 Rohit Das
Rohit Das Buddhiman Tamang
Buddhiman Tamang Ishfaq Nabi Najar1
Ishfaq Nabi Najar1 Nagendra Thakur
Nagendra Thakur Krishnendu Mondal
Krishnendu Mondal
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 12 April 2023
Sec. Food Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1158411
This article is part of the Research TopicMicrobiota Biodiversity of Traditional Fermented ProductsView all 14 articles
Moiya pansung, mileye amileye, moiya koshak, and midukeye are naturally fermented bamboo shoot foods of Tripura. The present study aimed to reveal the whole microbial community structure of naturally fermented moiya pangsung, mileye amileye, moiya koshak, and midukeye along with the prediction of microbial functional profiles by shotgun metagenomic sequence analysis. The metataxonomic profile of moiya pangsung, mileye amileye, moiya koshak, and midukeye samples showed different domains, viz., bacteria (97.70%) followed by the virus (0.76%), unclassified (0.09%), eukaryotes (1.46%) and archaea (0.05%). Overall, 49 phyla, 409 families, 841 genera, and 1,799 species were found in all the fermented bamboo shoot samples collected from different places of Tripura. Firmicutes was the most abundant phylum (89.28%) followed by Proteobacteria (5.13%), Bacteroidetes (4.38%), Actinobacteria (1.02%), and Fusobacteria (0.17%). Lactiplantibacillus plantarum was the most abundant species in moiya pangsung, mileye amileye, moiya koshak, and midukeye followed by Lactococcus lactis, Levilactobacillus brevis, Leuconostoc mesenteroides, Weissella paramesenteroides, Leuconostoc kimchii, Pediococcus pentosaceus, Leuconostoc gasicomitatum, and Lacticaseibacillus casei. A few phyla of fungus were found, viz., Ascomycota, Basidiomycota, and Glomeromycota, where Ascomycota was present in high abundance. Functional analysis of moiya pangsung, mileye amileye, moiya koshak, and midukeye metagenome revealed the genes for the synthesis and metabolism of a wide range of bioactive compounds including, various essential amino acids, and conjugated amino acids. The abundance profile and predictive analysis of fermented bamboo shoots revealed a huge plethora of essential microorganisms and KEGG analysis revealed genes for amino acid metabolism, pectin degradation, lipid metabolism, and many other essential pathways that can be essential for the improvement of nutritional and sensory qualities of the fermented bamboo shoot products.
Bamboo is a long-living, robust, adaptable, and highly renewable species of grass that grows as woody stems and is primarily found in moist deciduous, semi-evergreen, tropical, subtropical, and temperate regions of forests (Tewari et al., 2019). There are more than 75 genera and 1,250 species of bamboo documented on a global scale. Bamboo shoots are a healthy vegetable that is frequently harvested, consumed, and sold by the rural tribes of North Eastern India (Singhal et al., 2018). Bamboo shoots have a great deal of promise as a food resource with a high protein and fiber content but with little fat, making them a wonderful source of nourishment. They include vital antioxidants and therapeutic ingredients that can help delay the onset of metabolic problems in addition to being a storehouse of nutritious materials (Hussain et al., 2020). The product becomes acidic and digestive due to the action of lactic acid bacteria at large and some yeast species (Ferreira and Mendes-Faia, 2020). The fermentation of bamboo shoots not only makes them very nutritious and increases their shelf life but also makes them pleasant in terms of flavor, aroma, texture, and appearance (Behera and Balaji, 2021).
Fermented bamboo shoots are commonly consumed in North East India and go by a variety of regional names, including ekung and eup in Arunachal Pradesh (Tamang and Tamang, 2009), miyamikhri and khorisa in Assam, lung-siej, tuaithar, and tuairoi in Mizoram, soidon and soibum in Manipur, bastangapani in Nagaland and mesu in Sikkim (Tamang et al., 2008). Bamboo shoots are a rich source of nutrients, powerful antioxidants, and healing compounds that can postpone the onset of certain metabolic diseases (Yu et al., 2021). Colossal diversity of microorganisms has been reported from different types of fermented bamboo shoots, including the species of bacteria. Lactobacillus johnsonii, Lb. plantarum, Lb. brevis, Lb. fermentum, Lb. curvatus, Leuconostoc mesenteroides, Leuconostoc lactis, Pediococcus sp., Micrococcus sp., Bacillus sp., and the yeasts species, viz., Saccharomyces cerevisiae, Zygosaccharomyces rouxii, Yarrowia lipolytica, and candida sp. (Tamang et al., 2012; Nongdam, 2015; Voidarou et al., 2021).
A total of 21 species of bamboo are reported in Tripura among which muli bans [Meloncana. baccifera (Roxb.) Kurz] is the most important bamboo of the state while other species contribute only 5–7% of total production. About 708 metric tonnes of young bamboo shoots are harvested from Tripura, of which M. baccifera constitutes about 80–85% of the total harvest (Banik, 2010). Different ethnic communities, viz., Chakma, Debbarma, and Uchoi use young tender shoots of muli bans to prepare fermented products like moiya pangsung, mileye amileye, moiya koshak, and midukeye (Uchoi et al., 2015). All the fermented samples collected from Tripura are still prepared at a small household level by the ethnic group using rudimentary tools, so the microbiota of fermented foods is very essential to be studied to better understand the dominating microbes responsible for fermentation as well as to know the impact on the health of the consumers. This fermented bamboo shoot is usually prepared by aged women of the community.
The microbiology of fermented bamboo shoots has been rarely studied through culture-independent techniques except in tuaithar of Mizoram (Deka et al., 2021), khorisa of Assam (Behera et al., 2020), suansun of China (Hu et al., 2021). Moreover, there are no culture-independent studies present on the fermented bamboo shoot products of Tripura.
Since moiya pangsung, mileye amileye, moiya koshak, and midukeye are traditionally prepared from the young bamboo shoot by natural fermentation, diverse species of bacteria and fungi present in the local environment may appear in the final product. Moreover, there is no report on the predictive functionalities of microbial genes present in this fermented bamboo shoot. Predictive functional profiles of microbial communities in fermented foods are an appropriate approach to annotating the predictive metabolic pathways in gene sequences of bacteria and fungi (Sun et al., 2022) using different pipelines of bioinformatics tools such as Metagenomic Rapid Annotations using Subsystems Technology (MG-RAST) (Keegan et al., 2016), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto, 2000) for the bacterial gene. Hence, we aimed to study the bacterial and fungal community structure by shotgun sequencing tool during the natural fermentation of moiya pangsung, mileye amileye, moiya koshak, and midukeye of Tripura in India. We also aimed to predict the functional profiles of bacterial and fungal genes by using the bioinformatics pipelines, MG-RAST, during the natural fermentation of these fermented bamboo shoots.
A total of 6 samples corresponding to four different types of fermented bamboo shoots were sampled in duplicates from different households and marketplaces in Tripura. A sample of mileye amileye was collected from the Panisagar district (PBMA) market (24.263307, 92.152439), 2 samples of midukeye were collected from the Dharmanagar household (DBMD) and Panisagar district (PBMD) (24.380504, 92.166556, and 24.269956, 92.150036, respectively), 2 samples of moiya pangsung were collected from Manubazar (MBMP) market place and Dharmanagar household (DBMP) (24.393275, 92.166073, and 23.074567, 91.645094, respectively) and 1 sample of moiya koshak was collected from Dharmanagar household (DBMK) (24.379511, 92.165093). All collected samples were kept in an ice box carrier and immediately transported within 24 h, where they were stored at −20°C for further analysis. Before fermentation, the sheaths of the young tender shoots were removed and then washed with water. Moiya pangsung is prepared from a top tender shoot and fermented for 2 days at room temperature in a closed container completely submerged in water (Supplementary Figure 1A), while mileye amileye is also fermented in a similar way but the middle portion of the bamboo shoot is used (Supplementary Figure 1B). Similarly, for midukeye and moiya koshak, the middle portion of the tender shoot is cut into pieces, then wrapped in banana leaves and left at room temperature for 2 days (Supplementary Figure 1C). These bamboo shoot products are sour and with a sweet and earthy after-taste. The type of samples and code name assigned to the samples are mentioned in Supplementary Table 1.
In a stomacher (400 Circulator, Seward, UK) ten grams of each sample were homogenized with 90 mL of sterile 0.1 M phosphate buffer saline (pH 6.4). The homogenate was filtered after homogenization, and the filtrate was utilized to extract genomic DNA using the Maggenome XpressDNA plant kit (Maggenome, USA) as per the manufacturer’s protocol. Using 0.8% agarose gel electrophoresis, the DNA samples were separated to check for deterioration and contaminants and to assess quality (voltage 100 V, period 40 min). A precise measurement of the total DNA mass concentration was obtained utilizing nanodrop (Eppendorf Biospectrometer@ basic, Germany), to confirm that the parameters for the library creation adhered to. Following the manufacturer’s instructions, total DNA was submitted to DNA library construction and amplification using the TruSeqTM DNA Sample Prep Kit (Illumina, United States) and the cBOT TruSeq PE Cluster Kit v3 (Illumina, United States) reagents. The TruSeq SBS Kit v3-HS (Illumina, United States) was used to sequence the DNA libraries utilizing the Illumina Novaseq 6,000 technology. According to the established Illumina methods, the sample DNA libraries were submitted to a paired-end (2 × 150 bp) sequencing platform. Fastq format was used to store the raw reads.
High-quality filtered paired-end libraries of the samples of raw reads were assembled separately using MetaSPAdes assembler with default parameters (Nurk et al., 2017). The metagenomic data obtained from MetaSPAdes was run through the Galaxy platform1 (Afgan et al., 2018) using the default parameters.
The putative microbial population reads from six samples were classified using Kraken2 against the reference database containing all Refseq bacterial, archaeal, fungal, and viral genomes with a 0.1 confidence threshold (Marcelino et al., 2020). After the classification by Kraken2, Bracken was used to re-estimate the different microbial abundances at taxa levels from species to phylum using a read length parameter of 150 (Terrón-Camero et al., 2022).
The gene prediction for protein-coding genes (ORFs) was predicted using Prodigal (Hyatt et al., 2010). ORFs shorter than 180 nucleotides were excluded from further analysis by default. tRNA genes were predicted with the ARAGORN program (Laslett and Canback, 2004), and Ribosomal RNA genes (5S, 5.8S, 16S, 18S, 23S, 28S) were identified and classified using rRNAFinder (Dong and Strous, 2019). All ORFs were searched against the evolutionary genealogy of genes, non-supervised Orthologous Groups (eggnog) database (Muller et al., 2009), for Clusters of Orthologous Groups/Non-supervised Orthologous Groups (COG/NOG) annotation (Galperin et al., 2018) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto, 2000) for KEGG ID annotation.
Nucleotide diversity (pi) analysis and different indices of neutrality test based on sequence polymorphism in DnaSP software version 6 (Rozas et al., 2017) were performed to justify the pooling of DNA from each duplicate sample in terms of intra-sample (within the sample) diversity. Among the different indices of neutrality tests, Tajima’s D value and Fu’s Fs statistics were performed in the same software. To check the intra-sample diversity in terms of species level and functional profiles, one sample T-test was performed in IBM SPSS version 20 for all the samples. Statistical relations among the samples were performed using the Mann-Whitney test (Fong and Huang, 2019) in terms of species level and level 3 functional profiles in PAST version 4.0 software (Martino et al., 2019).
Non-parametric Shannon index, Simpson’s index of diversity (1-D), Chao-1, and evenness were calculated using PAST software version 4.0. The Bray-Curtis index of beta diversity was also calculated using past version 4.0 and visualized via a Principal Coordinates Analysis (PCoA) plot (Martino et al., 2019).
The clustering pattern among the samples was checked through the PCoA plot using the level 3 sub-pathways. The PCoA plot was constructed after the log transformation of data and then visualized in the ClustVis web tool (Metsalu and Vilo, 2015). Non-parametric Spearman’s rank correlation was performed using IBM SPSS software version 20 and PAST version 4.0 (Martino et al., 2019) software to check the correlation between the microorganisms in samples and different levels (super-pathways) functional profiles. Heatmap was constructed via the ClustVis web tool to visualize the correlation profiles. Correlation between different bacteria and the amino acid profiles obtained from KEGG annotation was also performed.
As mentioned earlier, each sample was collected in duplicates and was pooled on the basis of their geographical origin and we hypothesized that the samples from the same origin possess minimal diversity differences. Nucleotide diversity (pi) per site was 0.712 (PBMA), 0.713 (DBMD), 0.713 (PBMD), 0.733 (DBMK), 0.721 (DBMP), and 0.712 (MBMP), respectively. However, the theta values (nucleotide difference) found for each of the samples were 0.72, 0.73, 0.72, 0.72, and 0.73, respectively. First, we checked Tajima’s D value among the different indices of the neutrality test. Each sample showed a negative value for Tajima’s D, which were −0.21 (PBMA), −0.19 (DBMD), −0.19 (PBMD), −0.20 (DBMK), −0.21 (DBMP), and −0.19 (MBMP), respectively. The second neutrality test indices were Fu’s FS statistics (FST); FST values found for all the samples were −5.30, −3.24, −3.24, −3.88, −5.21, and −3.90.
A total of 1,570,915 reads were obtained from the samples with an average of 261,820 reads per sample. The average length of the reads was found 583 bp. The total number of bases recovered from the samples was 311,915,402 (DBMP), 285,708,731 (DBMD), 93,242,563 (MBMP), 71,456,540 (DBMD), 61,889,925 (PBMA), and 55,160,324 (DBMK), respectively. Shotgun metagenomic sequence analysis of moiya pangsung, mileye amileye, moiya koshak, and midukeye samples showed different domains, viz., bacteria, archaea, viruses, and eukaryotes. The total relative abundance was calculated in all the samples where bacteria were the most abundant domain (97.70%) followed by the virus (0.76%), unclassified (0.09%), eukaryotes (1.46%) and archaea (0.05%). Among samples, DBMD was seen to have the highest bacterial abundance (28.84%), PBMD had the highest archaeal abundance (24.46%), DBMP had the highest eukaryotic abundance of (0.41%), and DBMD had the highest viral abundance (31.34%) (Figure 1). A total of 49 phyla, 409 families, 841 genera, and 1,799 species were recorded after abundance analysis. At the bacterial phylum level, Firmicutes was the most abundant phylum (Figure 2). Across all the bacterial families, Lactobacillaceae was found the most abundant family followed by Enterobacteriaceae (Supplementary Table 2). Lactobacillus was found to be the most abundant genera of all, among them PBMA had the highest abundance (Figure 3). No cluster was found among the samples except PBMD and MBMP in PCoA analysis according to the abundance/distribution of bacterial genera from different samples (Figure 4). At the species level, Lactiplantibacillus plantarum, Levilactobacillus brevis, Lactococcus lactis, Pediococcus pentosaceus, Leuconostoc mesenteroides, and Leuconostoc citreum (Supplementary Figure 2), non-LAB, archaea, yeasts, filamentous moulds, other phages, microeukaryotes, and parasites were also detected in < 1% abundance from moiya pangsung, mileye amileye, moiya koshak, and midukeye samples. From the Eukaryota domain, 3 phyla of fungus were found Ascomycota, Basidiomycota, and Glomeromycota, where Ascomycota was present in abundance (Supplementary Figure 3A), Saccharomycetaceae was overall the most abundant family (Supplementary Figure 3B), Schizosaccharomyces was overall the most abundant fungal genera (Figure 5), and Schizosaccharomyces pombe was found to be the most abundant species on an overall basis (Supplementary Figure 3C).
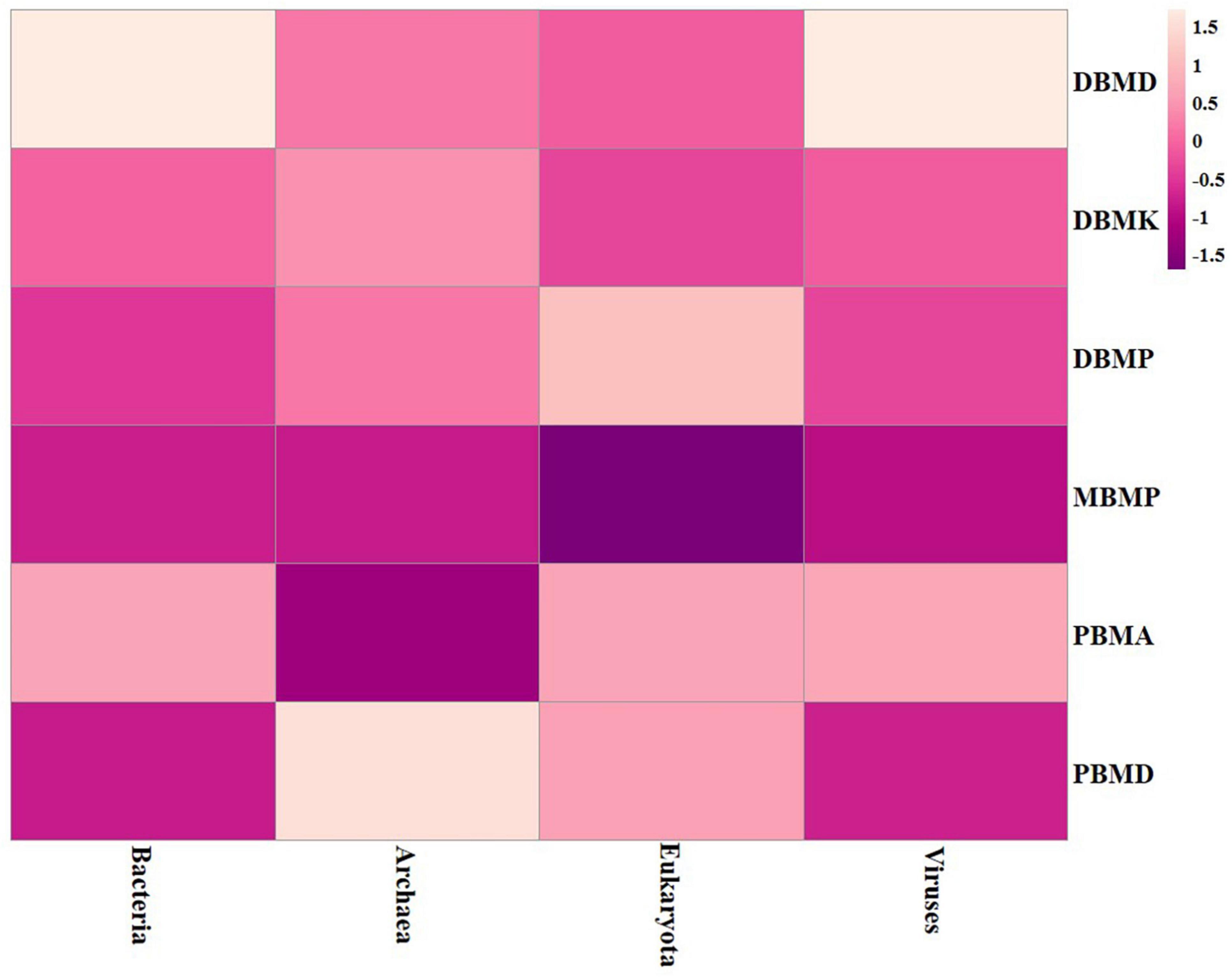
Figure 1. Heat map representation of major domain in moiya pangsung, mileye amileye, moiya koshak, and midukeye.

Figure 2. Bar plot for major phylum in bacteria.
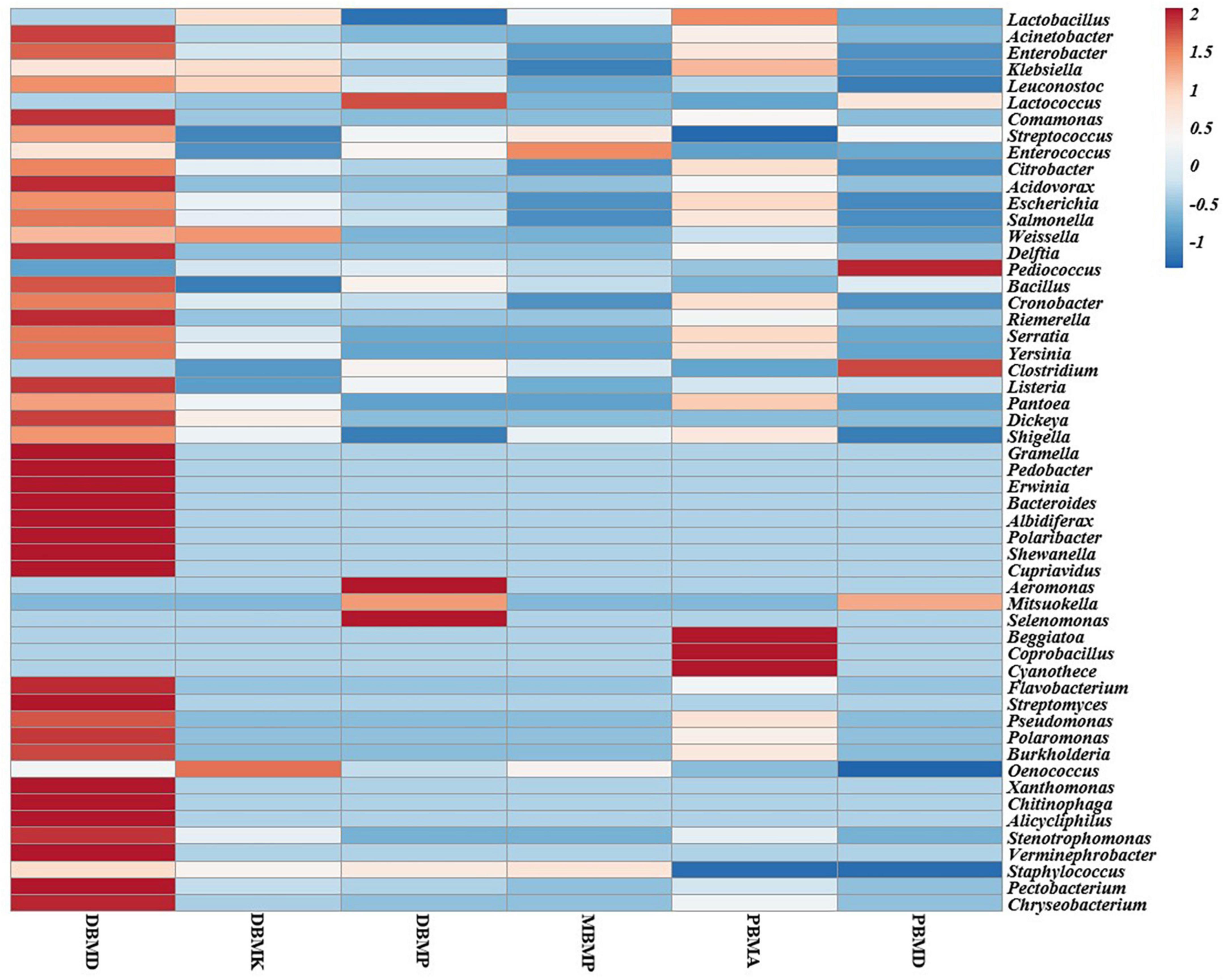
Figure 3. Heat map representation of major bacterial genera in moiya pangsung, mileye amileye, moiya koshak, and midukeye.
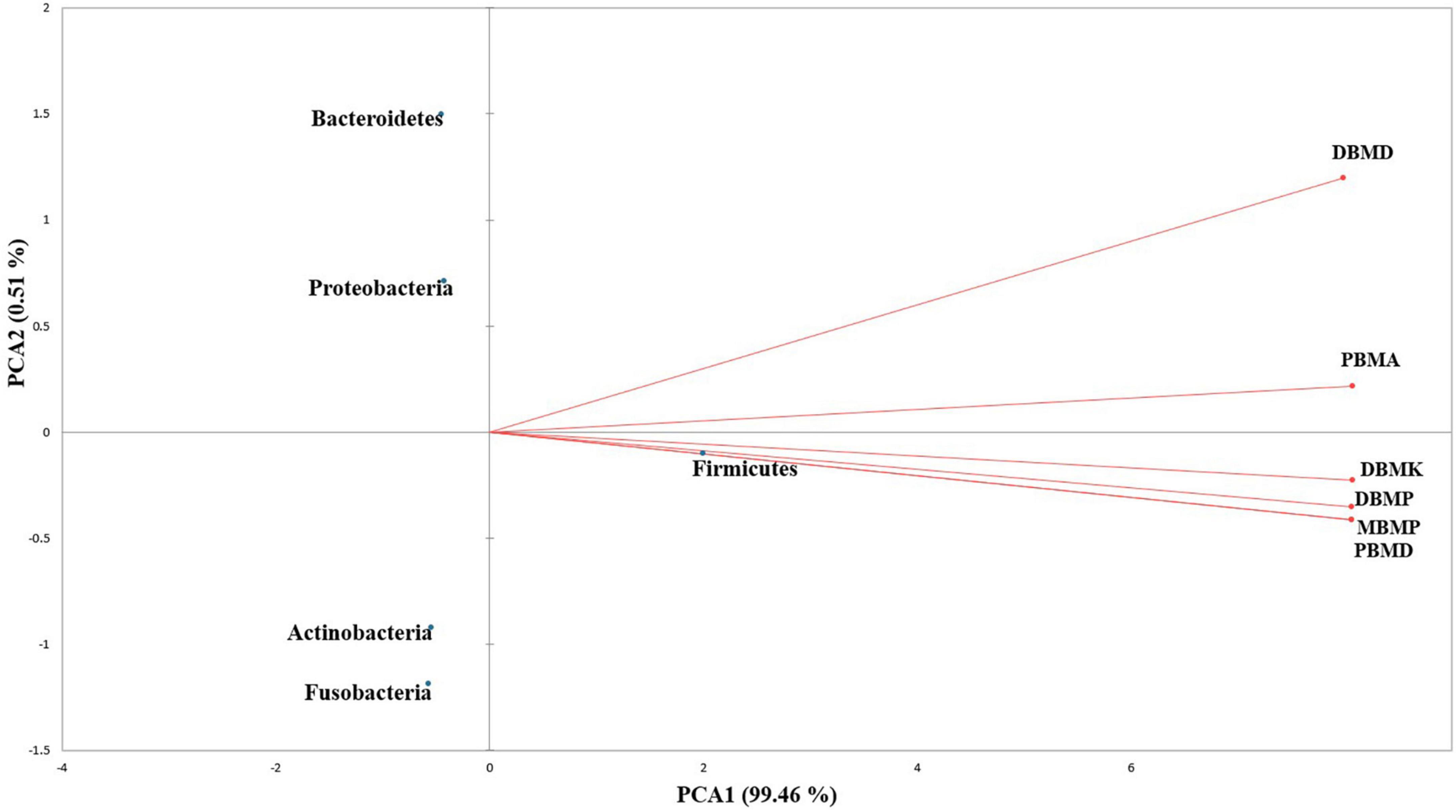
Figure 4. PCA biplot analysis of samples and microbial diversity at the genus level.

Figure 5. Heat map representation of major fungal genera communities in moiya pangsung, mileye amileye, moiya koshak, and midukeye.
The non-parametric Shannon, Simpson, Chao-1, and Evenness indices were highest in the DBMD sample (Table 1). At the genus level, the beta diversity of DBMP and PBMD were completely different from other samples as visualized in principal component analysis obtained through Jaccard indices (Figure 6). Similarly, Bray-Curti’s beta diversity at the genus level also revealed DBMD and PBMD were completely different from the other 4 samples (Figure 7). The shared and unique species were visualized between 4 samples using a venn diagram elaborated that 61, 8, 1, and 85 species were unique to moiya pangsung, moiya koshak, mileye amileye, and midukeye, respectively, whereas 306 species were common in all the fermented products (Supplementary Figure 4).
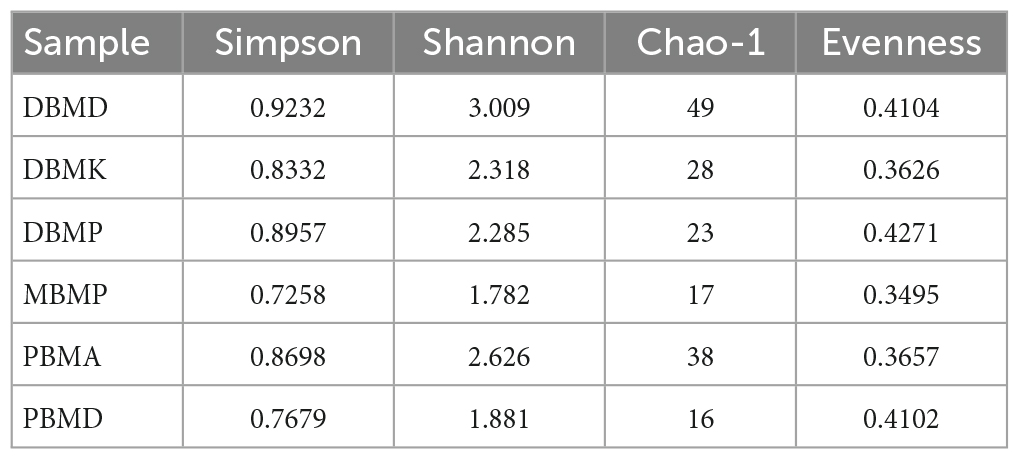
Table 1. Alpha diversity indexes.
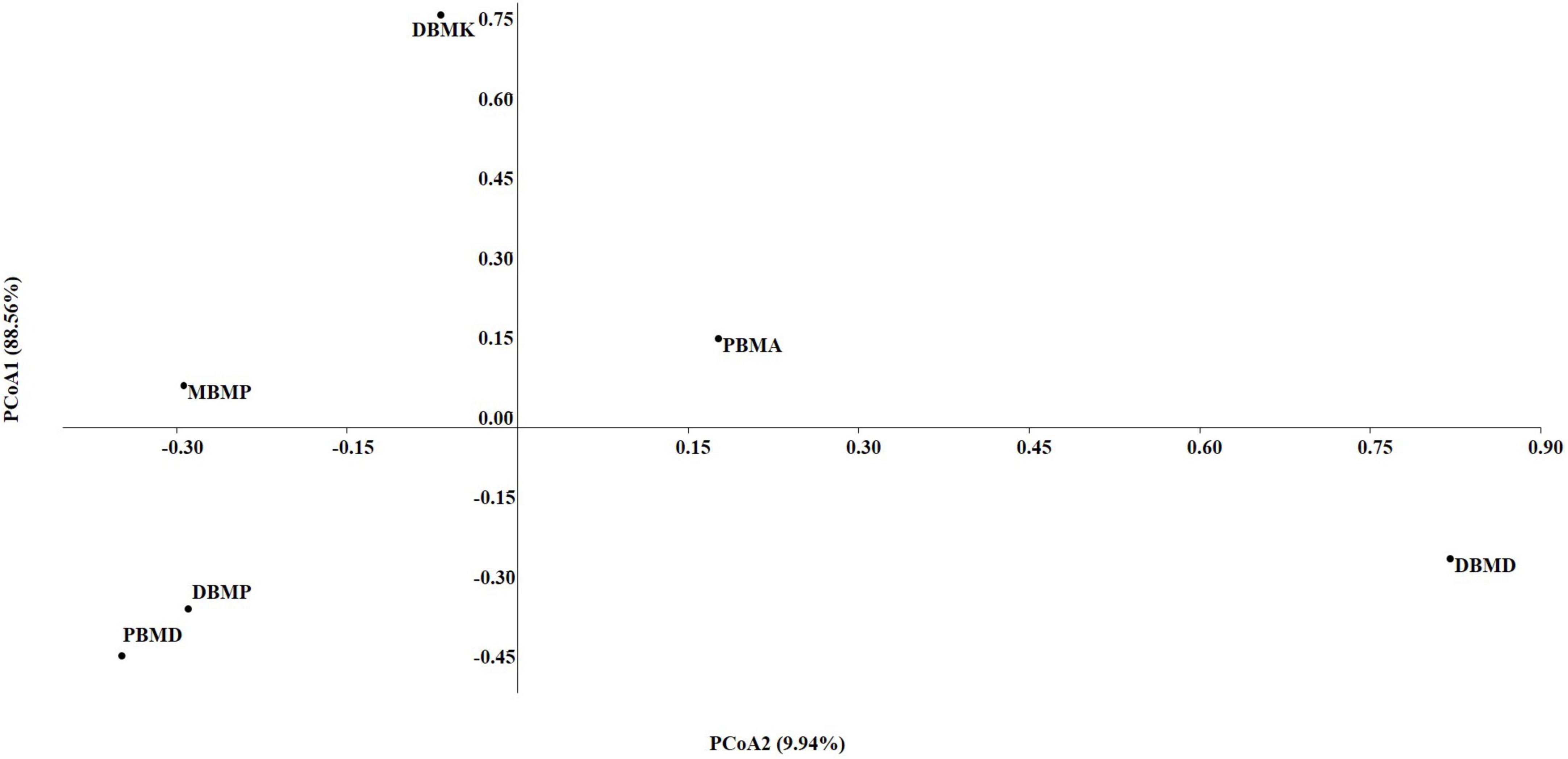
Figure 6. PCA plot for beta diversity analysis via Jaccard at the bacterial genus level.
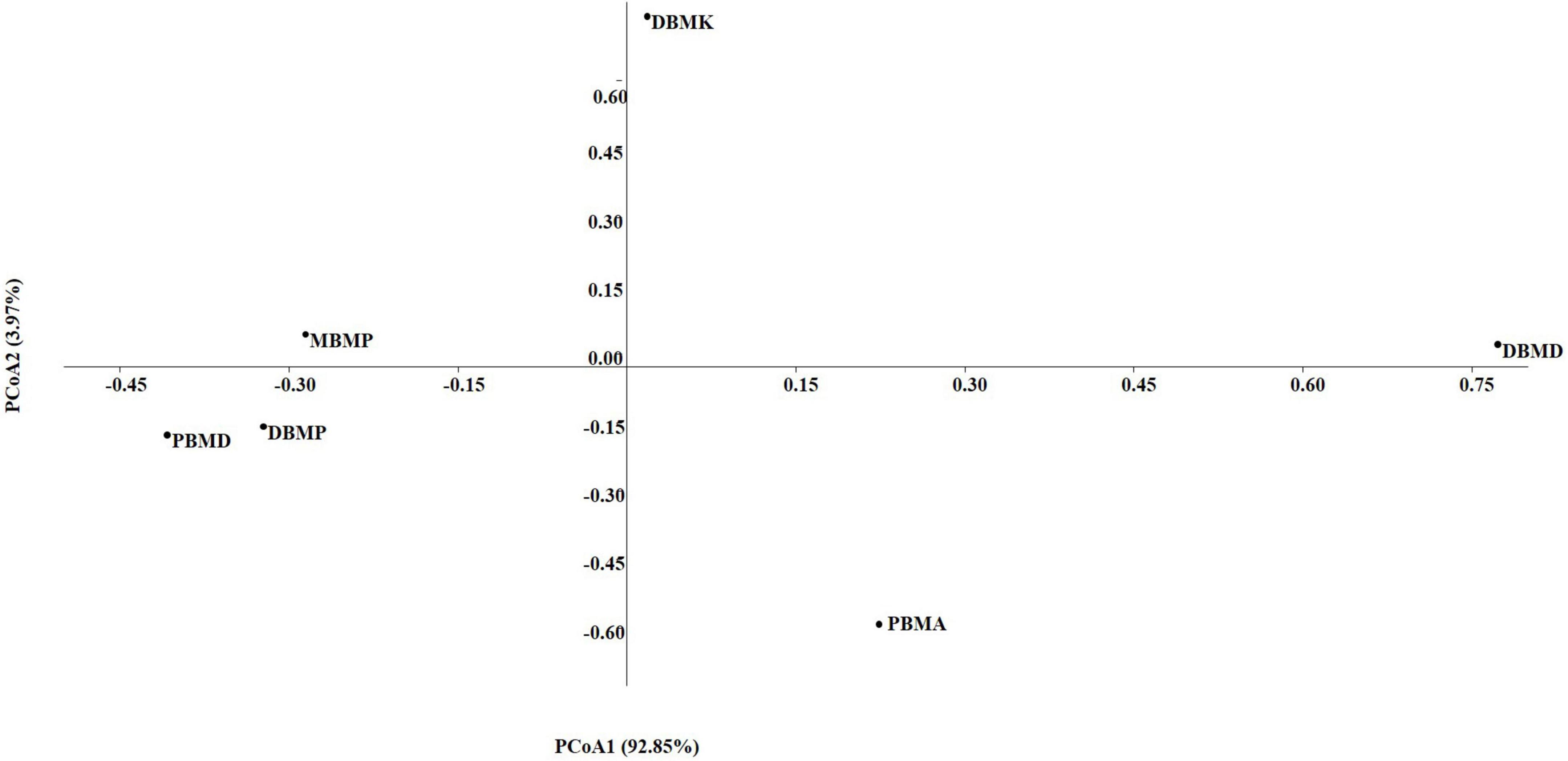
Figure 7. PCA plot for beta diversity analysis via Bray-Curtis at the bacterial genus level.
Different enhanced functional pathways were observed after mapping metagenomic ORFs against eggNOG and KEGG databases. The number of prodigal ORFs identified from moiya pangsung, moiya koshak, mileye amileye, and midukeye samples were 23,615 (DBMD), 14,447 (DBMK), 25,596 (DBMP), 13,483 (MBMP), 18,329 (PBMA), and 21,710 (PBMD), respectively. About 49.65% of the total mapped ORFs were assigned to the COG functional genes, 49.82% were assigned to the KEGG functional pathways, and the rest 0.53% were assigned to the NOG pathway. In this study, we focused more on the KEGG functional pathways, COG mapping, and NOG pathways. The functional pathways were categorized into three sub-classes via KEGG mapping. In level 1, metabolism was the most abundant category followed by genetic information processing, environmental information processing, cellular process, human disease, and organismal system (Figure 8). At level 2, 50 super-pathways, and at level 3, 238 sub-pathways (terminal) from different categories were identified (Supplementary Table 3). Spearman’s rank correlation was performed between some major microorganisms in moiya pangsung, moiya koshak, mileye amileye, and midukeye samples and different major level 2 super-pathways obtained from KO annotation. Lactiplantibacillus plantarum, Latilactobacillus curvatus, and Lentilactobacillus hilgardii showed a positive correlation with amino acid metabolism. While, Lactobacillus jensenii, and Lacticaseibacillus casei showed a positive correlation with lipid metabolism. Latilactobacillus curvatus showed the highest correlation with the membrane transport system (Figure 9).
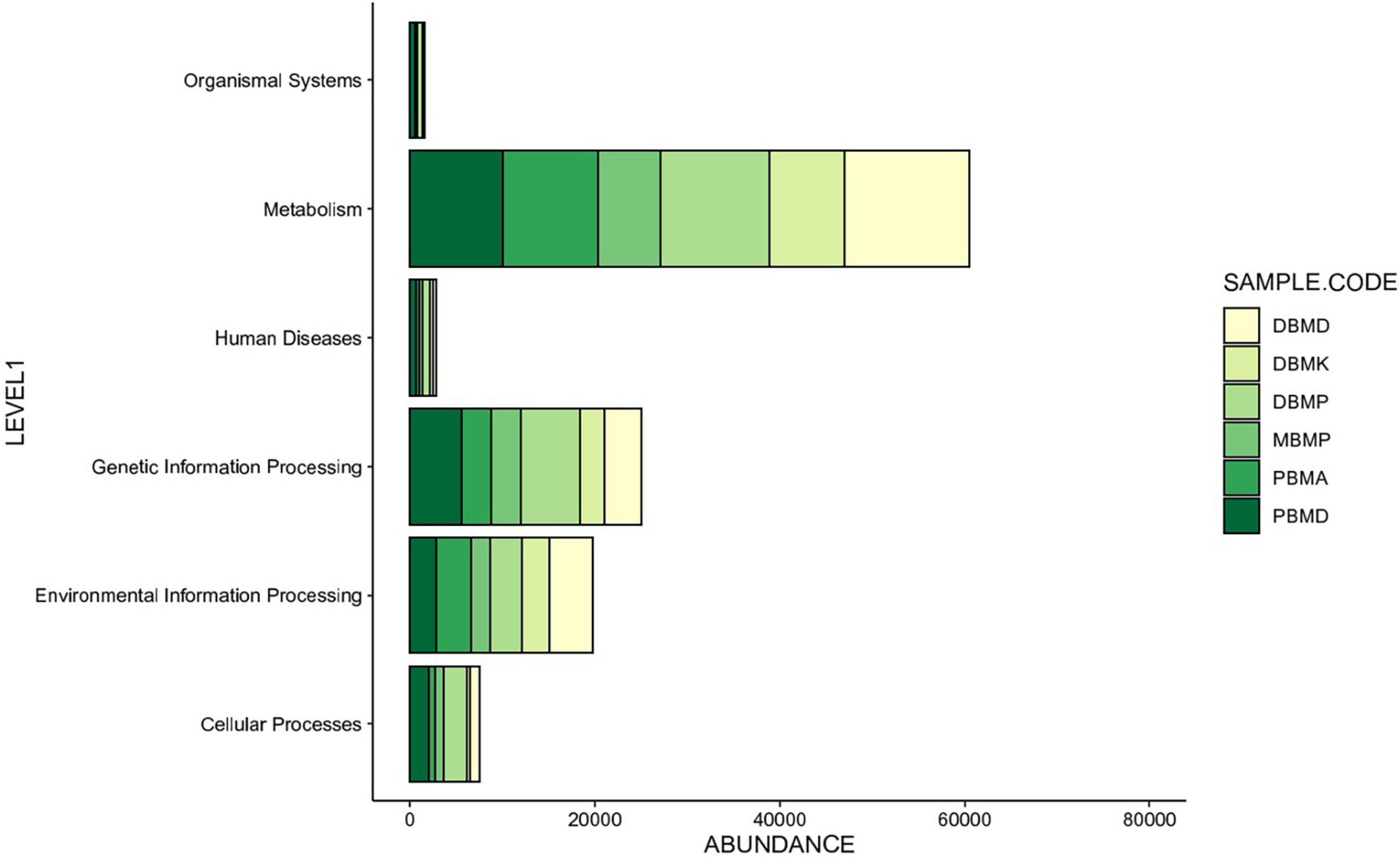
Figure 8. KEGG (level 1) showing the highest number of genes annotated to pathways.
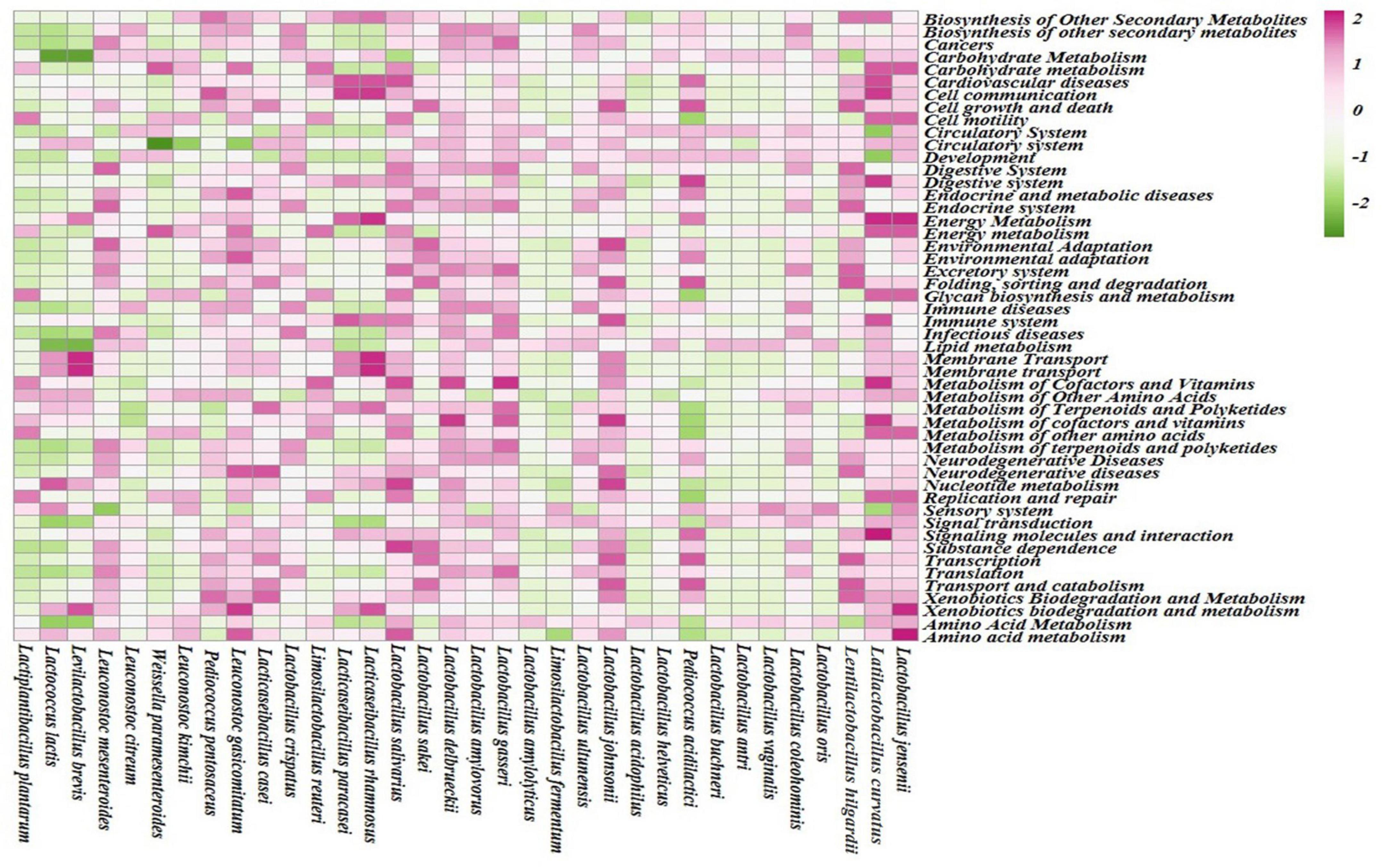
Figure 9. Spearman RS correlation of major bacterial species with KEGG (level 2) pathways.
One sample T-test was performed to check the intra-sample and inter-sample significance in terms of functional profiles of moiya pangsung, moiya koshak, mileye amileye, and midukeye samples metagenome. No intra-sample significance was found in any of the pooled samples in terms of functional profiles. The p-value for six samples were 0.98 (DBMD), 0.98 (DBMK), 1 (DBMP), 1 (MBMP), 0.99 (PBMA), and 0.99 (PBMA), respectively.
In total, 15 pathways for amino acid metabolism were visualized and annotated to microbial functional genes in the few microbial species in all samples. The most abundant amino acid metabolic pathway visualized in all the fermented bamboo shoot samples was Alanine, aspartate, and glutamate metabolism; ko00250 (16.98%), the next most abundant was glycine, serine, and threonine metabolism; ko00260 (15.97%) as well as cysteine and methionine metabolism; ko00270 (14.97%).
The second most abundant pathway found in all the fermented bamboo shoot samples related to the impartment of flavor and aroma were glycerolipid metabolism (ko00561) (25.33%), fatty acid biosynthesis (ko00061) (25.22%), and glycerophospholipid metabolism (ko00564) (24.02%) (Table 2). A few pathways like the metabolism of terpenoids and polyketides were related to the color and aroma of the fermented bamboo shoot such as carotenoid biosynthesis (ko00906). Pathways of secondary metabolism related to the impartment of flavor and aroma to the fermented bamboo shoots were also annotated such as phenylpropanoid biosynthesis (ko00940), flavonoid biosynthesis (ko00941), flavone and flavonol biosynthesis (ko00944), stilbenoid, diarylheptanoid and gingerol biosynthesis (ko00945), and tropane, piperidine, and pyridine alkaloid biosynthesis (ko00960), etc. Some metabolic pathway related to pectin degradation were identified within carbohydrate metabolisms such as pentose and glucuronate interconversions (ko00040). Pathways related to vitamins metabolism were also annotated such as thiamine metabolism (ko00730), riboflavin metabolism (ko00740), retinol biosynthesis (ko00830), nicotinate and nicotinamide metabolism (ko00760), pantothenate and CoA biosynthesis (ko00770), biotin biosynthesis (ko00780), folate biosynthesis (ko00790), and lipoic acid biosynthesis (ko00785).
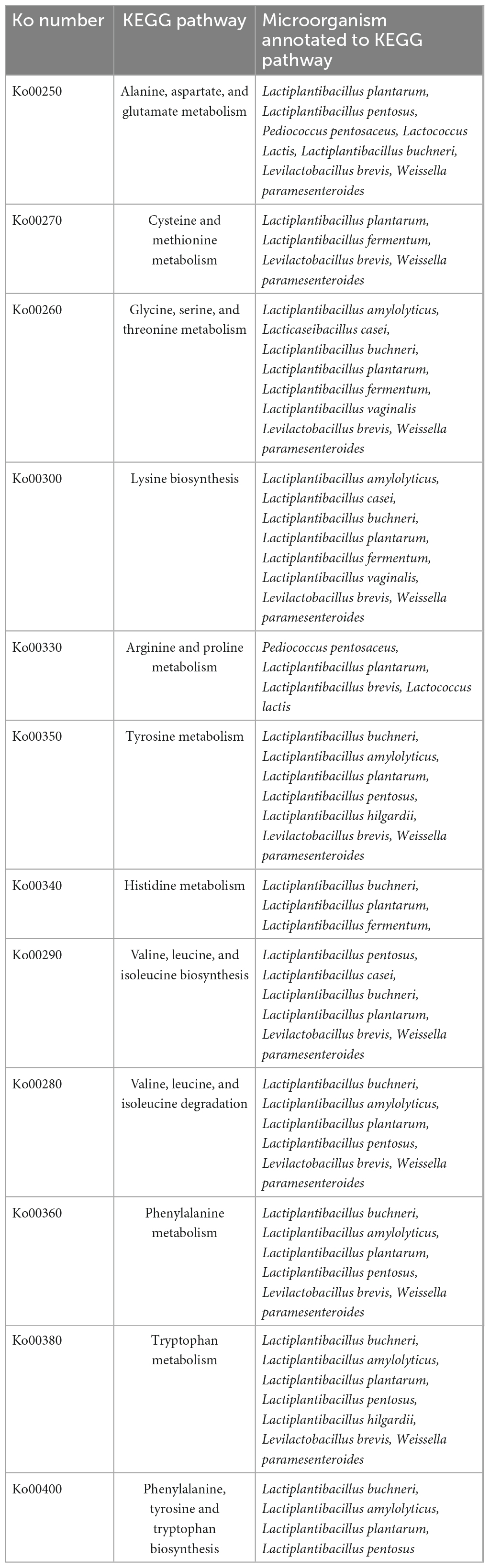
Table 2. KEGG metabolic pathways annotated to different microbial species.
Comparing the dataset of our samples with the NOG database, it was found that all the samples had the maximum number of amino acid transport metabolism unigenes, followed by cell wall/membrane/envelope biogenesis, transcription, and carbohydrate transport metabolism unigenes, respectively (Supplementary Figure 5). COG dataset yielded 97,635 unigenes, from which carbohydrate transport metabolism had the highest number of unigenes in all samples, followed by amino acid transport metabolism and replication, recombination, and repair, respectively. The sample bearing code DBMD has the highest number of unigenes as seen in the COG database than DBMK, PBMA, PBMD, MBMP, and DBMP, respectively (Supplementary Figure 6).
In our bamboo shoot samples, according to the KEGG pathway, there were 80 unigenes for cyano amino acid metabolism among which 46 unigenes from moiya pangsung, 3 unigenes moiya koshak, 1 from mileye amileye and 30 unigenes belonged to midukeye. Cyanogenic glycoside can be hypothesized to be metabolized by the cyano amino metabolic pathway via tyrosine metabolism.
Moiya pangsung, moiya koshak, mileye amileye, and midukeye are one of the oldest fermented bamboo shoot gastronomies of Tripura and are prepared by natural fermentation (Uchoi et al., 2015). Profiling of microbial community in naturally fermented moiya pangsung, moiya koshak, mileye amileye, and midukeye by the shotgun metagenomic method has not been reported yet. Hence, we applied the shotgun metagenomic method to profile the whole microbial community structure in naturally fermented moiya pangsung, moiya koshak, mileye amileye, and midukeye. In our study, we collected samples from three places in duplicate; mileye amileye and midukeye were collected from Panisagar and Dharmanagar, moiya pangsung was collected from Manubazzar and Dharmanagar, and moiya koshak was collected from Dharmanagar. The shotgun sequencing was performed after pooling the DNA of the samples which were all collected in duplicates, because the pooled sequence can also represent the actual sequences from the pooled DNA of the individual samples (Anand et al., 2016) to maintain adequate diversity within and between the samples. We anticipated the microbial community structure to vary within a single representative pool because these samples are naturally fermented. Nucleotide differences and the neutrality test were used to interpret the pooled sample using statistics, and the results clearly showed that there was little polymorphism and variation within the pooled sample, indicating that samples from the same origin have very little diversity differences (Hivert et al., 2018). Negative FST values are also unequivocal proof that there are more alleles than necessary in a single pooled sample, which is truly more than one sample (de Meeûs, 2018). These two tests demonstrate that each of our pooled samples truly has combined data from multiple samples and that there is very little intra-sample variations in the pooled sample, which can also support our pooling method.
Firmicutes was the most abundant phylum in moiya pangsung, moiya koshak, mileye amileye, and midukeye, like in other Asian fermented bamboo shoot foods like suansun of China (Hu et al., 2021), and tuaithar of Mizoram, India (Deka et al., 2021). Lactobacillaceae was the abundant family in moiya pangsung, moiya koshak, mileye amileye, and midukeye as reported earlier in other ethnic bamboo shoot fermented foods (Ghosh et al., 2019). Lactiplantibacillus plantarum was the most abundant species in all the samples followed by Lactococcus lactis, Levilactobacillus brevis, and Leuconostoc mesenteroides, which was consistent with other plant-based fermented foods (Lappe-Oliveras et al., 2022) L. plantarum attracted many researchers because of its wide applications in the medical field with antioxidant, anticancer, anti-inflammatory, antiproliferative, anti-obesity, and antidiabetic properties (Arasu et al., 2016). Lactococcus lactis has been found to be major bacterial species to be found in different fermented bamboo shoot foods as well as other fermented plant-based products (Swain et al., 2014). Lacticaseibacillus brevis has been seen to have probiotic and antifungal attributes (Somashekaraiah et al., 2021). Pediococcus pentosaceus which has been found in all the samples of the fermented bamboo shoots has been seen to pose a partial characterization of heat-stable, antilisterial, and cell lytic bacteriocin (Halami et al., 2011). Though in a minor abundance, the detection of amino acid-producing bacteria Corynebacterium glutamicum (Hahne et al., 2018) and Bacillus thuringiensis (Hibi et al., 2011) is an indication of nutritive significance in moiya pangsung, moiya koshak, mileye amileye, and midukeye.
Though the cumulative abundance of lactic acid bacterial species was > 1‘%, while other genera at lesser abundance than 1% were also detected such as Bacillus, Cronobacter, Riemerella, Serratia, Yersinia, Clostridium, Stenotrophomonas, Pseudomonas, Listeria, Burkholderia, Oenococcus, and Xanthomonas in all the samples. After bacteria, eukaryota was the most abundant domain found in the metagenome analysis of moiya pangsung, moiya koshak, mileye amileye, and midukeye. The most abundant eukaryota found in the samples were Ascomycota, Basidiomycota, and Glomeromycota in all the fermented bamboo shoot samples which is consistent with other samples like Suansun from China, tuaithar from Mizoram, India (Deka et al., 2021), fermented shrimp and sauerkraut (Park et al., 2020). We observed a co-existence of Lactobacillus and Saccharomyces in all the fermented bamboo shoot samples. Various non-Saccharomyces yeasts (Torulaspora, Lachancea, and Starmerella) were also found. These yeasts are responsible for the addition of aroma (Djeni et al., 2020), glycerol concentration, volatile compounds, and for increasing the concentration of terpenes in fermented foods (Das and Tamang, 2021). Acinetobacter phages were more abundant than other phages in moiya pangsung, moiya koshak, mileye amileye, and midukeye metagenome. Bacteriophages generally play a critical role in determining bacterial abundance over time (Shapiro et al., 2010). In our study, the abundance of lactobacillus phages was found much lower than the abundance of Lactobacillus species. However, the presence of phages against the hosts like Staphylococcus sp., Salmonella sp., and Streptococcus sp., indicates the bio-control activity of bacteriophages in food fermentation (Kumar et al., 2019).
Few species of haloarchaea were detected in moiya pangsung, moiya koshak, mileye amileye, and midukeye, though their abundance was very low. The exact role of archaea in moiya pangsung, moiya koshak, mileye amileye, and midukeye is still unknown. Halophilic archaea have been reported as the producer of halocin, a compound that infers anti-microbial activity (Corral et al., 2020). Hence the presence of haloarchaea again may be linked with the bio-enhancement of the fermented bamboo shoot food (Mota de Carvalho et al., 2018). Low abundances of the domain eukarya, which included yeasts, filamentous moulds, several species of algae, protozoa, and parasites, were observed in moiya pangsung, moiya koshak, mileye amileye, and midukeye. Protozoa and parasites are among the other microeukaryotes that are believed to have a detrimental effect on consumers’ health and wellbeing, albeit a comprehensive safety assessment of fermented bamboo shoot cuisine has not yet been conducted. A robust approach that can identify the whole microbial population in a sample with both high and low abundances is the shotgun metagenome sequence (Kumar et al., 2022), may help to understand the safety measure of foods on the basis of their abundances level.
The KEGG annotation showed that metabolic pathways were common in moiya pangsung, moiya koshak, mileye amileye, and midukeye. One of the predominant predictive functional super-pathways within the metabolism category was amino acid metabolism. During fermentation, alanine, aspartate, and glutamate metabolism and biosynthesis increase the creation of flavor and aroma compounds (Sulaiman et al., 2014). The abundance of genes involved in the synthesis of branched-chain amino acids (valine, leucine, and isoleucine) was also detected from the metagenome analysis of moiya pangsung, moiya koshak, mileye amileye, and midukeye, which are involved in energy metabolism of Lactobacillus sp. (Neinast et al., 2019). The functional study of the moiya pangsung, moiya koshak, mileye amileye, and midukeye metagenome was used to predict the synthesis and metabolism of glutathione. The cellular processes of anti-oxidant defense, drug detoxification, and cell signaling involved in the regulation of gene expression and apoptosis all involve the metabolism, production, and transport of glutathione (Kennedy et al., 2020). Additionally, computational methods were used to predict the presence of the taurine and hypotaurine production genes, which have been linked to a reduced risk of cardiovascular disease (Xu et al., 2008). According to the COG database, carbohydrate metabolism, RNA processing and modification, chromatin structure and dynamics and cytoskeleton were predominant in all the samples when correlated between COG data and major species which is consistent with the results obtained by Hu et al. (2021). KEGG pathway annotation of samples revealed the catabolism of cyanogenic glycosides, viz., dhurrin, and linamarin through the cyano amino metabolic pathway. It is possible that taxiphyllin may follow the same catabolic process thereby reducing its amount in the fermented bamboo shoots (Appenteng et al., 2021). Terpenoids are also known to be the aroma compound in wine fermentation (Belda et al., 2017). The presence of flavonoids and triterpenes in fermented bamboo shoots has been documented by Lin et al. (2018) to impart flavor to the bamboo shoots. The genes for flavonoid metabolism were also detected from our samples through the culture-independent method. The presence of polygalacturonase enzyme which is referred to as the key enzyme in the pectin degradation pathway may suggest or predict that the bacteria present in fermented bamboo shoots have pectinase degrading ability and soften the texture of the fermented bamboo shoots (Buergy et al., 2020).
The presence of the thiamine metabolic pathway not only indicates that thiamine acts like a coenzyme for various enzymes but also aids in the production of more flavoring substances as opposed to off-flavoring substances (Labuschagne and Divol, 2021). Lactic acid bacteria were the dominant microorganism in the fermented bamboo shoots and previously have been seen to produce riboflavin which is a strong candidate for food biofortification, besides its role in red blood cell production and also aids in the release of energy from proteins (Spacova et al., 2022). The vitamin A component retinol has become an increasingly sought-after cosmetic ingredient (Hu et al., 2022). The presence of a nicotinamide biosynthesis pathway ensures the good absorption of tryptophan and niacin when fermented food is consumed (Faivre et al., 2021). Pantothenate and biotin metabolic pathway help in the growth of various lactic acid bacteria (Yao et al., 2018). The water-soluble vitamin B group member folate, particularly in the forms of tetrahydrofolate (THF) and methyl-tetrahydrofolate (MTHF), is crucial for the carbon-1 transfer processes required for the creation of DNA, nucleic acids, amino acids, and other vitamins (Mahara et al., 2021). These metabolic pathways are attributed to microorganisms fermenting the food, lactic acid bacteria have been found to possess the above KEGG metabolic pathways as previously described in the correlation heat map of major lactic acid bacteria species and KEGG pathways (Cui et al., 2023).
This present study may also provide theoretical and technical support for search and application of the key enzymes produced by the fermenting microbes, and various essential amino acids with health benefits predicted in the fermentation of moiya pangsung, moiya koshak, mileye amileye, and midukeye for other Asian fermented bamboo shoot. The metataxonomic and predictive functional features of the moiya pangsung, moiya koshak, mileye amileye, and midukeye metagenome may be used to design the starter culture with enhanced nutritional quality under controlled and hygienic fermentation conditions.
Moiya pangsung, moiya koshak, mileye amileye, and midukeye is a delicacy of the Tripura ethnic tribe’s diets and presents one of the most consumed foods of Tripura. There have been no studies on microbiology and nutritional aspects of moiya pangsung, moiya koshak, mileye amileye, and midukeye. Our study revealed different beneficial microorganisms associated with moiya pangsung, moiya koshak, mileye amileye, and midukeye fermentation along with their beneficial predictive functional profiles. We hope that the information obtained from this study may help the commercial producers and consumers to be aware of the functional microbial community, the health benefits, and importance of hygiene and food safety during preparation and handling of moiya pangsung, moiya koshak, mileye amileye, and midukeye. We believe this is the first report on the shotgun-based metataxonomic profile of naturally fermented moiya pangsung, moiya koshak, mileye amileye, and midukeye.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
RD carried out sampling, investigation, data curation, formal analysis, and preparation of original draft. BT conceptualized the work and helped in writing and supervision. IN helped in data analysis. NT helped in writing and editing. KM helped in sampling. All authors contributed to the article and approved the submitted version.
The authors acknowledge the Department of Microbiology, Sikkim University for providing the research facilities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1158411/full#supplementary-material
Supplementary Figure 1A | Preparation of moiya pangsung a traditional fermented bamboo shoot of Tripura (a) tender shoots of Melanconna baccifera, (b) cutting, (c) top portion of the bamboo shoot, and (d) soaked in water for fermentation.
Supplementary Figure 1B | Preparation of mileye amileye a traditional fermented bamboo shoot of Tripura (a) tender shoots of Melanconna baccifera, (b) cutting, (c) middle portion of the bamboo shoot, (d) soaked in water for fermentation, and (e) fermented product.
Supplementary Figure 1C | Preparation of midukeye, and moiya koshak a traditional fermented bamboo shoot of Tripura (a) tender shoots of Melanconna baccifera, (b) cutting, (c) middle portion of the bamboo shoot, (d) washed with water, (e) wrapped in banana leaves for fermentation, and (f) fermented product.
Supplementary Figure 2 | Heat map representation of major species in moiya pangsung, mileye amileye, moiya koshak, and midukeye.
Supplementary Figure 3A | Stacked bar plot representation of major fungal phylum communities in moiya pangsung, mileye amileye, moiya koshak, and midukeye.
Supplementary Figure 3B | Heat map representation of major fungal family communities in moiya pangsung, mileye amileye, moiya koshak, and midukeye.
Supplementary Figure 3C | Heat map representation of the major fungal species communities in moiya pangsung, mileye amileye, moiya koshak, and midukeye.
Supplementary Figure 4 | The shared and unique species were visualized between samples using a venn diagram.
Supplementary Figure 5 | NOG pathways annotated.
Supplementary Figure 6 | COG pathways annotated.
Supplementary Table 1 | Fermented bamboo shoots samples collected from different locations in Tripura, India.
Supplementary Table 2 | Abundance of bacteria at family level in moiya pangsung, mileye amileye, moiya koshak, and midukeye.
Supplementary Table 3 | KEGG gene abundance in each sample in levels 2 and 3.
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Čech, M. et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Anand, S., Mangano, E., Barizzone, N., Bordoni, R., Sorosina, M., Clarelli, F., et al. (2016). Next generation sequencing of pooled samples: guideline for variants’ filtering. Sci. Rep. 6:33735. doi: 10.1038/srep33735
Appenteng, M. K., Krueger, R., Johnson, M. C., Ingold, H., Bell, R., Thomas, A. L., et al. (2021). Cyanogenic glycoside analysis in American elderberry. Molecules 26:1384. doi: 10.3390/molecules26051384
Arasu, M. V., Al-Dhabi, N. A., Ilavenil, S., Choi, K. C., and Srigopalram, S. (2016). In vitro importance of probiotic Lactobacillus plantarum related to medical field. Saudi J. Biol. Sci. 23, S6–S10. doi: 10.1016/j.sjbs.2015.09.022
Banik, R. L. (2010). Biology and Silviculture of Muli Bamboo Melocanna Baccifera. New Delhi: National Mission on Bamboo Applications (NBMA).
Behera, S. S., el Sheikha, A. F., Hammami, R., and Kumar, A. (2020). Traditionally fermented pickles: how the microbial diversity associated with their nutritional and health benefits? J. Funct Foods 70:103971. doi: 10.1016/j.jff.2020.103971
Behera, P., and Balaji, S. (2021). Health benefits of fermented bamboo shoots: the twenty-first century green gold of Northeast India. Appl. Biochem. Biotechnol. 193, 1800–1812. doi: 10.1007/s12010-021-03506-y
Belda, I., Zarraonaindia, I., Perisin, M., Palacios, A., and Acedo, A. (2017). From vineyard soil to wine fermentation: microbiome approximations to explain the “terroir”. Concept. Front. Microbiol. 8:821. doi: 10.3389/fmicb.2017.00821
Buergy, A., Rolland-Sabaté, A., Leca, A., and Renard, C. M. (2020). Pectin modifications in raw fruits alter texture of plant cell dispersions. Food Hydrocolloids 107:105962. doi: 10.1016/j.foodhyd.2020.105962
Corral, P., Amoozegar, M. A., and Ventosa, A. (2020). Halophiles and their biomolecules: recent advances and future applications in biomedicine. Mar. Drugs 18:33. doi: 10.3390/md18010033
Cui, Y., Wang, X., Yue, Y., Du, G., Chen, H., Ning, M., et al. (2023). Metagenomic features of Tibetan kefir grains and its metabolomics analysis during fermentation. LWT 175:114502. doi: 10.1016/j.lwt.2023.114502
Das, S., and Tamang, J. P. (2021). Changes in microbial communities and their predictive functionalities during fermentation of toddy, an alcoholic beverage of India. Microbiol. Res. 248:126769. doi: 10.1016/j.micres.2021.126769
de Meeûs, T. (2018). Revisiting FIS, FST, wahlund effects, and null alleles. J. Heredity 109, 446–456. doi: 10.1093/jhered/esx106
Deka, P., Mehetre, G. T., Lalnunmawii, E., Upadhyaya, K., Singh, G., Hashem, A., et al. (2021). Metagenomic analysis of bacterial diversity in traditional fermented foods reveals food-specific dominance of specific bacterial taxa. Fermentation 7:167. doi: 10.3390/fermentation7030167
Djeni, T. N., Kouame, K. H., Ake, F. D. M., Amoikon, L. S. T., Dje, M. K., and Jeyaram, K. (2020). Microbial diversity and metabolite profiles of palm wine produced from three different palm tree species in Côte d’Ivoire. Sci. Rep. 10:1715. doi: 10.1038/s41598-020-58587-2
Dong, X., and Strous, M. (2019). An integrated pipeline for annotation and visualization of metagenomic contigs. Front. Genet. 10:999. doi: 10.3389/fgene.2019.00999
Faivre, A., Katsyuba, E., Verissimo, T., Lindenmeyer, M., Rajaram, R. D., Naesens, M., et al. (2021). Differential role of nicotinamide adenine dinucleotide deficiency in acute and chronic kidney disease. Nephrol. Dial. Trans. 36, 60–68. doi: 10.1093/NDT/GFAA124
Ferreira, A. M., and Mendes-Faia, A. (2020). The role of yeasts and lactic acid bacteria on the metabolism of organic acids during winemaking. Foods 9:1231. doi: 10.3390/foods9091231
Fong, Y., and Huang, Y. (2019). Modified wilcoxon–mann–whitney test and power against strong null. Am. Statis. 73, 43–49. doi: 10.1080/00031305.2017.1328375
Galperin, M. Y., Kristensen, D. M., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2018). Microbial genome analysis: the COG approach. Brief Bioinform. 20, 1063–1070. doi: 10.1093/bib/bbx117
Ghosh, T., Beniwal, A., Semwal, A., and Navani, N. K. (2019). Mechanistic insights into probiotic properties of lactic acid bacteria associated with ethnic fermented dairy products. Front. Microbiol. 10:502. doi: 10.3389/fmicb.2019.00502
Hahne, J., Kloster, T., Rathmann, S., Weber, M., and Lipski, A. (2018). Isolation and characterization of Corynebacterium spp. from bulk tank raw cow’s milk of different dairy farms in Germany. PLoS One 13:e0194365. doi: 10.1371/journal.pone.0194365
Halami, P. M., Badarinath, V., Devi, S. M., and Vijayendra, S. V. N. (2011). Partial characterization of heat-stable, antilisterial and cell lytic bacteriocin of Pediococcus pentosaceus CFR SIII isolated from a vegetable source. Ann. Microbiol. 61, 323–330. doi: 10.1007/s13213-010-0145-x
Hibi, M., Kawashima, T., Kodera, T., Smirnov, S. V., Sokolov, P. M., Sugiyama, M., et al. (2011). Characterization of Bacillus thuringiensis L-isoleucine dioxygenase for production of useful amino acids. Appl. Environ. Microbiol. 77, 6926–6930. doi: 10.1128/AEM.05035-11
Hivert, V., Leblois, R., Petit, E. J., Gautier, M., and Vitalis, R. (2018). Measuring genetic differentiation from pool-seq data. Genetics 210, 315–330. doi: 10.1534/genetics.118.300900
Hu, Q., Zhang, T., Yu, H., and Ye, L. (2022). Selective biosynthesis of retinol in S. cerevisiae. Bioresour. Bioprocess 9, 399–410. doi: 10.1186/s40643-022-00512-8
Hu, Y., Chen, X., Zhou, J., Jing, W., and Guo, Q. (2021). Metagenomic analysis of suansun, a traditional chinese unsalted fermented food. Processes 9:1669. doi: 10.3390/pr9091669
Hussain, S., Jõudu, I., and Bhat, R. (2020). Dietary fiber from underutilized plant resources-A positive approach for valorization of fruit and vegetable wastes. Sustainability 12:5401. doi: 10.3390/su12135401
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27-30
Keegan, K. P., Glass, E. M., and Meyer, F. (2016). MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol. Biol. 1399, 207–233. doi: 10.1007/978-1-4939-3369-3_13
Kennedy, L., Sandhu, J. K., Harper, M. E., and Cuperlovic-culf, M. (2020). Role of glutathione in cancer: from mechanisms to therapies. Biomolecules 10, 1–27. doi: 10.3390/biom10101429
Kumar, J., Sharma, N., Kaushal, G., Samurailatpam, S., Sahoo, D., Rai, A. K., et al. (2019). Metagenomic insights into the taxonomic and functional features of kinema, a traditional fermented soybean product of Sikkim Himalaya. Front. Microbiol. 10:1744. doi: 10.3389/fmicb.2019.01744
Kumar, V., Naik, B., Sharma, S., Kumar, A., Khan, J. M., and Irfan, M. (2022). Molecular diversity of microbes associated with fermented bamboo shoots. Trop. Life Sci. Res. 33, 151–164. doi: 10.21315/tlsr2022.33.3.9
Labuschagne, P., and Divol, B. (2021). Thiamine: a key nutrient for yeasts during wine alcoholic fermentation. Appl. Microbiol. Biotechnol. 105, 953–973.
Lappe-Oliveras, P., Fonseca, H. C., Engevik, M., and Fava, F. (2022). Microbial and metabolic characterization of organic artisanal sauerkraut fermentation and study of gut health-promoting properties of sauerkraut brine. Front. Microbiol. 13:929738. doi: 10.3389/fmicb.2022.929738
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Lin, Z., Chen, J., Zhang, J., and Brooks, M. S. L. (2018). Potential for value-added utilization of bamboo shoot processing waste—recommendations for a biorefinery approach. Food Bioproc. Tech. 11, 901–912. doi: 10.1007/s11947-018-2088-3
Mahara, F. A., Nuraida, L., and Lioe, H. N. (2021). Folate in milk fermented by lactic acid bacteria from different food sources. Prev. Nutr. Food Sci. 26, 230–240. doi: 10.3746/pnf.2021.26.2.230
Marcelino, V. R., Clausen, P. T. L. C., Buchmann, J. P., Wille, M., Iredell, J. R., Meyer, W., et al. (2020). CCMetagen: comprehensive and accurate identification of eukaryotes and prokaryotes in metagenomic data. Genome Biol. 21:103. doi: 10.1186/s13059-020-02014-2
Martino, C., Morton, J. T., Marotz, C. A., Thompson, L. R., Tripathi, A., Knight, R., et al. (2019). A novel sparse compositional technique reveals microbial perturbations. mSystems 4:13. doi: 10.1128/msystems.00016-19
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Mota de Carvalho, N., Costa, E. M., Silva, S., Pimentel, L., Fernandes, T. H., et al. (2018). Fermented foods and beverages in human diet and their influence on gut microbiota and health. Fermentation 4:90. doi: 10.3390/fermentation4040090
Muller, J., Szklarczyk, D., Julien, P., Letunic, I., Roth, A., Kuhn, M., et al. (2009). eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 38, D190–D195. doi: 10.1093/nar/gkp951
Neinast, M., Murashige, D., and Arany, Z. (2019). Branched chain amino acids. Annu. Rev. Physiol. 81, 139–164. doi: 10.1146/annurev-physiol-020518-114455
Nongdam, P. (2015). “Traditional fermented bamboo shoot foods of North-East India and their characteristic natural microbial flora”, in Proceedings of the 10th World Bamboo Congress conference, Damyeng.
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). MetaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Park, Y. K., Jin, Y. H., Lee, J. H., Byun, B. Y., Lee, J., Jeong, K. C. C., et al. (2020). The role of enterococcus faecium as a key producer and fermentation condition as an influencing factor in tyramine accumulation in Cheonggukjang. Foods 9:915. doi: 10.3390/foods9070915
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Shapiro, O. H., Kushmaro, A., and Brenner, A. (2010). Bacteriophage predation regulates microbial abundance and diversity in a full-scale bioreactor treating industrial wastewater. ISME J. 4, 327–336. doi: 10.1038/ismej.2009.118
Singhal, P., Rudra, S. G., Singh, R. K., Satya, S., and Naik, S. N. (2018). Impact of drying techniques on physical quality of bamboo shoots: implications on tribal’s livelihoods. Ind. J. Trad. Knowledge 17, 353–359.
Somashekaraiah, R., Mottawea, W., Gunduraj, A., Joshi, U., Hammami, R., and Sreenivasa, M. Y. (2021). Probiotic and antifungal attributes of Levilactobacillus brevis MYSN105, isolated from an Indian traditional fermented food pozha. Front. Microbiol. 12:696267. doi: 10.3389/fmicb.2021.696267
Spacova, I., Ahannach, S., Breynaert, A., Erreygers, I., Wittouck, S., Bron, P. A., et al. (2022). Spontaneous riboflavin-overproducing Limosilactobacillus reuteri for biofortification of fermented foods. Front. Nutr. 9:916607. doi: 10.3389/fnut.2022.916607
Sulaiman, J., Gan, H. M., Yin, W. F., and Chan, K. G. (2014). Microbial succession and the functional potential during the fermentation of Chinese soy sauce brine. Front. Microbiol. 5:556. doi: 10.3389/fmicb.2014.00556
Sun, W., Shahrajabian, M. H., and Lin, M. (2022). Research progress of fermented functional foods and protein factory-microbial fermentation technology. Fermentation 8:688. doi: 10.3390/fermentation8120688
Swain, M. R., Anandharaj, M., Ray, R. C., and Parveen Rani, R. (2014). Fermented fruits and vegetables of asia: a potential source of probiotics. Biotechnol. Res. Int. 2014, 1–19. doi: 10.1155/2014/250424
Tamang, B., and Tamang, J. P. (2009). Lactic acid bacteria isolated from indigenous fermented bamboo products of Arunachal Pradesh in India and their functionality. Food Biotechnol. 23, 133–147. doi: 10.1080/08905430902875945
Tamang, B., Tamang, J. P., Schillinger, U., Franz, C. M., Gores, M., and Holzapfel, W. H. (2008). Phenotypic and genotypic identification of lactic acid bacteria isolated from ethnic fermented bamboo tender shoots of North East India. Int. J. Food Microbiol. 121, 35–40. doi: 10.1016/j.ijfoodmicro.2007.10.009
Tamang, J. P., Tamang, N., Thapa, S., Dewan, S., Tamang, B., Yonzan, H., et al. (2012). Microorganisms and nutritional value of Ethnic fermented foods and alcoholic beverages of North East India. Ind. J. Trad. Knowledge 11, 7–25.
Terrón-Camero, L. C., Gordillo-González, F., Salas-Espejo, E., and Andrés-León, E. (2022). Comparison of metagenomics and metatranscriptomics tools: a guide to making the right choice. Genes 13:2280. doi: 10.3390/genes13122280
Tewari, S., Negi, H., and Kaushal, R. (2019). Status of Bamboo in India. Int. J. Econ. Plants 6, 030–039. doi: 10.23910/IJEP/2019.6.1.0288
Uchoi, D., Roy, D., Majumdar, R. K., and Debbarma, P. (2015). Diversified traditional cured food products of certain indigenous tribes of Tripura. India. Ind. J. Trad. Knowledge 14, 440–446.
Voidarou, C., Antoniadou, M., Rozos, G., Tzora, A., Skoufos, I., Varzakas, T., et al. (2021). Fermentative foods: microbiology, biochemistry, potential human health benefits and public health issues. Foods 10:69. doi: 10.3390/foods10010069
Xu, Y.-J., Arneja, A. S., Tappia, P. S., Dhalla, and DSc, N. S. (2008). The potential health benefits of taurine in cardiovascular disease. Exp. Clin. Cardiol. 13, 57–65.
Yao, C., Chou, J., Wang, T., Zhao, H., and Zhang, B. (2018). Pantothenic acid, vitamin C, and biotin play important roles in the growth of Lactobacillus helveticus. Front. Microbiol. 9:1194. doi: 10.3389/fmicb.2018.01194
Keywords: fermented bamboo shoot, meloncana baccifera, shotgun metagenomics, microbial community analysis, functional gene analysis
Citation: Das R, Tamang B, Najar IN, Thakur N and Mondal K (2023) First report on metagenomics and their predictive functional analysis of fermented bamboo shoot food of Tripura, North East India. Front. Microbiol. 14:1158411. doi: 10.3389/fmicb.2023.1158411
Received: 03 February 2023; Accepted: 27 March 2023;
Published: 12 April 2023.
Edited by:
M. Y. Sreenivasa, University of Mysore, IndiaReviewed by:
Wen-Liang Xiang, Xihua University, ChinaCopyright © 2023 Das, Tamang, Najar, Thakur and Mondal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Buddhiman Tamang, Ym10YW1hbmczQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.