 Alba Kaci1
Alba Kaci1 Christine M. Jonassen1,2Steinar Skrede3,4Audun Sivertsen5 The Norwegian Study Group on Streptococcus dysgalactiaeMartin Steinbakk1†Oddvar Oppegaard3,4*
Christine M. Jonassen1,2Steinar Skrede3,4Audun Sivertsen5 The Norwegian Study Group on Streptococcus dysgalactiaeMartin Steinbakk1†Oddvar Oppegaard3,4*- 1Center for Laboratory Medicine, Østfold Hospital Trust, Grålum, Norway
- 2Department of Virology, Norwegian Institute of Public Health, Oslo, Norway
- 3Department of Clinical Science, University of Bergen, Bergen, Norway
- 4Department of Medicine, Haukeland University Hospital, Bergen, Norway
- 5Department of Microbiology, Haukeland University Hospital, Bergen, Norway
Background: Streptococcus dysgalactiae subspecies equisimilis (SDSE) is an emerging global pathogen, yet the epidemiology and population genetics of SDSE species have not been extensively characterized.
Methods: We carried out whole genome sequencing to characterize 274 SDSE isolates causing bloodstream infections obtained through national surveillance program in 2018. We conducted multilocus sequence typing (MLST), emm-typing, core genome phylogeny, as well as investigated key features associated with virulence. Moreover, comparison to SDSE from other geographic regions were performed in order to gain more insight in the evolutionary dynamics in SDSE.
Results: The phylogenetic analysis indicated a substantial diversity of emm-types and sequence types (STs). Briefly, 17 emm-types and 58 STs were identified that formed 10 clonal complexes (CCs). The predominant ST-types were ST20 (20%), ST17 (17%), and ST29 (11%). While CC17 and CC29 clades showed a substantial heterogeneity with well-separated emm-associated subclades, the CC20 clade harboring the stG62647 emm-type was more homogenous and the most prevalent in the present study. Moreover, we observed notable differences in the distribution of clades within Norway, as well as several disseminated CCs and also distinct geographic variations when compared to data from other countries. We also revealed extensive intra-species recombination events involving surface exposed virulence factors, including the emm gene important for phylogenetic profiling.
Conclusion: Recombination events involving the emm as well as other virulence genes in SDSE, are important mechanisms in shaping the genetic variability in the SDSE population, potentially offering selective advantages to certain lineages. The enhanced phylogenetic resolution offered by whole genome sequencing is necessary to identify and delimitate outbreaks, monitor and properly characterize emerging strains, as well as elucidate bacterial population dynamics.
Introduction
Streptococcus dysgalactiae subspecies equisimilis (SDSE) is a β-hemolytic streptococcal species harboring the Lancefield groups C or G antigen (rarely group A or L) (Brandt et al., 1999; Rantala, 2014). The clinical manifestations of SDSE in humans resemble those caused by Streptococcus pyogenes (Group A Streptococcus; GAS), ranging from tonsillitis or harmless superficial skin infections to life-threating necrotizing soft tissue infections or streptococcal toxic shock syndrome (Brandt and Spellerberg, 2009; Broyles et al., 2009; Rantala, 2014; Bruun et al., 2016; Oppegaard et al., 2016). Although SDSE was long considered a low-virulent or commensal organism, increasing rates of invasive infections have been reported worldwide the past decades, exceeding the incidence rates of invasive S. pyogenes disease in some geographic regions (Rantala et al., 2009; Wong et al., 2009; Harris et al., 2011; Lambertsen et al., 2014; Schwartz et al., 2014; Wajima et al., 2016). In western Norway, the annual incidence rates for SDSE bloodstream infections gradually rose from 1.4 to 7.6 per 100,000 inhabitants during 1999–2021, and it is currently the fifth most common pathogen identified in blood cultures in this region (Oppegaard et al., 2015, 2023).
The cell surface M protein, encoded by the emm-gene, is a major virulence factor in both SDSE and S. pyogenes, and important in inhibiting phagocytosis and inactivation of the complement system (Bisno et al., 1987; Brandt and Spellerberg, 2009). It is ubiquitous in SDSE of human origin, and its hypervariability in the 5’-terminal region forms the basis of classification into emm-types. Moreover, SDSE is often classified using multilocus sequence typing (MLST), and to date, more than 100 SDSE emm-types and 600 sequence types (ST) have been described (CDC Strep Lab, PubMLST).
Despite SDSE being an emerging human pathogen, the epidemiology and population genetics of SDSE species are still poorly understood. The majority of published population studies have involved molecular characterization and typing by PCR methods, providing a limited phylogenetic resolution. The substantial diversity of circulating emm-types and STs observed in such studies, combined with an incongruence between these two phylogenetic markers, have complicated the interpretation of evolutionary relationships of SDSE isolates (Ahmad et al., 2009; McMillan et al., 2011; McNeilly and Mcmillan, 2014).
Whole genome sequencing (WGS) of bacterial pathogens has emerged as a powerful tool, providing higher resolution than those of conventional methods (Reuter et al., 2013; Lytsy et al., 2017). Systematic large-scale population-based surveillance of SDSE isolates using whole genome sequencing may therefore contribute to enhance our understanding of molecular epidemiology and pathogenic mechanisms of SDSE.
In the present study, we employed WGS to explore the epidemiological, evolutionary and molecular characteristics of clinical SDSE isolates obtained from a nationwide surveillance in Norway in 2018. In addition, we compared our data to other SDSE isolate-sequences retrieved from other geographic regions in the recent years to gain a global perspective and increase our understanding of the epidemiology of SDSE.
Materials and methods
Surveillance and bacterial isolates
All SDSE isolates from blood cultures in Norway during 2018 were collected as part of the Norwegian surveillance program for antimicrobial resistance (NORM). Collection of data within NORM follows a standard protocol described in the yearly surveillance reports (NORM/NORM-VET, 2018). Primary laboratories performed species identification based on colony morphology (β-hemolytic reaction on 5% sheep-blood agar and colony size >0.5 mm after 24 h of incubation), serogroup specificity using rapid Lancefield agglutination test, and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). All isolates identified as S. dysgalactiae were submitted to Haukeland University Hospital, Bergen, or Østfold Hospital Trust, Grålum for susceptibility testing and genomic characterization. The patients’ residential postal code, as well as specimen type and date of collection were reported for each isolate. Annual incidence rates for SDSE bloodstream infections were calculated using population data from 2018 obtained from Statistics Norway.1
Whole genome sequencing
At Haukeland University Hospital, genomic DNA was extracted from 119 isolates (collected from regions North, Middle and West), followed by sequencing on an Illumina 4000 HiSeq system (Illumina, San Diego, CA) to produce 150 bp paired-end reads as previously described (Oppegaard et al., 2017b).
The Ion Torrent technology and Ion S5XL system (Thermo Fisher Scientific, Waltham, MA, USA) was used by Østfold Hospital Trust to WGS the remaining 157 isolates collected in the South and East regions. Briefly, genomic DNAs were extracted from an overnight culture pre-treated with lysozyme and proteinase K using the QIAmp DNA Mini Blood extraction kit (QIAGEN, Hilden, Germany). Genomic libraries (300-base reads) were prepared using the Ion Xpress Plus Fragment Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturers’ instructions. Prior to template preparation and sequencing, all libraries were amplified, quantified by qPCR and pooled together to a concentration of 50 pM. Amplified and pooled samples were then sequenced on the Ion S5XL platform as described in the Ion S5 and Ion S5XL instrument user guide, using Ion 530 chips (Thermo Fisher Scientific, Waltham, MA, USA), with 32 samples per chip.
In silico analysis
For data generated on the Illumina HiSeq system, reads were trimmed with Trimmomatic v0.39 (Bolger et al., 2014). For Ion Torrent-generated data, reads were processed with the incorporated S5 software plug-ins. All trimmed reads from the sequenced isolates were de novo assembled by SPAdes v5.14 (Bankevich et al., 2012), followed by evaluation of the assembled contigs using Quast v5.2.0 (Gurevich et al., 2013) and genome annotation using RAST v1.073 (Aziz et al., 2008). Species identity was confirmed by 16S rRNA analysis. Next, emm-types were determined by nucleotide BLAST-searches against the emm sequence database from the Center of Disease Control.2 MLST profiles were identified using the MLST-typing web-tool provided by the Center for Genomic Epidemiology (Larsen et al., 2012), and novel MLST alleles and profiles were submitted to pubMLST.3 STs were delineated into clonal complexes (CCs) based on single locus variants using Phyloviz (Ribeiro-Gonçalves et al., 2016). A core genome single-nucleotide polymorphism (SNPs) phylogeny was generated by Conserved Signature Indels (CSI) Phylogeny at the Center for Genomic Epidemiology4 using default settings and the SDSE type strain NCTC13762 as a reference.5 The resulting maximum likelihood phylogenetic trees were visualized using the Interactive Tree of Life platform, iTOL v6 (Letunic and Bork, 2021).
Geneious Prime v2022.2.2 was used to screen for the presence of key virulence factors and regulators, adapted and modified from the dataset of virulence factors of S. pyogenes in the Virulence Factors of Pathogenic Bacteria Database.6 In silico screening was performed with BLAST search using an E-value of 0.05 as cut-off. For identification of the hypervariable pilus islands we used BLAST to locate the conserved genes flanking these operons, and subsequently manually inspected interposing region for the presence or absence of pilus operon genes, including regulators, ancillary proteins, pilus tip adhesins and sortase genes. The genome of SDSE strains was further screened for bacteriophage sequences using PHASTER (Arndt et al., 2016) and Integrative Conjugative Elements (ICEs) were identified by manual inspection and BLAST-search of selected regions as previously described (Ambroset et al., 2015).
Whole genome sequences from epidemiological studies in Japan and Canada were downloaded from GenBank (PRJDB12179 and PRJNA325743). In order to match the disease severity and temporal characteristics of our study, only invasive isolates collected between 2012 and 2019 were included from GenBank. These genomes were de novo assembled (SPAdes), annotated (RAST) and MLST-typed (Center for Genomic Epidemiology). The geographical distribution of SDSE clonal complexes was visualized using Phyloviz.
Results
Patient characteristics and genome statistics of SDSE isolates
A total of 276 blood culture isolates were collected during the surveillance period in 2018 in Norway; 274 of the 276 blood culture isolates were confirmed as SDSE by whole genome sequencing and included for further analysis, whereas two isolates were identified as S. canis and S. equi and excluded from the study. The Lancefield group C antigen was detected in 81 isolates (30%) and 193 isolates were classified as Lancefield group G (70%). No isolates harbored group A or L-antigens. In general, a male predominance was observed (66%), and the age distribution ranged from 12 to 98 years; with a median age of 76 years.
The draft genomes of the 274 SDSE isolates had an average assembly length of 2.16 Mb; a GC content of approximately 39.2%; contigs >500 bp of 70; and protein encoding genes (CDSs) of 2,192 (genome annotation data and accession numbers are shown in Supplementary Table 1).
Molecular typing and phylogeny
Genotyping revealed substantial diversity, comprising 17 emm-types in total (Supplementary Table 1). The most prevalent emm-types were stG62647 (19%), stG485 (13%), stC74a (12%), stG6 (9.5%), stG643 (8.3%), and stG652 (5.8%). Among them, emm-types stG6, stG643, and stG652 were composed of two or more subtypes. Seven new emm-subtypes were identified (stC9431.1, stC485.19, stG6.23, stG6.24, stG6.25, stG2078.13, and stG652.21). The emm-type stG62647 was exclusively detected in group C isolates, whereas stC74a was found restricted to group G isolates. The other prevalent emm-types included a mix of isolates expressing group G or C antigens.
Exploring the phylogenetic relationships, the one strain belonging to the stC9431 emm-type was separated from all other isolates by >14,000 SNPs (Supplementary Figure 1). Comparison to previously characterized SDSE-strains of animal origin revealed that it clustered with canine-associated strains (Porcellato et al., 2021). This outlier affected the ability to discriminate the remaining isolates, and a decision was made to perform phylogenetic analyses without this genome. A phylogenetic tree excluding the canine-associated strain is depicted in Figure 1.
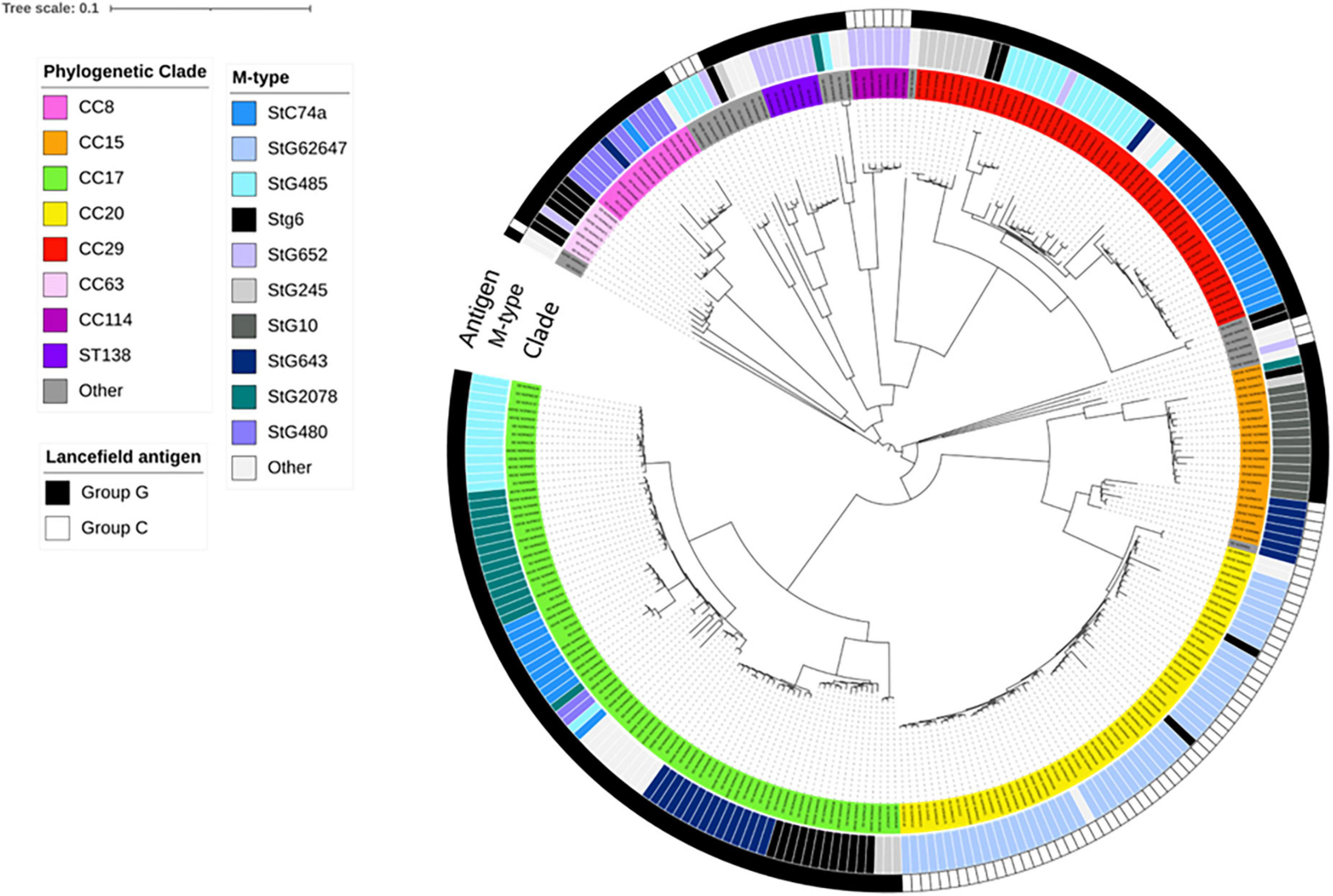
Figure 1. Whole genome single-nucleotide polymorphism (SNP)-derived phylogenetic tree of blood culture Streptococcus dysgalactiae subspecies equisimilis (SDSE) isolates obtained in Norway during 2018. This tree was constructed with CSI phylogeny website and metadata was added using iTOL, including information about antigen-type, emm-type and multilocus sequence typing clonal complex relatedness groups (CCs). Scale bar indicates the estimated evolutionary divergence between isolates measured in substitution per site. N = 273; excluding the canine-associated genome harboring the stC9431 emm gene. The first ring (Antigen) indicates the distribution of Lancefield group C or G antigen among the isolates. The second ring (M-type) indicates the distribution of SDSE isolates into emm-types, whereas the third ring (CC) indicates the most common clonal complexes and sequence types (STs).
Multilocus sequence typing performed for all SDSE isolates revealed a total of 58 STs, of which 16 new alleles and nine novel profiles. The predominant ST-types were ST20 (20%), ST17 (17%), and ST29 (11%). The ST-types clustered into ten clonal complexes and 13 singletons using single locus variants. The phylogenetic analysis indicated a substantial heterogeneity within the CC17 and CC29 clades, with CC17 isolates differing by a median of 1,337 SNPs (range 0–2,669), and CC29 isolates by a median of 1,070 SNPs (range 12–2,519). CC17 and CC29 were associated with numerous emm-types, but these were distributed into well-separated emm-associated subclades within the clonal complexes (Figure 1). The CC20 clade was more homogenous, and the isolates were separated by a median of 143 SNPs (range 0–1,006). Only five isolates were associated with other emm-types than stG62647. Inspecting the M- protein region in these five isolates we detected a recombinational replacement of the emm gene, whereas the surrounding region was almost identical to their phylogenetic background (Figure 2). We observed such emm switching events in CC20 isolates involving the acquisition of stG2574 (two isolates), stG6 (two isolates), and stG5420 (one isolate) emm genes. On the other hand, emm gene replacements in CC17 and CC29 clades were associated with more extensive phylogenetic discrepancies, reflecting more established subclades (data not shown).
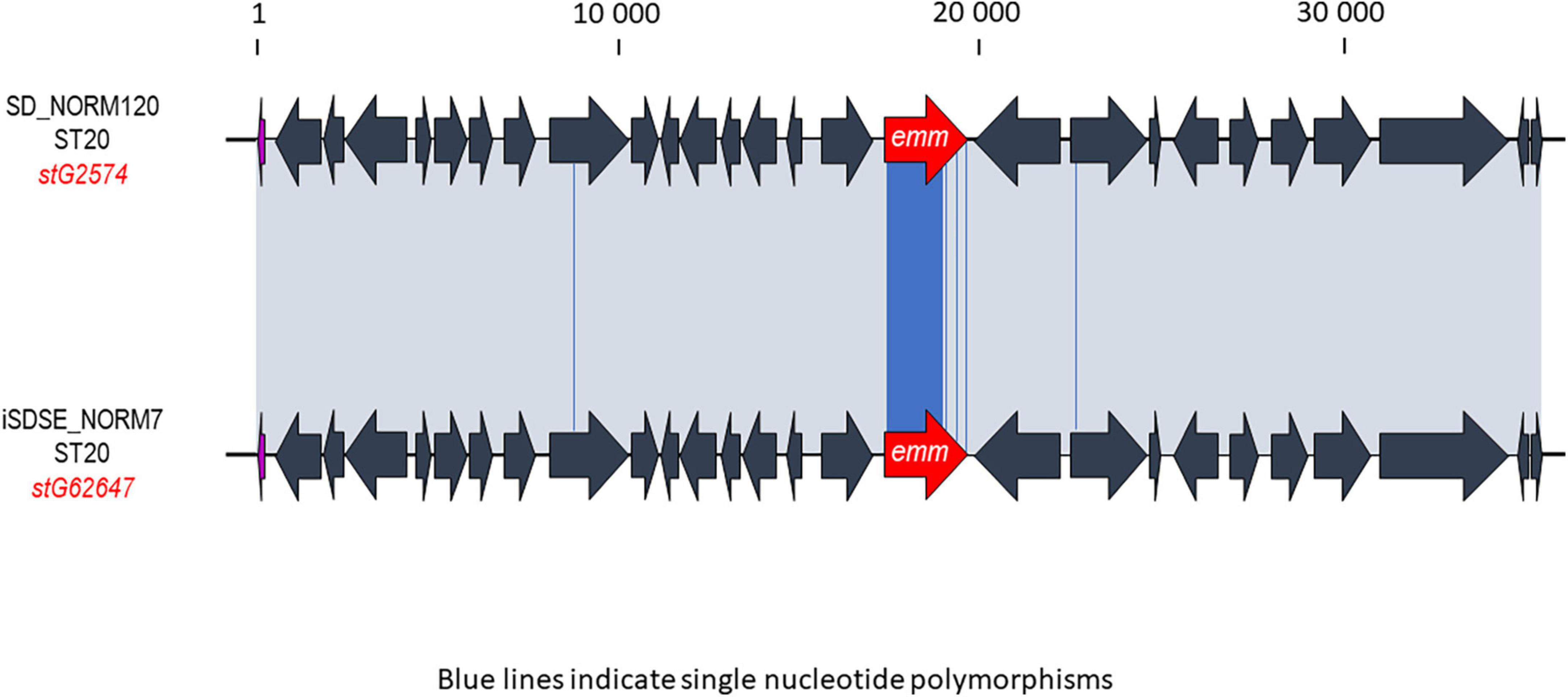
Figure 2. Graphic representation of a likely emm-switch event observed between two phylogenetically closely related isolates. The ST20 profile is usually associated with the stG62647 emm gene, but here the SD_NORM120 isolate harbors the stG2574 emm gene. The iSDSE_NORM7 strain was the closest relative in the single-nucleotide polymorphism (SNP)-analysis, and was chosen for comparison. The alignment was performed using Geneious. The emm gene in each Streptococcus dysgalactiae subspecies equisimilis (SDSE) isolate is highlighted in red. The gray shaded areas indicate 100% homology, and blue lines in the alignment indicate single nucleotide polymorphisms. The scale indicates number of base pairs.
In order to capture the heterogeneity in the CC17 and CC29 clades, we decided to further delineate these CCs into emm-specific subclades in the following phylogenetic analyses.
Phylogeography
The annual incidence rates for bloodstream infections varied geographically in Norway during 2018. Health Region North recorded the highest incidence rate of 7.6 cases per 100,000 inhabitants/year, followed by Health Region South with 6.2/100,000, Health Region Middle with 5.0/100,000, Health Region East with 4.9/100,000, and Health Region West with 4.2/100,000.
Interestingly, geographic variations in the distribution of clonal complexes were also noted (Figure 3). The CC20 clade was dominant in all geographic regions, apart from the northern health region where CC15 was most common. Notably, the ST138 clade seems to have emerged in the south, constituting the second most frequent ST-profile in this region, whereas it was completely absent from the western, middle and northern regions. The western health region lacked several of the most common clades and subclades circulating in Norway. Approximately a third of the isolates in this region (14/47) belonged to less common clades, comprising nine different ST-profiles.
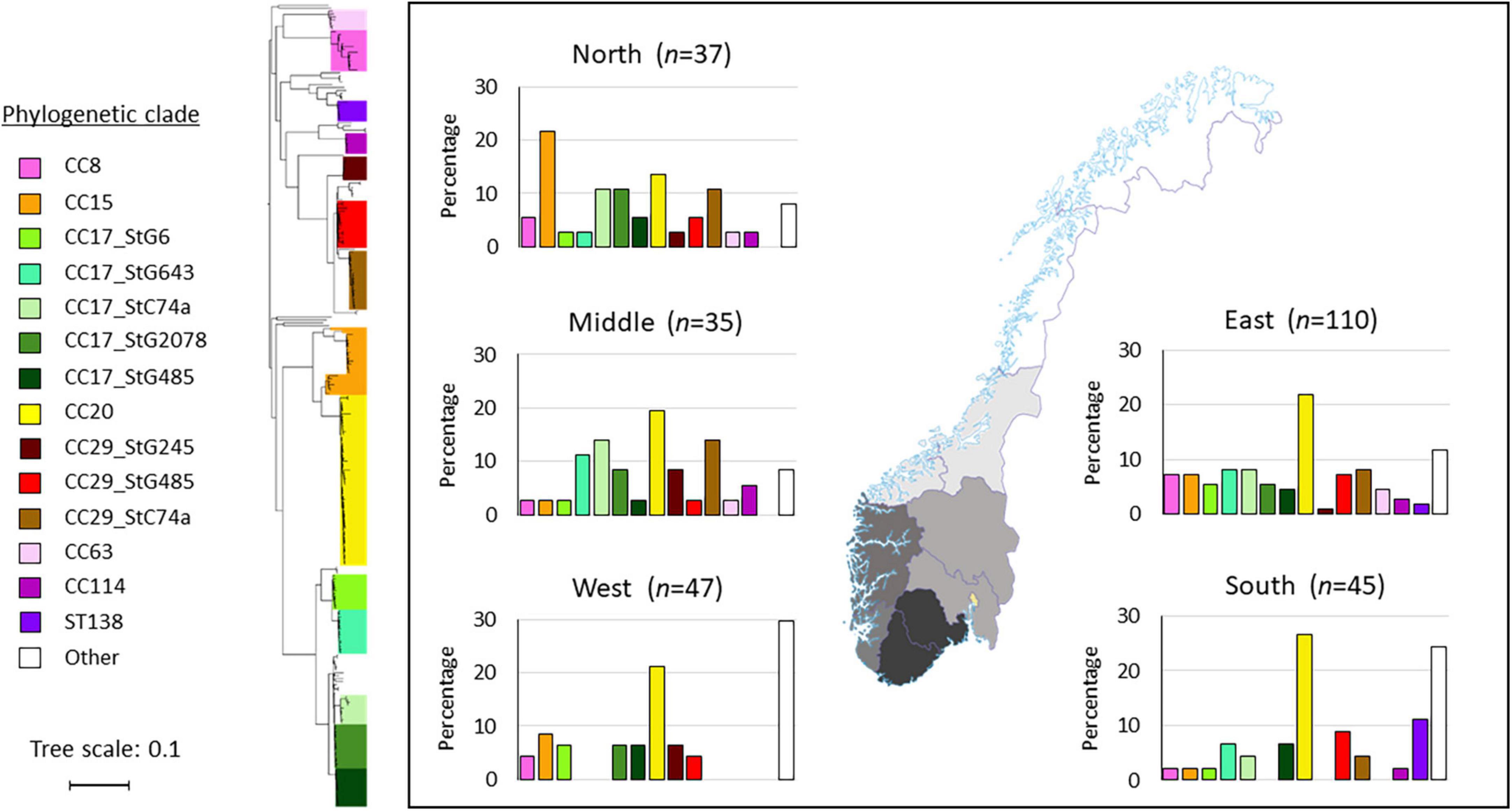
Figure 3. Distribution of Streptococcus dysgalactiae subspecies equisimilis (SDSE) clades and subclades in the five health regions in Norway. The geographic delineation of the regions is depicted on the map. In the histograms, the various clades and subclades are presented on the x-axis, and the y-axis represents the percentage of isolates within each region. The phylogenetic tree is constructed from single-nucleotide polymorphism (SNP)-analysis using the CSI-phylogeny website, and the clades and subclades were named based on multilocus sequence typing (MLST) and emm-type data. The tree scale bar indicates the estimated evolutionary divergence between isolates measured in substitutions per site.
Virulence profiling
To gain insight into genetic features promoting virulence, we investigated the presence of major SDSE-protein virulence factors (Table 1). All isolates harbored genes for the toxins Streptolysin O (slo), Streptolysin S (sagA-H), and Streptokinase (Skc), the adhesins lmb, fnbB, fbp54 and gapC, as well as the immune modulators C5a peptidase (scpA) and Protein G (prg). The superantigens ssa, smeZ, speA, speC, speH, speI, speJ, SpeK, speL, speM, speQ, and speR were universally absent among all isolates, and speG was the only streptococcal superantigen detected (present in 61% of the isolates). The canine-associated genome (stC9431) did not harbor slo, Skc, lmb, or scpA. However, it was equipped with the immunoglobulin cleaving enzyme EndoS2D (ndoS2D), which was absent from all the other SDSE isolates.
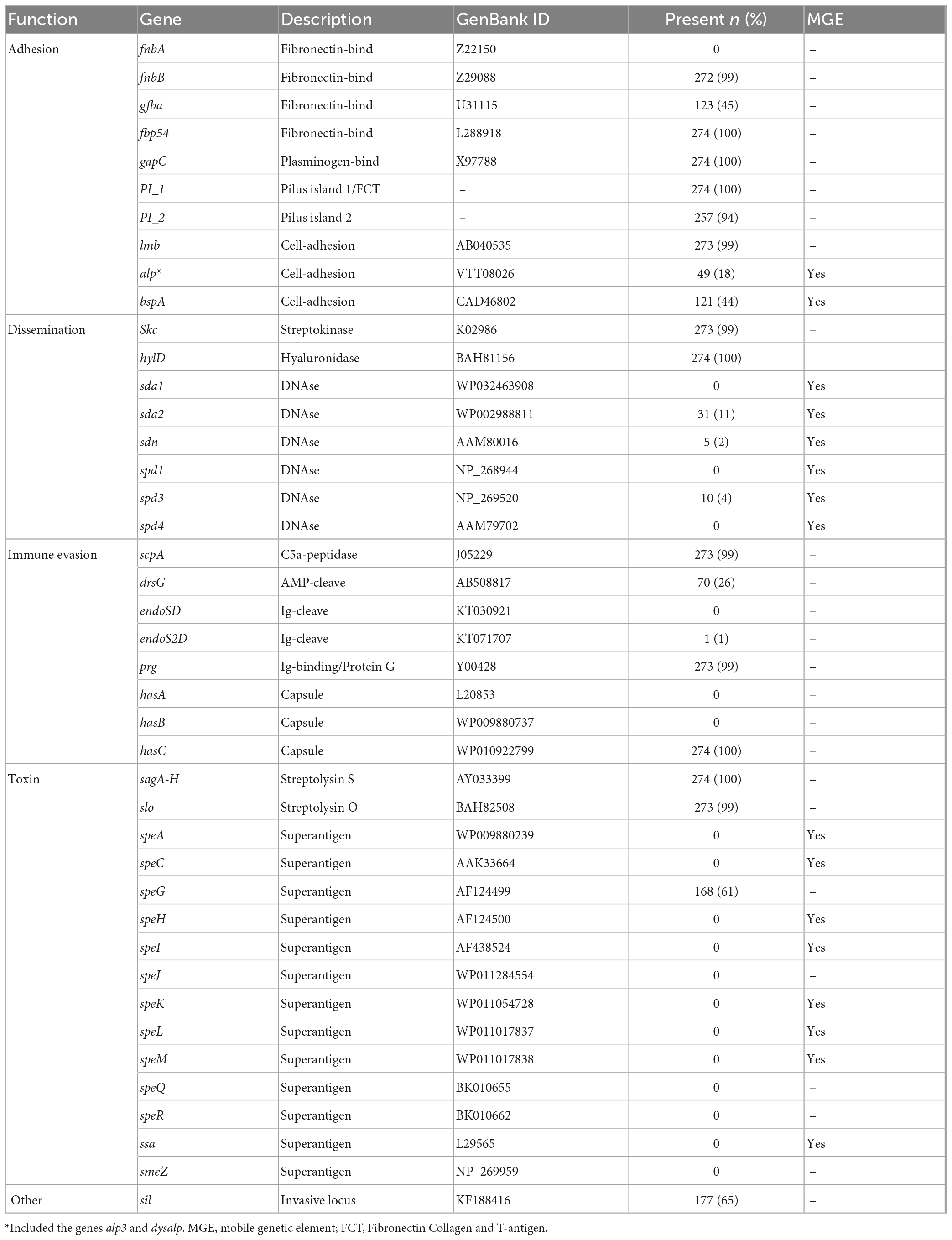
Table 1. Distribution of the virulence genes in 274 invasive Streptococcus dysgalactiae subspecies equisimilis (SDSE) isolates collected in Norway during 2018.
A few chromosomally located virulence factors had a variable presence, including drsG (28%), gfba (45%), Pilus island 2 (PI_2) (94%), and Streptococcus invasive locus (sil) (65%). Distribution of these genes was highly linked to phylogenetic origin. However, this was occasionally reflected at the subclade level, and not necessarily congruent with ST or emm-profiles (Figure 4A). Interestingly, the virulence gene drsG had a more variable presence even within phylogenetic subclades, in particular in isolates from CC17. Inspecting the C5a-peptidase regions in these closely related CC17 isolates, we detected an excision or insertion of the drsG gene in some genomes (Figure 5). The surrounding genetic context was virtually identical.
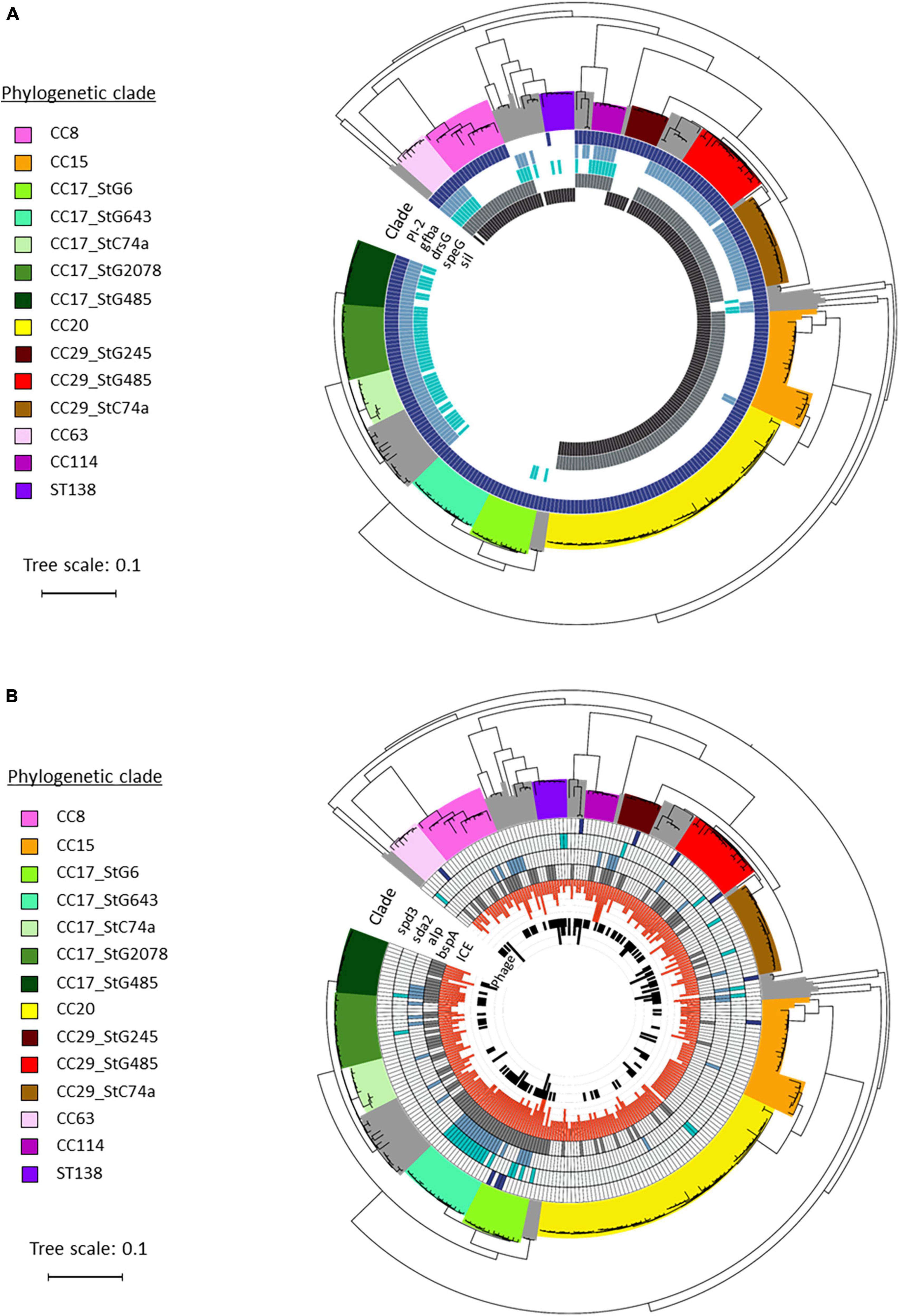
Figure 4. Distribution of virulence genes in blood culture Streptococcus dysgalactiae subspecies equisimilis (SDSE) isolates identified in Norway during 2018. Phylogenetic tree depicting the disposition of virulence genes among the SDSE isolates investigated in this study, and grouped in phylogenetic clades and subclades based on multilocus sequence typing (MLST) and emm-type data. The tree scale bar indicates the estimated evolutionary divergence between isolates measured in substitutions per site. (A) Distribution of chromosomal virulence factors, including Pilus island 2 PI_2, gfba, drsG, speG, and sil. (B) Distribution of virulence factors associated with mobile genetic elements, including phages and Integrative Conjugative Elements (ICEs). The phages associated DNAses spd3 and sda2, and ICEs associated adhesins alp and bspA were detected among the different SDSE isolates.
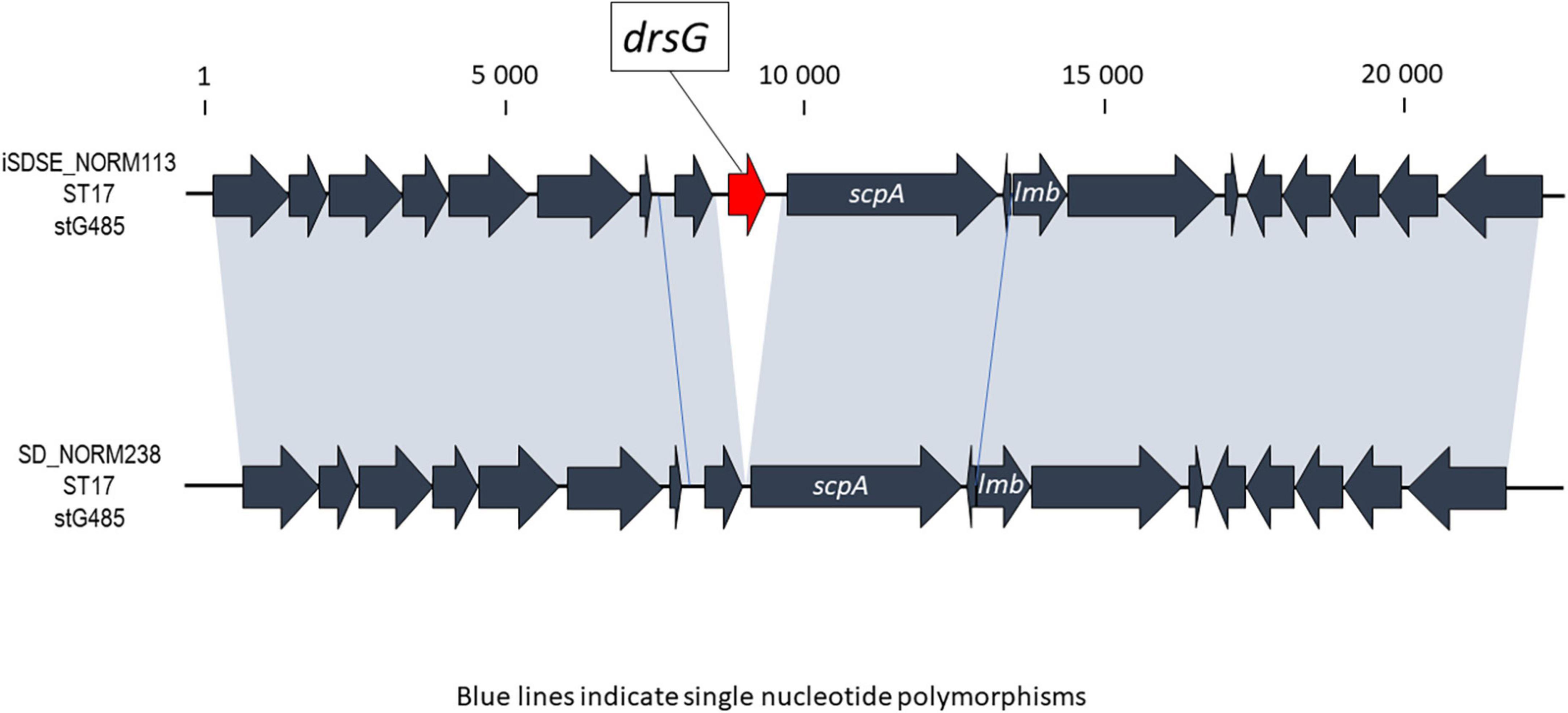
Figure 5. Graphic presentation of the event involving excision or insertion of the drsG gene in the C5a peptidase region of two phylogenetically closely related Streptococcus dysgalactiae subspecies equisimilis (SDSE) isolates. In the alignment, the drsG gene is marked in red, the gray shaded areas indicate 100% homology, and blue lines represent single nucleotide polymorphisms. The scale indicates number of base pairs.
Observed replacement of the Fibronectin, Collagen and T-antigen (FCT)-region (also known as the Pilus island-1 region) in addition to the emm gene (Supplementary Figure 2). These isolates had acquired a completely novel FCT-region harboring the gfba adhesin but were virtually identical to their phylogenetic ancestors in the surrounding genetic regions.
SDSE subspecies equisimilis isolates were also screened for mobile genetic elements (MGEs) and associated virulence genes (Table 1). The number of intact bacteriophages was low, on average 0.6 bacteriophages per genome (range 0–3). In line with this, we did not detect any phage-related superantigens, but some isolates harbored phages-associated DNAses, including sda2 (11%), spd3 (4%), and sdn (2%). Conversely, the prevalence of Integrative Conjugative Elements (ICEs) was relatively high among the SDSE isolates, with an average of 2.9 ICEs per genome (range 0–6). The ICE-associated virulence factors bspA and the alp-gene family, important for cell adhesion, were detected in 44% and 18% of the SDSE genomes, respectively.
Overall, these virulence genes carried by MGEs appeared to be randomly distributed, although an association with a few phylogenetic subclades was observed (Figure 4B).
Geographical impact of SDSE clonal complexes
Next, we examined the geographic distribution of invasive SDSE clades by comparing our ST profiles to whole genome sequenced isolate collections from Canada (comprising 89 invasive isolates from 2012 to 2014) (Lother et al., 2017) and Japan (111 invasive isolates from 2012 to 2019) (Shinohara et al., 2023). Among the SDSE isolates retrieved from Norway, Canada and Japan, 34 CCs were identified. The CC8, CC15, CC17, CC25, and CC29 clades were among the most prevalent in all three continents, indicating that several of the major genotypes of SDSE have an intercontinental distribution (Figure 6).
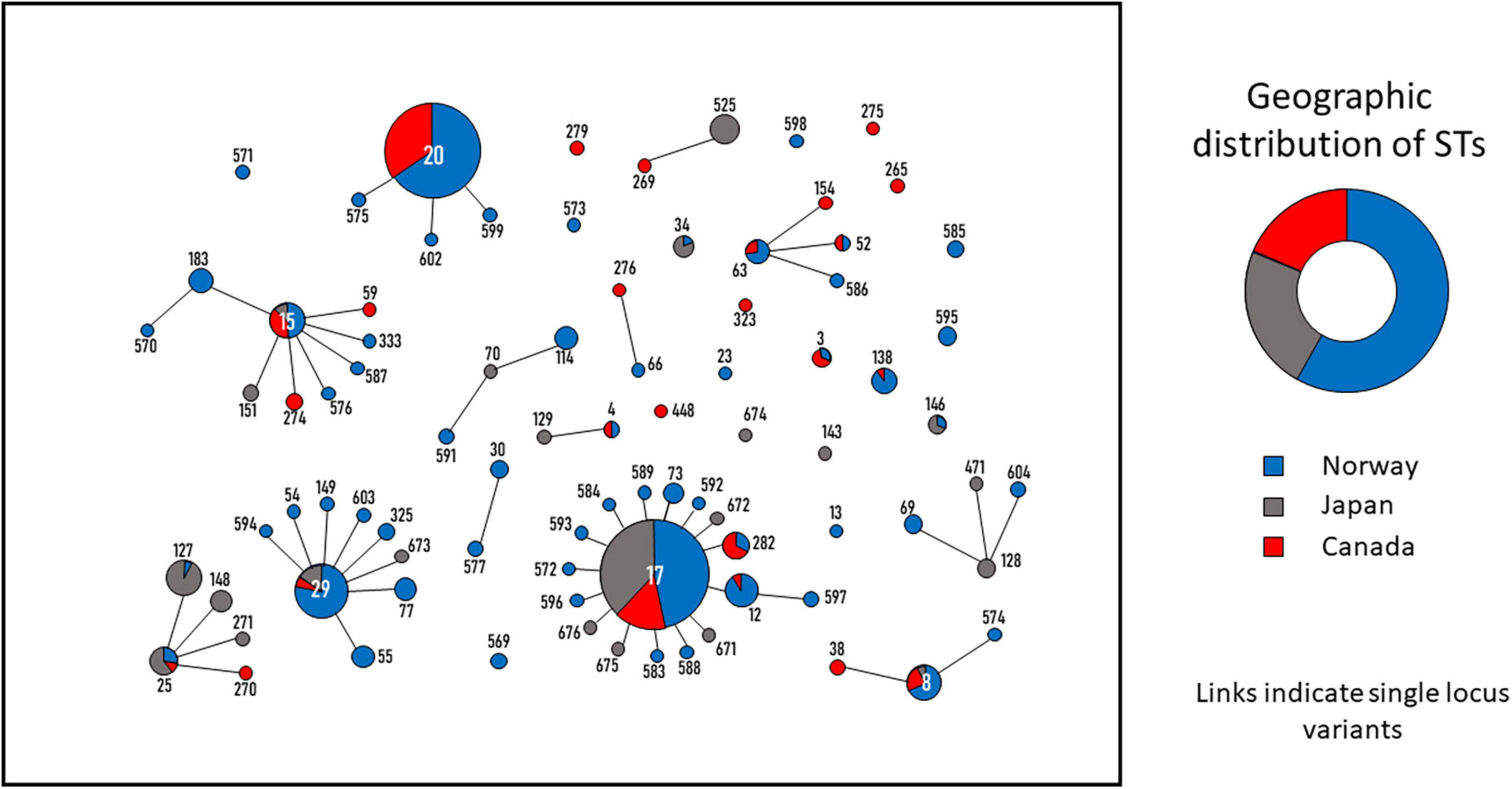
Figure 6. Minimum spanning tree depicting the genetic relatedness of Streptococcus dysgalactiae subspecies equisimilis (SDSE) blood culture isolates from Norway, Canada and Japan. The tree was constructed in Phyloviz. ST-profile and connections indicate single locus variants. The circles and numbers represent the individual sequence types (ST)-profiles. The size of the circles is proportional to the quantity of isolates belonging to each ST. The pie charts in the ST-circles indicate the percentage of isolates originating from the three countries. The donut chart shows the geographic distribution of all SDSE isolates included in the analysis. Blue = Norway; Red = Canada; Gray = Japan.
Interestingly, CC20, which had the highest prevalence in Norway and Canada, was not present in the invasive isolate collection from Japan. Conversely, CC25 is frequent in Japan, whereas very few isolates of this CC were recovered in Norway and Canada. In general, the distribution of ST-profiles circulating in Norway and Canada was quite similar, whereas Japan seemed to have a more diverging phylogenetic SDSE population (Figure 6 and Supplementary Table 2).
Discussion
Streptococcus dysgalactiae subspecies equisimilis (SDSE) is increasingly recognized as an emerging human pathogen implicated in invasive infections worldwide, yet the evolutionary mechanisms behind this development are still largely unknown. In this study, we characterized the genomic features of 274 invasive SDSE isolates recovered from a nationwide-based surveillance system in Norway during 2018. To the best of our knowledge, this is the first large in-depth genomic characterization by WGS of invasive SDSE isolates from a confined temporal and geographic population-based setting.
We observed a substantial diversity of emm-types and STs, as well as an imperfect association between emm-type and CC, indicating a high level of genomic plasticity in this species. Horizontal genetic transfer of the emm gene has been postulated to be the major driving force behind this population diversity in SDSE, but whole genome sequence data to support this has been lacking (Ahmad et al., 2009; McMillan et al., 2010, 2011). In the present study, we document several likely recombination events involving the emm gene in phylogenetically closely related SDSE isolates (Figure 2). Similar emm gene replacements have been noted in S. pyogenes in recent large-scale WGS studies (Chochua et al., 2017; Buckley et al., 2020), but the specific mechanisms involved in these horizontal genetic transfer events have not been elucidated. We did not detect any transposons or other mobile genetic elements in the vicinity, and transformation is the most likely mode of acquisition. However, an ability for natural transformation in SDSE and S. pyogenes remains to be shown (Mashburn-Warren et al., 2012).
Moreover, replacement events involving the FCT regions were also recorded between isolates. Recombination events in this locus have been reported, but predominantly in the form of intragenic mosaicism (McNeilly and Mcmillan, 2014). The FCT-region encodes the pilus apparatus (including the T-antigen) in S. pyogenes and SDSE, and mediates adhesion to host cells (Mora et al., 2005). Both the T-antigen and the M protein elicit strong antibody responses in humans, and replacement of these domains likely confers a selection advantage for the pathogen through immune escape (Mora et al., 2005). Recombination and exchange of these major immunogenic determinants thus appears to be important in shaping the evolution of the S. pyogenes and SDSE genomes.
In line with this, continuous replacement of circulating emm-types has been noted in S. pyogenes, and several outbreaks with novel virulent S. pyogenes clades have been characterized (Colman et al., 1993; Sanson et al., 2015; Zhu et al., 2015). The population dynamics of SDSE have been studied to a lesser extent, but the emergence of the CC20 clade harboring the stG62647 emm-type was reported in Norway few years ago (Oppegaard et al., 2017a). This clade has continued to expand, and it was the most prevalent genotype observed in the present study. The stG62647 emm-type appears to have gained a strong foothold also in other European countries, and recent studies in Austria (Leitner et al., 2015), Spain (Rojo-Bezares et al., 2021), Germany (Rößler et al., 2018), and Denmark (Lambertsen et al., 2014) all reported stG62647 as the predominant circulating emm-type. Moreover, the CC20_stG62647 clade comprised 31% of the SDSE isolates in Canada during 2012–2014, suggesting an intercontinental spread (Lother et al., 2017). In contrast, both CC20 and stG62647 are infrequent in Japan, and the CC17_stG6792 has been reported as the major clade in this region since 2003 (Wajima et al., 2016; Ikebe et al., 2018).
The CC20 was characterized by a high genetic homogeneity in our study, likely indicating a relatively novel clade. It was almost exclusively associated with the stG62647 emm-type, and only two isolates had single allele mutations giving rise to novel STs within the clonal complex. Differently, the CC17 and CC29 comprised several STs, and were associated with numerous emm-types, suggesting more established CCs (Figure 1). Nevertheless, the CC17 and CC29 isolates were distributed in well-separated emm-associated subclades within the clonal complexes, likely reflecting an emm switch event in a distant ancestor that has subsequently evolved into successful subclades over time. As such, neither emm-typing nor ST-profiling accurately captures the complexity of the phylogenetic relationships in SDSE. Insufficient phylogenetic resolution of these traditional typing tools has previously also been noted in S. pyogenes, highlighting the need for whole genome analyses to unravel the finer details of epidemiological trends in bacteriology (Davies et al., 2019).
Studying the distribution of clades within Norway, there were notable differences even in this confined geographic and temporal setting (Figure 3). Similarly, comparing the SDSE phylogeny in Norway to Japan and Canada, we observed several intercontinental disseminated CCs, but also distinct geographic variations (Figure 6). Taken together, our findings reflect the dynamic evolution of the SDSE population, where continuous rise and fall of novel lineages leads to dissemination of successful clones, but also large temporospatial differences in circulating SDSE strains.
Similar to previous reports, we found that the majority of virulence factors previously characterized in SDSE were ubiquitous, including genes involved in adhesion (lmb, fnbB, fbp54, and gapC), toxins (slo, sagA-H, and Skc) and immune invasion (scpA and prg) (Rantala, 2014; Wajima et al., 2016; Lother et al., 2017; Oppegaard et al., 2017b; Rößler et al., 2018; Rojo-Bezares et al., 2021). However, for the few chromosomally encoded virulence genes with variable presence, we observed clonally restricted virulence profiles in the SDSE isolates (Figure 4A), a finding that was also reported in a recent whole genome sequencing study from Canada (Lother et al., 2017).
Several studies have noted an association between the superantigen speG and certain emm-types, including stG62647 and stC74a (Kittang et al., 2011; Leitner et al., 2015; Oppegaard et al., 2017b). We found speG to be present in isolates of the stC74a emm-type belonging to CC29, but not to CC17 (Figure 4A). Moreover, speG was absent in CC29 isolates harboring the stG245 emm-type, indicating a distribution in accordance with the phylogenetic subclades, but not necessarily in accordance with emm-type nor CC. This infers that recombination events involving acquisition or loss of speG gene have occurred in the evolution of these subclades.
In line with this, we observed several events of insertion or deletion of the drsG gene in closely related CC17 isolates (Figure 5), highlighting that recombination events also are apparent in SDSE in virulence genes other than the emm gene. DrsG is a homolog of streptococcal inhibitor of complement (SIC) and distantly related SIC (DRS) proteins secreted by S. pyogenes, playing an important role in inhibiting complement-mediated lysis and/or the activity of certain antimicrobial peptides (McNeilly and Mcmillan, 2014). In SDSE, this gene resides next to the scpA gene, a region that has been demonstrated to have mobility (Franken et al., 2001). The drsG gene itself it is not surrounded by any insertion sequences or mobile elements, though, and the mechanisms promoting the observed excision or insertion events are unclear. Its role as virulence factors in in certain SDSE associated emm-types remains to be elucidated.
In contrast to the clonally restricted virulence profiles of chromosomally encoded genes, virulence factors transferred by mobile elements appeared to have a random distribution (Figure 4B). Nevertheless, transduction of phages and conjugation of ICEs between bacterial cells play an important role in bacterial evolution and adaption, both for escaping selective pressure and as an additional means of horizontal genetic exchange and virulence enhancement. Screening of genomes for such mobile elements in our study (Figure 4B and Table 1) revealed a higher presence of ICEs with an average of 2.9 per SDSE genome, in contrast to a low prevalence of bacteriophages among the SDSE isolates (here 0.6 bacteriophages/genome). This was reflected in the high prevalence of the ICE-associated virulence factors bspA and the alp-gene family among our strains, and fewer phage-associated virulence factors (Table 1). In line with our findings, a comprehensive search for ICEs in streptococcal species by Ambroset et al. (2015) showed also a relatively higher prevalence of ICEs among S. dysgalactiae isolates (2.0 per genome), while the prevalence of ICEs in S. pyogenes remained low with an average of only 0.3 ICEs per genome. According to previous studies, the presence of particular cargo genes in ICEs protects the cells against other mobile elements. More specifically, in Bacillus subtilis for instance, the presence of a single gene in the integrative and conjugative element ICEBs1 was shown to have an important role in repressing the adaption of bacteriophages, providing selective pressure for the host cells to maintain the presence of ICEs (Johnson et al., 2022). An attractive hypothesis is therefore that the strong presence of ICEs in SDSE genomes might reduce the number of bacteriophages in the cells. Nevertheless, more studies on the interplay between phage, ICE and microbe are needed to explore the evolutionary contribution of mobile genetic elements in SDSE.
The confined geographic and temporal setting of our study limits the ability to study temporal dynamics of the SDSE population. Moreover, the study period of the WGS studies from Japan and Canada did not completely overlap that in our study, and a potential impact of temporal shifts in circulating genotypes cannot be ruled out. However, our study included all blood culture isolates in Norway in 2018, and as such provides a very accurate and comprehensive snapshot of the SDSE epidemiology in our country. The geographical differences in the distribution of clades across Norway might be random since a limited sample size and insufficient power, did not allow for any statistical assessment. Yet, the extensive similarities with emm- and ST-profiles previously published indicated that our results also reflect the epidemiologic situation in other European countries.
Conclusion
The genetic landscape of SDSE appears to be continuously shaped by mutations and horizontal genetic transfer events, possibly driven by host-pathogen interactions and selection pressure. This genomic evolution fuels the temporal and geographic dynamics of the SDSE population, and gives rise to novel successful lineages such as the CC20_stG62647 clade. Traditional genotypic markers often lack sufficient phylogenetic resolution to capture these evolutionary events, and WGS has become a powerful tool for elucidating bacterial population dynamics.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving human participants were reviewed and approved by Western Norway Regional Ethics Committee (2021/63132). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
MS, OO, and CMJ: conceptualization and supervision. AK and OO: methodology, software, and formal analysis. OO: data curation and visualization. AK: writing—original draft preparation. AK, CMJ, SS, AS, and OO: writing—review and editing. MS, SS, CMJ, and OO: project administration. All authors have read and agreed to the published version of the manuscript.
The NORWEGIAN Study Group on Streptococcus dysgalactiae Members
Aasmund Fostervold, Department of Clinical Microbiology, Stavanger University Hospital, Stavanger, Norway; Aleksandra Jakovljev, Department of Microbiology, St. Olav’s University Hospital, Trondheim, Norway; Annette Onken, Department of Clinical Microbiology, Vestre Viken Hospital Trust, Baerum, Norway; Åshild Marvik, Department of Microbiology, Vestfold Hospital Trust, Tønsberg, Norway; Einar Nilsen, Department of Microbiology, Møre and Romsdal Hospital Trust, Ålesund, Norway; Fredrik Müller, Department of Microbiology, Oslo University Hospital, Oslo, Norway; Ghantous Milad Chedid, Laboratory for Clinical Microbiology, Fonna Hospital Trust, Haugesund, Norway; Gunnar Skov Simonsen, Department of Clinical Microbiology, University Hospital of North Norway, Tromsø, Norway; Kyriakos Zaragkoulias, Department of Clinical Microbiology, Nord-Trøndelag Hospital Trust, Levanger, Norway; Reidar Hjetland, Department of Microbiology, Division of Medicine, District General Hospital of Førde, Førde, Norway; Roar Magne Baevre-Jensen, Department of Clinical Microbiology, Vestre Viken Hospital Trust, Drammen, Norway; Sandra Åsheim, Unit for Clinical Microbiology, Norland Hospital Trust, Bodø, Norway; Ståle Tofteland, Department of Clinical Microbiology, Sørlandet Hospital Trust, Agder, Norway; Tine Smedsund Dons, Department of Clinical Microbiology, Innlandet Hospital Trust, Lillehammer, Norway; Truls Michael Leegaard, Department of Microbiology and Infection Control, Akerhus University Hospital, Lørenskog, Norway.
Funding
This work was supported by grants from the Norwegian surveillance program for antimicrobial resistance (NORM) (Grant number: 19/16 and 20/02 to CJ and MS, as well as OO) and Center for Laboratory Medicine at Østfold Hospital Trust. The genomics Core Facility (GCF) at the University of Bergen, which is part of the NorSeq consortium provided services on the whole genome sequencing in this study; GCF is supported by major grants from the Research Council of Norway (grant no. 245979/F50) and Trond Mohn Foundation (grant no. BFS2017TMT04/BFS2017TMT08). This study was received also financial contributions from the Western Norway Regional Health Authority (grant no. 912231). SS received grants from the Norwegian Research Council under the frames of NordForsk (Project: 90456, PerAID) and ERA PerMed (Project 2018-151, PerMIT).
Acknowledgments
We thank Anne Cathrine Hollekim particularly, and the rest of the staff at the Bacteriological Department at the Center for Laboratory Medicine at Østfold Hospital Trust for their technical support in reviving and culturing bacterial isolates. We thank the staff at the Microbiological Department at Haukeland University Hospital for excellent technical assistance and for providing access to their laboratory facilities. We acknowledge Haima Mylvaganam for her significant contribution to streptococcal research at Haukeland University Hospital during the past two decades, and for inspiration to continued scientific exploration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1171913/full#supplementary-material
Supplementary Figure 1 | Whole genome single-nucleotide polymorphism (SNP)-derived phylogenetic tree of invasive Streptococcus dysgalactiae subspecies equisimilis (SDSE) obtained in Norway during 2018. N = 274. Here the canine-associated genome stC9431 is included and highlighted in red. This tree was constructed with CSI phylogeny website. Scale bar indicates the estimated evolutionary divergence between isolates measured in substitution per site.
Supplementary Figure 2 | The Fibronectin, collagen and T-antigen (FCT) region, also known as Pilus island-1 region, harboring the gfba adhesin observed between two phylogenetically closely related isolates. The comparison was performed using Geneious alignment. The FCT-region in each Streptococcus dysgalactiae subspecies equisimilis (SDSE) isolate is highlighted in orange. The gray shaded areas indicate 100% homology, and blue lines in the alignment indicate single nucleotide polymorphisms. The scale indicates number of base pairs.
Footnotes
- ^ https://www.ssb.no/
- ^ https://www.cdc.gov/streplab/groupa-strep/emm-background.html
- ^ https://pubmlst.org/
- ^ https://cge.cbs.dtu.dk/services
- ^ http://www.ebi.ac.uk/ena/data/view/ERS1066625
- ^ www.mgc.ac.cn/VFs
References
Ahmad, Y., Gertz, R. E. Jr., Li, Z., Sakota, V., Broyles, L. N., Van Beneden, C., et al. (2009). Genetic relationships deduced from emm and multilocus sequence typing of invasive Streptococcus dysgalactiae subsp. equisimilis and S. canis recovered from isolates collected in the United States. J. Clin. Microbiol. 47, 2046–2054. doi: 10.1128/JCM.00246-09
Ambroset, C., Coluzzi, C., Guédon, G., Devignes, M. D., Loux, V., Lacroix, T., et al. (2015). New insights into the classification and integration specificity of Streptococcus integrative conjugative elements through extensive genome exploration. Front. Microbiol. 6:1483. doi: 10.3389/fmicb.2015.01483
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477.
Bisno, A. L., Craven, D. E., and Mccabe, W. R. (1987). M proteins of group G streptococci isolated from bacteremic human infections. Infect. Immun. 55, 753–757. doi: 10.1128/iai.55.3.753-757.1987
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120.
Brandt, C. M., Haase, G., Schnitzler, N., Zbinden, R., and Lutticken, R. (1999). Characterization of blood culture isolates of Streptococcus dysgalactiae subsp. equisimilis possessing Lancefield’s group A antigen. J. Clin. Microbiol. 37, 4194–4197. doi: 10.1128/JCM.37.12.4194-4197.1999
Brandt, C. M., and Spellerberg, B. (2009). Human infections due to Streptococcus dysgalactiae subspecies equisimilis. Clin. Infect. Dis. 49, 766–772. doi: 10.1086/605085
Broyles, L. N., Van Beneden, C., Beall, B., Facklam, R., Shewmaker, P. L., Malpiedi, P., et al. (2009). Population-based study of invasive disease due to beta-hemolytic streptococci of groups other than A and B. Clin. Infect. Dis. 48, 706–712. doi: 10.1086/597035
Bruun, T., Oppegaard, O., Kittang, B. R., Mylvaganam, H., Langeland, N., and Skrede, S. (2016). Etiology of cellulitis and clinical prediction of streptococcal disease: a prospective study. Open Forum Infect. Dis. 3:ofv181.
Buckley, S. J., Davies, M. R., and Mcmillan, D. J. (2020). In silico characterisation of stand-alone response regulators of Streptococcus pyogenes. PLoS One 15:e0240834. doi: 10.1371/journal.pone.0240834
Chochua, S., Metcalf, B. J., Li, Z., Rivers, J., Mathis, S., Jackson, D., et al. (2017). Population and whole genome sequence based characterization of invasive group a streptococci recovered in the United States during 2015. mBio 8:e01422-17. doi: 10.1128/mBio.01422-17
Colman, G., Tanna, A., Efstratiou, A., and Gaworzewska, E. T. (1993). The serotypes of Streptococcus pyogenes present in britain during 1980-1990 and their association with disease. J. Med. Microbiol. 39, 165–178. doi: 10.1099/00222615-39-3-165
Davies, M. R., Mcintyre, L., Mutreja, A., Lacey, J. A., Lees, J. A., Towers, R. J., et al. (2019). Atlas of group a streptococcal vaccine candidates compiled using large-scale comparative genomics. Nat. Genet. 51, 1035–1043.
Franken, C., Haase, G., Brandt, C., Weber-Heynemann, J., Martin, S., Lämmler, C., et al. (2001). Horizontal gene transfer and host specificity of beta-haemolytic streptococci: the role of a putative composite transposon containing scpB and lmb. Mol. Microbiol. 41, 925–935. doi: 10.1046/j.1365-2958.2001.02563.x
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075.
Harris, P., Siew, D. A., Proud, M., Buettner, P., and Norton, R. (2011). Bacteraemia caused by beta-haemolytic streptococci in North Queensland: changing trends over a 14-year period. Clin. Microbiol. Infect. 17, 1216–1222. doi: 10.1111/j.1469-0691.2010.03427.x
Ikebe, T., Okuno, R., Sasaki, M., Kanda, Y., Otsuka, H., Kawahara, R., et al. (2018). Molecular characterization and antibiotic resistance of Streptococcus dysgalactiae subspecies equisimilis isolated from patients with streptococcal toxic shock syndrome. J. Infect. Chemother. 24, 117–122. doi: 10.1016/j.jiac.2017.09.013
Johnson, C. M., Harden, M. M., and Grossman, A. D. (2022). Interactions between mobile genetic elements: an anti-phage gene in an integrative and conjugative element protects host cells from predation by a temperate bacteriophage. PLoS Genet. 18:e1010065. doi: 10.1371/journal.pgen.1010065
Kittang, B. R., Skrede, S., Langeland, N., Haanshuus, C. G., and Mylvaganam, H. (2011). emm gene diversity, superantigen gene profiles and presence of SlaA among clinical isolates of group A, C and G streptococci from western Norway. Eur. J. Clin. Microbiol. Infect. Dis. 30, 423–433. doi: 10.1007/s10096-010-1105-x
Lambertsen, L. M., Ingels, H., Schønheyder, H. C., and Hoffmann, S. (2014). Nationwide laboratory-based surveillance of invasive beta-haemolytic streptococci in Denmark from 2005 to 2011. Clin. Microbiol. Infect. 20, O216–O223. doi: 10.1111/1469-0691.12378
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Leitner, E., Zollner-Schwetz, I., Zarfel, G., Masoud-Landgraf, L., Gehrer, M., Wagner-Eibel, U., et al. (2015). Prevalence of emm types and antimicrobial susceptibility of Streptococcus dysgalactiae subsp. equisimilis in Austria. Int. J. Med. Microbiol. 305, 918–924. doi: 10.1016/j.ijmm.2015.10.001
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lother, S. A., Demczuk, W., Martin, I., Mulvey, M., Dufault, B., Lagace-Wiens, P., et al. (2017). Clonal clusters and virulence factors of group c and g streptococcus causing severe infections, Manitoba, Canada, 2012-2014. Emerg. Infect. Dis. 23, 1079–1088. doi: 10.3201/eid2307.161259
Lytsy, B., Engstrand, L., Gustafsson, A., and Kaden, R. (2017). Time to review the gold standard for genotyping vancomycin-resistant enterococci in epidemiology: comparing whole-genome sequencing with PFGE and MLST in three suspected outbreaks in Sweden during 2013-2015. Infect. Genet. Evol. 54, 74–80. doi: 10.1016/j.meegid.2017.06.010
Mashburn-Warren, L., Morrison, D. A., and Federle, M. J. (2012). The cryptic competence pathway in Streptococcus pyogenes is controlled by a peptide pheromone. J. Bacteriol. 194, 4589–4600. doi: 10.1128/JB.00830-12
McMillan, D. J., Bessen, D. E., Pinho, M., Ford, C., Hall, G. S., Melo-Cristino, J., et al. (2010). Population genetics of Streptococcus dysgalactiae subspecies equisimilis reveals widely dispersed clones and extensive recombination. PLoS One 5:e11741. doi: 10.1371/journal.pone.0011741
McMillan, D. J., Kaul, S. Y., Bramhachari, P. V., Smeesters, P. R., Vu, T., Karmarkar, M. G., et al. (2011). Recombination drives genetic diversification of Streptococcus dysgalactiae subspecies equisimilis in a region of streptococcal endemicity. PLoS One 6:e21346. doi: 10.1371/journal.pone.0021346
McNeilly, C. L., and Mcmillan, D. J. (2014). Horizontal gene transfer and recombination in Streptococcus dysgalactiae subsp. equisimilis. Front. Microbiol. 5:676. doi: 10.3389/fmicb.2014.00676
Mora, M., Bensi, G., Capo, S., Falugi, F., Zingaretti, C., Manetti, A. G., et al. (2005). Group a Streptococcus produce pilus-like structures containing protective antigens and lancefield T antigens. Proc. Natl. Acad. Sci. U S A. 102, 15641–15646. doi: 10.1073/pnas.0507808102
NORM/NORM-VET, (2018). Usage of Amtimicrobial Agents and Occurrence of Antimicrobial Resistance in Norway. Tromsø/Oslo 2019. ISSN:1502-2307 (print)/1890-9965 (electronic).
Oppegaard, O., Glambek, M., Skutlaberg, D. H., Skrede, S., Sivertsen, A., and Kittang, B. R. (2023). Streptococcus dysgalactiae bloodstream infections, Norway, 1999-2021. Emerg. Infect. Dis. 29, 260–267. doi: 10.3201/eid2902.221218
Oppegaard, O., Mylvaganam, H., and Kittang, B. R. (2015). Beta-haemolytic group A, C and G streptococcal infections in Western Norway: a 15-year retrospective survey. Clin. Microbiol. Infect. 21, 171–178. doi: 10.1016/j.cmi.2014.08.019
Oppegaard, O., Mylvaganam, H., Skrede, S., Jordal, S., Glambek, M., and Kittang, B. R. (2017a). Clinical and molecular characteristics of infective beta-hemolytic streptococcal endocarditis. Diagn. Microbiol. Infect. Dis. 89, 135–142. doi: 10.1016/j.diagmicrobio.2017.06.015
Oppegaard, O., Mylvaganam, H., Skrede, S., Lindemann, P. C., and Kittang, B. R. (2017b). Emergence of a Streptococcus dysgalactiae subspecies equisimilis stG62647-lineage associated with severe clinical manifestations. Sci. Rep. 7:7589. doi: 10.1038/s41598-017-08162-z
Oppegaard, O., Skrede, S., Mylvaganam, H., and Kittang, B. R. (2016). Temporal trends of β-haemolytic streptococcal osteoarticular infections in western Norway. BMC Infect. Dis. 16:535. doi: 10.1186/s12879-016-1874-7
Porcellato, D., Smistad, M., Skeie, S. B., Jørgensen, H. J., Austbø, L., and Oppegaard, O. (2021). Whole genome sequencing reveals possible host species adaptation of Streptococcus dysgalactiae. Sci. Rep. 11:17350. doi: 10.1038/s41598-021-96710-z
Rantala, S. (2014). Streptococcus dysgalactiae subsp. equisimilis bacteremia: an emerging infection. Eur. J. Clin. Microbiol. Infect. Dis. 33, 1303–1310. doi: 10.1007/s10096-014-2092-0
Rantala, S., Vuopio-Varkila, J., Vuento, R., Huhtala, H., and Syrjänen, J. (2009). Clinical presentations and epidemiology of beta-haemolytic streptococcal bacteraemia: a population-based study. Clin. Microbiol. Infect. 15, 286–288. doi: 10.1111/j.1469-0691.2008.02672.x
Reuter, S., Ellington, M. J., Cartwright, E. J., Köser, C. U., Török, M. E., Gouliouris, T., et al. (2013). Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern. Med. 173, 1397–1404. doi: 10.1001/jamainternmed.2013.7734
Ribeiro-Gonçalves, B., Francisco, A. P., Vaz, C., Ramirez, M., and Carriço, J. A. (2016). PHYLOViZ Online: web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 44, W246–W251. doi: 10.1093/nar/gkw359
Rojo-Bezares, B., Toca, L., Azcona-Gutiérrez, J. M., Ortega-Unanue, N., Toledano, P., and Sáenz, Y. (2021). Streptococcus dysgalactiae subsp. equisimilis from invasive and non-invasive infections in Spain: combining epidemiology, molecular characterization, and genetic diversity. Eur. J. Clin. Microbiol. Infect. Dis. 40, 1013–1021. doi: 10.1007/s10096-020-04119-9
Rößler, S., Berner, R., Jacobs, E., and Toepfner, N. (2018). Prevalence and molecular diversity of invasive Streptococcus dysgalactiae and Streptococcus pyogenes in a German tertiary care medical centre. Eur. J. Clin. Microbiol. Infect. Dis. 37, 1325–1332. doi: 10.1007/s10096-018-3254-2
Sanson, M., O’neill, B. E., Kachroo, P., Anderson, J. R., Flores, A. R., Valson, C., et al. (2015). A naturally occurring single amino acid replacement in multiple gene regulator of group a Streptococcus significantly increases virulence. Am. J. Pathol. 185, 462–471. doi: 10.1016/j.ajpath.2014.10.018
Schwartz, I. S., Keynan, Y., Gilmour, M. W., Dufault, B., and Lagacé-Wiens, P. (2014). Changing trends in β-hemolytic streptococcal bacteremia in Manitoba, Canada: 2007-2012. Int. J. Infect. Dis. 28, 211–213.
Shinohara, K., Murase, K., Tsuchido, Y., Noguchi, T., Yukawa, S., Yamamoto, M., et al. (2023). Clonal expansion of multidrug-resistant Streptococcus dysgalactiae subspecies equisimilis Causing Bacteremia, Japan, 2005-2021. Emerg. Infect. Dis. 29, 528–539. doi: 10.3201/eid2903.221060
Wajima, T., Morozumi, M., Hanada, S., Sunaoshi, K., Chiba, N., Iwata, S., et al. (2016). Molecular characterization of invasive Streptococcus dysgalactiae subsp. equisimilis, Japan. Emerg. Infect. Dis. 22, 247–254. doi: 10.3201/eid2202.141732
Wong, S. S., Lin, Y. S., Mathew, L., Rajagopal, L., and Sepkowitz, D. (2009). Increase in group G streptococcal infections in a community hospital, New York, USA. Emerg. Infect. Dis. 15, 991–993. doi: 10.3201/eid1506.080666
Keywords: whole genome sequencing (WGS), SDSE, surveillance studies, genomics, horizontal genetic transfer
Citation: Kaci A, Jonassen CM, Skrede S, Sivertsen A, The Norwegian Study Group on Streptococcus dysgalactiae, Steinbakk M and Oppegaard O (2023) Genomic epidemiology of Streptococcus dysgalactiae subsp. equisimilis strains causing invasive disease in Norway during 2018. Front. Microbiol. 14:1171913. doi: 10.3389/fmicb.2023.1171913
Received: 22 February 2023; Accepted: 31 March 2023;
Published: 17 April 2023.
Edited by:
Jean-Philippe Rasigade, Université Claude Bernard Lyon 1, FranceCopyright © 2023 Kaci, Jonassen, Skrede, Sivertsen, The Norwegian Study Group on Streptococcus dysgalactiae, Steinbakk and Oppegaard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oddvar Oppegaard, b2RkdmFyLm9wcGVnYWFyZEBoZWxzZS1iZXJnZW4ubm8=
†Deceased