 Huang-Jie Jia
Huang-Jie Jia Pan-Pan Jia3
Pan-Pan Jia3 De-Sheng Pei
De-Sheng Pei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 31 May 2023
Sec. Phage Biology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1172635
Bacteriophages, the most abundant organisms on earth, have the potential to address the rise of multidrug-resistant bacteria resulting from the overuse of antibiotics. However, their high specificity and limited host range can hinder their effectiveness. Phage engineering, through the use of gene editing techniques, offers a means to enhance the host range of bacteria, improve phage efficacy, and facilitate efficient cell-free production of phage drugs. To engineer phages effectively, it is necessary to understand the interaction between phages and host bacteria. Understanding the interaction between the receptor recognition protein of bacteriophages and host receptors can serve as a valuable guide for modifying or replacing these proteins, thereby altering the receptor range of the bacteriophage. Research and development focused on the CRISPR-Cas bacterial immune system against bacteriophage nucleic acids can provide the necessary tools to promote recombination and counter-selection in engineered bacteriophage programs. Additionally, studying the transcription and assembly functions of bacteriophages in host bacteria can facilitate the engineered assembly of bacteriophage genomes in non-host environments. This review highlights a comprehensive summary of phage engineering methods, including in-host and out-of-host engineering, and the use of high-throughput methods to understand their role. The main aim of these techniques is to harness the intricate interactions between bacteriophages and hosts to inform and guide the engineering of bacteriophages, particularly in the context of studying and manipulating the host range of bacteriophages. By employing advanced high-throughput methods to identify specific bacteriophage receptor recognition genes, and subsequently introducing modifications or performing gene swapping through in-host recombination or out-of-host synthesis, it becomes possible to strategically alter the host range of bacteriophages. This capability holds immense significance for leveraging bacteriophages as a promising therapeutic approach against antibiotic-resistant bacteria.
Phages, viruses that infect bacteria, are the most abundant and ubiquitous organisms on the planet (Balcazar, 2014; Islam et al., 2019), with an estimated number of 1031 phages worldwide (Czajkowski, 2019). The study of phages and their interactions with their bacterial hosts has been a driving force behind the development of molecular biology, from the initial experiments demonstrating that DNA is the genetic material of living cells, using phages as a model system (Hershey and Chase, 1952), to the discovery and application of temperate bacteriophage-based genetic integration tools, such as the lambda red recombinases (Fehér et al., 2012), and the development of the gene-editing system RM/CRISPR (Marraffini, 2015). Phages have now become a crucial model organism for understanding all aspects of modern molecular biology (Subedi and Barr, 2021).
The extensive usage of antibiotics globally has led to the emergence of multidrug-resistant (MDR) strains in many pathogenic bacteria (Sun et al., 2022; Ul-Hamid et al., 2022). Phage therapy, which involves the use of phages that specifically infect and kill the host bacteria without affecting other bacteria, is a promising strategy against MDR (Rodriguez-Gonzalez et al., 2020; Kim et al., 2021; Salazar et al., 2021; Jia et al., 2023). However, the high host specificity and narrow host range of most phages limit their effectiveness for use in phage therapy (Zhan et al., 2015; Jeon et al., 2016). The emergence of these limitations can be attributed primarily to two factors. Firstly, host bacteria can evade infection by narrow-host-range phages through mechanisms such as evolution or immunity. This evasive behavior leads to the development of phage resistance in the target pathogenic bacteria (Pires et al., 2017; Peng and Chen, 2021). Secondly, there exist significant variations among clinical isolates of bacterial pathogens within the same species. Consequently, if a phage is unable to infect and eliminate both bacterial strains, a phage that proves effective for one patient may not be suitable for another patient (Hatfull et al., 2022). The use of a phage cocktail containing multiple phages that infect the host bacteria through different mechanisms has proven to be an effective solution (Esteves et al., 2021; Zhang et al., 2021). Moreover, employing a combination of phages that target distinct receptors on the surface of the same host bacterium can effectively impede the emergence of phage resistance within the bacterial population. For example, when clinicians utilize a cocktail preparation consisting of two phages, it is crucial for these phages to possess minimal or no shared genes. This ensures that they are unlikely to rely on the same bacterial receptor, thereby reducing the risk of mutations in a single receptor that could confer resistance against both phages (Mutalik et al., 2020; Hatfull et al., 2022). The process of screening for phages with diverse invasion pathways from nature is a demanding and time-consuming task. However, an alternative approach to broaden the host range of phages and enhance their effectiveness in phage therapy is through phage engineering using gene editing techniques. Cell-free phage production can also facilitate the efficient production of phage drugs. To effectively modify phages for therapy, it is essential to have a comprehensive understanding of the mechanism underlying the interaction between phages and their host bacteria.
Previous reviews have systematically described the complex and diverse process of the interaction between bacteriophages and host bacteria (Hampton et al., 2020). Bacteria have evolved various antiviral defense systems against infection by their viruses and mobile genetic elements, including nine identified anti-phage systems that can provide specific or broad defense, such as RM, CRISPR, and Retron (Payne et al., 2022). Besides, bacteria can prevent bacteriophage infection by reducing attachment through mutations in surface bacteriophage receptors (Gurney et al., 2019). Under the defensive pressure of the host bacteria, bacteriophages continually evolve, with mutations allowing their DNA to evade systemic targeting defense system cleavage and mutations in their receptor-binding proteins enabling them to attach to mutated receptors on the host surface (Lee et al., 2016; Yuan et al., 2019; Broniewski et al., 2020). Furthermore, bacteriophage populations can exchange receptor-binding protein genes, leading to significant changes in their host range to adapt to environmental pressures (Xiao et al., 2018; Górniak et al., 2022). The discovery of these mechanisms provides insights into the application of gene editing systems, which have been widely used in biological organisms. Bacteriophages, as the origin of these tools, have been naturally utilized in engineering modifications.
This review presents an overview of the various methods for bacteriophage engineering, including both in-host and ex-host gene editing techniques. We focus on the strategies for modifying the host range of bacteriophages, specifically the alteration of their receptor-binding proteins. These engineering methods are all based on the understanding of the mechanisms underlying the interaction between bacteriophages and their host bacteria. Besides, we discuss high-throughput methods for studying this interaction, which can facilitate the development of more effective bacteriophage engineering approaches. In summary, the phage-bacteria interaction not only provides us with valuable tools for phage engineering but also offers guidance for the further improvement of these techniques.
The genome of a temperate bacteriophage can integrate into the host chromosome, forming a prophage. This integration allows for stable incorporation and makes the bacteriophage genome amenable to nucleic acid editing using the same methods employed for bacterial genomes. Besides, gene editing of virulent bacteriophages necessitates specialized methods. Traditional methods for modifying lytic phage genes involve the use of homologous recombination technology (Court et al., 2002), which takes place within the host bacteria. In this process, a DNA template sequence with homologous arms is transfected into the host cells, and the bacteriophage DNA is edited during subsequent infection of the host bacteria (Figure 1A). The edited bacteriophage DNA, packaged in a protein coat, forms the engineered bacteriophage progeny. However, it is worth noting that the generation of recombinant phages through this method results in only a small fraction of the progeny phages. The reported rates of recombination vary between 10−10 and 10−4 (Pires et al., 2016), primarily due to the inherent challenges posed by low recombination and transformation efficiency associated with this technique.
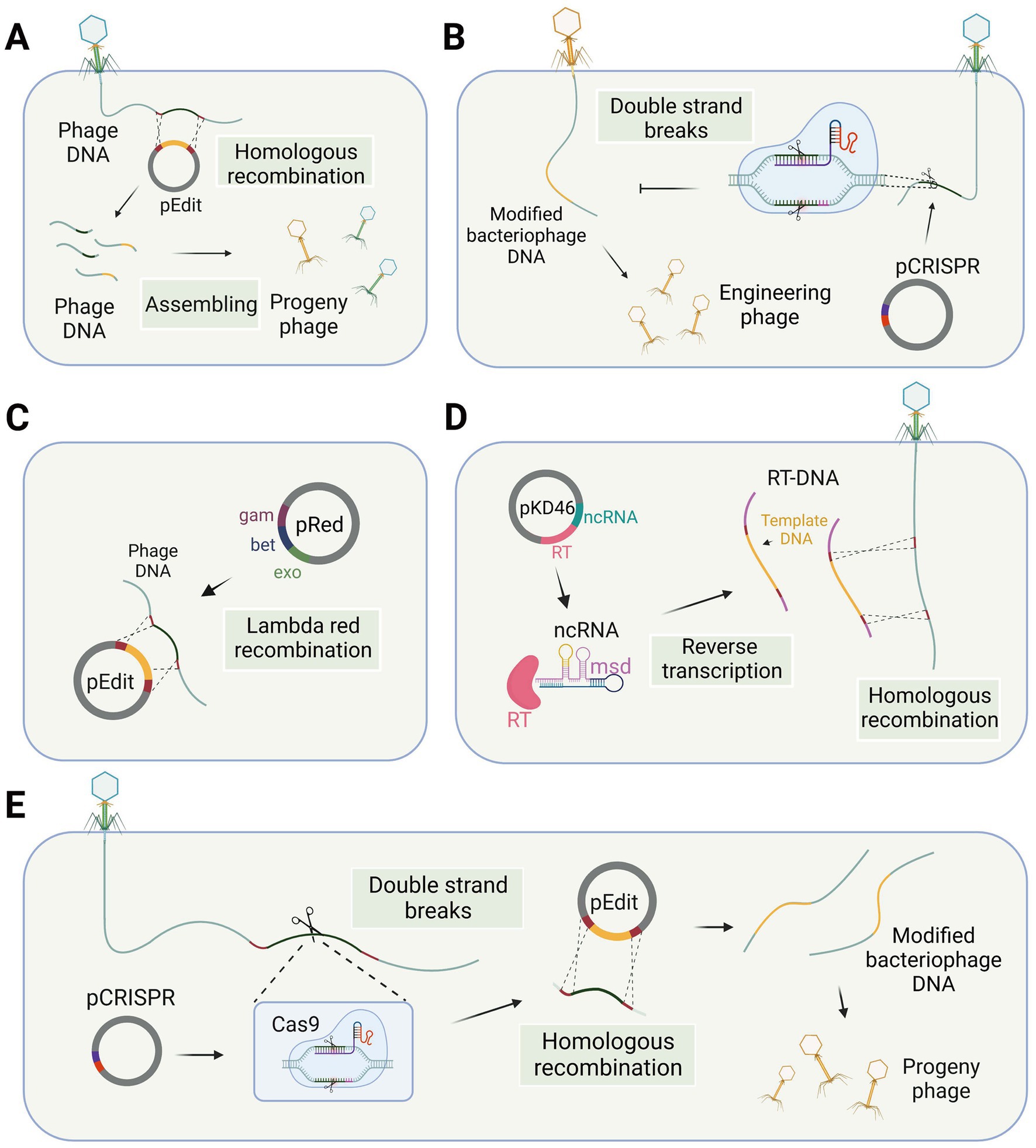
Figure 1. Phage gene editing technology in the host bacteria. (A) Phage genome editing through homologous recombination. (B) Differentiation of wild-type and engineered phages using the CRISPR-Cas system. (C) The lambda red recombination system facilitates recombination between editing templates and phage DNA in bacterial cells. (D) Modified retrons generate editing templates via reverse transcription in the host bacterium to facilitate homologous recombination. (E) CRISPR-Cas system utilized for modifying phage genes in host bacteria (single-plasmid-mediated asynchronous recombination, images are created with Biorender.com).
For gene editing of lytic bacteriophages, recombination needs to occur during the process of phage infection of the host. Shorter recombination times are a significant factor leading to low recombination efficiency. Researchers have made efforts to enhance the activity of the recombination enzyme to increase recombination efficiency. Some phage genomes contain their own recombinase enzymes, which exhibit superior performance in bacterial hosts compared to the RecA-mediated pathway and also possess better tolerance for mismatches (Manning and Dokland, 2020), although this process may result in the formation of recognition sequence scars. Consequently, in the case of this particular type of phage, the presence of a homologous template within the infected bacterial cell is adequate to generate recombinant phage progeny (Muniyappa and Radding, 1986). However, gene modification efficiency by recombination in some phages is not optimal, which may be due to the absence of self-contained recombinases in these phages. The lambda red recombinases, sourced from bacteriophages and comprising three proteins Exo, Beta, and Gam, can efficiently achieve recombination of exogenous fragments through the cooperation of these three proteins (Figure 1C). Therefore, the simultaneous introduction of the lambda red recombinase system and template DNA into the host bacterium during the editing process of Escherichia coli phages can facilitate recombination and enhance editing efficiency (Oppenheim et al., 2004; Marinelli et al., 2008; Thomason et al., 2009; Fehér et al., 2012).
The low transformation rate of host bacteria is a significant limiting factor when it comes to editing the phage genome within the host cell. This difficulty in importing template plasmids and exogenous recombination systems into the host cell hinders the improvement of recombination efficiency. Efficient transformation protocols have not been widely developed for many bacteria, particularly gram-positive bacteria with thicker cell walls. To overcome this issue, bacteriophage electroporation-based DNA recombination (BRED) has emerged as a highly efficient strategy. BRED employs electroporation to facilitate the transfer of phage DNA, template DNA, and exogenous recombination systems, such as the lambda red system or RecE/RecT-like proteins (Marinelli et al., 2008), into bacterial cells. A study conducted by Marinelli et al. (2008) demonstrated that this approach significantly enhances the level of phage gene recombination in Mycobacterium smegmatis, a gram-positive bacterium. The BRED technique achieves a high level of recombination efficiency by introducing suitable exogenous recombination systems into bacterial cells. However, it is essential for plasmid-based exogenous recombination systems to be efficiently transformed into cells and function effectively within them.
Efficient methods for selecting modified phages are essential due to the abundance of unmodified progeny phages produced in bacteriophage genetic engineering. For temperate phages, antibiotic resistance-genes can serve as markers, and conventional microbiological methods can be employed for screening. When the antibiotic resistance gene is recombined into the prophage, it imparts antibiotic resistance to the host bacteria, allowing for straightforward identification. Besides, specialized screening methods are necessary for virulent phages. One such method involves introducing marker genes, such as beta-galactosidase (Revel et al., 1961) or fluorescence protein (Yoichi et al., 2005), to facilitate plaque screening. The approach mentioned above still leads to the production of a considerable number of non-target progeny. However, an alternative method exists that can substantially decrease the occurrence of these non-target progeny, thus demonstrating a remarkable level of screening efficiency. This method leverages the utilization of genes from the bacterial host that are essential for phage invasion and replication but not crucial for host survival. For example, it has been established that the thioredoxin encoded by trxA and the dCMP kinase encoded by cmk gene are crucial during T7 bacteriophage infection of Escherichia coli (Qimron et al., 2006; Grigonyte et al., 2020). However, these genes are not essential for Escherichia coli’s growth. By inserting such genes into the template DNA and screening with host bacteria lacking these genes, one can select marked mutant bacteriophages (Qimron et al., 2006; Yosef et al., 2017). This method requires gene knockout of the host bacteria, and it can be challenging to identify appropriate screening genes for unknown bacteria and bacteriophages. Nevertheless, high-throughput methods like transposon sequencing and iCRISPR technology can study the genes involved in bacteriophage invasion in the host bacteria, and suitable screening genes can be selected from these research libraries. Both approaches for screening modified progeny demonstrate high efficiency, with the introduction of marker genes being the more convenient method. However, even with this method, a substantial number of unmodified phage progeny is still observed. On the other hand, the introduction of host genes requires modification of the host bacteria but eliminates the production of unmodified progeny. Additionally, an increasing number of studies have reported the utilization of the CRISPR-Cas system for counter-selecting modified progeny bacteriophages.
CRISPR-Cas was initially discovered as a component of the bacterial immune system and plays a crucial role in phage resistance (Barrangou et al., 2014; Lander et al., 2015; Fu et al., 2017). The mechanism has been extensively documented in various reports (Barrangou et al., 2014; Lander et al., 2015). The CRISPR-Cas system, which is based on adaptive immunity, RNA-guided DNA cleavage, and target specificity, has been developed into a powerful gene editing tool (Jinek et al., 2015a). Its gene editing potential has become more apparent over time, driving increased research and development in the field.
The CRISPR-Cas system is derived from the immune response of host bacteria against phages, making it an appropriate tool for engineering phages within bacterial cells. There are two main applications of the CRISPR-Cas system, specifically targeting the DNA strand, in the gene editing of bacteriophages. The first application involves introducing the CRISPR-Cas system into host bacteria, which are then modified to be resistant to wild-type bacteriophages rather than the modified genotype. After the recombination step, counter-selection is performed to identify the modified bacteriophages among the progeny. The second application involves co-transforming the CRISPR-Cas system and template DNA into host bacteria. The CRISPR-Cas system induces double-strand breaks at the target site, thereby enhancing recombination at the desired locus. Consequently, two gene editing strategies have been developed: single-plasmid-mediated asynchronous recombination (SPMAR) and dual-plasmid-mediated synchronous recombination (DPMSR) (Zhang X. et al., 2022). In the SPMAR approach, recombination (Figure 1A) and counter-selection (Figure 1B) are performed sequentially in different bacterial cells, while in DPMSR, the enhanced recombination process and counter-selection occur within a single bacterial cell (Figure 1E). Table 1 provides an overview of the existing literature on phage genome editing using various CRISPR-Cas systems. Among the 14 studies listed in the table, the majority (10 studies) employed the DPMSR strategy, which utilizes the CRISPR system to enhance the recombination process and achieve higher editing efficiency. Notably, the VI-A (Cas13a) CRISPR system, which targets RNA (Gootenberg et al., 2017), was utilized in two related studies employing the SPMAR strategy for counter-selection purposes (Adler et al., 2022; Guan et al., 2022). However, no significant differences in editing efficiency have been reported when using these two different strategies for DNA-targeting CRISPR systems. Zhang et al. successfully used both strategies to delete a 292 bp gene fragment in the bacteriophage genome using an exogenous CRISPR-Cas9 system in V. natriegens TT4. The editing efficiency obtained with both strategies was high and similar (Zhang X. et al., 2022).
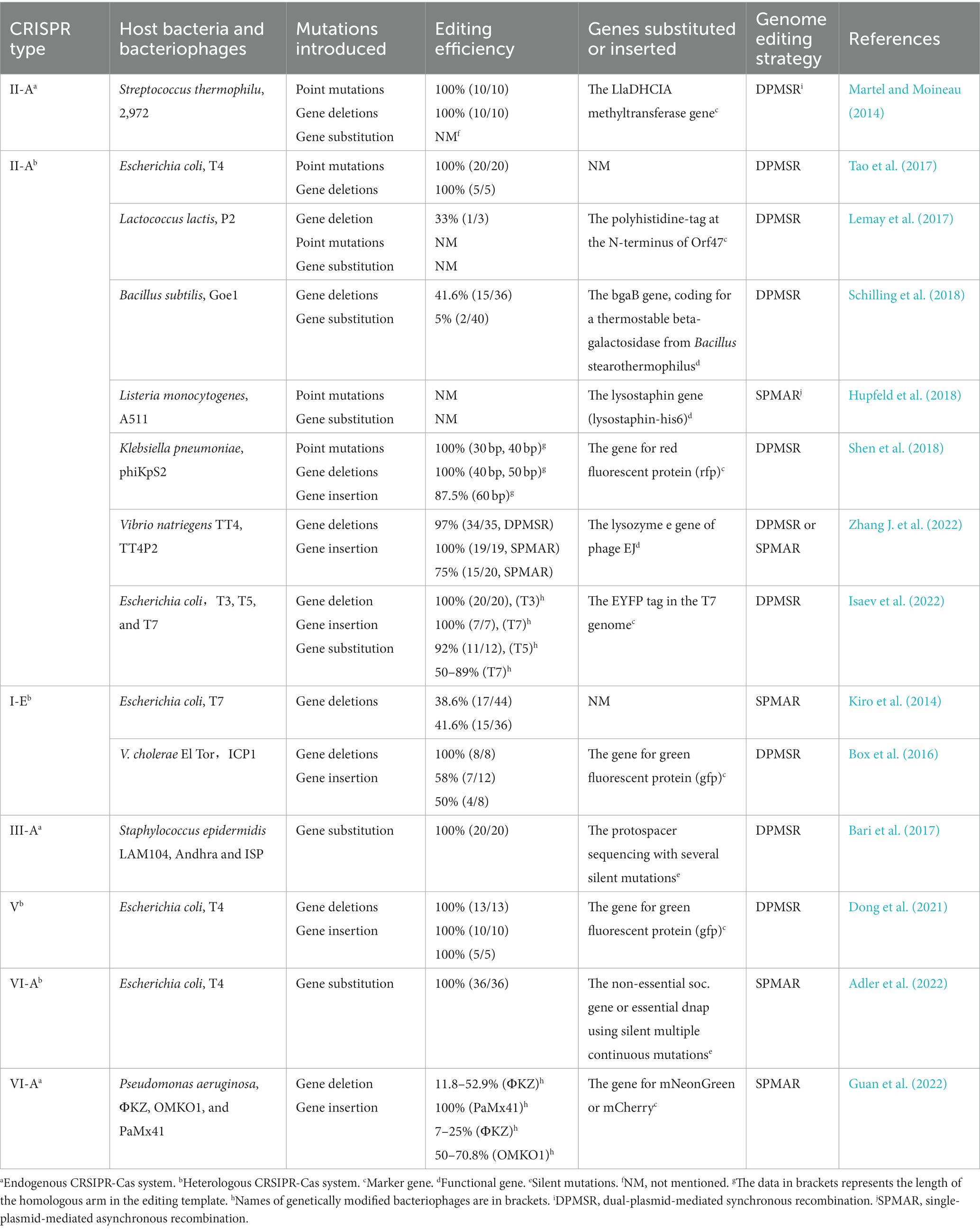
Table 1. Previous studies of phage genome using the CRSIPR-Cas system.
Five types of CRISPR systems, namely II-A (CRISPR-Cas9), I-E (CRISPR-Cas3), III-A (CRISPR-Cas10), V (CRISPR-Cas12a), and VI-A (CRISPR-Cas13a), have been utilized for phage genome modifications (Table 1). The CRISPR systems II-A (CRISPR-Cas9) and I-E (CRISPR-Cas3) represent early-stage CRISPR systems that have undergone extensive research and development. They were among the first CRISPR systems employed for gene editing in phages. The CRISPR-Cas3 system involves multiple genes and proteins that interact with each other (Yosef et al., 2015), and it is characterized by low efficiency and operational challenges. Consequently, research on its application for phage gene editing remains limited. Kiro et al. introduced the Type I-E CRISPR-Cas System into Escherichia coli for engineering bacteriophages using the counter-selection method (SPMAR), resulting in a relatively low editing efficiency of 17 out of 44 plaques exhibiting the desired gene deletion (Kiro et al., 2014). In contrast, Box et al. introduced a combination of the Type I-E CRISPR-Cas System and a homologous recombination template on a single plasmid into V. cholerae. This approach led to successful gene deletion (Editing efficiency: 7/12) and gene replacement (Editing efficiency: 4/8 relevant genes in the ICP1 bacteriophage genome) (Box et al., 2016). The utilization of the Type I-E CRISPR-Cas system in these two cases did not yield high editing efficiency, possibly due to the more complex composition of the CRISPR-Cas system. In comparison, Cas9 demonstrates superior gene-targeting efficiency, cost-effectiveness, and ease of use compared to CRISPR-Cas3 (Lu et al., 2022). As a result, Cas9 is the most commonly employed CRISPR system for editing bacteriophage genomes. Martel and Moineau (2014) first demonstrated that the endogenous CRISPR-Cas II-A system in the thermophilic bacterium Bacillus subtilis could selectively pressure and modify specific point mutations, large deletions, and gene exchange in the genome of virulent bacteriophage 2,972, thereby enhancing recombination efficiency. Following this, researchers introduced heterologous CRISPR-Cas type II-A systems into various host bacteria, including Escherichia coli (Tao et al., 2017), Klebsiella pneumoniae (Shen et al., 2018), Bacillus subtilis (Schilling et al., 2018), Vibrio natriegens (Zhang J. et al., 2022), and Listeria monocytogenes (Hupfeld et al., 2018), to enable specific phage fragment shearing. In summary, the CRISPR-Cas9 system has exhibited broad applicability in bacteriophage gene editing and has consistently demonstrated high editing efficiency across the majority of the cases listed in Table 1.
The primary challenge in editing bacteriophage genes using the commonly used CRISPR-Cas9 system lies in the development of bacteriophage resistance. Bacteriophages can acquire resistance to the CRISPR-Cas9 system through various mechanisms, including the emergence of escape mutants and hindering attachment (van Houte et al., 2016). To overcome this challenge, it is important to explore alternative CRISPR systems that are better suited to tackle the resistance mechanisms exhibited by bacteriophages. The immune response generated by the CRISPR-Cas9 system in bacteria against phages often leads to the generation of escape mutants, which is not desirable in phage genome editing. However, the III-A type system offers a potential solution as it does not require a PAM (Protospacer Adjacent Motif) or a seed sequence, thereby preventing wild-type phages from evading detection through single nucleotide replacements. This feature makes the III-A type system a promising approach to counter the emergence of escape mutants and enhance the effectiveness of phage genome editing. Bari et al. utilized the endogenous CRISPR-Cas10 system of Streptococcus pneumoniae to introduce targeted fragments and editing templates into bacterial cells, conducting bacteriophage gene engineering during the infection of the transformed bacteria. This approach confirmed a relatively high editing efficiency of 10/10 (Bari et al., 2017). The impeded attachment of phage nucleic acid protects it from being targeted by the CRISPR system. Choosing a suitable CRISPR system can counteract the protective effect of impeding attachment in some cases. Recently, Dong et al. introduced exogenous Type V CRISPR and Type II CRISPR-Cas9 into Escherichia coli separately to modify T4 phage. Their findings indicate that the T4 bacteriophage genome is modified by ghmc, conferring resistance to Type II CRISPR-Cas9 systems. The efficiency of the Cas12a editing system depends largely on crRNA, and the Cas12a system can effectively cleave the ghmc-modified genome to generate double-stranded DNA breaks (DSBs), facilitating efficient recombination between the phage DNA and the donor plasmid. The gene editing efficiency achieved in the experiment was 100% with the participation of Cas12a (Dong et al., 2021).
Recently, Type VI (CRISPR-Cas13) system has been employed for genome editing of bacteriophages (Adler et al., 2022; Guan et al., 2022). Type VI system can be utilized for RNA-targeted modification and regulation. During bacteriophage infection, the Type VI CRISPR system can remarkably induce host dormancy, effectively inhibiting the emergence of escape mutants (Meeske et al., 2019). Therefore, the Type VI system represents a highly advantageous tool for counter-selection when employing the SPMAR strategy. In support of this, Adler et al. conducted a study where they introduced Cas13a into E. coli to facilitate the counter-selection of modified bacteriophages in the progeny. Remarkably, all the phages selected through this screening process were subsequently confirmed to be modified as intended. The targeting of Cas13a also lacked the PAM requirement, indicating that virtually any position within or near the phage transcript can be edited and selected by Cas 13a counter-selection (Adler et al., 2022). Therefore, employing Type VI systems for counter-selection of modified bacteriophages is a precise and effective approach.
Among the different systems, CRISPR-Cas9 is the most commonly used due to its high efficiency and versatility. However, in cases where the CRISPR-Cas9 system is ineffective against certain phages, alternative CRISPR systems, such as Cas10 (Bari et al., 2017), Cas12 (Dong et al., 2021) have been explored. Additionally, Type VI system is a favorable tool for counter-selection using the SPMAR strategy. The use of CRISPR-Cas in phage genome editing relies on the innate or exogenous defense system of bacteria to target the wild-type gene fragment. However, bacteriophages possess the remarkable ability to develop resistance to the immune response induced by specific CRISPR systems. This resistance can arise through mechanisms, such as escape mutations or impeding attachment, leading to a decrease in the efficiency of gene editing. Therefore, it is crucial to understand the different characteristics of CRISPR systems to select the appropriate one based on the specific situation to achieve the desired editing effect.
Retrons are a type of bacterial retroelements that generate single-stranded reverse transcribed DNA (RT-DNA) and are composed of three essential components: the reverse transcriptase RT, a non-coding RNA (ncRNA), and one or more accessory proteins (Millman et al., 2020; Bobonis et al., 2022). Despite being discovered more than three decades ago, the function of retrons has remained elusive (Lampson et al., 1989). However, recent studies have shed light on some of the mechanisms and potential applications of retrons, which is an exciting development in this field.
Retrons are often found in the defense islands of bacteria (Hampton et al., 2020) and have been identified as important players in bacterial defense systems (Millman et al., 2020). The auxiliary protein RecB is a critical component of this defense system. The RT-ncRNA complex binds to RecB and acts as a “guard” to monitor its activity (Millman et al., 2020). When a phage protein invades and directly targets RecB, the RT-ncRNA complex detects this change, signaling the onset of a phage infection (Millman et al., 2020). This triggers the activation of the abortive infection mechanism. Some retrons are not found in bacterial defense islands or alongside known defense systems, and they appear to be adapted to perform functions other than phage defense, similar to the toxin-antitoxin system. Some retrons are involved in anti-phage defense, while others play a role in the bacterial stress response (Harms et al., 2018). Recent studies have reported the discovery of tripartite toxin-antitoxin systems composed of retron elements containing RcaT (Bobonis et al., 2022). RcaT-containing retrons are the most prevalent retron family in bacteria and have a high prevalence in proteobacteria (Mestre et al., 2020). RcaT encodes an auxiliary toxin protein that can form tripartite toxin-antitoxin systems (TAs) with the RT-msDNA antitoxin complex (Bobonis et al., 2022). Unlike the retrons systems containing RecB, phage-introduced trigger factors directly modify the msDNA, disrupting the stability of the TAs system and activating the RcaT toxin protein, leading to abortive infection (Millman et al., 2020; Bobonis et al., 2022). Based on these latest findings, it can be deduced that during the retron system’s defense against bacteriophage invasion, certain bacteriophage proteins can interact directly with auxiliary proteins or msDNA in the retron components, resulting in abortive infection and preventing bacteriophage replication and spread. To understand which phage proteins are involved in the defense system of retron, as these proteins impede the production and amplification of engineered phages, high-throughput methods can be utilized to identify these phage proteins.
In the absence of a natural manipulator, the expression of ncRNA and RT from plasmids lacking accessory proteins constitutes the minimal system for RT-DNA production. This system can be utilized for in-cell template gene amplification, enhancing the efficiency of gene editing (Sharon et al., 2018; Simon et al., 2019; Lopez et al., 2022). Furthermore, the RT-ncRNA system can be integrated with CRISPR-Cas for molecular tagging. In this process, the mRNA transcribed from the gene with the tagged promoter is reverse-transcribed into RT-DNA, which is then integrated into the host cell’s CRISPR array in sequence under the action of the CRISPR-Cas integrase, creating a permanent transcription record (Bhattarai-Kline et al., 2022).
Retrons have shown promise as a tool for gene editing in various studies, but the development of scalable retron-based gene editing technologies remains a challenge due to a limited understanding of retrons, particularly their retrotransposition mechanisms. The most commonly used retron in these studies is the engineered Ec86, also known as Eco1, which was the first retron discovered in E. coli (Wang et al., 2022). Retron engineering typically involves modifying non-coding RNAs (ncRNAs). For example, the msd sequence in Ec86’s ncRNA, which is reverse transcribed, can be replaced and used as a template for homology-directed repair (HDR) (Sharon et al., 2018). Additionally, extending the length of the a1/a2 complementary region in the msr sequence, which remains RNA in the final molecule and partially overlaps, can increase reverse transcriptase-mediated DNA production (Lopez et al., 2022). Cryo-electron microscopy (cryo-EM) analysis of Ec86 retron complexes in Escherichia coli has revealed that reverse transcriptase serves as an anchor point for auxin, facilitating direct interaction between msDNA and auxin (Wang et al., 2022). It may be a development trend to optimize the genome editing system based on transcriptase and msDNA structure for Retron-based gene editing.
Retron-based techniques for editing phage genes require careful consideration of various factors, such as the design of the template DNA in the msd sequence and the ability of modified transcripts to still mediate phage defense. To address these issues, Rossier et al. introduced an engineered Ec86 retron carrying a DNA template fragment into wild-type Escherichia coli, taking advantage of the bacterium’s reverse transcription capabilities to attain high concentrations of the editing template and facilitate homologous recombination (Figure 1D), resulting in the generation of T5 mutant phages. However, the editing efficiency of the phage genome editing using retron was suboptimal at 2% (2/100) (Ramirez-Chamorro et al., 2021), indicating a need for further optimization to improve the technique. Despite this, the editing template provided by the retron element offers several advantages over other methods of homologous editing templates. First, there is no need for targeted mutation of the cloned fragment, as the small size of the retron enables the cloning of annealed oligonucleotides carrying the desired mutation. Second, unlike classical HR templates, the retron-based approach does not require long sequences on either side of the mutation, which may encode toxic products for the host cell (Ramirez-Chamorro et al., 2021).
The process of synthesizing bacteriophages outside of their host bacteria involves the synthesis of the bacteriophage genome in alternative environments, including yeast cells or in vitro. Subsequently, the synthesized genome is transferred into an appropriate environment, such as a modified E. coli, L-form bacterium, or a cell-free TXTL platform, to initiate the engineered bacteriophage production (Figure 2). This approach offers several advantages for genetic modifications as the process occurs independently of the host bacteria. Consequently, it allows for a high degree of customization and facilitates more efficient genetic modifications without the need for extensive screening of engineered bacteriophages. Additionally, synthesizing bacteriophages outside of their host bacteria overcomes the challenge posed by the poor transformation capabilities of the host bacteria, which often hampers the editing process within the host bacteria. By utilizing alternative environments, scientists can optimize the genetic modification process, thereby opening up new possibilities for enhancing the capabilities and applications of bacteriophage engineering.
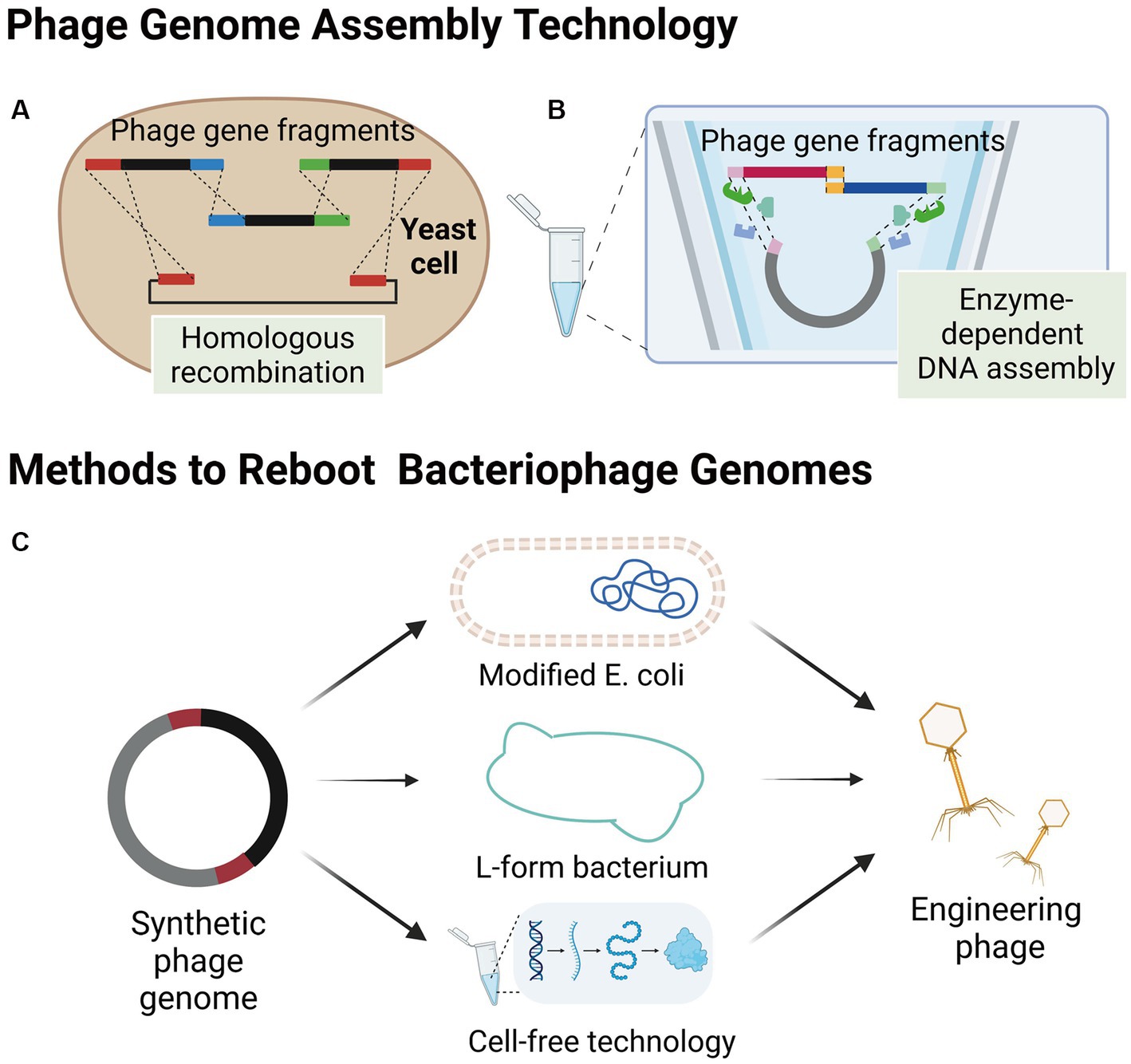
Figure 2. Assembly of bacteriophages outside host bacteria. (A) Smaller phage gene fragments are assembled in yeast cells by homologous recombination. (B) Enzyme-dependent DNA assembly, a cell-free genome assembly method for bacteriophage that utilizes exonucleases, polymerases, and ligases. (C) Several methods of engineering bacteriophage reboot (images are created with Biorender.com).
The yeast artificial chromosome (YAC) is a versatile tool for manipulating the design and construction of targeting vectors (Murray, 2006), as well as for assembling phage genomes. Small phage gene fragments can be assembled in yeast cells using transformation-associated recombination (TAR cloning) of complete DNA molecules (Figure 2A; Noskov et al., 2001; Gibson et al., 2008). Additionally, Enzyme-dependent DNA assemblies, such as the Gibson assembly and Golden gate assembly have been extensively used for synthesizing phage genomes in vitro (Figure 2B; Gibson et al., 2008, 2009; Kilcher et al., 2018). The in vitro synthesis approach provides a highly efficient and precise method for modifying specific gene sequences within bacteriophage genomes. Notable examples of this approach include the modular assembly and replacement of RBPs in bacteriophage genomes using yeast cell systems (Ando et al., 2015), as well as the construction of mutated bacteriophage gene libraries through golden gate assembly (Liang et al., 2022). One of the major advantages of in vitro synthesis is its superior efficiency compared to intracellular recombination within host cells. Furthermore, the rebooting of bacteriophages using synthesized genomes eliminates the need for phage screening and subsequent cloning, thereby significantly increasing the overall efficiency of engineering bacteriophages (Ando et al., 2015; Liang et al., 2022). By harnessing the power of in vitro synthesis, researchers can achieve precise and targeted modifications in bacteriophage genomes, paving the way for advancements in the field of bacteriophage engineering.
Electroporation has demonstrated a high transformation efficiency of E. coli (Marinelli et al., 2008, 2012). Thus, using electroporation for the transformation of synthetic genes of phages hosted by E. coli and other bacteria into E. coli for rebooting is a feasible approach. The success of this approach relies on whether the transcription and translation systems in E. coli meet the requirements for phage gene rebooting. To date, this method has only been reported to work with gram-negative phages, including E. coli phage phiX174 (Jaschke et al., 2012), T7-like (Ando et al., 2015), S13-like (Liu et al., 2012), and the large Salmonella Myovirus FelixO1 (Kilcher and Loessner, 2019). Recently, Cheng et al. developed the E. coli strain DH10B as a “stepping stone host” for phage rebooting and demonstrated its capability of reactivating 126 phages from T7 and non-T7 families that infected clinical multi-drug resistant Klebsiella pneumoniae, Streptococcus enterica, Pseudomonas aeruginosa, and Pseudomonas baumannii, indicating its potential application for the production of phages targeting drug-resistant bacteria (Cheng et al., 2022). Additionally, DH10B strains can uptake large amounts of DNA and protect exogenous DNA from restriction systems (Durfee et al., 2008), which enables Cheng et al. to transform bacteriophage DNA fragments into DH10B for bacteriophage assembly and reboot.
Gram-positive bacteria are challenging to transform due to their thick peptidoglycan layer, making it difficult to transform the phage genome. However, researchers have overcome this issue by generating L-form bacteria through long-term cultivation of gram-positive bacteria in the presence of cell wall-activating compounds, such as penicillin, which destroys the cell wall (White et al., 1981). This type of cell wall-deficient bacteria is more conducive to the transformation of large DNA molecules and is suitable for rebooting the corresponding bacteriophages. Furthermore, L-form bacteria with cell wall defects are susceptible to being permeabilized, releasing the engineered bacteriophages when subjected to dilution in an unstabilized medium (Kilcher et al., 2018).
L-form bacteria, which are characterized by their lack of a cell wall, present a unique advantage in the field of phage genome engineering. Their cell wall-deficient nature renders them highly susceptible to the uptake of foreign DNA and phage genomes, facilitating the genetic manipulation of these bacteria (Kilcher et al., 2018). Furthermore, the ability to cultivate L-form bacteria in liquid cultures offers a significant improvement in efficiency and speed compared to the conventional method of growth on solid media. This feature enhances the feasibility and effectiveness of rebooting phage genomes, making L-form bacteria a powerful tool in phage engineering research. Kilcher et al. (2018) also developed an L-type Listeria bacterium called Rev2L, which has been shown to reactivate eight Listeria monocytogenes phages, two Bacillus phages, and two Staphylococcus aureus phages, including mild (B025, B035, B056, PSA) and virulent phages (P35, P70, P100, A511), some of which have large genomes (>130 kb; P100, A511). These results demonstrate the versatility of the L-type bacterium as a reboot host.
To successfully reboot bacteriophages in a cell-free environment, it is essential to understand the mechanism of bacteriophage DNA expression within bacterial cells. When phage DNA enters the host cell, it hijacks the bacterium’s transcriptional system to express the associated enzymes in the phage gene, resulting in the generation of progeny phages and lysis of the host bacterium (Yang et al., 2014; Gogarten, 2022; Liao et al., 2022). The transcription of phage DNA in the host cell is divided into three stages: early, middle, and late (Melo et al., 2022). In the early stage, phage DNA expresses a set of genes responsible for hijacking the host transcription machinery, including the RNA polymerase, which is used by the original RNA polymerases in the host cell (Yang et al., 2014). After gaining control over the host, phage DNA expresses DNA enzymes that break down host cell DNA, DNA polymerase for replicating viral DNA, HMC synthetase, and mRNA polymerase for subsequent steps. During the final stage, a significant number of “components” are generated through mRNA translation, including head protein, tail protein, various assembly proteins, and lysozyme, which facilitate the assembly of progeny viruses (Roucourt and Lavigne, 2009).
The transcription mechanism of bacteriophages varies depending on the presence or absence of endogenous RNAP. For bacteriophages that lack endogenous RNAP, such as bacteriophage T4 and λ, gene expression is regulated through the encoding of specific protein factors, such as bacteriophage P23–45 gp39 protein (Berdygulova et al., 2011) and bacteriophage λ N protein (Archer et al., 2014), that modulate host RNAP specificity. In contrast, bacteriophages with endogenous RNAP, such as T7 and Xp10, utilize the host E. coli RNAP to transcribe early genes, but then switch to their own RNAP to activate middle and late promoters. Additionally, some small proteins of the bacteriophage inhibit the function of the host RNAP, such as T7 gp2 and Xp10 P7. Thus, protein factors encoded by bacteriophages play a critical role in bacteriophage expression. They alter RNAP specificity or function by interacting with RNAP, thereby transferring cellular resources to bacteriophage gene expression.
Our investigation focuses on the gene expression process of bacteriophages within bacteria and the involvement of host proteins, which are fundamental to our approach in restarting bacteriophages ex vivo. To achieve this, we rely on the cell-free gene expression (CFE) system, an essential tool for investigating these matters. The CFE system utilizes cell extracts to activate transcription and translation, preserving the native transcription and translation machinery of cells (Silverman et al., 2020). We can initiate protein synthesis in vitro using the CFE system by supplementing the extracts with exogenous resources, such as amino acids, nucleotides, and secondary energy substrates (Silverman et al., 2020). With the combined use of CFE and proteomics mass spectrometry, we can investigate the contents of various bacteria, particularly the gene regulatory elements, such as promoters and transcription factors. The transcriptional mechanisms of several bacteria, including Bacillus megaterium (Moore et al., 2018), Pseudomonas putida (Wang et al., 2018), Clostridium autoethanogenum (Kanehisa et al., 2020), and Vibrio natriegens (Bjorling et al., 2018; Djokic et al., 2018; Shen et al., 2018), have been investigated using this approach, leading to the development of corresponding CFE platforms. Moreover, Yim et al. (2019) used the Cell-Free Transcription and Sequencing (DRAFTS) method to investigate gene transcription activity in the cell-free lysates of 10 different bacteria from three phyla (Proteobacteria, Firmicutes, and Actinobacteria). They found both commonalities and differences in the transcriptional mechanisms of different bacteria, prompting the use of non-host-derived CFE systems or mixed CFE systems from multiple host sources to produce corresponding bacteriophages.
CFE can be utilized to directly examine bacteriophage transcription and translation. Emslander et al. employed a combination of CFE technology, time-resolved mass spectrometry, and plaque assay with T7 bacteriophage as verification, resulting in consistent expression of early, middle, and late-stage genes as previously proposed (Emslander et al., 2022). This approach is valuable for investigating the process, which can help improve the production of CFE.
The utilization of CFE technology in bacteriophage genome editing offers several advantages. Firstly, it eliminates the necessity of screening for engineered bacteriophages and assessing transformation efficiency. Instead, the emphasis shifts toward bacteriophage reboot, streamlining the editing process. This has led to increased attention toward cell-free bacteriophage synthesis, especially with the rapid advancements in CFE technology. Simple protein-based viral structures, such as phages, have been employed to validate cell-free biomanufacturing through in vitro transcription and translation of the entire viral genome. These efforts resulted in self-replicating phages, including E. coli phage T4 (Rustad et al., 2018), T7 (Shin et al., 2012), ФX174 (Rustad et al., 2017), phiX174 (Garamella et al., 2016), MS2 (Rustad et al., 2017), and K1F (Liyanagedera et al., 2022). Emslander et al. (2022) extended cell-free production to phages targeting the Gram-positive bacterium Bacillus subtilis by co-expressing appropriate host factors. In this example, highly similar RNA polymerases were utilized in both Escherichia coli and Bacillus subtilis.
In summary, the successful synthesis of bacteriophages through cell-free methods hinges on the proper assembly and reboot of the specific bacteriophage being studied. This entails investigating the transcription and translation systems of the bacteriophage, as well as the influence of host factors on the reboot process. The use of cell-free extracts (CFE) offers a means to acquire this knowledge and enhance CFE production efficiency. The development of CFE technology has made significant strides, with standardized methods for preparing extracts capable of transcription and translation from various bacteria having been established (Silverman et al., 2020). Furthermore, alternative CFE systems sourced from organisms other than Escherichia coli have been developed, indicating the feasibility of using different platforms for bacteriophage production with non-model hosts (Bjorling et al., 2018; Moore et al., 2018; Shen et al., 2018; Wang et al., 2018; Kanehisa et al., 2020; Xu et al., 2020).
The majority of phages that have been described so far have a tail morphology and contain a double-stranded DNA genome ranging from 15 to 500 Kbp, belonging to the order Caudovirales (Ackermann, 2007; Malik, 2019; Dion et al., 2020). These tail phages consist of a head that is made up of an icosahedral capsid and a tail that interacts with the surface receptors of host bacteria through surface receptor proteins (RBPs) (Malik, 2019). The initial step in bacteriophage infection of host bacteria is the binding of the RBPs to specific receptors present on the surface of bacteria. This interaction plays a vital role in guiding subsequent steps of the infection process and determining the range of host bacteria that the bacteriophage can infect (Chen et al., 2020; Wei et al., 2022).
Phage RBPs can be classified as tail fibers (TF) or tail spikes (TSP) based on their structural features. Morphologically, both TFs and TSPs consist of homotrimeric complexes, with TFs forming long, thin filaments that interact with protein and/or sugar receptors, whereas TSPs form more globular complexes (Klumpp et al., 2023). TFs are elongated fibrillar proteins lacking enzymatic activity and contain specific receptor binding sites at their distal ends that are involved in host binding. Besides, TSPs are shorter and more rigid with enzymatically active globular structures at their terminal ends, which are usually active against specific surface structures (such as the sugar fraction, phosphopeptides in Gram-positive hosts or lipopolysaccharides, and capsule polysaccharides in Gram-negative hosts) (Dunne et al., 2021; Klumpp et al., 2023). The presence of RBPs varies depending on the morphological characteristics of bacteriophages. For instance, Myoviridae phages (with icosahedral heads, retractable tails, and six short spines/long flagella that infect Gram-negative bacteria) and Siphoviruses (polyhedral heads with elongate, non-retractable tails, and short fibers, McCorquodale, 1999) typically have TFs as RBPs (Letarov and Kulikov, 2017). In contrast, TSPs are the more common RBPs in Podoviridae phages (with diverse polyhedra and short, non-contractile tails, Molineux, 1999; Letarov and Kulikov, 2017; Gonzalez and Scharf, 2021).
Phage receptor binding proteins (RBPs) play a critical role in initiating the infection process by binding to specific receptors on the surface of host bacteria. The receptors in Gram-negative bacteria are primarily lipopolysaccharides (LPS), outer membrane proteins (OMPs), and fimbria or capsule components, whereas Gram-positive bacteria have cell wall teichoic acid (WTA), lipoteichoic acid (LTA), and cell wall-associated polysaccharides as their primary receptors for phage RBPs (Ha et al., 2019). Recent studies on the receptors of bacteriophages for Escherichia coli or related gut bacteria have shown that Siphoviruses target porin proteins on the cell surface, Podoviridae phages target polysaccharides, and Myoviridae phages can target either of these two (Letarov and Kulikov, 2017). These differences in cell wall composition, thickness, lipid and lipoprotein content, and receptor specificity suggest that phages have evolved to infect specific bacterial hosts, highlighting the importance of understanding the molecular mechanisms underlying phage-host interactions.
The introductory information provides a general understanding of the RBPs in phages and bacterial receptors, which aids in the screening of phage RBPs for subsequent research. However, for unknown novel phages, bioinformatics methods are required to explore the RBPs of phages. The continuous mutation of phage RBPs genes resulting from the arms race between phage tails and host surface receptors has been extensively researched (Lee et al., 2016; Yuan et al., 2019). Moreover, in certain natural environments, the exchange of RBPs genes between different types of phages occurs, causing changes in host ranges to adapt to changing environments (Xiao et al., 2018; Górniak et al., 2022). These two naturally occurring processes result in some degree of homology between the RBPs of phages corresponding to the same or different host bacteria. Bioinformatics methods use this homology to help predict the RBPs of phages. In our study of the phage genome, we can select genomic annotation of TFs, TSPs or related genes that are vaguely annotated as “tail proteins” or “substrate proteins” for subsequent research (Boeckaerts et al., 2021; Phetruen et al., 2022). Furthermore, homology modeling of genes with high homology can be performed, selecting models with high coverage and confidence in the target sequence to analyze possible docking structural domains and associated sites (Yehl et al., 2019). The RBPs predicted through bioinformatics analysis can then be validated using various in vivo and ex vivo methods, such as affinity chromatography, knockout mutagenesis, phage plaque assays, and phage adsorption kinetic analysis (Chen et al., 2020).
Once bacteriophage RBPs are identified, we can engineer modifications to meet host range requirements, which mimic interactions between bacteriophages and bacteria in nature. Table 2 presents a synopsis of cases over the past 8 years in which bacteriophages were modified using evolutionary methods, phage chimeric methods, and RBPs gene swapping. The methods and outcomes of each study are briefly outlined in Table 2, and the following sections will elaborate on the significance of each approach.
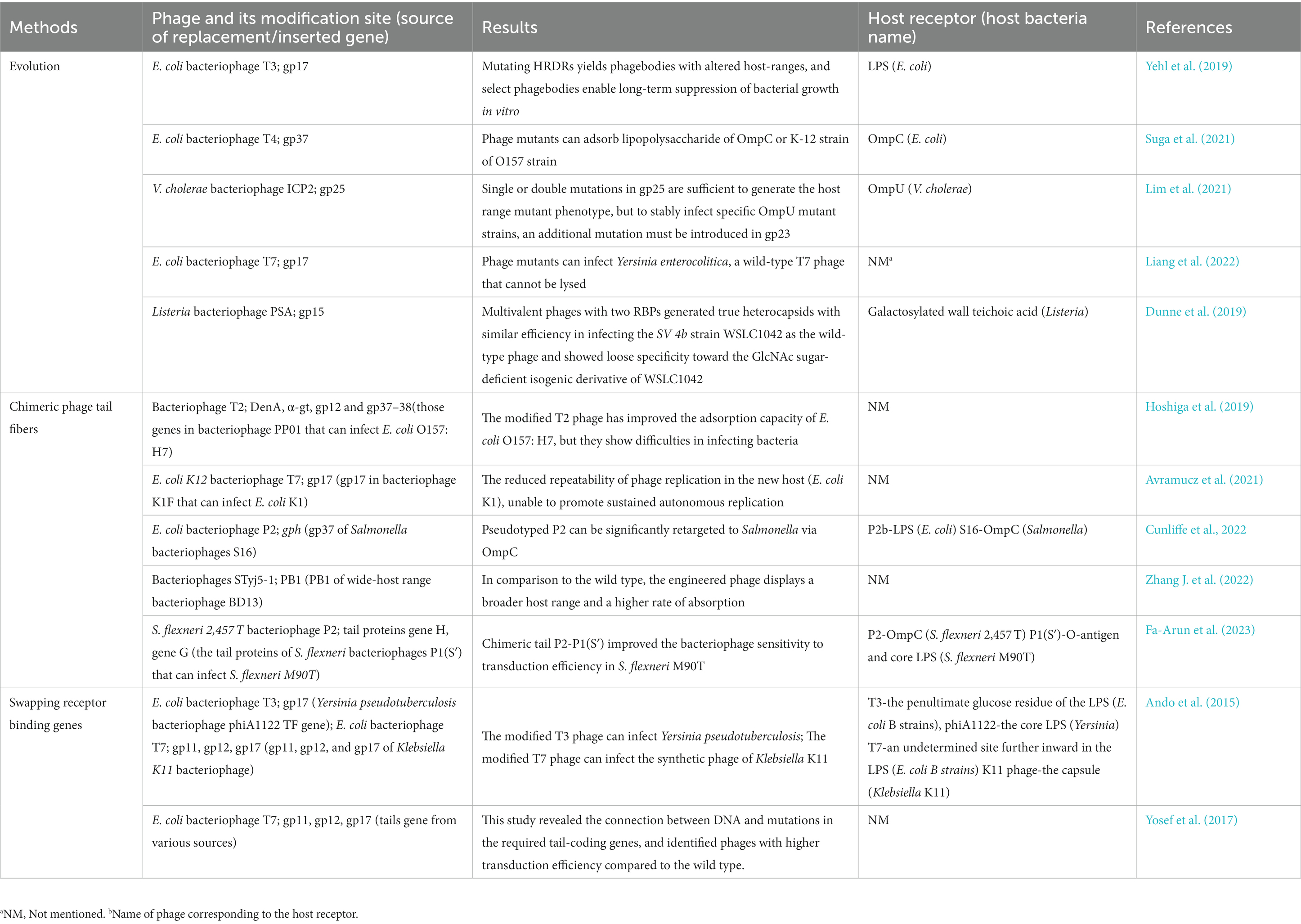
Table 2. Engineered modification of bacteriophage RBPs.
By utilizing evolutionary methods to modify bacteriophage RBPs, particularly the host range determining regions (HRDRs), it is possible to alter the host range and infectivity of bacteriophages. The method of evolution involves naturally evolved methods and artificially introducing mutations in the RBPs region of bacteriophages (Yehl et al., 2019; Lim et al., 2021). Naturally evolved methods refer to the repeated cultivation of wild-type phages with mutant strains, which promotes phage evolution. Artificially induced mutations are generated in vitro through the use of degenerate or random primers (Yehl et al., 2019), error-prone PCR (Dunne et al., 2019; Suga et al., 2021), and golden gate assembly (Liang et al., 2022) to create a random mutation library. The formation of mutant bacteriophages in RBPs is achieved through homologous recombination (Yehl et al., 2019; Lim et al., 2021; Suga et al., 2021), which introduces genes from the mutant library into RBPs, or by rebooting the synthesis of mutant bacteriophages outside the host, e.g., rebooting in L-form bacteria (Dunne et al., 2019) or cell-free platforms (Liang et al., 2022). The mutated bacteriophage is then screened using different host libraries to obtain the engineering bacteriophage. This method has been used to modify the host range of phages. For example, the T4 bacteriophage with gp37 mutation can attach to hosts, such as the K-12 strain of E. coli O157 strain, which has OmpC on its surface (Suga et al., 2021), and the Listeria phage PSA with gp15 mutation can infect SV 4d strain WSLC1033, which lacks WTA galactosidase function (Dunne et al., 2019). These are characteristics that are lacking in wild-type phages but can be acquired through evolutionary means. The research by Yehl et al. (2019) is significant because they investigate the phage-host attachment process from a more nuanced perspective. They generated substantial diversity through the substitution of every codon in the target RBPs’ ring with a random codon, resulting in the completely randomized DNA-level sequence of the RBPs gene. This was then screened through corresponding bacterial libraries to obtain phages with desired host ranges. This approach allows for the exploration of the relationship between the physical and chemical properties of each loop region in the receptor recognition area and the host range by using host libraries composed of different phenotypic bacteria to identify the infectivity of mutated phages. Yehl’s method is suitable for engineering modification of minor host range changes, especially in cases where wild-type phages have been rendered resistant by the bacterium, as it can quickly produce phages that are resistant to host bacterium resistance (Yehl et al., 2019).
Through multiple-site mutations and proper experimental design, evolutionary methods can also help us investigate the molecular mechanism by which phage RBPs determine host range. Lim et al. (2021) introduced mutations in gp23 and gp25 in the V. cholerae bacteriophage ICP2, either separately or simultaneously. They found that single or double mutations in gp25 were sufficient to produce host-range mutant phenotypes, but to stably infect specific OmpU mutant strains, an additional mutation must be introduced in gp23. Mutation in gp23 alone was insufficient to produce host-range mutation phenotypes.
Chimeric methods in bacteriophage (BRP) engineering can be utilized to enhance the host range of modified bacteriophages. This involves combining the tail proteins of different types of bacteriophages that share high homology. This approach can lead to a more significant expansion of the host range than evolutionary methods alone. For instance, one study integrated the E. coli bacteriophage ϕV10 into the tail of the S. flexneri bacteriophage T2, allowing T2 to transduce the non-host bacterium E. coli O157:H7 (Hoshiga et al., 2019). Another study indicated that the insertion of Salmonella bacteriophage S16 gene gp37 into the TF gene (gph) of E. coli bacteriophage P2 resulted in the redirection of host bacteria from E. coli to Salmonella (Cunliffe et al., 2022). However, chimeric receptor binding proteins (RBPs) may not guarantee stable infection of target bacteria. In a study conducted by Avramucz et al., the gp17 gene (TF gene) from bacteriophage K1F, which targets E. coli K1, was inserted into the gp17 gene of bacteriophage T7, which targets E. coli K12. The resulting engineered phage exhibited unstable infectivity toward E. coli K1, leading to insufficient replication capacity (Avramucz et al., 2021). Similarly, Hoshiga et al. (2019) integrated several RBPs genes from E. coli bacteriophage PP01 into the corresponding genes of bacteriophage T2 of E. coli. Although the engineered phage T2 demonstrated effective adsorption to E. coli, successful infection proved challenging (Hoshiga et al., 2019).
The host range of phages is directly influenced by the receptor recognition genes, making it possible to alter the host range by swapping the entire receptor gene of a phage. In vitro synthesis of phage genomes offers a practical approach for such gene swapping. Ando et al. (2015) developed a yeast-based platform for the production of synthetic phage genomes, assembled from fragments (Ando et al., 2015). This innovative approach enables the exchange of receptor-binding proteins (RBPs) and tail components among different phage scaffolds. To test the modification capabilities of the platform, Ando et al. (2015) utilized two E. coli phages, T3 and T7. By replacing the receptor recognition genes (gp17) of T3 bacteriophage with those from Yersinia phage TFs, they successfully engineered a T3 bacteriophage capable of infecting Yersinia pseudotuberculosis. Furthermore, they replaced the tail proteins (gp11 and gp12) and the receptor recognition protein (gp17) of T7 bacteriophage with their counterparts from Klebsiella phage K11, resulting in a synthetic bacteriophage capable of infecting Klebsiella. This study highlights the potential of in vitro phage genome synthesis methods to modify the host range of phages by replacing the entire receptor recognition genes (Ando et al., 2015).
Yosef et al. (2017) developed a platform called GOTraP for bacteriophage engineering. In their approach, a T7 phage lacking tail genes (gp11, gp12, gp17) was utilized to infect E. coli hosts containing a plasmid carrying tails sourced from various origins, along with an antibiotic resistance marker. Subsequently, the progeny phages were screened with the desired host, allowing only those phages with functional tails, resulting from homologous recombination, to transduce into the target host cells and confer antibiotic resistance. By sequencing the screened bacteria, Yosef et al. (2017) also established a connection between the observed phenotype, which was DNA transduction, and the required genotype, involving mutations in the tail-coding genes that facilitated this transduction process. This method enabled the selection of phages with enhanced transduction efficiency compared to the wild type, as demonstrated by Yosef et al. (2017). However, it should be noted that increased transduction efficiency does not necessarily imply higher infectivity. It is important to acknowledge that both methods, including the one discussed previously, have a common limitation, which is the requirement for homology at the sequence edges of the receptor recognition genes being replaced to meet the criteria for in vitro synthesis or homologous recombination (Yosef et al., 2017).
The interactions between bacteriophages and their host bacteria are complex and not yet fully understood. However, the development of high-throughput research methods has greatly improved our understanding of these interactions at the molecular level. In this review, we will provide an overview of several approaches, encompassing high-throughput methods for analyzing host bacterial genes (Figure 3) as well as high-throughput methods for analyzing bacteriophage genes (Figure 4).
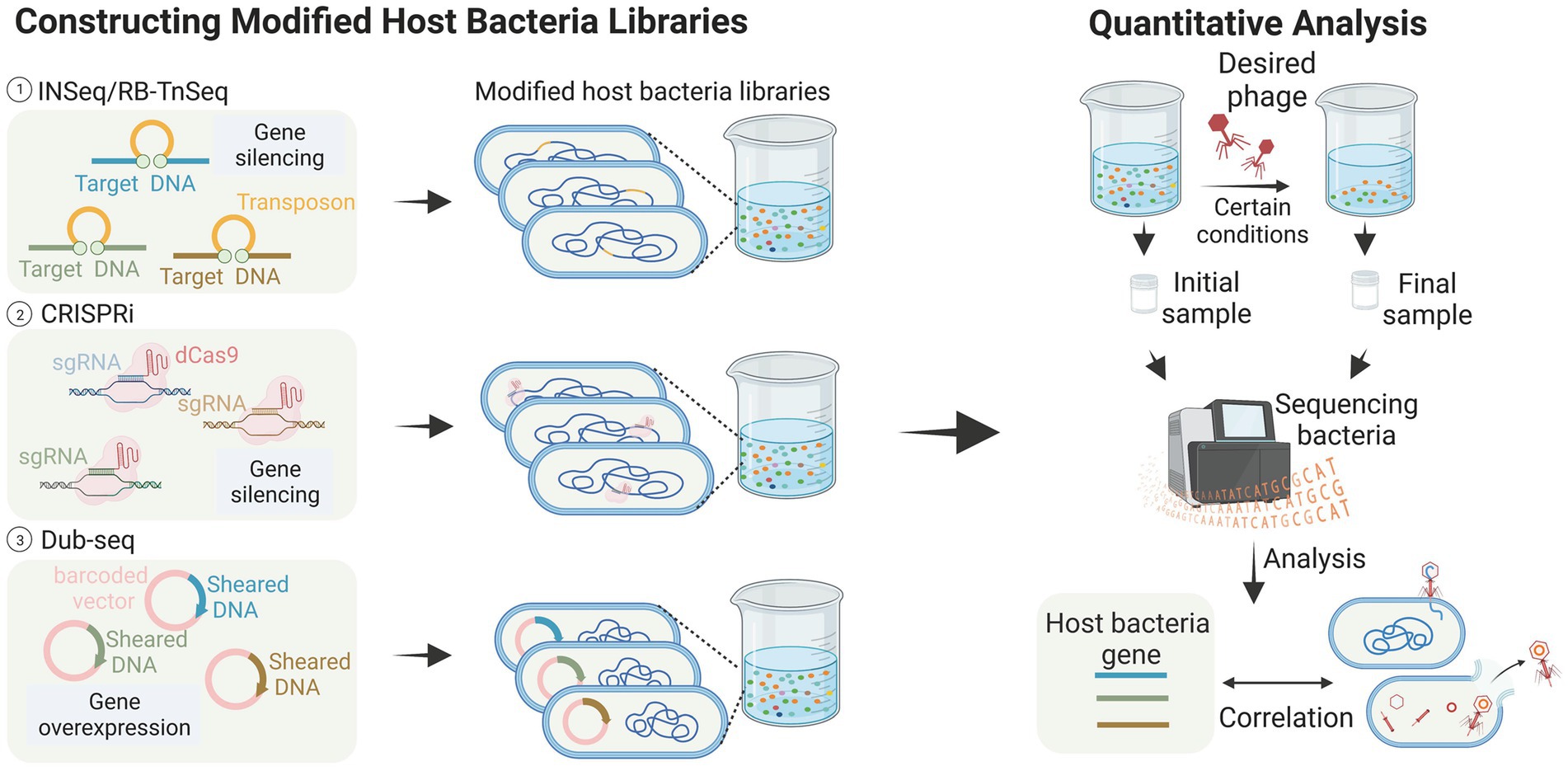
Figure 3. High-throughput analysis of host bacteria genes (images are created with Biorender.com).
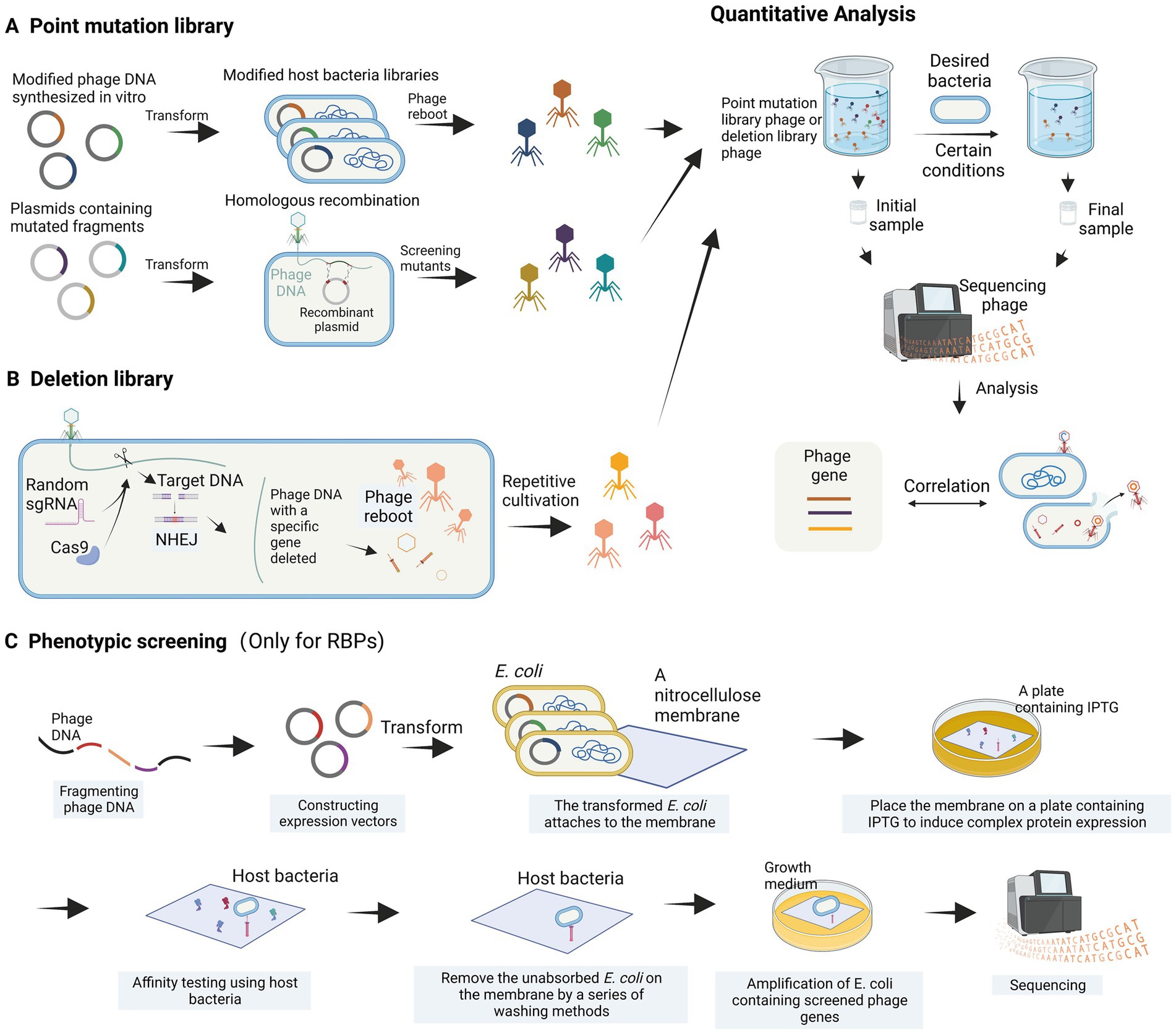
Figure 4. High-throughput analysis of phage genes. (A) Construction of a point mutation library. (B) Construction of a deletion library. (C) Phenotypic screening is employed to identify bacteriophage receptor recognition proteins (images are created with Biorender.com).
Transposon insertion sequencing (TIS) has been successfully used to detect genes essential for bacterial growth (Chao et al., 2016; Peters et al., 2016; Liu et al., 2017). This class of genome-wide loss-of-function (LOF) methods has also been applied to identify genes associated with phage infestation in the host bacteriophage genome. Kortright et al. (2020) explored the utility of Insertion Sequencing (INSeq) for identifying bacterial genes involved in bacteriophage binding. They created a transposon mutant library using a modified transposon, and enriched and differentiated the libraries. The method employed in this study successfully identified bacterial genes associated with receptor binding in T2, T4, T6, and T7 bacteriophages. These genes include outer membrane proteins, such as OmpA, OmpC, OmpX, LamB, and tsx, LPS-related genes like waaF, waaG, gmhB, and yaeI, as well as oxidoreductase proteins like trxA. These findings are consistent with previous studies (Yosef et al., 2017; Chen et al., 2020), and additionally demonstrated the successful identification of binding targets in five new bacteriophages, thereby proving the generalizability of the method (Kortright et al., 2020). The RB-TnSeq method, which is similar to INSeq, is also reported (Mutalik et al., 2020). These transposon insertion techniques greatly aid in identifying the receptors contained within bacteria, especially in the studies of new bacteriophages and new bacteria. For example, Mutalik et al. (2020) screened a transposon mutant library of the marine pathogenic bacterium Vibrio parahaemolyticus and discovered that the vp0980 mutant (encoding a predicted transmembrane protein) was not infected by bacteriophage OWB. Subsequent experiments confirmed that the binding of bacteriophage tail tube proteins A and B with Vp0980 mediated bacteriophage attachment and subsequent bacterial lysis.
Compared to transposon insertion methods, CRISPR-based methods do not directly modify DNA, making them more flexible in terms of operation. CRISPRi allows partial silencing of gene function through transcriptional repression to achieve LOF. Rousset et al. (2018) reported a CRISPR-dCas9 screening that utilized a library of approximately 17,220 sgRNAs targeting randomly assigned positions in the E. coli chromosome. In this experiment, bacteriophages dissolve bacteria unless they carry sgRNAs that allow them to resist infection. The results showed that phages λ, T4, and 186 employ different host genes when killing E. coli, and that three distinct host pathways may be involved in these bacteriophage infections. The advantages of CRISPRi technology over INSeq include the ability to examine the function of host-essential genes during phage infection. Unlike randomly inserted transposons, CRISPRi libraries can be designed in a targeted manner to focus on specific gene sites or subsets. Furthermore, CRISPRi allows for the attainment of moderate inhibition levels by targeting gene template strands or utilizing sgRNAs with varying levels of mismatches (Rousset et al., 2018).
Gain-of-function (GOF) genetic techniques have been utilized in phage-host interaction studies, where gene overexpression can effectively reveal dominant negative mutations, antisense RNAs, or other genes involved in phage resistance. Mutalik et al. (2020) employed dual-barcoded shotgun expression library sequencing (Dub-seq) to facilitate the overexpression of randomly selected genes in a model E. coli system and used this approach to screen and sequence 14 phages. The results showed a degree of similarity in the host genes identified by the two loss-of-function methods (Mutalik et al., 2020). However, Dub-seq diverged from these methods by identifying numerous multicopy repressor genes that encoded diverse functions, shedding light on how changes in host gene expression impact phage resistance differently. This study thus demonstrates the potential of Dub-seq as a useful tool in identifying genes involved in phage resistance.
The genomic modification of phages is often more complex than that of host bacteria, and it is challenging to use high-throughput methods to study the function of phage genes when they infect bacteria. Recently, a systematic review article introduced various high-throughput methods for investigating the relationship between bacteriophage sequences and their functions (Huss et al., 2023). The main concept of these methods involves constructing bacteriophage libraries, such as point mutation libraries and deletion libraries, through different bacteriophage engineering techniques, including gene editing using recombination and in vitro phage DNA synthesis. These libraries are then screened under different environmental and host conditions (Figure 4). Variants with essential genes for bacteriophage proliferation altered and resulting in functional deficiencies are depleted, while those with beneficial mutations are enriched. The depleted or enriched variants are identified using high-throughput sequencing methods (Figure 4). While this method is theoretically feasible, there have been limited reports on its application thus far. A partially analogous case is the study of T3 bacteriophage receptor-binding protein (RBP) gene recombination and screening (Yehl et al., 2019). In this study, recombination plasmids were constructed to contain the T3 tail fiber (TF) gene, where codons in the loops were substituted with NNK codons through site-directed mutagenesis. By homologous recombination with the bacteriophage genome within the host bacterium, a bacteriophage library with completely random sequences of the TF (loops) was generated (Figure 4A). Yehl et al. (2019) successfully screened and enriched mutant phages capable of infecting T3-resistant E. coli by repeatedly co-culturing the library with the host bacterium. Through high-throughput methods, this study provided evidence of the correlation between loop motifs in the T3 phage TF and its infectivity. For the construction of deletion libraries, the CRISPR-Cas system can be utilized. Yuan et al. developed a genome-scale top-down strategy involving the use of the CRISPR-Cas9 system in the host bacterium to randomly target cleavage and subsequently perform non-homologous end joining (NHEJ) of phage DNA, resulting in the random deletion of sequence fragments (Yuan et al., 2022). The progeny phage DNA obtained by repeating these steps several times showed significant simplification compared to the wild-type. This genome-scale top-down strategy, developed by Yuan et al., enables the deletion of non-essential genes in the phage genome. Moreover, by utilizing a random sgRNA library under appropriate conditions, this method can also be employed for the construction of bacteriophage deletion libraries (Figure 4B).
Additionally, Simpson et al. (2016) proposed a high-throughput phenotypic screening approach for RBP genes that entails fragmenting phage DNA, ligating it to an expression vector, transforming it into E. coli, and overexpressing the recombinant protein on a nitrocellulose membrane. The binding capability of the nitrocellulose membrane to the phage host bacterium is then assessed, allowing for the identification of phage proteins with high affinity for the host bacterium (Simpson et al., 2016). The in vitro phenotypic screening approach (Figure 4C) can help us find the genes of RBPs that interact with the host bacterium, but cannot detect the relevant genes involved in physiological processes.
In this review, we summarized the methods and applications of phage engineering that are based on the interaction between phages and their hosts. To further elucidate the connections between these mechanisms and methods, we have presented a conceptual diagram in Figure 5. The figure illustrates various mechanisms of interaction between phages and hosts, along with high-throughput methods for studying phage-host interactions. Additionally, it highlights the applications of engineered phages related to these interactions in light pink background boxes, and identifies relevant proteins that may be identified by high-throughput methods in light purple background boxes.
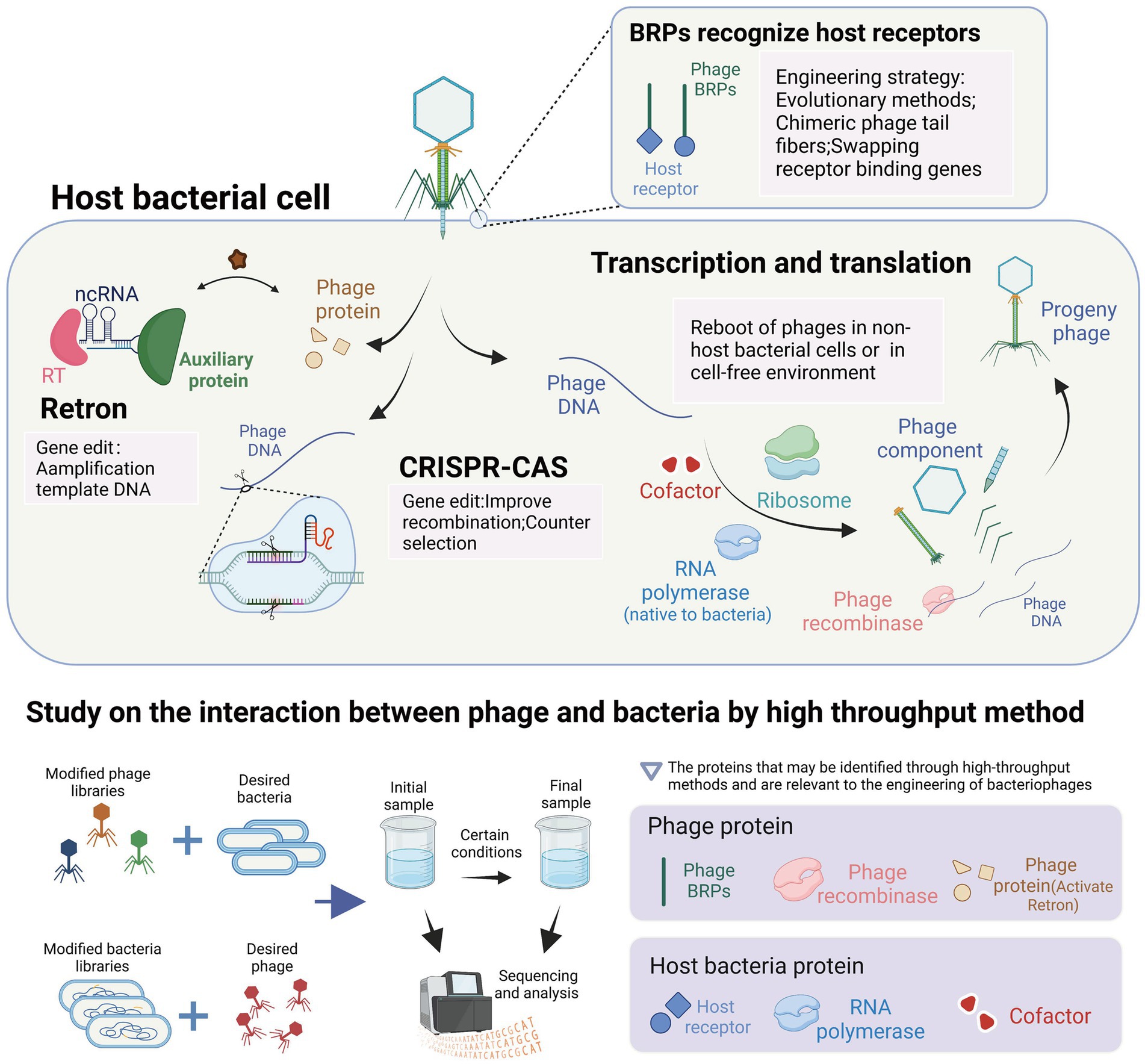
Figure 5. Interaction between bacteriophages and their host and its application in the engineering of bacteriophages. The highlighted content in light pink represents the utilization of interaction mechanisms in the context of phage engineering applications. The content highlighted with a light purple background represents the proteins that can be identified using high-throughput methods and are relevant to the engineering of bacteriophages. These proteins play a crucial role in understanding phage-host interactions and can be targeted for modification and optimization in phage engineering processes (images are created with Biorender.com).
The tools used for bacteriophage genome editing are derived from the bacterial anti-bacteriophage defense system within bacteria. These tools have been transferred and transformed to function in different bacteria, such as the CRISPR and retron systems. The CRISPR system is used during the editing process to induce double-stranded breaks in bacteriophage DNA and for screening engineered bacteriophages. In contrast, the retron system facilitates the amplification of template DNA in bacterial cells and promotes homologous recombination. Additionally, the lambda red recombinases system, which enhances recombination efficiency, was discovered in bacteriophage genes and has been developed. The reboot of phages outside the host bacteria requires the expression of phage genes. In the non-host bacterial environment, the reboot of phages requires the provision of corresponding enzymes by the bacteria to meet the transcriptional demands of the phages. In the cell-free environment, the reboot of phages directly utilizes the gene expression system from the bacterial cells. The expression of phage genes and the participation of host proteins in the development of cell-free extract (CFE) technology can be revealed, facilitating the development of CFE technology. A crucial goal of phage engineering is the remodeling of phage RNA-binding proteins (RBPs) as it can alter the host range of the phages to meet their therapeutic or other uses. The remodeling approach of RBPs mimics the evolution system of phages under bacterial defense pressure. For example, the introduction of artificial RBP mutations can allow the phages to retarget bacteria indicating receptor mutations, while the replacement or integration engineering of RBPs is similar to the gene exchange among phages in nature, allowing for a significant change in the host range of the phages. In conclusion, the interaction between phages and bacteria provides us with tools for phage engineering and guidance for their development. High-throughput methods offer valuable insights into the proteins involved in phage-host interactions and their application in phage engineering. These methods are instrumental in identifying phage receptor recognition proteins and bacterial receptors, essential for modifying phage receptor range and restoring infectivity against resistant host bacteria. Additionally, high-throughput methods can be used to discover phage recombinases, enzymes involved in phage DNA replication (Murphy, 2012), enabling enhanced recombination processes in phage gene engineering, such as lambda red recombinases (Fehér et al., 2012). Furthermore, the identification of phage proteins that activate the retron system can facilitate the engineering of phages with retron resistance. In terms of host bacteria, high-throughput methods assist in identifying RNA polymerases and cofactors involved in phage DNA transcription and assembly, crucial factors affecting the efficiency of in vitro phage rebooting (Yim et al., 2019; Silverman et al., 2020).
Retrons are a current topic of research that holds great promise due to their small size and immunological specificity. If we can fully understand the mechanism by which retrons specifically recognize bacteriophages, they may replace the CRISPR-Cas system for negative selection in bacteriophage engineering. Furthermore, when combined with reverse transcription editing templates, retrons could improve the efficiency of homologous recombination and offer a promising method for bacteriophage gene editing within the host bacterium. Besides, the development of cell-free expression (CFE) technology can provide us with more knowledge regarding the transcriptional mechanisms of bacteriophages. This, in turn, can assist us in synthesizing a greater number of engineered bacteriophages outside of host cells and reduce the reliance on engineering methods within host cells, which are limited by their lower transformation efficiency. CFE technology holds high prospects for application due to its efficiency and safety. In the process of CFE, the production of phages does not generate endotoxins produced by lysed bacteria, simplifying the purification process and reducing culturing time. As a result, the rapid development of CFE technology can effectively meet the demands of phage therapy. Furthermore, high-throughput methods for studying the interaction between phages and host bacteria are efficient and can help us identify genes that play a role in their interaction. This knowledge can effectively guide our modification of phages, leading to more efficient and effective phage therapy.
H-JJ: data curation, writing original draft, and software analysis. P-PJ, SY, L-KB, and GY: data curation and writing editing. D-SP: conceptualization, funding acquisition, project administration and writing editing. All authors contributed to the article and approved the submitted version.
This work was supported by the Chongqing Medical University Talent Project (Nos. R4014 to D-SP, and R4020 to P-PJ), National Natural Science Foundation (NSFC) of China (No. 32200386 to P-PJ), and China-Sri Lanka Joint Research and Demonstration Center for Water Technology, China-Sri Lanka Joint Center for Education and Research, Chinese Academy of Sciences, China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ackermann, H. W. (2007). 5500 phages examined in the electron microscope. Arch. Virol. 152, 227–243. doi: 10.1007/s00705-006-0849-1
Adler, B. A., Hessler, T., Cress, B. F., Lahiri, A., Mutalik, V. K., Barrangou, R., et al. (2022). Broad-spectrum CRISPR-Cas13a enables efficient phage genome editing. Nat. Microbiol. 7, 1967–1979. doi: 10.1038/s41564-022-01258-x
Ando, H., Lemire, S., Pires, D. P., and Lu, T. K. (2015). Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 1, 187–196. doi: 10.1016/j.cels.2015.08.013
Archer, P. K., Campbell, C. A., LaRock, D. L., Jarecke, D. J., Minnick, M. F., and Donohue, C. A. (2014). Bacteriophage λ N protein inhibits transcription slippage by Escherichia coli RNA polymerase. Nucleic Acids Res. 42, 5823–5829. doi: 10.1093/nar/gku203
Avramucz, Á., Møller-Olsen, C., Grigonyte, A. M., Paramalingam, Y., Millard, A., Sagona, A. P., et al. (2021). Analysing parallel strategies to alter the host specificity of bacteriophage T7. Biology 10:556. doi: 10.3390/biology10060556
Balcazar, J. L. (2014). Bacteriophages as vehicles for antibiotic resistance genes in the environment. PLoS Pathog. 10:e1004219. doi: 10.1371/journal.ppat.1004219
Bari, S. M. N., Walker, F. C., Cater, K., Aslan, B., and Hatoum-Aslan, A. (2017). Strategies for editing virulent staphylococcal phages using CRISPR-Cas10. ACS Synth. Biol. 6, 2316–2325. doi: 10.1021/acssynbio.7b00240
Barrangou, R., and Marraffini, L. A. (2014). CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell 54, 234–44. doi: 10.1016/j.molcel.2014.03.011
Berdygulova, Z., Westblade, L. F., Florens, L., Koonin, E. V., Chait, B. T., Ramanculov, E., et al. (2011). Temporal regulation of gene expression of the Thermus thermophilus bacteriophage P23-45. J. Mol. Biol. 405, 125–142. doi: 10.1016/j.jmb.2010.10.049
Bhattarai-Kline, S., Lear, S. K., Fishman, C. B., Lopez, S. C., Lockshin, E. R., Schubert, M. G., et al. (2022). Recording gene expression order in DNA by CRISPR addition of retron barcodes. Nature 608, 217–225. doi: 10.1038/s41586-022-04994-6
Bjorling, E. S., Devaraj, S., Whitney, M. T., Goers, G. W., and Jewett, M. C. (2018). Establishing a high-yielding cell-free protein synthesis platform derived from Vibrio natriegens. ACS Synth. Biol. 7, 2245–2255. doi: 10.1021/acssynbio.8b00252
Bobonis, J., Mitosch, K., Mateus, A., Karcher, N., Kritikos, G., Selkrig, J., et al. (2022). Bacterial retrons encode phage-defending tripartite toxin–antitoxin systems. Nature 609, 144–150. doi: 10.1038/s41586-022-05091-4
Boeckaerts, D., Stock, M., Criel, B., Gerstmans, H., De Baets, B., and Briers, Y. (2021). Predicting bacteriophage hosts based on sequences of annotated receptor-binding proteins. Sci. Rep. 11:1467. doi: 10.1038/s41598-021-81063-4
Box, A. M., Mcguffie, M. J., Ohara, B. J., and Seed, K. D. (2016). Functional analysis of bacteriophage immunity through a type I-E CRISPR-Cas system in vibrio cholerae and its application in bacteriophage genome engineering. J. Bacteriol. 198, 578–590. doi: 10.1128/JB.00747-15
Broniewski, J. M., Meaden, S., Paterson, S., Buckling, A., and Westra, E. R. (2020). The effect of phage genetic diversity on bacterial resistance evolution. ISME J. 14, 828–836. doi: 10.1038/s41396-019-0577-7
Chao, M. C., Abel, S., Davis, B. M., and Waldor, M. K. (2016). The design and analysis of transposon insertion sequencing experiments. Nat. Rev. Microbiol. 14, 119–128. doi: 10.1038/nrmicro.2015.7
Chen, P., Sun, H., Ren, H., Liu, W., Li, G., and Zhang, C. (2020). LamB, OmpC, and the core lipopolysaccharide of Escherichia coli K-12 function as receptors of bacteriophage Bp7. J. Virol. 94:e00325-20. doi: 10.1128/JVI.00325-20
Cheng, L., Deng, Z., Tao, H., Song, W., Xing, B., Liu, W., et al. (2022). Harnessing stepping-stone hosts to engineer, select, and reboot synthetic bacteriophages in one pot. Cell Rep. Methods 2:100217. doi: 10.1016/j.crmeth.2022.100217
Court, D. L., Sawitzke, J. A., and Thomason, L. C. (2002). Genetic engineering using homologous recombination. Annu. Rev. Genet. 36, 361–388. doi: 10.1146/annurev.genet.36.061102.093104
Cunliffe, T. G., Parker, A. L., and Jaramillo, A. (2022). Pseudotyping bacteriophage P2 tail fibers to extend the host range for biomedical applications. ACS Synth. Biol. 11, 3207–3215. doi: 10.1021/acssynbio.1c00629
Czajkowski, R. (2019). May the phage be with you? Prophage-like elements in the genomes of soft rot Pectobacteriaceae: Pectobacterium spp. and Dickeya spp. Front. Microbiol. 10:138. doi: 10.3389/fmicb.2019.00138
Dion, M. B., Oechslin, F., and Moineau, S. (2020). Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138. doi: 10.1038/s41579-019-0311-5
Djokic, W., Hidalgo-Lara, H., Nettleton, E. J., and Goodall, G. J. (2018). Establishing a cell-free Vibrio natriegens expression system. ACS Synth. Biol. 7, 2475–2479. doi: 10.1021/acssynbio.8b00222
Dong, J., Chen, C., Liu, Y., Zhu, J., Li, M., Rao, V. B., et al. (2021). Engineering T4 bacteriophage for in vivo display by type V CRISPR-Cas genome editing. ACS Synth. Biol. 10, 2639–2648. doi: 10.1021/acssynbio.1c00251
Dunne, M., Prokhorov, N. S., Loessner, M. J., and Leiman, P. G. (2021). Reprogramming bacteriophage host range: design principles and strategies for engineering receptor binding proteins. Curr. Opin. Biotechnol. 68, 272–281. doi: 10.1016/j.copbio.2021.02.006
Dunne, M., Rupf, B., Tala, M., Qabrati, X., Ernst, P., Shen, Y., et al. (2019). Reprogramming bacteriophage host range through structure-guided design of chimeric receptor binding proteins. Cell Rep. 29, 1336–1350.e4. doi: 10.1016/j.celrep.2019.09.062
Durfee, T., Nelson, R., Baldwin, S., Plunkett, G., Burland, V., Mau, B., et al. (2008). The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J. Bacteriol. 190, 2597–2606. doi: 10.1128/JB.01695-07
Emslander, Q., Vogele, K., Braun, P., Stender, J., Willy, C., Joppich, M., et al. (2022). Cell-free production of personalized therapeutic phages targeting multidrug-resistant bacteria. Cell Chem. Biol. 29, 1434–1445.e7. doi: 10.1016/j.chembiol.2022.06.003
Esteves, N. C., Porwollik, S., Mcclelland, M., and Scharf, B. E. (2021). The multi-drug efflux system AcrABZ-TolC is essential for infection of Salmonella Typhimurium by the flagellum-dependent bacteriophage chi. J. Virol. 95:e00394-21. doi: 10.1128/JVI.00394-21
Fa-Arun, J., Huan, Y. W., Darmon, E., and Wang, B. (2023). Tail-engineered phage P2 enables delivery of antimicrobials into multiple gut pathogens. ACS Synth. Biol. 12, 596–607. doi: 10.1021/acssynbio.2c00615
Fehér, T., Karcagi, I., Blattner, F. R., and Pósfai, G. (2012). Bacteriophage recombineering in the lytic state using the lambda red recombinases. Microb. Biotechnol. 5, 466–476. doi: 10.1111/j.1751-7915.2011.00292.x
Fu, Q., Li, S., Wang, Z., Shan, W., Ma, J., Cheng, Y., et al. (2017). H-NS mutation-mediated CRISPR-Cas activation inhibits phage release and toxin production of Escherichia coli Stx2 phage lysogen. Front. Microbiol. 8:652. doi: 10.3389/fmicb.2017.00652
Garamella, J., Marshall, R., Rustad, M., and Noireaux, V. (2016). The all E. coli TX-TL toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth. Biol. 5, 344–355. doi: 10.1021/acssynbio.5b00296
Gibson, D. G., Benders, G. A., Andrews-Pfannkoch, C., Denisova, E. A., Baden-Tillson, H., Zaveri, J., et al. (2008). Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319, 1215–1220. doi: 10.1126/science.1151721
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A. 3rd, and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi: 10.1038/nmeth.1318
Gogarten, J. F. (2022). Roles for non-human primate-associated phage diversity in improving medicine and public health. Evol. Med. Public Health 10, 123–129. doi: 10.1093/emph/eoac006
Gonzalez, F., and Scharf, B. E. (2021). Identification of receptor binding proteins in flagellotropic Agrobacterium phage 7-7-1. Viruses 13:1267. doi: 10.3390/v13071267
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. doi: 10.1126/science.aam9321
Górniak, M., Zalewska, A., and Jurczak-Kurek, A. (2022). Recombination events in putative tail fibre gene in Litunavirus phages infecting Pseudomonas aeruginosa and their phylogenetic consequences. Viruses 14:2669. doi: 10.3390/v14122669
Grigonyte, A. M., Harrison, C., Macdonald, P. R., Montero-Blay, A., Tridgett, M., Duncan, J., et al. (2020). Comparison of CRISPR and marker-based methods for the engineering of phage T7. Viruses 12:193. doi: 10.3390/v12020193
Guan, J., Oromí-Bosch, A., Mendoza, S. D., Karambelkar, S., Berry, J. D., and Bondy-Denomy, J. (2022). Bacteriophage genome engineering with CRISPR-Cas13a. Nat. Microbiol. 7, 1956–1966. doi: 10.1038/s41564-022-01243-4
Gurney, J., Pleška, M., and Levin, B. R. (2019). Why put up with immunity when there is resistance: an excursion into the population and evolutionary dynamics of restriction-modification and CRISPR-Cas. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 374:20180096. doi: 10.1098/rstb.2018.0096
Ha, E., Chun, J., Kim, M., and Ryu, S. (2019). Capsular polysaccharide is a receptor of a Clostridium perfringens bacteriophage CPS1. Viruses 11:1002. doi: 10.3390/v11111002
Hampton, H. G., Watson, B. N. J., and Fineran, P. C. (2020). The arms race between bacteria and their phage foes. Nature 577, 327–336. doi: 10.1038/s41586-019-1894-8
Harms, A., Brodersen, D. E., Mitarai, N., and Gerdes, K. (2018). Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell 70, 768–784. doi: 10.1016/j.molcel.2018.01.003
Hatfull, G. F., Dedrick, R. M., and Schooley, R. T. (2022). Phage therapy for antibiotic-resistant bacterial infections. Annu. Rev. Med. 73, 197–211. doi: 10.1146/annurev-med-080219-122208
Hershey, A. D., and Chase, M. (1952). Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 36, 39–56. doi: 10.1085/jgp.36.1.39
Hoshiga, F., Yoshizaki, K., Takao, N., Miyanaga, K., and Tanji, Y. (2019). Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol. Lett. 366:fnz041. doi: 10.1093/femsle/fnz041
Hupfeld, M., Trasanidou, D., Ramazzini, L., Klumpp, J., Loessner, M. J., and Kilcher, S. (2018). A functional type II-A CRISPR-Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic Acids Res. 46, 6920–6933. doi: 10.1093/nar/gky544
Huss, P., Chen, J., and Raman, S. (2023). High-throughput approaches to understand and engineer bacteriophages. Trends Biochem. Sci. 48, 187–197. doi: 10.1016/j.tibs.2022.08.012
Isaev, A., Andriianov, A., Znobishcheva, E., Zorin, E., Morozova, N., and Severinov, K. (2022). Editing of phage genomes - recombineering-assisted SpCas9 modification of model Coliphages T7, T5, and T3. Mol. Biol. 56, 801–815. doi: 10.1134/S0026893322060073
Islam, M. Z., Fokine, A., Mahalingam, M., Zhang, Z., Garcia-Doval, C., Van Raaij, M. J., et al. (2019). Molecular anatomy of the receptor binding module of a bacteriophage long tail fiber. PLoS Pathog. 15:e1008193. doi: 10.1371/journal.ppat.1008193
Jaschke, P. R., Lieberman, E. K., Rodriguez, J., Sierra, A., and Endy, D. (2012). A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology 434, 278–284. doi: 10.1016/j.virol.2012.09.020
Jeon, J., Ryu, C. M., Lee, J. Y., Park, J. H., Yong, D., and Lee, K. (2016). In vivo application of bacteriophage as a potential therapeutic agent to control OXA-66-like carbapenemase-producing Acinetobacter baumannii strains belonging to sequence type 357. Appl. Environ. Microbiol. 82, 4200–4208. doi: 10.1128/AEM.00526-16
Jia, P. P., Yang, Y. F., Junaid, M., Jia, H. J., Li, W. G., and Pei, D. S. (2023). Bacteriophage-based techniques for elucidating the function of zebrafish gut microbiota. Appl. Microbiol. Biotechnol. 107, 2039–2059. doi: 10.1007/s00253-023-12439-x
Kanehisa, A., Morita, M., Goto, R., Noguchi, E., Yokota, Z., Tajima, T., et al. (2020). Development of a clostridia-based cell-free system for prototyping genetic parts and metabolic pathways. Metab. Eng. 62, 95–105. doi: 10.1016/j.ymben.2020.06.004
Kilcher, S., and Loessner, M. J. (2019). Engineering bacteriophages as versatile biologics. Trends Microbiol. 27, 355–367. doi: 10.1016/j.tim.2018.09.006
Kilcher, S., Studer, P., Muessner, C., Klumpp, J., and Loessner, M. J. (2018). Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc. Natl. Acad. Sci. U. S. A. 115, 567–572. doi: 10.1073/pnas.1714658115
Kim, J., Hur, J. I., Ryu, S., and Jeon, B. (2021). Bacteriophage-mediated modulation of bacterial competition during selective enrichment of Campylobacter. Microbiol. Spectr. 9:e0170321. doi: 10.1128/Spectrum.01703-21
Kiro, R., Shitrit, D., and Qimron, U. (2014). Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 11, 42–44. doi: 10.4161/rna.27766
Klumpp, J., Dunne, M., and Loessner, M. J. (2023). A perfect fit: bacteriophage receptor-binding proteins for diagnostic and therapeutic applications. Curr. Opin. Microbiol. 71:102240. doi: 10.1016/j.mib.2022.102240
Kortright, K. E., Chan, B. K., and Turner, P. E. (2020). High-throughput discovery of phage receptors using transposon insertion sequencing of bacteria. Proc. Natl. Acad. Sci. U. S. A. 117, 18670–18679. doi: 10.1073/pnas.2001888117
Lampson, B. C., Inouye, M., and Inouye, S. (1989). Reverse transcriptase with concomitant ribonuclease H activity in the cell-free synthesis of branched RNA-linked msDNA of Myxococcus xanthus. Cells 56, 701–707. doi: 10.1016/0092-8674(89)90592-8
Lander, N., Li, Z.-H., Niyogi, S., and Docampo, R. (2015). CRISPR/Cas9-Induced Disruption of Paraflagellar Rod Protein 1 and 2 Genes in Trypanosoma cruzi Reveals Their Role in Flagellar Attachment. mBio 6:e01012. doi: 10.1128/mBio.01012-15
Lee, G., Chakraborty, U., Gebhart, D., Govoni, G. R., Zhou, Z. H., and Scholl, D. (2016). F-type bacteriocins of Listeria monocytogenes: a new class of phage tail-like structures reveals broad parallel coevolution between tailed bacteriophages and high-molecular-weight bacteriocins. J. Bacteriol. 198, 2784–2793. doi: 10.1128/JB.00489-16
Lemay, M.-L., Tremblay, D. M., and Moineau, S. (2017). Genome engineering of virulent Lactococcal phages using CRISPR-Cas9. ACS Synth. Biol. 6, 1351–1358. doi: 10.1021/acssynbio.6b00388
Letarov, A. V., and Kulikov, E. E. (2017). Adsorption of bacteriophages on bacterial cells. Biochem. Mosc. 82, 1632–1658. doi: 10.1134/S0006297917130053
Liang, J., Zhang, H., Tan, Y. L., Zhao, H., and Ang, E. L. (2022). Directed evolution of replication-competent double-stranded DNA bacteriophage toward new host specificity. ACS Synth. Biol. 11, 634–643. doi: 10.1021/acssynbio.1c00319
Liao, Y. T., Zhang, Y., Salvador, A., Harden, L. A., and Wu, V. C. H. (2022). Characterization of a T4-like bacteriophage vB_EcoM-Sa45lw as a potential biocontrol agent for Shiga toxin-producing Escherichia coli O45 contaminated on Mung bean seeds. Microbiol. Spectr. 10:e0222021. doi: 10.1128/spectrum.02220-21
Lim, A. N. W., Yen, M., Seed, K. D., Lazinski, D. W., and Camilli, A. (2021). A tail Fiber protein and a receptor-binding protein mediate ICP2 bacteriophage interactions with Vibrio cholerae OmpU. J. Bacteriol. 203:e0014121. doi: 10.1128/JB.00141-21
Liu, X., Gallay, C., Kjos, M., Domenech, A., Slager, J., Van Kessel, S. P., et al. (2017). High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 13:931. doi: 10.15252/msb.20167449
Liu, Y., Han, Y., Huang, W., Duan, Y., Mou, L., Jiang, Z., et al. (2012). Whole-genome synthesis and characterization of viable S13-like bacteriophages. PLoS One 7:e41124. doi: 10.1371/journal.pone.0041124
Liyanagedera, S. B. W., Williams, J., Wheatley, J. P., Biketova, A. Y., Hasan, M., Sagona, A. P., et al. (2022). SpyPhage: a cell-free TXTL platform for rapid engineering of targeted phage therapies. ACS Synth. Biol. 11, 3330–3342. doi: 10.1021/acssynbio.2c00244
Lopez, S. C., Crawford, K. D., Lear, S. K., Bhattarai-Kline, S., and Shipman, S. L. (2022). Precise genome editing across kingdoms of life using retron-derived DNA. Nat. Chem. Biol. 18, 199–206. doi: 10.1038/s41589-021-00927-y
Lu, D., Gong, X., Fang, Y., Guo, X., Chen, Y., Yang, F., et al. (2022). Correction of RNA splicing defect in β(654)-thalassemia mice using CRISPR/Cas9 gene-editing technology. Haematologica 107, 1427–1437. doi: 10.3324/haematol.2020.278238
Malik, D. J. (2019). “Chapter 19 - targeted delivery of bacteriophages to the gastrointestinal tract and their controlled release: unleashing the therapeutic potential of phage therapy” in Microbiome and Metabolome in diagnosis, therapy, and other strategic applications. eds. J. Faintuch and S. Faintuch (San Diego, CA, USA: Academic Press), 185–194.
Manning, K. A., and Dokland, T. (2020). The gp44 ejection protein of Staphylococcus aureus bacteriophage 80α binds to the ends of the genome and protects it from degradation. Viruses 12:563. doi: 10.3390/v12050563
Marinelli, L. J., Hatfull, G. F., and Piuri, M. (2012). Recombineering. Bacteriophage 2, 5–14. doi: 10.4161/bact.18778
Marinelli, L. J., Piuri, M., Swigonova, Z., Balachandran, A., Oldfield, L. M., Van Kessel, J. C., et al. (2008). BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 3:8. doi: 10.1371/journal.pone.0003957
Marraffini, L. A. (2015). CRISPR-Cas immunity in prokaryotes. Nature 526, 55–61. doi: 10.1038/nature15386
Martel, B., and Moineau, S. (2014). CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 42, 9504–9513. doi: 10.1093/nar/gku628
McCorquodale, D. J. (1999). “T5-like phages (Siphoviridae)” in Encyclopedia of virology. eds. A. Granoff and R. G. Webster. 2nd ed (Oxford: Elsevier), 1716–1722.
Meeske, A. J., Nakandakari-Higa, S., and Marraffini, L. A. (2019). Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature 570, 241–245. doi: 10.1038/s41586-019-1257-5
Melo, L. D. R., Monteiro, R., Pires, D. P., and Azeredo, J. (2022). Phage-host interaction analysis by flow cytometry allows for rapid and efficient screening of phages. Antibiotics 11:164. doi: 10.3390/antibiotics11020164
Mestre, M. R., González-Delgado, A., Gutiérrez-Rus, L. I., Martínez-Abarca, F., and Toro, N. (2020). Systematic prediction of genes functionally associated with bacterial retrons and classification of the encoded tripartite systems. Nucleic Acids Res. 48, 12632–12647. doi: 10.1093/nar/gkaa1149
Millman, A., Bernheim, A., Stokar-Avihail, A., Fedorenko, T., Voichek, M., Leavitt, A., et al. (2020). Bacterial retrons function in anti-phage defense. Cells 183, 1551–1561.e12. doi: 10.1016/j.cell.2020.09.065
Molineux, I. J. (1999). “T7-like phages (Podoviridae)” in Encyclopedia of virology. eds. A. Granoff and R. G. Webster. 2nd ed (Oxford: Elsevier), 1722–1729.
Moore, S. J., MacDonald, J. T., Wienecke, S., Ishwarbhai, A., Tsipa, A., Aw, R., et al. (2018). Rapid acquisition and model-based analysis of cell-free transcription-translation reactions from nonmodel bacteria. Proc. Natl. Acad. Sci. U. S. A. 115, E4340–E4349. doi: 10.1073/pnas.1715806115
Muniyappa, K., and Radding, C. M. (1986). The homologous recombination system of phage lambda. Pairing activities of beta protein. J. Biol. Chem. 261, 7472–7478. doi: 10.1016/S0021-9258(17)38416-8
Murphy, K. C. (2012). Phage recombinases and their applications. Adv. Virus Res. 83, 367–414. doi: 10.1016/B978-0-12-394438-2.00008-6
Murray, P. J. (2006). “Targeting vector construction by yeast artificial chromosome modification” in YAC protocols. ed. A. Mackenzie (Totowa, NJ: Humana Press), 127–137.
Mutalik, V. K., Adler, B. A., Rishi, H. S., Piya, D., Zhong, C., Koskella, B., et al. (2020). High-throughput mapping of the phage resistance landscape in E. coli. PLoS Biol. 18:e3000877. doi: 10.1371/journal.pbio.3000877
Noskov, V. N., Koriabine, M., Solomon, G., Randolph, M., Barrett, J. C., Leem, S. H., et al. (2001). Defining the minimal length of sequence homology required for selective gene isolation by TAR cloning. Nucleic Acids Res. 29, 32e–332e. doi: 10.1093/nar/29.6.e32
Oppenheim, A. B., Rattray, A. J., Bubunenko, M., Thomason, L. C., and Court, D. L. (2004). In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology 319, 185–189. doi: 10.1016/j.virol.2003.11.007
Payne, L. J., Meaden, S., Mestre, M. R., Palmer, C., Toro, N., Fineran, P. C., et al. (2022). PADLOC: a web server for the identification of antiviral defence systems in microbial genomes. Nucleic Acids Res. 50, W541–W550. doi: 10.1093/nar/gkac400
Peng, H., and Chen, I. A. (2021). Phage engineering and the evolutionary arms race. Curr. Opin. Biotechnol. 68, 23–29. doi: 10.1016/j.copbio.2020.09.009
Peters, J. M., Colavin, A., Shi, H., Czarny, T. L., Larson, M. H., Wong, S., et al. (2016). A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cells 165, 1493–1506. doi: 10.1016/j.cell.2016.05.003
Phetruen, T., Chanarat, S., Janvilisri, T., Phanchana, M., Charoensutthivarakul, S., Phothichaisri, W., et al. (2022). Receptor binding protein of prophage reversibly recognizes the low-molecular weight subunit of the surface-layer protein SlpA in Clostridioides difficile. Front. Microbiol. 13:998215. doi: 10.3389/fmicb.2022.998215
Pires, D. P., Cleto, S., Sillankorva, S., Azeredo, J., and Lu, T. K. (2016). Genetically engineered phages: a review of advances over the last decade. Microbiol. Mol. Biol. Rev. 80, 523–543. doi: 10.1128/MMBR.00069-15
Pires, D. P., Dötsch, A., Anderson, E. M., Hao, Y., Khursigara, C. M., Lam, J. S., et al. (2017). A genotypic analysis of Five P. aeruginosa strains after biofilm infection by phages targeting different cell surface receptors. Front. Microbiol. 8:1229. doi: 10.3389/fmicb.2017.01229
Qimron, U., Marintcheva, B., Tabor, S., and Richardson, C. C. (2006). Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. U. S. A. 103, 19039–19044. doi: 10.1073/pnas.0609428103
Ramirez-Chamorro, L., Boulanger, P., and Rossier, O. (2021). Strategies for bacteriophage T5 mutagenesis: expanding the toolbox for phage genome engineering. Front. Microbiol. 12:667332. doi: 10.3389/fmicb.2021.667332
Revel, H. R., Luria, S. E., and Young, N. L. (1961). Biosynthesis of beta-D-galactosidase controlled by phage-carried genes. III. Dereprssion of beta-d-galactosidase synthesis following induction of phage development in lysogenic bacteria. Proc. Natl. Acad. Sci. U. S. A. 47, 1974–1980. doi: 10.1073/pnas.47.12.1974
Rodriguez-Gonzalez, R. A., Leung, C. Y., Chan, B. K., Turner, P. E., and Weitz, J. S. (2020). Quantitative models of phage-antibiotic combination therapy. mSystems 5:5. doi: 10.1128/mSystems.00756-19
Roucourt, B., and Lavigne, R. (2009). The role of interactions between phage and bacterial proteins within the infected cell: a diverse and puzzling interactome. Environ. Microbiol. 11, 2789–2805. doi: 10.1111/j.1462-2920.2009.02029.x
Rousset, F., Cui, L., Siouve, E., Becavin, C., Depardieu, F., and Bikard, D. (2018). Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLoS Genet. 14:e1007749. doi: 10.1371/journal.pgen.1007749
Rustad, M., Eastlund, A., Jardine, P., and Noireaux, V. (2018). Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth. Biol. 3:ysy002. doi: 10.1093/synbio/ysy002
Rustad, M., Eastlund, A., Marshall, R., Jardine, P., and Noireaux, V. (2017). Synthesis of Infectious Bacteriophages in an E. coli-based Cell-free Expression System. J. Vis. Exp. :56144. doi: 10.3791/56144
Salazar, K. C., Ma, L., Green, S. I., Zulk, J. J., Trautner, B. W., Ramig, R. F., et al. (2021). Antiviral resistance and phage counter adaptation to antibiotic-resistant Extraintestinal pathogenic Escherichia coli. MBio 12:e00211-21. doi: 10.1128/mBio.00211-21
Schilling, T., Dietrich, S., Hoppert, M., and Hertel, R. (2018). A CRISPR-Cas9-based toolkit for fast and precise in vivo genetic engineering of Bacillus subtilis phages. Viruses 10:241. doi: 10.3390/v10050241
Sharon, E., Chen, S. A., Khosla, N. M., Smith, J. D., Pritchard, J. K., and Fraser, H. B. (2018). Functional genetic variants revealed by massively parallel precise genome editing. Cells 175, 544–557.e16. doi: 10.1016/j.cell.2018.08.057
Shen, J., Zhou, J., Chen, G.-Q., and Xiu, Z.-L. (2018). Efficient genome engineering of a virulent Klebsiella bacteriophage using CRISPR-Cas9. J. Virol. 92:e00534-18. doi: 10.1128/JVI.00534-18
Shin, J., Jardine, P., and Noireaux, V. (2012). Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth. Biol. 1, 408–413. doi: 10.1021/sb300049p
Silverman, A. D., Karim, A. S., and Jewett, M. C. (2020). Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet. 21, 151–170. doi: 10.1038/s41576-019-0186-3
Simon, A. J., Ellington, A. D., and Finkelstein, I. J. (2019). Retrons and their applications in genome engineering. Nucleic Acids Res. 47, 11007–11019. doi: 10.1093/nar/gkz865
Simpson, D. J., Sacher, J. C., and Szymanski, C. M. (2016). Development of an assay for the identification of receptor binding proteins from bacteriophages. Viruses 8:17. doi: 10.3390/v8010017
Subedi, D., and Barr, J. J. (2021). Temporal stability and genetic diversity of 48-year-old T-series phages. mSystems 6:e00990–20. doi: 10.1128/mSystems.00990-20
Suga, A., Kawaguchi, M., Yonesaki, T., and Otsuka, Y. (2021). Manipulating interactions between T4 phage long tail fibers and Escherichia coli receptors. Appl. Environ. Microbiol. 87:e0042321. doi: 10.1128/AEM.00423-21
Sun, D. S., Kissler, S. M., Kanjilal, S., Olesen, S. W., Lipsitch, M., and Grad, Y. H. (2022). Analysis of multiple bacterial species and antibiotic classes reveals large variation in the association between seasonal antibiotic use and resistance. PLoS Biol. 20:e3001579. doi: 10.1371/journal.pbio.3001579
Tao, P., Wu, X., Tang, W.-C., Zhu, J., and Rao, V. (2017). Engineering of bacteriophage T4 genome using CRISPR-Cas9. ACS Synth. Biol. 6, 1952–1961. doi: 10.1021/acssynbio.7b00179
Thomason, L. C., Oppenheim, A. B., and Court, D. L. (2009). Modifying bacteriophage lambda with recombineering. Methods Mol. Biol. 501, 239–251. doi: 10.1007/978-1-60327-164-6_21
Ul-Hamid, A., Dafalla, H., Hakeem, A. S., Haider, A., and Ikram, M. (2022). In vitro catalytic and antibacterial potential of Green synthesized CuO nanoparticles against prevalent multiple drug resistant bovine mastitogen Staphylococcus aureus. Int. J. Mol. Sci. 23:2335. doi: 10.3390/ijms23042335
van Houte, S., Buckling, A., and Westra, E. R. (2016). Evolutionary ecology of prokaryotic immune mechanisms. Microbiol. Mol. Biol. Rev. 80, 745–763. doi: 10.1128/MMBR.00011-16
Wang, Y., Guan, Z., Wang, C., Nie, Y., Chen, Y., Qian, Z., et al. (2022). Cryo-EM structures of Escherichia coli Ec86 retron complexes reveal architecture and defence mechanism. Nat. Microbiol. 7, 1480–1489. doi: 10.1038/s41564-022-01197-7
Wang, H., Li, J., and Jewett, M. C. (2018). Development of a Pseudomonas putida cell-free protein synthesis platform for rapid screening of gene regulatory elements. Synth. Biol. 3:ysy003. doi: 10.1093/synbio/ysy003
Wei, Z. L., Yang, F., Li, B., Hou, P., Kong, W. W., Wang, J., et al. (2022). Structural insights into the chaperone-assisted assembly of a simplified tail fiber of the myocyanophage Pam3. Viruses 14:2260. doi: 10.3390/v14102260
White, T. B., Doyle, R. J., and Streips, U. N. (1981). Transformation of a Bacillus subtilis L-form with bacteriophage deoxyribonucleic acid. J. Bacteriol. 145, 878–883. doi: 10.1128/jb.145.2.878-883.1981
Xiao, Y., Luo, M., Dolan, A. E., Liao, M., and Ke, A. (2018). Structure basis for RNA-guided DNA degradation by cascade and Cas3. Science 361:eaat0839. doi: 10.1126/science.aat0839
Xu, H., Liu, W. Q., and Li, J. (2020). Translation related factors improve the productivity of a Streptomyces-based cell-free protein synthesis system. ACS Synth. Biol. 9, 1221–1224. doi: 10.1021/acssynbio.0c00140
Yang, H., Ma, Y., Wang, Y., Yang, H., Shen, W., and Chen, X. (2014). Transcription regulation mechanisms of bacteriophages: recent advances and future prospects. Bioengineered 5, 300–304. doi: 10.4161/bioe.32110
Yehl, K., Lemire, S., Yang, A. C., Ando, H., Mimee, M., Torres, M. T., et al. (2019). Engineering phage host-range and suppressing bacterial resistance through phage tail Fiber mutagenesis. Cells 179, 459–469.e9. doi: 10.1016/j.cell.2019.09.015
Yim, S. S., Johns, N. I., Park, J., Gomes, A. L., McBee, R. M., Richardson, M., et al. (2019). Multiplex transcriptional characterizations across diverse bacterial species using cell-free systems. Mol. Syst. Biol. 15:e8875. doi: 10.15252/msb.20198875
Yoichi, M., Abe, M., Miyanaga, K., Unno, H., and Tanji, Y. (2005). Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157:H7. J. Biotechnol. 115, 101–107. doi: 10.1016/j.jbiotec.2004.08.003
Yosef, I., Goren, M. G., Globus, R., Molshanski-Mor, S., and Qimron, U. (2017). Extending the host range of bacteriophage particles for DNA transduction. Mol. Cell 66, 721–728.e3. doi: 10.1016/j.molcel.2017.04.025
Yosef, I., Manor, M., Kiro, R., and Qimron, U. (2015). Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. U. S. A. 112, 7267–7272. doi: 10.1073/pnas.1500107112
Yuan, Y., Peng, Q., Zhang, S., Liu, T., Yang, S., Yu, Q., et al. (2019). Phage reduce stability for regaining infectivity during antagonistic coevolution with host bacterium. Viruses 11:118. doi: 10.3390/v11020118
Yuan, S., Shi, J., Jiang, J., and Ma, Y. (2022). Genome-scale top-down strategy to generate viable genome-reduced phages. Nucleic Acids Res. 50, 13183–13197. doi: 10.1093/nar/gkac1168
Zhan, Y., Buchan, A., and Chen, F. (2015). Novel N4 bacteriophages prevail in the cold biosphere. Appl. Environ. Microbiol. 81, 5196–5202. doi: 10.1128/AEM.00832-15
Zhang, J., Ning, H., Lin, H., She, J., Wang, L., Jing, Y., et al. (2022). Expansion of the Plaquing host range and improvement of the absorption rate of a T5-like Salmonella phage by altering the long tail fibers. Appl. Environ. Microbiol. 88:e0089522. doi: 10.1128/aem.00895-22
Zhang, X., Wang, S., Li, T., Zhang, Q., Zhang, R., and Zhang, Z. (2021). Bacteriophage: a useful tool for studying gut bacteria function of housefly larvae, Musca domestica. Microbiol Spectr 9:e0059921. doi: 10.1128/Spectrum.00599-21
Keywords: multidrug resistant bacteria, phage engineering, gene editing, phage-host interaction, high-throughput methods
Citation: Jia H-J, Jia P-P, Yin S, Bu L-K, Yang G and Pei D-S (2023) Engineering bacteriophages for enhanced host range and efficacy: insights from bacteriophage-bacteria interactions. Front. Microbiol. 14:1172635. doi: 10.3389/fmicb.2023.1172635
Edited by:
Hany Anany, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Jun-Seob Kim, Incheon National University, Republic of KoreaCopyright © 2023 Jia, Jia, Yin, Bu, Yang and Pei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: De-Sheng Pei, cGVpZHNAY3FtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.