 Abdul Samad
Abdul Samad Dani Degenhardt
Dani Degenhardt Armand Séguin
Armand Séguin Marie-Josée Morency1
Marie-Josée Morency1 Christine Martineau
Christine Martineau
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 July 2023
Sec. Microbiotechnology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1168653
This article is part of the Research Topic Trends of Microbial Technologies in Rehabilitation of Contaminated Environments View all 6 articles
The oil sands mining operations in Alberta have produced billions of m3 of tailings which must be reclaimed and integrated into various mine closure landforms, including terrestrial landforms. Microorganisms play a central role in nutrient cycling during the reclamation of disturbed landscapes, contributing to successful vegetation restoration and long-term sustainability. However, microbial community succession and response in reconstructed and revegetated tailings remain largely unexplored. This study aimed to monitor the structural and functional responses of microbial communities in tailings subjected to different capping and vegetation strategies over two growing seasons (GS). To achieve this, a column-based greenhouse experiment was conducted to investigate microbial communities in tailings that were capped with a layer (10 or 30 cm) of peat-mineral mix (PMM) and planted with either upland or wetland communities. DNA metabarcoding analysis of the bacterial 16S rRNA gene and fungal ITS2 region as well as shotgun metagenomics were used to asses the impact of treatments on microbial taxonomy and functions, respectively. Results showed that tailings microbial diversity and community composition changed considerably after two GS compared to baseline samples, while communities in the PMM capping layer were much more stable. Likewise, several microbial functions were significantly enriched in tailings after two GS. Interestingly, the impact of capping on bacterial communities in tailings varied depending on the plant community, leading to a higher number of differentially abundant taxa and to a decrease in Shannon diversity and evenness in the upland treatment but not in the wetland treatment. Moreover, while capping in the presence of wetland vegetation increased the energy-related metabolic functions (carbon, nitrogen, and sulfur), these functions were depleted by capping in the upland treatment. Fungi represented a small proportion of the microbial community in tailings, but the relative abundance of several taxa changed over time, while the capping treatments favored the growth of some beneficial taxa, notably the root endophyte Serendipita, in both upland and wetland columns. The results suggest that selecting the right combination of capping material and vegetation type may contribute to improve below-ground microbial processes and sustain plant growth in harsh environments such as oil sands tailings.
Oil sands surface mining in northern Alberta has generated over a billion m3 of a waste stream called tailings stored temporarily in tailings ponds which must be reclaimed to an equivalent land capability to the pre-disturbance landscape, as directed by environmental regulations (Chalaturnyk et al., 2002; Parrott et al., 2019; Cossey et al., 2021). Fluid tailings are composed of water and solids, typically consisting of sand, silt, clay and residual bitumen. The terrestrial reclamation can begin after sufficient consolidation of fluid tailings streams. Oil sands mine operators use various technologies to facilitate dewatering and consolidation of fluid tailings (Wang et al., 2014). During tailings thickening process, fluid tailings are treated with flocculants such as polyacrylamide (PAM) which bind the smaller particles together to create thickened tailings (TT) of about 45% solids content (Wang et al., 2014; Lin et al., 2015). Once TT deposits are placed in their final landscape position and meet specified performance criteria, they can be reclaimed into terrestrial landscapes (Alberta Energy Regulator, 2020, 2022). Terrestrial landform construction on tailings (i.e., revegetation on tailings after covering with a layer of suitable substrate) using native plants is an appropriate strategy for reclaiming oil sands tailings (Lalonde et al., 2020; Samad et al., 2022). Plant growth in tailings is limited by the inherent ecotoxicity from residual hydrocarbon, salts, alkalinity, and the low levels of organic matter and nutrients leading to the necessity of covering tailings with a suitable reclamation cover material before revegetation. Peat-based reclamation material, including peat mineral mix (PMM), has been shown to improve plant growth (Lalonde et al., 2020; Degenhardt et al., 2023) and reduce stresses associated with tailings in various plant communities, including different upland and wetland species (Samad et al., 2022). Native boreal upland and wetland plant communities are considered appropriate for revegetation and reclamation operations in the oil sands mining area (Dhar et al., 2018; Lalonde et al., 2020; Samad et al., 2022).
Microbial communities play pivotal roles in soil nutrient cycling and plant growth and have been shown to shift in response to physicochemical and biological variables during the ecological restoration of disturbed sites (Wu et al., 2020; Gazitúa et al., 2021). Tailings streams with different age, processing, and management histories can host distinct microbial communities which may influence the long-term reclamation efforts (Foght et al., 2017). Previous research on the microbiology of tailings has mainly focused on the indigenous microbial communities of tailings ponds and has found that these communities can impact tailings management by affecting tailings chemistry and greenhouse gas emissions (Wilson et al., 2016; Foght et al., 2017). However, microbial responses to thickened tailings in a terrestrial reclamation setting with native boreal plant species remain largely unknown. Some soil microorganisms, including plant growth-promoting bacteria (PGPB) can promote plant growth through several mechanisms such as: fixing atmospheric nitrogen, solubilizing phosphates, secreting phytohormones (auxin, cytokinin, gibberellin), lowering plant ethylene levels by ACC deaminase production, and sequestrating iron through siderophores production (Olanrewaju et al., 2017; Poria et al., 2022). In addition to PGPB, some fungi, such as arbuscular mycorrhizal fungi (AMF) can enhance plant nutrient uptake and growth (Gamalero et al., 2004; del Barrio-Duque et al., 2019). Indeed, it has been reported that arbuscular mycorrhizal fungi can increase root branching, resulting in increased plant nutrient uptake (Berta et al., 2002; Gamalero et al., 2004). There is increasing evidence that microorganisms are critical to improving plant productivity in harsh environments (Harris, 2009; Vimal et al., 2017; Gazitúa et al., 2021) and play a central role in soil mineral nutrient availability in soils of the oil sands region (MacKenzie and Quideau, 2010). Moreover, tailings associated microorganisms can metabolize toxic and recalcitrant constituents found in oil sands tailings (Chegounian et al., 2020; Saborimanesh, 2021).
Various studies have shown that vegetation influences the development of microbial communities in disturbed sites (Dimitriu et al., 2010; Gagnon et al., 2020b). Furthermore, plants can influence plant-microbial relationship by altering the quantity and quality of organic matter entering the soil via root organic matter, exudates, and leaf litter (Hargreaves and Hofmockel, 2014). Thus, the variation in plant communities has important implications for microbial communities and nutrient cycling. Additionally, the placement of organic capping (e.g., PMM) over tailings can modulate the response of microorganisms by modifying soil properties and plant growth (Jamro et al., 2014; Lalonde et al., 2020). Microbial communities can be affected by various factors, including soil amendments such as forest floor-mineral mix (FFMM) and PMM (Ma et al., 2017; Mitter et al., 2017; Stefani et al., 2018), vegetation (Gagnon et al., 2020b), soil rehabilitation time (Birnbaum et al., 2017) and other physiochemical properties including pH, salinity, moisture, aeration, etc. (Xue et al., 2018; Gschwend et al., 2021). In fact, soil pH is considered one of the key drivers of microbial community composition (Rousk et al., 2010b). Salinity has also been demonstrated as a dominant factor affecting soil microbial populations (Rath et al., 2019; Yang and Sun, 2020). In addition, soil moisture and aeration have been reported to impact microbial community structure (Meisner et al., 2021). For instance, high soil aeration was shown to stimulate microbial biomass and shift the composition of microbial communities (Picek and Simek, 2000).
Previous studies concerning oil sands tailings reclamation primarily looked at plant community establishment and assemblage rather than microbial community dynamics (Rowland et al., 2009; Errington and Pinno, 2015; Dhar et al., 2018, 2020). However, microbial communities can contribute considerably to tailings restoration outcomes through nutrient cycling and complex mutualistic interactions with plants (Harris, 2009; Gazitúa et al., 2021). The primary goal of this study was therefore to understand the response of tailings bacterial and fungal communities to different capping and vegetation strategies over time. For this purpose, a mesoscale greenhouse experiment was sampled for two growing seasons to evaluate the response of tailings and PMM-associated microbial communities during the establishment of boreal upland and wetland species grown directly in tailings or in tailings capped with PMM. The combined knowledge of aboveground biomass and belowground soil–plant-microorganism interactions will guide the development of effective strategies to reclaim sustainable landscapes in the oil sands mining area.
For this experiment, columns (plastic cylindrical barrels of 208 L volume, 54 cm in diameter and 90 cm in height) containing thickened tailings (TT) alone or TT capped with two different depths (10 or 30 cm) of peat mineral mix (PMM) were assembled in 2019 and planted with two distinct plant communities (upland and wetland). The upland community was composed of four different upland plant species (a total of 12 plants per column) including Populus tremuloides Michx (aspen), Elymus trachycaulus ssp. trachycaulus (Link) Malte (slender wheat grass), Pinus banksiana Lamb (jack pine), and Cornus sericea L. also referred to as Cornus stolonifera Michx. (red-osier dogwood). The wetland community was composed of five different plant species (a total of 14 plants per column) including Salix bebbiana Sarg. (Bebb’s willow), Rumex salicifolius Weinm. (willow dock), Scirpus microcarpus J. Presl and C. Presl (Small-fruited bulrush), Carex aquatilis Wahlenb (water sedge) and Triglochin maritima L. (sea-side arrow grass). Four replicate columns were assembled for each combination of plant community and capping treatment, for a total of 24 columns. More details about the treatments can be found in Figure 1. TT and capping material (PMM) were received from Imperial Oil Ltd., Calgary, Canada. TT has a loamy sand texture with a sand to fines ratio (SFR) of 2.8. The basic physicochemical properties of tailings and PMM are described in Table 1. The columns were placed in a greenhouse set at a temperature of 19–21°C with 65–75% humidity and a photoperiod of 12 h. Overhead irrigation using reverse osmosis water was applied consistently to all columns to mimic precipitation, and the only loss of water was through evapotranspiration since the columns were an enclosed system with no drainage. Moreover, moisture conditions in the top 20 cm for each column were assessed weekly using a TDR 100 Soil Moisture Tester (Spectrum Technologies, Inc., United States); these values were used to inform if additional hand watering was required to maintain a range of volumetric water contents (VWCs) between 19–24% and 25–30% for the upland and the wetland columns, respectively. No fertilization was applied. Soil temperature logging sensors were installed during column setup in three of the total four replicates for each treatment. The EM50 loggers recorded hourly moisture and temperature data from three 5TM sensors installed at 20, 40, and 60 cm below the surface (METER Environment, United States), and average temperature (Supplementary Figure S1) and average moisture (Supplementary Figure S2) were recorded at each depth. Plant growth and survival were recorded before sampling (Supplementary Figure S3). The tailings and PMM (capping) were sampled (n = 4) using a sterile core sampler at the start of the experiment pre-planting (May 2019; Baseline), after the first growing season (Sep 2019; 5 months after plantation; GS1), and after the second growing season (Oct 2020; 18 months after plantation; GS2). The samples were collected using a small soil corer, transferred to a 50 mL Falcon tube, immediately placed on ice, and stored at −20°C until further processing.
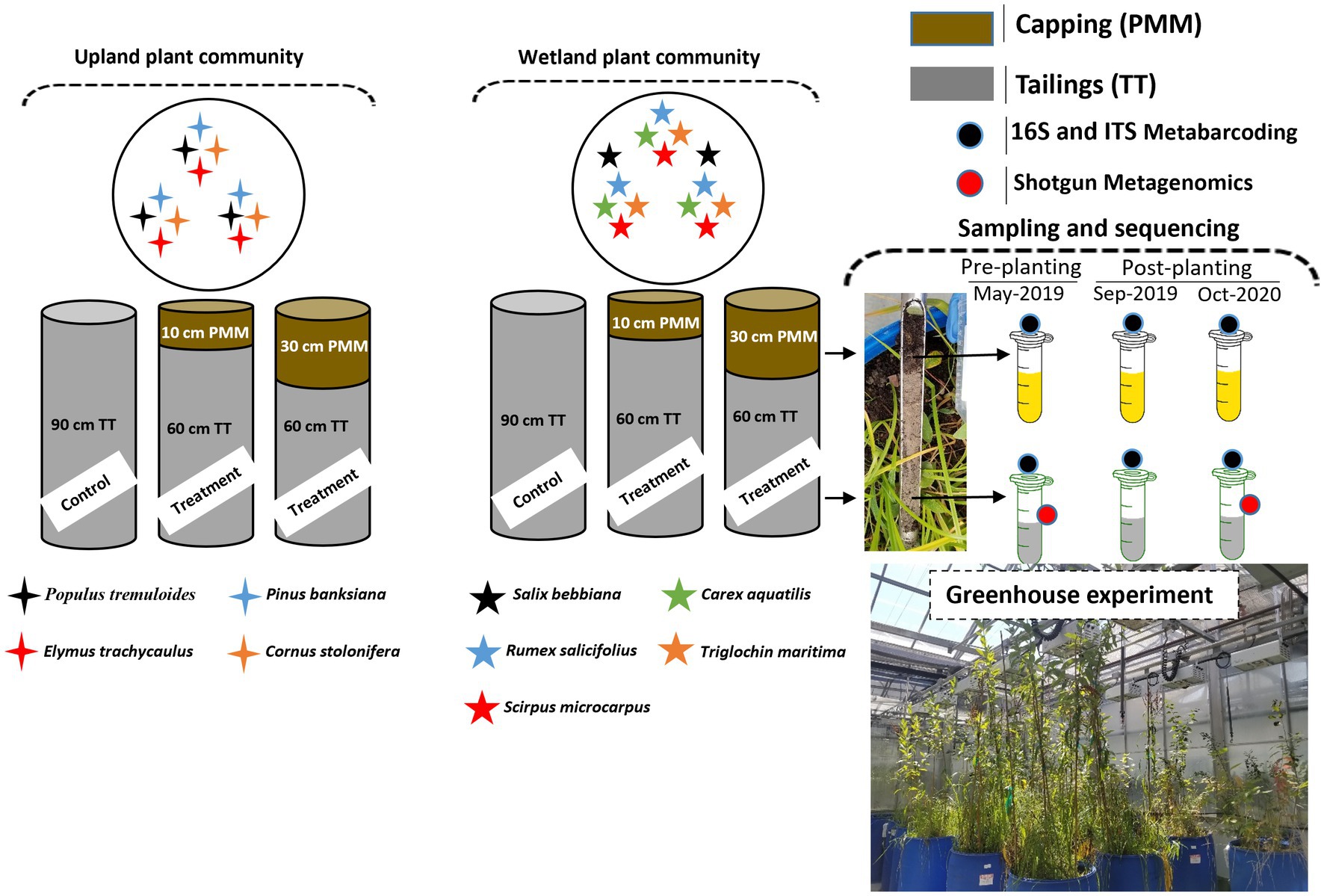
Figure 1. Diagram of experimental design in which upland and wetland plant communities were grown on thickened tailings with or without PMM capping of different depths (10 and 30 cm). A total of 12 plants (indicated by 4-point stars) per column belonging to four different upland plant species and 14 plants (indicated by 5-point stars) per column belonging to five different wetland plant species were planted in upland and wetland columns, respectively. Four columns were assembled for each treatment (n = 4). The tailings and PMM layers were sampled at the start of the experiment (May 2019; Pre-planting baseline), after the first growing season (Sep 2019; GS1), and after the second growing season (Oct 2020; GS2). All samples were subjected to 16S and ITS metabarcoding (indicated by black circles). Tailings samples from baseline and GS2 were also sequenced by shotgun metagenomics (indicated by red circles). TT, thickened tailings; PMM, peat mineral mix.
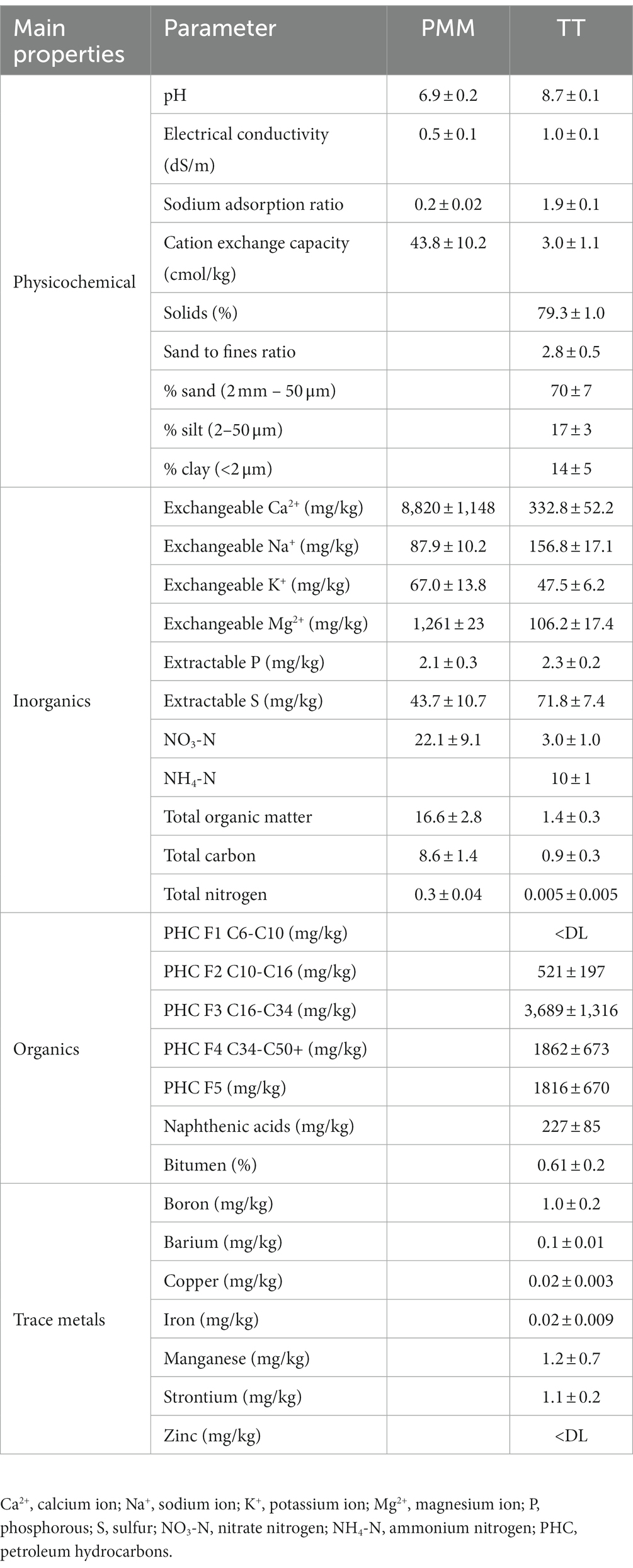
Table 1. Basic properties of thickened tailings (TT) and peat mineral mix (PMM); mean ± one standard deviation of the mean.
A metabarcoding approach was used to characterize the microbial communities in TT and in PMM capping material. DNA was extracted from the samples (0.25 g for TT and 0.1 g for PMM) using the DNeasy PowerSoil kit on a QIAcube automated platform (Qiagen, Toronto, ON) following the manufacturer’s instructions. DNA from each sample was quantified with the Qubit dsDNA HS (for tailings) or BR (for PMM) assay kit (Thermo Fisher Scientific, United States) using a Qubit Fluorometer 2.0 (Life Technologies, Burlington, ON, Canada). Primers 515F-Y (5′-GTGYCAGCMGCCGCGGTAA-3′) and 926R (5′-CCGYCAATTYMTTTRAGTTT-3′) targeting the V4-V5 regions of the 16S ribosomal RNA gene of bacteria and archaea (Parada et al., 2016) and primer pairs ITS9 (5′-GAACGCAGCRAAIIGYGA-3′) (Menkis et al., 2012) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) targeting the ITS2 region of fungi (White et al., 1990; Rivers, 2016) were used in this study. Library preparation for Illumina sequencing was performed using the above-mentioned primers according to the manufacturer’s instructions (Illumina, 2013). The reactions for amplicon PCR were set up by mixing 25 μL of HotStarTaq Plus Master Mix (QIAGEN, Inc., Germantown, MD, United States), 19 μL RNase-free water, 0.5 μL of each 10 μM primer and 5 μL of gDNA normalized at 5 ng μL−1 Thermal cycler (C1000 Touch Thermal Cycler, BIO-RAD) conditions were as follows: 95°C for 5 min; 95°C for 45 s; 50°C for 45 s (16S primers, 35 Cycles and ITS2 primers, 40 cycles); 72°C for 1 min; and final elongation at 72°C for 10 min. PCR products were purified using magnetic beads solution (81 μL/sample; Agencourt AMPure XP), then unique indexes were added to each sample using the Nextera XT Index Kit v2, in accordance with Illumina’s protocol (Illumina, 2013). After index PCR, the amplicons were purified again with magnetic beads (56 μL/sample), quantified using the Synergy™ Mx Microplate Reader (BioTek Instruments, Inc., Winooski, VT, United States), and combined at equimolar concentration. Paired-end sequencing (2 × 300 bp) was performed on an Illumina MiSeq platform at the Next Generation Sequencing Platform of the CHU de Québec-Université Laval Research Centre. Sequences were deposited in the NCBI Sequence Read Archive under BioProject number PRJNA894999 (accession number SAMN31487590 to SAMN31487742).
A metagenomics approach was applied to a subset of tailings samples to investigate the treatment effects on microbial functions. Shotgun metagenomic libraries were prepared using the Nextera XT DNA library preparation kit and the Nextera XT Index kit v2 (Illumina) following the manufacturer’s instructions. The resulting DNA libraries were purified with Agencourt AMPure XP beads (Beckman Coulter, Inc., United States) and fragment size distribution within the range of 250–1,000 bp was verified by running the libraries on the 2,100 Bioanalyzer using the Agilent high sensitivity DNA kit. Libraries were quantified with the Qubit BR dsDNA assay kit and manually normalized to ensure equal library representation in the pool prior to sequencing. Normalized libraries were sequenced on an Illumina NovaSeq 6,000 using a S4 PE 2 × 150 bp flow cell at the Centre d’Expertise et de Services Génome Québec. Sequences were deposited in the NCBI Sequence Read Archive under BioProject number PRJNA894999 (accession number SAMN31487743 to SAMN31487757).
Analysis of metabarcoding sequences was performed with QIIME 2 (Bolyen et al., 2019). Raw sequences were imported with the “Casava 1.8 paired-end demultiplexed fastq” function, and the files were stored into Qiime2 artifacts (demux.qza). Data were demultiplexed and quality filtered using the q2-demux plugin, followed by denoising with DADA2 using the “qiime dada2 denoise-paired” function (Callahan et al., 2016). Taxonomy was assigned to ASVs using the naïve Bayes classifier (q2-feature-classifier) (Bokulich et al., 2018) using SILVA 138 (Yilmaz et al., 2014) and UNITE 8.2 (Abarenkov et al., 2010) databases for 16S and ITS, respectively. The classifier Qiime2-compatible database files were obtained from Qiime2 data resources.1 Then taxonomy-based filtering of the feature table and feature data was performed to remove sequences and counts corresponding to non-target amplification such as chloroplasts, mitochondria, and sequences unidentified at the kingdom level. About one-third of ITS reads in tailings samples were assigned to ciliated protists Ciliophora and were removed from the ITS dataset before further analysis. The “qiime tools export” function was used to export ASV tables generated by QIIME2, which were further imported into R version 4.2.1 (R Core Team, 2022) for taxonomic composition, alpha diversity and beta diversity analysis. The R package phyloseq and qiime2R were used for data handling (McMurdie and Holmes, 2013; Bisanz, 2022), and all plots were created using the ggplot2 package (Wickham, 2016).
To analyze alpha diversity (Observed ASVs, Shannon index and Pielou’s evenness), datasets were rarefied to the minimum sample size, 6,402 ASVs per sample for bacteria and 2,527 ASVs per sample for fungi (Weiss et al., 2017). Prior to statistical analyses, parametric assumptions of data normality were verified by Shapiro–Wilk test of normality using the “shapiro.test” function. Homoscedasticity was checked using the Bartlett test (“bartlett.test” function). Akaike’s Information Criterion (AIC) was used for the model selection and the model with lowest AIC value was used for forecasting. Generalized least squares (gls) linear models (package nlme) were fitted, with weights allowing different variances for treatment in case of heteroscedasticity. Three-way ANOVA (function: anova, package: stats) was applied to determine how capping treatments (0, 10, 30 cm), plant communities (upland, wetland), post-planting time points (Sep 2019, Oct 2020) and their interaction influence alpha diversity indices. When the main effects were significant but interaction effects were not, multiple comparisons of the means were done on the main effects only, while in the presence of significant interaction effects, multiple comparisons were done within factors involved in the significant interactions. Additionally, one-way ANOVAs were applied on three sampling time points including pre-planting baseline samples (May 2019), GS1 (Sep 2019), and GS2 (Oct 2020) to test how alpha diversity changes after planting when compared to baseline samples. The gls models were fitted (nlme package), taking temporal autocorrelations into account using corCAR1 function (Pinheiro et al., 2022). The R package emmeans was used for post hoc multiple comparisons among groups and FDR-adjusted p < 0.05 were considered statistically significant. For beta diversity analysis, ASV tables (non-rarefied) were normalized using the geometric mean of pairwise ratios (GMPR) method (Chen et al., 2018). Beta-diversity was calculated as the Bray–Curtis dissimilarity index (function: distance, package: phyloseq) and visualized using a non-metric multidimensional scaling plot (NMDS, “metaMDS” function, “vegan” package). Permutational Multivariate Analysis of Variance was performed using Bray–Curtis dissimilarity matrices with 9,999 permutations (PERMANOVA function: adonis, package: vegan). Comparisons between groups were checked by a multilevel pairwise comparison test (“pairwise.adonis2” function, “pairwiseAdonis” package). Variance heterogeneity between groups was tested with the function “betadisper” in the “vegan” package. To identify the taxa whose relative abundances differed between the capping treatments (10 and 30 cm) and control (0 cm), differential abundance analysis (DAA) was conducted using ANCOM-BC (Analysis of compositions of microbiomes with bias correction) package in R (Lin and Peddada, 2020). Rare taxa with less than 10 counts were removed, and taxa were considered as differentially abundant only if Benjamini-Hochberg adjusted p-values were significant (p < 0.05). A total of 80 samples, 48 from the TT layer and 32 samples from the PMM layer, were sequenced. Six samples of the PMM layer from GS1 sampling were removed due to low read abundance.
Metagenomics data were processed through a previously described bioinformatic pipeline (Tremblay et al., 2017). Briefly, Sequencing adapters in all reads were removed, and bases with low-quality scores (<30) at the end of reads were cut using Trimmomatic 0.39 (Bolger et al., 2014) to generate quality-controlled (QC) reads. QC-passed reads from each sample were co-assembled using Megahit 1.2.9. Genes were predicted from contigs, and good quality reads used for co-assembly input were mapped back onto contigs to evaluate contig and gene abundances. The counts of QC-passed reads are summarized in Supplementary Table S1 for all metagenomic sequencing libraries. Gene prediction on the obtained contigs was performed by calling genes on each assembled contig using Prodigal v2.6.3 (Hyatt et al., 2010). Genes were annotated following the JGI’s guidelines (Huntemann et al., 2016) using six different databases: (1) RPSBLAST (v2.2.29+; Camacho et al., 2009) against COG database (v3.11); (2) RPSBLAST (v2.2.29+) against KOG database (v3.11); (3) HMMSCAN (v3.1b1; Eddy, 2011) against PFAM-A v27.0 database (Finn et al., 2014); (4) TIGRFAM database v15.0; (5) BLASTP (v2.2.29+) against KEGG database (v71.0) and (6) BLASTN (v2.2.29+) against NCBI’s nucleotide (nt) database (Li and Durbin, 2010). For each database comparison, the best hit having at least an e-value ≥0.01 was kept for each query. Alignment files in bam format were sorted by reads coordinates using SAMtools v1.1,2 and only properly aligned read pairs were kept for downstream steps (Supplementary Table S1). Each bam file (containing properly aligned paired-reads only) was analyzed for coverage of called genes and contigs by bedtools 2.23 (Quinlan and Hall, 2010) using a custom bed file representing gene coordinates on each contig. Only paired reads overlapping their contigs or genes were considered for gene counts.
Contigs (not genes) sequences were also blasted against NCBI’s nt database for taxonomic assignment. Taxonomy of each contig was assigned using the NCBI taxonomy database (Sayers et al., 2011).3 GenInfo Identifier (GIs) resulting from BLASTN against nt was used to retrieve full taxonomic lineage (when available) from the NCBI taxonomy database. Taxonomic lineages were integrated into the contig abundance of read counts matrix to generate an OTU table format file (with contigs replacing OTUs as rows). Taxonomic summaries were performed using a combination of in-house Perl, R scripts, and Qiime v.1.9.0 (Caporaso et al., 2010). The R software package Deseq2 (Love et al., 2014) was used for differential abundance analysis of genes (log fold-change = 2, False Discovery Rate < 0.05) to identify significantly enriched or depleted potential genes and pathways in different capping treatments and control comparisons. Differentially abundant genes were grouped according to KEGG database functional categories (Kanehisa, 2000).
Bacterial and fungal alpha diversity was measured by Observed ASVs (richness), Shannon’s diversity index (Shannon), and Pielou’s evenness index (evenness). Time was identified as the most important driver of bacterial alpha diversity (Figure 2), with overall lower bacterial alpha diversity recorded in the tailings at the beginning of the experiment (May 2019; pre-planting baseline samples). Bacterial richness, Shannon diversity and evenness increased significantly after the first growing season (Sep 2019; GS1) when compared to baseline samples (Figure 2). Moreover, Shannon and evenness significantly increased between GS1 and GS2.
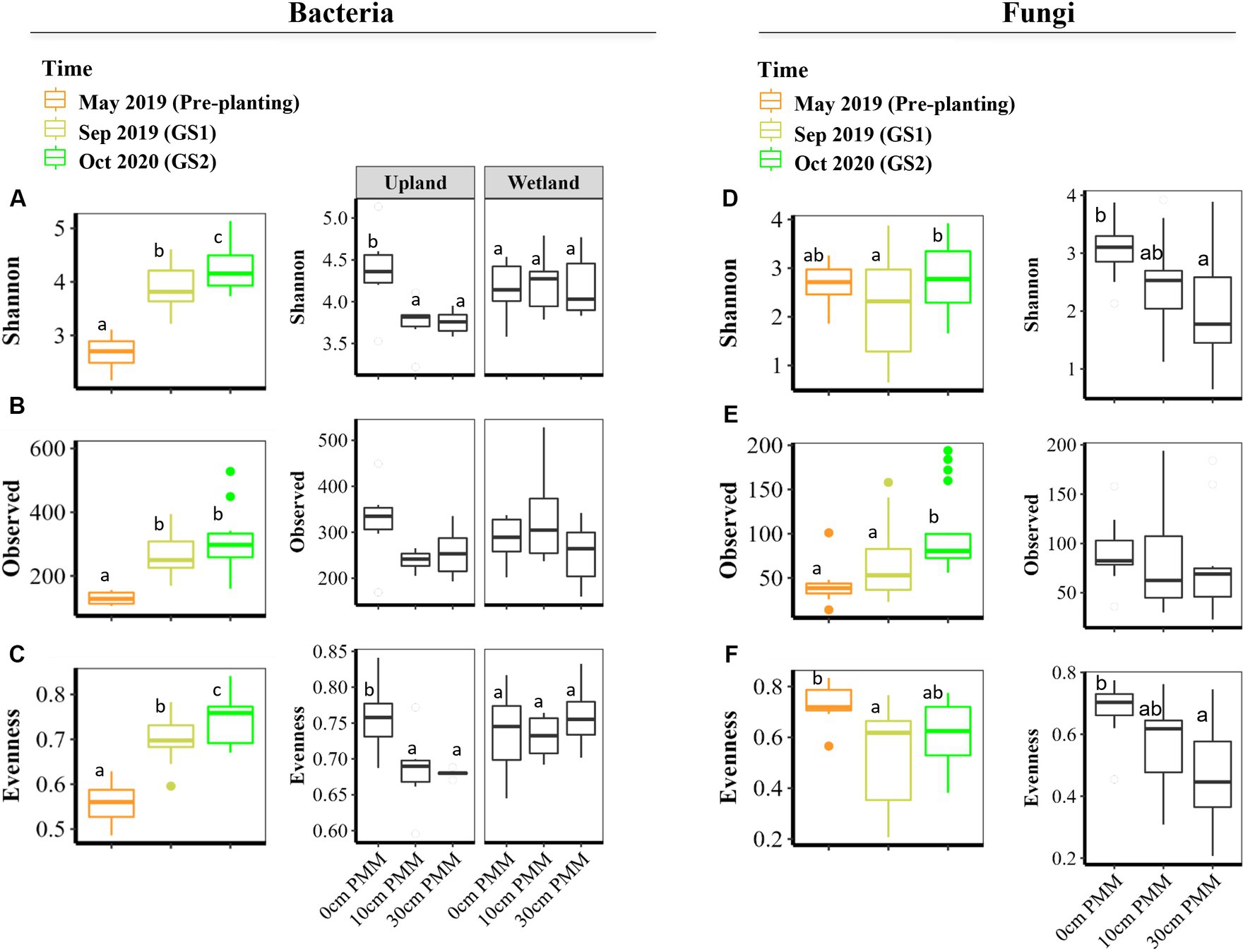
Figure 2. Alpha diversity indices of bacterial and fungal communities in thickened tailings (TT) samples, determined by Observed ASVs (i.e., richness), Shannon diversity, and Pielou’s evenness for bacteria (A–C) and fungi (D–F). Generalized least squares (gls) models and three-way ANOVA was applied (see Supplementary Table S2) to determine how capping treatments (0, 10, 30 cm), plant communities (upland, wetland) and time points (Sep 2019, Oct 2020) and their interaction influence alpha diversity indices (n = 3). One-way ANOVA was applied to test how microbial communities changed after each growing season compared to pre-planting baseline samples (May 2019). FDR, false discovery rate; May 2019, baseline or pre-planting; Sep 2019, GS1; Oct 2020, GS2.
The main effects of capping treatments on bacterial alpha diversity were not significant. However, a significant interaction effect between plant community and capping treatment was detected for Shannon and evenness (Figure 2 and Supplementary Table S2). Further testing of the impact of capping treatment within each plant community type showed a negative impact of capping on bacterial alpha diversity of the tailings in the presence of the upland plant community, while no significant effect of capping was detected for the wetland plant community. The plant community itself (upland vs. wetland) only affected the evenness but did not affect the richness and Shannon (Supplementary Table S2). Capping depth (10 cm vs. 30 cm) had no significant effect on bacterial alpha diversity (Figures 2A–C).
Fungal alpha diversity showed a slightly different response than bacterial alpha diversity (Figures 2D–F and Supplementary Table S2). While the fungal richness and Shannon diversity index significantly increased between GS1 and GS2 (post-planting), fungal evenness was not significantly changed (Figures 2D–F). Unlike bacterial evenness, fungal evenness significantly decreased after GS1 compared to pre-planting baseline samples. Fungal richness was not significantly affected by capping treatment nor by plant community. On the other hand, both Shannon and evenness were significantly affected by capping treatment, with lower values detected in columns with capping compared to the controls with no cap. Capping depth as well as plant community had no significant effect on fungal alpha diversity (Figures 2D–F and Supplementary Table S2).
Similarly, beta diversity after planting was significantly changed compared to pre-planting baseline for both bacterial and fungal communities (Figure 3 and Table 2). GS1 and GS2 samples were significantly different compared to the baseline samples for both groups, but the difference between GS1 and GS2 samples was significant only for bacteria in wetland columns (Table 2 and Figure 3). While the main effects of the capping treatments on bacterial and fungal beta diversity were not significant, the main effects of plant community and the interaction effects between capping treatment and plant community were significant (Table 2). Further pairwise comparisons within each plant community showed that capping treatment had a significant effect on bacterial and fungal beta diversity. The effect of capping depth (10 cm vs. 30 cm) on beta diversity was only observed in upland columns.
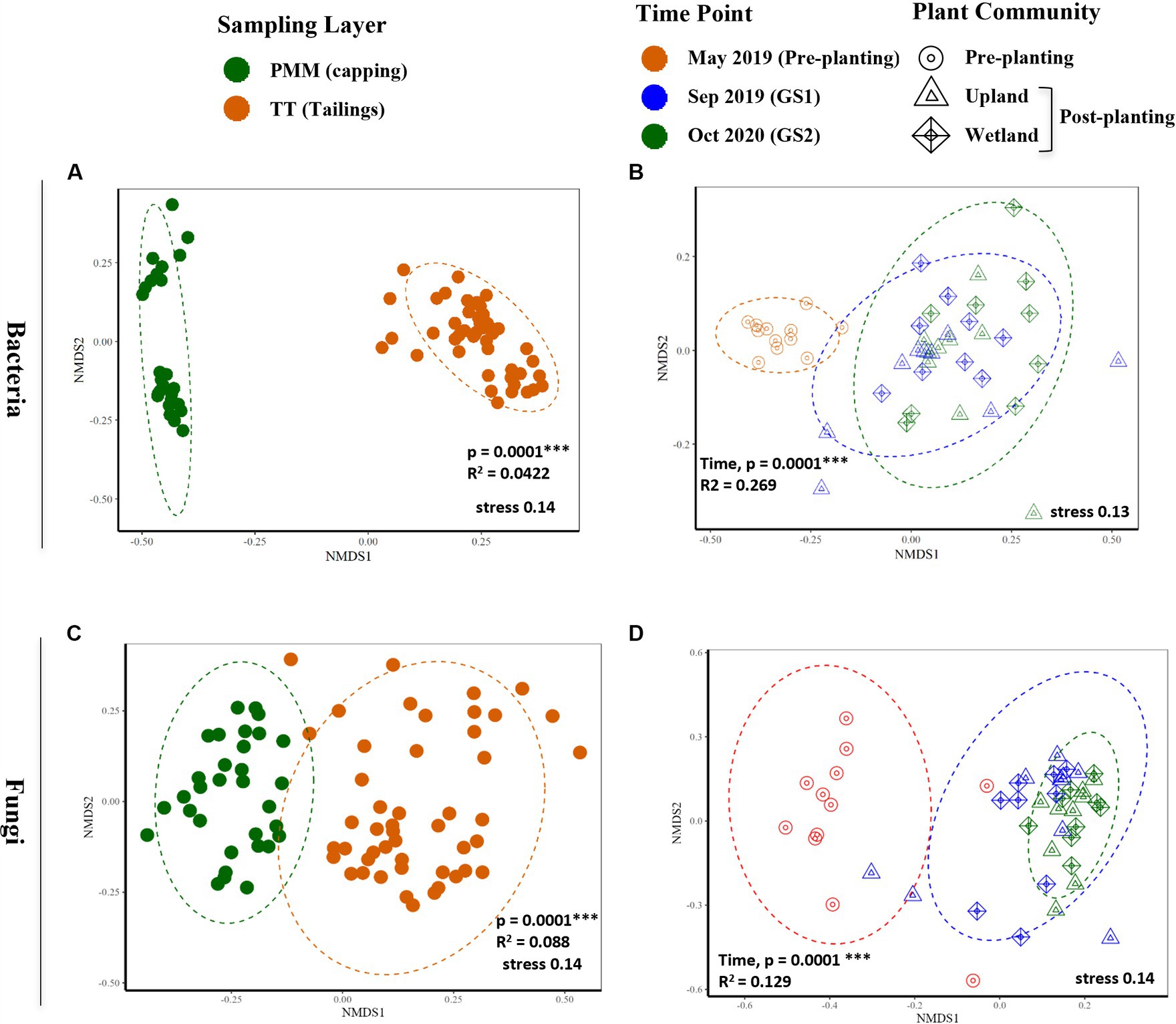
Figure 3. Non-metric multidimensional scaling (NMDS) based on Bray-Curtis dissimilarities of bacterial (A,B) and fungal (C,D) communities. (A,C) Differences between capping (PMM) and thickened tailings (TT) samples for bacterial and fungal communities, respectively. (B,D) Effect of time on TT samples for bacterial and fungal communities, respectively. p-values indicate statistical differences between capping layers (A,C) and time points (B,D) according to PERMANOVA. FDR-adjusted p < 0.05 was considered statistically significant, as indicated by asterisk, * < 0.05, ** < 0.005, *** < 0.0005. FDR, false discovery rate. Detailed results of PERMANOVA are provided in Table 2.
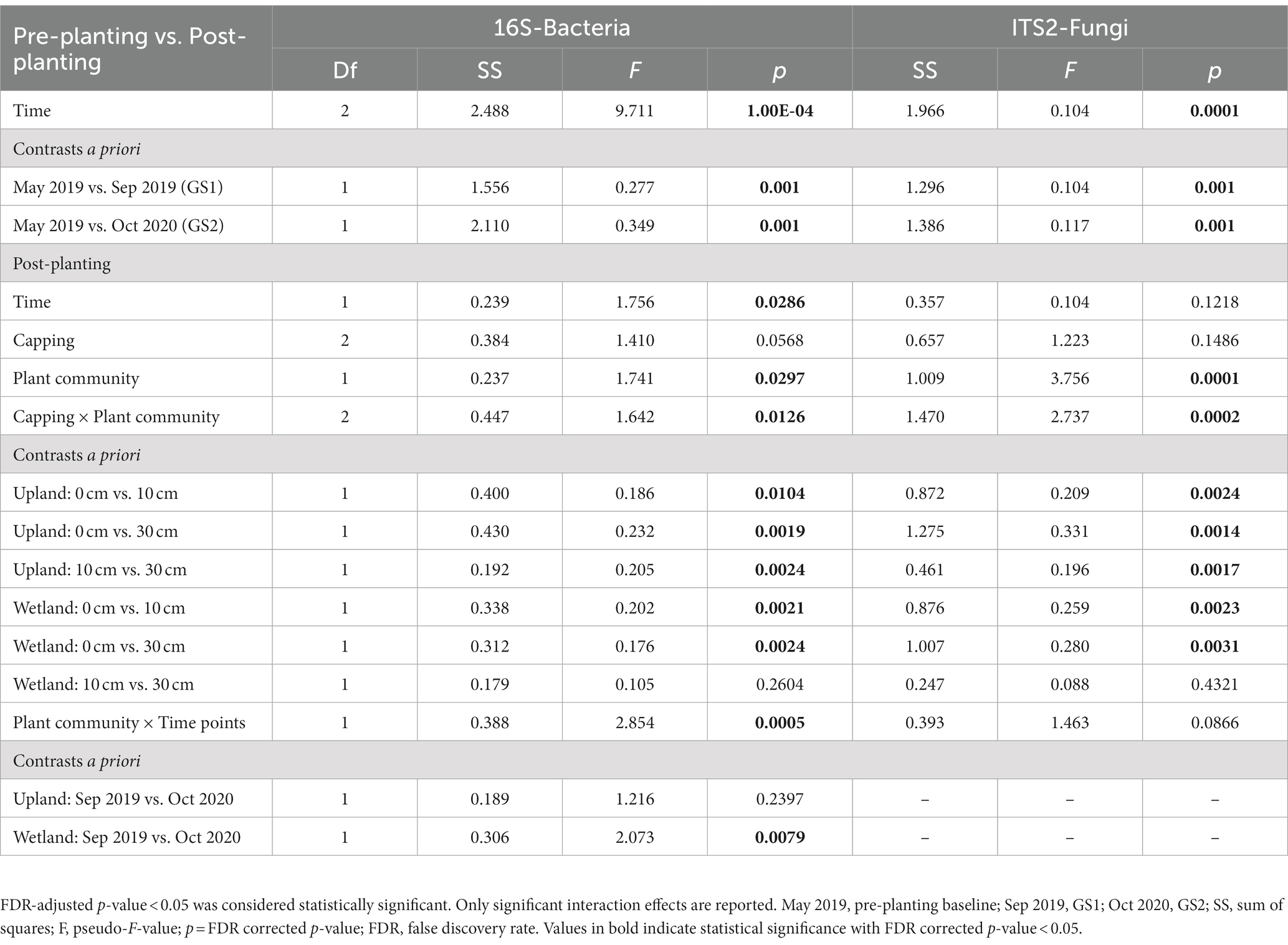
Table 2. Results of permutational multivariate analysis of variance (PERMANOVA, 9999 permutations) on Bray–Curtis dissimilarity of bacterial and fungal communities as a function of capping treatments (0, 10, 30 cm), plant communities (upland, wetland), post-planting time points (Sep 2019, Oct 2020), and their interactions (n ≥ 3).
Bacterial and fungal alpha diversity (Observed ASVs and Shannon) was significantly higher in capping material (PMM) than in tailings (Supplementary Figure S4). Alpha diversity was not significantly impacted by time in the capping material and remained relatively stable temporally when compared to the tailings layer (Supplementary Figure S4). Likewise, the beta diversity of the capping material was also significantly different from the tailings layer for both bacteria and fungi, with capping samples showing a clear separation from the tailings samples in ordinations (Figures 3A,B). However, time (Sep 2019 and Oct 2020), capping depth (10 and 30 cm) and plant communities (upland and wetland) had no impact on the beta diversity of capping material (Supplementary Table S3).
Bacterial taxonomic profiles in capping material and tailings were clearly distinct but dominated mainly by four bacterial phyla: the Proteobacteria, Bacteroidota, Acidobacteriota, and Actinobacteria (Figure 4A and Supplementary Figure S5). At a lower taxonomic level, the order Burkholderiales (48%; mainly Hydrogenophilaceae and Comamonadaceae families) was predominant in the tailings layer, while Rhizobiales (14%) was dominant in the capping layer (Figure 4B). In line with results obtained for alpha and beta diversity, significant variations in relative abundances were observed over time for bacterial communities in the tailings layer but not in the capping layer (Figure 4B and Supplementary Figure S5). In the tailings layer, the order Burkholderiales (particularly family Comamonadaceae) was predominant in the baseline samples, but its relative abundance decreased after GS1. In contrast, the relative abundances of Acidithiobacillales, Rhizobiales, and Micrococcales (f_Microbacteriaceae) increased with time compared to baseline samples (Figure 4B). Taxonomic profiles at the genus level (Figure 4C), and further differential abundance (DA) analysis (Figure 5) showed that the relative abundance of genera Hydrogenophaga and Polaromonas (family Comamonadaceae) significantly decreased with time. Other taxa related to sulfur oxidation, such as the Acidithiobacillaceae KCM-B-112 and Sulfuritalea, were strongly enriched after GS1 and GS2 compared to baseline tailings (Figure 5). Moreover, the relative abundance of several dominant taxa (at the genus level) such as the uncultured Holophagae Subgroup 7 of the Acidobacteriota and Nitrosomonadaceae GOUTA6 was significantly increased in tailings layer after planting (GS1 and GS2) compared to baseline samples (Figure 5). Post-planting, the relative abundance of dominant taxa was not significantly changed between GS1 and GS2, apart from Holophagae and Nitrosomonadaceae which significantly increased over time (Figure 5).
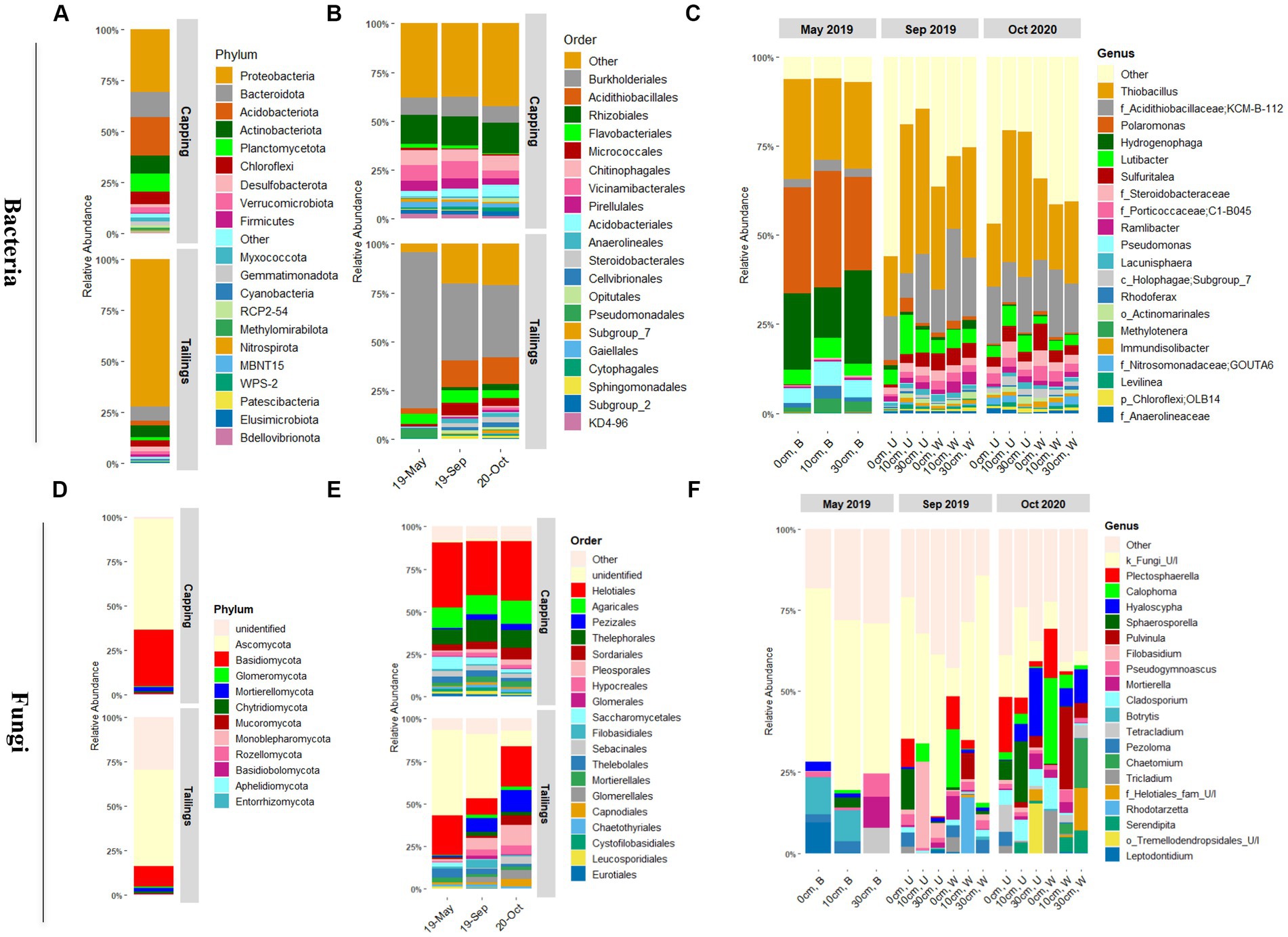
Figure 4. Taxonomic profiles of microbial communities in thickened tailings and capping material (PMM) as assessed by sequencing the 16S rRNA gene for bacteria (A–C), and ITS2 region for fungi (D–F). (A,D), taxonomic profiles at the phylum level across all samples for the capping (PMM) and tickened tailings (TT) layers. (B,E), taxonomic profiles at the order level as a function of time for the capping and tailing layers. (C,F), taxonomic profiles at the genus level as a function of time, vegetation type, and capping treatment for the tailings layer only. Only 20 most abundant classified bacterial and fungal taxa are shown (n ≥ 3). B, baseline; W, wetland; U, upland; 0 cm, control; U/I, unidentified.
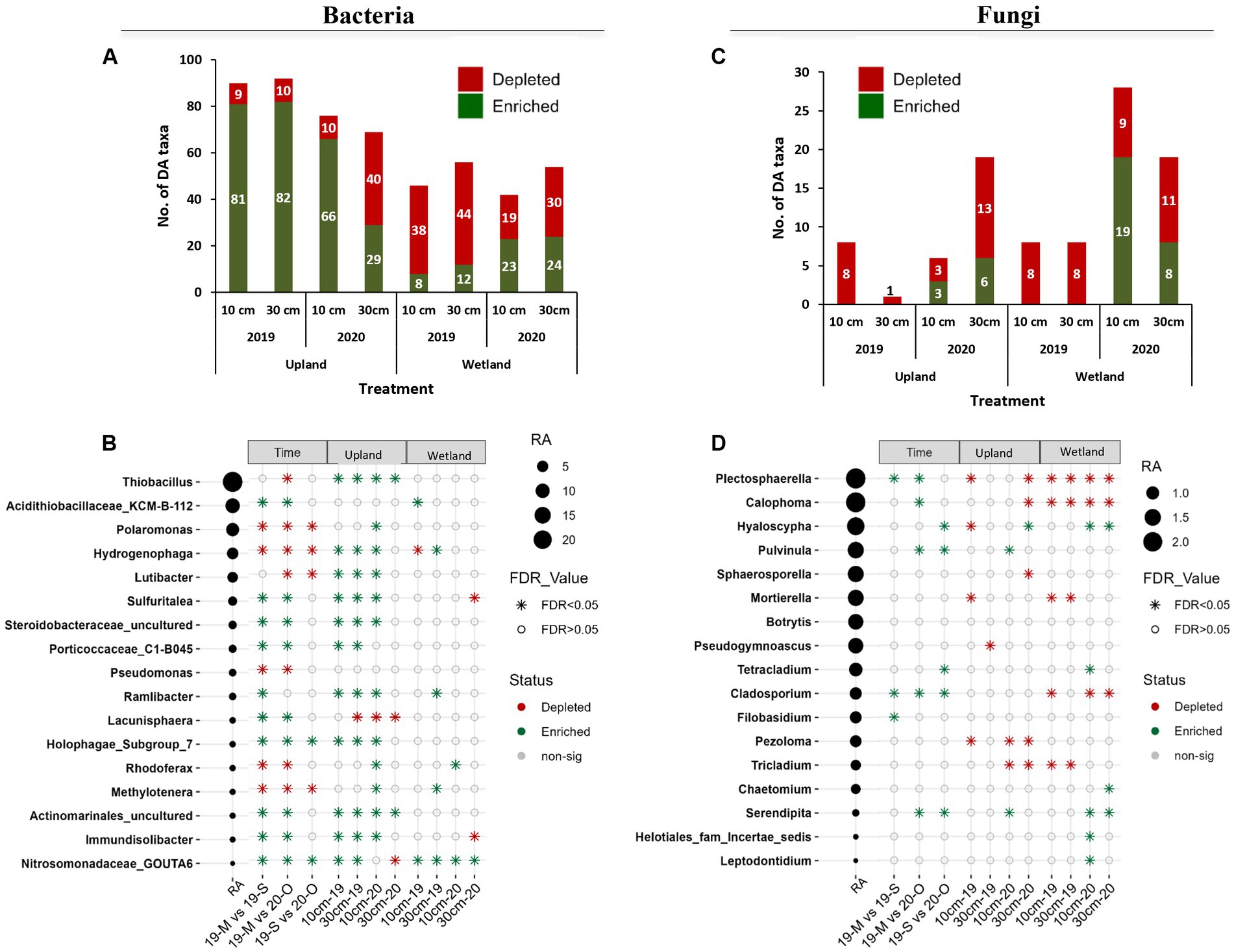
Figure 5. Differential abundance analysis as determined by the ANCOM-BC method for bacteria (A,B) and fungi (C,D). (A,C) Total number of significantly enriched or depleted genera for capping treatments (10 cm vs. control and 30 cm vs. control). (B,D) List of significantly enriched or depleted genera for time point (May 2019 vs. Sep 2019, May 2019 vs. Oct 2020, and Sep 2019 vs. Oct 2020) and capping treatments (10 cm vs. control and 30 cm vs. control). Taxa with more than 0.05% relative abundance (RA) are sorted with decreasing RA. Taxa were considered as differentially abundant only if FDR adjusted p-values were significant (p < 0.05). 19-M, May 2019 (baseline); 19-S, Sep 2019 (GS1); 20-O, Oct 2020 (GS2); 0 cm, control; DA, differentially abundant; FDR, false discovery rate; AN-COMBC, analysis of the composition of microbiomes with bias correction.
The capping treatment affected the relative abundance of dominant bacterial taxa differently depending on the plant community (Figures 4C, 5A). Indeed, the number of taxa significantly affected by the capping treatment (i.e., number of DA taxa) was much higher in the upland plant community than in the wetland plant community (Figure 5A). For example, while the relative abundance of Thiobacillus and Lutibacter was significantly higher in 10 and 30 cm capping treatments than in control (0 cm) in the presence of the upland community, no effect of capping on these taxa was detected in the presence of the wetland community (Figure 5B). In fact, in the wetland community, Nitrosomonadaceae_ GOUTA6 was the only taxon to be consistently affected by capping, with higher relative abundance in both capped treatments (10 and 30 cm) compared to uncapped controls in both GS1 and GS2. It is noteworthy that several of the taxa that were significantly impacted by capping treatments are involved in sulfur and nitrogen cycling, including sulfur-oxidizers (e.g., Thiobacillus and Sulfuritalea that can oxidize reduced inorganic sulfur compounds such as thiosulfate and sulfide into sulfate), ammonia-oxidizer (Nitrosomonadaceae_GOUTA6) and nitrogen-reducer (Lutibacter) (Figure 5B). Like beta diversity, the influence of capping depth on relative abundance (RA) was mainly observed in the upland columns. For example, in GS2, the RA of Polaromonas, Hydrogenophaga and Lutibacter was significantly higher in the 10 cm PMM capping but not in the 30 cm PMM capping treatment compared to the uncapped control (Figure 5B). Overall, the RA of individual bacterial taxa changed over time and responded to capping treatments depending upon the plant community, growing season and, to some extent, the capping depth. Nitrosomonadaceae was the only taxa which significantly increased with both time and capping.
Ascomycota and Basidiomycota were the predominant fungal phyla detected in both capping material and tailings (Figure 4D). In tailings however, a total of 28% of the sequences could not be identified at the phylum level, indicating the presence of a high proportion of uncharacterized fungi in this sample type. The Ascomycota relative abundance increased over time, especially in the tailings layer (Supplementary Figure S6), while the proportion of unidentified fungi decreased over time (Figure 4E and Supplementary Figure S6). At the order level, the Helotiales remained dominant at all time point in tailings, while the relative abundance of the Pleosporales and Pezizales increased with time (Figure 4E). In the PMM layer, the order Helotiales was also dominant, followed by the Agaricales and the Thelephorales, with limited changes over time (Figure 4E). Many fungal ASVs from tailings were not identified at the genus level, especially in baseline and GS1 samples. Fungal taxonomic profiles were also much more variable between samples than bacterial profiles. Overall, Plectosphaerella and Calophoma were the most dominant fungal genera in the tailings layer (Figure 4F and Supplementary Figure S6). In the capping layer, Hyaloscypha and Inocybe were the most prevalent fungal genera (Supplementary Figure S6).
Differential abundance (DA) analysis showed that a much lower number of fungal taxa than of bacterial taxa were impacted by time and by the capping treatments (Figure 5C). Like bacterial taxa, fungal taxa responded to capping mainly according to the plant community, but in this case the number of DA taxa was higher in wetland than in upland communities (Figure 5C). Plectosphaerella and Calophoma were enriched compared to baseline samples, but they were generally depleted by capping (Figures 4F, 5D). The abundance of taxa known to form close associations with plant roots, including Hyaloscypha and Serendipita, generally increased with capping and time (Figure 5D). The RA of some abundant fungal taxa, such as Plectosphaerella and Calophoma was significantly decreased by capping, mainly in the wetland treatments (Figure 5D).
Shotgun metagenomics analysis was performed on selected samples (15 samples) from the tailings layer to assess the impact of capping treatments and plant communities on microbial functions. Samples from baseline (May 2019; 3 samples) and GS2 (Oct 2020; 3 samples × 4 treatments) were selected for metagenomic analysis. About 96.5% of reads of quality-controlled shotgun metagenomic data were taxonomically assigned to Bacteria and Archaea and only 0.05% to Fungi, indicating a very low biomass of fungi in the thickened tailings microbiome (Supplementary Table S4). Moreover, 0.07% of reads were assigned to each of Metazoa and Viridiplantae, 0.4% to Viruses, and 2.8% remained unidentified at the kingdom level. Like microbial diversity, the functional profile was significantly changed after two growing seasons compared to baseline samples (Figure 6A). Differential abundance analysis showed that 9% of total genes, mainly including energy metabolism and nutrient cycling, were significantly (log fold change>2, p < 0.05) enriched in GS2 samples when compared to baseline samples. However, only 2.2% of total genes were depleted after GS2 (Figure 6B). Further functional analysis specifically focused on the microbial pathways related to energy metabolism (KEGG pathway m09102, methane, sulfur, and nitrogen) and xenobiotics biodegradation and metabolism (m09111). Genes related to methane metabolism, benzoate degradation, nitrogen, and sulfur metabolism were significantly enriched between baseline and GS2 (Figure 7A). Interestingly, similarily to the 16S metabarcoding data, capping influenced the functional profile depending on the plant community (Figure 7B). Indeed, as previously observed for 16S-based alpha diversity and DA of individual taxa, more microbial functions were affected by capping in the upland treatment than in the wetland treatment. Moreover, upland and wetland treatments showed a contrasting response at the functional level (Figure 7B and Supplementary Table S5). Most of the differentially abundant genes belonging to methane metabolism [ko00680], benzoate degradation [ko00362], metabolism of xenobiotics by cytochrome P450 [ko00980], sulfur and nitrogen metabolism pathways [ko00920, ko00910] were found enriched by capping in wetland treatment. In contrast, most of the genes belonging to the above-stated pathways were depleted by capping in upland treatments. In methane metabolism pathways, acetyl-CoA synthetase (K01895; ACSS1_2, acs) was the most abundant enzyme (Figure 7 and Supplementary Table S5), which is involved in carbon metabolism and methanogenesis (acetate to methane). The second most abundant enzyme was glycine hydroxymethyltransferase (K00600; glyA, SHMT; glycine hydroxymethyltransferase) which converts formaldehyde (the product of methane oxidation) to C2 or C3 for energy source and assimilation for biosynthesis. In benzoate degradation, KEGG Orthology (KO) K00626 (ACAT, atoB; acetyl-CoA C-acetyltransferase) and K07516 (fadN; 3-hydroxyacyl-CoA dehydrogenase) involved in the conversion of benzoyl-CoA to acetyl-CoA were abundant. In Sulfur metabolism, genes belonging to K01011 (TST, MPST, sseA; thiosulfate/3-mercaptopyruvate sulfurtransferase) were dominant. Moreover, genes related to sulfur oxidation (dsrABC, aprAB, and sat), and pathways for thiosulfate oxidation (soxABCXYZ genes) were also found to be differentially affected by time and capping treatments. For nitrogen metabolism, genes encoding for glutamine synthetase, nitrate reductase, including dissimilatory nitrate reduction (K00370; narG, narZ, nxrA) and nitrogen fixation (nifDKH) were enriched by time and wetland capping treatments (Supplementary Table S5).
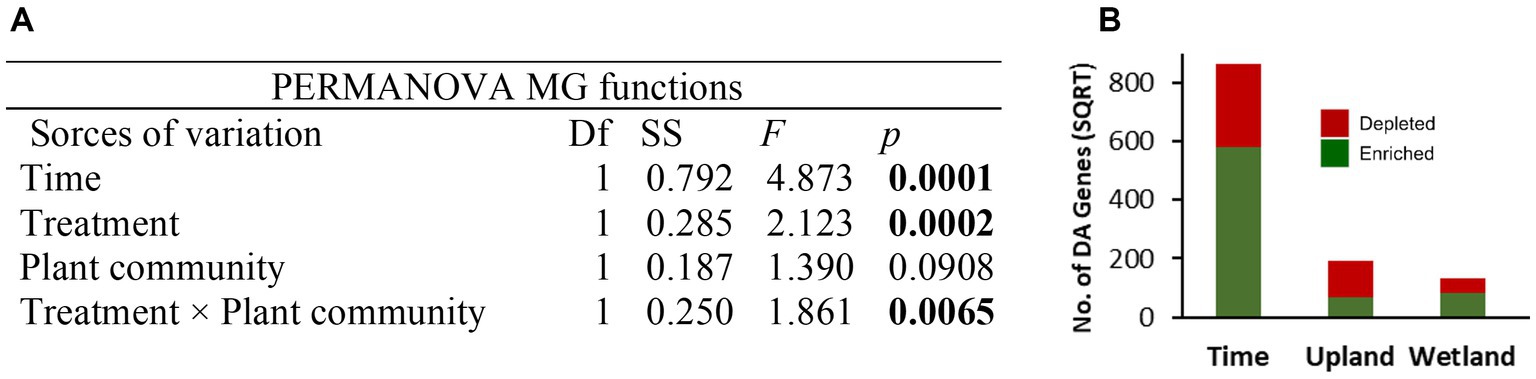
Figure 6. Permutational multivariate analysis of variance (PERMANOVA, 9,999 permutations) on Bray–Curtis dissimilarity matrix based on shotgun metagenomic data after normalization of gene abundance table using the geometric mean of pairwise ratios (GMPR) method (A). The total number of differentially abundant (DA) genes are shown for each group comparison between different time points (May 2019 vs. Oct 2020) and caping treatments (30 cm vs. control) of upland and wetland community (B), details of the DA genes are provided in Figure 7. PERMANOVA, permutational multivariate analysis of variance.
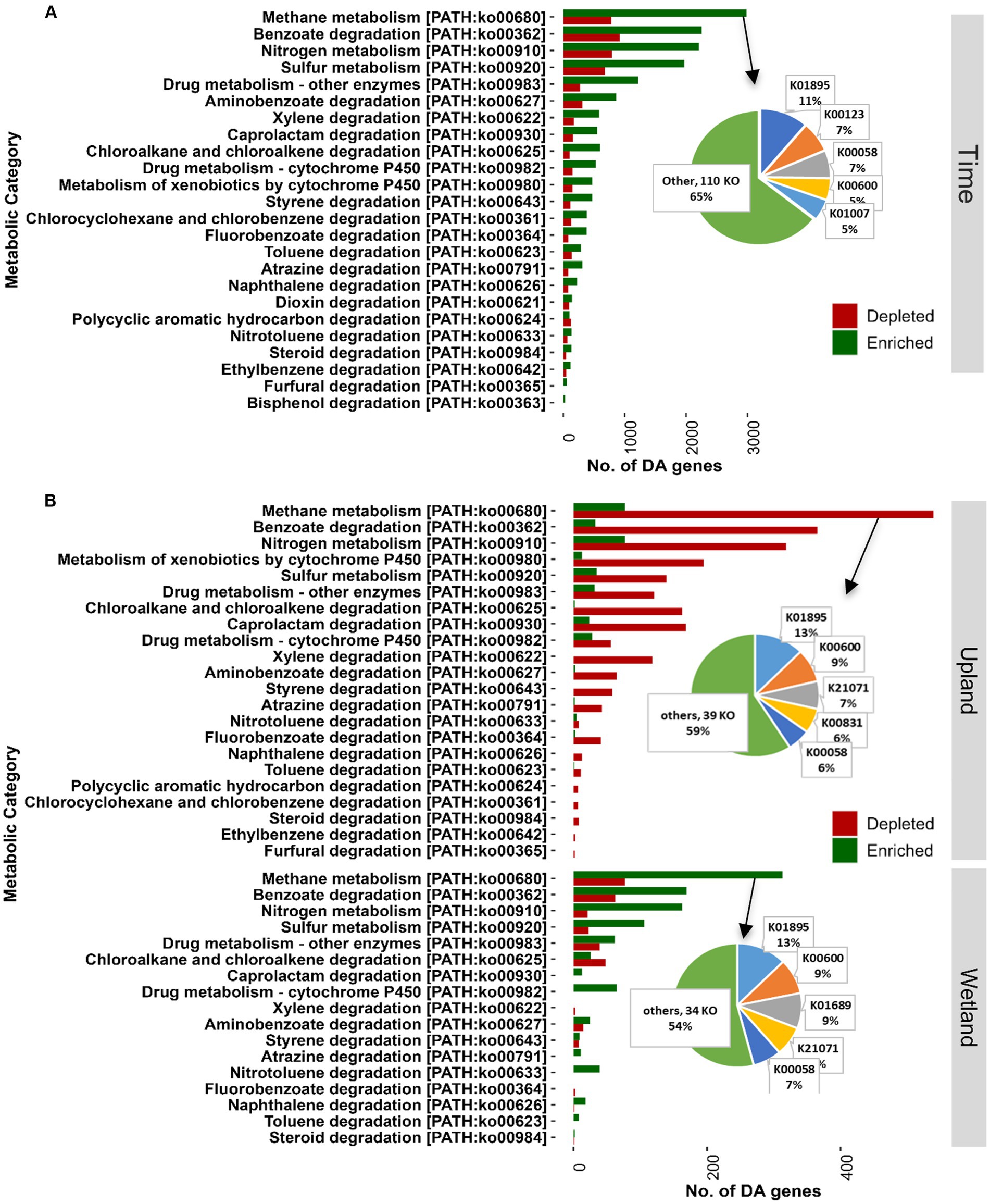
Figure 7. Shotgun metagenomics analysis showing KEGG functional categories for energy metabolism (09102, methane, sulfur, and nitrogen) and xenobiotics biodegradation (09111). Differentially abundant genes were identified between different time points (May 2019 vs. Oct 2020; A) and the caping treatments (30 cm vs. control, B). Genes were considered as differentially abundant only if FDR-adjusted p-values were significant (p < 0.05). The list of differentially abundant genes assigned to KEGG Orthology and KEGG functional categories is provided in Supplementary Table S5. 0 cm, control; DA, differentially abundant; FDR, false discovery rate; KEGG, Kyoto Encyclopedia of Genes and Genomes.
In the present study, prokaryotic and fungal communities were monitored for two growing seasons (GS) in columns composed of thickened oil sands tailings (TT) without capping (0 cm) or with a thin layer of PMM capping (10 or 30 cm) and planted with one of two distinct native boreal plant communities (upland or wetland). Bacterial diversity greatly increased after planting compared to baseline, with a more significant effect in GS1 than in GS2. Likewise, several microbial taxa as well as critical microbial functions in the tailings were significantly enriched after two growing seasons. These changes correlated with the aboveground plant growth, which increased over time (Supplementary Figure S3). The variations in microbial diversity and functions with time were expected as the establishment of plants and the increase in vegetation cover lead to an increase in root biomass and to rhizodeposition-driven changes in soil chemical, physical, and biological properties (Masse et al., 2017; Mohapatra et al., 2021). Most of the bacterial taxa that were enriched over time were potential sulfur-oxidizing bacteria (SOB), ammonia-oxidizing bacteria (AOB), and heterotrophic microorganisms which can contribute to nutrient cycling, plant litter decomposition, and xenobiotic degradation in soils (Mohapatra et al., 2021). Of note, an increase in the relative abundances of Acidobacteria, Actinobacteriota, Chloroflexi, and Planctomycetota was observed over time in tailings when compared to baseline samples. These taxa are among the most abundant bacterial phyla across soil habitats, which positively correlate with the mineralization of organic matter and nutrient cycling (Fierer et al., 2010; Delgado-Baquerizo et al., 2018; Heo et al., 2020). The increase in plant growth and resulting carbon input and nutrient turnover in soil likely influenced the tailings microbial community, favoring taxa more adapted to conditions created by vegetation establishment (Rousk et al., 2010a; Semchenko et al., 2022). Importantly, the uncultured bacteria Acidithiobacillaceae KCM-B-112 was significantly increased after two growing seasons when compared to baseline samples. Another study found Acidithiobacillaceae KCM-B-112 as abundant taxa in oil-contaminated soil during phytoremediation with poplars (Lopez-Echartea et al., 2020). Here, the significant enrichment of such sulfur-oxidizing acidophilic taxa of the Acidithiobacillaceae family and other SOB like Sulfuritalea suggests acidification of the alkaline tailings (initial pH of 8.7) over time. In fact, it has been shown that Acidithiobacillus thiooxidans decreased the pH in alkaline dolomitic tailings, which further increased nutrient solubilization, especially for phosphorus (Dhakar et al., 2015). On the other hand, typical tailings-associated bacteria (Wilson et al., 2016), particularly Burkholderiales (Comamonadaceae, Hydrogenophilaceae) were outcompeted over time. Similarly, a study found Comamonadaceae in young barren soil, which decreased over time with soil age and was completely depleted after about 20 years (Nemergut et al., 2007). In general, Polaromonas is found in association with unvegetated soil due to its metabolic versatility and capability to survive in a dormant state (Darcy et al., 2011).
The presence of a PMM cap mainly influenced bacterial alpha diversity and evenness (not richness), beta diversity, and the relative abundance of several dominant bacterial taxa depending upon vegetation type, with a generally limited impact of the thickness of the capping layer (10 cm vs. 30 cm). PMM cover can create micro-level conditions which can impact plant growth, microbial interactions as well as soil properties in the underlying tailings layer, resulting in the enrichment or depletion of specific taxa and functions (Jamro et al., 2014; Duan et al., 2015; Samad et al., 2022). As an example, capping could favor the growth in tailings of some opportunistic taxa that can metabolize the refractory carbon or other nutrients provided by the PMM layer. PMM capping can also alter microbial growth due to changes in oxygen supplies, moisture, pH, pore and particle size, and nutrient fluxes compared to the underlying tailings layer, all of which have been shown to influence the bacterial community composition in soils (Rousk et al., 2010a; Khodadad et al., 2011; Gschwend et al., 2021). Here, the observed changes in microbial communities due to capping were primarily related to changes in the relative contribution of specific bacterial taxa and were mostly observed in the upland plant community. For instance, abundant taxa assigned to Thiobacillus and Nitrosomonadaceae involved in sulfur and/or nitrogen cycling were enriched in tailings with a PMM cap. Thiobacillus (enriched with capping in upland treatments) is a neutrophilic bacteria that can oxidize sulfur in oxic environment through a sulfur-oxidizing multienzyme system (Sox) and in semi-oxic or anoxic environments through the reverse dissimilatory sulfite reductase (rDSR) pathways (Whaley-Martin et al., 2023). Higher relative abundances of other organisms involved in the oxidation of reduced compounds, like Sulfuritalea and Hydrogenophaga, were observed with capping, again mostly in the upland vegetation treatment, possibly due to the generally dryer conditions (Supplementary Figure S2), root-driven transport of O2 to tailings layers, the presence of alternative electron acceptor such as nitrate, or the use of oxygen-independent rDSR pathways for sulfur oxidation. In fact, sox and dsr genes were differentially abundant with capping. Overall, while the mechanisms remain unclear, the enrichment of such a broad diversity of oxidizing microorganisms indicate that capping did not slow down mineral oxidation processes in the tailing layer.
Metagenomics analysis of key biogeochemical processes, including carbon, nitrogen, and sulfur cycling, showed that the dynamics of energy utilization and transformation also changed with time and, to a lower extent, with capping treatment depending upon the vegetation type. Several genes associated with important soil functions were enriched over time, which can be linked to the development of a win/win partnership between microbes and plants (Liu et al., 2014; Hannula et al., 2019; Wu et al., 2020). Indeed, vegetation growth can supply energy and resources for heterotrophic microorganisms, therefore influencing metabolic functions (Li et al., 2017), and prior studies have shown that plant species influence the microbial composition and associated functions (Wu et al., 2020; Gagnon et al., 2020a). Interestingly here, while capping in the presence of wetland vegetation increased the energy-related metabolic functions (carbon, nitrogen, and sulfur metabolism), the upland treatments showed a contrasting effect, with most of the energy-related metabolic functions being depleted with capping. Similarly, capping also significantly decreased bacterial evenness in the upland but not in the wetland treatment. Previous studies have shown that evenness rather than richness responded rapidly in different stress conditions (Wittebolle et al., 2009; Yi et al., 2022). When microbial communities are highly uneven or exhibit extreme dominance by one or a few species, their functioning is less resilient to environmental stress (Wittebolle et al., 2009; Junkins et al., 2022). For example, manipulation in microbial evenness revealed that resistance to salinity stress was lower when initial microbial communities were uneven (Wittebolle et al., 2009), which suggests that functionally redundant species can better contribute to the ecosystem functioning under stressful conditions. This indicates that aspects of diversity other than microbial richness can play a critical role in community functional stability and may be of interest for further investigation of the effectiveness of reclamation treatments (Shade et al., 2012).
Fungal communities in tailings were dominated by Ascomycetes, with a high proportion of the order Helotiales, but also included a basidiomycetous component dominated by Agaricales. Based on shotgun metagenomics, fungal biomass was very low in tailings (<0.01% of total sequences), and most of the fungal microbiome (especially in the baseline samples) was unclassifiable below the phylum level, which makes it difficult to infer the role of tailings fungal community. Past surveys using 18S rRNA gene sequencing showed that more than 40% of fungal operational taxonomic units (OTUs) were unclassified below the phylum level in the oil sands environment (Richardson et al., 2020). Nevertheless, in the present study, the fungal richness and relative abundance of several dominant taxa significantly increased with time. Capping suppressed some potential plant pathogenic fungi such as Plectosphaerella and Calophoma (Raimondo and Carlucci, 2018; Hou et al., 2020) while substantially increasing the relative abundance of the beneficial plant-associated fungi Serendipta and Hyaloscypha in upland and wetland treatments (Vohník et al., 2016; Fehrer et al., 2019; Bzdyk et al., 2022). Serendipta and other beneficial fungal taxa from the Helotiales (Jean et al., 2021) and Sebacinales (Vohník et al., 2016) orders were also detected in PMM, indicating that capping material can serve as a source of potentially beneficial microorganisms to help vegetation establishment. These fungi may promote plant growth and alleviate environmental stress through enhanced phytohormone synthesis, nutrient cycling and uptake, and contaminant biodegradation (Gonzalez et al., 2018; del Barrio-Duque et al., 2019; Jean et al., 2021). Moreover, Serendipta can positively and synergistically interact with bacteria, including Rhizobium (also detected in PMM), and positive impacts on host plants of these intimate inter-kingdom associations have been observed (del Barrio-Duque et al., 2019; Mahdi et al., 2022). Such beneficial interactions are likely to increase reclamation success and further support the use of PMM as capping material for tailings.
Different responses of bacterial and fungal communities were observed in the presence of wetland and upland vegetation (Hannula et al., 2019; Gschwend et al., 2021). Earlier investigations have confirmed that tree or plant species can contribute to differences in soil properties and, in turn, microbial communities (Samad et al., 2017; Vuong et al., 2020). Indeed, different plant species can produce various microbial resources through root exudates, root cap sloughing, and root turnover, and can be colonized by distinct populations of more or less diverse microorganisms (Zhang et al., 2017; Gagnon et al., 2020b). The plant’s physiological ability to provide oxygen to the rhizosphere also differs among plant species (Callaway, 1995, 1997), and different vegetation types may therefore affect oxygen availability in the underline layer, which is an important driver of microbial composition and functions (Callaway, 1995; Junkins et al., 2022). Upland and wetland species have heterogeneous distributions of shallow and deep roots and vertical stratification of soil resources that can create finely partitioned niches with different microbial resources (Phillips et al., 2014; Vergani et al., 2017). Therefore, the differences in root architecture and growth can have a contrasting effect on soil resources and microbial assembly (Steinauer et al., 2020). Indeed, plant species and pH have been reported as the main factors influencing alpha diversity in PMM-capped and reconstructed oil sand soils (Masse et al., 2017; Mitter et al., 2017). Additionally, the abundance of wetland species like Salix bebbiana was positively correlated with bacterial alpha diversity, while the abundance of an upland species, Pinus banksiana, was inversely related to alpha diversity (Masse et al., 2017). A similar trend was observed in this study, where alpha diversity (Shannon) was inversely correlated with P. banksiana survival [Pearson’s correlation coefficient (r) = −0.90] and growth (r = −0.21) but was positively correlated with Salix bebbiana growth (r = 0.77). Wetland species such as willows have better capability to adapt to oxygen shortage conditions and reconstructed environments and concomitant microbial facilitation in such soil environments (Vartapetian and Jackson, 1997). The compatibility of peat-mineral mix (PMM) capping with upland and wetland vegetation and its impact on belowground physicochemical and biological interactions need to be further explored.
The results from this study provide valuable insights into the succession patterns of microbial communities following the application of organic capping and plantation in oil sands thickened tailings. Depending on the plant community, the microbial structural and functional features varied across the capping treatments. Noteworthy, wetland capping treatments enhanced critical functions related to nutrient cycling and xenobiotic biodegradation. These results suggest that selecting the right combination of capping material and vegetation type may improve microbially-driven processes during the reclamation of oil sands tailings. In line with plant establishment, soil microbial communities have also undergone important changes over two growing seasons, indicating the importance of tracking microbial communities and associated services over extended periods of time during the management and reclamation of tailings. Still, it is difficult to fully disentangle the effect of capping substrate on underline tailings-associated microbial communities due to the complex interplay between multiple biotic and abiotic factors that can change across a few microns in layered soils with distinct textural differences. Further long-term research is required to gain a more comprehensive understanding of microbial responses in the context of oil sands tailings reclamation, with a particular focus on compartments that are more closely associated with plants, such as the rhizosphere and endosphere. A thorough comprehension of soil–plant-microorganism interactions can assist in determining the optimal conditions for capping placement and plant establishment on mine tailings to develop better reclamation practices in such harsh environments.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
CM, ASé, and DD contributed to the conception and design of the study. DD conducted greenhouse experiments. ASa contributed to the lab experiments, performed bioinformatics analysis, prepared figures, and wrote the manuscript. M-JM contributed to lab experiments. PG contributed to the bioinformatic analysis of shotgun metagenomic data. All authors contributed to the manuscript’s revision and read and approved the submitted version.
Funding for this research came from the Natural Resources Canada Office of Energy Research and Development.
We gratefully acknowledge Imperial Oil Resources Limited for their support and contribution for this work. We would also like to acknowledge Jessica Hudson for assistance in sample collection and Angeline Van Dongen for reviewing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1168653/full#supplementary-material
Abarenkov, K., Henrik Nilsson, R., Larsson, K.-H., Alexander, I. J., Eberhardt, U., Erland, S., et al. (2010). The UNITE database for molecular identification of fungi--recent updates and future perspectives. New Phytol. 186, 281–285. doi: 10.1111/j.1469-8137.2009.03160.x
Alberta Energy Regulator (2020). State of fluid tailings management for mineable oil sands. Calgary, AB: Alberta Energy Regulator.
Alberta Energy Regulator (2022). Directive 085: fluid tailings management for oil sands mining projects. Calgary, AB: Alberta Energy Regulator.
Berta, G., Fusconi, A., and Hooker, J. E. (2002). “Arbuscular mycorrhizal modifications to plant root systems: scale, mechanisms and consequences” in Mycorrhizal technology in agriculture: from genes to bioproducts. eds. S. Gianinazzi, H. Schüepp, J. M. Barea, and K. Haselwandter (Basel: Birkhäuser), 71–85.
Birnbaum, C., Bradshaw, L. E., Ruthrof, K. X., and Fontaine, J. B. (2017). Topsoil stockpiling in restoration: impact of storage time on plant growth and symbiotic soil biota. Ecol. Res. 35, 237–245. doi: 10.3368/er.35.3.237
Bisanz, J. (2022). Tutorial: integrating QIIME2 and R for data visualization and analysis using qiime2R (v0.99.6). Available at: https://github.com/jbisanz/qiime2R.
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bzdyk, R. M., Sikora, K., Studnicki, M., and Aleksandrowicz-Trzcińska, M. (2022). Communities of mycorrhizal fungi among seedlings of scots pine (Pinus sylvestris L.) growing on a clearcut in microsites generated by different site-preparation methods. Forests 13:353. doi: 10.3390/f13020353
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Callaway, R. M. (1995). Positive interactions among plants. Bot. Rev. 61, 306–349. doi: 10.1007/BF02912621
Callaway, R. M. (1997). Positive interactions in plant communities and the individualistic-continuum concept. Oecologia 112, 143–149. doi: 10.1007/s004420050293
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chalaturnyk, R. J., Don Scott, J., and Özüm, B. (2002). Management of oil sands tailings. Pet. Sci. Technol. 20, 1025–1046. doi: 10.1081/LFT-120003695
Chegounian, P., Zerriffi, H., and Yadav, V. G. (2020). Engineering microbes for remediation of oil sands tailings. Trends Biotechnol. 38, 1192–1196. doi: 10.1016/j.tibtech.2020.04.007
Chen, L., Reeve, J., Zhang, L., Huang, S., Wang, X., and Chen, J. (2018). GMPR: a robust normalization method for zero-inflated count data with application to microbiome sequencing data. PeerJ 6:e4600. doi: 10.7717/peerj.4600
Cossey, H. L., Batycky, A. E., Kaminsky, H., and Ulrich, A. C. (2021). Geochemical stability of oil sands tailings in mine closure landforms. Fortschr. Mineral. 11:830. doi: 10.3390/min11080830
Darcy, J. L., Lynch, R. C., King, A. J., Robeson, M. S., and Schmidt, S. K. (2011). Global distribution of polaromonas phylotypes - evidence for a highly successful dispersal capacity. PLoS One 6:e23742. doi: 10.1371/journal.pone.0023742
Degenhardt, D., Van Dongen, A., Hudson, J. J., Utting, N., and Schreiber, S. G. (2023). Growth and survival of native wetland species in shallow capped centrifuged tailings and co-mixed tailings: a meso-scale greenhouse study. Can. J. Soil. Sci. doi: 10.1139/CJSS-2022-0129
del Barrio-Duque, A., Ley, J., Samad, A., Antonielli, L., Sessitsch, A., and Compant, S. (2019). Beneficial endophytic bacteria-Serendipita indica interaction for crop enhancement and resistance to phytopathogens. Front. Microbiol. 10:2888. doi: 10.3389/fmicb.2019.02888
Delgado-Baquerizo, M., Oliverio, A. M., Brewer, T. E., Benavent-González, A., Eldridge, D. J., Bardgett, R. D., et al. (2018). A global atlas of the dominant bacteria found in soil. Science 359, 320–325. doi: 10.1126/science.aap9516
Dhakar, S., Ali, N., Chauhan, R. S., and Yadav, V. (2015). Potentiality of Acidithiobacillus thiooxidans in microbial solubilization of phosphate mine tailings. J. Degrad. Min. Lands Manag. 2, 355–360. doi: 10.15243/jdmlm.2015.023.355
Dhar, A., Comeau, P. G., Karst, J., Pinno, B. D., Chang, S. X., Naeth, A. M., et al. (2018). Plant community development following reclamation of oil sands mine sites in the boreal forest: a review. Environ. Rev. 26, 286–298. doi: 10.1139/er-2017-0091
Dhar, A., Comeau, P. G., Naeth, M. A., Pinno, B. D., and Vassov, R. (2020). Plant community development following reclamation of oil sands mines using four cover soil types in northern Alberta. Restor. Ecol. 28, 82–92. doi: 10.1111/rec.13039
Dimitriu, P. A., Prescott, C. E., Quideau, S. A., and Grayston, S. J. (2010). Impact of reclamation of surface-mined boreal forest soils on microbial community composition and function. Soil Biol. Biochem. 42, 2289–2297. doi: 10.1016/j.soilbio.2010.09.001
Duan, M., House, J., and Chang, S. X. (2015). Limiting factors for lodgepole pine (Pinus contorta) and white spruce (Picea glauca) growth differ in some reconstructed sites in the Athabasca oil sands region. Ecol. Eng. 75, 323–331. doi: 10.1016/j.ecoleng.2014.12.010
Eddy, S. R. (2011). Accelerated Profile HMM Searches. PLoS Comput Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Errington, R. C., and Pinno, B. D. (2015). Early successional plant community dynamics on a reclaimed oil sands mine in comparison with natural boreal forest communities. Écoscience 22, 133–144. doi: 10.1080/11956860.2016.1169385
Fehrer, J., Réblová, M., Bambasová, V., and Vohník, M. (2019). The root-symbiotic Rhizoscyphus ericae aggregate and Hyaloscypha (Leotiomycetes) are congeneric: phylogenetic and experimental evidence. Stud. Mycol. 92, 195–225. doi: 10.1016/j.simyco.2018.10.004
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–230. doi: 10.1093/nar/gkt1223
Fierer, N., Nemergut, D., Knight, R., and Craine, J. M. (2010). Changes through time: integrating microorganisms into the study of succession. Res. Microbiol. 161, 635–642. doi: 10.1016/j.resmic.2010.06.002
Foght, J. M., Gieg, L. M., and Siddique, T. (2017). The microbiology of oil sands tailings: past, present, future. FEMS Microbiol. Ecol. 93. doi: 10.1093/femsec/fix034
Gagnon, V., Rodrigue-Morin, M., Tremblay, J., Wasserscheid, J., Champagne, J., Bellenger, J.-P., et al. (2020a). Life in mine tailings: microbial population structure across the bulk soil, rhizosphere, and roots of boreal species colonizing mine tailings in northwestern Québec. Ann. Microbiol. 70, 1–18. doi: 10.1186/s13213-020-01582-9
Gagnon, V., Rodrigue-Morin, M., Tremblay, J., Wasserscheid, J., Champagne, J., Bellenger, J.-P., et al. (2020b). Vegetation drives the structure of active microbial communities on an acidogenic mine tailings deposit. PeerJ 8:e10109. doi: 10.7717/peerj.10109
Gamalero, E., Trotta, A., Massa, N., Copetta, A., Martinotti, M. G., and Berta, G. (2004). Impact of two fluorescent pseudomonads and an arbuscular mycorrhizal fungus on tomato plant growth, root architecture and P acquisition. Mycorrhiza 14, 185–192. doi: 10.1007/s00572-003-0256-3
Gazitúa, M. C., Morgante, V., Poupin, M. J., Ledger, T., Rodríguez-Valdecantos, G., Herrera, C., et al. (2021). The microbial community from the early-plant colonizer (Baccharis linearis) is required for plant establishment on copper mine tailings. Sci. Rep. 11:10448. doi: 10.1038/s41598-021-89769-1
Gonzalez, E., Pitre, F. E., Pagé, A. P., Marleau, J., Guidi Nissim, W., St-Arnaud, M., et al. (2018). Trees, fungi and bacteria: tripartite metatranscriptomics of a root microbiome responding to soil contamination. Microbiome 6:53. doi: 10.1186/s40168-018-0432-5
Gschwend, F., Hartmann, M., Mayerhofer, J., Hug, A.-S., Enkerli, J., Gubler, A., et al. (2021). Site and land-use associations of soil bacteria and fungi define core and indicative taxa. FEMS Microbiol. Ecol. 97:fiab165. doi: 10.1093/femsec/fiab165
Hannula, S. E., Kielak, A. M., Steinauer, K., Huberty, M., Jongen, R., Long, J. R. D., et al. (2019). Time after time: temporal variation in the effects of grass and forb species on soil bacterial and fungal communities. MBio. 10:e02635-19. doi: 10.1128/mBio.02635-19
Hargreaves, S. K., and Hofmockel, K. S. (2014). Physiological shifts in the microbial community drive changes in enzyme activity in a perennial agroecosystem. Biogeochemistry 117, 67–79. doi: 10.1007/s10533-013-9893-6
Harris, J. (2009). Soil microbial communities and restoration ecology: facilitators or followers. Science 325, 573–574. doi: 10.1126/science.1172975
Heo, Y.-M., Lee, H., Kwon, S.-L., Yoo, Y., Kim, D., Han, S.-I., et al. (2020). Influence of tree vegetation on soil microbial communities in temperate forests and their potential as a proactive Indicator of vegetation shift due to climate change. Sustainability 12:10591. doi: 10.3390/su122410591
Hou, L. W., Groenewald, J. Z., Pfenning, L. H., Yarden, O., Crous, P. W., and Cai, L. (2020). The phoma-like dilemma. Stud. Mycol. 96, 309–396. doi: 10.1016/j.simyco.2020.05.001
Huntemann, M., Ivanova, N. N., Mavromatis, K., Tripp, H. J., Paez-Espino, D., Tennessen, K., et al. (2016). The standard operating procedure of the DOE-JGI Metagenome Annotation Pipeline (MAP v.4). Standards Genom. Sci. 11:17. doi: 10.1186/s40793-016-0138-x
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Illumina., (2013). 16S metagenomic sequencing library preparation. Preparing 16S ribosomal RNA gene amplicons for the illumina MiSeq (Part #15044223 Rev. B). San Diego, CA, USA: Illumina.
Jamro, G. M., Chang, S. X., and Naeth, M. A. (2014). Organic capping type affected nitrogen availability and associated enzyme activities in reconstructed oil sands soils in Alberta, Canada. Ecol. Eng. 73, 92–101. doi: 10.1016/j.ecoleng.2014.09.005
Jean, R., Khasa, D. P., and Boudreau, S. (2021). Effects of one dark septate endophytic fungal and two Helotiales strains on the growth of plane-leaved willow (Salix planifolia) cuttings on iron ore waste rock. Botany 99, 725–733. doi: 10.1139/cjb-2021-0043
Junkins, E. N., McWhirter, J. B., McCall, L.-I., and Stevenson, B. S. (2022). Environmental structure impacts microbial composition and secondary metabolism. ISME Commun. 2, 1–10. doi: 10.1038/s43705-022-00097-5
Kanehisa, M. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Khodadad, C. L. M., Zimmerman, A. R., Green, S. J., Uthandi, S., and Foster, J. S. (2011). Taxa-specific changes in soil microbial community composition induced by pyrogenic carbon amendments. Soil Biol. Biochem. 43, 385–392. doi: 10.1016/j.soilbio.2010.11.005
Lalonde, R. S., Pinno, B. D., MacKenzie, M. D., and Utting, N. (2020). Capping dewatered oil sands fluid fine tailings with salvaged reclamation soils at varying depths to grow woody plants. Can. J. Soil Sci. 100, 1–12. doi: 10.1139/cjss-2019-0120
Li, H., and Durbin, R. (2010). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Y., Sun, Q., Zhan, J., Yang, Y., and Wang, D. (2017). Soil-covered strategy for ecological restoration alters the bacterial community structure and predictive energy metabolic functions in mine tailings profiles. Appl. Microbiol. Biotechnol. 101, 2549–2561. doi: 10.1007/s00253-016-7969-7
Lin, C., Esmaeili, P., and Myers, R. D. (2015). Fluid tailings flocculation and dewatering using chemically-induced micro-agglomerates. Available at: https://patents.google.com/patent/CA2767510C/en (Accessed January 25, 2023).
Lin, H., and Peddada, S. D. (2020). Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11:3514. doi: 10.1038/s41467-020-17041-7
Liu, Y., Yang, H., Li, X., and Xing, Z. (2014). Effects of biological soil crusts on soil enzyme activities in revegetated areas of the Tengger Desert, China. Appl. Soil Ecol. 80, 6–14. doi: 10.1016/j.apsoil.2014.03.015
Lopez-Echartea, E., Strejcek, M., Mukherjee, S., Uhlik, O., and Yrjälä, K. (2020). Bacterial succession in oil-contaminated soil under phytoremediation with poplars. Chemosphere 243:125242. doi: 10.1016/j.chemosphere.2019.125242
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Ma, B., Li, X., and Chang, S. X. (2017). Capping material type affects rhizosphere bacteria community structure in the cover soil in oil sands reclamation. J. Soils Sediments 17, 2516–2523. doi: 10.1007/s11368-017-1696-2
MacKenzie, M. D., and Quideau, S. A. (2010). Microbial community structure and nutrient availability in oil sands reclaimed boreal soils. Appl. Soil Ecol. 44, 32–41. doi: 10.1016/j.apsoil.2009.09.002
Mahdi, L. K., Miyauchi, S., Uhlmann, C., Garrido-Oter, R., Langen, G., Wawra, S., et al. (2022). The fungal root endophyte Serendipita vermifera displays inter-kingdom synergistic beneficial effects with the microbiota in Arabidopsis thaliana and barley. ISME J. 16, 876–889. doi: 10.1038/s41396-021-01138-y
Masse, J., Prescott, C. E., Renaut, S., Terrat, Y., and Grayston, S. J. (2017). Plant community and nitrogen deposition as drivers of alpha and beta diversities of prokaryotes in reconstructed oil sand soils and natural boreal forest soils. Appl. Environ. Microbiol. 83, e03319–e03316. doi: 10.1128/AEM.03319-16
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an r package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meisner, A., Snoek, B. L., Nesme, J., Dent, E., Jacquiod, S., Classen, A. T., et al. (2021). Soil microbial legacies differ following drying-rewetting and freezing-thawing cycles. ISME J. 15, 1207–1221. doi: 10.1038/s41396-020-00844-3
Menkis, A., Burokienė, D., Gaitnieks, T., Uotila, A., Johannesson, H., Rosling, A., et al. (2012). Occurrence and impact of the root-rot biocontrol agent Phlebiopsis gigantea on soil fungal communities in Picea abies forests of northern Europe. FEMS Microbiol. Ecol. 81, 438–445. doi: 10.1111/j.1574-6941.2012.01366.x
Mitter, E. K., de Freitas, J. R., and Germida, J. J. (2017). Bacterial root microbiome of plants growing in oil sands reclamation covers. Front. Microbiol. 8:849. doi: 10.3389/fmicb.2017.00849
Mohapatra, M., Yadav, R., Rajput, V., Dharne, M. S., and Rastogi, G. (2021). Metagenomic analysis reveals genetic insights on biogeochemical cycling, xenobiotic degradation, and stress resistance in mudflat microbiome. J. Environ. Manag. 292:112738. doi: 10.1016/j.jenvman.2021.112738
Nemergut, D. R., Anderson, S. P., Cleveland, C. C., Martin, A. P., Miller, A. E., Seimon, A., et al. (2007). Microbial community succession in an unvegetated, recently deglaciated soil. Microb. Ecol. 53, 110–122. doi: 10.1007/s00248-006-9144-7
Olanrewaju, O. S., Glick, B. R., and Babalola, O. O. (2017). Mechanisms of action of plant growth promoting bacteria. World J. Microbiol. Biotechnol. 33:197. doi: 10.1007/s11274-017-2364-9
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples: primers for marine microbiome studies. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Parrott, J. L., Raine, J. C., McMaster, M. E., and Hewitt, L. M. (2019). Chronic toxicity of oil sands tailings pond sediments to early life stages of fathead minnow (Pimephales promelas). Heliyon 5:e02509. doi: 10.1016/j.heliyon.2019.e02509
Phillips, C. J., Marden, M., and Suzanne, L. M. (2014). Observations of root growth of young poplar and willow planting types. N. Z. J. For. Sci. 44:15. doi: 10.1186/s40490-014-0015-6
Picek, T., and Simek, M. (2000). Microbial responses to fluctuation of soil aeration status and redox conditions. Biol. Fertil. Soils 31, 315–322. doi: 10.1007/s003740050662
Pinheiro, J., Bates, D., DebRoy, S., Sarkar, D., Heisterkamp, S., Van Willigen, B., et al. (2022). Package ‘nlme.’ Linear and nonlinear mixed effects models, version 3. Available at: https://CRAN.R-project.org/package=nlme.
Poria, V., Dębiec-Andrzejewska, K., Fiodor, A., Lyzohub, M., Ajijah, N., Singh, S., et al. (2022). Plant growth-promoting bacteria (PGPB) integrated phytotechnology: a sustainable approach for remediation of marginal lands. Front. Plant Sci. 13:999866. doi: 10.3389/fpls.2022.999866
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
R Core Team (2022). R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Raimondo, M. L., and Carlucci, A. (2018). Characterization and pathogenicity assessment of Plectosphaerella species associated with stunting disease on tomato and pepper crops in Italy. Plant Pathol. 67, 626–641. doi: 10.1111/ppa.12766
Rath, K. M., Fierer, N., Murphy, D. V., and Rousk, J. (2019). Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 13, 836–846. doi: 10.1038/s41396-018-0313-8
Richardson, E., Bass, D., Smirnova, A., Paoli, L., Dunfield, P., and Dacks, J. B. (2020). Phylogenetic estimation of community composition and novel eukaryotic lineages in base mine Lake: an oil sands tailings reclamation site in northern Alberta. J. Eukaryot. Microbiol. 67, 86–99. doi: 10.1111/jeu.12757
Rivers, A. R. (2016). iTag amplicon sequencing for taxonomic identification at JGI Joint Genome Institute Available at: https://jgi.doe.gov/wp-content/uploads/2013/05/iTagger-methods.pdf.
Rousk, J., Bååth, E., Brookes, P. C., Lauber, C. L., Lozupone, C., Caporaso, J. G., et al. (2010a). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340–1351. doi: 10.1038/ismej.2010.58
Rousk, J., Brookes, P. C., and Bååth, E. (2010b). The microbial PLFA composition as affected by pH in an arable soil. Soil Biol. Biochem. 42, 516–520. doi: 10.1016/j.soilbio.2009.11.026
Rowland, S. M., Prescott, C. E., Grayston, S. J., Quideau, S. A., and Bradfield, G. E. (2009). Recreating a functioning forest soil in reclaimed oil sands in northern Alberta: an approach for measuring success in ecological restoration. J. Environ. Qual. 38, 1580–1590. doi: 10.2134/jeq2008.0317
Saborimanesh, N. (2021). Toward sustainable remediation of oil sands fine tailings-a review. J. Environ. Manag. 288:112418. doi: 10.1016/j.jenvman.2021.112418
Samad, A., Pelletier, G., Séguin, A., Degenhardt, D., Muench, D. G., and Martineau, C. (2022). Understanding willow transcriptional response in the context of oil sands tailings reclamation. Front. Plant Sci. 13:857535. doi: 10.3389/fpls.2022.857535
Samad, A., Trognitz, F., Compant, S., Antonielli, L., and Sessitsch, A. (2017). Shared and host-specific microbiome diversity and functioning of grapevine and accompanying weed plants. Environ. Microbiol. 19, 1407–1424. doi: 10.1111/1462-2920.13618
Sayers, E. W., Barrett, T., Benson, D. A., Bolton, E., Bryant, S. H., Canese, K., et al. (2011). Database resources of the national center for biotechnology information. Nucleic Acids Res. 39, D38–D51. doi: 10.1093/nar/gkq1172
Semchenko, M., Barry, K. E., de Vries, F. T., Mommer, L., Moora, M., and Maciá-Vicente, J. G. (2022). Deciphering the role of specialist and generalist plant–microbial interactions as drivers of plant–soil feedback. New Phytol. 234, 1929–1944. doi: 10.1111/nph.18118
Shade, A., Peter, H., Allison, S., Baho, D., Berga, M., Buergmann, H., et al. (2012). Fundamentals of microbial community resistance and resilience. Front. Microbiol. 3:417. doi: 10.3389/fmicb.2012.00417
Stefani, F., Isabel, N., Morency, M.-J., Lamothe, M., Nadeau, S., Lachance, D., et al. (2018). The impact of reconstructed soils following oil sands exploitation on aspen and its associated belowground microbiome. Sci. Rep. 8:2761. doi: 10.1038/s41598-018-20783-6
Steinauer, K., Heinen, R., Hannula, S. E., De Long, J. R., Huberty, M., Jongen, R., et al. (2020). Above-belowground linkages of functionally dissimilar plant communities and soil properties in a grassland experiment. Ecosphere 11:e03246. doi: 10.1002/ecs2.3246
Tremblay, J., Yergeau, E., Fortin, N., Cobanli, S., Elias, M., King, T. L., et al. (2017). Chemical dispersants enhance the activity of oil- and gas condensate-degrading marine bacteria. ISME J. 11, 2793–2808. doi: 10.1038/ismej.2017.129
Vartapetian, B. B., and Jackson, M. B. (1997). Plant adaptations to anaerobic stress. Ann. Bot. 79, 3–20. doi: 10.1093/oxfordjournals.aob.a010303
Vergani, C., Giadrossich, F., Buckley, P., Conedera, M., Pividori, M., Salbitano, F., et al. (2017). Root reinforcement dynamics of European coppice woodlands and their effect on shallow landslides: a review. Earth Sci. Rev. 167, 88–102. doi: 10.1016/j.earscirev.2017.02.002
Vimal, S. R., Singh, J. S., Arora, N. K., and Singh, S. (2017). Soil-plant-microbe interactions in stressed agriculture management: a review. Pedosphere 27, 177–192. doi: 10.1016/S1002-0160(17)60309-6
Vohník, M., Pánek, M., Fehrer, J., and Selosse, M.-A. (2016). Experimental evidence of ericoid mycorrhizal potential within Serendipitaceae (Sebacinales). Mycorrhiza 26, 831–846. doi: 10.1007/s00572-016-0717-0
Vuong, T.-M.-D., Zeng, J.-Y., and Man, X.-L. (2020). Soil fungal and bacterial communities in southern boreal forests of the greater Khingan Mountains and their relationship with soil properties. Sci. Rep. 10:22025. doi: 10.1038/s41598-020-79206-0
Wang, C., Harbottle, D., Liu, Q., and Xu, Z. (2014). Current state of fine mineral tailings treatment: a critical review on theory and practice. Miner. Eng. 58, 113–131. doi: 10.1016/j.mineng.2014.01.018
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5:27. doi: 10.1186/s40168-017-0237-y
Whaley-Martin, K. J., Chen, L.-X., Nelson, T. C., Gordon, J., Kantor, R., Twible, L. E., et al. (2023). O2 partitioning of sulfur oxidizing bacteria drives acidity and thiosulfate distributions in mining waters. Nat. Commun. 14:2006. doi: 10.1038/s41467-023-37426-8
White, T., Bruns, T., Lee, S., Taylor, J., Innis, M., Gelfand, D., et al. (1990). “Amplification and direct sequencing of fungal ribosomal rna genes for phylogenetics” in PCR protocols: a guide to methods and applications. eds. M. A. Innis, D. H. Gelfand, J. J. Sninsky and T. J. White (New York: Academic Press), 315–322.
Wickham, H. (2016). Package ‘ggplot2’: elegant graphics for data analysis. New York, NY: Springer-Verlag.
Wilson, S. L., Li, C., Ramos-Padrón, E., Nesbø, C., Soh, J., Sensen, C. W., et al. (2016). Oil sands tailings ponds harbour a small core prokaryotic microbiome and diverse accessory communities. J. Biotechnol. 235, 187–196. doi: 10.1016/j.jbiotec.2016.06.030
Wittebolle, L., Marzorati, M., Clement, L., Balloi, A., Daffonchio, D., Heylen, K., et al. (2009). Initial community evenness favours functionality under selective stress. Nature 458, 623–626. doi: 10.1038/nature07840
Wu, Z., Sun, L., Li, Y., and Sun, Q. (2020). Shifts in vegetation-associated microbial community in the reclamation of coal mining subsidence land. Environ. Eng. Sci. 37, 838–848. doi: 10.1089/ees.2019.0491
Xue, P.-P., Carrillo, Y., Pino, V., Minasny, B., and McBratney, A. B. (2018). Soil properties drive microbial community structure in a large scale transect in south eastern Australia. Sci. Rep. 8:11725. doi: 10.1038/s41598-018-30005-8
Yang, C., and Sun, J. (2020). Soil salinity drives the distribution patterns and ecological functions of Fungi in saline-alkali land in the Yellow River Delta, China. Front. Microbiol. 11:594284. doi: 10.3389/fmicb.2020.594284
Yi, M., Zhang, L., Li, Y., and Qian, Y. (2022). Structural, metabolic, and functional characteristics of soil microbial communities in response to benzo[a]pyrene stress. J. Hazard. Mater. 431:128632. doi: 10.1016/j.jhazmat.2022.128632
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Keywords: oil sands tailings, reclamation, metabarcoding, metagenomics, ecological restoration, soil microbiology, boreal forest
Citation: Samad A, Degenhardt D, Séguin A, Morency M-J, Gagné P and Martineau C (2023) Microbial community structural and functional differentiation in capped thickened oil sands tailings planted with native boreal species. Front. Microbiol. 14:1168653. doi: 10.3389/fmicb.2023.1168653
Edited by:
Hari Prasanna Deka Boruah, North East Institute of Science and Technology (CSIR), IndiaReviewed by:
Karolina Furtak, Institute of Soil Science and Plant Cultivation, PolandCopyright © 2023 Samad, Degenhardt, Séguin, Morency, Gagné and Martineau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine Martineau, Y2hyaXN0aW5lLm1hcnRpbmVhdUBucmNhbi1ybmNhbi5nYy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.