 Emilisa Frirdich
Emilisa Frirdich Jenny Vermeulen1
Jenny Vermeulen1 Jacob Biboy
Jacob Biboy- 1Department of Microbiology and Immunology, University of British Columbia, Vancouver, BC, Canada
- 2The Centre for Bacterial Cell Biology, Institute for Cell and Molecular Biosciences, Newcastle University, Newcastle upon Tyne, United Kingdom
Campylobacter jejuni is a Gram-negative helical bacterium. Its helical morphology, maintained by the peptidoglycan (PG) layer, plays a key role in its transmission in the environment, colonization, and pathogenic properties. The previously characterized PG hydrolases Pgp1 and Pgp2 are important for generating C. jejuni helical morphology, with deletion mutants being rod-shaped and showing alterations in their PG muropeptide profiles in comparison to the wild type. Homology searches and bioinformatics were used to identify additional gene products involved in C. jejuni morphogenesis: the putative bactofilin 1104 and the M23 peptidase domain-containing proteins 0166, 1105, and 1228. Deletions in the corresponding genes resulted in varying curved rod morphologies with changes in their PG muropeptide profiles. All changes in the mutants complemented except 1104. Overexpression of 1104 and 1105 also resulted in changes in the morphology and in the muropeptide profiles, suggesting that the dose of these two gene products influences these characteristics. The related helical ε-Proteobacterium Helicobacter pylori has characterized homologs of C. jejuni 1104, 1105, and 1228 proteins, yet deletion of the homologous genes in H. pylori had differing effects on H. pylori PG muropeptide profiles and/or morphology compared to the C. jejuni deletion mutants. It is therefore apparent that even related organisms with similar morphologies and homologous proteins can have diverse PG biosynthetic pathways, highlighting the importance of studying PG biosynthesis in related organisms.
1. Introduction
The Gram-negative bacterium Campylobacter jejuni is an important zoonotic pathogen that can cause severe disease in humans while asymptomatically colonizing the intestinal tracts of birds and other animals (Young et al., 2007; Bolton, 2015; Burnham and Hendrixson, 2018; Tegtmeyer et al., 2021). Campylobacter jejuni is a major cause of bacterial gastroenteritis worldwide, often outnumbering the more widely known and studied enteric pathogens Escherichia coli, Salmonella, and Shigella in the developed world (Sherman et al., 2010; Silva et al., 2011; Kaakoush et al., 2015; Sheppard and Maiden, 2015; Hansson et al., 2018). Evidence also suggests a global rise in C. jejuni infections over the last decade (Kaakoush et al., 2015).
The name Campylobacter means “curved rod,” derived from the Greek campylos (curved) and baktron (rod) describing the predominant morphology of C. jejuni in its most pathogenic form. As C. jejuni ages, and under stress and adverse growth conditions, it can adopt different morphologies such as that of a filamented helical rod (Justice et al., 2008; Cameron et al., 2012; Frirdich and Gaynor, 2013) and/or a coccoid form (Svensson et al., 2008; Jackson et al., 2009; Ikeda and Karlyshev, 2012; Frirdich et al., 2019). Rod-shaped variants have also been detected in C. jejuni populations by passage through the chicken gut or chick embryos (Field et al., 1993; Hänel et al., 2009), by laboratory passage (Esson et al., 2016; Frirdich et al., 2017), and by selection through laboratory exposure to the compound calcofluor white (CFW; Frirdich et al., 2017). Changes to the C. jejuni helical shape affect its biological properties and pathogenic potential (Cameron et al., 2012; Frirdich et al., 2012, 2014, 2019; Ha et al., 2016; Stahl et al., 2016), altering its ability to be transmitted, colonize, and interact with its host, all of which ultimately impact disease outcome. Therefore, studying C. jejuni morphology determination is a key aspect of understanding C. jejuni pathogenesis and survival.
Bacterial cell shape is maintained by the peptidoglycan (PG) sacculus (Vollmer et al., 2008a). PG is a macromolecular structure essential in most bacteria, including C. jejuni. It surrounds the cytoplasmic membrane to form a rigid mesh-like layer that protects the cell from lysis due to turgor, as well as maintaining morphology. The basic composition of PG is generally conserved among bacteria, consisting of glycan chains of β-1-4 linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues cross-linked by short peptides. Glycan strands terminate with 1,6-anhydro MurNAc (anhMurNAc) residues. In Gram-negative bacteria, the peptides primarily consist of L-Ala1-D-iso-glutamate2-mesodiaminopimelic acid (mDAP)3-D-Ala4-D-Ala5 residues and are attached to the MurNAc residue. Most cross-links between peptides of adjacent strands occur between the mDAP3 of one stem peptide and D-Ala4 of another. No other cross-links were detected in C. jejuni (Frirdich et al., 2012). PG structure shows a high diversity between bacterial species but is relatively well conserved within a species (Vollmer et al., 2008a; Pazos and Peters, 2019). However, variations within a species do occur depending on the strain, growth conditions, and growth phase (Vollmer, 2008; Pazos and Peters, 2019). In addition, bacteria can modulate their PG, and these structural modifications can serve as a protection strategy against environmental threats such as those posed by predatory PG hydrolytic proteins and antimicrobial compounds (Vollmer et al., 2008a; Yadav et al., 2018; Pazos and Peters, 2019). These variations can include differences in the length of the glycan strand, length and composition of the stem peptide, degree and type of cross-linking, and the presence or absence of modifications (Vollmer et al., 2008a; Yadav et al., 2018; Pazos and Peters, 2019).
Growth of the PG layer involves the highly coordinated activity of synthases and hydrolases regulated by factors such as cytoskeletal elements (Typas et al., 2012; Egan et al., 2020). PG subunits are incorporated into the growing glycan chain by transglycosylases; then, inter-strand peptide cross-linking is formed by transpeptidases. C. jejuni has a homolog of the PBP1a (CJJ811876_0536) synthetase, as well as homologs of the transpeptidases PBP2 (CJJ811876_0680; involved in cell elongation) and PBP3 (CJJ811876_0550; involved in cell division). PG hydrolases play an important role in PG biosynthesis by cleaving PG bonds and allowing insertion of new PG subunits during growth and elongation, ensuring cell separation during division, and remodeling the PG to generate shape (Vollmer et al., 2008b; Uehara and Bernhardt, 2011; Wyckoff et al., 2012; Frirdich and Gaynor, 2013; Cava and de Pedro, 2014; Do et al., 2020). This process is key in bacterial differentiation, enabling bacteria to adapt to different environmental conditions (Vollmer et al., 2008b; Wyckoff et al., 2012; Frirdich and Gaynor, 2013; Cava and de Pedro, 2014; Do et al., 2020). PG hydrolases also have numerous other functions such as spore maturation, roles in PG recycling, and as phage and predator lysins (Vollmer et al., 2008b; Wyckoff et al., 2012; Frirdich and Gaynor, 2013; Cava and de Pedro, 2014; Do et al., 2020). Differences in the complement of PG hydrolases between bacterial species contribute to variations in the muropeptide profile between organisms, can influence the morphology, and can impact overall bacterial fitness and pathogenesis.
Peptidoglycan hydrolases are classified based on their cleavage specificity (Vollmer et al., 2008b; Vermassen et al., 2019). Glycosidases cleave the PG backbone. Amidases cleave the peptide from the glycan backbone, while peptidases cleave within the peptides. One amidase, AmiA, has been identified in C. jejuni (Frirdich et al., 2019). Peptidases can be further classified into carboxypeptidases that cleave the C-terminal peptide and endopeptidases that cleave within the peptides. Three PG peptidases have been characterized in C. jejuni: Pgp1, Pgp2, and Pgp3, which are all involved in helical morphology generation (Frirdich et al., 2012, 2014; Frirdich and Gaynor, 2013; Min et al., 2020). Pgp1 is a DL carboxypeptidase that cleaves PG tripeptides to dipeptides. We identified Pgp1 as a hypo-reactive mutant in a transposon mutant screen using calcofluor white (CFW), a fluorescent dye binding β-1-3 and β-1-4 linked sugars such as those found in PG (Frirdich et al., 2012). Altered CFW reactivity correlates with changes in C. jeuni pathogenic properties (McLennan et al., 2008). We identified Pgp2 bioinformatically by STRING analysis which suggested Pgp2 as a potential interaction partner for Pgp1. Further analyses showed that Pgp2 is an LD-carboxypeptidase cleaving PG tetrapeptides to tripeptides, providing the substrate for Pgp1 (Frirdich et al., 2014). No additional PG hydrolases were identified using these methods. Deletion mutants in pgp1 and pgp2 were rod-shaped and had an altered PG muropeptide profile. Pgp3, characterized by Min et al. (2020), was shown to have DD-carboxypeptidase (cleaving penta- to tetra-peptides) and DD-endopeptidase activity (cleaving tetra-tri peptide cross-links) using synthetic PG substrates, with the DD-endopeptidase activity also having been demonstrated on E. coli sacculi (Min et al., 2020). A ∆pgp3 mutant has a curved rod appearance, but changes in the mutant PG profile have yet to be determined.
In this study, bioinformatic approaches were used to identify additional C. jejuni PG hydrolases CJJ81176_0166 (henceforth referred to by the gene number, 0166), CJJ81176_1105 (1105), CJJ81176_1228 (1228), and the bactofilin CJJ81176_1104 (1104). The roles of these proteins in the maintenance of C. jejuni helical morphology and in PG biosynthesis were analyzed by examining the morphology and muropeptide profiles of mutant, complement, and overexpressing strains. Deletion of 0166, 1104, 1105, and 1228 produced strains with morphologies varying in curvature in comparison to the wild type. Unlike mutants in C. jejuni PG hydrolases pgp1 and pgp2, analysis of the mutant muropeptide profile of ∆0166, ∆1105, and ∆1228 displayed complex changes. In addition, overexpression with 1104 resulted in a more pronounced loss of curvature, while overexpression with 1105 resulted in a gain in curvature, both accompanied by muropeptide changes. The broad effects that the loss and/or overexpression of 0166, 1104, 1105, and 1228 have on the muropeptide composition, highlighted by analyses of double and triple mutants, support the idea that the presence and levels of these gene products have effects on each other and/or other PG biosynthetic proteins.
Like C. jejuni, Helicobacter pylori is a member of the ε-Proteobacteria and has a helical morphology. H. pylori has characterized homologs of 1104 (CcmA), 1105 (Csd1), and 1228 (Csd3/HdpA; henceforth referred to as Csd3 unless the specific hdpA mutant or HdpA protein is being referred to; Bonis et al., 2010; Sycuro et al., 2010). However, deletions in these homologs had differing morphological and/or muropeptide changes to the C. jejuni proteins (Sycuro et al., 2010). This indicates that the morphogenesis program of C. jejuni and H. pylori has unique differences, despite being related organisms with similar helical morphology and several homologous enzymes. Therefore, it is important to study the PG biosynthetic program in related bacterial pathogens and the biological role of homologous PG enzymes.
2. Materials and methods
2.1. Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study and their construction are described in the Supplementary Information. All strains were verified by PCR and sequencing. Unless otherwise indicated, C. jejuni strains were grown at 38°C in Mueller–Hinton (MH; Oxoid) broth or on 1.7% (w/v) agar plates supplemented with vancomycin (10 μg/ml) and trimethoprim (5 μg/ml), denoted MH-TV, under microaerobic/capnophilic conditions (6% O2, 12% CO2) in a Sanyo tri-gas incubator for plates or using the Oxoid CampyGen system for broth cultures. Growth media were supplemented with chloramphenicol (Cm; 20 μg/ml), kanamycin (Km; 50 μg/ml), or apramycin (Apr; 60 μg/ml) where appropriate. E. coli strains used for plasmid construction were grown at 38°C in Luria–Bertani (LB; Sigma) broth or 1.5% agar (w/v) agar plates and supplemented with ampicillin (Ap, 100 μg/ml), chloramphenicol (15 μg/ml), kanamycin (25 μg/ml), or apramycin (60 μg/ml) as necessary.
For growth analyses, C. jejuni strains were streaked from 16- to 18-h plate cultures and grown again on plates for 7–8 h. Bacteria were harvested in MH-TV broth and inoculated at an OD600 of 0.0005 into MH-TV broth and grown shaking for 18 h. Strains were subcultured to an OD600 of 0.005, and samples were taken at various timepoints for the colony-forming unit (CFU) and microscopic analysis. For morphological analysis, overnight cultures were subcultured to an OD600 of 0.05 and incubated for 4 h to generate mid-exponential phase cultures.
2.2. Microscopy and morphological analysis
Visualization under DIC microscopy and TEM was carried out as described (Frirdich et al., 2019). In brief, for visualization under DIC microscopy, 1 μl of a mid-exponential phase broth culture was immobilized on a thin 1% agar (w/v in H20) slab and overlayed with a cover slip. Images were captured with a Nikon Eclipse TE2000-U microscope equipped with 100 × objective and a Hamamatsu Orca camera system. DIC images were taken from multiple fields of view (totaling approximately 300 cells/strain). Images were converted to an 8-bit gray scale and processed using FIJI by applying a median filter of radius 1. Binary images were thresholded (Pincus and Theriot, 2007) using a pixel block size of 102 and subtracting −4. Artifacts and cells that were cut off on the sides of the image were manually removed. Morphological analysis was carried out using the MicrobeJ plug-in for Fiji (Ducret et al., 2016) and the CellTool shape analysis program (Pincus and Theriot, 2007; Sycuro et al., 2010; Ha et al., 2016). Both programs enable automated high throughput processing of binary bacterial images to detect bacterial cell contours at sub-pixel resolution and analyze bacterial cell shape parameters. Contours were generated in MicrobeJ using the medial axis method. These were analyzed, and a result table of the values of the shape descriptors was generated. The mean values and standard deviations were calculated, and significant differences were determined using multiple comparisons with Dunn’s correction after a Kruskal–Wallis test using GraphPad Prism 8. CellTool was used to compare differences in population morphology using the principal component analysis (PCA), as well as differences in the distributions of axis length, normalized axis curvature, and side curvature. CellTool analysis was carried out as previously described (Pincus and Theriot, 2007). For PCA, an average C. jejuni wild-type cell shape was generated by aligning the contours of the wild-type population. PCA was performed to generate a shape model based on principal components called shape modes. The shape modes combined describe at least 94.9% of the variation in the wild-type population. Contours of other strains were then aligned using the wild-type PCA shape model as a reference. The Kolmogorov–Smirnov statistics were calculated to determine whether changes between the different strains and wild type were significantly different with significance denoted by p-values of < 0.05.
2.3. Peptidoglycan isolation and muropeptide analysis
Campylobacter jejuni strains were passaged once from frozen stocks and then passaged to 20–25 MH plates and grown for 18–20 h. Cells were collected into cold MH broth by scraping to a total amount equivalent to an OD of 200–600, harvested by centrifugation at 8000 ×g for 15 min, and then resuspended in 6 ml ice-cold H2O. Cells were lysed by dropwise addition to an equivalent amount of 8% SDS to that of the cell suspension boiling under reflux. PG was purified from the cell lysate, digested with the muramidase cellosyl (kindly provided by Hoechst, Frankfurt, Germany), and the resulting muropeptides were reduced with sodium borohydride and separated by HPLC as described (Glauner, 1988). Muropeptide structures were assigned based on a comparison with retention times of known muropeptides from C. jejuni (Frirdich et al., 2012).
3. Results and discussion
3.1. Identification and bioinformatics analysis of proteins with a role in Campylobacter jejuni morphology
Gene products with a potential role in C. jejuni cell shape determination were identified by two bioinformatic approaches: (1) homology searches with characterized H. pylori PG hydrolases and cytoskeletal elements (Sycuro et al., 2010), and (2) searching the genome of C. jejuni strain 81-176 for proteins with peptidase domains. Those identified by approach (2) were further narrowed down to peptidases within the MEROPS M23B metallopeptidase family, some of which have a known role in PG biosynthesis (Vermassen et al., 2019). Approach (1) identified the genes cjj81176_1104 (1104), cjj81176_1105 (1105), and cjj81176_1228 (1228), and approach (2) identified cjj81176_0166 (0166) in addition to 1105 and 1228 (Figure 1).
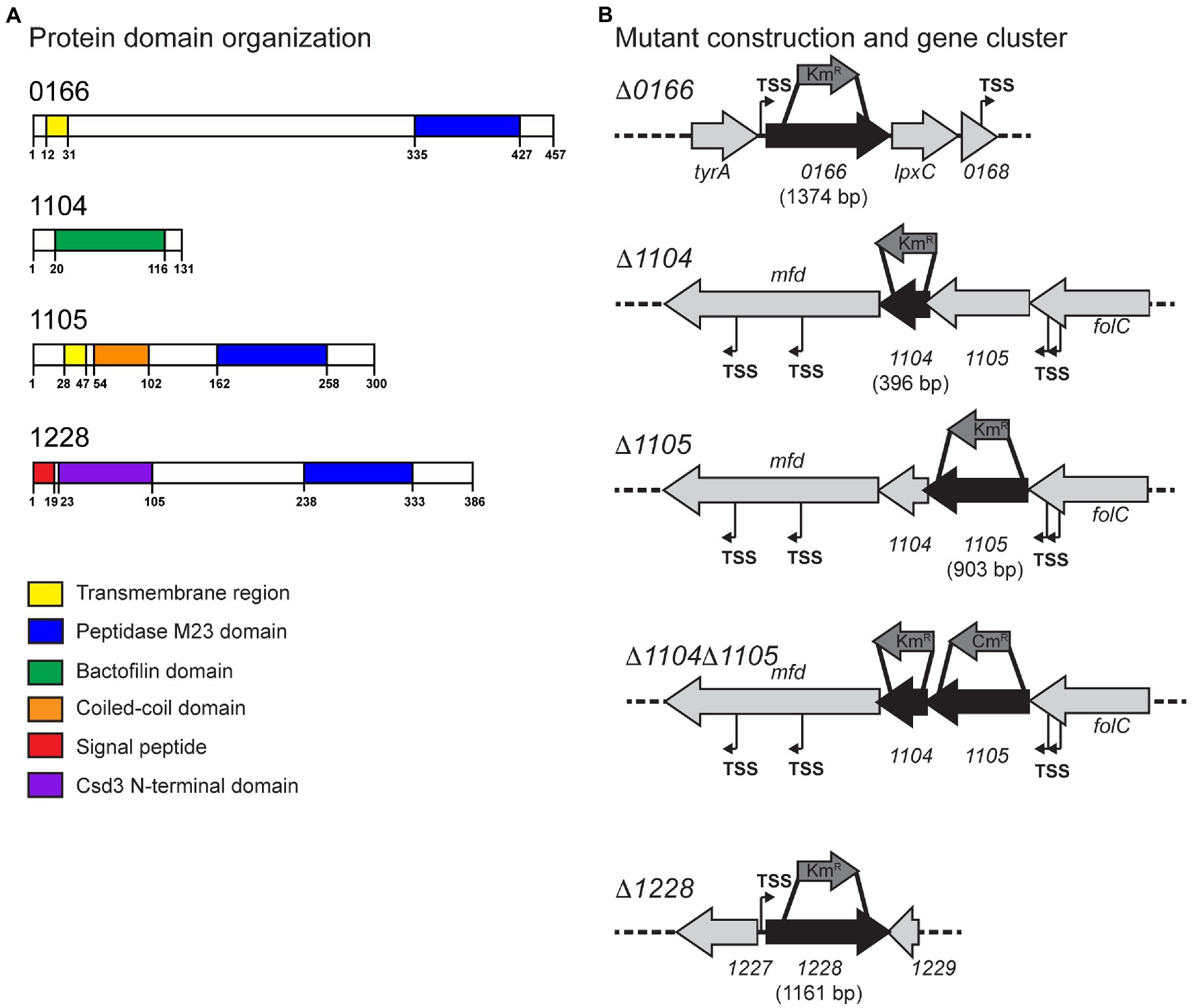
Figure 1. Protein domains of Campylobacter jejuni 0166, 1104, 1105, and 1228 and the corresponding gene loci. (A) The protein domains in the 0166, 1104, 1105, and 1228 gene products predicted by jackhmmer. (B) The gene loci of 0166, 1104, 1105, 1104–1105, and 1228 and the approximate location of the internal fragments deleted and replaced with a non-polar KmR cassette or a CmR cassette for mutant construction. Putative promoters (transcription start sites, TSS) predicted by RNA-seq are indicated (Dugar et al., 2013).
3.1.1. M23 peptidase domain-containing protein CJJ81176_0166
Conserved domain analysis of the 0166 gene product (457 amino acids) by jackhmmr (Potter et al., 2018) identified a transmembrane region (amino acids 12-31) and an M23 peptidase domain (PF01551; amino acids 335-427) with the conserved metalloprotease HxxxD and HxH motifs (Figure 1A). The N-terminal domain exhibited no similarities to other proteins or conserved domains. 0166 showed 27% identity/46% similarity to the uncharacterized H. pylori 26695 protein HP1054. Uncharacterized homologs of 0166 are well conserved in species of the δ- and ε-bacteria and Spirochaetia, all of which primarily consist of bacteria with curved or helical morphologies.
3.1.2. CJJ81176_1104 bactofilin homolog
Thousand hundred and four has 36.4% identity/59.3% similarity to CcmA of H. pylori 2695 HP1542.
As with H. pylori CcmA (Sycuro et al., 2010; Sichel et al., 2022), 1104 (131 amino acids) has a bactofilin domain (PF04519, amino acids 20-116; Figure 1A; Supplementary Figure S1). Bactofilins are small β-helical proteins that form cytoskeletal filaments (Kühn et al., 2010). They are found in a wide range of bacteria and have diverse functions, including modulating cell shape in H. pylori (Sycuro et al., 2010; Taylor et al., 2020; Sichel et al., 2022). H. pylori CcmA localizes to the major axis of curvature and helps to recruit the PG synthesis machinery and coordinate the modifications required to generate helical morphology (Taylor et al., 2020; Sichel et al., 2022). The bactofilin domain is flanked by an N-terminal and C-terminal region in H. pylori, an organization that is conserved in 1104 (Supplementary Figure S1). The N-terminal region contains a highly conserved short membrane binding motif (Deng et al., 2019; Supplementary Figure S1). This motif was shown to mediate membrane binding in the Thermus thermophilus bactofilin but not in H. pylori CcmA (Deng et al., 2019; Sichel et al., 2022). The H. pylori CcmA N-terminal region is important in generating helical cell shape and regulating interactions with the transmembrane scaffolding protein Csd7 (Sichel et al., 2022). The C. jejuni 1104 N-terminal domain will function differently, as C. jejuni lacks a Csd7 homolog, and may thus have more of a membrane binding role. The bioinformatic STRING analysis showed a potential interaction between the 1104 and the 1105 putative peptidase (described below) and vice versa based on their co-occurrence. H. pylori CcmA and Csd1/2 (1105 homologs) do not interact directly, but CcmA does regulate Csd1 activity indirectly (Yang et al., 2019; Sichel et al., 2022). The C. jejuni 1104 gene overlaps by 64 bp with and is directly downstream of the M23 peptidase gene 1105. This genomic arrangement of having a bactofilin gene overlapping with and downstream of an M23 peptidase domain-containing gene is conserved in most curved and helical organisms (Figure 1B; Sycuro et al., 2010; Sichel and Salama, 2020).
3.1.3. M23 peptidase domain-containing protein CJJ81176_1105
A previous study by our group described a reduction of cell curvature resulting from a frameshift mutation in 1105 isolated by a passage on CFW (Frirdich et al., 2017). Another group identified 1105 in a visual screen for cell shape mutants in a Tn mutant library, with a Tn insertion in 1105 resulting in a decrease in cell curvature (Esson et al., 2017). The 1105 gene product has 35.0% identity/55.8% similarity to H. pylori 26695 Csd1 (HP1543) and 30.0% identity/50.0% similarity to H. pylori 26695 Csd2 (HP1544). Csd1 is an endopeptidase, and Csd2 is a homolog of Csd1 but has an inactive catalytic site (An et al., 2016). Csd2 and the Csd7 scaffolding protein are required for Csd1 stability (Yang et al., 2019). Neither an inactive homolog of 1105/Csd1 (similar to Csd2) nor a homolog of the scaffolding protein Csd7 have been identified in C. jejuni. Csd2 homologs have only been found in H. pylori and H. hepaticus (Sycuro et al., 2010).
Conserved domain analysis of 1105 (300 amino acids) by jackhmmr identified a transmembrane region (amino acids 28-47), a coiled-coil domain (amino acids 54-102), and an M23 peptidase domain (PF01551; amino acids 162-258) containing the conserved metalloprotease HxxxD and HxH motifs (Figure 1; Supplementary Figure S2). Interestingly, Csd1 and Csd2 do not have the coiled-coil domain found in C. jejuni 1105 and conserved among other Campylobacter spp. and members of the LmdC clade of M23 peptidases to which 1105, Csd1, and Csd2 belong (Figueroa-Cuilan et al., 2021). Coiled-coil domains are ubiquitous and highly versatile repetitive peptide motifs consisting of two or more α-helices wound around each other into a supercoil with a hydrophobic core (Burkhard et al., 2001). They can have various biological functions. For example, they are widely involved in oligomerization, many have been proposed to act as molecular spacers separating functional domains, and they have been shown to act as molecular rulers confining enzymatic activity to certain subcellular locations and less commonly act as a scaffold in protein complexes (Truebestein and Leonard, 2016).
3.1.4. M23 peptidase domain-containing protein CJJ81176_1228
The 1228 gene product and its role in determining the degree of C. jejuni cell curvature were first described as part of a previous study (Stahl et al., 2016). 1228 shows 45% identity/65% similarity to the H. pylori 26695 Csd3 DD-carboxypeptidase and DD-endopeptidase (HP0506; Sycuro et al., 2010). H. pylori Csd3 has a predicted N-terminal transmembrane region and was crystallized without this transmembrane region in its latent, inactive form (An et al., 2015). The structure can be divided into three domains: domain 1 which occludes the active site of the M23 (LytM) domain, domain 2 of unknown function, and the C-terminal M23 (LytM) domain containing the catalytic Zn2+ binding site making up domain 3 (An et al., 2015; Supplementary Figure S3). The C. jejuni 1228 protein (386 amino acids) is predicted by jackhmmr to have a signal peptide (amino acids 1-19), the Csd3 N-terminal inhibitory domain (PF18059; Csd3 domain 1; amino acids 23-105), and the M23 peptidase domain (PF01551; Csd3 domain 3; amino acids 238-333) with the conserved active site HxxxD and HxH motifs (Figure 1A; Supplementary Figure S3). Activation of Csd3 was suggested to occur by removal of the inhibitory domain 1 from the active site by either autoproteolysis or cleavage by another endopeptidase (An et al., 2015). This may be similar to 1228. Proteins with a similar architecture are unique to members of the ε-Proteobacteria.
3.2. The 0166, 1104, 1105, and 1228 proteins are required for maintaining the degree of Campylobacter jejuni cell curvature
The methods used to examine the morphology of strains are described here followed by the results and discussion of the analyses in separate sections organized by gene name. To determine the roles of the 0166, 1104, 1105, and 1228 gene products in C. jejuni morphology, deletion mutants were constructed in each of the genes using a non-polar kanamycin resistance (KmR) cassette (Menard et al., 1993; Figure 1B; Supplementary material). Since 1104 and 1105 may interact, a ∆1104∆1105 double mutant was also generated to examine the effect of losing both genes. This mutant was constructed by deleting 1105 with a chloramphenicol resistance (CmR) cassette in an ∆1104 (KmR) background (Yao et al., 1993; Figure 1B; Supplementary material). The ∆1105 (KmR) and ∆1105 (CmR) mutants had similar effects on morphology (Figure 2; data not shown). Complementing and overexpressing strains were constructed using pRRC (Karlyshev and Wren, 2005) or pRRA (Cameron and Gaynor, 2014) integrative vectors (Supplementary material). All mutants, complements, and overexpressing strains showed wild-type growth characteristics (data not shown). The cell morphologies of the ∆0166, ∆1104, ∆1105, ∆1104∆1105, and ∆1228 mutants, complementing, and overexpressing strains were assessed by microscopy. Unlike ∆pgp1 and ∆pgp2 that were uniformly rod-shaped (Frirdich et al., 2012, 2014), the ∆0166, ∆1104, ∆1105, and ∆1228 mutants all retained a degree of cell curvature but differed in the amount of curvature [Figure 2 (DIC) and S4 (TEM)]. The roles of 1105 (Frirdich et al., 2017) and 1228 (Stahl et al., 2017) in maintaining cell curvature were previously described microscopically and followed up by quantification in this study.

Figure 2. Morphology of Campylobacter jejuni 0166, 1104, 1105, 1104–1105, and 1228 mutant, complement, and overexpressor strains. DIC microscope images of the helical Campylobacter jejuni 81-176 strain, the ∆0166, ∆1104, ∆1105, ∆1104, ∆1105, and ∆1228 mutant strains and corresponding complemented (∆0166c, ∆1104c, ∆1105c, ∆1104, ∆1105c, and ∆1228c) and overexpression strains (denoted + 0166, + 1104, + 1105, + 1104–1105, and + 1228). The scale bar indicates 2 μm.
To compare the subtle curvature changes between strains, morphological parameters were assessed by both MicrobeJ (Ducret et al., 2016) and Celltool (Pincus and Theriot, 2007; Sycuro et al., 2010; Ha et al., 2016; Liu et al., 2016) software programs. These two independent yet complementary analysis tools were used to maximize the description of curvature changes as they have different outputs: MicrobeJ displays mean values, while CellTool provides graphical representations of curvature changes. For quantification, DIC images of mid-exponential phase cells were taken from multiple fields of view totaling approximately 300 cells/strain and converted to binary images. Each with its own algorithms, the MicrobeJ and Celltool analysis programs detect the contour of each cell in the binary image. Then, they generate and record these as polygonal outlines along with their corresponding central axis. The central axis (referred to as the medial axis in MicrobeJ) runs from pole to pole through the middle of the cell and can be used to determine the morphological attributes of the cell.
We used MicrobeJ to quantify differences in cell length, width, and angularity (Table 1). The length is defined as the length of the central axis, the width is the mean width along the central axis, and angularity is defined as the mean curvature (the reciprocal of the radius of a circle tangent to a curve) values measured along the central axis. Within a wild-type population, variation in cell length accounts for most of the morphological variation in that population as shown by the larger standard deviation associated with the length, in contrast to the very small standard deviation in width and angularity. The variation in cell length in a population is expected since the population of cells will be at different stages of growth and division.
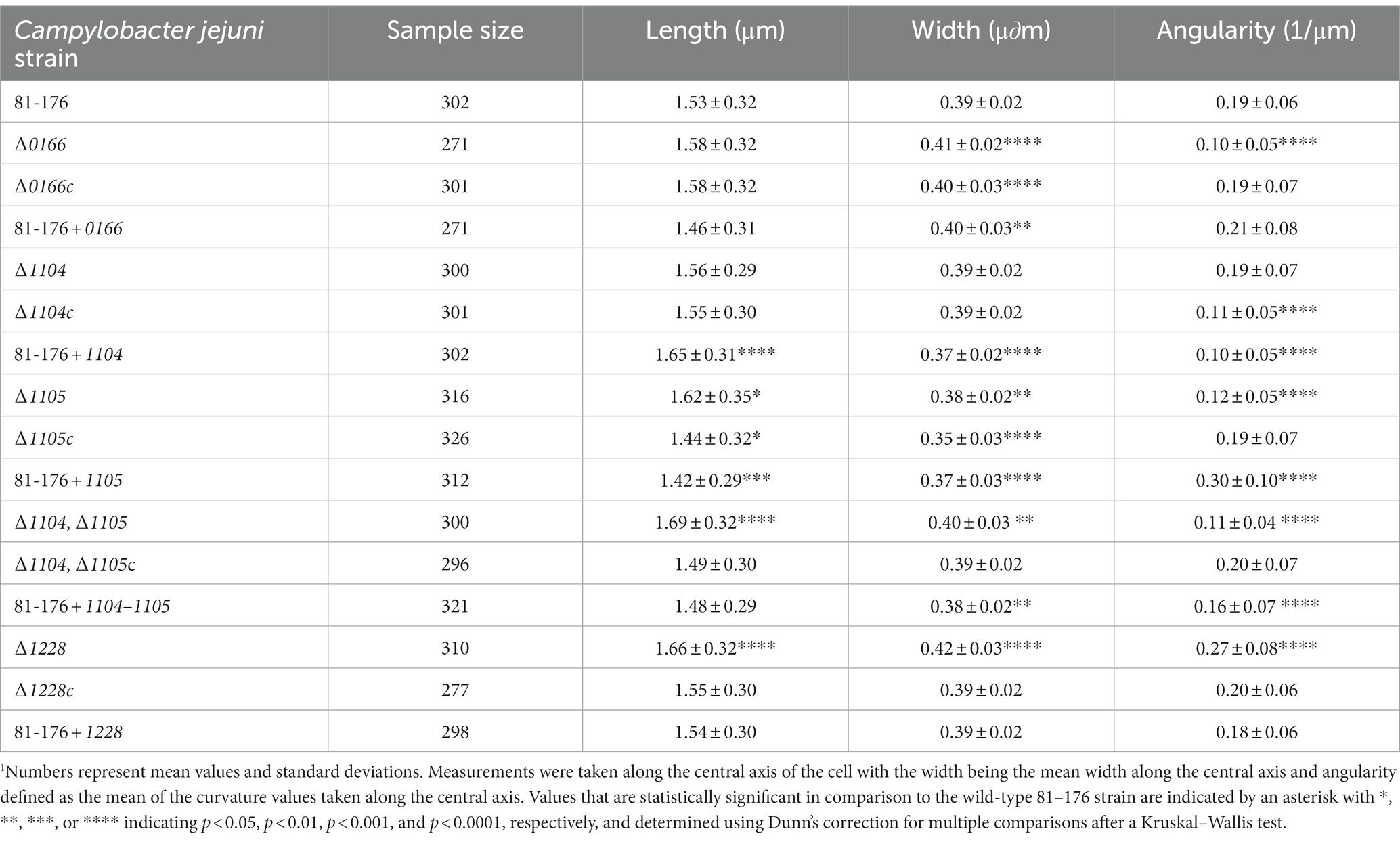
Table 1. MicrobeJ quantitative analysis of cell morphology parameters of Campylobacter jejuni wild-type 81-176, ∆0166, ∆1104, ∆1105, ∆1104∆1105, and ∆1228 mutant strains, complemented strains ∆0166c, ∆1104c, ∆1105c, ∆1104, ∆1105c, and ∆1228c, and overexpression strains 81-176 + 0166, 81–176 + 1104, 81–176 + 1105, 81–176 + 1104–1105, and 81-176 + 1228.1
CellTool was used to compare differences in population morphology using the principal component analysis (PCA; Figure 3), as well as differences in the distributions of axis length (length of the central axis), normalized axis curvature (a measure of the normalized curvature of the central axis), and side curvature (a measure of the normalized curvatures along the contour itself excluding the poles; Supplementary Figure S5). As mentioned earlier, the curvature is the reciprocal of the radius of a circle tangent to a curve at any point. CellTool normalizes curvature to the arc length represented by the radius of curvature. The average of the absolute values of the pointwise curvature is computed over a specified range and multiplied by the contour length over the same range. The CellTool analysis was carried out as previously described (Pincus and Theriot, 2007; Sycuro et al., 2010; Ha et al., 2016; Liu et al., 2016). For PCA, an average C. jejuni wild-type cell shape was generated by aligning the contours of the wild-type population. PCA was performed to generate a shape model based on principal components called shape modes that describe changes in the population. Three shape modes describe 95% of the morphological variation within the wild-type C. jejuni 81-176 population (Figure 3A). Shape mode 1 describes the variation in cell length and represents 91% of the variance within the population, as seen with the MicrobeJ analysis. Shape mode 2 describes the variation in helical pitch ranging from a large (stretched out helix; 2 s.d. in shape mode 2; Figure 3) to a smaller pitch (compressed helix; − 2 s.d. in shape mode 2; Figure 3) and represents 3.0% of the variance. Shape mode 3 describes the variation in the helical radius ranging from straight (2 s.d. in shape mode 3; Figure 3) to curved (−2 s.d. in shape mode 3; Figure 3) and represents 1.9% of the variance. Contours of other strains were then aligned using the wild-type PCA shape model as a reference.
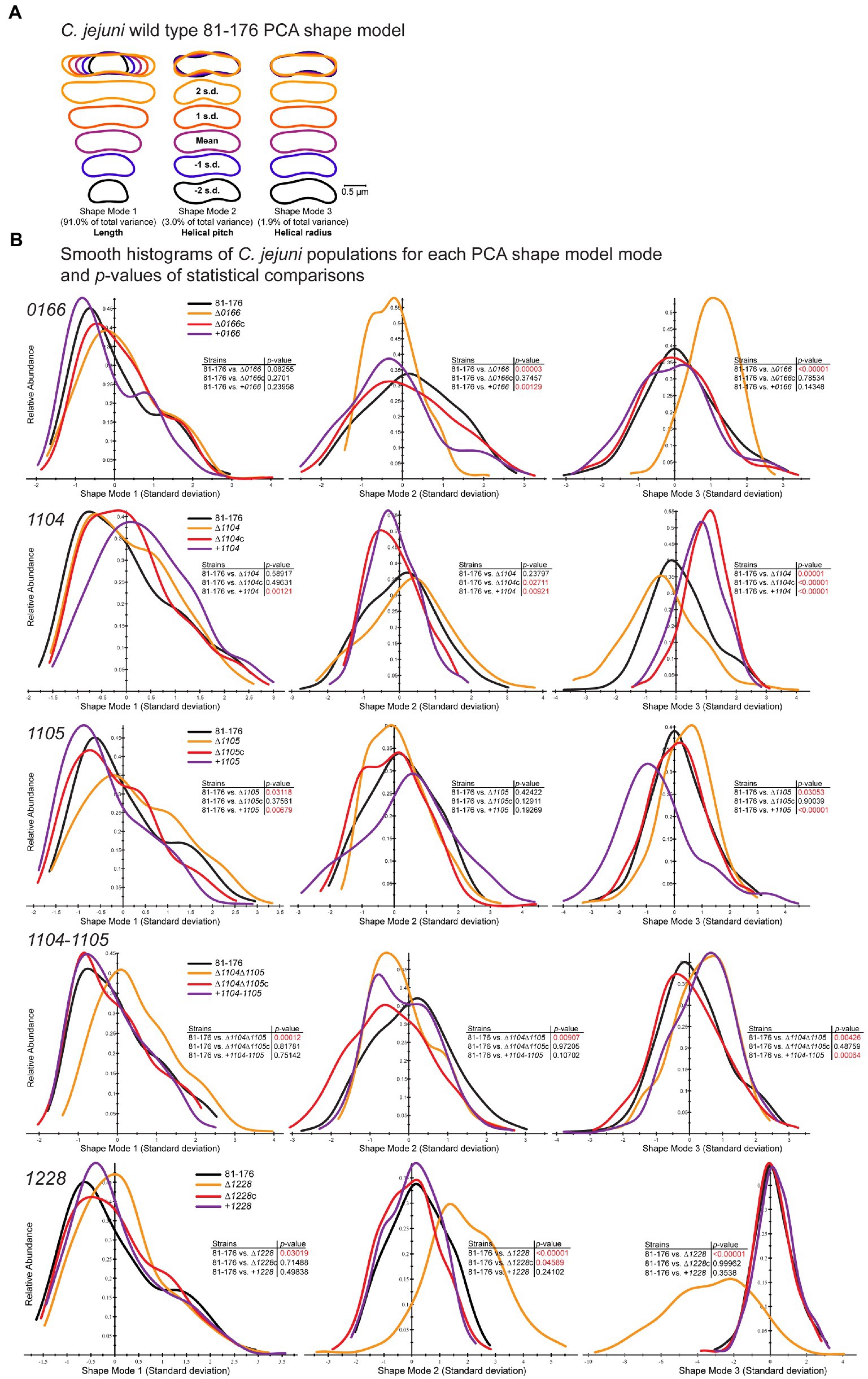
Figure 3. CellTool PCA analysis of population morphology for Campylobacter jejuni 0166, 1104, 1105, 1104–1105, and 1228 mutant, complement, and overexpressor strains. Campylobacter jejuni strains grown to the mid-exponential phase were imaged by DIC microscopy. The images were converted to binary, and cell contours were extracted and aligned. (A) A C. jejuni wild-type shape model was generated from aligned contours by PCA analysis to explain 94.9% of the population variation with the principal component shape modes 1–3. The mean shape mode ±2 standard deviations (s.d.) are shown. Shape mode 1 represents length, shape mode 2 helical pitch, and shape mode 3 helical radius. CellTool was then used to align the contours of mutant and associated complemented and overexpression strain population morphologies to the wild-type shape model. (B) Measurements representing the normalized standard deviations from each wild-type shape mode relative to abundance are represented as a histogram for each mutant (∆0166, ∆1104, ∆1105, ∆1104, ∆1105, and ∆1228), complement (∆0166c, ∆1104c, ∆1105c, ∆1104, ∆1105c, and ∆12285), and overexpressor (+0166, +1104, +1105, +1104–1105, and + 1228). The analyses are grouped by gene: 0166, 1104, 1105, 1104–1105, and 1228. The p-values comparing each strain to the wild type were determined by the Kolmogorov–Smirnov statistics and are shown as an inset to each graph. Values in red highlight statistically significant comparisons with p-values of < 0.05.
3.2.1. Morphological analysis of ∆0166, complement, and overexpressor strains
The ∆0166 mutant displayed a pleomorphic morphology, with some straight cells and some cells with varying decreased curvature (Figure 2; Supplementary Figure S4) as indicated by the reduction in mean angularity (Table 1) and the shifted distribution of normalized axis curvature and side curvature (Supplementary Figure S5). The PCA analysis (Figure 3) showed no variation in cell length (shape mode 1) but statistically significant variation in helical pitch (shape mode 2) and helical radius (shape mode 3). The curvature defects were complemented by wild-type morphology in all morphological attributes (Table 1; Figures 2, 3; Supplementary Figure S5). Overexpression of 0166 had a subtle effect on morphology, displaying a slight change only in the distribution of the helical pitch (Figure 3, shape mode 2).
3.2.2. Morphological analysis of ∆1104, ∆1105, ∆1104∆1105, and corresponding complement and overexpressor strains
The ∆1104 mutant morphology was similar to that of the wild type both visually (Figure 2; Supplementary Figure S4) and in all quantitative parameters (Table 1; Figure 3; Supplementary Figure S5) examined except for PCA shape mode 3 (helical radius), which showed a very slight increase in curvature that was not reflected in the MicrobeJ angularity values (Table 1). Complementation and 1104 overexpression resulted in a decrease in angularity (Table 1), with an increase in helical pitch (Figure 3, shape mode 2) and a decrease in helical radius (Figure 3, shape mode 3). The overexpressing strain also showed an increase in cell length and a decrease in width (Table 1; Figure 3, shape mode 1). The changes in the morphology of the complement are likely due to higher than wild-type levels of 1104 resulting from the expression at the non-native locus from the CmR promoter, as these changes were further accentuated in the overexpressing strain. Therefore, a dose of 1104 appears to affect morphology.
The ∆1105 mutant morphology showed a decrease in curvature that was consistent across the population and not pleomorphic like in ∆0166 (Figure 2; Supplementary Figure S4). In comparison to the wild type, there was a reduction in mean angularity and normalized axis curvature and side curvature (Table 1; Supplementary Figure S5) like that of ∆1104c and 81-176 + 1104. The complement restored the changes in angularity, axis curvature, and side curvature but showed a larger decrease in width (Table 1; Supplementary Figure S5). Overexpression of 1105 resulted in the opposite effect as the deletion, with an increase in angularity (Table 1) which was also evident from the DIC images (Figure 2). The width of the overexpressing strain was more similar to that of the mutant (Table 1). PCA analysis demonstrated changes in shape mode 3 (helical radius) in the mutant and overexpressing strains, with the mutant having less of a helical radius (less curvature) than the wild type, and the overexpressor having a greater helical radius (more curvature; Figure 3), supporting the changes in angularity values. There were no changes in shape mode 2 (helical pitch). The mean length values (Table 1) and distribution of length (Figure 3, shape mode 1) showed an increase in length in the mutant, a slight decrease with the complemented strain, and a more pronounced decrease with the overexpressing strain. Thus, like 1104, a dose of 1105 also appears to affect morphology.
A ∆1104∆1105 double mutant displayed a loss of cell curvature similar to the ∆1105 mutant (Figure 2) with a reduction in angularity (Table 1), normalized axis curvature and side curvature (Supplementary Figure S5), an increase in cell radius (Figure 3, shape mode 3), and a lengthening in helical pitch (Figure 3, shape mode 2). The length of ∆1104∆1105 cells was longer than that of a single ∆1105 mutant (Table 1; Figure 3, shape mode 1). Complementation of ∆1104∆1105 with 1104–1105 restored all wild-type morphological characteristics. The 1104–1105 overexpressing strain showed a decrease in angularity in comparison to the wild type that was not as pronounced as the ∆1104∆1105 mutant (the 1104 overexpressing strain also showed a decrease in angularity while the 1105 overexpressing strain displayed an increase over wild-type levels; Table 1). The helical radius (Figure 3, shape mode 3) of the 1104–1105 overexpressing strain was similar to that of the ∆1104∆1105 mutant and the 1104 overexpressing strain. Overall, the morphology of ∆1104∆1105 mutant resembled that of ∆1105. Overexpression of 1104–1105 resulted in morphological characteristics similar to 1104 but not 1105 overexpression.
3.2.3. Morphological analysis of ∆1228, complement, and overexpressor strains
The ∆1228 mutant displayed variable curved rod morphologies primarily consisting of C- and S-shaped cells (Figure 2 and Supplementary Figure S4). Unlike ∆0166 and ∆1105 which had a reduction in angularity and curvature in comparison to the wild type, the angularity of ∆1228 was increased compared to the wild type (Table 1), as was the normalized axis curvature and side curvature (Supplementary Figure S5). The ∆1228 helical pitch (shape mode 2) and helical radius (shape mode 3) were both increased and had a broader distribution in comparison to the wild type (Figure 3). The ∆1228 mutant also was longer and slightly wider than the wild type (Table 1; Figure 3, shape mode 1). The length and width were restored in the complemented strain (Table 1). The angularity (Table 1), helical radius (Figure 3, shape mode 3), and normalized curvature (Supplementary Figure S5) were restored in the complemented strain; however, the side curvature distribution was between that of the wild type and the mutant and not completely complemented (Supplementary Figure S5). The mean helical pitch (Figure 3, shape mode 2) was restored to the wild type but with a slightly altered distribution. The overexpressing strain was similar to the wild type (Table 1; Figure 3; Supplementary Figure S5) indicating that the lack of complementation with some parameters was not due to the overexpression but could be due to the expression from only the non-native promoter.
3.2.4. Comparison of Campylobacter jejuni morphology to that of Helicobacter pylori homologs
A mutant in H. pylori csd1 (1105 homolog) showed a similar decrease in curvature to that of ∆1105 (Sycuro et al., 2010). The ∆csd1 mutant was very slightly wider than the wild type (Sycuro et al., 2010), while ∆1105 was narrower (Table 1). H. pylori ∆ccmA (1104 homolog) was less curved than H. pylori csd1 (Sycuro et al., 2010), unlike ∆1104 which was very similar to the wild type except for a very slight increase in helical radius (Table 1; Figure 3; Supplementary Figure S5). The morphology of mutants in the homologs of C. jejuni 1228 in H. pylori (csd3/hdpA) varied depending on the strain of H. pylori. The H. pylori strain G27 csd3 mutant displayed variable curved morphologies, with the majority of the cells being highly curved or C-shaped and a small amount having little to no curvature (Sycuro et al., 2010). These cells were slightly wider and shorter than the wild type. A mutant in the gene corresponding to the Csd3 homolog in H. pylori strain N6, ∆hdpA, was significantly wider and shorter than the wild type, forming stocky cells (Bonis et al., 2010). In addition, a portion of the ∆hdpA population displayed branched cells with additional poles. Note that the morphology of the H. pylori N6 strain shows very little curvature, unlike that of G27 (Bonis et al., 2010). The morphology of C. jejuni ∆1228 consisted of cells with variable curvature, which as noted were primarily C- and S-shaped but with less curvature than those of ∆csd3 (Table 1; Figure 3; Supplementary Figure S5; Sycuro et al., 2010). The ∆1228 cells were longer than the wild type (Table 1) unlike ∆csd3 (Sycuro et al., 2010) and ∆hdpA (Bonis et al., 2010) which were shorter. The widths of all the ∆1228, ∆csd3, and ∆hdpA mutants were greater than that of their corresponding wild type. Of note, the double mutant ∆pgp2∆1228 (described below) displayed some branched cells like ∆hdpA and cells with additional non-polar flagella as seen by TEM (data not shown).
3.3. Peptidoglycan analyses of ∆0166, ∆1104, ∆1105, and ∆1228 reveal changes from the wild type and differences to the Helicobacter pylori homologs (where applicable)
As morphology is maintained by the PG layer, changes in the cell curvature are usually accompanied by changes in the muropeptide profile. PG was isolated from wild-type, mutant, complemented, and overexpressor strains to identify how 0166, 1104, 1105, and 1228 affect the muropeptide composition of C. jejuni in vivo. Muropeptide profiles were analyzed by HPLC as done previously (Frirdich et al., 2012, 2014, 2017, 2019; Ha et al., 2016; Table 2; Supplementary Figure S3, with the structures of the PG species identified shown for reference in Supplementary Figure S6). Changes described are in comparison to the wild type analyzed in parallel unless otherwise noted. Generally, only changes in muropeptide species that differed by 20% or more are discussed below. It is important to keep in mind when analyzing muropeptide profiles that even small changes in muropeptides present in small amounts, such as total pentapeptides in C. jejuni, have a high percent change, while larger changes in muropeptides present in larger amounts, such as total tetrapeptides, can have a low percent change.
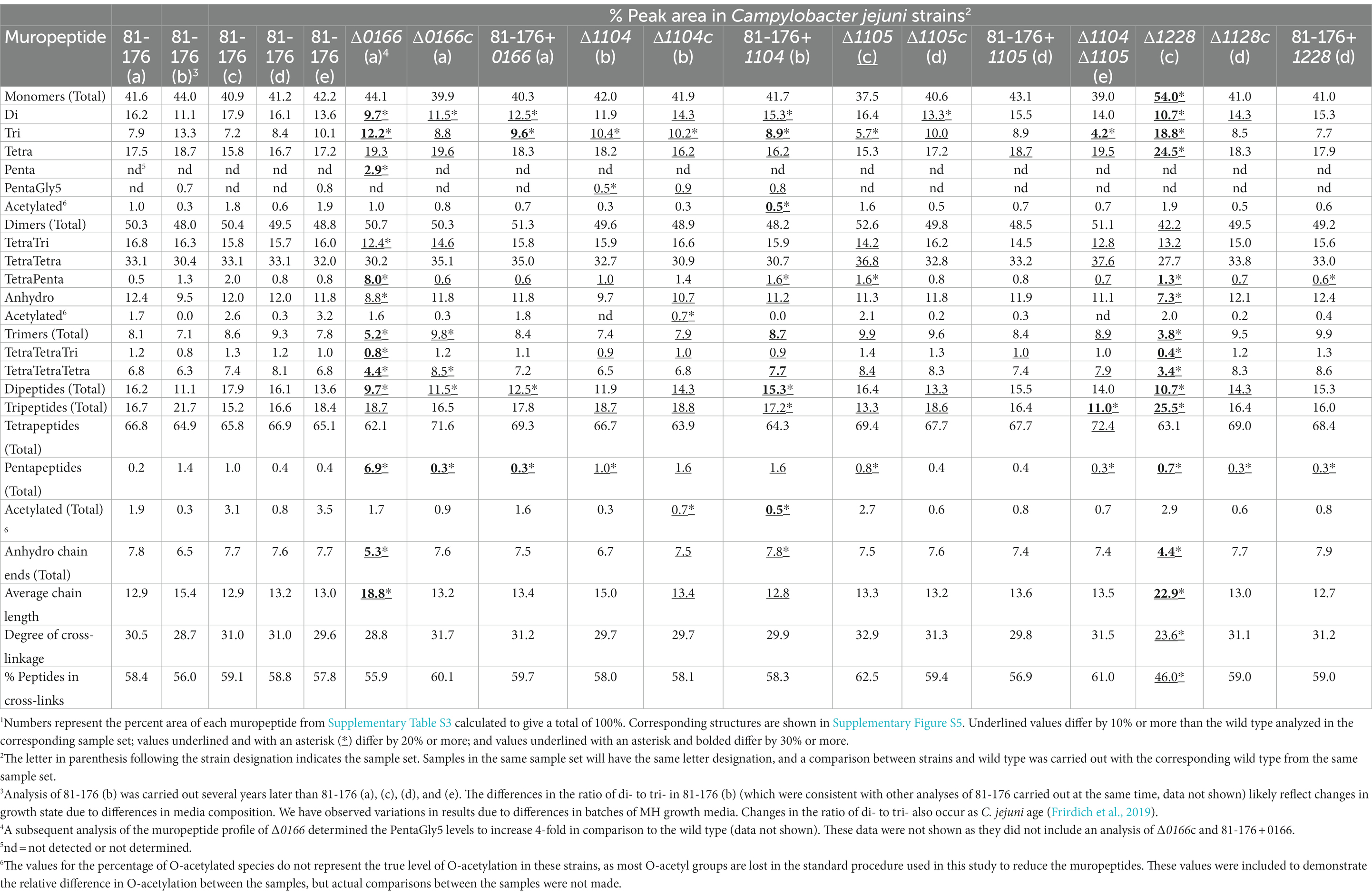
Table 2. Summary of the muropeptide composition of C. jejuni wild-type 81-176, ∆0166, ∆1104, ∆1105, ∆1104, ∆1105, and ∆1228 mutant strains, complemented strains ∆0166c, ∆1104c, ∆1105c, ∆1104, ∆1105c, and ∆1228c, and overexpression strains 81–176 + 0166, 81–176 + 1104, 81–176 + 1105, 81–176 + 1104–1105, and 81–176 + 1228.1
3.3.1. Muropeptide profile of ∆0166, complement, and overexpressor strains
The ∆0166 muropeptide profile displayed numerous significant changes compared to the wild type and was the only mutant muropeptide profile to show a relatively large increase in pentapeptides both in the monomeric and dimeric forms, with the largest increase occurring in the dimeric form (Table 2). The overall total dipeptides decreased and monomeric tripeptides increased. The TetraTri dimer levels were reduced which may be a result of the lack of processing of TetraPenta species. There was a decrease in overall levels of trimers but no change in the total amount of monomers and dimers. The amount of anhydro chain ends was reduced which was accompanied by a corresponding increase in average glycan strand length but no change in cross-linking. The complemented strain showed near complementation of most but not all of the muropeptide changes, such as the decrease in dipeptides. The number of trimers in the complement was more than the wild type, while that of the mutant was less than in the wild type. The 0166 overexpressing strain showed wild-type trimers as well as a decrease in dipeptides and an increase in monomeric tripeptides, with more tripeptides than in the complemented strain. Differences between the complement and overexpressing strain can result from the expression of 0166 from a non-native promoter. As such, this muropeptide analysis suggests that 0166 plays a role in processing pentapeptides. Both the 0166 deletion and 0166 overexpressing strains had increased the amounts of tripeptides and decreased dipeptides, indicating that the levels of 0166 in the cell could have an effect on Pgp1, which cleaves tri- to di-peptides. The decrease in the anhydro chain ends also suggests a possible effect on lytic transglycosylase activity, which is involved in generating anhydro sugars. There are four predicted lytic transglycosylases in C. jejuni: MltG, MltD, Slt, and RlpA. Interestingly, the bioinformatic STRING analysis indicated a potential interaction between 0166 and the membrane-bound murein lytic transglycosylase MltD.
3.3.2. Muropeptide profile of ∆1104, ∆1105, ∆1104∆1105, and corresponding complement and overexpressor strains
There were similar changes in the muropeptide profiles of ∆1104 and ∆1105 (Table 2), with the changes accentuated in the ∆1104∆1105 mutant, suggesting that these genes may function in a similar pathway. This is also supported by the STRING analysis and data on the H. pylori homologs, CcmA and Csd1, that show CcmA indirectly regulates Csd1 activity (Yang et al., 2019). Since 1104 does not have a peptidase domain, it likely affects muropeptide changes through interactions with the 1105 peptidase, enhancing and/or localizing 1105 activity. The most significant muropeptide changes observed in ∆1104, ∆1105, and ∆1104∆1105 are a decrease in monomeric tripeptides, which was most pronounced in ∆1104∆1105. The complemented strain ∆1105c showed a small increase in monomeric tripeptides (complementing the decrease in the mutant) and a decrease in dipeptides. These changes were not seen in an 1105 overexpressing strain, which was similar to the wild type. This was intriguing considering that the morphology of the 1105 overexpressing strain was more curved than the wild type (described earlier in Section 3.2.2). Changes in the morphology are therefore not always reflected in the muropeptide analysis. The ∆1104c strain showed no complementation of the reduction of tripeptides and a slight increase in dipeptides. The increase in dipeptides and decrease in tripeptides observed in ∆1104c were more pronounced in the 1104 overexpressing strain, indicating that these changes are likely due to the overexpression of 1104. These changes are reflected in changes in the morphology of both ∆1104c and the 1104 overexpressing strain, which were less curved that the wild type and the mutant.
Another group analyzed the muropeptide profile of ∆1105 in their 81-176 background and reported a lack of changes in comparison to the wild type (Esson et al., 2017). That work presented the HPLC chromatogram of the muropeptide profile but did not report the percent peak area of each muropeptide species in the chromatogram in relation to the area of all the peaks identified. Quantitative analyses, as shown here, highlight the subtle changes in muropeptide species such as those in ∆1105 (Table 2).
The muropeptide profiles of H. pylori ∆csd1, ∆csd2 (both homologs of C. jejuni 1105), and ∆ccmA (C. jejuni 1104 homolog) showed an increase in TetraPenta dimers and a decrease in tetrapeptide monomers, characteristic of a loss of endopeptidase activity required for the cleavage of TetraPenta cross-links (Sycuro et al., 2010). This was not seen with ∆1104 and ∆1105. Potentially, another DD-endopeptidase is substituting for 1105 activity in its absence, such as Pgp3 which was shown to have both DD-endopeptidase and DD-carboxypeptidase activity against synthetic peptides (Min et al., 2020). The muropeptide profiles of H. pylori ∆csd1, ∆csd2, and ∆ccmA mutants all show an increase in cross-linking, which did not occur in C. jejuni ∆1104 and ∆1105.
3.3.3. Muropeptide profile of ∆1228, complement, and overexpressor strains
There were many significant changes in the muropeptide profile of ∆1228 in comparison to the wild type (Table 2). There was an increase in the total amount of monomers, with a concomitant decrease in dimers and trimers. The overall total dipeptides and pentapeptides (although this was minor) decreased and tripeptides increased, with no change in overall tetrapeptides. However, both monomeric tri- and tetra-peptide levels increased. The muropeptide profile of ∆1228 showed a decrease in anhydroMurNAc-containing chain ends with an increase in average glycan strand length, as well as a decrease in cross-linking in comparison to the wild type. To date, this is the only C. jejuni mutant observed to have a change in cross-linking. As with ∆0166, the decrease in anhydroMurNAc chain ends indicates a possible effect on one of the lytic transglycosylases (MltG, MltD, Slt, and RlpA). The bioinformatic STRING analysis showed a potential interaction between 1228 and MltG. STRING also showed a potential interaction between 1228 and the PG synthase PbpA, which has glycosyltransferase and transpeptidase domains and which could potentially explain the changes in cross-linking. As well, STRING showed a potential interaction with other proteins involved with PG biosynthesis: the M23 peptidase and putative hydrolase 1105 and the amidase AmiA (Frirdich et al., 2019). All changes in the deletion strain were complemented. No additional significant changes were detected in the complemented strain or the 1228 overexpressing strain.
Deletion of the C. jejuni 1228 homolog HdpA of H. pylori strain N6 resulted in a muropeptide profile with an increase in monomeric and dimeric pentapeptides and a reduction in tetrapeptides (Bonis et al., 2010), while in an hdpA overexpressing strain, there was a decrease in pentapeptide species and an increase in tetrapeptides and tripeptides with an increase in monomers and a decrease in dimers (Bonis et al., 2010). The profile of the hdpA overexpressing strain is similar to that of ∆1228. The muropeptide profiles of ∆hdpA and the hdpA overexpressing strain suggested that HdpA possesses DD-carboxypeptidase and DD-endopeptidase activity, which was confirmed in vitro with purified protein (Bonis et al., 2010). The H. pylori csd3 gene is a homolog of hdpA in H. pylori G27. The muropeptide profile of a csd3 mutant also indicated that this protein has DD-carboxypeptidase and DD-endopeptidase activity, although the muropeptide profile was slightly different from that of ∆hdpA (Bonis et al., 2010; Sycuro et al., 2010). Of note, in the muropeptide profiles of both H. pylori hdpA and csd3 mutants, the glycan strand length, the number of anhydro chain ends, and the degree of cross-linking were unchanged, unlike in ∆1228. This again highlights that PG hydrolase homologs, even in related bacteria, do not necessarily have the same effect on the muropeptide profile and role in PG biosynthesis.
3.4. The effects of multiple hydrolase deletions: Double and triple PG hydrolase mutant analyses show a complex interdependency between 0166, 1105, and 1228
Combinations of double mutants [∆1104∆1105 (discussed earlier), ∆0166∆1105, ∆0166∆1228, and ∆1105∆1228], and a triple mutant (∆0166∆1105∆1228) were constructed to examine the relationships between these different peptidases and the effects of multiple hydrolase deletions. Each mutant was examined by DIC microscopy, and the muropeptide profiles were determined for the single mutants (Figure 4; Table 3; Supplementary Figure S4).
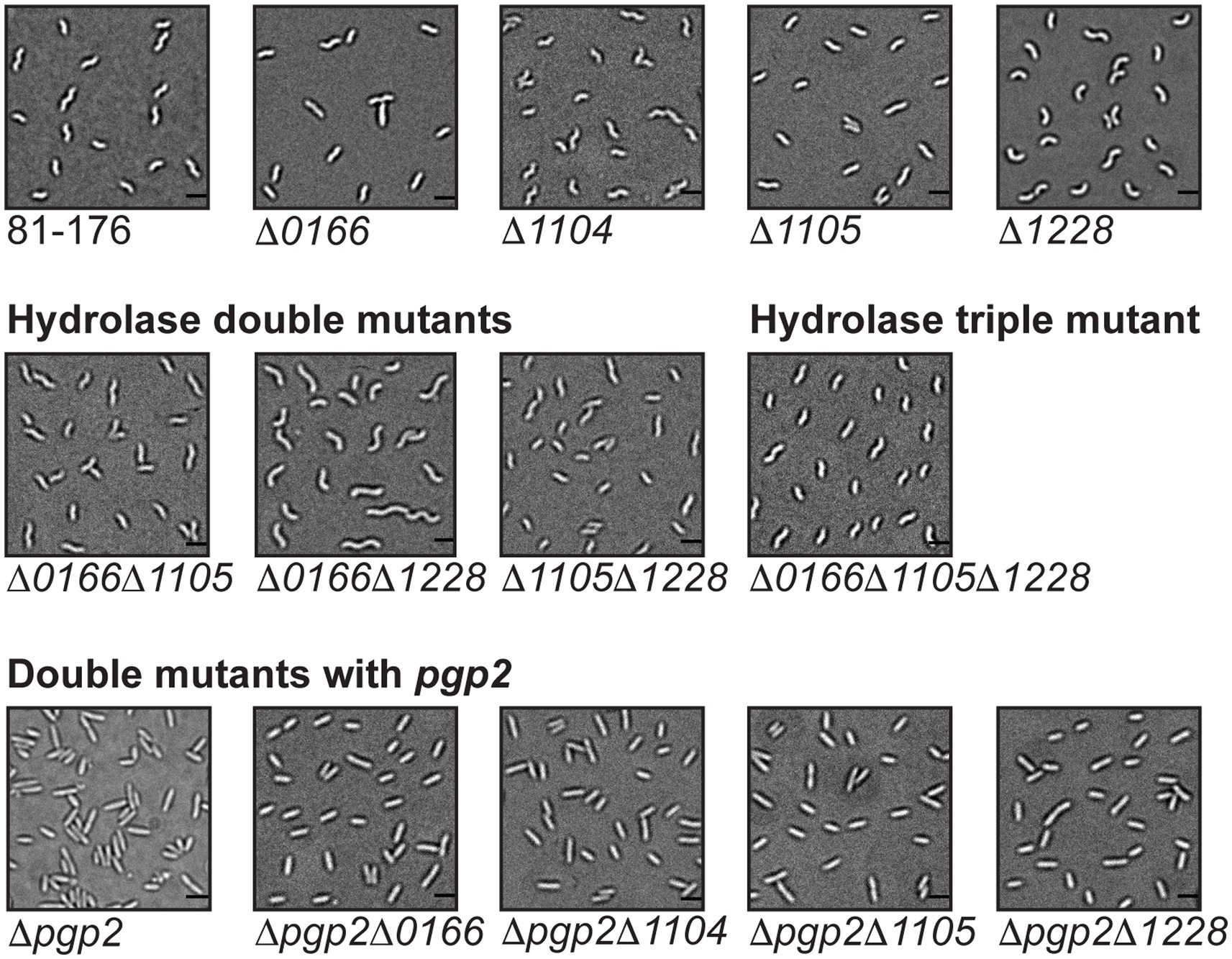
Figure 4. Mutant morphology of C. jejuni hydrolase double and triple mutants and double mutants with pgp2. DIC microscope images of hydrolase double mutants ∆0166, ∆1105, ∆0166, ∆1228, and ∆1105, ∆1228 and the triple mutant ∆0166, ∆1105, ∆1228, as well as double mutants with pgp2, including images of the wild-type 81-176 strain and the single mutants for comparison. The scale bar indicates 2 μm.
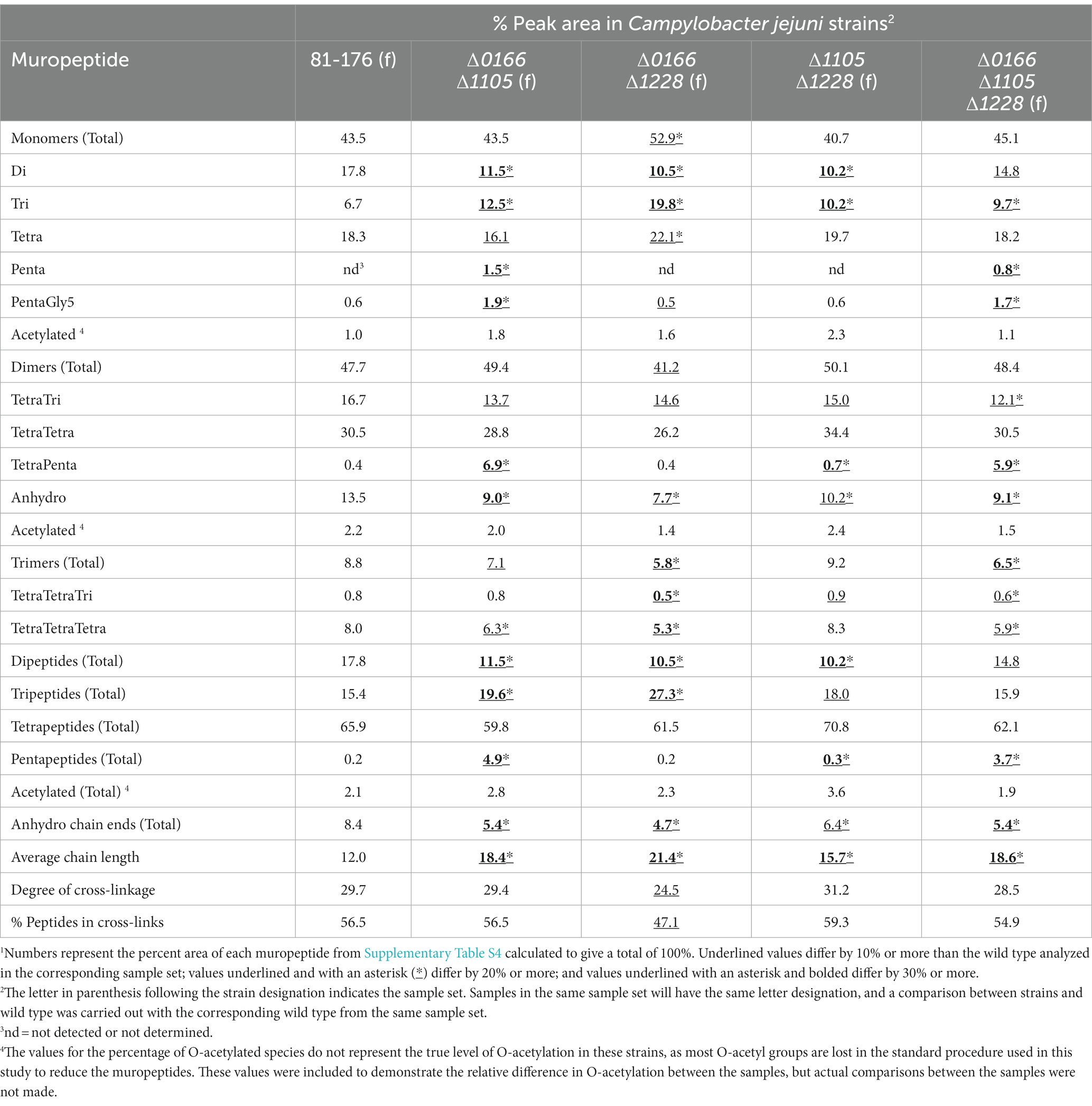
Table 3. Summary of the muropeptide composition of 0166, 1105, and 1228 double and triple mutants.1
The morphology of the ∆0166∆1228 mutant resembled that of ∆1228 with a possible increase in variability in cell length (Figure 4). Following from this, the muropeptide profile of ∆0166∆1228 (Table 3) was identical to ∆1228 (Table 2), with no increase in pentapeptide characteristic of an 0166 deletion (Table 2). From these data, it would appear that 1228 is epistatic to 0166.
Unlike ∆0166∆1228, the morphology of the ∆1105∆1228 mutant displayed the relaxed curvature of ∆1105 and not the C- and S-shaped cell characteristics of ∆1228 (Figure 4). In contrast to ∆1228, the muropeptide profile of ∆1105∆1228 showed no change in the levels of monomers, dimers, and trimers, no increase in tetrapeptides, and an elevation in tripeptides that was at an intermediate level between the wild type and that of ∆1228 (Tables 2, 3). An ∆1105 mutant displayed decreased tripeptides in comparison to the wild type; as such, loss of 1105 may counteract the increase in tripeptides of ∆1228. Only the decrease in dipeptides and anhydro chain ends with a corresponding increase in average chain length of ∆1105∆1228 was similar to that of ∆1228. As explained earlier, the effect on the anhydro chain ends and chain length would be due to an effect on lytic transglycosylase activity. From these results, it appears that 1105 may be required for 1228 function, be it either 1105 enzymatic activity or the presence of the protein itself.
The ∆0166∆1105 double mutant also displayed a relaxed curvature (Figure 4). The ∆0166∆1105 muropeptide profile was quite similar to that of ∆0166 except for a slightly more pronounced increase in tripeptides and a less pronounced decrease in trimers (Tables 2, 3). The ∆1105 mutant did have a slight increase in trimers, which may have an antagonistic effect on the overall level of trimers in the double mutant.
The triple mutant ∆0166∆1105∆1228 had a similarly relaxed curvature to ∆0166∆1105 and ∆0166∆1228 (Figure 4). The ∆0166∆1105∆1228 mutant still retained a degree of curvature, indicating that the effects of these three genes on curvature are not additive. The morphology of the ∆0166∆1228 mutant resembled ∆1228, but when ∆1105 was also deleted to generate strain ∆1105∆0166∆1228, the morphology was similar to ∆1105. Based on morphology, 1105 would appear to be epistatic to 1228 which is epistatic to 0166. This is more difficult to conclude based on the muropeptide profile. The muropeptide profile of the triple mutant had a less pronounced decrease in dipeptides and an increase in tripeptides than any of the double mutants, which was surprising, but a more pronounced increase in TetraTri dimers. The triple mutant did show an increase in pentapeptides similar to ∆0166∆1105 but not seen with ∆0166∆1228 and ∆1105∆1,228.
In sum, the double and triple mutant muropeptide analyses revealed a complex interdependency between 0166, 1105, and 1228. Ongoing work to establish enzymatic activity and protein–protein interactions should provide increased insight into the relationships between these peptidases. Nonetheless, both the morphological characteristics shown in Figure 4, and the muropeptide analyses in Table 3 suggest that 1228 is required for the 0166 function, and to some extent, 0166 is required for the 1105 function and 1105 for 1228.
3.5. The LD-carboxypeptidase gene pgp2 functions in a different pathway from 0166, 1104, 1105, and 1228
To determine the relationship among the 0166, 1104, 1105, and 1228 genes involved in determining the degree of cell curvature and the pgp2 gene required for generating helical cell shape (Frirdich et al., 2014), double mutants were constructed with pgp2. The ∆pgp2∆0166, ∆pgp2∆1104, ∆pgp2∆1105, and ∆pgp2∆1228 double mutants were all rod-shaped similar to a ∆pgp2 single mutant (Figure 4). However, despite their morphologies resembling that of ∆pgp2, the muropeptide profiles of the double mutants did show muropeptide differences characteristic of ∆0166, ∆1104, ∆1105, and ∆1228 single mutants (Table 4; Supplementary Figure S5; described below) in addition to those characteristics of ∆pgp2 (very little to no tripeptides and elevated tetrapeptides; Frirdich et al., 2014). This suggests that the proteins function independently and in different pathways, with Pgp2 activity not necessarily being required for their activity [unlike Pgp1 which requires Pgp2 to generate its substrate (Frirdich et al., 2014)].
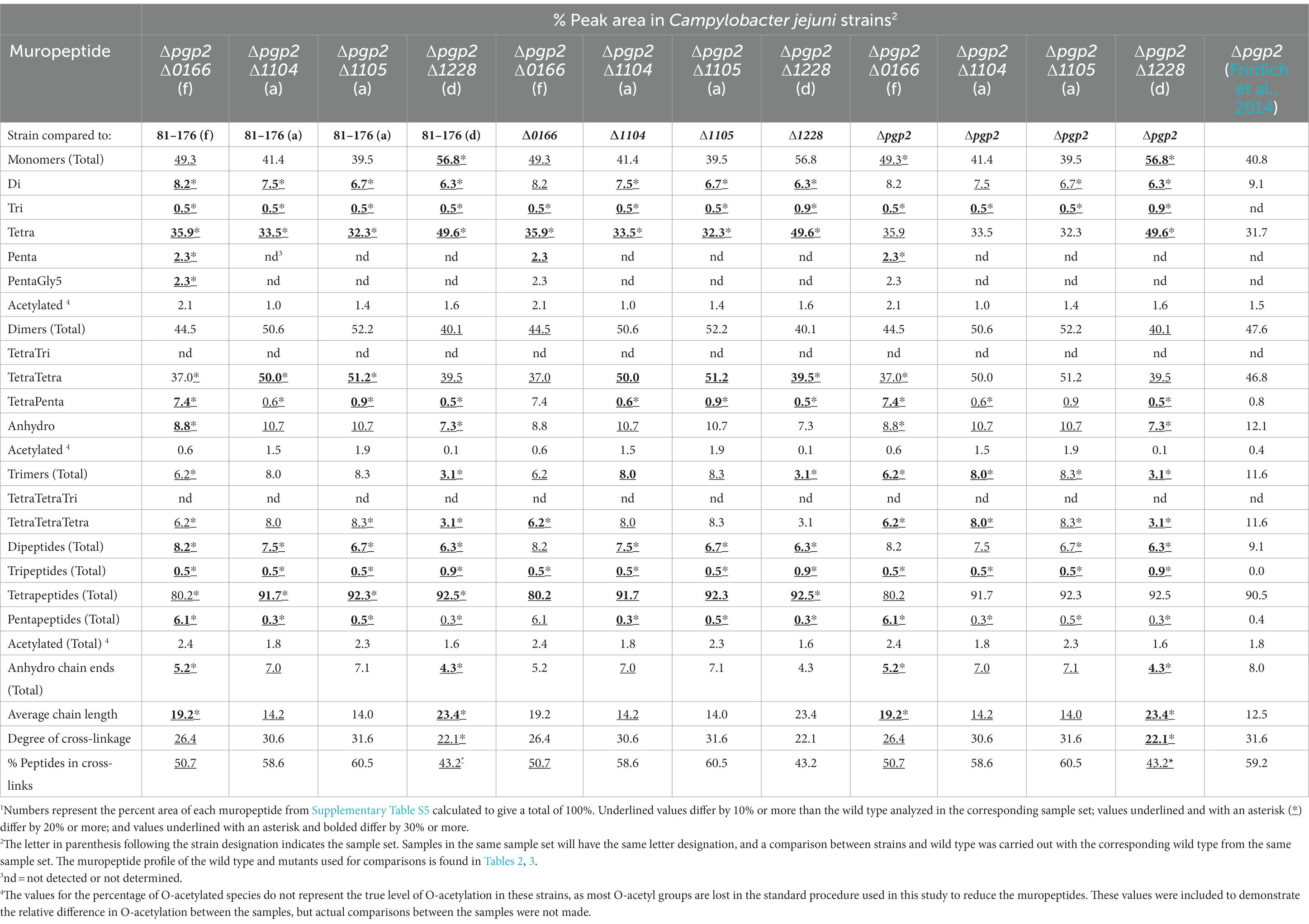
Table 4. Summary of the muropeptide composition of double mutants with pgp2 and 0166, 1104, 1105, or 1228.1
The ∆pgp2∆0166 double mutant showed an increase in total pentapeptides, as well as a decrease in the anhydro chain ends and an increase in chain length characteristic of ∆0166.
The muropeptide profile of the ∆pgp2∆1105 double mutant showed a further reduction in dipeptides in comparison to ∆pgp2. This is a change not present in ∆1105. There was an increase in TetraTetraTetra trimers in comparison to the wild type as seen in ∆1105 and a decrease in comparison to ∆pgp2, with a decrease in total trimers in comparison to ∆pgp2. The ∆pgp2∆1104 double mutant profile was similar to that of ∆pgp2∆1105 except that there was less of a reduction in dipeptides. A H. pylori ∆csd1∆csd6 double mutant (homologous to ∆pgp2∆1105) showed changes that were characteristic of each of the single mutants with elevated TetraPenta dimers similar to ∆csd1 and increased tetrapeptides and reduced tri- and di-peptides similar to ∆csd6 (Sycuro et al., 2013). In addition, the morphology of ∆csd1∆csd6 was intermediate to both single mutants, unlike that of ∆pgp2∆1105 which resembled ∆pgp2.
The ∆pgp2∆1228 mutant showed some muropeptide changes that were accentuated in the double mutant in comparison to the single mutants, most notably a decrease in dipeptides and an increase in tetrapeptides. Some changes noted were characteristic of ∆1228: an increase in monomers, a decrease in dimers and trimers, a reduction in the number of anhydro chain ends, an increase in chain length, and a decrease in cross-linking.
4. Conclusion
Several genes in the C. jejuni genome encode proteins with predicted functions in PG biosynthesis, with only a few to date having been characterized as having a role in C. jejuni bacterial morphogenesis: Pgp1 (Frirdich et al., 2012), Pgp2 (Frirdich et al., 2014), Pgp3 (Esson et al., 2017), and AmiA (Frirdich et al., 2019). These enzymes are involved in generating the helical shape of C. jejuni (Pgp1, Pgp2, and Pgp3) and/or the transition to the coccoid form in response to stress (Pgp1 and AmiA), both of which influence pathogenesis and/or pathogenic properties. AmiA also plays a role in cell separation during cell division (Frirdich et al., 2019). This study describes the identification of four additional C. jejuni genes with a role in morphology generation, 0166, 1104, 1105, and 1228, and determined the effect of these genes on morphology and PG muropeptide composition. The 1104 gene encodes a putative bactofilin, a cytoskeletal protein with reported roles in bacterial morphology maintenance in other bacteria (Ramos-Leon and Ramamurthi, 2022). The 0166, 1105, and 1228 genes encoding M23 domain-containing proteins are likely PG hydrolases with endopeptidase and/or carboxypeptidase activity. The enzymatic activity of these gene products is currently being established. The influence of deleting 0166, 1104, 1105, and 1228 on C. jejuni pathogenic attributes will be described in a subsequent study.
Deletion of 0166, 1104, 1105, and 1228 produced strains with varying curvature in comparison to the wild type: The ∆1104 mutant had a very slight increase in cellular helical radius, ∆1105 was less curved, ∆0166 was less curved but pleomorphic with morphologies ranging from straight rods to slightly curved, and ∆1228 displayed variable curved rod morphologies of C- and S-shaped cells. The expression of 1104 from a non-native promoter in the complemented and overexpressing strains resulted in a decrease in cell curvature more pronounced than in the 1104 mutant. In contrast, 1105 deletion resulted in reduced curvature and 1105 overexpression in increased curvature. Therefore, a dose of both 1104 and 1105 appears to play an important role in determining wild-type helical morphology.
Unlike with C. jejuni ∆pgp1 and ∆pgp2, specific assignments of 0166, 1105, and 1228 hydrolase function from the mutant muropeptide profiles of ∆0166, ∆1105, and ∆1228 were not possible. There are several potential explanations for this. (1) PG hydrolases can have more than one enzymatic function, such as C. jejuni Pgp3 which was shown to have endopeptidase and carboxypeptidase activity in vitro (Min et al., 2020). (2) Different hydrolases can have redundant functions and can substitute for each other. For example, in E. coli, there are seven proteins with DD-endopeptidase activity (Pazos et al., 2017; Vermassen et al., 2019). (3) Changes in the levels of the 0166, 1104, 1105, and 1228 proteins (through deletion or overexpression) could affect other proteins in the PG biosynthetic complex by disruption of protein–protein interactions, resulting in numerous additional muropeptide changes. (4) PG hydrolases that further trim muropeptide species generated by the loss or overexpression of a hydrolase could also complicate the interpretation of the muropeptide profiles. The in vitro enzyme activities of 0166, 1105, and 1228 and the identification of their protein interaction partners will help establish the concrete roles these proteins play in C. jejuni PG biosynthesis and how they interact with each other and/or other PG biosynthetic enzymes such as the PG synthases, lytic transglycosylases, and the amidase AmiA.
Like C. jejuni, Helicobacter pylori is a member of the ε-Proteobacteria and has a helical morphology. H. pylori has homologs of Pgp1 (Csd4) and Pgp2 (Csd6) that have similar enzymatic functions and roles in helical shape generation as Pgp1 and Pgp2, respectively (Frirdich et al., 2012, 2014; Sycuro et al., 2012, 2013). However, we have observed that increases in Pgp1 levels also affect helical morphology which was not seen in H. pylori (Frirdich et al., 2012). Deletions in the H. pylori homologs for 1104 (ccmA), 1105 (csd1), and 1228 (csd3; Sycuro et al., 2010) had different morphological effects (except for csd1) and muropeptide changes compared to the C. jejuni gene products (note that an H. pylori 0166 homolog has not been studied). It was suggested that trimming of PG cross-links enables wild-type helical cell curvature and twisting in H. pylori, as mutants in ccmA, csd1, and csd3 with curved rods but not helical morphologies had increased levels of cross-linking (Sycuro et al., 2010). In contrast, deletions in 0166, 1104, and 1105 had no effect on total cross-linking, while a 1228 mutant had decreased levels of cross-linking. Cross-linking may still play a role in generating C. jejuni helical cell morphology, but these data suggest that it may be mechanistically different than in H. pylori.
Collectively, this study provides novel and detailed morphology and muropeptide analyses for four new C. jejuni proteins involved in PG modification. In addition, it also highlights that, despite their similar helical morphology and the presence of several homologous proteins, some of which have similar activities, the morphogenesis programs of C. jejuni and H. pylori have striking differences. These include differences in effects on the muropeptide profiles of homologous enzymes, in the complement of morphogenesis proteins (i.e., H. pylori has several proteins lacking homologs in C. jejuni, such as Csd2, Csd5, and Csd7), and possibly how they generate the helical shape. These similarities and differences will continue to be discovered as our understanding of how helical shape is generated in C. jejuni and H. pylori evolves. In addition, this study emphasizes the importance of studying the PG biosynthetic program in related but distinct bacterial pathogens.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
EF conceived and designed the study. EF and JV constructed the strains used in the study and carried out the morphological analysis. JB carried out the muropeptide analysis. Data analysis and interpretation was carried out by all authors. Funding was provided by EG and WV. EF wrote the manuscript which was revised in depth with EG. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Canadian Institutes of Health Research grants 68981 and 461767 to EG. Work in WV’s laboratory was supported by the UKRI Strategic Priorities Fund (EP/T002778/1) and the BBSRC (BB/W005557/1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1162806/full#supplementary-material
References
An, D. R., Im, H. N., Jang, J. Y., Kim, H. S., Kim, J., Yoon, H. J., et al. (2016). Structural basis of the heterodimer formation between cell shape-determining proteins Csd1 and Csd2 from Helicobacter pylori. PLoS One 11:e0164243. doi: 10.1371/journal.pone.0164243
An, D. R., Kim, H. S., Kim, J., Im, H. N., Yoon, H. J., Yoon, J. Y., et al. (2015). Structure of Csd3 from Helicobacter pylori a cell shape-determining metallopeptidase. Acta Crystallogr. D Biol. Crystallogr. 71, 675–686. doi: 10.1107/S1399004715000152
Bolton, D. J. (2015). Campylobacter virulence and survival factors. Food Microbiol. 48, 99–108. doi: 10.1016/j.fm.2014.11.017
Bonis, M., Ecobichon, C., Guadagnini, S., Prevost, M. C., and Boneca, I. G. (2010). A M23B-family metallopeptidase of Helicobacter pylori required for cell shape, pole formation and virulence. Mol. Microbiol. 78, 809–819. doi: 10.1111/j.1365-2958.2010.07383.x
Burkhard, P., Stetefeld, J., and Strelkov, S. V. (2001). Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 11, 82–88. doi: 10.1016/s0962-8924(00)01898-5
Burnham, P. M., and Hendrixson, D. R. (2018). Campylobacter jejuni collective components promoting a successful enteric lifestyle. Nat. Rev. Microbiol. 16, 551–565. doi: 10.1038/s41579-018-0037-9
Cameron, A., Frirdich, E., Huynh, S., Parker, C. T., and Gaynor, E. C. (2012). Hyperosmotic stress response of Campylobacter jejuni J. Bacteriol. 194, 6116–6130. doi: 10.1128/JB.01409-12
Cameron, A., and Gaynor, E. C. (2014). Hygromycin B and apramycin antibiotic resistance cassettes for use in Campylobacter jejuni PLoS One 9:e95084. doi: 10.1371/journal.pone.0095084
Cava, F., and de Pedro, M. A. (2014). Peptidoglycan plasticity in bacteria: emerging variability of the murein sacculus and their associated biological functions. Curr. Opin. Microbiol. 18, 46–53. doi: 10.1016/j.mib.2014.01.004
Deng, X., Gonzalez Llamazares, A., Wagstaff, J. M., Hale, V. L., Cannone, G., McLaughlin, S. H., et al. (2019). The structure of bactofilin filaments reveals their mode of membrane binding and lack of polarity. Nat. Microbiol. 4, 2357–2368. doi: 10.1038/s41564-019-0544-0
Do, T., Page, J. E., and Walker, S. (2020). Uncovering the activities, biological roles, and regulation of bacterial cell wall hydrolases and tailoring enzymes. J. Biol. Chem. 295, 3347–3361. doi: 10.1074/jbc.REV119.010155
Ducret, A., Quardokus, E. M., and Brun, Y. V. (2016). MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat. Microbiol. 1:16077. doi: 10.1038/nmicrobiol.2016.77
Dugar, G., Herbig, A., Förstner, K. U., Heidrich, N., Reinhardt, R., Nieselt, K., et al. (2013). High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 9:e1003495. doi: 10.1371/journal.pgen.1003495
Egan, A. J. F., Errington, J., and Vollmer, W. (2020). Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 18, 446–460. doi: 10.1038/s41579-020-0366-3
Esson, D., Gupta, S., Bailey, D., Wigley, P., Wedley, A., Mather, A. E., et al. (2017). Identification and initial characterisation of a protein involved in Campylobacter jejuni cell shape. Microb. Pathog. 104, 202–211. doi: 10.1016/j.micpath.2017.01.042
Esson, D., Mather, A. E., Scanlan, E., Gupta, S., de Vries, S. P., Bailey, D., et al. (2016). Genomic variations leading to alterations in cell morphology of Campylobacter spp. Sci. Rep. 6:38303. doi: 10.1038/srep38303
Field, L. H., Underwood, J. L., Payne, S. M., and Berry, L. J. (1993). Characteristics of an avirulent Campylobacter jejuni strain and its virulence-enhanced variants. J. Med. Microbiol. 38, 293–300. doi: 10.1099/00222615-38-4-293
Figueroa-Cuilan, W. M., Randich, A. M., Dunn, C. M., Santiago-Collazo, G., Yowell, A., and Brown, P. J. B. (2021). Diversification of LytM protein functions in polar elongation and cell division of Agrobacterium Tumefaciens. Front. Microbiol. 12:729307. doi: 10.3389/fmicb.2021.729307
Frirdich, E., Biboy, J., Adams, C., Lee, J., Ellermeier, J., Gielda, L. D., et al. (2012). Peptidoglycan-modifying enzyme Pgp1 is required for helical cell shape and pathogenicity traits in Campylobacter jejuni. PLoS Pathog. 8:e1002602. doi: 10.1371/journal.ppat.1002602
Frirdich, E., Biboy, J., Huynh, S., Parker, C. T., Vollmer, W., and Gaynor, E. C. (2017). Morphology heterogeneity within a Campylobacter jejuni helical population: the use of calcofluor white to generate rod-shaped C. Jejuni 81-176 clones and the genetic determinants responsible for differences in morphology within 11168 strains. Mol. Microbiol. 104, 948–971. doi: 10.1111/mmi.13672
Frirdich, E., Biboy, J., Pryjma, M., Lee, J., Huynh, S., Parker, C. T., et al. (2019). The Campylobacter jejuni helical to coccoid transition involves changes to peptidoglycan and the ability to elicit an immune response. Mol. Microbiol. 112, 280–301. doi: 10.1111/mmi.14269
Frirdich, E., and Gaynor, E. C. (2013). Peptidoglycan hydrolases, bacterial shape, and pathogenesis. Curr. Opin. Microbiol. 16, 767–778. doi: 10.1016/j.mib.2013.09.005
Frirdich, E., Vermeulen, J., Biboy, J., Soares, F., Taveirne, M. E., Johnson, J. G., et al. (2014). Peptidoglycan LD-carboxypeptidase Pgp2 influences Campylobacter jejuni helical cell shape and pathogenic properties and provides the substrate for the DL-carboxypeptidase Pgp1. J. Biol. Chem. 289, 8007–8018. doi: 10.1074/jbc.M113.491829
Glauner, B. (1988). Separation and quantification of muropeptides with high-performance liquid chromatography. Anal. Biochem. 172, 451–464. doi: 10.1016/0003-2697(88)90468-X
Ha, R., Frirdich, E., Sychantha, D., Biboy, J., Taveirne, M. E., Johnson, J. G., et al. (2016). Accumulation of peptidoglycan O-acetylation leads to altered cell wall biochemistry and negatively impacts pathogenesis factors of Campylobacter jejuni. J. Biol. Chem. 291, 22686–22702. doi: 10.1074/jbc.M116.746404
Hänel, I., Borrmann, E., Müller, J., Müller, W., Pauly, B., Liebler-Tenorio, E. M., et al. (2009). Genomic and phenotypic changes of Campylobacter jejuni strains after passage of the chicken gut. Vet. Microbiol. 136, 121–129. doi: 10.1016/j.vetmic.2008.10.018
Hansson, I., Sandberg, M., Habib, I., Lowman, R., and Engvall, E. O. (2018). Knowledge gaps in control of Campylobacter for prevention of campylobacteriosis. Transbound. Emerg. Dis. 65, 30–48. doi: 10.1111/tbed.12870
Ikeda, N., and Karlyshev, A. V. (2012). Putative mechanisms and biological role of coccoid form formation In Campylobacter jejuni. Eur. J. Microbiol. Immunol. 2, 41–49. doi: 10.1556/EuJMI.2.2012.1.7
Jackson, D. N., Davis, B., Tirado, S. M., Duggal, M., van Frankenhuyzen, J. K., Deaville, D., et al. (2009). Survival mechanisms and culturability of Campylobacter jejuni under stress conditions. Antonie Van Leeuwenhoek 96, 377–394. doi: 10.1007/s10482-009-9378-8
Justice, S. S., Hunstad, D. A., Cegelski, L., and Hultgren, S. J. (2008). Morphological plasticity as a bacterial survival strategy. Nat. Rev. Microbiol. 6, 162–168. doi: 10.1038/nrmicro1820
Kaakoush, N. O., Castano-Rodriguez, N., Mitchell, H. M., and Man, S. M. (2015). Global epidemiology of Campylobacter infection. Clin. Microbiol. Rev. 28, 687–720. doi: 10.1128/CMR.00006-15
Karlyshev, A. V., and Wren, B. W. (2005). Development and application of an insertional system for gene delivery and expression in Campylobacter jejuni. Appl. Environ. Microbiol. 71, 4004–4013. doi: 10.1128/AEM.71.7.4004-4013.2005
Kühn, J., Briegel, A., Mörschel, E., Kahnt, J., Leser, K., Wick, S., et al. (2010). Bactofilins, a ubiquitous class of cytoskeletal proteins mediating polar localization of a cell wall synthase in Caulobacter Crescentus. EMBO J. 29, 327–339. doi: 10.1038/emboj.2009.358
Liu, Y., Frirdich, E., Taylor, J. A., Chan, A. C., Blair, K. M., Vermeulen, J., et al. (2016). A bacterial cell shape-determining inhibitor. ACS Chem. Biol. 11, 981–991. doi: 10.1021/acschembio.5b01039
McLennan, M. K., Ringoir, D. D., Frirdich, E., Svensson, S. L., Wells, D. H., Jarrell, H., et al. (2008). Campylobacter jejuni biofilms up-regulated in the absence of the stringent response utilize a calcofluor white-reactive polysaccharide. J. Bacteriol. 190, 1097–1107. doi: 10.1128/JB.00516-07
Menard, R., Sansonetti, P. J., and Parsot, C. (1993). Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J. Bacteriol. 175, 5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993
Min, K., An, D. R., Yoon, H. J., Rana, N., Park, J. S., Kim, J., et al. (2020). Peptidoglycan reshaping by a noncanonical peptidase for helical cell shape in Campylobacter jejuni. Nat. Commun. 11:458. doi: 10.1038/s41467-019-13934-4
Pazos, M., and Peters, K. (2019). Peptidoglycan. Subcell. Biochem. 92, 127–168. doi: 10.1007/978-3-030-18768-2_5
Pazos, M., Peters, K., and Vollmer, W. (2017). Robust peptidoglycan growth by dynamic and variable multi-protein complexes. Curr. Opin. Microbiol. 36, 55–61. doi: 10.1016/j.mib.2017.01.006
Pincus, Z., and Theriot, J. A. (2007). Comparison of quantitative methods for cell-shape analysis. J. Microsc. 227, 140–156. doi: 10.1111/j.1365-2818.2007.01799.x
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., and Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. doi: 10.1093/nar/gky448
Ramos-Leon, F., and Ramamurthi, K. S. (2022). Cytoskeletal proteins: lessons learned from bacteria. Phys. Biol. 19:021005. doi: 10.1088/1478-3975/ac4ef0
Sheppard, S. K., and Maiden, M. C. (2015). The evolution of Campylobacter jejuni and Campylobacter Coli. Cold Spring Harb. Perspect. Biol. 7:a018119. doi: 10.1101/cshperspect.a018119
Sherman, P. M., Ossa, J. C., and Wine, E. (2010). Bacterial infections: new and emerging enteric pathogens. Curr. Opin. Gastroenterol. 26, 1–4. doi: 10.1097/MOG.0b013e328333d73b
Sichel, S. R., Bratton, B. P., and Salama, N. R. (2022). Distinct regions of H. pylori’s bactofilin CcmA regulate protein-protein interactions to control helical cell shape. elife 11:e80111. doi: 10.7554/eLife.80111
Sichel, S. R., and Salama, N. R. (2020). Bacterial cell biology: it takes two to tango. Curr. Biol. 30, R1258–R1260. doi: 10.1016/j.cub.2020.08.040
Silva, J., Leite, D., Fernandes, M., Mena, C., Gibbs, P. A., and Teixeira, P. (2011). Campylobacter Spp. as a foodborne pathogen: a review. Front. Microbiol. 2:200. doi: 10.3389/fmicb.2011.00200
Stahl, M., Frirdich, E., Vermeulen, J., Badayeva, Y., Li, X., Vallance, B. A., et al. (2016). The helical shape of Campylobacter jejuni promotes in vivo pathogenesis by aiding transit through intestinal mucus and colonization of crypts. Infect. Immun. 84, 3399–3407. doi: 10.1128/IAI.00751-16
Stahl, M., Graef, F. A., and Vallance, B. A. (2017). Mouse models for Campylobacter jejuni colonization and infection. Methods Mol. Biol. 1512, 171–188. doi: 10.1007/978-1-4939-6536-6_15
Svensson, S. L., Frirdich, E., and Gaynor, E. C. (2008). “Survival strategies of Campylobacter jejuni: stress responses, the viable but non-culturable state, and biofilms” in Campylobacter. eds. C. M. Szymanski, I. Nachamkin, and M. J. Blaser. 3rd ed (Washington, DC: ASM Press), 62–85.
Sycuro, L. K., Pincus, Z., Gutierrez, K. D., Biboy, J., Stern, C. A., Vollmer, W., et al. (2010). Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cells 141, 822–833. doi: 10.1016/j.cell.2010.03.046
Sycuro, L. K., Rule, C. S., Petersen, T. W., Wyckoff, T. J., Sessler, T., Nagarkar, D. B., et al. (2013). Flow cytometry-based enrichment for cell shape mutants identifies multiple genes that Influence Helicobacter pylorimorphology. Mol. Microbiol. 90, 869–883. doi: 10.1111/mmi.12405
Sycuro, L. K., Wyckoff, T. J., Biboy, J., Born, P., Pincus, Z., Vollmer, W., et al. (2012). Multiple peptidoglycan modification networks modulate Helicobacter pylori’s cell shape, motility, and colonization potential. PLoS Pathog. 8:e1002603. doi: 10.1371/journal.ppat.1002603
Taylor, J. A., Bratton, B. P., Sichel, S. R., Blair, K. M., Jacobs, H. M., DeMeester, K. E., et al. (2020). Distinct cytoskeletal proteins define zones of enhanced cell wall synthesis in Helicobacter pylori. elife 9, e52482. doi: 10.7554/eLife.52482
Tegtmeyer, N., Sharafutdinov, I., Harrer, A., Soltan Esmaeili, D., Linz, B., and Backert, S. (2021). Campylobacter virulence factors and molecular host-pathogen interactions. Curr. Top. Microbiol. Immunol. 431, 169–202. doi: 10.1007/978-3-030-65481-8_7
Truebestein, L., and Leonard, T. A. (2016). Coiled-coils: the long and short of it. BioEssays 38, 903–916. doi: 10.1002/bies.201600062
Typas, A., Banzhaf, M., Gross, C. A., and Vollmer, W. (2012). From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10, 123–136. doi: 10.1038/nrmicro2677
Uehara, T., and Bernhardt, T. G. (2011). More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr. Opin. Microbiol. 14, 698–703. doi: 10.1016/j.mib.2011.10.003
Vermassen, A., Leroy, S., Talon, R., Provot, C., Popowska, M., and Desvaux, M. (2019). Cell Wall hydrolases in bacteria: insight on the diversity of cell wall amidases, glycosidases and peptidases toward peptidoglycan. Front. Microbiol. 10:331. doi: 10.3389/fmicb.2019.00331
Vollmer, W. (2008). Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol. Rev. 32, 287–306. doi: 10.1111/j.1574-6976.2007.00088.x
Vollmer, W., Blanot, D., and de Pedro, M. A. (2008a). Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167. doi: 10.1111/j.1574-6976.2007.00094.x
Vollmer, W., Joris, B., Charlier, P., and Foster, S. (2008b). Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32, 259–286. doi: 10.1111/j.1574-6976.2007.00099.x
Wyckoff, T. J., Taylor, J. A., and Salama, N. R. (2012). Beyond growth: novel functions for bacterial cell wall hydrolases. Trends Microbiol. 20, 540–547. doi: 10.1016/j.tim.2012.08.003
Yadav, A. K., Espaillat, A., and Cava, F. (2018). Bacterial strategies to preserve cell wall integrity against environmental threats. Front. Microbiol. 9:2064. doi: 10.3389/fmicb.2018.02064
Yang, D. C., Blair, K. M., Taylor, J. A., Petersen, T. W., Sessler, T., Tull, C. M., et al. (2019). A genome-wide Helicobacter pylori morphology screen uncovers a membrane-spanning helical cell shape complex. J. Bacteriol. 201, e00724–18. doi: 10.1128/JB.00724-18
Yao, R., Alm, R. A., Trust, T. J., and Guerry, P. (1993). Construction of new Campylobacter cloning vectors and a new mutational cat cassette. Gene 130, 127–130. doi: 10.1016/0378-1119(93)90355-7
Keywords: Campylobacter jejuni, bacterial cell shape, helical morphology, morphological quantitation, peptidoglycan, bactofilin, M23 peptidase domain, peptidoglycan hydrolase
Citation: Frirdich E, Vermeulen J, Biboy J, Vollmer W and Gaynor EC (2023) Multiple Campylobacter jejuni proteins affecting the peptidoglycan structure and the degree of helical cell curvature. Front. Microbiol. 14:1162806. doi: 10.3389/fmicb.2023.1162806
Edited by:
Stuart A. Thompson, Augusta University, United StatesReviewed by:
Ivo Gomperts Boneca, Institut Pasteur, FranceNicolae Corcionivoschi, Agri-Food and Biosciences Institute, United Kingdom
Copyright © 2023 Frirdich, Vermeulen, Biboy, Vollmer and Gaynor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emilisa Frirdich, ZW1pbGlzYUBtYWlsLnViYy5jYQ==