 Xiao Xiong
Xiao Xiong Christopher J. Geden3
Christopher J. Geden3 Dan T. Bergstralh
Dan T. Bergstralh Xu Wang
Xu Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 April 2023
Sec. Microbial Symbioses
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1152586
This article is part of the Research Topic Genomic and Evolutionary Analysis of Microsporidian Parasites View all 7 articles
Introduction: Nosema is a diverse genus of unicellular microsporidian parasites of insects and other arthropods. Nosema muscidifuracis infects parasitoid wasp species of Muscidifurax zaraptor and M. raptor (Hymenoptera: Pteromalidae), causing ~50% reduction in longevity and ~90% reduction in fecundity.
Methods and Results: Here, we report the first assembly of the N. muscidifuracis genome (14,397,169 bp in 28 contigs) of high continuity (contig N50 544.3 Kb) and completeness (BUSCO score 97.0%). A total of 2,782 protein-coding genes were annotated, with 66.2% of the genes having two copies and 24.0% of genes having three copies. These duplicated genes are highly similar, with a sequence identity of 99.3%. The complex pattern suggests extensive gene duplications and rearrangements across the genome. We annotated 57 rDNA loci, which are highly GC-rich (37%) in a GC-poor genome (25% genome average). Nosema-specific qPCR primer sets were designed based on 18S rDNA annotation as a diagnostic tool to determine its titer in host samples. We discovered high Nosema titers in Nosema-cured M. raptor and M. zaraptor using heat treatment in 2017 and 2019, suggesting that the remedy did not completely eliminate the Nosema infection. Cytogenetic analyses revealed heavy infections of N. muscidifuracis within the ovaries of M. raptor and M. zaraptor, consistent with the titer determined by qPCR and suggesting a heritable component of infection and per ovum vertical transmission.
Discussion: The parasitoids-Nosema system is laboratory tractable and, therefore, can serve as a model to inform future genome manipulations of Nosema-host system for investigations of Nosemosis.
Nosema (Microsporidia: Nosematidae) is one of the most diverse genera of microsporidian parasites, and they are widely distributed unicellular parasites of insects and other arthropods (Didier et al., 1998; Becnel and Andreadis, 1999). Nosema infection, a disease known as Nosemosis, occurs throughout the world and leads to agricultural economic losses through detrimental effects on pollinators due to N. apis infections found in European honey bee Apis mellifera and N. ceranae infections originally detected in Asian honey bee Apis cerana (Fries et al., 1996; Chen et al., 2013; Pelin et al., 2015; Stanimirović et al., 2019). Another Nosema species, N. bombycis, infects the silk moth Bombyx mori, causing a lethal disease called Pébrine, which is a serious threat to silk production (Pan et al., 2013). The consequences of Nosema infection were extensively studied due to its economic importance.
Nosema parasitism has destructive effects on honey bees, including digestive disorders, poor colony development, and reduction in metabolism and reproduction (Rinderer and Sylvester, 1978; Malone et al., 1995; Higes et al., 2009). Nosema ceranae is more prevalent, impairing bee health and declining bee colonies. It damages the physical and immune barriers of honey bees, making them more vulnerable to other pathogenic factors, which is related to colony collapse (Antúnez et al., 2009; Chen et al., 2009; Costa et al., 2011; Dussaubat et al., 2012; Gómez-Moracho et al., 2021). Despite the low prevalence of N. apis compared to N. ceranae (Cox-Foster et al., 2007), there is no substantial difference in virulence between the two species, and the negative impacts of N. apis on the infected colonies cannot be neglected (Forsgren and Fries, 2010; Botías et al., 2013). Nosema apis was reported to be associated with reduced lifespan and increased winter mortality in infected bees, and its negative effects on colony strength and productivity have been described in several studies (Fries et al., 1984; Fries, 1988; Anderson and Giacon, 1992; Farrar, 2014). Nosema bombycis-infected larvae are inactive and develop slowly, with black spots spreading all over the bodies, eventually leading to death (Pan et al., 2013). Currently, there is no effective treatment for the disease.
The mechanisms of Nosema transmission are directly related to the control of Nosemosis. Nosema bombycis can be transmitted from the mother host to their eggs vertically and gradually invades the whole body of the larvae, including muscles, intestines, silk glands, and Malpighian tubules. Nosema transmission in honey bees is primarily through the mouth to uninfected bees. Nosema apis tends to restrict its life cycle to gut epithelial cells after infection, while N. ceranae is mainly distributed in the midgut but also spread in other tissues (Chen et al., 2009; Dussaubat et al., 2012). Due to the limitations of conducting Nosema infection experiments in bees, another Hymenoptera-Nosema model system is needed for research in controlled laboratory settings to determine the infection, distribution, and transmission of Nosema and inform the control of Nosemosis.
Nosema also infects other beneficial insects, including the parasitoid wasp genus Muscidifurax (Hymenoptera: Pteromalidae). The genus Muscidifurax is a natural biocontrol agent of the dipteran filth flies with nine identified species, all of which are pupal parasitoid wasps. Muscidifurax raptor Girault and Sanders was the first species characterized in 1910 (Girault and Sanders, 1910). In 1970, four sibling species in this genus were described: M. zaraptor Kogan and Legner, from the southwestern United States; M. raptoroides Kogan and Legner from Central America and Mexico; M. raptorellus Kogan and Legner from Uruguay and Chile; and a thelytokous species M. uniraptor Kogan and Legner from Puerto Rico (Kogan and Legner, 1970). Muscidifurax uniraptor only produces a female offspring, and the parthenogenesis is caused by an intracellular bacterium which is an A-group Wolbachia wUni (Zchori-Fein et al., 2000; Newton et al., 2016). The microsporidium was first found infecting M. raptor collected from New York dairy farms in 1990 (Zchori-Fein et al., 1992), and was later described as N. muscidifuracis (Becnel and Geden, 1994). Nosemosis significantly affects the development, longevity, and fecundity of M. raptor. Muscidifurax raptor infected by N. muscidifuracis takes longer to complete development, lives about half as long, and produces about 10% as many progeny as uninfected individuals (Geden et al., 1995).
Nosema disease reduction methods have been explored to cure the N. muscidifuracis infection. Exposure of parasitoid eggs within host pupae at several temperatures (45°C, 47°C, and 50°C) was shown to be effective in managing Nosema disease and increasing parasitoid fecundity (Boohene et al., 2003). A 100% cure rate was achieved at 50°C for 45 min with a relative survival of 18%. To completely eliminate the pathogen, heat treatment in combination with the Pasteur method was applied: heat treatment minimizes disease prevalence and ensures an adequate genetic base for healthy parasitoids, and the Pasteur method (based on visual examination for patent infections) isolates uninfected wasps as parents for rearing their progeny (Geden et al., 1995; Steinhaus, 2012). However, the efficacy of this approach depends on the efficiency of visual detection, which may not detect low-level infections.
Understanding the Nosema disease transmission mechanism is critical for developing new control methods, but the transmission of Nosema in parasitoid wasps is not fully understood yet. The following is known about transmission patterns: (1) maternal transmission is highly efficient; (2) adults can acquire infection by feeding on spore suspensions or infected parasitoid immatures within hosts; (3) infected male adults do not transmit infections to healthy females; (4) house fly hosts do not become infected; and (5) horizontal transmission occurs when healthy immatures feed on infected larvae in superparasitized hosts (Geden et al., 1995). The intracellular nature of Nosema suggested that transmission can occur vertically, either within the egg (cytoplasmic), and/or on the egg surface (per ovum). If per ovum or through ingestion by feeding larvae, parasitoid wasp lines can be cured of the infection by surface sterilization or egg transfer experiments. To establish a parasitoid-Nosema model for Nosemosis research, a high-quality reference genome is essential for studying gene expression changes and gene manipulations in Nosema. In this study, we sequenced and assembled the N. muscidifuracis reference genome in the parasitoid wasp species M. zaraptor using PacBio long-read sequencing, developed qPCR assays for accurate quantification of Nosema titer, and explored the vertical transmission mechanisms using cytogenetic analyses.
We sequenced the M. zaraptor genome using a combination of PacBio long-read and Illumina 10× Genomics linked-read sequencing technologies in Nosema-infected M. zaraptor samples (see Materials and methods). A total of 23.7 Gb PacBio Sequel II HiFi reads (61.2-fold coverage of M. zaraptor genome), and 63.6 Gb of Illumina linked-reads (54.5-fold coverage) were generated (Supplementary Table S1), resulting in a high-quality assembly of M. zaraptor genome. Bioinformatic analyses revealed that symbiotic microbes are among the assembled contigs, including Nosema muscidifuracis, a known microsporidian species infecting M. zaraptor. Nosema muscidifuracis contigs were separated from the host contigs based on a much higher sequencing depth (258.2×; Supplementary Table S2) compared to M. zaraptor (61.2×) and a much lower GC content (22.6% compared to 42% in M. zaraptor). The drastic differences in sequence coverage and GC content allowed the complete separation of N. muscidifuracis contigs from host sequences (Supplementary Figure S1). In addition, N. muscidifuracis contigs were aligned to a closely-related, Nosema-free M. raptorellus genome (Xiong et al., 2021) to confirm the absence of host contaminations. The final assembly of the N. muscidifuracis genome contains 14,397,169 bp in 28 contigs, with contig length ranging from 299,473 to 982,164 bp (Figure 1A).
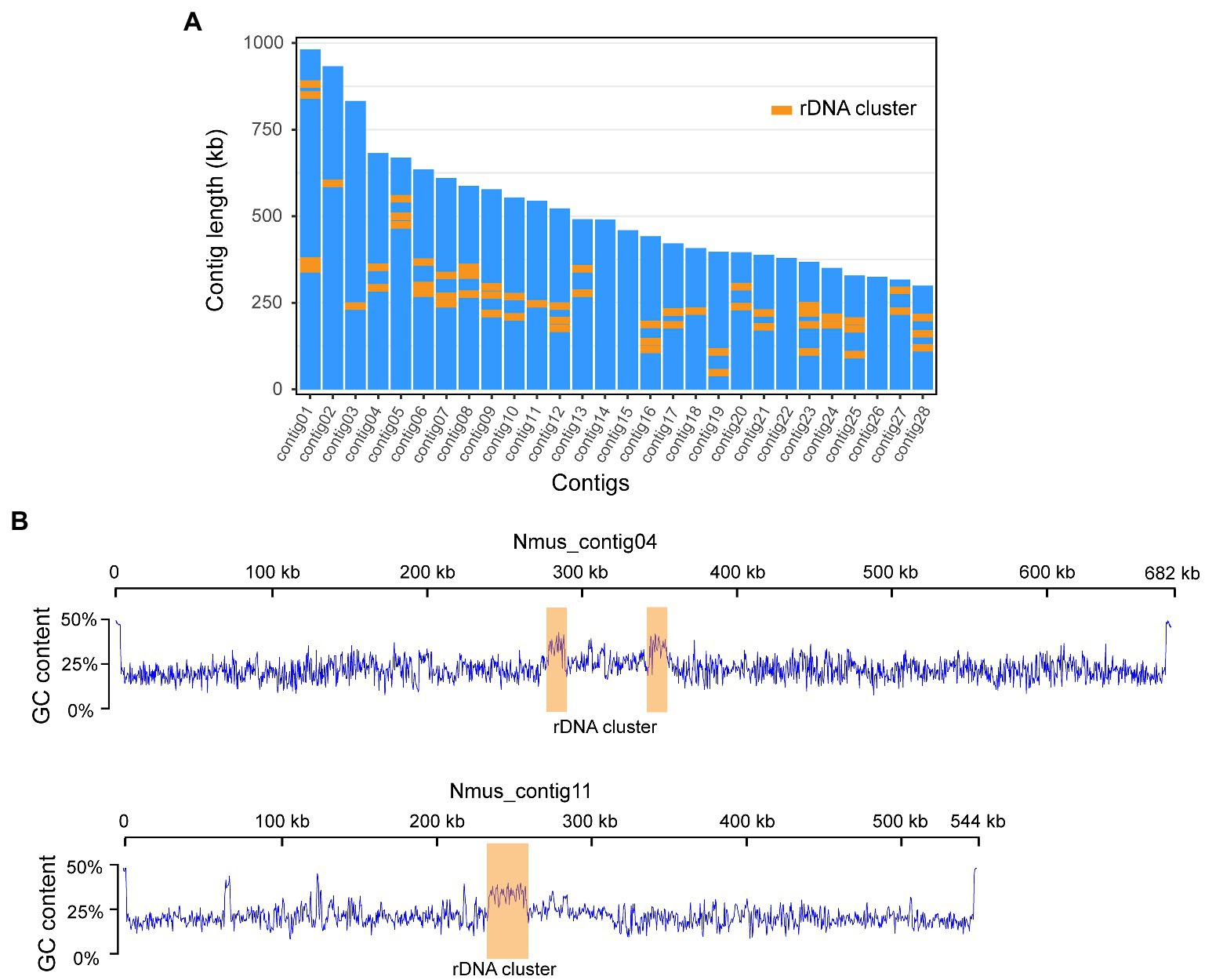
Figure 1. Chromosome level genome assembly of Nosema Muscidifuracis showing GC content and rDNA clusters. (A) The length of all 28 contigs in N. Muscidifuracis genome. A total of 57 rDNA clusters are plotted using orange boxes. (B) Plots of GC content along N. Muscidifuracis contig04 and contig11 show low average GC-content of the N. Muscidifuracis genome and elevated GC content at rDNA clusters.
The assembled N. muscidifuracis contigs had an N50 of 544,348 bp, which is much larger than all published Nosema genomes, indicating excellent continuity (Table 1). Nine contigs have regions at termini with elevated GC content (>50%), which are putative telomeric regions, suggesting chromosome-level assembly. To evaluate the completeness of the genome assembly, we aligned 10× Genomics short-read reads using BWA, and 99.42% of the assembled contigs were covered (Supplementary Table S1). The BUSCO completeness score is 97.0%, which is the same as N. ceranae assemblies and much higher than other Nosema assemblies (Table 1). The assembly statistics indicate that the quality of our assembly is high in both genome continuity and completeness.
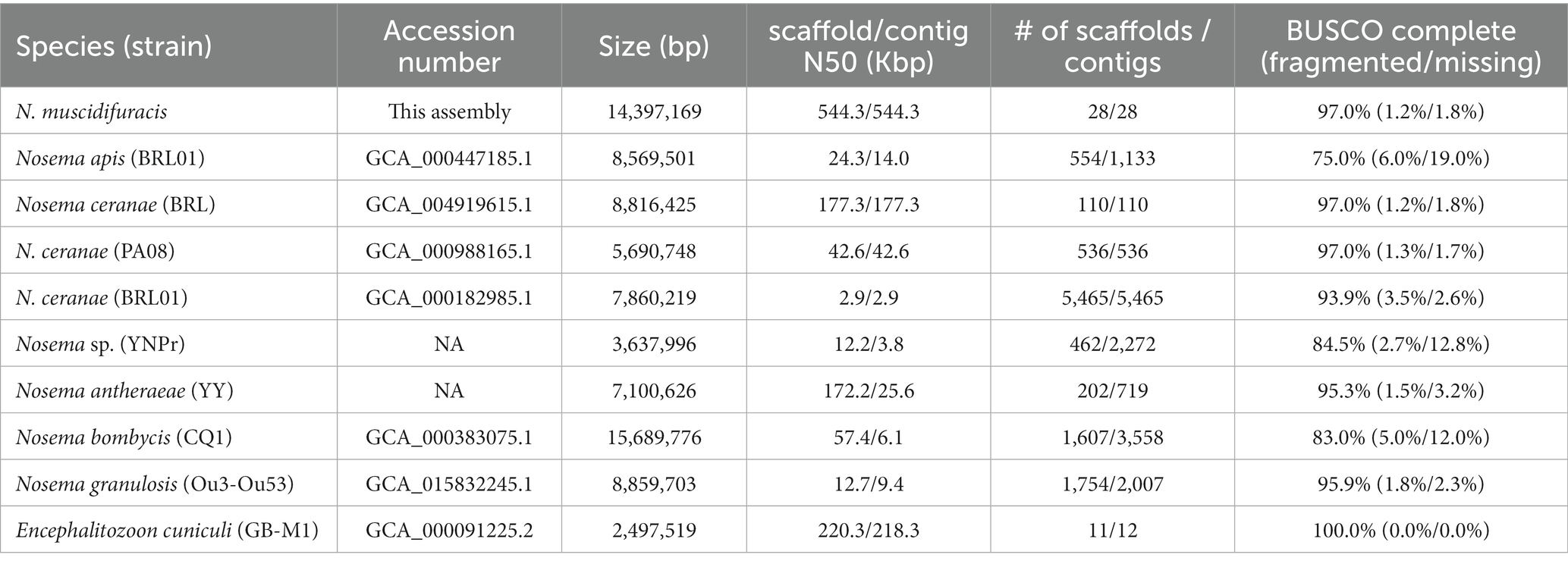
Table 1. Genome assembly statistics for Nosema muscidifuracis and comparison with other microsporidian genomes.
In the N. muscidifuracis genome, a total of 4,078,013 bp repetitive regions (28.3% of the genome) were identified (Supplementary Table S3). Most repeats belong to the unclassified category (58.8% of total repeats). Among classified repetitive elements, the Gypsy/DIRS1 LTR element is the most abundant, accounting for 37% of all known repeats, and 4.32% of the whole genome. The following classes of repetitive elements account for more than 1% of the N. muscidifuracis genome: DNA transposons (3.49%), LINEs (1.68%), and simple repeats (1.71%).
A total of 327 noncoding RNA (ncRNA) genes were predicted and annotated in N. muscidifuracis genome based on the Rfam (Nawrocki et al., 2014) database of RNA families by using the Infernal software package (Nawrocki and Eddy, 2013). The 170 tRNA genes account for 0.09% of the entire genome. We also found seven snRNA associated with U2/U4/U6 small nuclear ribonucleoproteins, and two CD-box, small nucleolar RNA U3, suggesting that the splicing function may be present in N. muscidifuracis. The most abundant ncRNA genes in N. muscidifuracis are the rDNA genes. We identified 57 complete rDNA clusters encoding 18S/28S ribosomal RNAs (Supplementary Table S4). A hallmark of N. muscidifuracis rDNA region is increased GC context (~37%) compared to the genome average (~25%; see Figure 1B). Collectively, the rDNA clustered are more than 206 kbp in length, accounting for 1.4% of the N. muscidifuracis genome (Supplementary Table S4).
The genome assembly used M. zaraptor in the AUB colony, which has never been treated for Nosema (Figure 2A). In 2019, treatment to eliminate Nosema was performed at the USDA colony, using a heat incubation approach (Boohene et al., 2003). The M. raptor USDA colony was established in 2015 from Nosema-free founders based on microscopic examination (Zchori-Fein et al., 1992). The recurrence of Nosema infection was cured in 2017 using an extensive treatment procedure described in Materials and methods (summarized in Figure 2A). To determine whether the treatment has effectively eliminated or reduced the Nosema infection, we designed PCR primer sets (Supplementary Table S5) to target the 18S rDNA genes with sequence information in our genome assembly. The Nosema titers in the AUB (Ct values 10.2~11.7) and USDA (Ct value 12.1~13.8) M. zaraptor samples were extremely high compared to the control uninfected Muscidifurax uniraptor samples (Ct value >38; p < 0.001, t-test), indicating heavy infection in both colonies (Figure 2B). The relative abundance of Nosema in AUB samples is significantly higher than the USDA colony for both females (2.8-fold; p < 0.01, t-test) and males (3.3-fold; p < 0.001, t-test), suggesting a significant reduction of the Nosema load in treated wasps (Figure 2B). The results indicated that heat treatment reduced the Nosema titer in a short time but failed to eradicate the Nosema, and the infection came back and reached high titer rapidly. For the M. raptor samples, the relative abundance of Nosema (Ct values 10.8~11.7) was also high compared to the Nosema-free M. uniraptor samples (p < 0.001, t-test), which is comparable to AUB M. zaraptor samples (Figure 2B). It is clear the Nosema has rebounded in the heat-treated line, either due to incomplete elimination or cross-infection from infected wasps.
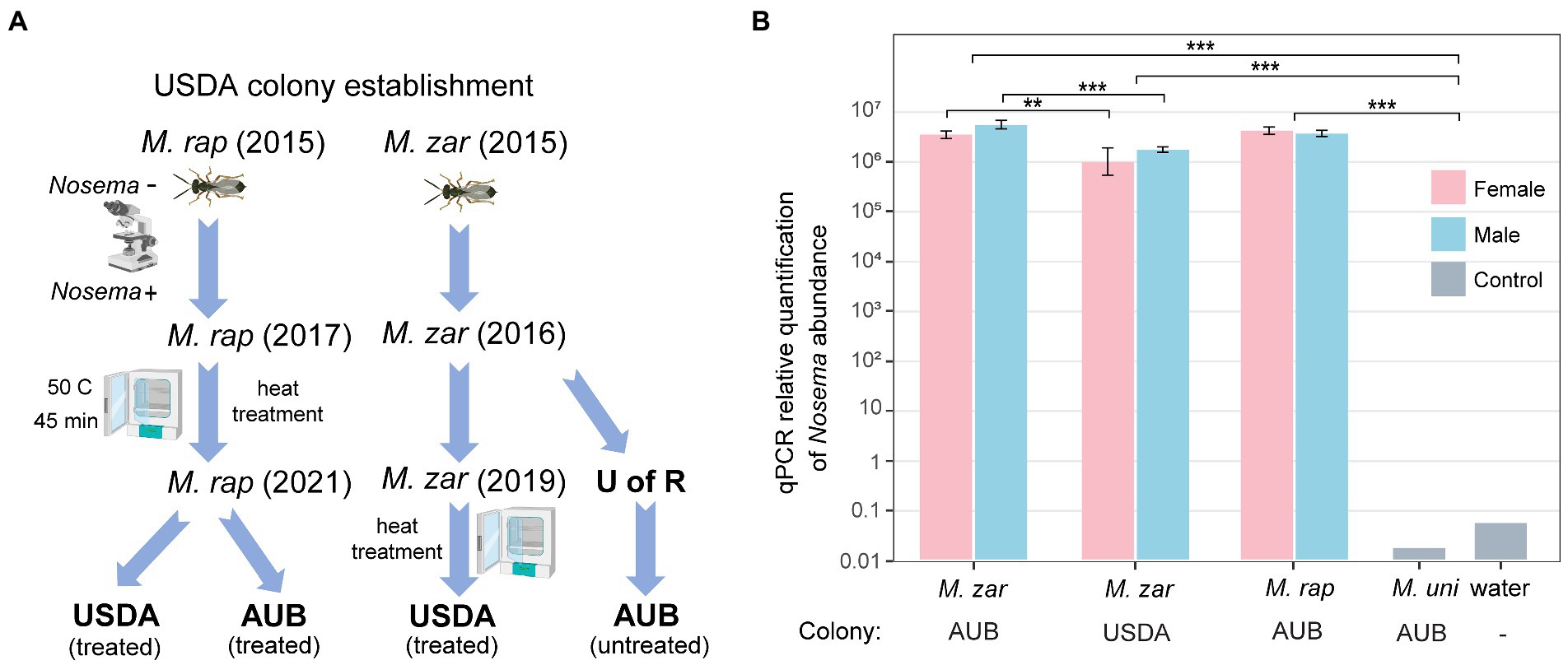
Figure 2. Quantification of the Nosema titer in AUB and USDA Muscidifurax zaraptor colony. (A) The USDA M. zaraptor and M. raptor colonies were founded in 2015 in Dr. Geden’s laboratory. The USDA M. raptor colony was treated using a combination of heat treatment and Pasteur method in 2017, resulting in a Nosema-free colony confirmed by microscopic examination. The AUB M. zaraptor was derived from a colony from the University of Rochester (U of R), which was never treated. The USDA M. zaraptor colony was cured for Nosema infection using a heat treatment approach in 2019 (Geden et al., 2019). (B) Results of qPCR quantifications of Nosema muscidifuracis in M. zaraptor and M. raptor. Female samples were plotted using pink bars and male samples were plotted in blue. Negative control (water) and Nosema-free wasp M. uniraptor were included as controls. Statistical significance was assessed by t-test (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
We identified 2,782 protein-coding gene models using Fungal Genome Annotation Pipeline (FunGAP; Min et al., 2017), with RNA-seq read support (Supplementary Table S6). The number of protein-coding genes we annotated for N. muscidifuracis is close to that of N. ceranae (N = 2,905; Cornman et al., 2009) and N. apis (N = 2,764). Comparative genomic analysis revealed that 26/28 N. muscidifuracis contigs have syntenic regions in the N. ceranae genome based on protein-coding genes (Figure 3). There is a moderate level of conservation in gene order, with many genome rearrangement events (Figure 3). When an outgroup microsporidian species was compared, all 11 chromosomes in E. cuniculi (Katinka et al., 2001) have syntenic regions in N. muscidifuracis, which mapped to 24 N. muscidifuracis contigs (Supplementary Figure S2).
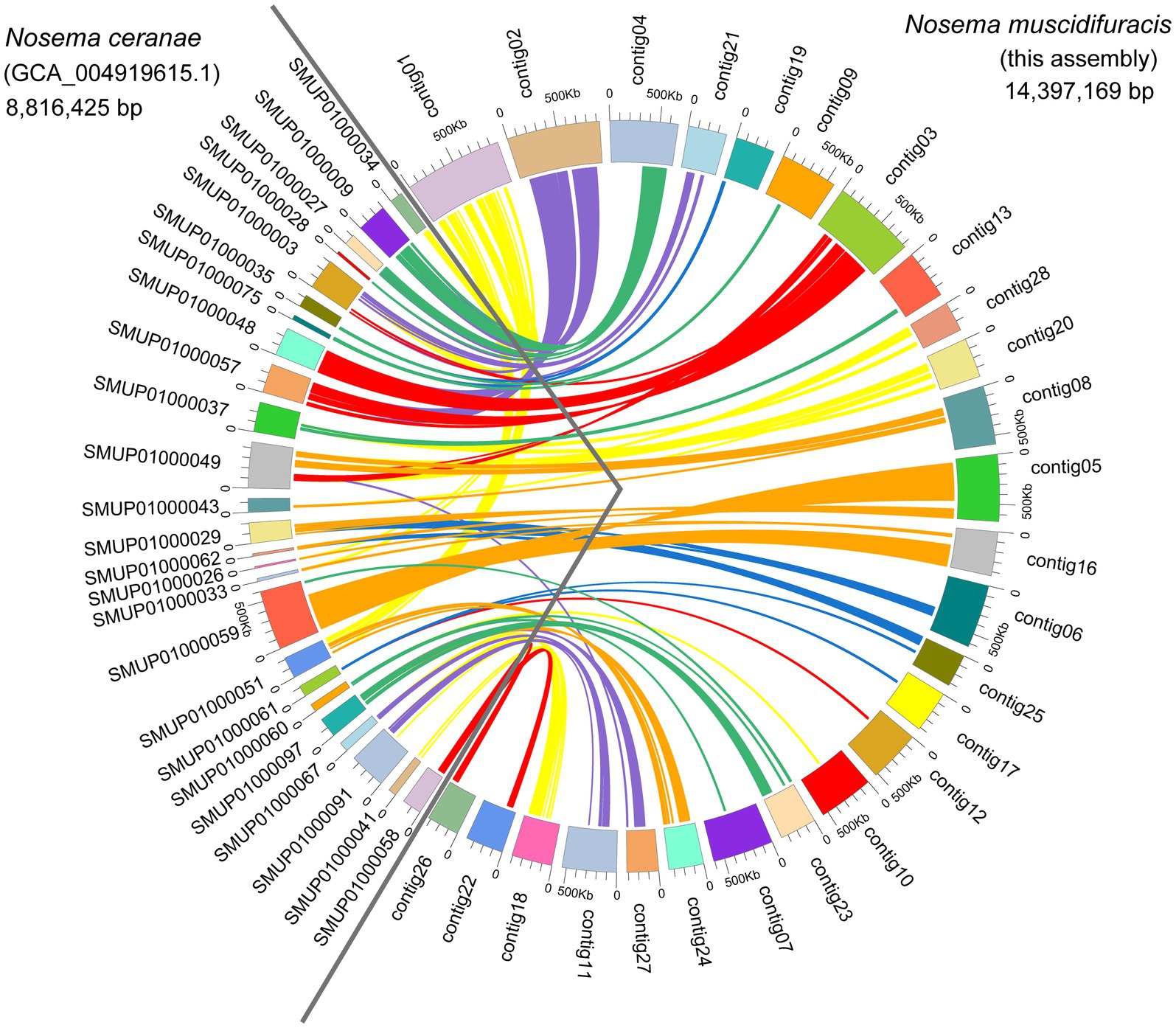
Figure 3. Genome comparisons between Nosema muscidifuracis and Nosema ceranae. A total of 26 contigs of N. muscidifuracis (93.4% of assembly in this research) show a one-to-one relationship with 25 scaffolds in the N. ceranae genome (54.1% of assembly GCA_004919615). The scaffolds on the left of the circle represent N. ceranae scaffolds, and the contigs on the right represent N. muscidifuracis contigs.
In the N. muscidifuracis genome, the majority of genes were duplicated with two or more copies (N = 2,526 genes in 1,147 paralogous groups). In sharp contrast, only a small number of genes (N = 256) are single-copy (Figure 4A). This is not the case in N. ceranae or E. cuniculi genomes (Figure 4A). The pattern is very complex (Figure 4B), indicating extensive duplications and rearrangements within the genome. For N. muscidifuracis with two copies, we aligned all 919 pairs and found that 859 of them are the same length, and only 60 pairs have different gene sizes. The average pairwise identity for the 859 gene pairs with the same length is 99.7%, and that of the remaining 60 gene pairs is 94.1%. This indicates relative recent duplications in N. muscidifuracis. We computed the number of substitutions for the 859 gene pairs with the same size (average gene length = 1,027 bp), and on average, 1.66 synonymous and 1.09 nonsynonymous substitutions were observed (2.75 total). We next examined the syntenic pattern between homologous genes within N. muscidifuracis. A self-circos plot (syntenic relationship among contigs determined by homolog of gene models) did not reveal whole contig duplications, but rather extensive duplications and rearrangements across the genome (Figure 4B). The majority of the duplicated gene pairs are on different contigs, except for 66 gene pairs on contig01 (Figure 4B). Contig01 is the largest contig/chromosome (982 kb in length), with duplicated regions on contig16, contig21, and contig24, with short to moderate stretches of synteny (Figure 4B). To check whether the duplication events have an impact on the genome size, we estimated the genome size of N. muscidifuracis using a k-mer approach (see Materials and methods), and the estimated genome size is approximately half of the assembled size (7,040,619 bp) with an estimated heterozygosity of 0.9%, which is consistent with the extensive duplication events we observed.
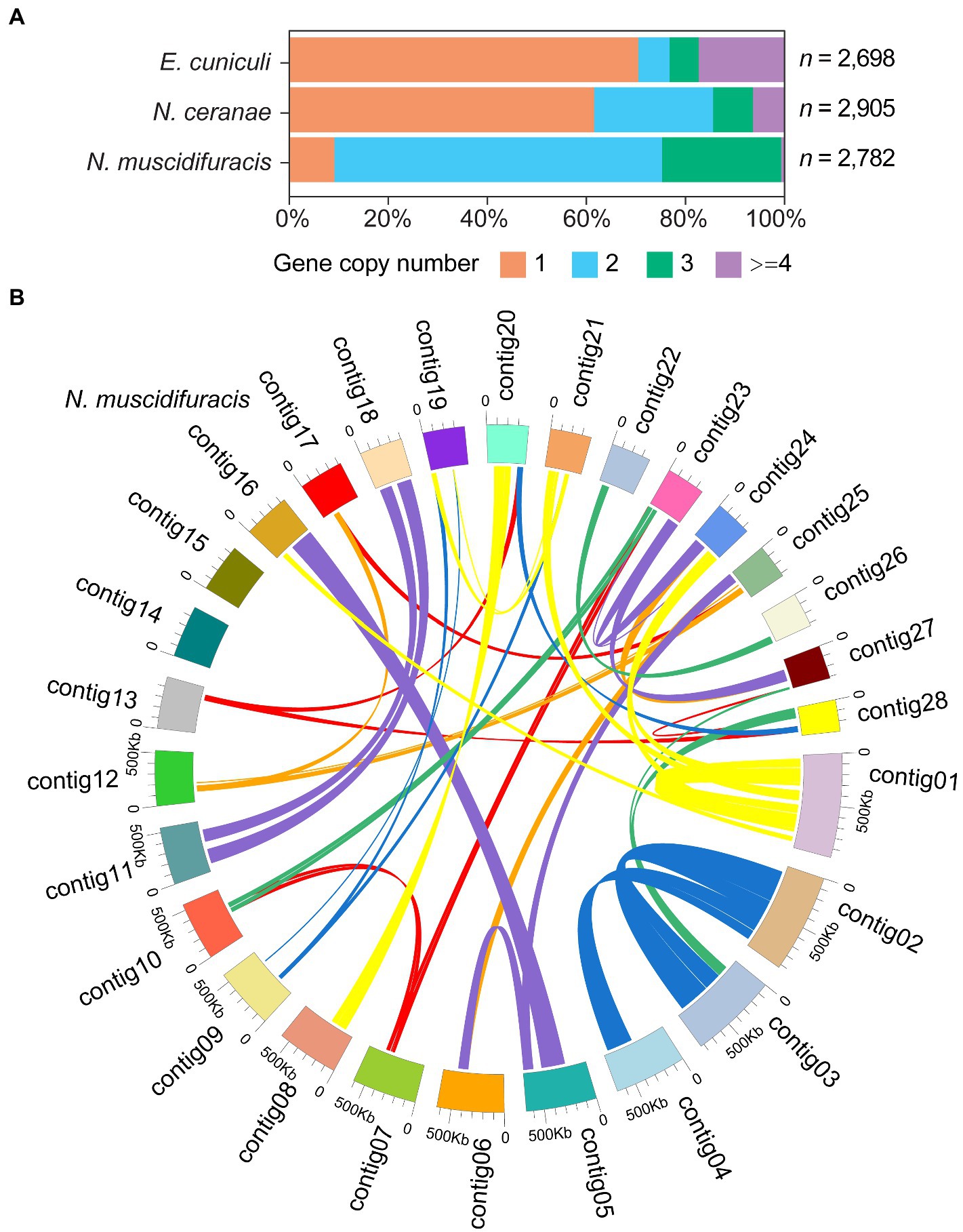
Figure 4. Histograms of gene copy number in Nosema muscidifuracis, Nosema ceranae, and Encephalitozoon cuniculi genomes and self-circos plot of 28 contigs in N. muscidifuracis. (A) Proportion of annotated genes (x-axis) with different gene copy number in the genomes of N. muscidifuracis, N. ceranae, and E. cuniculi (y-axis). (B) The self-circos of N. muscidifuracis genome shows the linked relationship of 28 contigs.
We examined the read depth in single-copy gene regions and duplicated gene regions. There are significant differences in depth between single-copy genes and genes with two copies. For single-copy genes, the average coverage depth is 44.7 with a standard error of 1.6. In duplicated gene regions, the average coverage total depth is 72.0 with a standard error of 0.7, which is significantly higher than single-copy genes (p < 2.2 × 10−16, Mann–Whitney U test). To further confirm the gene duplication events, we quantified the relative abundance of a single-copy gene bim1 (microtubule binding protein BIM1), a two-copy genes mfs1 (major facilitator superfamily 1 nucleoside transporter), and a three-copy gene tef1 (translation elongation factor 1 alpha), and compared them with the 18S rDNA gene (Figure 5). The tef1 relative abundance is significantly higher than mfs1 (p < 0.05, t-test), and mfs1 has significantly higher abundance that the one-copy bim1 gene (p < 0.05, t-test; Figure 5). The read depth and qPCR results confirmed the copy number differences of the N. muscidifuracis genes.
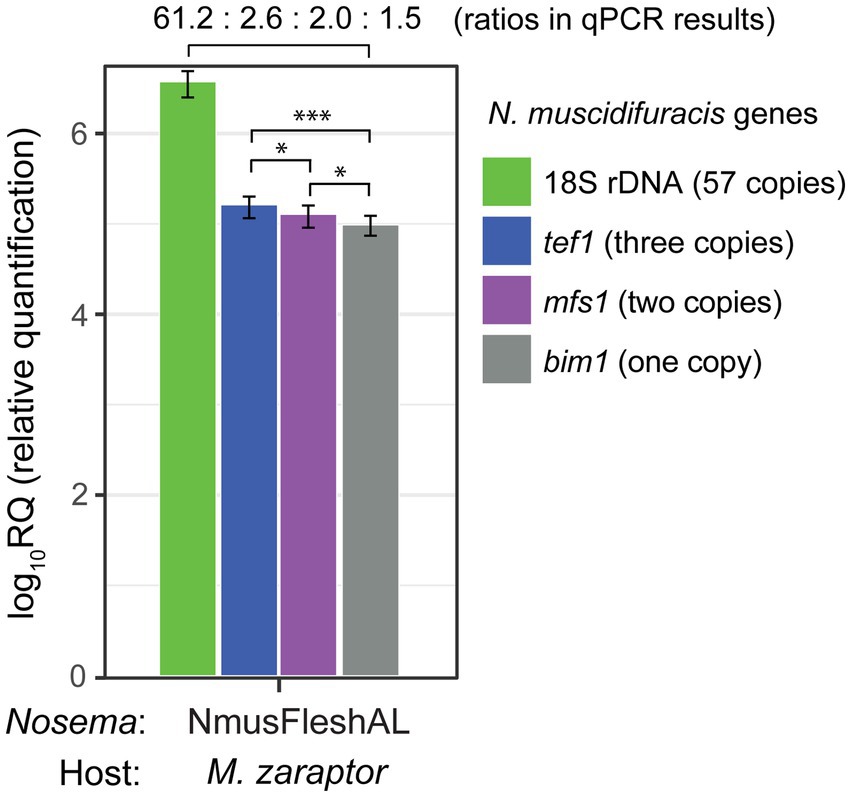
Figure 5. Quantification and confirmation of gene copy number in Nosema muscidifuracis using quantitative PCR approach. Results of qPCR analysis of 18S rDNA gene (57 copies in the genome, in green), protein-coding genes tef1 (three copies, in blue), mfs1 (two copies, in purple), and bim1 (single-copy, in gray). NmusFleshAL: Nosema-infected Muscidifurax zaraptor reared using flesh fly pupae at Auburn, Alabama. Statistical significance was assessed by t-test (*p < 0.05; ***p < 0.001).
Nosema muscidifuracis shows evidence of vertical transmission. We therefore investigated the ovaries of three species, Muscidifurax zaraptor and M. raptor, which are infected with Nosema, and the closely-related species M. uniraptor, which is Nosema-free despite being reared in the same research environment. Staining with the DNA intercalating agent DAPI revealed puncta of ~2 μM in diameter, much smaller than nurse cell or follicle cell nuclei, in ovarioles from M. zaraptor and M. raptor but not M. uniraptor. These puncta, which are consistent in appearance with previous observations of N. muscidifuracis (Pei et al., 2021), are associated with developing egg chambers at all stages and can be readily distinguished inside late-stage oocytes (Figure 6). Infected ovarioles also demonstrate signs of infection in other cell types, including nurse cells and follicle epithelial cells (Figure 6). This confirms heavy infections of microsporidia in the ovaries, and likely supports cytoplasmic transmission of the microbe through eggs.
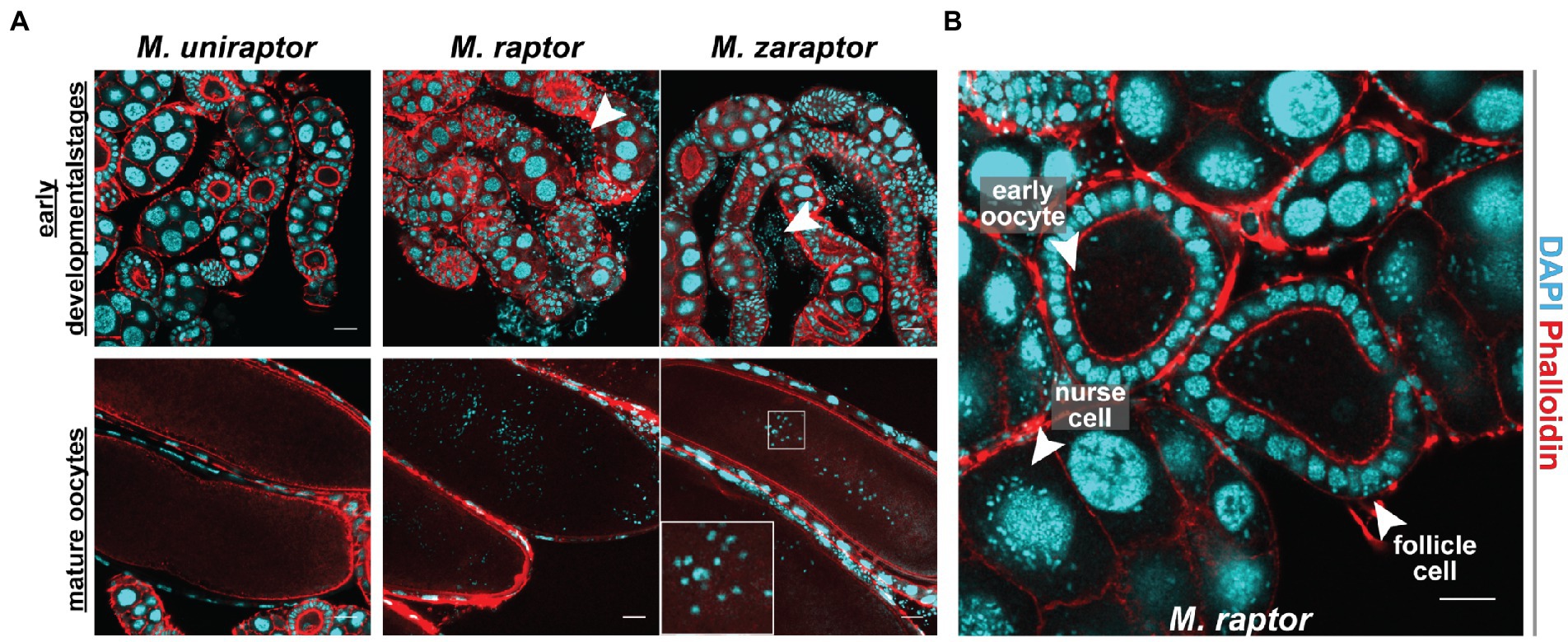
Figure 6. Cytogenetic analysis of Nosema muscidifuracis distribution in developing and mature oocytes of three Muscidifurax species. (A) Staining of ovaries from adult female Muscidifurax uniraptor, Muscidifurax raptor, and Muscidifurax zaraptor (from left to right) showing early developmental stages (top) and mature ovaries (bottom). Red, actin staining using Rhodamine Phalloidin. Blue, DNA staining using DAPI. The length of the scale bar is 20 μm. (B) Staining showing the distribution of N. muscidifuracis in M. raptor early oocyte, follicle cells, and nurse cells (white arrows). The length of the scale bar is 20 μm.
Nosema (Microsporidia: Nosematidae) is one of the most widespread unicellular parasites belonging to the microsporidia group. Previously, microsporidia were classified as protozoan, but recently they were recognized as a group of fungi. As a genus in microsporidia, Nosema infect insects and other arthropods using a specialized organelle known as the polar filament, which is coiled inside its spores. Compared to other microsporidians, Nosema has a much lower GC content, which makes it very difficult to assemble its genome at high levels of completeness and continuity. In this study, we sequenced and annotated the genome of Nosema muscidifuracis, which is the Nosema species infecting the parasitoid wasp species Muscidifurax zaraptor and M. raptor. This is the first genome assembly of N. muscidifuracis, with the highest completeness (97.0% BUSCO score) and continuity (contig N50 = 544 kb) among all available Nosema genomes (Table 1). The high quality and well annotated genome of N. muscidifuracis is crucial for the comparative genomic analysis and evolutionary study in Nosema, and will facilitate future genome manipulation in this fungal pathogen to control Nosemosis.
Microsporidia usually have a severely reduced genome, such as a 2.5 Mb genome in E. cuniculi (Katinka et al., 2001; Peyretaillade et al., 2009). In Nosema, the smallest reported genome is Nosema sp. YNPr (3.6 Mb), which infects the cabbage butterfly Pieris rapae (Xu et al., 2016). However, the BUSCO completeness score was 84.5%, raising the question of whether sizeable numbers of genes were missed in that assembly (Table 1). The smaller genome may be partly due to incomplete genome assembly, or loss of some conserved genes in microsporidia. Four Nosema species have a reported genome size of 7~9 Mb, including 8.8 Mb in N. ceranae BRL (Huang et al., 2021), 8.6 Mb in N. apis (Chen et al., 2013), 7.8 Mb in N. antheraeae infecting the Chinese tussar moth Antheraea pernyi (Li et al., 2017), and 8.6 Mb N. granulosis infecting the Amphipod Gammarus duebeni (Cormier et al., 2021). The 15.7 Mb N. bombycis (Pan et al., 2013) and 14.4 Mb N. muscidifuracis (this research) assemblies are a bit less than doubled genome size of other Nosema species (Table 1), indicating changes in the genome size in Nosema. A larger genome harbors more genes for adapting to different conditions and host environments, but this increase in capacity could also be achieved by polyploidy, which is quite common in intracellular parasites (Nakabachi and Moran, 2022). The evolution of genome size in Nosema and other microsporidia warrants further study. Furthermore, little is known about the sexual cycle of Nosema, and how this may relate to life cycle changes in ploidy levels.
We observed a unique feature in the N. muscidifuracis genome, which is the majority of protein-coding genes have two copies (66.2% of genes) or multiple copies (8.1%). One possible reason for this observation is polyploidy, which is a phenomenon that organisms possess more than two complete sets of chromosomes in individual cells (Van de Peer et al., 2017). However, the duplication of genomic regions in N. muscidifuracis is not consistent with complete chromosome duplication (Figure 4). Based on our results, the most plausible scenario would be extensive gene duplications affecting multiple regions in the genome. In fungi, alternating haploid and diploid phases are present in many species (Valero et al., 1992), and it is not clear whether such a process exists in Nosema. Polyploidy has been suggested in natural N. ceranae population (4 N or more), based on the distribution of genetic variation (Pelin et al., 2015). A recent study of multiple fungal genomes identified that most zoosporic fungi, including microsporidia, are diplontic (diploid-dominant) based on SNP density (Amses et al., 2022). The density of pairwise substitutions in N. muscidifuracis duplicated genes is 2.68 × 10−3 in this study, which would be classified as diplontic according to the criteria for diploid genomes (mean = 2.04 × 10−3) in Amses et al. (2022). However, not all N. muscidifuracis genes are duplicated (only 66%), and our long-read assembled results do not support complete chromosome duplication. It is likely to be a complex history of gene duplication and rearrangement in N. muscidifuracis evolution, which expanded its genome size and resulted in the gene copy number profile we observed in this study.
Transovarial transmission of unicellular parasites has been observed and reported in microsporidia (Becnel, 1994; Terry et al., 1997; Dunn et al., 2001; Pei et al., 2021). In this study, we established vertical transmission of N. muscidifuracis in the parasitoid wasps Muscidifurax zaraptor and M. raptor through straining experiments in the ovaries of infected females. However, whether this is the exclusive or primary mode of transmission remains to be determined. In addition, maternal transmission by injection of microsporidia during stinging of the host is also possible, which could also result in transfer between lineages when two females parasitized the same host. Additional studies are needed to characterize the transmission mechanism(s), which will be relevant to attempts to maintain Nosema-free colonies of these biological control agents.
Pest flies can cause a huge economic loss on livestock operations. The impact of stable flies on feeder cattle dropped feed efficiency by 10% to 15%, and gains were reduced by 0.2 to 0.5 pounds per day (Taylor et al., 2012). When dairy cattle spend their energy getting the flies off their legs, bunching in a corner, or not eating, milk production can be reduced by almost 2.2 pounds per day. It is estimated that stable flies can cost the livestock industry $2.2 billion per year. Parasitoid wasp species in the genus of Muscidifurax are excellent biological control agents for dipteran filth flies, including house fly, stable fly, horn fly, black dump fly, and flesh fly (Petersen and Currey, 1996; Geden and Hogsette, 2006; Geden and Moon, 2009; Machtinger and Geden, 2018). These parasitoids are more environment-friendly and sustainable, and host flies are not known to develop resistance to them, as they do with chemical insecticides (Oyarzun et al., 2008; Scott et al., 2013). The production of parasitoids is affected by the Nosema disease caused by persistent infection of N. muscidifuracis, which significantly reduces fitness and fecundity (Geden et al., 1995). A heat shock treatment approach is effective in controlling Nosema disease, resulting in a 100% cure rate and significantly improved fitness (Boohene et al., 2003). In 2019, the USDA M. zaraptor colony was cured by heat treatment. However, the Nosema bounced back to a high level within a short period of time, based on microscopic inspection, and qPCR quantification. The Nosema titer in the USDA M. zaraptor is about a third of the untreated lines (AUB), although the titer is high in both colonies. The USDA M. raptor colony was started from Nosema-free founders based on microscopic examinations. However, infection was observed after 1 year, and the M. raptor colony was treated using a series of heat treatment procedures in 2017. Parasitoids from the colony appeared to still be uninfected after 3 months, but infection reappeared after several years of rearing without regular inspection or further treatment for infection. These results demonstrate that visual inspection for patent infections is an insufficiently sensitive metric for eliminating Nosema disease from colonies of M. raptor. In the qPCR results, the average Ct value was 11.23, which is orders of magnitude higher than uninfected controls (Ct value = 38; Figure 2B). Therefore, the carefully designed extensive heat treatment approach failed to eradicate Nosema, and the infection came back and reached high titer. The vertical transmission we showed may play a role in the rapid reoccurrence of Nosema because some spores could escape the treatment and be transmitted in the eggs. To combat this highly infectious parasite, molecular approaches, such as RNA interference or Nosema genome manipulation, need to be considered. Our high-quality genome assembly and annotation serve as a first step to providing the necessary genome toolkit for these approaches, and for a better understanding mechanisms and etiology of Nosemosis.
Three Muscidifurax species, M. zaraptor, M. raptor, and M. uniraptor, were used in this study. The source of M. zaraptor was from two independent colonies, and both of them were derived from the same USDA colony maintained by Dr. Chris Geden’s laboratory, which was originally collected in 2015 from dairy farms in Minnesota, Nebraska, and California. The USDA M. zaraptor colony is maintained in the Geden laboratory at the Center for Medical, Agricultural and Veterinary Entomology, USDA Agricultural Research Service (USDA-ARS, Gainesville, FL, United States). An attempt was made to cure the colony for Nosema infection in 2019 using a heat shock treatment approach (Geden et al., 2019). The M. zaraptor colony maintained in the Wang laboratory (AUB colony) at Auburn University College of Veterinary Medicine (Auburn, AL, United States) was obtained from the University of Rochester in 2019, which was derived from the USDA colony infected with N. muscidifuracis. The M. uniraptor colony at AUB was also obtained from the University of Rochester in 2019, and it is Nosema-free. The M. raptor colony maintained in the Wang laboratory at AUB was derived from a Nosema-cured colony treated in 2017 in the Geden laboratory (see Materials and methods). All AUB colonies were maintained on commercial flesh fly (Sarcophaga bullata) pupae (Ward’s Science, Rochester, NY, United States) at a constant temperature of 25°C and 24 h constant light in the Wang laboratory. The USDA colonies were maintained on housefly (Musca domestica) pupae in the Geden laboratory (Gainesville, FL).
High molecular weight (HMW) genomic DNA (gDNA) was extracted from M. zaraptor whole-body samples collected 24 h after eclosion from the AUB colony using Genomic-tip 20/G kit (Qiagen, MD, United States). The DNA concentration was measured on a Qubit 3.0 Fluorometer instrument (Thermo Fisher Scientific, MD, United States). The gDNA quality and the size distribution were assessed on an Agilent TapeStation 4200 machine (Agilent Technologies, CA, USA) with the genomics screentapes. A total of 10 μg high-quality M. zaraptor HMW gDNA was sheared into 20 Kb fragments. After end-repair and ligating the specific adapter oligos, the DNA fragments were annealed with sequencing primer v2 and Sequel II DNA Polymerase, bound to the SMRTbell templates, and the library was prepared using the SMRTbell Template Prep kit v2 with the CCS HiFi Library construction protocol (Pacific Biosciences, CA, United States) at the HudsonAlpha Genome Sequencing Center (HGSC, Huntsville, AL, United States). The concentration and the size distribution for the prepared library were determined on LabChip GX Touch HT (PerkinElmer, MA, United States), and sequenced on a PacBio Sequel II System at HGSC (Supplementary Table S1).
HMW gDNA from AUB M. zaraptor was diluted to ~0.8 ng/uL with EB buffer through a series of dilutions, with concentrations determined by Qubit 3.0 Fluorometer (Thermo Fisher Scientific, United States). The Chromium Genome Reagent Kit v2 (10× Genomics Inc., CA, United States) was used for linked-read library preparation, according to the manufacturer’s instructions. The genome chip was loaded with diluted denatured gDNA, sample master mix, and gel beads following the protocol. Gel Bead-In-EMulsions (GEMs) were generated using a 10× Chromium Controller. After the incubation and cleanup of the obtained GEMs, Chromium i7 Sample Index was ligated and served as the library barcode to provide linked information. The size distribution of the prepared library was assessed using Agilent TapeStation 4200 (Agilent Technologies, CA, United States), final library quantity was checked with Qubit 3.0 Fluorometer (Thermo Fisher Scientific, United States). After quality control, the 10× Genomic library was sequenced on an Illumina NovaSeq 6000 machine.
De novo genome assembly for the M. zaraptor genome was performed with 23.7 Gb PacBio HiFi reads (377.2 Gb raw reads) using dedicated long-read assemblers hifiasm v0.13 (Cheng et al., 2021). The 10× Genomics reads were aligned to the assembled contigs using the LongRanger pipeline v2.1.6 (McKenna et al., 2010). The identity of M. zaraptor contigs was determined by coverage depth (54.5X Illumina read depth) and homology to closely-related M. raptorellus with a high-quality reference genome available (Xiong et al., 2021). Contigs from six microbial species were also detected in the hifiasm assembly, including five bacterial species and N. muscidifuracis. A total of 30 N. muscidifuracis PacBio HiFi contigs were identified based on coverage depth, GC content, and CpG percentages (Wang et al., 2019). De novo assembly of 10× Genomics data was performed using Supernova v2.1.1 with default parameters (Weisenfeld et al., 2017). Overlap of HiFi contigs and 10× scaffolds were detected by quickmerge v0.3.0 (Chakraborty et al., 2016). The 30 N. muscidifuracis contigs were merged into 28 contigs based on manual inspection of the contig overlap.
The final genome completeness of N. muscidifuracis was assessed by BUSCO (Benchmarking Universal Single-Copy Orthologs) v5.3.2 (Seppey et al., 2019). The BUSCO scores were computed using microsporidia_odb10 with a total of 600 orthologs. The BUSCO scores were also computed for an outgroup microsporidian species, Encephalitozoon cuniculi (Katinka et al., 2001; Peyretaillade et al., 2009), as well as eight other Nosema species/strains, including N. ceranae BRL 01 (Cornman et al., 2009), N. ceranae PA08 (Pelin et al., 2015), N. ceranae BRL (Huang et al., 2021), N. apis BRL 01 (Chen et al., 2013), N. bombycis CQ1 (Pan et al., 2013), N. granulosis Ou3-Ou53 (Cormier et al., 2021), N. antheraeae YY (Li et al., 2017), and Nosema sp. YNPr (Xu et al., 2016; Table 1).
Illumina short-reads aligned to N. muscidifuracis assembly were utilized to estimate the genome size. Low-quality bases and adapter sequences were trimmed using Trimmomatic version 0.39 (Bolger et al., 2014), with the parameters “ILLUMINACLIP:adapter:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:60.” High-quality trimmed reads were used for multiple k-mer counting using Jellyfish version 2.3.0 (Amoroso, 1968) with parameters count “-m 25 -s 20G -t 48.” The genome size and heterozygosity were estimated using GenomeScope (Vurture et al., 2017).
Before gene prediction, repeat annotation was performed to identify the repetitive elements in N. muscidifuracis genome. We first constructed a de novo N. muscidifuracis repeat database using RepeatModeler v2.0.1 (Flynn et al., 2020), which provided a list of repeat family sequences. The repeat-identifying was implemented by three complementary computational programs, RECON v1.0.8 (Bao and Eddy, 2002), RepeatScout v1.0.5 (Price et al., 2005), and Tandem Repeats Finder (TRF; Benson, 1999). Based on the transposon element library, the homologous repeats and low-complexity DNA sequences were masked using RepeatMasker v4.1.1 (Tarailo-Graovac and Chen, 2009) with RMBlast v2.10.0 sequence search engine (Supplementary Table S3).1
Noncoding RNAs (ncRNAs) were predicted using the Infernal software version 1.1.2 (Nawrocki and Eddy, 2013) based on the multiple sequence alignments using the covariance models in the Rfam database (Nawrocki et al., 2014). Before prediction, the esl-seqstat program was used to determine the total database size for the N. muscidifuracis genome. The ncRNAs in the N. muscidifuracis genome were predicted and annotated using the cmscan program in Infernal software (Nawrocki and Eddy, 2013) with RNA families in the Rfam (Nawrocki et al., 2014) database (Supplementary Table S4).
Colonies of M. raptor were established in 2015 from parasitoids collected from dairy farms in Florida, Minnesota, Nebraska, and California, and samples of parasitoids were examined visually for Nosema infection by crushing wasps in a drop of sterile water on a microscope slide and examining at 400X for the presence of spores (Zchori-Fein et al., 1992). None of the specimens examined at the time showed patent infections. After 1 year in colony, 100% of examined parasitoids had patent infections, indicating that low-level infections had escaped detection at the time of colony founding. In 2017, colonies were heat-treated by exposing multiple cohorts of newly parasitized house fly pupae to 50°C for 45 min and holding them for adult emergence (Boohene et al., 2003). Emerged adults were examined visually for infection as before, and progeny from cohorts that were Nosema-free based on visual inspections were used to start new colonies. Examination of these colonies after three additional generations indicated a resurgence of infection in three out the four strains. A final attempt was made to eliminate Nosema disease from the colonies by first subjecting three successive generations of parasitoids from the four strains to heat treatment as before and holding them for adult emergence. This procedure was conducted with three separate lineages of each of the four strains. Parasitized pupae were then held in individual gelatin capsules for emergence. Pairs of parasitoids (n = 100 pairs for each strain) were then held with 70 house fly pupae/pair for 3 days, then given another set of 70 for three more days. Each female was examined individually for the presence of spores; it was expected that 6 days would be sufficient time for infections to be evident by visual inspection. Progeny of any females with patent infections were discarded, and the parasitized pupae from females without patent infections were pooled to start a new colony.
To determine the levels of Nosema infection in the Nosema-cured M. raptor AUB colony, Nosema-infected M. zaraptor AUB colony and Nosema-cured M. zaraptor USDA colony, we extracted genomic DNA from 24-h adult male and female samples with three replicates per sex using AllPrep DNA/RNA Mini Kit (Qiagen, MD). Three primer sets were designed to target the different regions of the 18S rDNA gene in N. Muscidifuracis using Oligo 7 primer analysis software (Molecular Biology Insights Inc., Cascade, CO, United States; Supplementary Table S5). Primers were synthesized at Eurofins Genomics LLC (Louisville, KY, United States). All primer sets were evaluated by PCR using the M. zaraptor DNA template, with Nosema-free DNA as a negative control, followed by electrophoresis on 2% agarose gel (Supplementary Figure S3). The PCR experiments were performed with Q5 High-Fidelity 2x Master Mix on an Eppendorf Mastercycler Pro PCR machine (Eppendorf North America, Enfield, CT, United States). The thermal cycling protocol for the primers was as follows: initial denaturation step at 98°C for 30 s, followed by 30 cycles of initial denaturation at 98°C for 10 s, annealing at 55°C for 30 s, and extension at 72°C for 15 s. The qPCR reaction was conducted in a 20 μL system using Luna® Universal qPCR Master Mix (New England BioLabs, Ipswich, MA, United States). Each reaction contained 10 μL of Luna Universal qPCR Master Mix, 8 μL of nuclease-free water, 0.5 μL of forward primer and 0.5 μL reverse primer (10 μmol/L), and 1 μL of DNA template. The Bio-Rad C1000 Touch Thermal Cycler with CFX96 Real-Time PCR Detection Systems (Bio-Rad Laboratories, Hercules, CA, United States) was used to conduct qPCR experiments with the SYBR scan mode using the Nosema 18S NP2 primer set (Supplementary Table S5). The relative quantification method was applied to determine the Nosema abundance in M. raptor AUB colony and M. zaraptor from both AUB and USDA colonies. The thermocycling conditions for the qPCR assays were 95°C for 60 s, followed by 40 cycles at 95°C for 15 s and 50°C for 30 s.
Adult male and female M. zaraptor were collected 24 h after eclosion from both the USDA colony and AUB colony in the Wang laboratory. Total RNA was extracted from the adult whole-body samples in three biological replicates for each sex. RNA extractions were performed with AllPrep DNA/RNA Mini Kit (Qiagen, MD, United States) following the manufacturer’s protocol. The RNA yield was quantified using a Qubit 3.0 Fluorometer instrument (Thermo Fisher Scientific, MD, United States), followed by a quality check on an Agilent TapeStation 4,200 Bioanalyzer (Agilent Technologies, CA, United States). For RNA-seq library preparation, 1 μg of total RNA was used as input for all samples. To remove the abundant rRNA (ribosomal RNA) in the sample, an rRNA removal protocol was performed using the NEBNext rRNA Depletion Kit (New England Biolabs, MA, United States). The remaining mRNA was used for RNA-seq library construction using NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs, MA, United States) with the manufacturer-provided protocol. After quality control, the RNA-seq library was sequenced on an Illumina NovaSeq6000 platform (Supplementary Table S6).
On average, 128 million 150-bp reads were generated for each of the 12 M. zaraptor RNA-seq libraries. The raw RNA-seq data was checked for sequencing quality using FastQC (Andrews et al., 2010). Adapter sequences and low-quality bases in the paired-end RNA-seq reads were trimmed with Trimmomatic v0.39 (Bolger et al., 2014). A total of 6.2 million non-rDNA reads mapped uniquely to the N. muscidifuracis were used for de novo transcriptome assembly by Trinity v2.4.0 (Haas et al., 2013). Protein-coding genes were predicted and annotated in the N. muscidifuracis genome assembly using Fungal Genome Annotation Pipeline (FunGAP; Min et al., 2017), and filtered RNA-seq reads as input. FunGAP masked the repeats in the genome and assembled the RNA-seq reads. Augustus (Stanke et al., 2006) was used for gene model prediction with species parameter “--augustus_species encephalitozoon_cuniculi_GB,” which is the closest to Nosema. The microsporidian protein database used by FunGAP was downloaded by “download_sister_orgs.py” script in FunGAP.
To compare the genomes of N. muscidifuracis and its closely-related species N. ceranae, MCscanX (Wang et al., 2012) was used to perform synteny analysis and identify syntenic blocks between these genomes based on core orthologous gene sets identified using BlastP with default settings (E-value ≤ 1e−5; minimum number of genes in a syntenic block ≥ 5). The genome and annotation files of N. ceranae (BRL strain) were downloaded at NCBI Assembly with the accession number GCA_004919615.1. The genomic circle of collinearity was visualized in Circos (Krzywinski et al., 2009). To check the location of duplicated genes in N. muscidifuracis, we detected and plotted the collinear blocks within the N. muscidifuracis genome using MCscanX (Wang et al., 2012). The duplicated genes were aligned using mafft software (version 7.475; Katoh and Standley, 2013). The identity of the two sequences for each gene pair was computed by the Needle program with the default parameters (Gap penalty = 10, Extend penalty = 0.5) using the Needleman-Wunsch algorithm (Needleman and Wunsch, 1970). The numbers of synonymous (dS) and non-synonymous (dN) substitutions between two sequences were calculated using the alignments by KaKs_Calculator software (version 2.0) with γ-YN method (Yang and Nielsen, 2000; Wang D. et al., 2009; Wang D.-P. et al., 2009; Wang et al., 2010).
We selected one single-copy gene bim1, mfs1 with two homologous, and tef1 with three homologous copies in N. muscidifuracis genome for qPCR validation. The Oligo 7 primer analysis software (Molecular Biology Insights Inc., Cascade, CO, USA) was used to design the primer sets targeting the three selected protein-coding genes. The primers used for bim1 are 5′-GTAGAAGAGAATTGCTTGAATG-3′ and 5′-ACTCATACTCTGATGAAGGATTT-3′, the primers used for mfs1 are 5′-TTTAGCCACAAAATTATGTCC-3′ and 5′-ATGTTAAATACTTGTGCTCT-3′, and the primers used for tef1 are 5′-GCTGCTGAAAATAACAAGTCT-3′ and 5′-GCTGGTACAATAACTACACCT-3′. All primers were synthesized by Eurofins Genomics LLC (Louisville, KY, United States). Before the qPCR experiments, we checked the size of PCR products and the amplification efficiency using 2% agarose gel electrophoresis. The qPCR experiments were performed using Luna® Universal qPCR Master Mix (New England BioLabs, Ipswich, MA, United States) on a Bio-Rad C1000 Touch Thermal Cycler with CFX96 Real-Time PCR Detection Systems (Bio-Rad Laboratories, Hercules, CA, United States). The 20 μL reaction system consisted of 10 μL of Luna Universal qPCR Master Mix, 0.5 μL of forward primer (10 μmol/L), 0.5 μL of reverse primer (10 μmol/L), 8 μL of nuclease-free water, and 1 μL DNA template (M. zaraptor DNA samples extracted from AUB colony). The qPCR reaction conditions were 95°C for 60 s, followed by 40 cycles at 95°C for 15 s and 50°C for 30 s.
The ovaries of infected M. zaraptor and M. raptor were compared with the closely-related Nosema-free species M. uniraptor. Ovaries were fixed for 15 min in 10% Formaldehyde and 0.2% Tween in Phosphate Buffered Saline (PBS-T). Actin staining (Rhodamine Phalloidin, Invitrogen) was performed overnight or longer at 4°C in PBS-T. Ovaries were then washed 3X in PBS-T and moved to Vectashield with DAPI (Vector Labs) for at least 16 h before mounting. Microscopy was performed using a Leica SP5 point scanning confocal (63x/1.4 HCX PL Apo CS oil lens). Images were collected with LAS AF. Minor processing (Gaussian blur) was performed using FIJI.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, JAIOKI000000000.
XW and JW contributed to the conception and design of the study. XX and CG performed the insect rearing and sample collection. CG and RW performed the heat treatment experiments. XX performed the DNA and RNA sequencing experiments. XX and XW performed the genomic data analysis and wrote the first draft of the manuscript. DB performed cytogenetic experiments and analysis. XW, CG, DB, and JW provided samples, resources, and analysis tools. JW, CG, RW, and DB wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by the Alabama Agricultural Experiment Station and the Hatch program (ALA05-2-18041) of the National Institute of Food and Agriculture, U.S. Department of Agriculture. XW was supported by the National Science Foundation award (1928770) and a laboratory start-up fund from Auburn University College Veterinary Medicine. XX was supported by the Auburn University Presidential Graduate Research Fellowship and the College of Veterinary Medicine Dean’s Fellowship. JW thanks for support from NSF 1950078.
We thank the HudsonAlpha Genome Sequencing Center for assistance with the PacBio sequencing. We also thank Charles Vossbrinck for providing preliminary Nosema muscidifuracis 18S rDNA sequence information for the initial confirmation of Nosema species’ identity. Justin Fay was thanked for discussions on ploidy variation in fungal species. We acknowledge the Auburn University Easley Cluster for support of this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1152586/full#supplementary-material
Amoroso, E. (1968). The evolution of viviparity. Proc. R. Soc. Med. 61, 1188–1200. doi: 10.1177/003591576806111P202
Amses, K. R., Simmons, D. R., Longcore, J. E., Mondo, S. J., Seto, K., Jerônimo, G. H., et al. (2022). Diploid-dominant life cycles characterize the early evolution of fungi. Proc. Natl. Acad. Sci. U. S. A. 119:e2116841119. doi: 10.1073/pnas.2116841119
Anderson, D. L., and Giacon, H. (1992). Reduced pollen collection by honey bee (hymenoptera: Apidae) colonies infected with Nosema apis and sacbrood virus. J. Econ. Entomol. 85, 47–51. doi: 10.1093/jee/85.1.47
Andrews, S., Krueger, F., Segonds-Pichon, A., Biggins, L., Krueger, C., and Wingett, S. (2010) FastQC. Available online at http:// www.bioinformatics.babraham.ac.uk/projects/fastqc
Antúnez, K., Martín-Hernández, R., Prieto, L., Meana, A., Zunino, P., and Higes, M. (2009). Immune suppression in the honey bee (Apis mellifera) following infection by Nosema ceranae (Microsporidia). Environ. Microbiol. 11, 2284–2290. doi: 10.1111/j.1462-2920.2009.01953.x
Bao, Z., and Eddy, S. R. (2002). Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276. doi: 10.1101/gr.88502
Becnel, J. J. (1994). Life cycles and host-parasite relationships of Microsporidia in culicine mosquitoes. Folia Parasitol. 41, 91–96.
Becnel, J. J., and Andreadis, T. G. (1999). “Microsporidia in insects” in The microsporidia and microsporidiosis, eds. M. Wittner and L. M. Weiss (Hoboken, New Jersey: Wiley), 447–501. doi: 10.1002/9781118395264.ch21
Becnel, J. J., and Geden, C. (1994). Description of a new species of microsporidia from Muscidifurax raptor (hymenoptera: Pteromalidae), a pupal parasitoid of muscoid flies. J. Eukaryot. Microbiol. 41, 236–243. doi: 10.1111/j.1550-7408.1994.tb01504.x
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boohene, C., Geden, C., and Becnel, J. (2003). Evaluation of remediation methods for Nosema disease in Muscidifurax raptor (hymenoptera: Pteromalidae). Environ. Entomol. 32, 1146–1153. doi: 10.1603/0046-225X-32.5.1146
Botías, C., Martín-Hernández, R., Barrios, L., Meana, A., and Higes, M. (2013). Nosema spp. infection and its negative effects on honey bees (Apis mellifera iberiensis) at the colony level. Vet. Res. 44, 25–15. doi: 10.1186/1297-9716-44-25
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D., and Emerson, J. (2016). Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44:e147. doi: 10.1093/nar/gkw654
Chen, Y. P., Evans, J. D., Murphy, C., Gutell, R., Zuker, M., Gundensen-Rindal, D., et al. (2009). Morphological, molecular, and phylogenetic characterization of Nosema ceranae, a microsporidian parasite isolated from the European honey bee, Apis mellifera. J. Eukaryot. Microbiol. 56, 142–147. doi: 10.1111/j.1550-7408.2008.00374.x
Chen, Y., Evans, J. D., Zhou, L., Boncristiani, H., Kimura, K., Xiao, T., et al. (2009). Asymmetrical coexistence of Nosema ceranae and Nosema apis in honey bees. J. Invertebr. Pathol. 101, 204–209. doi: 10.1016/j.jip.2009.05.012
Chen, Y. P., Pettis, J. S., Zhao, Y., Liu, X., Tallon, L. J., Sadzewicz, L. D., et al. (2013). Genome sequencing and comparative genomics of honey bee microsporidia, Nosema apis reveal novel insights into host-parasite interactions. BMC Genomics 14, 1–16. doi: 10.1186/1471-2164-14-451
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Cormier, A., Chebbi, M. A., Giraud, I., Wattier, R., Teixeira, M., Gilbert, C., et al. (2021). Comparative genomics of strictly vertically transmitted, feminizing microsporidia endosymbionts of amphipod crustaceans. Genome Biol. Evol. 13:evaa245. doi: 10.1093/gbe/evaa245
Cornman, R. S., Chen, Y. P., Schatz, M. C., Street, C., Zhao, Y., Desany, B., et al. (2009). Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathog. 5:e1000466. doi: 10.1371/journal.ppat.1000466
Costa, C., Tanner, G., Lodesani, M., Maistrello, L., and Neumann, P. (2011). Negative correlation between Nosema ceranae spore loads and deformed wing virus infection levels in adult honey bee workers. J. Invertebr. Pathol. 108, 224–225. doi: 10.1016/j.jip.2011.08.012
Cox-Foster, D. L., Conlan, S., Holmes, E. C., Palacios, G., Evans, J. D., Moran, N. A., et al. (2007). A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318, 283–287. doi: 10.1126/science.1146498
Didier, E., Snowden, K., and Shadduck, J. (1998). Biology of microsporidian species infecting mammals. Adv. Parasitol. 40, 283–320. doi: 10.1016/S0065-308X(08)60125-6
Dunn, A. M., Terry, R. S., and Smith, J. E. (2001). Transovarial transmission in the microsporidia. Adv. Parasitol. 48, 57–100. doi: 10.1016/S0065-308X(01)48005-5
Dussaubat, C., Brunet, J.-L., Higes, M., Colbourne, J. K., Lopez, J., Choi, J.-H., et al. (2012). Gut pathology and responses to the microsporidium Nosema ceranae in the honey bee Apis mellifera. PLoS One 7:e37017. doi: 10.1371/journal.pone.0037017
Farrar, C. (2014). Nosema losses in package bees as related to queen supersedure and honey yields. J. Econ. Entomol. 40, 333–338. doi: 10.1093/jee/40.3.333
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Forsgren, E., and Fries, I. (2010). Comparative virulence of Nosema ceranae and Nosema apis in individual European honey bees. Vet. Parasitol. 170, 212–217. doi: 10.1016/j.vetpar.2010.02.010
Fries, I. (1988). Comb replacement and Nosema disease (Nosema apis Z.) in honey bee colonies. Apidologie 19, 343–354. doi: 10.1051/apido:19880402
Fries, I., Ekbohm, G., and Villumstad, E. (1984). Nosema apis, sampling techniques and honey yield. J. Apic. Res. 23, 102–105. doi: 10.1080/00218839.1984.11100617
Fries, I., Feng, F., da Silva, A., Slemenda, S. B., and Pieniazek, N. J. (1996). Nosema ceranae n. sp.(Microspora, Nosematidae), morphological and molecular characterization of a microsporidian parasite of the Asian honey bee Apis cerana (hymenoptera, Apidae). Eur. J. Protistol. 32, 356–365. doi: 10.1016/S0932-4739(96)80059-9
Geden, C. J., Biale, H., Chiel, E., and Johnson, D. M. (2019). Effect of fluctuating high temperatures on house flies (Diptera: Muscidae) and their principal parasitoids (Muscidifurax spp. and Spalangia spp. [hymenoptera: Pteromalidae]) from the United States. J. Med. Entomol. 56, 1650–1660. doi: 10.1093/jme/tjz080
Geden, C. J., and Hogsette, J. A. (2006). Suppression of house flies (Diptera: Muscidae) in Florida poultry houses by sustained releases of Muscidifurax raptorellus and Spalangia cameroni (hymenoptera: Pteromalidae). Environ. Entomol. 35, 75–82. doi: 10.1603/0046-225X-35.1.75
Geden, C. J., Long, S. J., Rutz, D. A., and Becnel, J. J. (1995). Nosema disease of the parasitoid Muscidifurax raptor (hymenoptera: Pteromalidae): prevalence, patterns of transmission, management, and impact. Biol. Control 5, 607–614. doi: 10.1006/bcon.1995.1072
Geden, C. J., and Moon, R. D. (2009). Host ranges of gregarious muscoid fly parasitoids: Muscidifurax raptorellus (hymenoptera: Pteromalidae), Tachinaephagus zealandicus (hymenoptera: Encyrtidae), and Trichopria nigra (hymenoptera: Diapriidae). Environ. Entomol. 38, 700–707. doi: 10.1603/022.038.0321
Girault, A., and Sanders, G. E. (1910). The chalcidoid parasites of the common house or typhoid fly (Musca domestica Linn.) and its allies. Psyche 17, 9–28. doi: 10.1155/1910/17925
Gómez-Moracho, T., Durand, T., Pasquaretta, C., Heeb, P., and Lihoreau, M. (2021). Artificial diets modulate infection rates by nosema ceranae in bumblebees. Microorganisms 9:158. doi: 10.3390/microorganisms9010158
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Higes, M., Martín-Hernández, R., Garrido-Bailón, E., González-Porto, A. V., García-Palencia, P., Meana, A., et al. (2009). Honeybee colony collapse due to Nosema ceranae in professional apiaries. Environ. Microbiol. Rep. 1, 110–113. doi: 10.1111/j.1758-2229.2009.00014.x
Huang, Q., Wu, Z. H., Li, W. F., Guo, R., Xu, J. S., Dang, X. Q., et al. (2021). Genome and evolutionary analysis of Nosema ceranae: a microsporidian parasite of honey bees. Front. Microbiol. 12:1303. doi: 10.3389/fmicb.2021.645353
Katinka, M. D., Duprat, S., Cornillot, E., Méténier, G., Thomarat, F., Prensier, G., et al. (2001). Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414, 450–453. doi: 10.1038/35106579
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kogan, M., and Legner, E. (1970). A biosystematic revision of the genus Muscidifurax (hymenoptera: Pteromalidae) with descriptions of four new species. Can Entomol 102, 1268–1290. doi: 10.4039/Ent1021268-10
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Li, T., Pan, G.-Q., Vossbrinck, C. R., Xu, J.-S., Li, C.-F., Chen, J., et al. (2017). SilkPathDB: a comprehensive resource for the study of silkworm pathogens. Database bax0001. doi: 10.1093/database/bax0001
Machtinger, E. T., and Geden, C. J. (2018). “Biological control with parasitoids” in Pests and vector-borne diseases in the livestock industry, eds. C. Garros, J. Bouyer, W. Takken, and R. C. Smallegange (Wageningen, The Netherlands: Wageningen Academic Publishers), 299–335.
Malone, L., Giacon, H., and Newton, M. (1995). Comparison of the responses of some New Zealand and Australian honey bees (Apis mellifera L) to Nosema apis Z. Apidologie 26, 495–502. doi: 10.1051/apido:19950606
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Min, B., Grigoriev, I. V., and Choi, I.-G. (2017). FunGAP: fungal genome annotation pipeline using evidence-based gene model evaluation. Bioinformatics 33, 2936–2937. doi: 10.1093/bioinformatics/btx353
Nakabachi, A., and Moran, N. A. (2022). Extreme polyploidy of Carsonella, an organelle-like bacterium with a drastically reduced genome. Microbiol Spectr 10:e0035022. doi: 10.1128/spectrum.00350-22
Nawrocki, E. P., Burge, S. W., Bateman, A., Daub, J., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137. doi: 10.1093/nar/gku1063
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Needleman, S. B., and Wunsch, C. D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453. doi: 10.1016/0022-2836(70)90057-4
Newton, I. L. G., Clark, M. E., Kent, B. N., Bordenstein, S. R., Qu, J., Richards, S., et al. (2016). Comparative genomics of two closely related Wolbachia with different reproductive effects on hosts. Genome Biol. Evol. 8, 1526–1542. doi: 10.1093/gbe/evw096
Oyarzun, M. P., Quiroz, A., and Birkett, M. A. (2008). Insecticide resistance in the horn fly: alternative control strategies. Med. Vet. Entomol. 22, 188–202. doi: 10.1111/j.1365-2915.2008.00733.x
Pan, G., Xu, J., Li, T., Xia, Q., Liu, S.-L., Zhang, G., et al. (2013). Comparative genomics of parasitic silkworm microsporidia reveal an association between genome expansion and host adaptation. BMC Genomics 14, 1–14. doi: 10.1186/1471-2164-14-186
Pei, B., Wang, C., Yu, B., Xia, D., Li, T., and Zhou, Z. (2021). The first report on the Transovarial transmission of Microsporidian Nosema bombycis in Lepidopteran crop pests Spodoptera litura and Helicoverpa armigera. Microorganisms 9:9. doi: 10.3390/microorganisms9071442
Pelin, A., Selman, M., Aris-Brosou, S., Farinelli, L., and Corradi, N. (2015). Genome analyses suggest the presence of polyploidy and recent human-driven expansions in eight global populations of the honeybee pathogen Nosema ceranae. Environ. Microbiol. 17, 4443–4458. doi: 10.1111/1462-2920.12883
Petersen, J., and Currey, D. (1996). Reproduction and development of Muscidifurax raptorellus (hymenoptera: Pteromalidae), a parasite of filth flies. J Agricult Entomol. 13, 99–107.
Peyretaillade, E., Gonçalves, O., Terrat, S., Dugat-Bony, E., Wincker, P., Cornman, R. S., et al. (2009). Identification of transcriptional signals in Encephalitozoon cuniculi widespread among Microsporidia phylum: support for accurate structural genome annotation. BMC Genomics 10, 1–13. doi: 10.1186/1471-2164-10-607
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Rinderer, T. E., and Sylvester, H. A. (1978). Variation in response to Nosema apis, longevity, and hoarding behavior in a free-mating population of the honey Bee1, 2. Ann. Entomol. Soc. Am. 71, 372–374. doi: 10.1093/aesa/71.3.372
Scott, J. G., Leichter, C. A., Rinkevihc, F. D., Harris, S. A., Su, C., Aberegg, L. C., et al. (2013). Insecticide resistance in house flies from the United States: resistance levels and frequency of pyrethroid resistance alleles. Pestic. Biochem. Physiol. 107, 377–384. doi: 10.1016/j.pestbp.2013.10.006
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14
Stanimirović, Z., Glavinić, U., Ristanić, M., Aleksić, N., Jovanović, N., Vejnović, B., et al. (2019). Looking for the causes of and solutions to the issue of honey bee colony losses. Acta Vet. Brno 69, 1–31. doi: 10.2478/acve-2019-0001
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Steinhaus, E. (2012). Insect pathology V1: An advanced treatise. (Amsterdam, the Netherlands: Elsevier).
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 4, 4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s25
Taylor, D. B., Moon, R. D., and Mark, D. R. (2012). Economic impact of stable flies (Diptera: Muscidae) on dairy and beef cattle production. J. Med. Entomol. 49, 198–209. doi: 10.1603/ME10050
Terry, R. S., Dunn, A. M., and Smith, J. E. (1997). Cellular distribution of a feminizing microsporidian parasite: a strategy for transovarial transmission. Parasitology 115, 157–163. doi: 10.1017/S0031182097001236
Valero, M., Richerd, S., Perrot, V., and Destombe, C. (1992). Evolution of alternation of haploid and diploid phases in life cycles. Trends Ecol. Evol. 7, 25–29. doi: 10.1016/0169-5347(92)90195-H
Van de Peer, Y., Mizrachi, E., and Marchal, K. (2017). The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424. doi: 10.1038/nrg.2017.26
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, D.-P., Wan, H.-L., Zhang, S., and Yu, J. (2009). γ-MYN: a new algorithm for estimating Ka and Ks with consideration of variable substitution rates. Biol. Direct 4, 1–18. doi: 10.1186/1745-6150-4-20
Wang, X., Xiong, X., Cao, W., Zhang, C., Werren, J. H., and Wang, X. (2019). Genome assembly of the A-group Wolbachia in Nasonia oneida using linked-reads technology. Genome Biol. Evol. 11, 3008–3013. doi: 10.1093/gbe/evz223
Wang, D., Zhang, S., He, F., Zhu, J., Hu, S., and Yu, J. (2009). How do variable substitution rates influence Ka and Ks calculations? Genomics Proteomics Bioinformatics 7, 116–127. doi: 10.1016/S1672-0229(08)60040-6
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M., and Jaffe, D. B. (2017). Direct determination of diploid genome sequences. Genome Res. 27, 757–767. doi: 10.1101/gr.214874.116
Xiong, X., Kelkar, Y. D., Geden, C. J., Zhang, C., Wang, Y., Jongepier, E., et al. (2021). Long-read assembly and annotation of the parasitoid wasp Muscidifurax raptorellus, a biological control agent for filth flies. Front. Genet. 12:748135. doi: 10.3389/fgene.2021.748135
Xu, J., He, Q., Ma, Z., Li, T., Zhang, X., Debrunner-Vossbrinck, B. A., et al. (2016). The genome of Nosema sp. isolate YNPr: a comparative analysis of genome evolution within the Nosema/Vairimorpha clade. PLoS One 11:e0162336. doi: 10.1371/journal.pone.0162336
Yang, Z., and Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Zchori-Fein, E., Geden, C. J., and Rutz, D. A. (1992). Microsporidioses of Muscidifurax raptor (hymenoptera: Pteromalidae) and other pteromalid parasitoids of muscoid flies. J. Invertebr. Pathol. 60, 292–298. doi: 10.1016/0022-2011(92)90011-R
Keywords: fungal genomics, hymenopteran fungal pathogens, parasitoid wasps, biological control, Nosema disease, microsporidiosis
Citation: Xiong X, Geden CJ, Bergstralh DT, White RL, Werren JH and Wang X (2023) New insights into the genome and transmission of the microsporidian pathogen Nosema muscidifuracis. Front. Microbiol. 14:1152586. doi: 10.3389/fmicb.2023.1152586
Edited by:
Hamed Mirjalali, Shahid Beheshti University of Medical Sciences, IranReviewed by:
Tian Li, Southwest University, ChinaCopyright © 2023 Xiong, Geden, Bergstralh, White, Werren and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Wang, eHp3MDA3MEBhdWJ1cm4uZWR1
†ORCID: John H. Werren https://orcid.org/0000-0001-9353-2070
Xu Wang https://orcid.org/0000-0002-7594-5004
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.