
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 05 May 2023
Sec. Virology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1161265
 Xinran Zhang1,2
Xinran Zhang1,2 Yantao Liang1,3*Kaiyang Zheng1Ziyue Wang1Yue Dong1Yundan Liu1Linyi Ren1Hongmin Wang1Ying Han1
Yantao Liang1,3*Kaiyang Zheng1Ziyue Wang1Yue Dong1Yundan Liu1Linyi Ren1Hongmin Wang1Ying Han1 Andrew McMinn1,4
Andrew McMinn1,4 Yeong Yik Sung3,5
Yeong Yik Sung3,5 Wen Jye Mok3,5
Wen Jye Mok3,5 Li Lian Wong3,5
Li Lian Wong3,5 Jianfeng He2,6*
Jianfeng He2,6* Min Wang1,3,7,8*
Min Wang1,3,7,8*Introduction: Vibrio is an important bacterial genus containing many pathogenic species. Although more and more Vibrio phages were isolated, the genome, ecology and evolution of Vibrio phages and their roles in bacteriophage therapy, have not been fully revealed.
Methods: Novel Vibrio phage vB_ValR_NF infecting Vibrio alginolyticus was isolated from the coastal waters of Qingdao during the Ulva prolifera blooms, Characterization and genomic feature of phage vB_ValR_NF has been analysed using phage isolation, sequencing and metagenome method.
Results and Discussion: Phage vB_ValR_NF has a siphoviral morphology (icosahedral head 114±1 nm in diameter; a tail length of 231±1 nm), a short latent period (30 minutes) and a large burst size (113 virions per cell), and the thermal/pH stability study showed that phage vB_ValR_NF was highly tolerant to a range of pHs (4-12) and temperatures (-20 - 45 °C), respectively. Host range analysis suggests that phage vB_ValR_NF not only has a high inhibitory ability against the host strain V. alginolyticus, but also can infect 7 other Vibrio strains. In addition, the phage vB_ValR_NF has a double-stranded 44, 507 bp DNA genome, with 43.10 % GC content and 75 open reading frames. Three auxiliary metabolic genes associated with aldehyde dehydrogenase, serine/threonine protein phosphatase and calcineurin-like phosphoesterase were predicted, might help the host V. alginolyticus occupy the survival advantage, thus improving the survival chance of phage vB_ValR_NF under harsh conditions. This point can be supported by the higher abundance of phage vB_ValR_NF during the U. prolifera blooms than in other marine environments. Further phylogenetic and genomic analysis shows that the viral group represented by Vibrio phage vB_ValR_NF is different from other well-defined reference viruses, and can be classified into a new family, named Ruirongviridae. In general, as a new marine phage infecting V. alginolyticus, phage vB_ValR_NF provides basic information for further molecular research on phage–host interactions and evolution, and may unravel a novel insight into changes in the community structure of organisms during the U. prolifera blooms. At the same time, its high tolerance to extreme conditions and excellent bactericidal ability will become important reference factors when evaluating the potential of phage vB_ValR_NF in bacteriophage therapy in the future.
Vibrio species are heterotrophic bacteria with genetic and metabolic diversity (de Souza Valente and Wan, 2021). They are the natural inhabitants as free-living or related communities in the marine and brackish water environment worldwide, usually accounting for 0.5%–5% of the total number of marine bacteria (Frans et al., 2013; Thompson and Polz, 2014). Several species of Vibrio have a wide range of virulence related genes, which are pathogenic for humans and animals. For example, Vibrio anguillarum can infect more than 50 kinds of fishes, molluscs and crustaceans (Vasquez et al., 2020). Vibrio vulnificus can cause severe wound infection and primary sepsis, which are usually due to humans eating raw seafood or seawater contaminated wounds (López-Pérez et al., 2019). Vibrio alginolyticus, the dominant Vibrio species in the ocean infect a variety of aquaculture species, such as sea bream, kuruma prawn, shellfish species and so on, it is also a serious pathogen that infects humans (Dong et al., 2020). The characteristics of easier mass reproduction in summer enable V. alginolyticus to spread in the food chain through various kinds of contaminated seafood, which eventually leads to the outbreak of human epidemic bacterial diseases (Emam et al., 2019; Liu et al., 2020).
Although Vibrio is a great threat to food and public safety even human health, the limited available treatment options for Vibrio infection are still antibiotics (Wong et al., 2015). However, the abuse of antibiotics has led to pathogenic bacteria developing resistance to antibiotics, which greatly reduces the therapeutic effect of Vibrio infection (Mok et al., 2019). In this context, bacteriophages as biological control agents that are antagonistic to Vibrio have appeared on the horizon.
Bacteriophages are viruses that attack bacteria and lyse sensitive bacterial cells (Zhang et al., 2021). Due to their high host specificity, phages are also considered to be an ideal tool to deal with specific pathogens. In fact, several cases of phage therapy successfully treating bacterial infections have been reported recently, including resistant Acinetobacter baumannii (Tan et al., 2021; Wu et al., 2021), Mycobacterium (Pamela Rodríguez et al., 2020), and Pseudomonas (Oechslin et al., 2017; Yang et al., 2021). These spectacular results indicate that bacteriophages have an extraordinary prospect in the treatment of bacterial infection. In addition, phages have also been shown to play an important role in maintaining ecosystem coordination: they occur in high abundance (to 108/mL) in the oceans and top-down control of microbial growth across ecosystems by infecting and lysing their host cells (Zhao et al., 2009; Xu et al., 2018; Bartlau et al., 2021). Some bacteriophages also affect the metabolic activities of the host through horizontal gene transfer (HGT) or the expression of auxiliary metabolism genes (AMGs) generated by their own coding, thus participating in the global geochemical cycle (Rohwer and Thurber, 2009; Lang et al., 2012).
Due to the potential application of Vibrio phages in bacteriophage therapy, the genome information of about 400 Vibrio phages has been uploaded to GenBank (Liu et al., 2019). Most of them infect pathogenic Vibrio, including V. cholerae, V. parahaemolyticus, and V. vulnificus. Such as Vibrio anguillarum phage CHOED, isolated from Chilean mussels, has been proved that it can effectively inhibit V. anguillarum, thus protect fish from vibriosis in fish farms (Romero et al., 2014). Phage KVP40 has a wide host range, including V. cholerae, V. parahaemolyticus and other non-pathogenic species (Miller et al., 2003). Lytic phage pVa-21infecting several fish pathogenic Harveyi clade bacteria, showed potential at biofilm eradication mechanism (Kim et al., 2019). In addition, previous literature has verified the presence of enzymatic functional domains in uncharacterized proteins of many Vibrio phages, their roles can be used for the understanding of phage-host interaction and pathogenicity (Sanmukh et al., 2010). Thus, more Vibrio phages should be isolated to expand the genetic database of bacteriophages, so as to ensure that Vibrio phages can be successfully applied to the antimicrobial treatment of Vibrio infection. In this study, a novel Vibrio phage vB_ValR_NF, capable of inhibiting V. alginolyticus growth was isolated and characterized. This study not only expands the scientific understanding of phage biology, and genomic information, but represents a good model for the exploitation of marine host-phage interactions. The potential usefulness of this phage against V. alginolyticus, may provide insight into the best combination of various phages for use in a phage cocktail.
The V. alginolyticus strain ATCC 17749 was used as the host for bacteriophage, isolated from the seawater off the coast of Qingdao (36°06′N, 120°34′E) during the U. prolifera blooms in July 2019 and were inoculated in liquid Zobell medium at 28°C (Wang Q. et al., 2020).
The genome sequence of the V. alginolyticus strain ATCC 17749 is available under the GenBank accession number NZ_BATK00000000. The phylogenetic tree based on 16S rRNA sequence of the V. alginolyticus strain ATCC 17749 was constructed with the 16S rRNA sequences of other relative hosts using IQ-TREE (Maximum Likelihood method with 1,000 bootstraps; Supplementary Figure S1).
The Vibrio phage vB_ValR_NF was prepared following the standard virus enrichment and double-layer agar methods (Zhang et al., 2021). Briefly, the host cell (1 mL) was added to the seawater sample (25 mL) collected from the coastal waters of Qingdao where the U. prolifera blooms in 2019, incubated overnight at 28°C with 10% liquid Zobell medium, then the mixture was centrifuged at 6,000 × g for 10 min. Supernatants that filtered through 0.22 m–pore-size membranes (Millipore), were concentrated with 100KDa Amincon centrifugal filters (Millipore), then 200 μL concentrated samples were co-cultured with the equivalent host cell, plaques were detected by double-layer plate method. Three isolation rounds were performed by picking single plaques to ensure that phages are purified. Then samples were placed in SM buffer [100 mM NaCl, 81.2 mM MgSO4.7H2O, 50 mM Tris–HCl (pH7.5), 0.01% gelatin] for new plaque assays.
20 μL phage suspension was negatively stained with 2% uranyl acetate, then the morphology of the Vibrio phage vB_ValR_NF was examined by energy-filtering transmission electron microscopy (TEM, JEOL Model JEM-1200EX) at 100 kV at NICEM (Liu et al., 2018; Li H. et al., 2019). The average size was taken as the reference diameter by measuring three phage particles in the same field of vision.
The one-step growth curve assay of the Vibrio phage vB_ValR_NF was performed following the previous method with some modifications (Middelboe et al., 2010; Wang et al., 2021). Generally, phage suspension (MOI = 0.01) was inoculated into exponentially growing cultures of V. alginolyticus strain ATCC 17749 (2.13 × 1013 cfu/mL). The mixture was allowed to adsorb for 15 min at 28°C, cells were pelleted by centrifugation (13,000 × g, 1 min), then re-suspended and transferred to 50 mL liquid Zobell medium. Samples were taken at 5 min intervals for 1 h and at 10 min intervals for the following hour, at 30 min intervals in the last hour. Each sample was performed in triplicate, using double-layer agar methods for plaque assays and calculating phage titer to draw a standard curve. Burst size was calculated as the ratio of the final count of liberated phage particles to the initial number of infected host cells at the beginning of the test (Zhang et al., 2020; Liu et al., 2021).
To gain more information on the biological properties of the Vibrio phage vB_ValR_NF, we tested its pH / thermal stability (Thammatinna et al., 2020). Briefly, 100 μL phage suspension was incubated with 900 μL SM buffer under a range of pH conditions from 3 to 12 at 30°C for 2 h. For thermal stability, phage suspension (pH = 7) was incubated for 2 h at −20°C, 4°C, 25°C, 35°C, 45°C, 55°C, 65°C, and 75°C. Both tests were determined using the double-layer plate method under room conditions (30°C; pH = 7). Each sample was performed in triplicate.
Twelve Vibrio strains and other genus strains were used in this study to detect the host of phage vB_ValR_NF. Briefly, dropping 10 μL of phage suspension (MOI = 0.01) on double-layer agar plates was incubated with bacteria. Repeat the above operation three times for each plate, and then samples were incubated overnight at 28°C, to check whether plaques in the plates (Liu et al., 2018).
In addition, in order to test the ability of phage vB_ValR_NF to kill the host V. alginolyticus, an in vitro lysis assay was determined: V. alginolyticus strain ATCC 17749 in exponential phase was co-cultured with phage suspensions at various MOIs (10−1, 10−2, 10−3, 10−4, and 10−5). The absorbance (OD600nm) was determined at 2 h intervals for 12 h to reflect the infection trend of the host cells. Bacteria without phage inoculation were used as a control (Lu et al., 2017; Chen et al., 2020). After the in vitro lysis assay end, we re-measured the final absorbance (OD600nm) value 24 h after infection at different MOIs, and the SPSS software package (version 12.0) was used for determining statistically significant differences using the Kruskal-Wallis test. Each sample was performed in triplicate.
Phage DNA was extracted by Virus DNA Kit (OMEGA) and sequenced by the Illumina HiSeq PE150 paired terminal sequence method in Beijing Novogene company (Liu et al., 2021). The data gaps between the remaining contigs were filled using GapCloser and GapFiller (Gong et al., 2017). Open reading frames (ORFs) were predicted by GeneMarkS1 and RAST2 (Besemer and Borodovsky, 2005; Aziz et al., 2008). The protein function and homology of ORFs were predicted using the BLASTP (http://blast.ncbi.nlm.nih.gov/, E-value ≤10−3), Pfam3 and HHpred tool (E-value ≤10−3), which can fit the results of several databases [NCBI4 conserved domain database (CDD), Swiss-Prot5 and PDB6] (Sayers et al., 2009; Gabler et al., 2020; Mistry et al., 2021). The genome map was performed using the CLC Main Workbench 20 (Tohidi and Javanmard, 2020). tRNAscan-SE7 was used to test whether the tRNA sequence is in the phage genome (Lowe and Eddy, 1997). The genome sequence of the phage vB_ValR_NF is available under the GenBank accession number MN812722.1.
In order to explore the position of phage vB_ValR_NF in taxonomy and the homology between phage vB_ValR_NF and other phages, we have done the following work. First, the sequence of phage vB_ValR_NF was searched as a query against the NCBI database through tBLASTx (E-value ≤ 10−5; query cover ≥ 50%) and the Integrated Microbial Genomes/Virus (IMG/VR) through BLASTp (E-value ≤ 10−5; qurey cover ≥ 50%; Paez-Espino et al., 2017; Roux et al., 2021); Then, the sequence of phage vB_ValR_NF and the screened sequences from IMG/VR and the NCBI database were analyzed together with a total of 4,469 Caudoviricetes phages in the NCBI RefSeq database [NCBI Virus (nih.gov)] by vConTACT 2.0, Viral clusters (VCs) were identified using ClusterONE with default parameters, network diagram was visualized by Gephi (Bolduc et al., 2017; Bin Jang et al., 2019). According to the results of vConTACT 2.0, phylogenetic and genome comparison analyses of the Vibrio phage vB_ValR_NF and its closest relatives were conducted by viptree8 (Nishimura et al., 2017). Meanwhile, we use VIRIDIC to calculate intergenomic similarities between phage vB_ValR_NF and other related phages (Moraru et al., 2020).
Considering that phage vB_ValR_NF was isolated from the U. prolifera blooms, it is necessary to discuss the impact of phage vB_ValR_NF in the marine ecosystem. To this end, we selected 154 viral metagenomes from five viral ecological zones (VEZs) of the Global Ocean Viromes (GOV2.0) data set [Arctic (ARC), Antarctic (ANT), bathy-pelagic (BATHY), temperate and tropical epipelagic (EPI), temperate and tropical mesopelagic (MES)] (Gregory et al., 2019), and supplemented 17 viral metagenomes data sets from the U. prolifera blooms in summer 2017 in Qingdao offshore (Bioproject number: PRJNA797266) to see the abundance of phage vB_ValR_NF in the global system. To be more rigorous, we also calculated the abundance of other representative phages like Pelagibacter phages (Chen et al., 2019), Cyanophages including Prochlorococcus phages and Synechococcus phages (Labrie et al., 2013), and other V. alginolyticus phages. The relative abundances of these viral genomes calculate by CoverM (v0.3.1; Wang et al., 2021), and the normalized data were log10 transformed based on TPM (transcripts per million).
A novel Vibrio phage vB_ValR_NF was isolated from the coast of Qingdao during the Ulva prolifera blooms. As observed by transmission electron microscope (TEM), the phage vB_ValR_NF has the typical morphology of siphovirus, consisting of an icosahedral capsid (~114 ± 1 nm in diameter) with a long non-contractile tail (~231 ± 1 nm; Figure 1A).
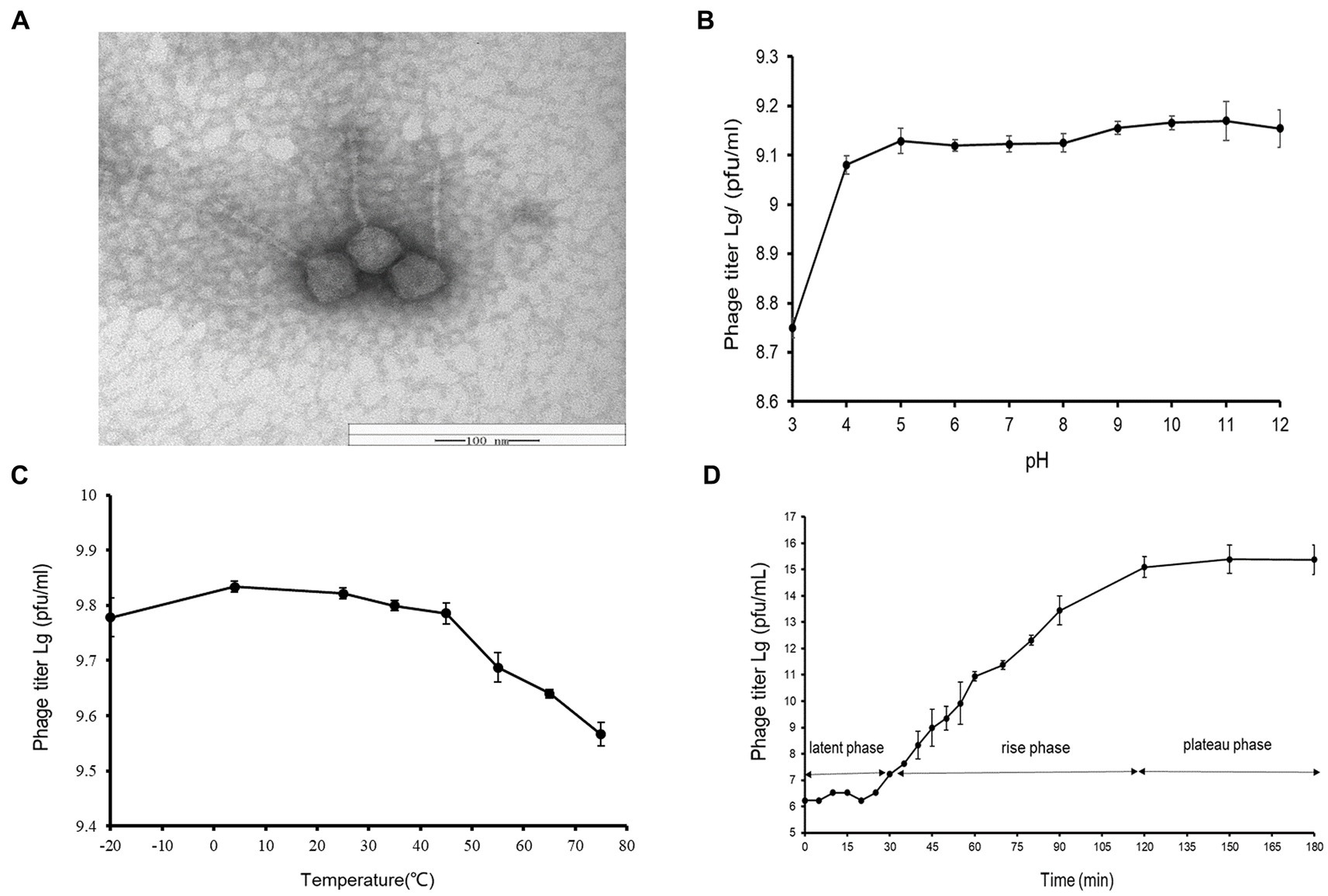
Figure 1. (A) Transmission electron micrograph of Vibrio phage vB_ValR_NF. Scale bar, 100 nm. (B) pH stability of Vibrio phage vB_ValR_NF. Y-axis shows the log of plaque-forming units per milliliter (PFU/mL). (C) Temperature stability of Vibrio phage vB_ValR_NF. Y-axis shows the log of plaque-forming units per milliliter (PFU/mL). (D) One-step growth curve of Vibrio phage vB_ValR_NF. Y-axis shows the log of plaque-forming units per milliliter (PFU/mL).
By setting different pH/thermal gradients to treat phage vB_ValR_NF, and using the double-layer plate method under room conditions (30°C; pH = 7), we obtained experimental results on the stability of phage vB_ValR_NF to pH /temperature. As shown in Figure 1B, the phage titer was the lowest at pH 3, subsequently, the phage vB_ValR_NF maintained stable infectivity at the range of pH 4-pH 12, and the highest infectivity to infect the host at pH 11. The thermal stability study showed that phage vB_ValR_NF was able to active between −20°C and 45°C and its infectivity gradually weakens over 45°C, while the phage titer was highest when the temperature was at 5°C (Figure 1C).
The one-step growth curve showed that the latent period of phage vB_ValR_NF was about 30 min and reached the plateau stage after 90 min. The final count of liberated phage particles was 2.42 × 1015 PFU/mL (Figure 1D). The burst size was approximately 113 virions per cell. The short latent period and a large burst size indicated that this phage has high lytic activity and robust propagation.
As shown in Table 1, phage vB_ValR_NF is infectious to several Vibrio strains of the Vibrio genus. The ability of phages to infect multiple hosts usually means that the genes encoding the tail fibers of phages can recognize specifically related proteins on the surface of many hosts (Ma et al., 2021). In the future, more Vibrio strains, especially Vibrio cholerae and others related to human diseases, will be used to test the host range of phage vB_ValR_NF. We will also further explore the tail coding genes of phage vB_ValR_NF, better understanding the interaction mechanism between phage and host.
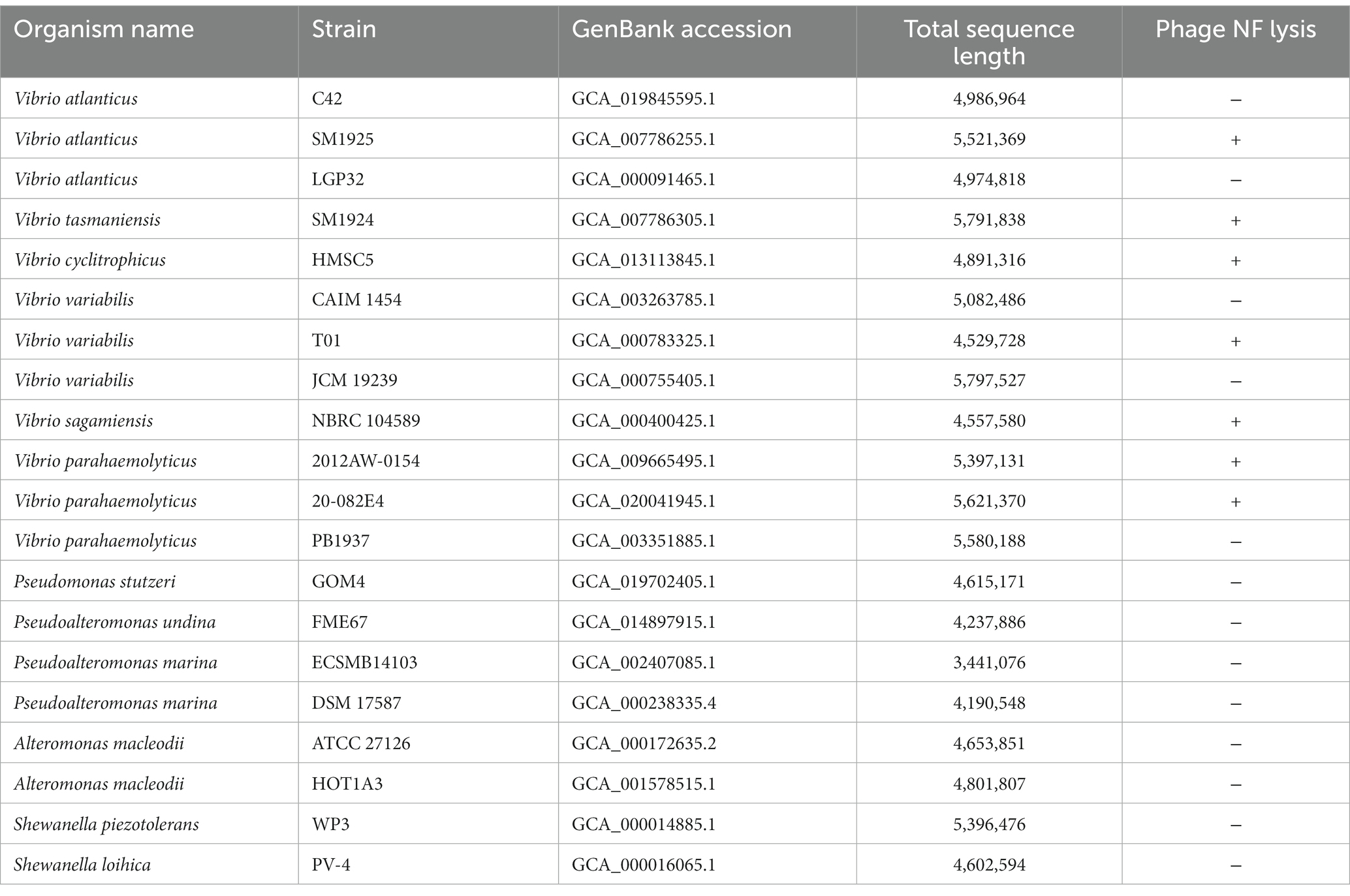
Table 1. Host range of Vibrio phage NF.
In addition, the curves of the host lysis test (Figure 2A) indicated that the growth of the bacterial strains was inhibited after phage vB_ValR_NF inoculation at different MOIs, and the phage vB_ValR_NF had the best infection efficiency to the host at the MOI = 10−3. The re-determination of OD600 value at the end of the host lysis test (Figure 2B) also showed that there was a significant difference (p < 0.05) in the number of microorganisms between the experimental groups with phage vB_ValR_NF inoculation and the control group, which indicated that phage vB_ValR_NF had great potential in the treatment of Vibrio infection.
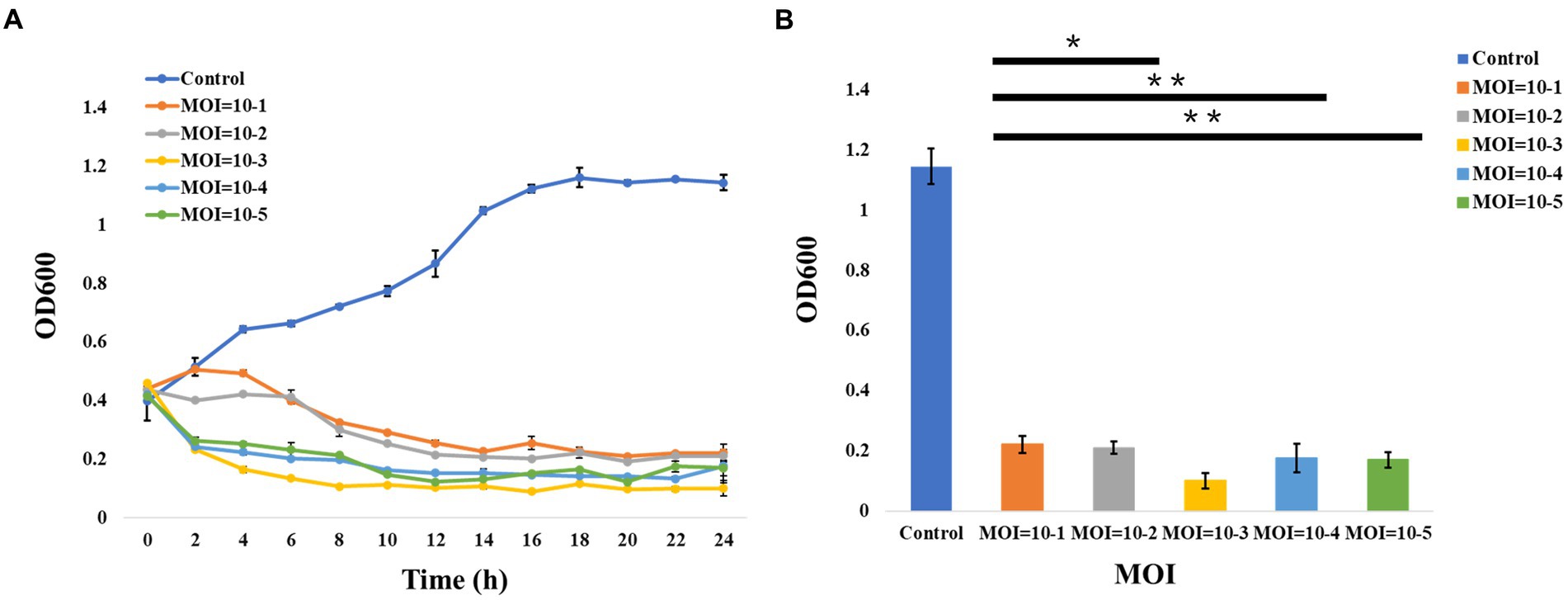
Figure 2. Host lysis investigation. (A) The curve of host (Vibrio alginolyticus strain) lysis by Vibrio phage vB_ValR_NF inoculation at different MOIs (10−1, 10−2, 10−3, 10−4, and 10−5). (B) The final OD600 value after 24 h infection at different MOIs. “*” shows the level of p<0.05, “**” shows the level of p<0.01, indicating that there is a significant difference between control and the experimental groups. Each point represents average from three replicate experiments.
The phage vB_ValR_NF contains a double-stranded DNA genome with a total length of 44,507 bp, 43.10% GC content and there are no tRNAs. Seventy five ORFs were scattered throughout the phage vB_ValR_NF genome with different gene arrangements, among them, the protein functions of 46 ORFs (61%) are unknown, which are called hypothetical proteins. The protein functions of 29 ORFs (39%) can be significantly hit according to the results of searches of the databases, including three auxiliary metabolic genes (AMGs; ORF10, ORF16, ORF 45; Table 2; Figure 3).
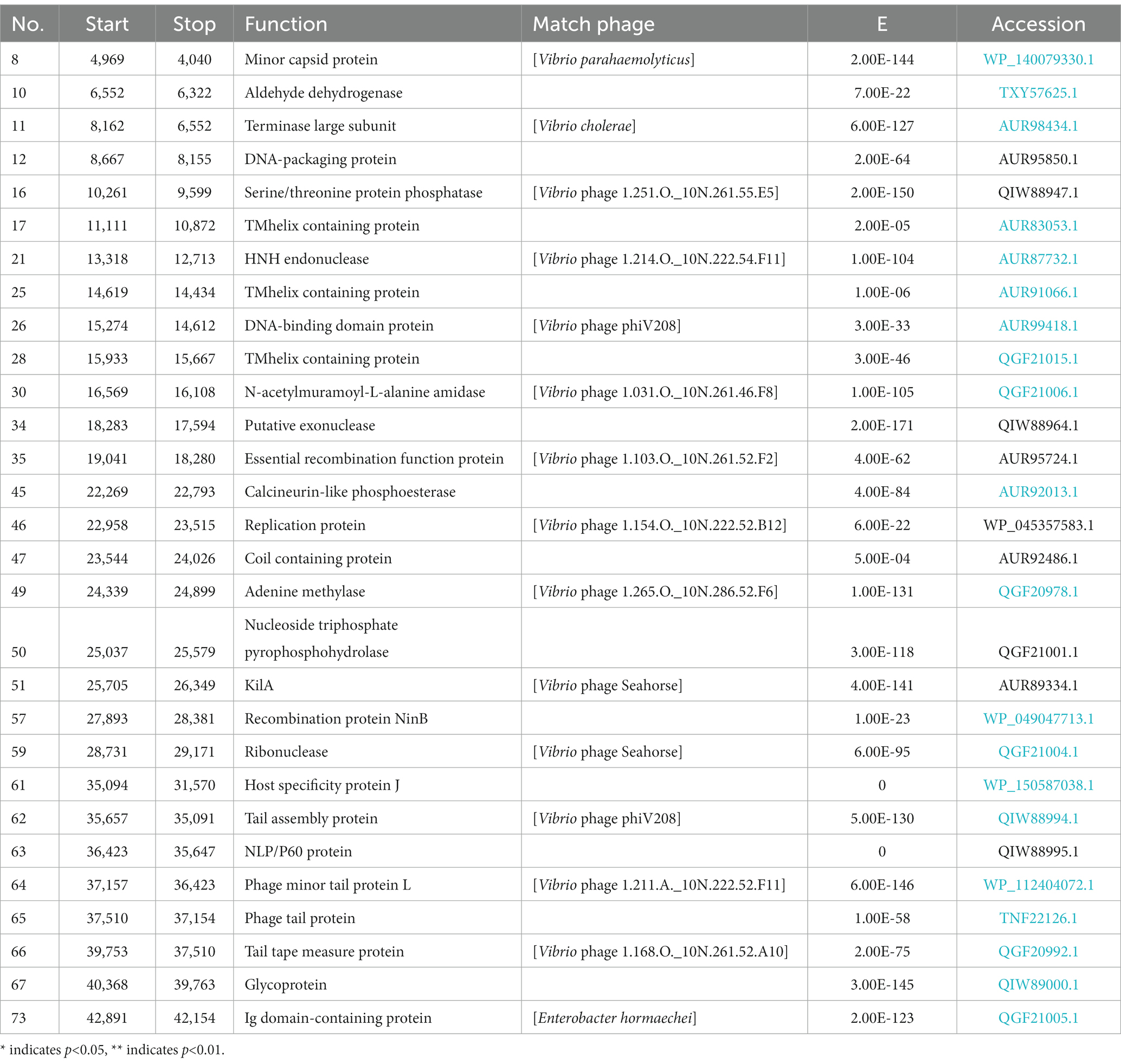
Table 2. The conserved domains information of Vibrio phage NF genome.
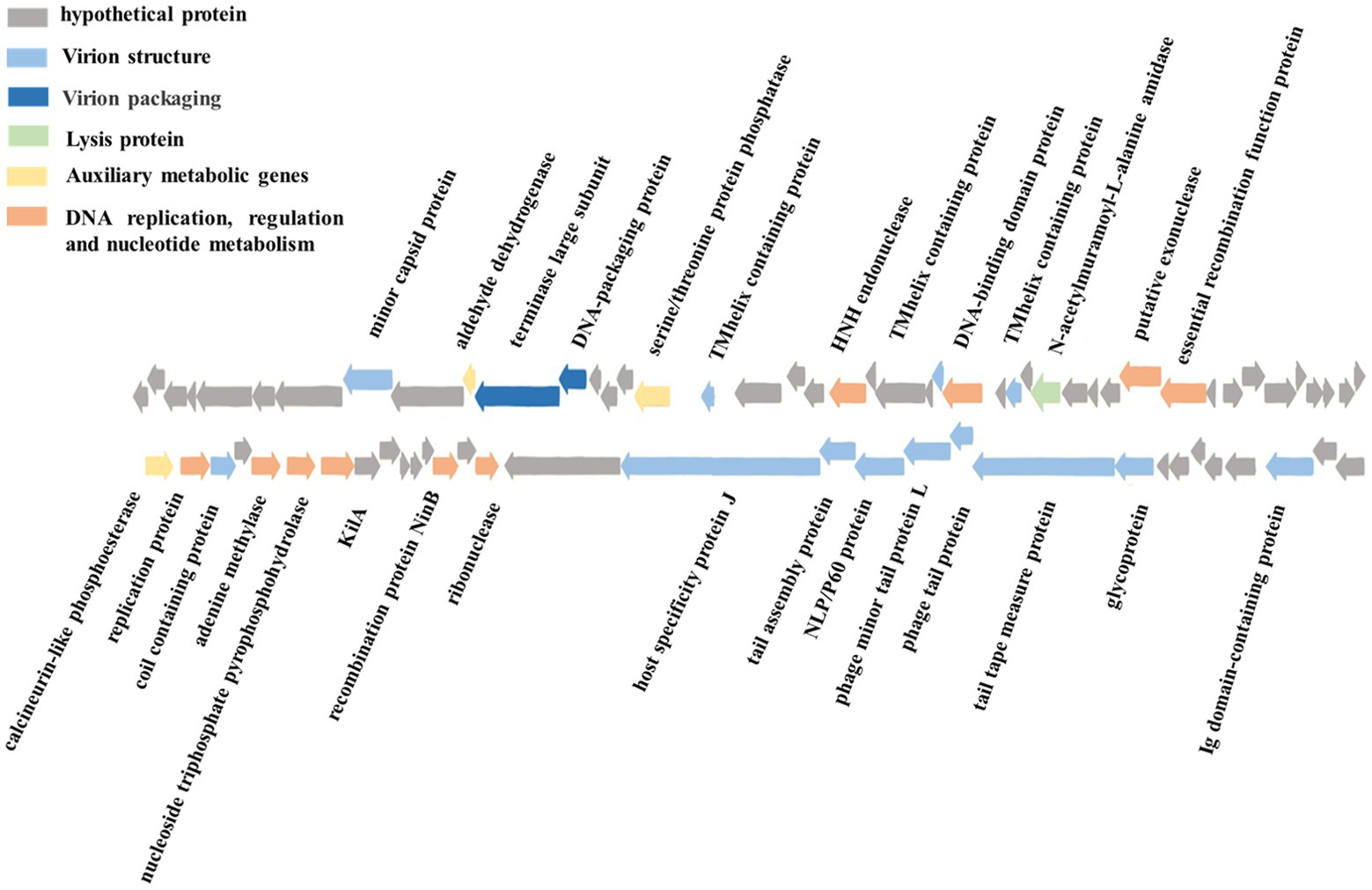
Figure 3. Genome map and functional annotation of the predicted proteins of Vibrio phage vB_ValR_NF (gray: hypothetical protein; blue: virion structure module; dark blue: virion packaging module; Green: lysis protein module; Yellow: auxiliary metabolic genes; Orange: DNA replication, regulation and nucleotide metabolism module).
One of the most interesting features of in structure module in phage vB_ValR_NF is the existence of genes for tail assembly protein (ORF 62) and NLP/P60 protein (ORF 63). The tail assembly protein contains a domain that belongs to the family of Lambda_tail_I (PF06805), called bacteriophage lambda tail assembly protein I (TAPI; Iyer et al., 2006; North et al., 2019). TAPI is related to ubiquitin (Ub)-like mediated signaling systems in prokaryotes and a large of phages (Makarova et al., 2000; Li Y. et al., 2019). Similar to the ubiquitin (Ub)-related signal system of eukaryotes, the Ub-like signal system is considered to be widely involved in protein localization, metabolism, function, regulation and degradation (Wu et al., 2018; Valerdi et al., 2021). NlpC/P60 is a ubiquitous cell-wall-associated and papain-like deubiquitinating peptidases (DUBs), it plays an important role in maintaining the dynamic balance between ubiquitination and deubiquitination in eukaryotes, and further participating in the regulation of protein degradation and the regulation of life activities of hosts (Bannantine et al., 2016; Duchêne et al., 2019). Previous works have confirmed many Ub-like proteins in phages and prokaryotes were the precursors to the eukaryotic Ub-signaling system (Iyer et al., 2006; Berk et al., 2020). Consequently, the Vibrio phage vB_ValR_NF containing tail assembly protein and NLP/P60 protein may provide an extraordinary model for further studying the evolutionary process of the complete signaling system between viruses, prokaryotes and eukaryotes.
A total of 10 genes were detected to encode proteins related to DNA replication, regulation and nucleotide metabolism, which to some extent explained that the replication system of phage vB_ValR_NF was independent of the host. Interestingly, KilA encoded by ORF 51 is a conserved DNA binding domain, it widely exists in the proteins of large bacteria and eukaryotic DNA viruses (Pritham et al., 2007), considering that the abundance of viruses infected with eukaryotic algae generally increases during the U. prolifera blooms, so we speculate that the ORF encoding kilA was detected in phage vB_ValR_NF, which may indicate that there may be complex horizontal gene transfer between microbe during this period (Guo et al., 2011; Jung et al., 2018; Bhatnagar et al., 2020; Wang J. et al., 2020). Meanwhile, ORF 57, coding DNA adenine methylase (Dam), was detected in the genome of phage vB_ValR_NF. Methylases are ubiquitous in prokaryotes and form Restriction-Modification (RM) systems with endonucleases to resist phage infection (Nathans and Smith, 1975). However, about 20% of phages have coding genes related to methylase. They have been proven to help phages avoid the crisis removed by the RM system in hosts and improve their infection efficiency (Pleška and Guet, 2017). In addition, Dams were found to be essential for the virulence of some members of the Vibrio, like Vibrio cholerae. Dams overproduction can lead to the attenuation of virulence in Vibrio, and then induce protective an immune response in the organism infected by Vibrio (Murphy et al., 2013; Kalatzis et al., 2017). We may be able to use phage vB_ ValR_ NF to manipulate the virulence of the host V. alginolyticus by encoding Dam, then stimulate the immune defense mechanism of the infected body to resist the attack of Vibrio. Therefore, the interaction mechanism between phage vB_ValR_NF and the host will also be the focus of our future research.
Most strikingly, three AMGs were found in the genome of phage vB_ValR_NF, including aldehyde dehydrogenase (ORF 10), serine/threonine protein phosphatase (ORF 16), calcineurin-like phosphoesterase (ORF 45).
Vibrio alginolyticus produces many aldehydes in its life cycle. In turn, these toxic substances will cause serious harm to the survival of V. alginolyticus itself under the limited living environment (such as the U. prolifera blooms; Baburam and Feto, 2021). The aldehyde dehydrogenases (ALDHs) are ubiquitous in all organisms and indispensable intermediate reactants in the biological reaction of toxic aldehyde conversion in non-toxic acid, furthermore, the resulting non-toxic acids mediated by ALDHs are also beneficial for promoting plant growth (Ahvazi et al., 2000; Cho et al., 2021). Therefore, we speculate that phage vB_ValR_NF may help host cells improve the degradation ability of toxic aldehydes by encoding ALDHs to alleviate the energetic and bioconversion bottleneck of hosts during the U. prolifera blooms. At the same time, it might maintain the dynamic balance of the micro-ecosystem by indirectly affecting the growth of eukaryotes so as to improve the survival chance of phage vB_ValR_NF under harsh conditions.
Serine/threonine protein phosphatase and calcineurin-like phosphoesterase both catalyze the dephosphorylation of phosphorylated protein molecules to regulate protein expression or protect cells from damage when misfolded proteins are accumulated in the periplasm (Lamp et al., 2013). It is likely that serine/threonine protein phosphatase and calcineurin-like phosphoesterase encoded by phage vB_ValR_NF may catalyze the dephosphorylation process of the hosts and alter hosts activities to allow more effective production of phage DNA or protein under some conditions or hinder the synthesis of some important proteins in the hosts.
The functional proteins found in phage vB_ValR_NF are not only essential to maintain the survival of the phage itself, but also widely involved in the interaction with the host, even having a profound impact on the clinical treatment of Vibrio infection. However, most of the proteins encoded by phage vB_ValR_NF are labeled as hypothetical proteins, which is due to the small number of phages isolated and identified, the functions of some important viral proteins cannot be successfully hit in the databases. Therefore, the isolation of more Vibrio phages has a profound impact on the supplement of the database related viruses, even the clinical treatment of Vibrio infection.
Based on search results in the NCBI and the IMG/VR database, only two metagenomic assembled uncultured viral sequences (IMGVR_UViG_3300029182_000002, IMGVR_UViG_3300029182_000009; E-value ≤ 10−5; qurey cover ≥ 50%) were matched with phage vB_ValR_NF. Then, vConTACT 2.0 was used to compare the genomic similarity at the protein level between these three sequences and a total of 4,469 Caudoviricetes viruses in the NCBI RefSeq database. Only IMGVR_UViG_3300029182_000009 was classified into Viral Cluster 24 (VC_24) together with phage vB_ValR_NF (Figure 4A). The results of phylogenetic (Figure 4B) and genomic comparative analysis (Figure 4C) using viptree also showed that VC_24 is relatively independent of other viral clusters and the level of genes encoding homologous proteins between VC_24 and other phages varies greatly, which may suggest that phage vB_ValR_NF may represent a unique group. To confirm this hypothesis, nucleotide-based intergenomic similarities of the total of 14 phages from the phylogenetic tree of Figure 4B were calculated with VIRIDIC, the result showed that the nucleotide-based intergenomic similarities between vB_ValR_NF and other phages were low (0–27.4%; Figure 5A). Considering that phages are regarded by the ICTV as being members of the same genus when their nucleotide sequence identities are greater than 70% (Moraru et al., 2020), phage vB_ValR_NF is likely to represent a new family under the Caudoviricetes viruses. In order to fully verify the reliability of the conclusions, we also used IQ-TREE to build a phylogenetic tree based on the conservative proteins including the TerL (ORF 11) and TMP (ORF 66) of phage vB_ValR_NF with the most matching 10 phage proteins, respectively. The results indicated that phage vB_ValR_NF has conservative proteins distinguished from other phages and form a novel sub-branch in the phylogenetic tree (Figures 5B,C). Therefore, we can propose that phage vB_ValR_NF represents a new family within Caudoviricetes, named here as Ruirongviridae.
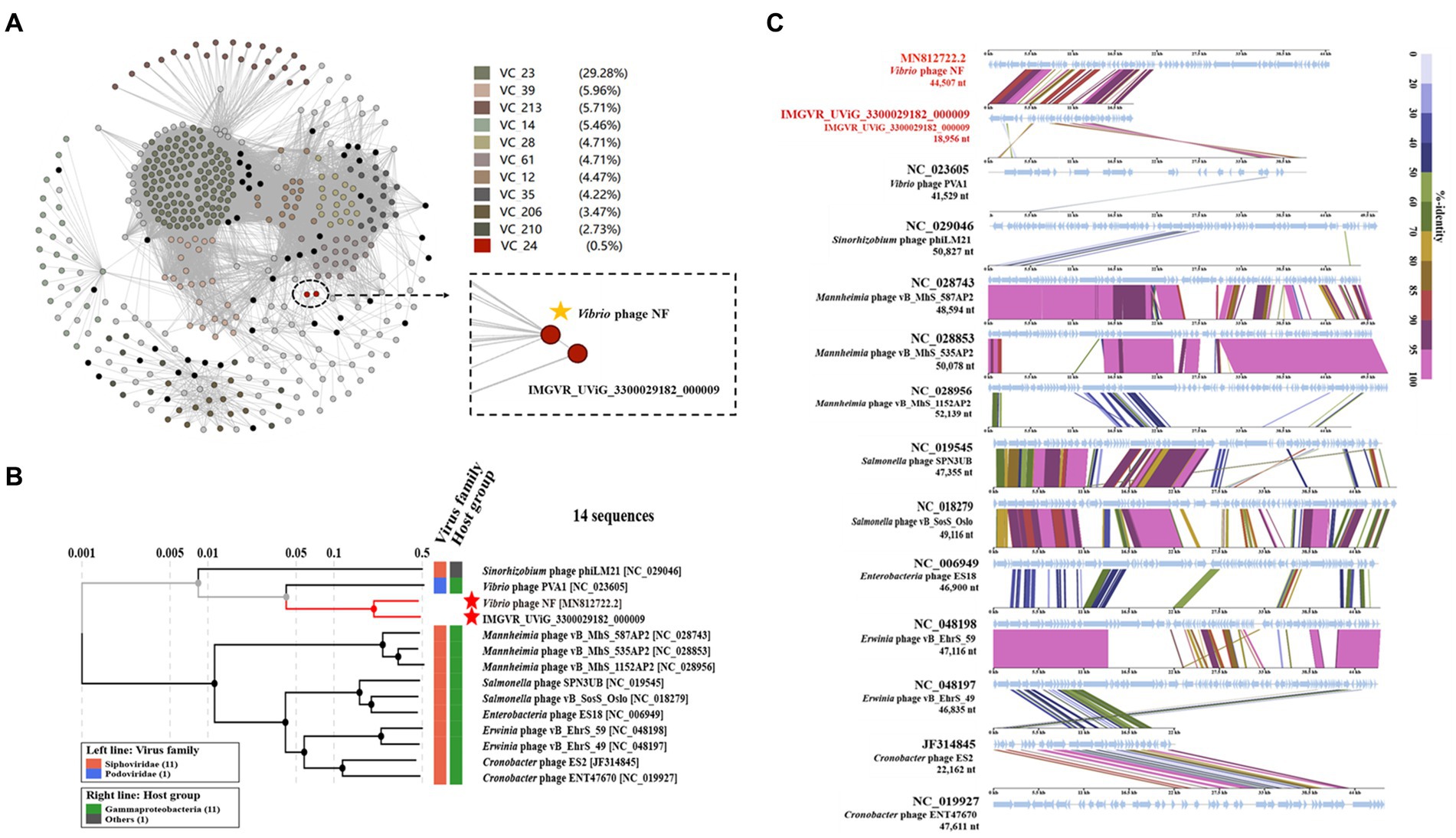
Figure 4. (A) Gene-content-based viral network of Vibrio phage vB_ValR_NF with related Caudoviricetes viruses in the NCBI RefSeq database, and related uncultured viral genome sequences (UViGs) from IMG/VR dataset. The Vibrio phage vB_ValR_NF and 1 sequence named IMGVR_UViG_3300029182_000009 belong to the Viral Cluster_24 (VC_24). (B) Phylogenetic analysis based on the genome-wide sequence of Vibrio phage vB_ValR_NF with other dsDNA viruses in viptree database. (C) Genome-wide comparison of Vibrio phage vB_ValR_NF and the most related viruses in the phylogenetic tree based on the genome-wide sequence. Genome regions showing similarity were searched using tBLASTX and the matches satisfying length and E-value (<10−5) cutoffs were indicated by the rectangle according to the color scale on the right.
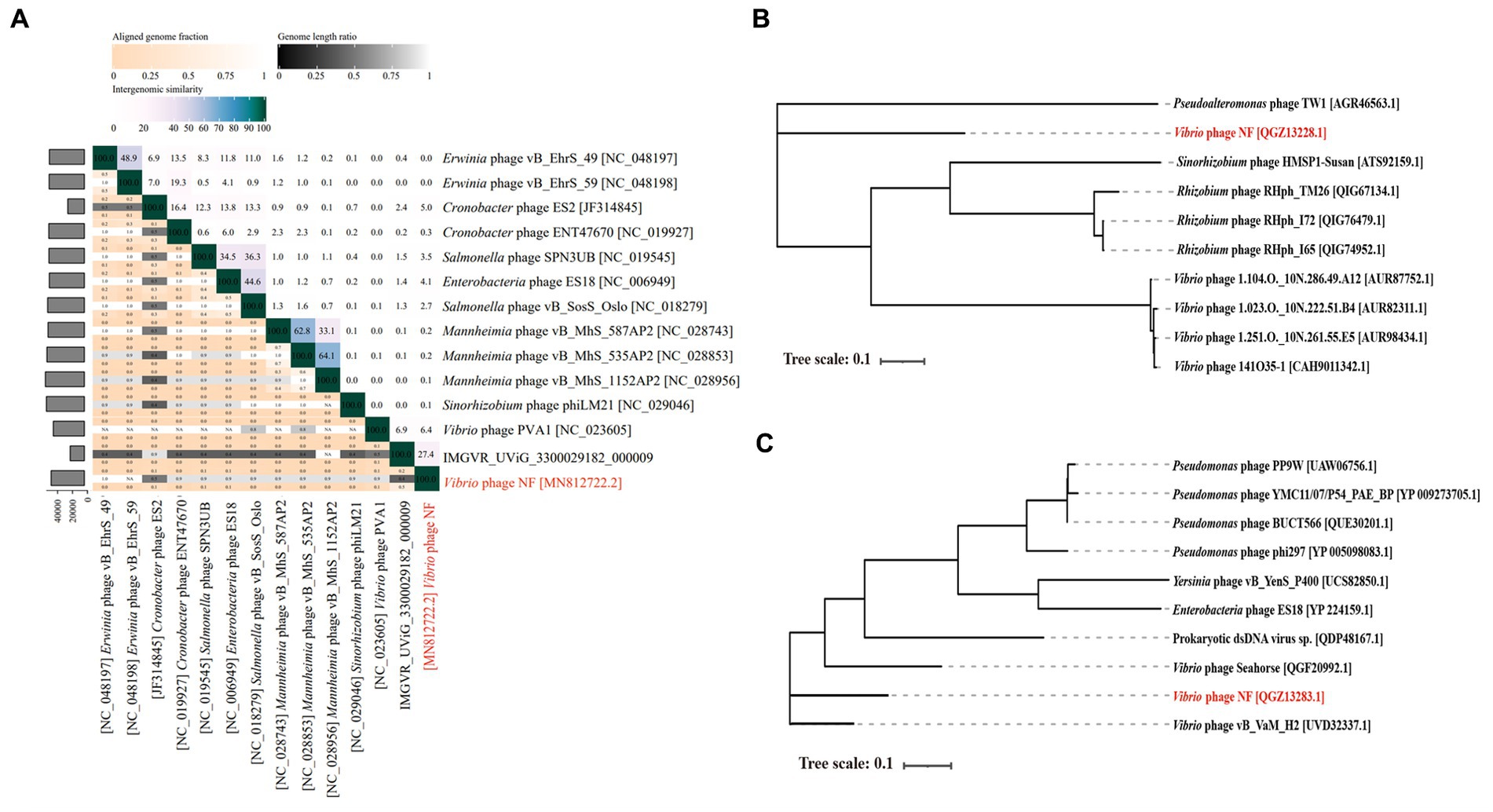
Figure 5. (A) The heat map shows nucleotide-based intergenomic similarities of the total of 14 phages from the phylogenetic tree of Figure 4B calculated with VIRIDIC. Values represent Average Nucleotide Identity (ANI). (B) The phylogenetic tree is based on terminase large subunits (TerL) of Vibrio phage vB_ValR_NF and homolog proteins from other phages. Amino acid sequences were aligned based on the Maximum Likelihood method with 1,000 bootstrap replications by the software IQ-TREE. (C) The phylogenetic tree is based on tape measure protein (TMP) of Vibrio phage vB_ValR_NF and homolog proteins from other phages. Amino acid sequences were aligned based on the Maximum Likelihood method with 1,000 bootstrap replications by the software IQ-TREE.
As expected, the relative abundance of Vibrio phage vB_ValR_NF is high in the area where the U. prolifera blooms off the coast of Qingdao, far exceeding the proportion in these data sets of global ocean viromes (GOV2.0; Figure 6). We also noticed that some other phages, such as Synechococcus phage, Prochlorococcus phage, etc., were also active during the outbreak of the U. prolifera, which also confirmed that viruses might be played a pivotal role in the process of the green tide. Although the microbial community structure during the U. prolifera blooms has been studied to some extent (Lin et al., 2017; Wang et al., 2017; Fan et al., 2022), there is no report on the single phage isolated during this period. Therefore, Vibrio phage vB_ValR_NF will be an ideal model to help us understand how a single virus affects the host, the microbial loop, and even the whole ecosystem. At present, we have only compared the Vibrio phage vB_ValR_NF with viral metagenomes data sets from the U. prolifera blooms in the summer of 2017 in Qingdao offshore, which is not enough to help us further study the impact of Vibrio phage vB_ValR_NF on Qingdao offshore environment. Therefore, we will also collect more viral metagenomes data sets during the U. prolifera blooms in the future for a more in-depth discussion on the functions of phage vB_ValR_NF.
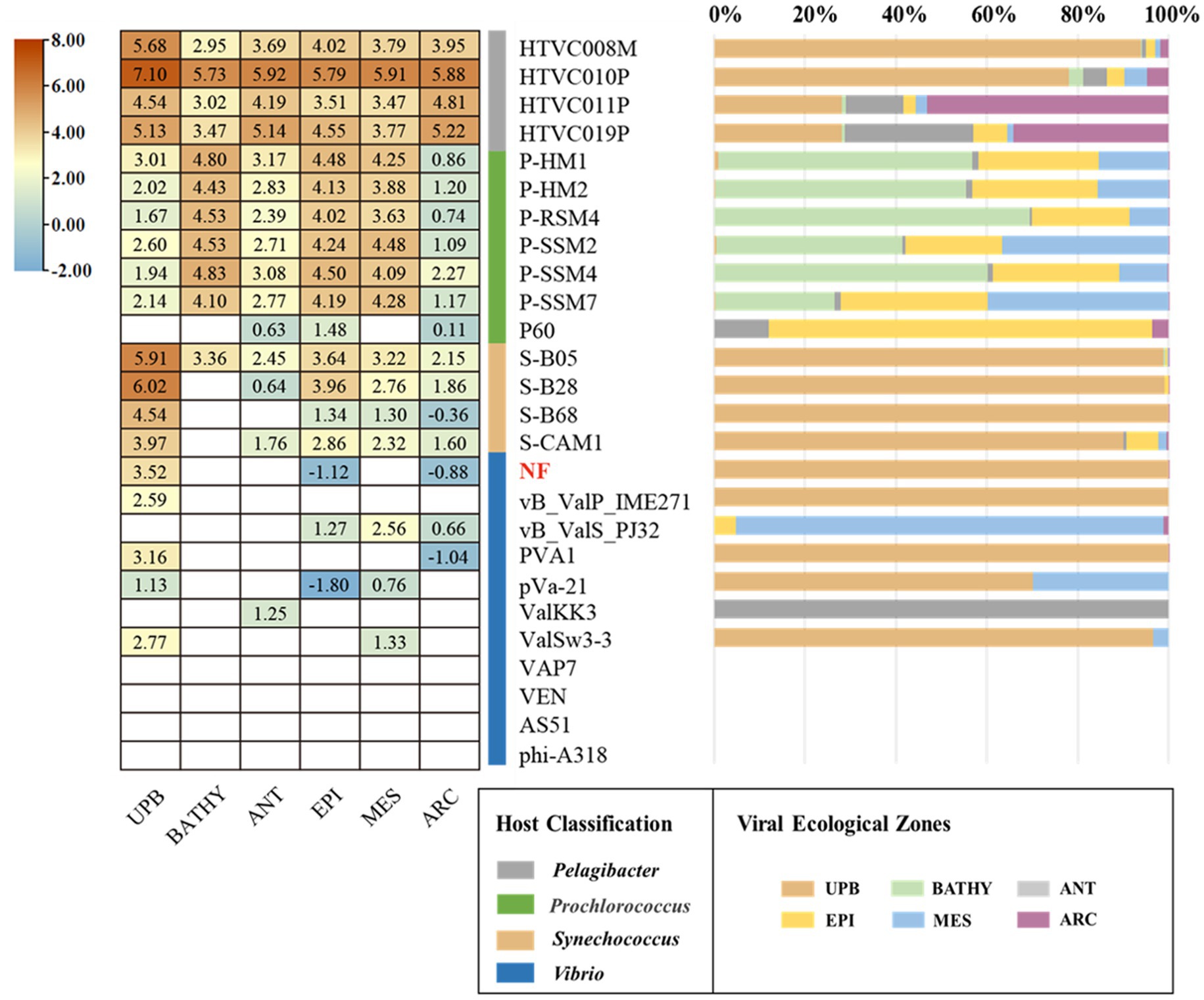
Figure 6. Relative abundance of the Vibrio phage vB_ValR_NF in marine environments. Left, the heatmap reflects the relative abundances of the Vibrio phage vB_ValR_NF and other bacteriophages in five marine viral ecological zones (VEZs) defined by the GOV2.0. (ARC, Arctic; ANT, Antarctic; BATHY, bathypelagic; EPI, temperate and tropical epipelagic; MES, temperate and tropical mesopelagic.) and 2017 viral metagenomes data sets from the Ulva prolifera blooms in the summer of 2017 in Qingdao offshore (UPB). The normalized data were log10 transformed based on TPM (transcripts per million). Right, the bar graph shows the percentage of bacteriophages in the five VEZs and UPB.
In this study, Vibrio phage vB_ValR_NF was isolated from the coastal area of Qingdao, China in 2019 (36°06′N, 120°34′E), where the U. prolifera blooms. Genomics analysis shows that Vibrio phage vB_ValR_NF is distanced from other reference viruses in terms of evolutionary relationship, and has unique conservative protein genes far from other phages. We propose that it represents a new family in the Caudoviricetes viruses, called Ruirongviridae. The stability, growth curve and host range analysis suggest that phage vB_ValR_NF could be active in a high range of pH and temperature, with a short latent period and a large burst size, and has a high inhibitory ability against the host V. alginolyticus and other strains of Vibrio genus, these characteristics will become important reference factors when evaluating the potential of phage vB_ValR_NF in bacteriophage therapy in the future. In addition, Vibrio phage vB_ValR_NF was more abundant during the U. prolifera blooms than in other marine environments, the existence of three AMGs and RM modification system also seems to mean that it can promote the host and itself to occupy the survival advantage during the U. prolifera blooms by influencing some metabolic mechanisms of the host, suggest it may play a key role in affecting the microbial community structure and the micro-environment during the occurrence and decline of U. prolifera blooms. In view of the great potential of Vibrio phage vB_ValR_NF in bacteriophage therapy and its important role in the U. prolifera blooms ecosystem, we will focus on these aspects to discuss and analyze phage vB_ValR_NF in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, MN812722.1.
YaL, JH, AM, and MW: conceptualization, revision, project administration, supervision, and funding acquisition. XZ, KZ, YuL, YH, and LR: methodology, formal analysis, and writing and original draft preparation. XZ, YD, ZW, and HW: sample collection and expedition organization. YS, WM, and LW: review and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the Laoshan Laboratory (no. LSKJ202203201), National Key Research and Development Program of China (2022YFC2807500), Natural Science Foundation of China (nos. 41976117, 42120104006, 42176111, and 42188102), and the Fundamental Research Funds for the Central Universities (202172002, 201812002, and 202072001 to AM).
We sincerely appreciate the Center for High Performance Computing and System Simulation, Pilot National Laboratory for Marine Science and Technology (Qingdao). We appreciate the computing resources provided by IEMB-1, a high-performance computing cluster operated by the Institute of Evolution and Marine Biodiversity, and Marine Big Data Center of Institute for the Advanced Ocean Study of the Ocean University of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1161265/full#supplementary-material
1. ^http://topaz.gatech.edu/GeneMark/
3. ^http://pfam.xfam.org/search/sequence
4. ^http://www.ncbi.nlm.nih.gov/
5. ^https://web.expasy.org/docs/swiss-prot_guideline.html
Ahvazi, B., Coulombe, R., Delarge, M., Vedadi, M., Zhang, L., Meighen, E., et al. (2000). Crystal structure of the NADP+-dependent aldehyde dehydrogenase from Vibrio harveyi: structural implications for cofactor specificity and affinity. Biochem. J. 349, 853–861. doi: 10.1042/bj3490853
Aziz, R. K., Bartels, D., Best, A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Baburam, C., and Feto, N. A. (2021). Mining of two novel aldehyde dehydrogenases (DHY-SC-VUT5 and DHY-G-VUT7) from metagenome of hydrocarbon contaminated soils. BMC Biotechnol. 21:18. doi: 10.1186/s12896-021-00677-8
Bannantine, J. P., Lingle, C. K., Adam, P. R., Ramyar, K. X., McWhorter, W. J., Stabel, J. R., et al. (2016). NlpC/P60 domain-containing proteins of Mycobacterium avium subspecies paratuberculosis that differentially bind and hydrolyze peptidoglycan. Protein Sci. 25, 840–851. doi: 10.1002/pro.2884
Bartlau, N., Wichels, A., Krohne, G., Adriaenssens, E. M., Heins, A., Fuchs, B. M., et al. (2021). Highly diverse flavobacterial phages isolated from North Sea spring blooms. ISME J. 16, 555–568. doi: 10.1038/s41396-021-01097-4
Berk, J. M., Lim, C., Ronau, J. A., Chaudhuri, A., Chen, H., Beckmann, J. F., et al. (2020). A deubiquitylase with an unusually high-affinity ubiquitin-binding domain from the scrub typhus pathogen Orientia tsutsugamushi. Nat. Commun. 11:2343. doi: 10.1038/s41467-020-15985-4
Besemer, J., and Borodovsky, M. (2005). GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451–W454. doi: 10.1093/nar/gki487
Bhatnagar, S., Cowley, E. S., Kopf, S. H., Pérez Castro, S., Kearney, S., Dawson, S. C., et al. (2020). Microbial community dynamics and coexistence in a sulfide-driven phototrophic bloom. Environ Microbiomes 15:3. doi: 10.1186/s40793-019-0348-0
Bin Jang, H., Bolduc, B., Zablocki, O., Kuhn, J. H., Roux, S., Adriaenssens, E. M., et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 37, 632–639. doi: 10.1038/s41587-019-0100-8
Bolduc, B., Jang, H. B., Doulcier, G., You, Z. Q., Roux, S., and Sullivan, M. B. (2017). vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect archaea and bacteria. PeerJ 5:e3243. doi: 10.7717/peerj.3243
Chen, L., Liu, Q., Fan, J., Yan, T., Zhang, H., Yang, J., et al. (2020). Characterization and genomic analysis of ValSw3-3, a new Siphoviridae bacteriophage infecting vibrio alginolyticus. J. Virol. 94:e0066-20. doi: 10.1128/jvi.00066-20
Chen, L.-X., Zhao, Y., McMahon, K. D., Mori, J. F., Jessen, G. L., Nelson, T. C., et al. (2019). Wide distribution of phage that infect freshwater SAR11 bacteria. mSystems 4:e00410-19. doi: 10.1128/msystems.00410-19
Cho, S., Kim, G., Song, J. J., and Cho, C. (2021). Cryo-EM structure of vibrio cholerae aldehyde-alcohol dehydrogenase spirosomes. Biochem. Biophys. Res. Commun. 536, 38–44. doi: 10.1016/j.bbrc.2020.12.040
de Souza Valente, C., and Wan, A. H. L. (2021). Vibrio and major commercially important vibriosis diseases in decapod crustaceans. J. Invertebr. Pathol. 181:107527. doi: 10.1016/j.jip.2020.107527
Dong, Y., Zhao, P., Chen, L., Wu, H., Si, X., Shen, X., et al. (2020). Fast, simple and highly specific molecular detection of vibrio alginolyticus pathogenic strains using a visualized isothermal amplification method. BMC Vet. Res. 16:76. doi: 10.1186/s12917-020-02297-4
Duchêne, M. C., Rolain, T., Knoops, A., Courtin, P., Chapot-Chartier, M. P., Dufrêne, Y. F., et al. (2019). Distinct and specific role of NlpC/P60 endopeptidases LytA and LytB in cell elongation and division of lactobacillus plantarum. Front. Microbiol. 10:713. doi: 10.3389/fmicb.2019.00713
Emam, A. M., Hashem, M., Gadallah, A. O., and Haridy, M. (2019). An outbreak of vibrio alginolyticus infection in aquarium-maintained dark-spotted (Himantura uarnak) and Tahitian (H. fai) stingrays. Egypt. J. Aquat. Res. 45, 153–158. doi: 10.1016/j.ejar.2019.05.003
Fan, Q., Shi, K., Zhan, M., Xu, Q., Liu, X., Li, Z., et al. (2022). Acute damage from the degradation of Ulva prolifera on the environmental microbiota, intestinal microbiota and transcriptome of Japanese flounder Paralichthys olivaceus. Environ. Pollut. 302:119022. doi: 10.1016/j.envpol.2022.119022
Frans, I., Dierckens, K., Crauwels, S., van Assche, A., Leisner, J., Larsen, M. H., et al. (2013). Does virulence assessment of vibrio anguillarum using sea bass (Dicentrarchus labrax) larvae correspond with genotypic and phenotypic characterization? PLoS One 8:e70477. doi: 10.1371/journal.pone.0070477
Gabler, F., Nam, S. Z., Till, S., Mirdita, M., Steinegger, M., Söding, J., et al. (2020). Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinformatics 72:e108. doi: 10.1002/cpbi.108
Gong, Z., Wang, M., Yang, Q., Li, Z., Xia, J., Gao, Y., et al. (2017). Isolation and complete genome sequence of a novel Pseudoalteromonas phage PH357 from the Yangtze River estuary. Curr. Microbiol. 74, 832–839. doi: 10.1007/s00284-017-1244-8
Gregory, A. C., Zayed, A. A., Conceição-Neto, N., Temperton, B., Bolduc, B., Alberti, A., et al. (2019). Marine DNA viral macro- and microdiversity from pole to pole. Cells 177, 1109–1123.e14. doi: 10.1016/j.cell.2019.03.040
Guo, C., Li, F., Jiang, P., Liu, Z., and Qin, S. (2011). Bacterial diversity in surface water of the Yellow Sea during and after a green alga tide in 2008. Chinese J. Oceanol. Limnol. 29, 1147–1154. doi: 10.1007/s00343-011-0264-7
Iyer, L. M., Burroughs, A. M., and Aravind, L. (2006). The prokaryotic antecedents of the ubiquitin-signaling system and the early evolution of ubiquitin-like β-grasp domains. Genome Biol. 7, R60–R23. doi: 10.1186/gb-2006-7-7-r60
Jung, H., Kim, J., and Lee, C. (2018). Biomethanation of harmful macroalgal biomass in leach-bed reactor coupled to anaerobic filter: effect of water regime and filter media. Int. J. Environ. Res. Public Health 15. doi: 10.3390/ijerph15050866
Kalatzis, P. G., Rørbo, N., Castillo, D., Mauritzen, J. J., Jørgensen, J., Kokkari, C., et al. (2017). Stumbling across the same phage: comparative genomics of widespread temperate phages infecting the fish pathogen vibrio anguillarum. Viruses 9:122. doi: 10.3390/v9050122
Kim, S. G., Jun, J. W., Giri, S. S., Yun, S., Kim, H. J., Kim, S. W., et al. (2019). Isolation and characterisation of pVa-21, a giant bacteriophage with anti-biofilm potential against vibrio alginolyticus. Sci. Rep. 9:6284. doi: 10.1038/s41598-019-42681-1
Labrie, S. J., Frois-Moniz, K., Osburne, M. S., Kelly, L., Roggensack, S. E., Sullivan, M. B., et al. (2013). Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ. Microbiol. 15, 1356–1376. doi: 10.1111/1462-2920.12053
Lamp, J., Weber, M., Cingöz, G., de Orué Lucana, D. O., and Schrempf, H. (2013). A Streptomyces-specific member of the metallophosphatase superfamily contributes to spore dormancy and interaction with aspergillus proliferans. FEMS Microbiol. Lett. 342, 89–97. doi: 10.1111/1574-6968.12121
Lang, A. S., Zhaxybayeva, O., and Beatty, J. T. (2012). Gene transfer agents: phage-like elements of genetic exchange. Nat. Rev. Microbiol. 10, 472–482. doi: 10.1038/nrmicro2802
Li, H., Liu, Z., Wang, M., Liu, X., Wang, Q., Liu, Q., et al. (2019). Isolation and genome sequencing of the novel marine phage PHS3 from the Yellow Sea, China. Mar. Genomics 44, 70–73. doi: 10.1016/j.margen.2018.10.001
Li, Y., Shi, F., Hu, J., Xie, L., Bode, A. M., and Cao, Y. (2019). The role of Deubiquitinases in Oncovirus and host interactions. J. Oncol. 2019, 1–9. doi: 10.1155/2019/2128410
Lin, G., Sun, F., Wang, C., Zhang, L., and Zhang, X. (2017). Assessment of the effect of Enteromorpha prolifera on bacterial community structures in aquaculture environment. PLoS One 12:e0179792. doi: 10.1371/journal.pone.0179792
Liu, Z., Li, H., Wang, M., Jiang, Y., Yang, Q., Zhou, X., et al. (2018). Isolation, characterization and genome sequencing of the novel phage SL25 from the Yellow Sea, China. Mar. Genomics 37, 31–34. doi: 10.1016/j.margen.2017.09.008
Liu, H., Wang, Y., Cao, J., Jiang, H., Yao, J., Gong, G., et al. (2020). Antimicrobial activity and virulence attenuation of citral against the fish pathogen vibrio alginolyticus. Aquaculture 515:734578. doi: 10.1016/j.aquaculture.2019.734578
Liu, Y., Zhao, L., Wang, M., Wang, Q., Zhang, X., Han, Y., et al. (2019). Complete genomic sequence of bacteriophage P23: a novel vibrio phage isolated from the Yellow Sea, China. Virus Genes 55, 834–842. doi: 10.1007/s11262-019-01699-3
Liu, Y., Zheng, K., Liu, B., Liang, Y., You, S., Zhang, W., et al. (2021). Characterization and genomic analysis of Marinobacter phage vB_MalS-PS3, representing a new lambda-like temperate Siphoviral genus infecting algae-associated bacteria. Front. Microbiol. 12, 1–12. doi: 10.3389/fmicb.2021.726074
López-Pérez, M., Jayakumar, J. M., Haro-Moreno, J. M., Zaragoza-Solas, A., Reddi, G., Rodriguez-Valera, F., et al. (2019). Evolutionary model of cluster divergence of the emergent marine pathogen vibrio vulnificus: from genotype to ecotype. MBio 10:e02852-18. doi: 10.1128/mBio.02852-18
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lu, L., Cai, L., Jiao, N., and Zhang, R. (2017). Isolation and characterization of the first phage infecting ecologically important marine bacteria Erythrobacter. Virol. J. 14, 104–115. doi: 10.1186/s12985-017-0773-x
Ma, R., Lai, J., Chen, X., Wang, L., Yang, Y., Wei, S., et al. (2021). A novel phage infecting Alteromonas represents a distinct group of Siphophages infecting diverse aquatic Copiotrophs. mSphere 6:e00454-21. doi: 10.1128/msphere.00454-21
Makarova, K. S., Aravind, L., and Koonin, E. (2000). A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and chlamydia pneumoniae. Trends Biochem. Sci. 25, 50–52. doi: 10.1016/S0968-0004(99)01530-3
Middelboe, M., Chan, A. M., and Bertelsen, S. K. (2010). “Isolation and life cycle characterization of lytic viruses infecting heterotrophic bacteria and cyanobacteria” in Manual of aquatic viral ecology
Miller, E. S., Heidelberg, J. F., Eisen, J. A., Nelson, W. C., Durkin, A. S., Ciecko, A., et al. (2003). Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J. Bacteriol. 185, 5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Mok, J. S., Ryu, A., Kwon, J. Y., Park, K., and Shim, K. B. (2019). Abundance, antimicrobial resistance, and virulence of pathogenic vibrio strains from molluscan shellfish farms along the Korean coast. Mar. Pollut. Bull. 149:110559. doi: 10.1016/j.marpolbul.2019.110559
Moraru, C., Varsani, A., and Kropinski, A. M. (2020). VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 12. doi: 10.3390/v12111268
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A., and van Sinderen, D. (2013). Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 79, 7547–7555. doi: 10.1128/AEM.02229-13
Nathans, D., and Smith, H. O. (1975). Restriction endonucleases in the analysis and restructuring of DNA molecules. Annu. Rev. Biochem. 44, 273–293. doi: 10.1146/annurev.bi.44.070175.001421
Nishimura, Y., Yoshida, T., Kuronishi, M., Uehara, H., Ogata, H., and Goto, S. (2017). ViPTree: the viral proteomic tree server. Bioinformatics 33, 2379–2380. doi: 10.1093/bioinformatics/btx157
North, O. I., Sakai, K., Yamashita, E., Nakagawa, A., Iwazaki, T., Büttner, C. R., et al. (2019). Phage tail fibre assembly proteins employ a modular structure to drive the correct folding of diverse fibres. Nat. Microbiol. 4, 1645–1653. doi: 10.1038/s41564-019-0477-7
Oechslin, F., Piccardi, P., Mancini, S., Gabard, J., Moreillon, P., Entenza, J. M., et al. (2017). Synergistic interaction between phage therapy and antibiotics clears pseudomonas aeruginosa infection in endocarditis and reduces virulence. J Infect Dis 215, 703–712. doi: 10.1093/infdis/jiw632
Paez-Espino, D., Chen, I. M. A., Palaniappan, K., Ratner, A., Chu, K., Szeto, E., et al. (2017). IMG/VR: a database of cultured and uncultured DNA viruses and retroviruses. Nucleic Acids Res. 45, D457–D465. doi: 10.1093/nar/gkw1030
Pamela Rodríguez, H., Angie Changuán, C., and Quiroz, L. X. (2020). Phage therapy with mycobacteriophage as an alternative against antibiotic resistance produced by mycobacterium tuberculosis. Bionatura 5, 1078–1081. doi: 10.21931/RB/2020.05.01.12
Pleška, M., and Guet, C. C. (2017). Effects of mutations in phage restriction sites during escape from restriction–modification. Biol. Lett. 13:20170646. doi: 10.1098/rsbl.2017.0646
Pritham, E. J., Putliwala, T., and Feschotte, C. (2007). Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 390, 3–17. doi: 10.1016/j.gene.2006.08.008
Rohwer, F., and Thurber, R. V. (2009). Viruses manipulate the marine environment. Nature 459, 207–212. doi: 10.1038/nature08060
Romero, J., Higuera, G., Gajardo, F., Castillo, D., Middleboe, M., García, K., et al. (2014). Complete genome sequence of vibrio anguillarum phage CHOED successfully used for phage therapy in aquaculture. Genome Announc. 2. doi: 10.1128/genomeA.00091-14
Roux, S., Páez-Espino, D., Chen, I. M. A., Palaniappan, K., Ratner, A., Chu, K., et al. (2021). IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 49, D764–D775. doi: 10.1093/nar/gkaa946
Sanmukh, S. G., Paunika, W. N., Ghosh, T. K., and Tapan, C. (2010). Structure and function predictions of 646 hypothetical proteins in vibrio phages. Int. J. Biom. Bioinforma. 4, 161–175.
Sayers, E. W., Barrett, T., Benson, D. A., Bryant, S. H., Canese, K., Chetvernin, V., et al. (2009). Database resources of the National Center for biotechnology information. Nucleic Acids Res. 37, D5–D15. doi: 10.1093/nar/gkn741
Tan, X., Chen, H., Zhang, M., Zhao, Y., Jiang, Y., Liu, X., et al. (2021). Clinical experience of personalized phage therapy against Carbapenem-resistant Acinetobacter baumannii lung infection in a patient with chronic obstructive pulmonary disease. Front. Cell. Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.631585
Thammatinna, K., Egan, M. K. E., Htoo, H. H., Khanna, K., Sugie, J., Nideffer, J. F., et al. (2020). A novel vibriophage exhibits inhibitory activity against host protein synthesis machinery. Sci. Rep. 10:2347. doi: 10.1038/s41598-020-59396-3
Thompson, J. R., and Polz, M. F. (2014). “Dynamics of vibrio populations and their role in environmental nutrient cycling” in The biology of Vibrios. eds. F. L. Thompson, B. Austin, and J. Swings.
Tohidi, R., and Javanmard, A. (2020). Identification of novel mutations in IL-2 gene in khorasan native fowls. Poultry Sci J 8. doi: 10.22069/psj.2020.16789.1465
Valerdi, K. M., Hage, A., van Tol, S., Rajsbaum, R., and Giraldo, M. I. (2021). The role of the host ubiquitin system in promoting replication of emergent viruses. Viruses 13:369. doi: 10.3390/v13030369
Vasquez, I., Cao, T., Chakraborty, S., Gnanagobal, H., O’Brien, N., Monk, J., et al. (2020). Comparative genomics analysis of vibrio anguillarum isolated from lumpfish (Cyclopterus lumpus) in newfoundland reveal novel chromosomal organizations. Microorganisms 8:1666. doi: 10.3390/microorganisms8111666
Wang, Q., Liu, Y., Liu, Q., Liu, X., Yang, F., Wang, M., et al. (2020). Isolation and complete genome of the marine Pseudoalteromonas phage C7 from coastal seawater of Yellow Sea, China. Curr. Microbiol. 77, 279–285. doi: 10.1007/s00284-019-01815-4
Wang, J., Lu, J., Zhang, Y., and Wu, J. (2020). Microbial ecology might serve as new indicator for the influence of green tide on the coastal water quality: assessment the bioturbation of Ulva prolifera outbreak on bacterial community in coastal waters. Ecol. Indic. 113:106211. doi: 10.1016/j.ecolind.2020.106211
Wang, C., Sun, F., Hou, D., Xiao, Y., Lin, G., and Yan, T. (2017). Study on the microbial characteristics of seawater pond based on Enteromorpha bloom. Haiyang Xuebao 39. doi: 10.3969/j.issn.0253-4193.2017.04.011
Wang, Z., Zhang, F., Liang, Y., Zheng, K., Gu, C., Zhang, W., et al. (2021). Genome and ecology of a novel Alteromonas Podovirus, ZP6, representing a new viral genus, Mareflavirus. Microbiol Spectr 9, 1–15. doi: 10.1128/spectrum.00463-21
Wong, K. C., Brown, A. M., Luscombe, G. M., Wong, S. J., and Mendis, K. (2015). Antibiotic use for vibrio infections: important insights from surveillance data. BMC Infect. Dis. 15:226. doi: 10.1186/s12879-015-0959-z
Wu, N., Dai, J., Guo, M., Li, J., Zhou, X., Li, F., et al. (2021). Pre-optimized phage therapy on secondary Acinetobacter baumannii infection in four critical COVID-19 patients. Emerg Microbes Infect 10, 612–618. doi: 10.1080/22221751.2021.1902754
Wu, Y., Kang, J., Zhang, L., Liang, Z., Tang, X., Yan, Y., et al. (2018). Ubiquitination regulation of inflammatory responses through Nf-κB pathway. Am. J. Transl. Res. 10:4094.
Xu, Y., Zhang, R., Wang, N., Cai, L., Tong, Y., Sun, Q., et al. (2018). Novel phage-host interactions and evolution as revealed by a cyanomyovirus isolated from an estuarine environment. Environ. Microbiol. 20, 2974–2989. doi: 10.1111/1462-2920.14326
Yang, X., Haque, A., Matsuzaki, S., Matsumoto, T., and Nakamura, S. (2021). The efficacy of phage therapy in a murine model of Pseudomonas aeruginosa pneumonia and sepsis. Front. Microbiol. 12:682255. doi: 10.3389/fmicb.2021.682255
Zhang, X., Liu, Y., Wang, M., Wang, M., Jiang, T., Sun, J., et al. (2020). Characterization and genome analysis of a novel marine Alteromonas phage P24. Curr. Microbiol. 77, 2813–2820. doi: 10.1007/s00284-020-02077-1
Zhang, X., Zhang, F., Mi, Y., Liu, Y., Zheng, K., Zhou, Y., et al. (2021). Characterization and genome analysis of phage AL infecting Pseudoalteromonas marina. Virus Res. 295:198265. doi: 10.1016/j.virusres.2020.198265
Keywords: Vibrio phage , siphovirus, Ulva prolifera blooms, genomic and phylogenetic analysis, bacteriophage therapy
Citation: Zhang X, Liang Y, Zheng K, Wang Z, Dong Y, Liu Y, Ren L, Wang H, Han Y, McMinn A, Sung YY, Mok WJ, Wong LL, He J and Wang M (2023) Characterization and genomic analysis of phage vB_ValR_NF, representing a new viral family prevalent in the Ulva prolifera blooms. Front. Microbiol. 14:1161265. doi: 10.3389/fmicb.2023.1161265
Edited by:
Julianne H. Grose, Brigham Young University, United StatesReviewed by:
Robert Schooley, University of California, San Diego, United StatesCopyright © 2023 Zhang, Liang, Zheng, Wang, Dong, Liu, Ren, Wang, Han, McMinn, Sung, Mok, Wong, He and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yantao Liang, bGlhbmd5YW50YW9Ab3VjLmVkdS5jbg==; Jianfeng He, aGVqaWFuZmVuZ0BwcmljLm9yZy5jbg==; Min Wang, bWluZ3dhbmdAb3VjLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.