater network complexity than abundant microbial networks due to their higher numbers of nodes and links. The keystone species differed between the abundant and rare microbial networks and consisted of 1 and 48 keystone taxa in the abundant and rare microbial networks, respectively. Soil TP was the most important influencing factor of abundant and rare bacterial communities. Successional stages and plant richness had the most important influences on abundant and rare fungal communities, respectively. C:P, SM and N:P were mainly related to abundant and rare microbial network topological properties. Our study indicates that abundant and rare microbial communities, interactions and driving factors respond differently to secondary forest succession.
Introduction
Secondary succession refers to a sequential replacement of plant species over time or due to a natural or human forces disturbance (Horn, 1974). In recent years, large tracts of forest have undergone artificial or natural restoration stages due to the disturbance of human activities (i.e., logging and abandoned agricultural fields) and natural factors (i.e., insect outbreaks and wildfire) (Chai et al., 2019; Rodriguez-Ramos et al., 2021; Zhang X. et al., 2022). Previous studies have found that secondary forest succession is an effective way to improve soil conditions and restore degraded environments (Yan et al., 2020; Hu et al., 2022). Plant communities change during secondary forest succession, resulting in variations in soil microbial communities (Cline and Zak, 2015; Chai et al., 2019; Liu et al., 2020). Thus, elucidating the responses of soil microorganisms to secondary forest succession is essential for understanding the mechanism of forest development (Jiang et al., 2021). Numerous studies have found that the diversity and composition of soil microbial communities differ with secondary forest succession (Cline and Zak, 2015; Liu et al., 2020; Yan et al., 2020; Jiang et al., 2021; Zhang X. et al., 2022; Zhang et al., 2023). The variations in soil microbial community composition are connected to plant composition (Li et al., 2020), soil nutrient cycling (Li et al., 2020; Jiang et al., 2021; Zhang X. et al., 2022; Zhang et al., 2023), litter decomposition (Cline and Zak, 2015), plant disease resistance (Hu et al., 2020) and other ecosystem processes.
It is known that the distribution of microbial communities is usually skewed in natural ecosystems; there are relatively few abundant species with high relative abundance and numerous rare species with low relative abundance (Shade et al., 2014; Lynch and Neufeld, 2015; Jousset et al., 2017). The changes in abundant species can obscure low-abundance taxa dynamics; thus, abundant species are traditionally thought to participate in major ecological functions, while rare microorganisms are usually ignored (Lynch and Neufeld, 2015; Zhou and Wu, 2021). However, recent studies have indicated that rare microorganisms serve as vital contributors to microbial αdiversity and βdiversity (Lynch and Neufeld, 2015) and play a major role in driving the multifunctional stability of ecosystems (Jousset et al., 2017; Shu et al., 2021; Wu et al., 2021; Zhang Z. et al., 2022). For example, Wu et al. (2021) found that rare microbial communities indirectly regulate the impact of land degradation on ecosystem functions. Shu et al. (2021) reported that rare microorganisms are the main drivers of the soil nutrient cycle (i.e., C, N, and S cycling). Recently, numerous studies have focused on the responses of abundant and rare microbial communities to environmental change (Jiang et al., 2019; Du et al., 2020; Jiao and Lu, 2020; He Z. et al., 2022; Zhang Z. et al., 2022). The results of these studies showed that abundant and rare microbial communities exhibited distinct changes along environmental gradients and demonstrated different contributions to soil ecosystem multifunctionality. However, knowledge of how the abundant and rare microbial communities respond to secondary forest succession is still unclear.
Network analysis has been extensively adopted for investigating the interactions between microbial communities, which can also provide more information about the correlations between microbial interactions and ecosystem functions (de Vries et al., 2018; Wagg et al., 2019; Shi et al., 2020). In microbial networks, positive and negative correlations represent cooperative and competitive interactions between microbial communities, respectively (Faust and Raes, 2012; Xue et al., 2022). Population dynamics can be stabilized by a moderate mixture of cooperative and competitive interactions (Mougi and Kondoh, 2012). Furthermore, the increase in microbial interactions represents higher network complexity and microbial community stability (Mougi and Kondoh, 2012; Guo et al., 2019) and reveals more resistance to disturbance in the external environment (i.e., drought) (Wang et al., 2018; Yu et al., 2021). Recent studies have found that microbial network complexity varied in salt marsh succession (Dini-Andreote et al., 2014), abandoned cropland succession (Morrien et al., 2017), grassland succession (Guo et al., 2019) and forest succession (de Araujo et al., 2018; Hu et al., 2022). Moreover, sporadic research has also focused on abundant and rare microbial networks. For example, Jiang et al. (2019) found that abundant bacterial taxa exhibited closer relationships than rare bacterial taxa in bacterial networks in glacier forefield succession. The nodes of the abundant bacterial network mainly consisted of Proteobacteria and Acidobacteria, while Planctomycetia, Sphingobacteriia, and Phycisphaerae were dominant in the bacterial network. In addition, network analysis has also been used to identify potential keystone species by network-based scores (He et al., 2017; Wang et al., 2018; Yu et al., 2021). These keystone species have been reported to play unique and important roles in maintaining microbial community structures and ecosystem functions (Faust and Raes, 2012; Banerjee et al., 2018a; Zheng et al., 2021).
Both abundant and rare microbial communities were affected by biotic (i.e., plant richness) and abiotic factors (Jiang et al., 2019; He Z. et al., 2022; Pan et al., 2022). For instance, Jiang et al. (2019) found that plant richness, soil organic carbon (SOC) and pH mainly affected abundant bacterial communities, while soil total nitrogen (TN) and pH primarily affected rare bacterial communities during glacier forefield succession. He Z. et al. (2022) found that soil electrical conductivity (EC), available nitrogen (AN), nitrate (NO3−-N), available phosphorus (AP) and land-use type had extremely significant influences on abundant bacteria, while soil pH, EC, soil water content (SM), AP and land-use type had extremely significant influences on rare bacteria in reforestation succession soil. In addition, abiotic factors have also been reported to have important influences on microbial interactions. For example, soil bulk density, altitude, soil moisture (Ji et al., 2022), SOC (Dong et al., 2022), pH (Banerjee et al., 2018b; Dong et al., 2022; Ji et al., 2022), NH4+-N, C:N and organic phosphorus (Banerjee et al., 2018b) have been reported to exert major functions in regulating soil microbial interactions. Understanding the driving factors of soil microbial communities and interactions during succession is crucial. However, how the changes in environmental factors (i.e., soil properties and plant richness) affect abundant and rare microbial communities and interactions during secondary forest succession remains unclear.
The subalpine forest in the southwest is regarded as the major body of alpine forests on the eastern Qinghai-Tibet Plateau, which is situated in the upper reaches of the Yangtze River. This area has a vital effect on water resource conservation, regional economy and ecological shelter (Liu et al., 2001). This area gradually displayed different recovery stages of secondary forest (grassland–shrubland–secondary coniferous forest) after large-scale deforestation, which provided an ideal platform for studying the succession of subalpine forests (Qiang et al., 2021). Sporadic studies in this area have revealed changes in microbial community compositions (Cao et al., 2020; Qiang et al., 2021; Xu et al., 2021; Zhang X. et al., 2022; Zhang et al., 2023), functions (Zhang X. et al., 2022; Zhang et al., 2023), and interactions (Qiang et al., 2021; Zhang et al., 2023). Our previous studies in this area also revealed that soil microbial communities and functions change significantly during secondary forest succession (Zhang X. et al., 2022; Zhang et al., 2023). However, it is still unknown how the abundant and rare microbial communities and their interactions respond to secondary forest succession, which limits our understanding of the mechanism of secondary forest succession. As a result, the present study was aimed at exploring the variations in abundant and rare microbial community structures, interactions and driving factors during secondary forest succession. We hypothesized that 1) abundant and rare microbial communities respond differently to secondary forest succession; 2) abundant and rare microbial interactions are inconsistent in response to secondary forest succession; 3) the influencing factors of abundant and rare microbial communities and interactions are different during secondary forest succession.
Materials and Methods
Site description and sampling
The study site is located at the Miyaluo Nature Reserve of Sichuan Province (ranged from 31°42′ to 31°51’N, ranged from 102°41′ to 102°44′E, 2200 to 5,500 m a.s.l.). The region is characterized by a warm summer and a dry winter with a snowy climate. With the annual rainfall precipitation ranging from 600 mm/y to 1,100 mm/y, the mean annual temperature is shown to be 8.7°C. In addition, the soil type in this area is mountain brown soil. This area gradually formed a natural recovery sequence (from grassland to secondary forest) after being extensively deforested from 1918 to 1998 (Liu et al., 2001; Liu, 2002). In this study, the early, middle, and late successional stages of the natural recovery sequence were selected to investigate the responses of abundant and rare microbial communities and interactions to secondary forest succession. Specifically, the grassland stage represents the early stage of forest recovery (approximately 0–20 years) and is dominated by Poa annua; the shrubland stage represents the middle stage of forest recovery (approximately 30 years) and is dominated by Quercus aquifolioides, followed by Picea asperata, Sorbus koehneana and Betula albosinensis; the secondary forest represents the late stage of forest recovery (approximately 60 years) and is dominated by Picea asperata, followed by Quercus aquifolioides and Betula platyphylla. The three selected successional stages were similar in climate, slope and geology.
In August 2018, we set up approximately 100 m × 100 m sampling areas in grassland, shrubland and secondary forest. For sampling rhizosphere and bulk soil, this study established five plots of grassland (1 m × 1 m), shrubland (5 m × 5 m) and secondary forest (10 m × 10 m). In order to avoid edge effects and spatial autocorrelation, the plots of each successional stage were more than 20 m apart (Chen et al., 2019). Detailed sampling information and sampling methods have been published in our recent papers (Zhang X. et al., 2022; Zhang et al., 2023). Briefly, the rhizosphere and bulk soils were collected by root shaking methods and soil drilling (Zhang X. et al., 2022), respectively. The 5 rhizosphere and bulk soil samples collected from each plot were combined as one composite soil. Finally, 30 soil samples were collected at the three successional stages. These soil samples were classified into the following two parts after passing through a 2 mm sieve: one part was maintained at 4°C for soil physical and chemical property analysis; besides, the other part was kept at −40°C for soil DNA extraction.
Soil physicochemical properties
Soil EC and pH were determined with a soil-to-water ratio of 1:2.5 by a DDS307 conductivity meter and Mettler-Toledo FE28 digital pH meter, respectively. We calculated SM by drying soil samples for 72 h at 65°C. SOC and TN were measured with a 2400II CHN elemental analyser (PerkinElmer, Boston, MA, United States). The measurement of ammonium (NH4+-N) and NO3−-N was performed using the indophenol blue method as well as the phenol disulfonic acid method following extraction with 2 M KCl, respectively. Total phosphorus (TP) was calculated by molybdenum antimony blue colorimetry after digestion with HClO4-H2SO4. Using the ratio of SOC and TN, the soil C:N was calculated; C:P was calculated by the ratio of SOC and TP, and N:P was calculated by the ratio of TN and TP. All of the soil properties were previously published in Zhang X. et al. (2022).
DNA extraction, high-throughput sequencing, and data processing
Using MoBio PowerSoil DNA Isolation Kits (MoBio Laboratories, CA, United States), total DNA was extracted from each soil sample. We evaluated the integrity of the extracted DNA by 1.0% (w/v) agarose gel electrophoresis. In addition, with a NanoDrop NC 2000 spectrophotometer (Thermo Scientific), the quality of the DNA extracts was calculated. To amplify the ITS2 region of fungi (Ihrmark et al., 2012), the primer set gITS7F (5′-GTGARTCATCGARTCTTTG-3′) and ITS4R (5′-TCCTCCGCTTATTGATATGC-3′) was used, and the primer set 515\u00B0F 5′-TATCGCCTCCCTCGCGCCATCAG-3′ and 909 R 5′-CTATGCGCCTTGCCAGCCCGCT-3′ was applied to amplify the V4-V5 region of the bacterial 16S rRNA gene (Tamaki et al., 2011). More details regarding the PCR system and conditions for fungal and bacterial PCR amplification were given by Zhang X. et al. (2022) and Zhang et al. (2023), respectively. Then, the amplicon samples were subjected to paired-end sequencing on the Illumina HiSeq (PE 250 × 250) platform at Biomarker Technologies Corporation (Beijing, China). The high quality sequences were clustered at the 100% similarity level. To avoid biases of the different sequencing depths, random resampling was carried out at the number of sequences 10,065 and 8,000 for bacteria and fungi, respectively. On the basis of the Silva v138.1 database and UNITE1 or fungal ITS database (Nilsson et al., 2019), the representative sequences of bacterial and fungal species were classified. The raw sequencing data were submitted to the National Center for Biotechnology Information (NCBI) SRA database (accession number: PRJNA835217 and PRJNA733908) and have been reported in our previous studies (Zhang X. et al., 2022; Zhang et al., 2023).
Network construction with a random matrix theory-based approach
Molecular Ecological Networks Pipeline (MENAP)2 was used to illustrate the ecological interactions within microbial communities (Zhou et al., 2010; Deng et al., 2012). Initially, following the operational standards of the MENAP, the standardized ASV table was uploaded. ASVs with a relative abundance >0.1% were regarded as abundant ASVs, while ASVs with a relative abundance <0.01% were determined to be rare ASVs (Jiao and Lu, 2020; Wan et al., 2021). More than 50% of the total samples of abundant and rare microbial taxa were used for microbial network analysis at each successional stage. The missing values were filled with 0.01 in blanks with paired valid values. This correlation matrix was transformed into a similarity matrix by taking the absolute values. Then, thresholds from 0.30 to 1.00 (with an interval of 0.01) were adopted until an optimal similarity threshold was identified where significant non-random patterns were detected in the microbial networks (Wang et al., 2018). To compare abundant and rare microbial networks among different successional stages, an identical cut-off of 0.88 was used to construct the microbial networks. For the purpose of depicting the characteristic parameters of the microbial network, nodes, links, path length, the clustering coefficient and modularity were used in this study. A detailed description of these parameters can be found in (Deng et al., 2012). The visualization of networks was conducted in Cytoscape 3.8.0 (Shannon et al., 2003). The nodes with Zi ≤ 2.5 and Pi ≤ 0.62 can be regarded as peripheral nodes; connectors were determined by Zi ≤ 2.5 and Pi > 0.62; module hubs were characterized by Zi > 2.5 and Pi ≤ 0.62; besides, and network hubs were identified as Zi > 2.5 and Pi > 0.62.
Statistical analysis
One-way analysis of variance (ANOVA) and Tukey’s HSD test were conducted to analyse the differences (p < 0.05) in plant richness and soil properties among different successional stages by SPSS software (Version 19.0, SPSS Inc., Chicago, IL, United States). The Wilcoxon test was used to identify the differences in abundant and rare microbial α-diversities among different successional stages. Principal coordinate analysis (PCoA) based on Bray–Curtis distance was conducted to compare the abundant and rare microbial communities among different successional stages. One-way analysis of similarity (ANOSIM) was used to identify the differences in abundant and rare microbial communities between different successional stages. The partial Mantel test with 999 permutations was used to calculate the relationships between abundant and rare microbial communities and environmental factors based on Pearson correlation (p < 0.05) with the “vegan” package. The Pearson correlation analysis was used to analyse the correlations between environmental factors and microbial network topological properties. Partial least squares path modeling (PLS-PM) was conducted to identify the complex relationships between forest succession, plant richness, soil properties and microbial communities with SmartPLS 4 software.
Results
Abundant and rare microbial diversities and community compositions among different successional stages
Abundant bacterial Chao 1 and Shannon index and the rare bacterial Chao 1 index showed no significant changes during secondary forest succession (p > 0.05), while the rare bacterial Shannon index of shrubland and secondary forest was significantly higher than that in the grassland (p < 0.05) (Figures 1A,C). The abundant fungal Shannon index and rare fungal Chao 1 and Shannon index revealed no significant difference (p > 0.05), while the abundant fungal Chao 1 index of shrubland and secondary forest was significantly higher than that in the grassland (p < 0.05) (Figures 1B,D). Moreover, the Chao 1 and Shannon index of abundant bacteria and fungi were significantly lower than those of rare bacteria and fungi (p < 0.05) (Figures 1A-D). The β-diversities of abundant bacteria and fungi (R = 0.595, p = 0.001; R = 0.813, p = 0.001) and rare bacteria and fungi (R = 0.785, p = 0.001; R = 0.776, p = 0.001) changed significantly with secondary forest succession (Figure 2).
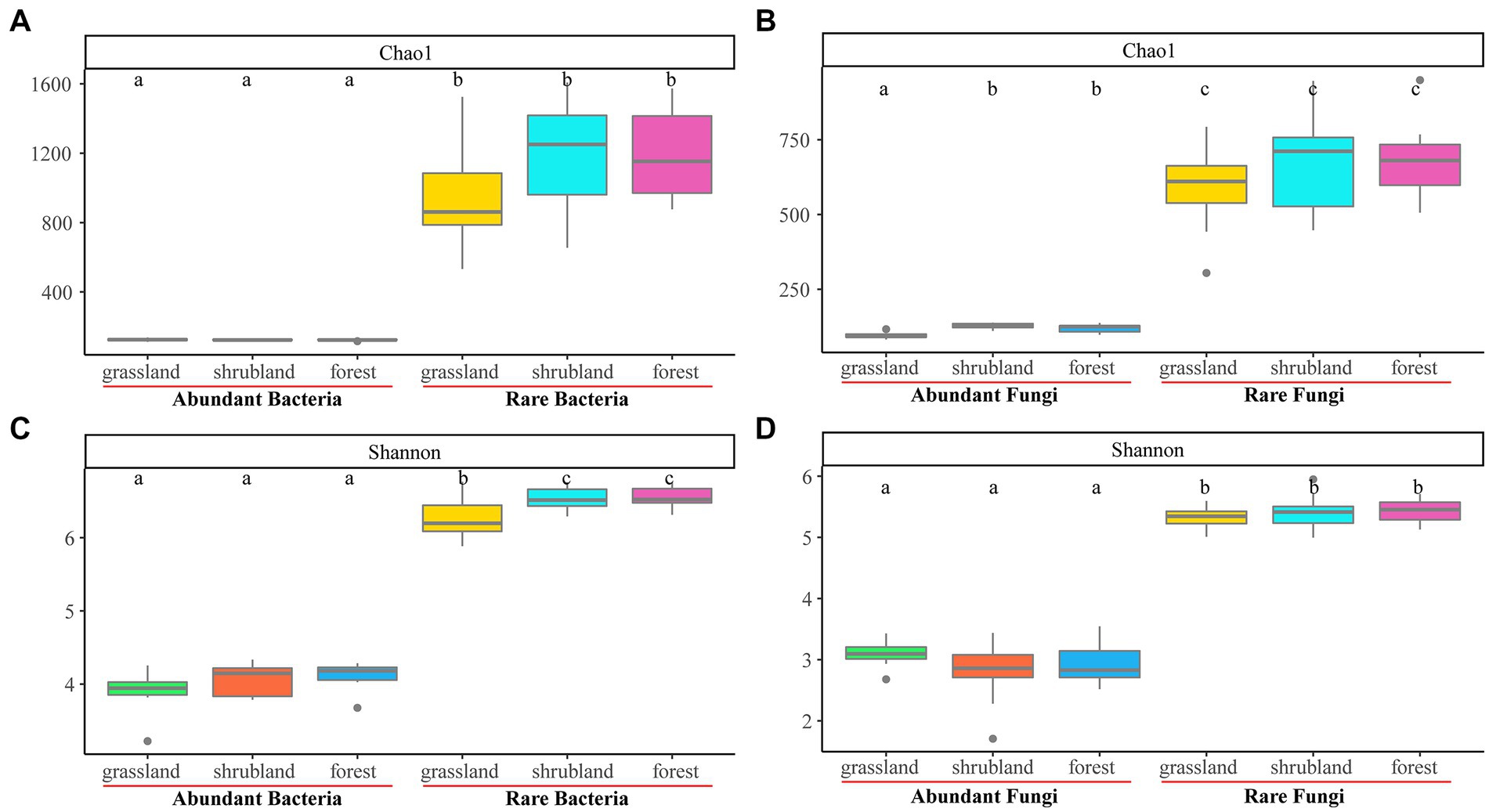
Figure 1. Boxplot of the Chao1 and Shannon index of abundant and rare bacterial communities (A, C), and abundant and rare fungal communities (B, D) during secondary forest succession.
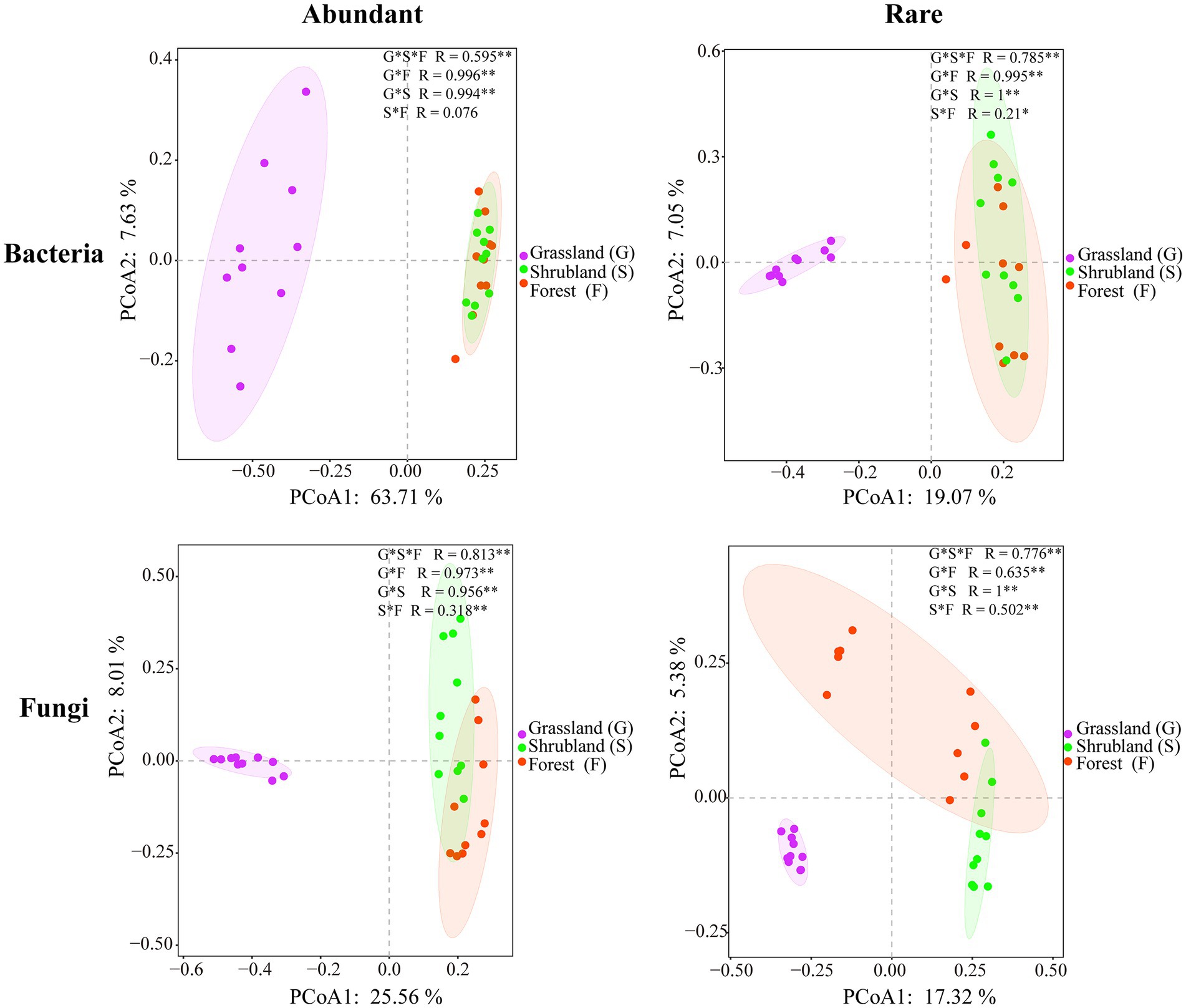
Figure 2. Principal coordinate analysis according to the Bray–Curtis distances showing the abundant and rare microbial communities.
A total of 7,263 bacterial ASVs were detected in all samples, and abundant and rare bacterial taxa included 166 ASVs and 5,671 ASVs, respectively. The numbers of abundant and rare bacterial phyla were 10 and 29 at the three successional stages, respectively (Figure 3). The abundant and rare bacterial communities were dominated by Proteobacteria (18.97–20.18%, 4.86–5.41%), Acidobacteria (4.82–17.20%, 2.43–2.78%) and Actinobacteria (4.16–11.80%, 2.00–4.07%) (Figure 3). A total of 3,971 fungal ASVs were identified in all samples, and abundant and rare fungal taxa included 181 ASVs and 3,043 ASVs among the three successional stages, respectively. The numbers of abundant and rare fungal phyla were 4 and 10 at the three successional stages, respectively (Figure 3). Ascomycota (26.34–42.31%, 3.99–5.71%) and Basidiomycota (15.93–38.58%, 1.12–2.30%) were the dominant phyla of abundant and rare fungal communities (Figure 3).
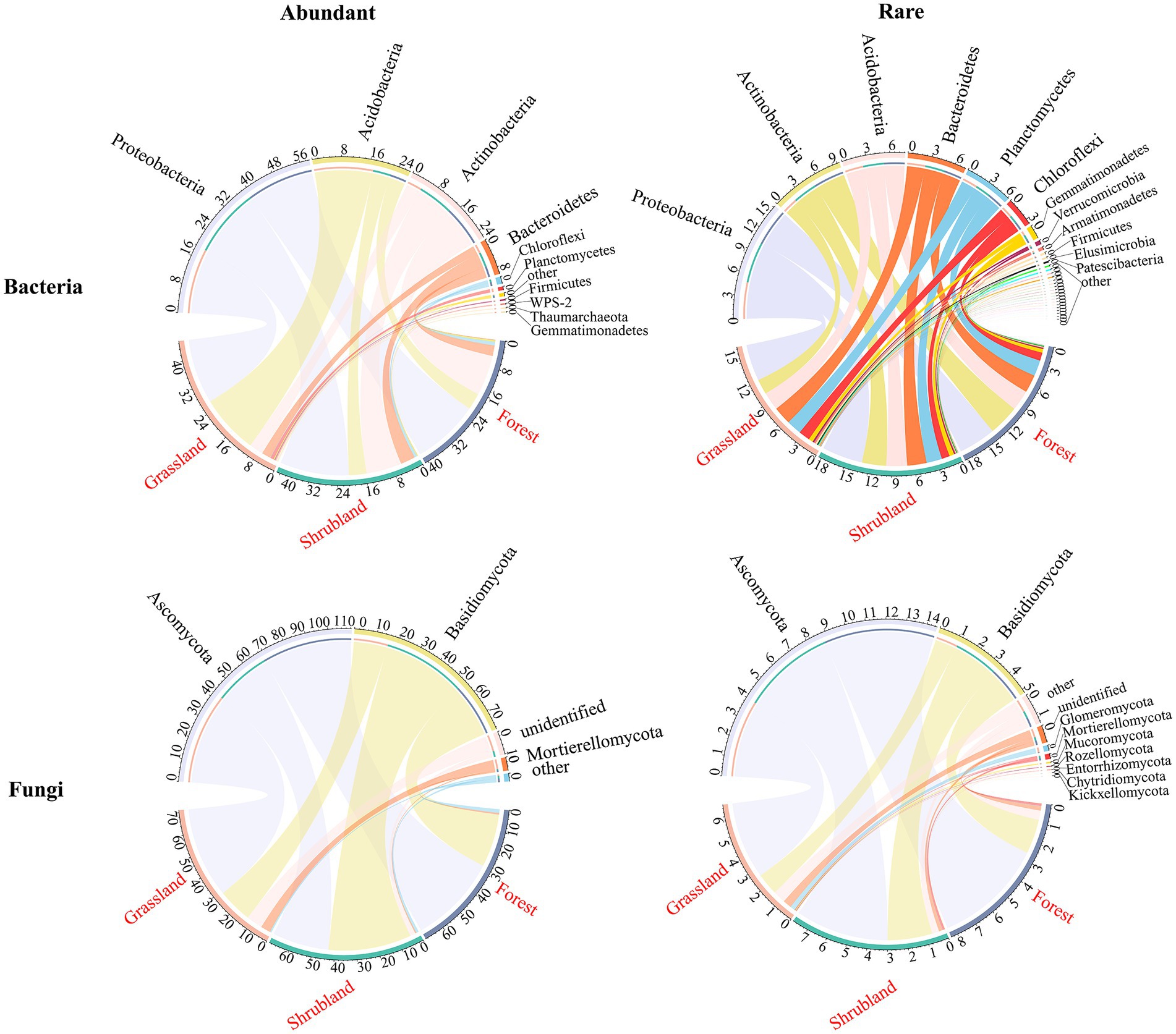
Figure 3. Chord diagram of abundant and rare bacterial and fungal community compositions at the phylum level among the three successional stages.
Co-occurrence networks of abundant and rare microbial communities among different successional stages
According to the network analysis results, the modularity of abundant and rare microbial networks was >0.4 (Table 1). The nodes, links, and average degree of the abundant microbial network showed no obvious changes with succession (Table 1). In contrast, the nodes, links, transitivity, density, and average degree of the rare microbial network in the secondary forest were higher than those in the grassland and shrubland, while the modularity of the rare microbial network revealed a contrary trend (Table 1). Furthermore, the numbers of nodes and links, average degree, average path distance, and harmonic geodesic distance of the abundant microbial networks were lower than those of the rare microbial networks at the three successional stages (Table 1). Additionally, our findings demonstrated that the transitivity of the abundant microbial network was higher than that of the rare microbial network at the three successional stages (Table 1). Furthermore, the nodes of abundant and rare microbial networks were dominated by bacteria (except for in the abundant microbial network in the shrubland) rather than fungi (Figure 4).
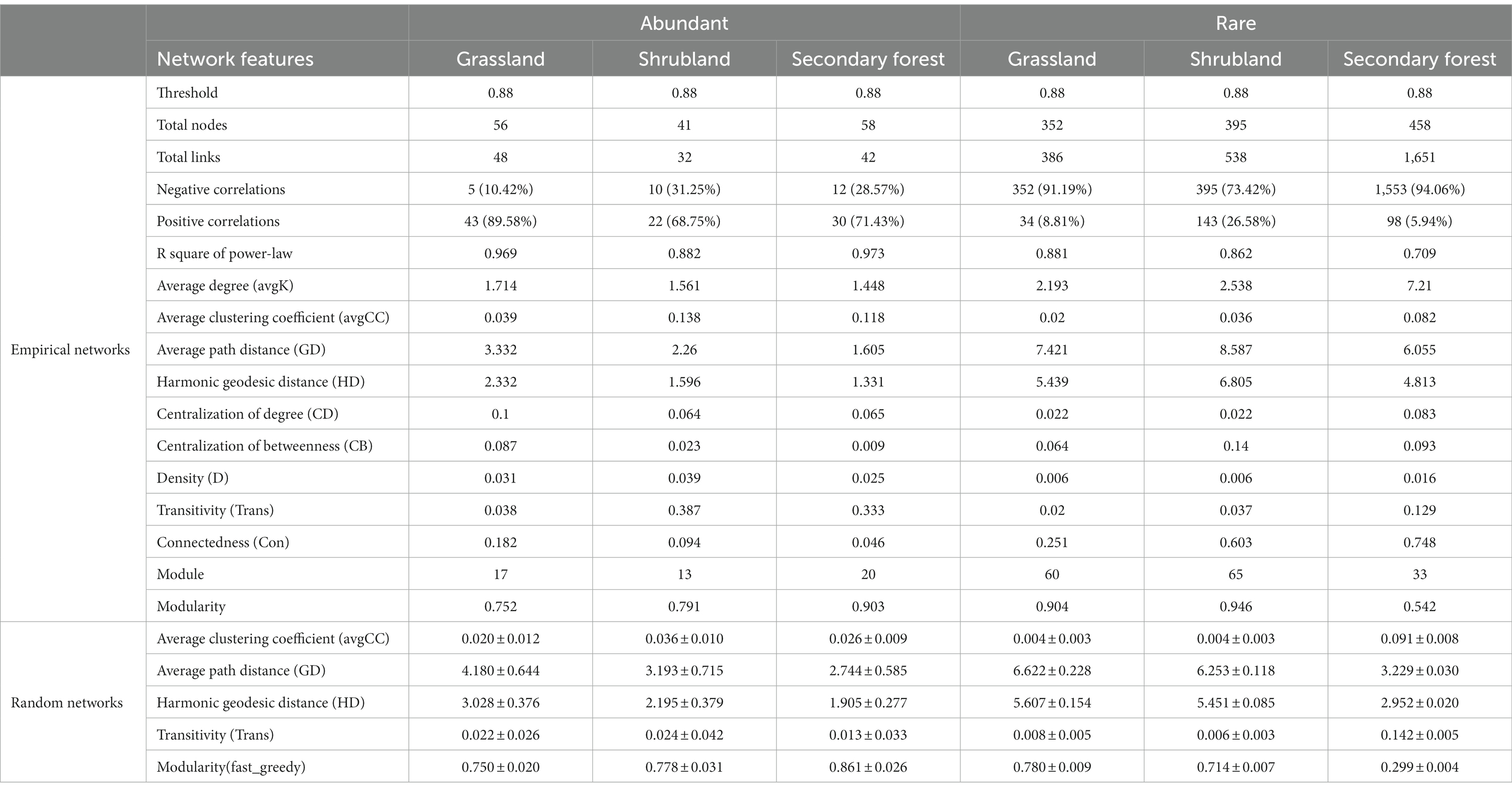
Table 1. Topologicacl properties of the abundant and rare microbial networks as well as their random networks during different successional stages.
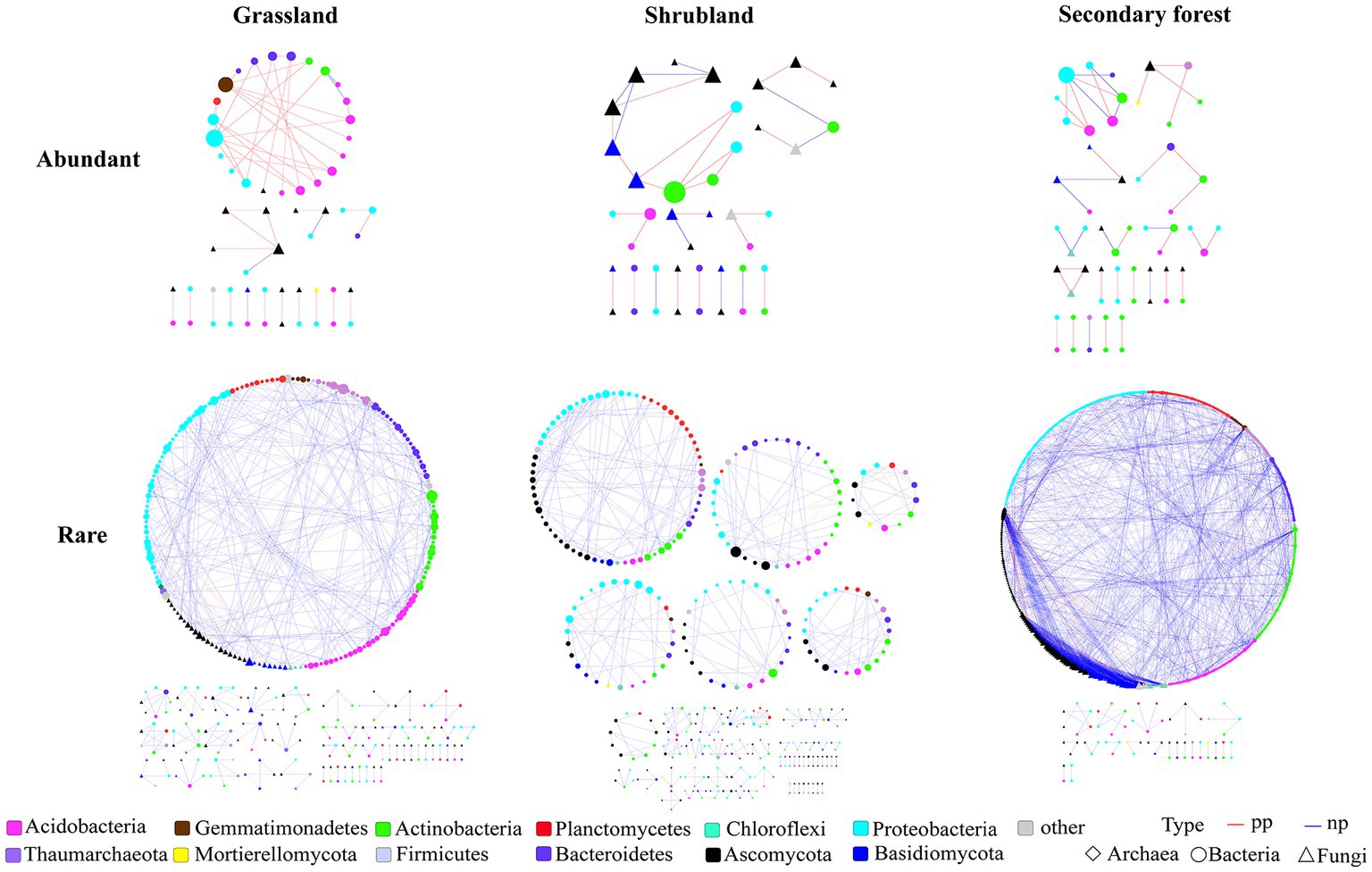
Figure 4. Co-occurrence network analysis of the abundant and rare microbial communities during forest successional stages.
Keystone species of abundant and rare microbial networks among different successional stages
Previous studies have noted that module hubs, connectors and network hubs could play the role of putative keystone species (He et al., 2017; Wang et al., 2018; Yu et al., 2021). In the current study, we found that the numbers of keystone species in the abundant microbial networks were lower than those in the rare microbial networks. Specifically, the abundant microbial networks consisted of 1 keystone taxa (Gemmatimonadetes, Gemmatimonas). The rare microbial networks consisted of 49 keystone taxa, including 5 in Acidobacteria (Acidobacteriales, Acidobacteria bacterium SCN 69–37, Subgroup 6 and Bryobacter), 4 in Actinobacteria (Ilumatobacteraceae, uncultured actinobacterium, IMCC26256 and Jatrophihabitans), 2 in Bacteroidetes (Microscillaceae and uncultured Flexibacter sp.), 1 in Chloroflexi (Elev-1,554), 1 in Gemmatimonadetes (Gemmatimonadaceae), 2 in Planctomycetes (Gemmataceae and Zavarzinella), 10 in Proteobacteria (Acetobacteraceae, Diplorickettsiaceae, R7C24, Betaproteobacteriales, SC-I-84, Rhizobiales, Myxococcaceae, Caulobacter and Bradyrhizobium), 1 in Verrucomicrobia (Opitutus), 17 in Ascomycota (Helotiales, Helotiaceae, GS34, Capnodiales, Venturia, Knufia, Leohumicola, Oidiodendron, Phialocephala and Podospora), 2 in Basidiomycota (Hymenogaster and Mycena) and 4 king taxa (Figure 5).
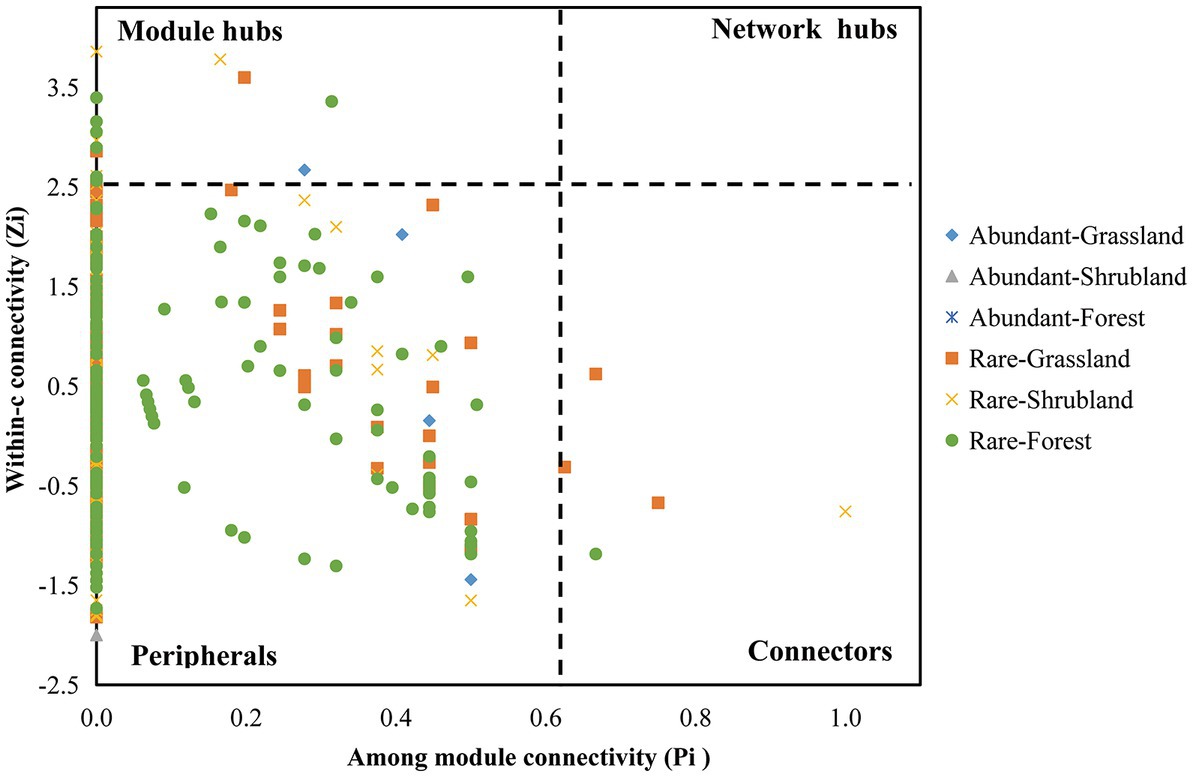
Figure 5. Putative keystone species of abundant and rare microbial networks within forest succession. Based on the scatter plot of among-module connectivity (Pi) and within-module connectivity (Zi), the topological role of each ASV was identified. In addition, the threshold values of Zi and Pi for categorizing ASVs were found to be 2.5 and 0.62, respectively.
Driving factors of abundant and rare microbial community compositions and network topological properties
A partial Mantel test was used to analyse the correlations between environmental factors (successional stages, plant richness and soil properties) and abundant and rare bacterial and fungal communities. As shown in the results, all the soil factors (except for NO3−-N) had significant influences on the abundant and rare bacterial communities (p < 0.05) (Figure 6A). Plant richness had a significant influence on the rare bacterial community (p < 0.05) but no significant influence on the abundant bacterial community (p > 0.05). Among them, soil TP was the most important factor affecting abundant (r = 0.89, p = 0.001) and rare (r = 0.79, p = 0.001) bacterial communities (Figure 6A). For the fungal community, soil properties (SOC, TN, TP, SM, C:P and N:P), successional stage and plant richness were significantly correlated with abundant and rare fungal communities (p < 0.05) (Figure 6B). NH4+-N, pH and C:N presented strong associations with abundant fungal communities rather than rare fungal communities. Successional stage (r = 0.42, p = 0.001) and plant richness (r = 0.44, p = 0.001) were the most vital influencing factors of the abundant and rare fungal communities, respectively (Figure 6B).
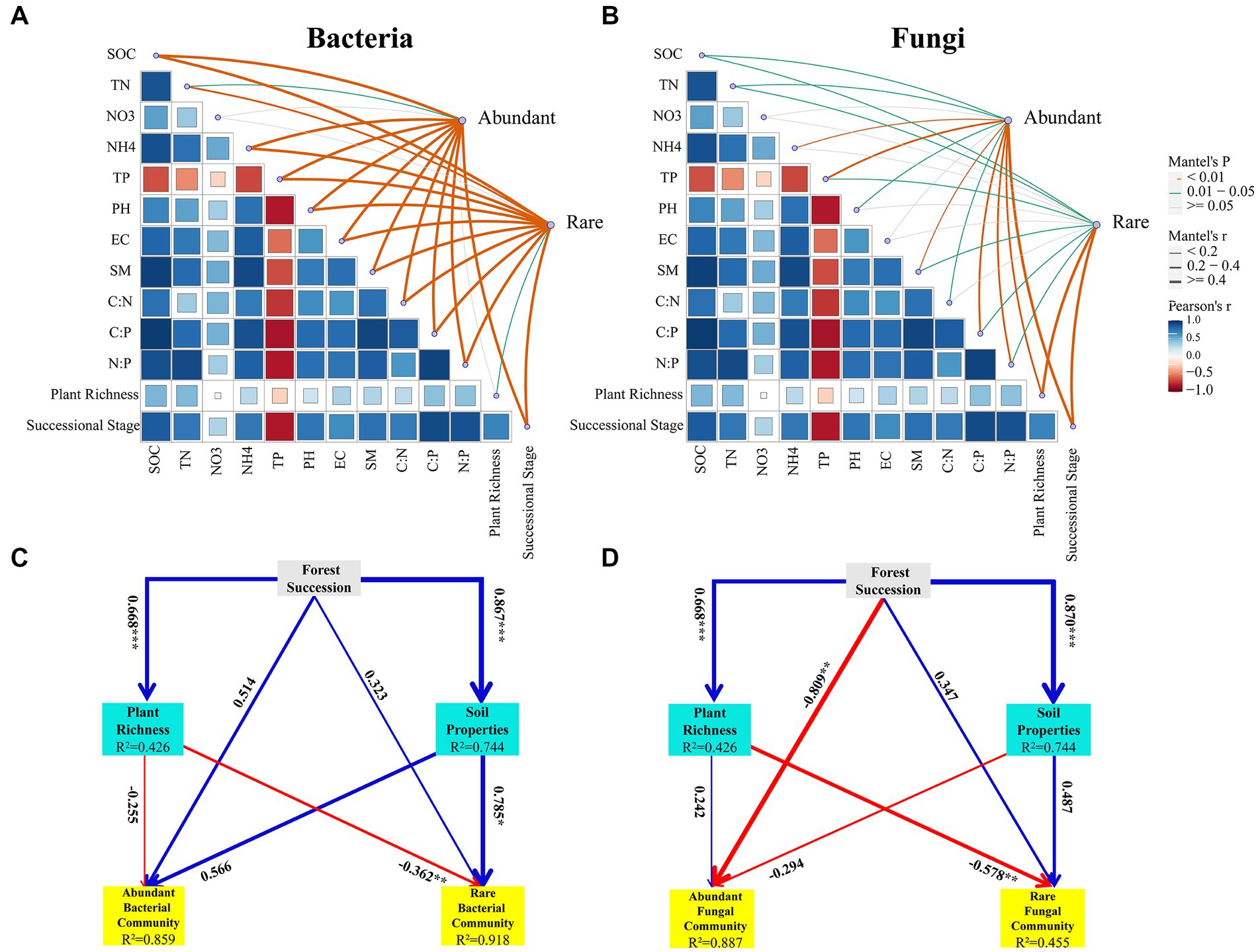
Figure 6. Pairwise comparisons between the abundant and rare bacterial (A) and fungal communities (B) in response to environmental factors during secondary forest succession. Pearson’s correlation coefficients of abundant and rare bacterial and fungal taxa and environmental factors in accordance with Mantel tests. Edge width is consistent with Mantel’s r value, and the colour of the edge suggests the statistical significance on the basis of 999 permutations. Partial least-squares path model (PLS-PM) of selected environmental factors for abundant and rare bacterial (C) and fungal (D) communities during different successional stages. The value of path coefficients are shown arrow width, blue and red colors indicate positive and negative effects, respectively. SOC, soil organic carbon; TN, total nitrogen; NO3, nitrate nitrogen; NH4, ammonium nitrogen; TP, total phosphorus; EC, electrical conductivity; SM, soil moisture content; C:N, soil organic carbon to total nitrogen; C:P, soil organic carbon to total phosphorus; N:P, total nitrogen to total phosphorus.
The PLS-PM results revealed that the path model explained 85.9 and 91.8% of the variances in the abundant and rare bacterial β-diversities, respectively (Figure 6C). Plant richness had a negative effect on the abundant (path coefficient = −0.255) and rare (path coefficient = −0.362) bacterial β-diversities (Figure 6C). Soil properties had a positive effect on the abundant (path coefficient = 0.566) and rare (path coefficient = 0.785) bacterial β-diversities (Figure 6C). For soil fungi, the path model explained 88.7 and 45.5% of the variances in the abundant and rare fungal β-diversities, respectively (Figure 6D). Plant richness had a positive effect on the abundant fungal β-diversity (path coefficient = 0.242), but had a negative effect on the rare fungal β-diversity (path coefficient = −0.578) (Figure 6D). Soil properties had a negative effect on the abundant (path coefficient = −0.294) fungal β-diversity, while it had a positive effect on the rare fungal β-diversity (path coefficient = 0.487) (Figure 6D).
The results of Pearson correlation analysis showed that C:P, SM, and N:P were significantly positively correlated with negative correlations of the abundant microbial networks but significantly negatively correlated with the harmonic geodesic distance (p < 0.05) (Figure 7A). SOC was negatively related to the average degree and average path distance of the abundant microbial networks (p < 0.05) (Figure 7A). C:P, SM and N:P were positively correlated with connectedness and total nodes of the rare microbial networks (p < 0.05) (Figure 7B). Plant richness was negatively related to the module of the rare microbial networks (p < 0.05) (Figure 7B).
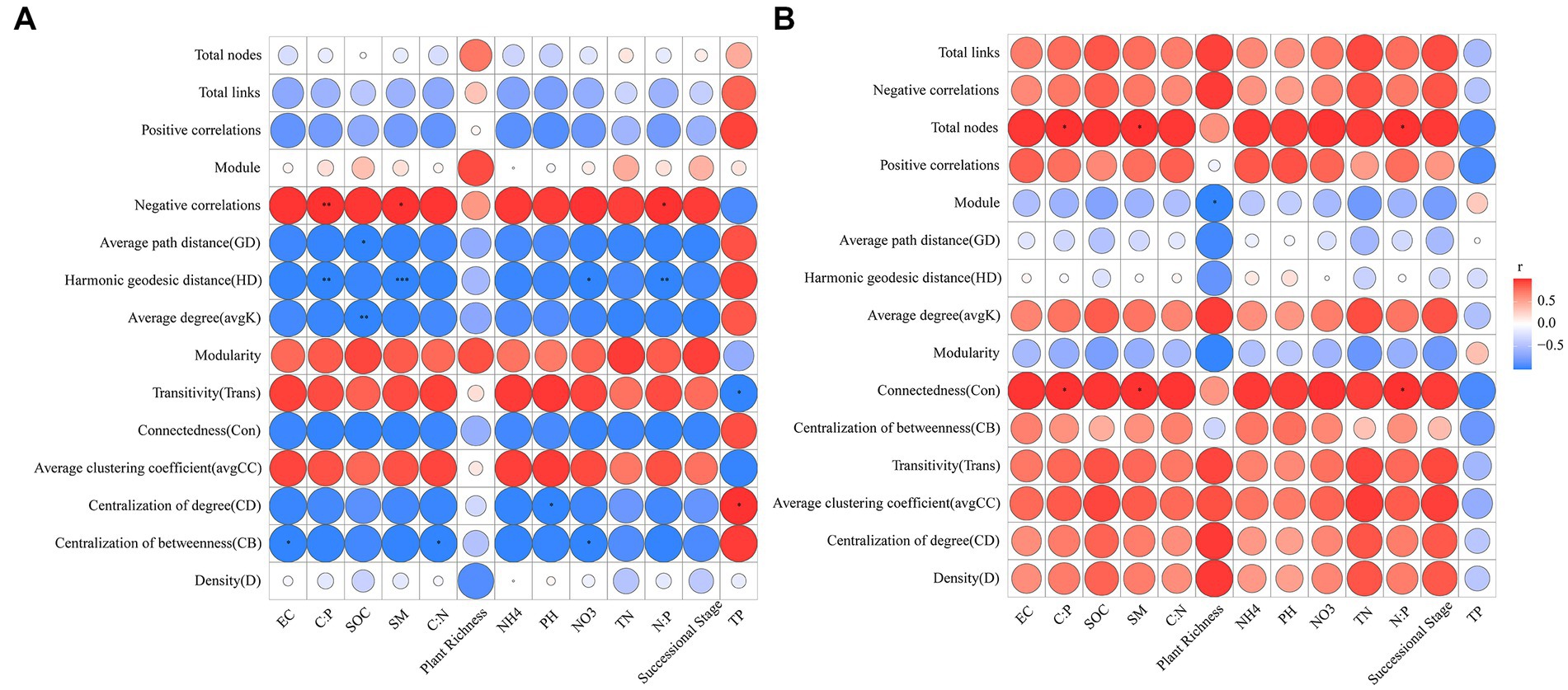
Figure 7. Pearson correlations between abundant (A) and rare (B) microbial network topological properties and environmental factors. SOC, soil organic carbon; TN, total nitrogen; NO3, nitrate nitrogen; NH4, ammonium nitrogen; TP, total phosphorus; EC, electrical conductivity; SM, soil moisture content; C:N, soil organic carbon to total nitrogen; C:P, soil organic carbon to total phosphorus; N:P, total nitrogen to total phosphorus.
Discussion
Abundant and rare microbial communities exhibit distinct changes during secondary forest succession
Previous studies have found that biotic factors (i.e., plant traits and plant richness) (Chai et al., 2019; Qiang et al., 2021) and soil factors (Qiang et al., 2021; Zhang X. et al., 2022; Zhang et al., 2023) drive changes in microbial diversity and community composition during secondary forest succession. In this study, we found significant changes in the abundant and rare bacterial and fungal β-diversities during secondary forest succession (p = 0.001). This was in line with previous studies in different ecosystems (Jiang et al., 2019; Du et al., 2020; Shu et al., 2021). This could be related to the significant changes in plant richness and soil factors (i.e., SOC, TN, NH4+-N, NO3−-N, C:N, C:P and N:P) (Table S1, S2). Moreover, the α-diversities of the abundant bacterial and fungal communities were significantly lower than those of the rare communities, which was consistent with previous studies in reforestation succession (He Z. et al., 2022), salt marsh ecosystems (Du et al., 2020) and glacier forefield succession (Jiang et al., 2019). This result also supports the previous idea that rare taxa were the main contributors to microbial diversity (Lynch and Neufeld, 2015). In this study, the numbers oSf rare bacterial and fungal phyla were obviously higher than those of the abundant bacterial and fungal phyla. Proteobacteria was the main dominant bacterial phyla of abundant and rare bacteria. Proteobacteria species have high resource utilization and adaptability to external conditions (Goldfarb et al., 2011), thus showing a higher competitive advantage in the process of secondary forest succession. Ascomycota and Basidiomycota were dominant abundant and rare fungal phyla and were also the main soil fungi in forest soil (Jiang et al., 2018, 2021; Hu et al., 2022); they can participate in the decomposition of plant litter (Štursová et al., 2020; Dong et al., 2021; Zheng et al., 2021) and play significant roles in soil nutrient cycling during forest succession.
Rare microbial co-occurrence networks are more responsive to forest succession than abundant microbial networks
Network analysis has been extensively adopted for elucidating microbial interactions as well as their responses to environmental changes (Deng et al., 2012; Dini-Andreote et al., 2014; Hu et al., 2022). In the current work, we first analysed the interactions of abundant and rare microbial communities within secondary forest succession in the subalpine area. Our results showed that the modularity of abundant and rare microbial networks during different successional stages was >0.4, indicating that microbial networks have modular structures during secondary forest succession (Xue et al., 2017; Hu et al., 2022). Some recent studies have found that abundant and rare microbial networks respond differently to environmental changes (Jiang et al., 2019; He Z. et al., 2022; Zhang X. et al., 2022). This is also supported by our study, we found that the network topological properties (i.e., nodes, links, and average degree) of the abundant microbial networks showed no obvious changes with secondary forest succession, while these properties of the rare microbial network in the secondary forest were higher than those in the grassland and shrubland, indicating that the rare microbial networks are more responsive to secondary forest succession than the abundant microbial networks. In addition, our results found that positive correlations were dominant in abundant microbial networks at different successional stages (Table 1), indicating stronger mutual cooperation interactions between abundant microorganisms, which is conducive to wider ecological niches and better environmental adaptation (Jiang et al., 2019; Du et al., 2020; Jiao and Lu, 2020; He G. et al., 2022). However, the rare microbial community was mainly negatively correlated, indicating that the competition among rare taxa was strong during the succession process, which resulted in a narrow niche of rare microbial communities (He Z. et al., 2022). In addition, we found that the numbers of nodes and links of the abundant microbial network were lower than those of the rare microbial networks at the three successional stages (Table 1). This suggests that rare microbial communities have more interactions and more complex microbial networks than abundant microbial communities. This result is similar to the findings obtained from reforestation succession soil (He Z. et al., 2022). Previous studies have confirmed that microbial network complexity is positively related to the versatility of ecosystems (Wagg et al., 2019). Therefore, the abundant and rare microbial community might have different influences on ecosystem function during secondary succession. Notably, in line with previous studies in a woodland-grassland ecotone (Banerjee et al., 2018b) and forest soil (Nakayama et al., 2019), we found that the nodes of the microbial networks were dominated by bacteria rather than fungi, which suggested that bacteria might play more important roles in maintaining the microbial network structure than fungi during secondary forest succession.
Keystone species were different between the abundant and rare microbial networks during secondary forest succession
Keystone species are usually highly connected taxonomic groups and are important for microbial structure and function (Banerjee et al., 2018a; Zheng et al., 2021). The removal of keystone species leads to the decomposition of the microbial network (Zheng et al., 2021). Keystone communities have a vital effect on ecosystem functioning (Banerjee et al., 2018a; Zheng et al., 2021; Jin et al., 2022). For example, Zheng et al. (2021) and Jin et al. (2022) confirmed that keystone species certified by network analysis were involved in litter degradation through in vitro culturing experiments. In this study, the abundant keystone taxon Gemmatimonas, which plays a major role in nitrous oxide reduction due to product-specific nitrous oxide reductases (Park et al., 2017; Oshiki et al., 2022), was reported to be the keystone taxon in Cunninghamia Lanceolata-Phoebe chekiangensis mixed plantations (Ding et al., 2023). Moreover, Gemmatimonas could be the indicator bacteria in soil nutrient content changes (Liu et al., 2022) and is able to mineralize organophosphate and dissolve inorganic nitrogen, phosphorus and potassium (Xu et al., 2023). In addition, most of the rare keystone taxa have effects on plant growth and the nutrient cycle. In general, Gemmatimonadaceae, Bryobacter (Wang et al., 2022) and Zavarzinella (Ivanova et al., 2018) contribute to organic matter decomposition and nutrient cycling. Acetobacteraceae (Reis and dos Santos Teixeira, 2015) and Bradyrhizobium (Shahrajabian et al., 2021) are nitrogen fixation bacteria that can improve the organic nitrogen content to promote plant production. Caulobacter was also reported to enhance plant biomass (Berrios, 2022). Oidiodendron is an ericoid mycobiont that can form mycorrhizae with Rhododendron sp. and prompts plant growth by improving plant nitrogen absorption (Wei et al., 2016; Lin et al., 2020). Mycena is a mycorrhizal fungus and can enhance the growth of seedlings and roots (Zhang et al., 2012). Phialocephala is a widespread plant endophyte that can improve plant phosphorus nutrition (Mikheev et al., 2022). In contrast, Venturia has been widely reported as a plant pathogenic fungus, causing plant diseases such as apple scab (Bowen et al., 2011), olive leaf spot (Buonaurio et al., 2023), and pecan scab (Standish et al., 2021), which can adversely affect the health and growth of plants. Notably, some rare keystone taxa (i.e., Jatrophihabitans, Knufia, Leohumicola, Hymenogaster and Podospora) have rarely been studied. Thus, the ecological functions of these microbes are still unknown and need to be focused on in the future. Overall, our study explores the keystone species of abundant and rare microbial communities, providing a theoretical basis for an in-depth understanding of microbial community composition and interactions. Notably, network analysis does not always represent actual microbial interactions, so the keystone species identified by network analysis are statistical. The ecological function and importance of keystone taxa still require further experimental verification (Freilich et al., 2018; Banerjee et al., 2018a; Zheng et al., 2021).
Driving factors of the abundant and rare microbial community compositions and co-occurrence networks
Investigating the driving factors of microbial community compositions and networks during secondary forest succession is crucial for understanding forest succession mechanisms. In this study, we considered the joint effects of successional stages, plant richness and soil properties on the abundant and rare microbial communities and networks during secondary forest succession. The results showed that secondary forest succession had both direct and indirect influences on the abundant and rare microbial community compositions (Figures 6C,D). Secondary forest succession was positively related to plant richness (p < 0.001) (Figures 6C,D). Plant richness increased with secondary forest succession and was conducive to improving soil carbon availability (Lange et al., 2015) and diversifying plant-derived available resources (Reynolds et al., 2003) for soil microorganisms, thereby regulating abundant and rare soil microbial community compositions during secondary forest succession. In addition, secondary forest succession had significantly positive influences on soil properties (i.e., SOC, TN, NH4+-N, NO3−-N, SM, EC, pH, C:N, N:P, and N:P) (p < 0.001) (Figures 6C,D), and the changed soil properties also drove the changes in abundant and rare microbial communities (Jiang et al., 2019; He Z. et al., 2022). The PLS-PM results showed that the interpretation rate of the joint effects of forest succession, plant richness and soil properties for the abundant fungal community was obviously higher than that of the rare fungal community, and most of the variations in the rare fungal community could not be explained by the selected environmental variables. A possible explanation for this result is that rare fungi were driven not only by environmental factors but also by stochastic population fluctuation processes and and fitness trade-offs (Jousset et al., 2017). Soil TP was the most important driving factor of abundant and rare bacterial communities during succession (r = 0.89, p = 0.001; r = 0.79, p = 0.001) (Figure 6A). This could be attributed to extensive P limitation in this study area (Yin et al., 2021). Previous studies in forest soils (Wan and He, 2020; Yan et al., 2021) also found that TP had an important impact on soil bacterial communities. In contrast, successional stages and plant richness were the most important influencing factors for the abundant and rare fungal communities, respectively (Figure 6B). The different influencing factors of bacteria and fungi during succession might be related to the following reasons: Soil fungi (i.e., mycorrhizal fungi) could form symbioses with specific plant species (Bai et al., 2020) and participate in lignin and cellulose decomposition of litter (i.e., white-rot fungi) (Rodríguez-Couto, 2017). The changes in plant richness during secondary forest succession result in changes in litter properties, which further lead to diversification of fungal litter decomposers. Thus, plant richness had a more important influence on the soil fungal community than on the soil bacterial community. Different from the driving factors of abundant and rare microbial communities during succession, soil C:P, SM, and N:P revealed significant correlations with the abundant and rare microbial network topological properties. These results suggest that soil C:P, SM, and N:P might play major roles in regulating the abundant and rare microbial networks. Taken together, our results confirm that the driving factors of abundant and rare microbial communities and interactions are different during secondary forest succession.
Conclusion
In conclusion, our results showed that both the β-diversities of abundant and rare bacterial and fungal communities changed significantly during secondary forest succession. The α-diversities of rare bacterial and fungal communities were significantly higher than those of the abundant communities, indicating that rare taxa might be the main contributors to microbial α-diversities. In addition, rare microbial networks showed more microbial interactions and greater network complexity, and were more responsive to secondary forest succession than abundant microbial communities, indicating that abundant and rare microbial co-occurrence networks responded differently to secondary forest succession. The keystone species varied in abundant and rare microorganisms at the three successional stages, and the numbers of keystone taxa of rare microorganisms were higher than those of abundant microorganisms. These keystone species might participate in plant growth and nutrient cycle during secondary forest succession. Moreover, plant richness and soil properties jointly drove changes in abundant and rare microbial communities and co-occurrence networks during secondary forest succession. However, the main influencing factors of abundant and rare microbial communities and co-occurrence networks were different. This finding provides theoretical knowledge for deeply understanding the mechanism driving microbial dynamics in the process of secondary forest succession.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
QL, YK, and WZ designed this study. QL, YK, WZ, YL, and HH collected the soil samples. HH carried out the soil property measurement. XZ performed the data analysis and wrote the manuscript. QL, YK, and KF revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant numbers 41930645 and 32171550); the Second Tibetan Plateau Scientific Expedition and Research Program (2019QZKK0303); the Youth Innovation Promotion Association of the Chinese Academy of Sciences (grant numbers 2021371); the Open Fund of Ecological Restoration and Biodiversity Conservation Key Laboratory of Sichuan Province, Chengdu Institute of Biology, Chinese Academy of Sciences (KXYSWS2202).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1177239/full#supplementary-material
Footnotes
References
Bai, Z., Yuan, Z. Q., Wang, D. M., Fang, S., Ye, J., Wang, X. G., et al. (2020). Ectomycorrhizal fungus-associated determinants jointly reflect ecological processes in a temperature broad-leaved mixed forest. Sci. Total Environ. 703:135475. doi: 10.1016/j.scitotenv.2019.135475
Banerjee, S., Schlaeppi, K., and van der Heijden, M. G. A. (2018a). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576. doi: 10.1038/s41579-018-0024-1
Banerjee, S., Thrall, P. H., Bissett, A., van der Heijden, M. G. A., and Richardson, A. E. (2018b). Linking microbial co-occurrences to soil ecological processes across a woodland-grassland ecotone. Ecol. Evol. 8, 8217–8230. doi: 10.1002/ece3.4346
Berrios, L. (2022). The genus Caulobacter and its role in plant microbiomes. World J Microb Biot 38:43. doi: 10.1007/s11274-022-03237-0
Bowen, J. K., Mesarich, C. H., Bus, V. G. M., Beresford, R. M., Plummer, K. M., and Templeton, M. D. (2011). Venturia inaequalis: the causal agent of apple scab. Mol. Plant Pathol. 12, 105–122. doi: 10.1111/J.1364-3703.2010.00656.X
Buonaurio, R., Almadi, L., Famiani, F., Moretti, C., Agosteo, G. E., and Schena, L. (2023). Olive leaf spot caused by Venturia oleaginea: An updated review. Front. Plant Sci. 13:1061136. doi: 10.3389/fpls.2022.1061136
Cao, J., Pan, H., Chen, Z., and Shang, H. (2020). Bacterial, fungal, and archaeal community assembly patterns and their determining factors across three subalpine stands at different stages of natural restoration after clear-cutting. J. Soils Sediments 20, 2794–2803. doi: 10.1007/s11368-020-02608-0
Chai, Y., Cao, Y., Yue, M., Tian, T., Yin, Q., Dang, H., et al. (2019). Soil abiotic properties and plant functional traits mediate associations between soil microbial and plant communities during a secondary forest succession on the Loess Plateau. Front. Microbiol. 10:895. doi: 10.3389/fmicb.2019.00895
Chen, L., Xiang, W., Wu, H., Ouyang, S., Zhou, B., Zeng, Y., et al. (2019). Tree species identity surpasses richness in affecting soil microbial richness and community composition in subtropical forests. Soil Biol. Biochem. 130, 113–121. doi: 10.1016/j.soilbio.2018.12.008
Cline, L. C., and Zak, D. R. (2015). Soil microbial communities are shaped by plant-driven changes in resource availability during secondary succession. Ecology 96, 3374–3385. doi: 10.1890/15-0184.1
de Araujo, A. S. F., Mendes, L. W., Lemos, L. N., Antunes, J. E. L., Beserra, J. E. A. Jr., de Lyra, M. D. C. C. P., et al. (2018). Protist species richness and soil microbiome complexity increase towards climax vegetation in the Brazilian Cerrado. Commun Biol 1:135. doi: 10.1038/s42003-018-0129-0
de Vries, F. T., Griffiths, R. I., Bailey, M., Craig, H., Girlanda, M., Gweon, H. S., et al. (2018). Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 9:3033. doi: 10.1038/s41467-018-05516-7
Deng, Y., Jiang, Y. H., Yang, Y., He, Z., Luo, F., and Zhou, J. (2012). Molecular ecological network analyses. BMC Bioinformatics 13:113. doi: 10.1186/1471-2105-13-113
Ding, K., Zhang, Y., Liu, H., Yang, X., Zhang, J., and Tong, Z. (2023). Soil bacterial community structure and functions but not assembly processes are affected by the conversion from monospecific Cunninghamia lanceolata plantations to mixed plantations. Appl. Soil Ecol. 185:104775. doi: 10.1016/j.apsoil.2022.104775
Dini-Andreote, F., de Cássia Pereira e Silva, M., Triadó-Margarit, X., Casamayor, E. O., van Elsas, J. D., and Salles, J. F. (2014). Dynamics of bacterial community succession in a salt marsh chronosequence: evidences for temporal niche partitioning. ISME J. 8, 1989–2001. doi: 10.1038/ismej.2014.54
Dong, X., Gao, P., Zhou, R., Li, C., Dun, X., and Niu, X. (2021). Changing characteristics and influencing factors of the soil microbial community during litter decomposition in a mixed Quercus acutissima Carruth. and Robinia pseudoacacia L. forest in Northern China. Catena 196:104811. doi: 10.1016/j.catena.2020.104811
Dong, K., Yu, Z., Kerfahi, D., Lee, S. S., Li, N., Yang, T., et al. (2022). Soil microbial co-occurrence networks become less connected with soil development in a high Arctic glacier foreland succession. Sci. Total Environ. 813:152565. doi: 10.1016/j.scitotenv.2021.152565
Du, S., Dini-Andreote, F., Zhang, N., Liang, C., Yao, Z., Zhang, H., et al. (2020). Divergent co-occurrence patterns and assembly processes structure the abundant and rare bacterial communities in a salt marsh ecosystem. Appl Environ Microb 86, e00322–e00320. doi: 10.1128/aem.00322-20
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Freilich, M. A., Wieters, E., Broitman, B. R., Marquet, P. A., and Navarrete, S. A. (2018). Species co-occurrence networks: Can they reveal trophic and non-trophic interactions in ecological communities? Ecology 99, 690–699. doi: 10.1002/ecy.2142
Goldfarb, K. C., Karaoz, U., Hanson, C. A., Santee, C. A., Bradford, M. A., Treseder, K. K., et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol. 2:94. doi: 10.3389/fmicb.2011.00094
Guo, Y., Hou, L., Zhang, Z., Zhang, J., Cheng, J., Wei, G., et al. (2019). Soil microbial diversity during 30 years of grassland restoration on the Loess Plateau, China: Tight linkages with plant diversity. Land Degrad. Dev. 30, 1172–1182. doi: 10.1002/ldr.3300
He, Z., Liu, D., Shi, Y., Wu, X., Dai, Y., Shang, Y., et al. (2022). Broader environmental adaptation of rare rather than abundant bacteria in reforestation succession soil. Sci. Total Environ. 828:154364. doi: 10.1016/j.scitotenv.2022.154364
He, G., Peng, T., Guo, Y., Wen, S., Ji, L., and Luo, Z. (2022). Forest succession improves the complexity of soil microbial interaction and ecological stochasticity of community assembly: Evidence from Phoebe bournei-dominated forests in subtropical regions. Front. Microbiol. 13:1021258. doi: 10.3389/fmicb.2022.1021258
He, D., Shen, W., Eberwein, J., Zhao, Q., Ren, L., and Wu, Q. L. (2017). Diversity and co-occurrence network of soil fungi are more responsive than those of bacteria to shifts in precipitation seasonality in a subtropical forest. Soil Biol. Biochem. 115, 499–510. doi: 10.1016/j.soilbio.2017.09.023
Horn, H. S. (1974). The ecology of secondary succession. Annu. Rev. Ecol. Systemat.SSS 5, 25–37. doi: 10.1146/annurev.es.05.110174.000325
Hu, L., Li, Q., Yan, J., Liu, C., and Zhong, J. (2022). Vegetation restoration facilitates belowground microbial network complexity and recalcitrant soil organic carbon storage in southwest China karst region. Sci. Total Environ. 820:153137. doi: 10.1016/j.scitotenv.2022.153137
Hu, J., Wei, Z., Kowalchuk, G. A., Xu, Y., Shen, Q., and Jousset, A. (2020). Rhizosphere microbiome functional diversity and pathogen invasion resistance build up during plant development. Environ. Microbiol. 22, 5005–5018. doi: 10.1111/1462-2920.15097
Ihrmark, K., Bodeker, I. T. M., Cruz-Martinez, K., Friberg, H., Kubartova, A., Schenck, J., et al. (2012). New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 82, 666–677. doi: 10.1111/j.1574-6941.2012.01437.x
Ivanova, A. A., Philippov, D. A., Kulichevskaya, I. S., and Dedysh, S. N. (2018). Distinct diversity patterns of Planctomycetes associated with the freshwater macrophyte Nuphar lutea (L.) Smith. Anton Leeuw 111, 811–823. doi: 10.1007/s10482-017-0986-4
Ji, L., Shen, F., Liu, Y., Yang, Y., Wang, J., Purahong, W., et al. (2022). Contrasting altitudinal patterns and co-occurrence networks of soil bacterial and fungal communities along soil depths in the cold-temperate montane forests of China. Catena 209:105844. doi: 10.1016/j.catena.2021.105844
Jiang, Y., Lei, Y., Yang, Y., Korpelainen, H., Niinemets, U., and Li, C. (2018). Divergent assemblage patterns and driving forces for bacterial and fungal communities along a glacier forefield chronosequence. Soil Biol. Biochem. 118, 207–216. doi: 10.1016/j.soilbio.2017.12.019
Jiang, Y., Song, H., Lei, Y., Korpelainen, H., and Li, C. (2019). Distinct co-occurrence patterns and driving forces of rare and abundant bacterial subcommunities following a glacial retreat in the eastern Tibetan Plateau. Biol. Fert. Soils 55, 351–364. doi: 10.1007/s00374-019-01355-w
Jiang, S., Xing, Y., Liu, G., Hu, C., Wang, X., Yan, G., et al. (2021). Changes in soil bacterial and fungal community composition and functional groups during the succession of boreal forests. Soil Biol. Biochem. 161:108393. doi: 10.1016/j.soilbio.2021.108393
Jiao, S., and Lu, Y. (2020). Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob. Chang. Biol. 26, 4506–4520. doi: 10.1111/gcb.15130
Jin, X., Wang, Z., Wu, F., Li, X., and Zhou, X. (2022). Litter mixing alters microbial decomposer community to accelerate tomato root litter decomposition. Microbiol. Spectr. 10:3. doi: 10.1128/spectrum.00186-22
Jousset, A., Bienhold, C., Chatzinotas, A., Gallien, L., Gobet, A., Kurm, V., et al. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862. doi: 10.1038/ismej.2016.174
Lange, M., Eisenhauer, N., Sierra, C. A., Bessler, H., Engels, C., Griffiths, R. I., et al. (2015). Plant diversity increases soil microbial activity and soil carbon storage. Nat. Commun. 6:6707. doi: 10.1038/ncomms7707
Li, S., Huang, X., Shen, J., Xu, F., and Su, J. (2020). Effects of plant diversity and soil properties on soil fungal community structure with secondary succession in the Pinus yunnanensis forest. Geoderma 379:114646. doi: 10.1016/j.geoderma.2020.114646
Lin, L. C., Lin, W. R., Hsu, Y. C., and Pan, H. Y. (2020). Influences of three Oidiodendron maius isolates and two inorganic nitrogen sources on the growth of Rhododendron kanehirae. Hortic. Sci. Technol. 38, 742–753. doi: 10.7235/HORT.20200067
Liu, Q (2002) Ecological Research on Subalpine Coniferous Forests. Sichuan University Press, Chengdu (In Chinese).
Liu, J., Jia, X., Yan, W., Zhong, Y., and Shangguan, Z. (2020). Changes in soil microbial community structure during long-term secondary succession. Land Degrad. Dev. 31, 1151–1166. doi: 10.1002/ldr.3505
Liu, Q., Wu, Y., and He, H. (2001). Ecological problems of subalpine coniferous forest in the southwest of China. World Scientif. Technol Res. Dev. 2, 63–69. doi: 10.16507/j.issn.1006-6055.2001.02.024
Liu, C., Zhuang, J., Wang, J., Fan, G., Feng, M., and Zhang, S. (2022). Soil bacterial communities of three types of plants from ecological restoration areas and plantgrowth promotional benefits of Microbacterium invictum (strain X-18). Front. Microbiol. 13:926037. doi: 10.3389/fmicb.2022.926037
Lynch, M. D. J., and Neufeld, J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Mikheev, V. S., Struchkova, I. V., Ageyeva, M. N., Brilkina, A. A., and Berezina, E. V. (2022). The role of Phialocephala fortinii in improving plants’ phosphorus nutrition: New puzzle pieces. J. Fungi 8:1225. doi: 10.3390/jof8111225
Morrien, E., Hannula, S. E., Snoek, L. B., Helmsing, N. R., Zweers, H., de Hollander, M., et al. (2017). Soil networks become more connected and take up more carbon as nature restoration progresses. Nat. Commun. 8:14349. doi: 10.1038/ncomms14349
Mougi, A., and Kondoh, M. (2012). Diversity of interaction types and ecological community stability. Science 337, 349–351. doi: 10.1126/science.1220529
Nakayama, M., Imamura, S., Taniguchi, T., and Tateno, R. (2019). Does conversion from natural forest to plantation affect fungal and bacterial biodiversity, community structure, and co-occurrence networks in the organic horizon and mineral soil? Forest Ecol. Manag. 446, 238–250. doi: 10.1016/j.foreco.2019.05.042
Nilsson, R. H., Larsson, K. H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Oshiki, M., Toyama, Y., Suenaga, T., Terada, A., Kasahara, Y., Yamaguchi, T., et al. (2022). N2O reduction by Gemmatimonas aurantiaca and potential involvement of Gemmatimonadetes bacteria in N2O reduction in agricultural soils. Microbes Environ. 37:ME21090. doi: 10.1264/jsme2.ME21090
Pan, C., Feng, Q., Li, Y., Li, Y., Liu, L., Yu, X., et al. (2022). Rare soil bacteria are more responsive in desertification restoration than abundant bacteria. Environ. Sci. Pollut. R. 29, 33323–33334. doi: 10.1007/s11356-021-16830-x
Park, D., Kim, H., and Yoon, S. (2017). Nitrous oxide reduction by an obligate aerobic bacterium, Gemmatimonas aurantiaca strain T-27. Appl. Environ. Microbiol. 83, e00502–e00517. doi: 10.1128/AEM.00502-17
Qiang, W., He, L., Zhang, Y., Liu, B., Liu, Y., Liu, Q., et al. (2021). Aboveground vegetation and soil physicochemical properties jointly drive the shift of soil microbial community during subalpine secondary succession in southwest China. Catena 202:105251. doi: 10.1016/j.catena.2021.105251
Reis, V. M., and dos Santos Teixeira, K. R. (2015). Nitrogen fixing bacteria in the family Acetobacteraceae and their role in agriculture. J. Basic Microbiol. 55, 931–949. doi: 10.1002/jobm.201400898
Reynolds, H. L., Packer, A., Bever, J. D., and Clay, K. (2003). Grassroots ecology: Plant-microbe-soil interactions as drivers of plant community structure and dynamics. Ecology 84, 2281–2291. doi: 10.1890/02-0298
Rodríguez-Couto, S. (2017). Industrial and environmental applications of white-rot fungi. Mycosphere 8, 456–466. doi: 10.5943/mycosphere/8/3/7
Rodriguez-Ramos, J. C., Cale, J. A., Cahill, J. F. Jr., Simard, S. W., Karst, J., and Erbilgin, N. (2021). Changes in soil fungal community composition depend on functional group and forest disturbance type. New Phytol. 229, 1105–1117. doi: 10.1111/nph.16749
Shade, A., Jones, S. E., Caporaso, J. G., Handelsman, J., Knight, R., Fierer, N., et al. (2014). Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. MBio 5, e01371–e01314. doi: 10.1128/mBio.01371-14
Shahrajabian, M. H., Sun, W., and Cheng, Q. (2021). The importance of Rhizobium, Agrobacterium, Bradyrhizobium, Herbaspirillum, Sinorhizobium in sustainable agricultural production. Not. Bot. Horti. Agrobo. 49:12183. doi: 10.15835/nbha49312183
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shi, Y., Delgado-Baquerizo, M., Li, Y., Yang, Y., Zhu, Y. G., Peñuelas, J., et al. (2020). Abundance of kinless hubs within soil microbial networks are associated with high functional potential in agricultural ecosystems. Environ. Int. 142:105869. doi: 10.1016/j.envint.2020.105869
Shu, D., Guo, Y., Zhang, B., Zhang, C., Van Nostrand, J. D., Lin, Y., et al. (2021). Rare prokaryotic sub-communities dominate the complexity of ecological networks and soil multinutrient cycling during long-term secondary succession in China's Loess Plateau. Sci. Total Environ. 774:145737. doi: 10.1016/j.scitotenv.2021.145737
Standish, J. R., Brenneman, T. B., Bock, C. H., and Stevenson, K. L. (2021). Fungicide resistance in Venturia effusa, cause of pecan scab: current status and practical implications. Phytopathology 111, 244–252. doi: 10.1094/PHYTO-06-20-0221-RVW
Štursová, M., Šnajdr, J., Koukol, O., Tláskal, V., Cajthaml, T., and Baldrian, P. (2020). Long-term decomposition of litter in the montane forest and the definition of fungal traits in the successional space. Fungal Ecol. 46:100913. doi: 10.1016/j.funeco.2020.100913
Tamaki, H., Wright, C. L., Li, X., Lin, Q., Hwang, C., Wang, S., et al. (2011). Analysis of 16S rRNA amplicon sequencing options on the Roche/454 next-generation titanium sequencing platform. PLoS One 6:e25263. doi: 10.1371/journal.pone.0025263
Wagg, C., Schlaeppi, K., Banerjee, S., Kuramae, E. E., and van der Heijden, M. G. A. (2019). Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 10:4841. doi: 10.1038/s41467-019-12798-y
Wan, P., and He, R. (2020). Soil microbial community characteristics under different vegetation types at the national nature reserve of Xiaolongshan Mountains. Northwest China. Ecol. Inform. 55:101020. doi: 10.1016/j.ecoinf.2019.101020
Wan, W., Liu, S., Li, X., Xing, Y., Chen, W., and Huang, Q. (2021). Bridging rare and abundant bacteria with ecosystem multifunctionality in salinized agricultural soils: from community diversity to environmental adaptation. mSystems. 6, e01221–e01220. doi: 10.1128/mSystems.01221-20
Wang, K., Qiu, Z., Zhang, M., Li, X., Fang, X., Zhao, M., et al. (2022). Effect of afforestation mode on rhizosphere soil physicochemical properties and bacterial community structure of two major tree species in Xiong’an New Area. Forest Ecol. Manag. 520:120361. doi: 10.1016/j.foreco.2022.120361
Wang, S., Wang, X., Han, X., and Deng, Y. (2018). Higher precipitation strengthens the microbial interactions in semi-arid grassland soils. Glob. Ecol. Biogeogr. 27, 570–580. doi: 10.1111/geb.12718
Wei, X., Chen, J., Zhang, C., and Pan, D. (2016). A New Oidiodendron maius strain isolated from Rhododendron fortunei and its effects on nitrogen uptake and plant growth. Front. Microbiol. 7:1327. doi: 10.3389/fmicb.2016.01327
Wu, Y., Chen, D., Saleem, M., Wang, B., Hu, S., Delgado-Baquerizo, M., et al. (2021). Rare soil microbial taxa regulate the negative effects of land degradation drivers on soil organic matter decomposition. J. Appl. Ecol. 58, 1658–1669. doi: 10.1111/1365-2664.13935
Xu, Y., Chen, Z., Li, X., Tan, J., Liu, F., and Wu, J. (2023). The mechanism of promoting rhizosphere nutrient turnover for arbuscular mycorrhizal fungi attributes to recruited functional bacterial assembly. Mol. Ecol. 32, 2335–2350. doi: 10.1111/mec.16880
Xu, G., Chen, H., Shi, Z., Liu, S., Cao, X., Zhang, M., et al. (2021). Mycorrhizal and rhizospheric fungal community assembly differs during subalpine forest restoration on the eastern Qinghai-Tibetan Plateau. Plant Soil 458, 245–259. doi: 10.1007/s11104-019-04400-7
Xue, P., Minasny, B., and McBratney, A. B. (2022). Land-use affects soil microbial co-occurrence networks and their putative functions. Appl. Soil Ecol. 169:104184. doi: 10.1016/j.apsoil.2021.104184
Xue, L., Ren, H., Li, S., Leng, X., and Yao, X. (2017). Soil bacterial community structure and co-occurrence pattern during vegetation restoration in karst rocky desertification area. Front. Microbiol. 8:2377. doi: 10.3389/fmicb.2017.02377
Yan, K., Dong, Y., Gong, Y., Zhu, Q., and Wang, Y. (2021). Climatic and edaphic factors affecting soil bacterial community biodiversity in different forests of China. Catena 207:105675. doi: 10.1016/j.catena.2021.105675
Yan, B., Sun, L., Li, J., Liang, C., Wei, F., Xue, S., et al. (2020). Change in composition and potential functional genes of soil bacterial and fungal communities with secondary succession in Quercus liaotungensis forests of the Loess Plateau, western China. Geoderma 364:114199. doi: 10.1016/j.geoderma.2020.114199
Yin, M., Guo, W., Zhu, X., Zhang, Z., Liu, Q., and Yin, H. (2021). Nutrient limiting characteristics of subalpine coniferous forests under conditions of nitrogen deposition in the southwest mountains of China (In Chinese). Chin. J. Appl. Environ. Biol. 27, 1–7. doi: 10.19675/j.cnki.1006-687x.2020.07040
Yu, Q., Hanif, A., Rao, X., He, J., Sun, D., Liu, S., et al. (2021). Long-term restoration altered edaphic properties and soil microbial communities in forests: evidence from four plantations of southern China. Restor. Ecol. 29:e13354. doi: 10.1111/rec.13354
Zhang, L., Chen, J., Lv, Y., Gao, C., and Guo, S. (2012). Mycena sp., a mycorrhizal fungus of the orchid Dendrobium officinale. Mycol. Prog. 11, 395–401. doi: 10.1007/s11557-011-0754-1
Zhang, Z., Lu, Y., Wei, G., and Jiao, S. (2022). Rare species-driven diversity-ecosystem multifunctionality relationships are promoted by stochastic community assembly. MBio 13:e0044922. doi: 10.1128/mbio.00449-22
Zhang, X., Zhao, W., Kou, Y., Liu, Y., He, H., and Liu, Q. (2023). Secondary forest succession drives differential responses of bacterial communities and interactions rather than bacterial functional groups in the rhizosphere and bulk soils in a subalpine region. Plant Soil 484, 293–312. doi: 10.1007/s11104-022-05788-5
Zhang, X., Zhao, W., Liu, Y., He, H., Kou, Y., and Liu, Q. (2022). Dominant plant species and soil properties drive differential responses of fungal communities and functions in the soils and roots during secondary forest succession in the subalpine region. Rhizosphere 21:100483. doi: 10.1016/j.rhisph.2022.100483
Zheng, H., Yang, T., Bao, Y., He, P., Yang, K., Mei, X., et al. (2021). Network analysis and subsequent culturing reveal keystone taxa involved in microbial litter decomposition dynamics. Soil Biol. Biochem. 157:108230. doi: 10.1016/j.soilbio.2021.108230
Zhou, J., Deng, Y., Luo, F., He, Z., Tu, Q., and Zhi, X. (2010). Functional molecular ecological networks. MBio 1, e00169–e00110. doi: 10.1128/mBio.00169-10
Keywords: subalpine secondary forest succession, abundant microbial community, rare microbial community, co-occurrence network, keystone taxa
Citation: Zhang X, Zhao W, Kou Y, Fang K, Liu Y, He H and Liu Q (2023) The contrasting responses of abundant and rare microbial community structures and co-occurrence networks to secondary forest succession in the subalpine region. Front. Microbiol. 14:1177239. doi: 10.3389/fmicb.2023.1177239
Edited by:
Sandhya Mishra, Key Laboratory of Tropical Forest Ecology, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Li Ji, Helmholtz Association of German Research Centres (HZ), GermanyDaquan Sun, Academy of Sciences of the Czech Republic (ASCR), Czechia
Pu Jia, South China Normal University, China
Copyright © 2023 Zhang, Zhao, Kou, Fang, Liu, He and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongping Kou, a291eXBAY2liLmFjLmNu; Qing Liu, bGl1cWluZ0BjaWIuYWMuY24=
†ORCID: Qing Liu, https://orcid.org/0000-0002-7046-0307