 Qi Ning
Qi Ning Lin Chen
Lin Chen Fang Li
Fang Li Guixiang Zhou
Guixiang Zhou Congzhi Zhang
Congzhi Zhang Donghao Ma1
Donghao Ma1 Jiabao Zhang
Jiabao Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 March 2023
Sec. Terrestrial Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1141436
This article is part of the Research Topic Insights in Terrestrial Microbiology: 2022 View all 7 articles
Stable soil organic carbon (SOC) formation in coastal saline soils is important to improve arable land quality and mitigate greenhouse gas emissions. However, how microbial life-history strategies and metabolic traits regulate SOC turnover in coastal saline soils remains unknown. Here, we investigated the effects of microbial life history strategy tradeoffs on microbial carbon use efficiency (CUE) and microbial-derived SOC formation using metagenomic sequencing technology in different salinity soils. The results showed that high-salinity is detrimental to microbial CUE and microbial-derived SOC formation. Moreover, the regulation of nutrients stoichiometry could not mitigate adverse effects of salt stress on microbial CUE, which indicated that microbial-derived SOC formation is independent of stoichiometry in high-salinity soil. Low-salinity soil is dominated by a high growth yield (Y) strategy, such as higher microbial biomass carbon and metabolic traits which are related to amino acid metabolism, carbohydrate metabolism, and cell processes. However, high-salinity soil is dominated by stress tolerance (S) (e.g., higher metabolic functions of homologous recombination, base excision repair, biofilm formation, extracellular polysaccharide biosynthesis, and osmolytes production) and resource acquisition (A) strategies (e.g., higher alkaline phosphatase activity, transporters, and flagellar assembly). These trade-offs of strategies implied that resource reallocation took place. The high-salinity soil microbes diverted investments away from growth yield to microbial survival and resource capture, thereby decreasing biomass turnover efficiency and impeding microbial-derived SOC formation. Moreover, altering the stoichiometry in low-salinity soil caused more investment in the A-strategy, such as the production of more β-glucosidase and β-N-acetyl-glucosaminidase, and increasing bacterial chemotaxis, which thereby reduced microbial-derived SOC formation. Our research reveals that shift the microbial community from S- and A- strategies to the Y-strategy is important to increase the microbial CUE, and thus enhance SOC turnover in coastal saline soils.
Soil salinization seriously restricts the development of agricultural production. Of concern is that 25% of the total land is affected by salinity globally, and the saline-alkali soil area is growing at a rate of more than 1 million ha per year (Xia et al., 2019). The area of salinization soil reached 9.21 × 106 ha, which accounted for 6.62% of the arable soil in China (Yang, 2008). Coastal saline soil is one of the primary types of saline-alkali soils, which is primarily distributed in coastal areas. The Yellow River Delta is a typical coastal saline soil area in China, and soil salinization has expanded rapidly from the coast to inland in this region during the past 20 years (Zhao et al., 2020), which has caused high soil salinity, poor soil structure, and low soil fertility (Xia et al., 2019). Saline soil is an important terrestrial carbon (C) sink with a high capacity for absorbing CO2 (Xie et al., 2009), which indicates an enormous potential for carbon sequestration. Therefore, enhancing the carbon sequestration capacity of coastal saline soils is highly important for mitigating greenhouse gas emissions and improving soil fertility.
The enhancement of soil organic carbon (SOC) is an effective measure to reduce salinity, improve fertility, and maintain sustainability in saline soils (Dong et al., 2022). Recently, numerous studies have revealed that microbial growth and its turnover products are major components of mineral-associated SOC (MASOC), which is a key fraction of stable SOC pools (Kallenbach et al., 2016; Liang et al., 2017; Sokol and Bradford, 2019; Georgiou et al., 2022). Living microbial biomass comprises only a small fraction of the total SOC, the persistent SOC accumulation primarily occurs through the rapid turnover of microbes to produce microbial-derived materials (Liang et al., 2017). The contribution of microbial-derived carbon to the SOC was reported to comprise more than 50% of the stable SOC and even up to 80% in some soils (Simpson et al., 2007; Sokol and Bradford, 2019). The process of mediation of the transformation of SOC by microorganisms is seriously affected by soil salinity. Typically, the toxic ions and high osmotic pressure in saline soils inhibit microbial growth and activity, which subsequently reduces the microbial biomass and metabolites, and is ultimately detrimental to SOC accumulation (Rath and Rousk, 2015). In addition, salinity affects the metabolic function and genes of microorganisms that are related to carbohydrate metabolism by altering the microbial community diversity and composition (Yang et al., 2021), which consequently exacerbates the loss of SOC. Microorganisms drive the generation and accumulation of SOC by catabolism and anabolism (Liang et al., 2017), and microbial carbon use efficiency (CUE) can be used to predict the balance between catabolic and anabolic metabolism (Rath and Rousk, 2015; Mo et al., 2021).
Recent literature proposed that the distinct prevailing microbial life history strategies (Y-A-S) could affect the microbial CUE and SOC accumulation under stress conditions (Malik et al., 2020). Specifically, high growth yield (Y) strategy microorganisms facilitate SOC accumulation and stabilization by increasing the turnover of microbial biomass. Resource acquisition (A) strategy microorganisms invest more energy in producing extracellular enzymes to acquire nutrient substrates, which contribute to the decomposition and losses of SOC. Stress tolerance (S) strategists divert resources away from growth in response to stress and therefore, reduce the microbial CUE and SOC formation. Metagenomic sequencing showed advantages in analyzing the microbial taxonomic community, as well as its functions and genes (Yang et al., 2021), and can provide a deeper understanding of the mechanisms of microbial-mediated SOC formation under stress habitats. Previous researchers have revealed the metabolic efficiency, microbial traits, and their underlying consequences on soil C dynamics under abiotic stress, such as cold (Feng et al., 2021) and drought stress (Li C. N. et al., 2022; Malik and Bouskill, 2022). However, the effect of salt stress on how microorganisms regulate their CUE and further affect SOC formation through the alternation of microbial life history strategies is rarely studied.
In addition, microbial CUE is also affected by homeostatic stoichiometry (Dong et al., 2022). Microorganisms require not only C sources for energy when converting substrate into microbial biomass but also an input of balanced nutrients, such as nitrogen (N) and phosphorus (P), to maintain a homeostasis of elemental stoichiometry (Mo et al., 2021). It is estimated that the microbial biomass C:N:P molar ratio of 60:7:1 is optimal for microbial growth (Cleveland and Liptzin, 2007). To reveal the microbial-mediated mechanisms of SOC formation under salt stress and imbalanced stoichiometry from the perspective of microbial life history strategy, we hypothesized that the microorganisms in highly saline soil are dominated by S-strategists, and microorganisms in low-salinity soil are dominated by Y-strategists; while the adjustment of stoichiometric relationships would mitigate the effect of salt stress (high- or low-salinity) on microbial CUE, and thereby alleviate the negative effect on soil SOC formation. In this study, coastal saline soils with different levels of salinity in the Yellow River Delta region were selected to analyze the differences in microbial CUE and microbial-derived SOC formation, and metagenomic sequencing technology was used to reveal the microbial trait-based strategies that drive SOC formation, aimed to provide new insights for carbon sequestration in coastal saline soils.
Coast saline soils for this study were collected from the Yellow River Delta, which is located in the Kenli District, Dongying, Shandong Province, China (118°54′31″E, 37°39′58″N). This area has a continental monsoon climate, with an average annual temperature, precipitation, and evaporation of 12.3°C, 542 mm, and 1926 mm, respectively (Liu et al., 2020). The natural vegetation types in this area are halophytic plants, such as Suaeda salsa and Phragmites australis, and the farm field was planted with wheat (Triticum aestivum) for years. The soils are loamy clay that developed from the alluvial sediments and are classified as a Salic Fluvisol (Xia et al., 2019). We selected the typical Suaeda salsa community habitats and nearby farm fields as high-salinity (HS, soil salinity >8 g kg−1) and low-salinity (LS, soil salinity <2 g kg−1) soils, respectively. The two saline soils have four independent replicates. In specific, four plots of 30 m × 30 m were set in both Suaeda salsa community habitats and farm fields. Nine cores of bulk soils in each plot were sampled from the surface soil (0–15 cm) and mixed evenly into one soil sample. All soil samples without plant residues were air-dried and passed through a 2-mm mesh, and were used to determine background soil physicochemical properties. HS and LS soils are with 2,274 and 436 μS cm−1 of electrical conductivity (EC), and with 8.66 and 1.50 g kg−1 of total salt content, respectively. Other soil properties, such as the pH, C, N, P contents, C:N:P ratio, and particle composition, are shown in Table 1.
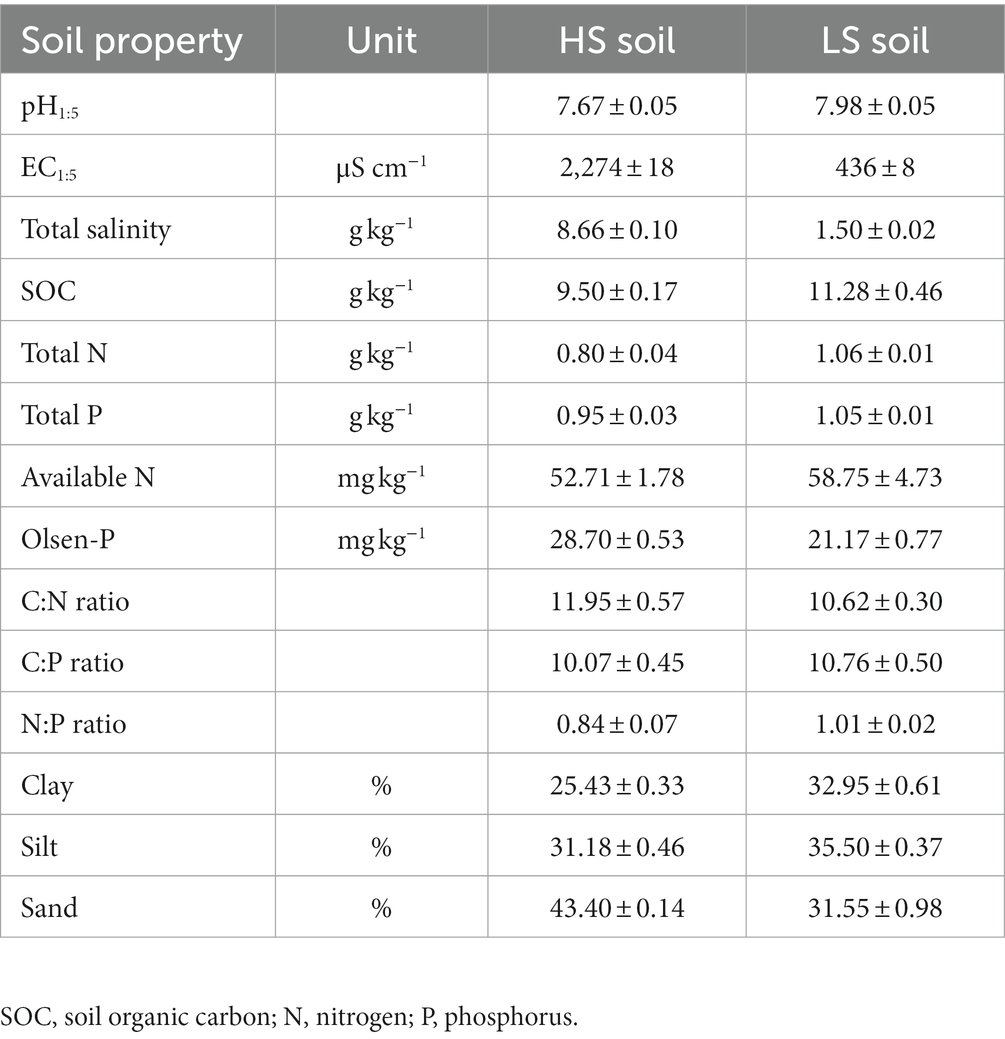
Table 1. Basic soil properties in high-salinity (HS) and low-salinity (LS) soil.
The experiment included four treatments: addition with 13C-glucose, addition with 13C-glucose and NP nutrients, addition with NP nutrients, and non-addition (without 13C-glucose or NP nutrients). Thus, there are 32 microcosms in total (2 saline soils × 4 treatments × 4 replicates), and half of microcosms without 13C-glucose added were used to quantify 13C fractions derived from glucose but microbial metagenomic sequencing analysis was not performed. To eliminate the sampling and sieving interferences and activate the soil microbes, the air-dried soils were pre-incubated for a week at 25°C at 30% of water-holding capacity (WHC) before the experiments were conducted. Each microcosm was established by placing 200 g (dry weight) of soil into a 1 L glass jar, with the addition of 0.5 mg C per g dry soil per week using 13C-glucose (10 atom% labelled) to simulate root exudations (Sokol and Bradford, 2019). N (0.068 mg g−1 soil) and P nutrients (0.022 mg g−1 soil) were inputted to regulate the intrinsic stoichiometry of the soil. 13C-glucose and inorganic nutrients were added as an aqueous solution. An equivalent amount of deionized water was added to the control soil (without glucose or nutrients), and all the microcosms were maintained with a water content at 55% WHC.
Each glass jar contained two 50 ml vials, one with 20 ml of 1.0 M NaOH to absorb the CO2, and the other with 10 ml of deionized water to maintain a moist atmosphere (Auwal et al., 2021). An additional four blank jars that contained only water and NaOH were established to account for the headspace CO2. All the microcosms were sealed and incubated at 25°C in the dark. The NaOH vials were exchanged at 1, 3, 5, and 7 days to analyze the CO2 concentration and its isotopes and the microbial CUE in a week. Approximately 50 g of soil was collected after 1 week. One portion was stored at 4°C to analyze the dissolved organic C (DOC), microbial biomass C (MBC) and extracellular enzymes, while another portion was stored at −80°C for a metagenomics sequencing analysis. The remaining soil was incubated for 8 weeks and collected to analyze the MASOC, DOC, MBC, and extracellular enzymes.
The amount of CO2 was determined by titrating the NaOH solution with 1.0 M of standardized HCl. The stable 13C isotope ratios of CO2-C were determined as previously described (Auwal et al., 2021; Chen et al., 2022). Briefly, 10 ml of 1.0 M NaOH was added with 10 ml of 1.5 M BaCl2 to precipitate the carbonates. The BaCO3 precipitate was filtered, washed several times, and oven-dried at 60°C.
The soil MBC was measured using the chloroform fumigation-K2SO4 extraction method and calculated with a correction factor of 0.45 (Vance et al., 1987). The DOC was extracted from the non-fumigated soil sample. The contents of MBC and DOC were measured using a total organic carbon (TOC) analyzer (Analytik Jena AG, Jena, Germany), and the analysis of δ13C (‰) values of MBC and DOC used a persulfate digestion method according to Fang et al. (2020).
The soil samples were separated by a solution of sodium hexametaphosphate, and the dispersed soil was passed through a 53 μm sieve and collected as the MASOC fraction. The MASOC fraction was oven-dried at 60°C, and the inorganic C was removed with 6.0 M of HCl before the elemental and isotope analyses were conducted (Sokol and Bradford, 2019). The δ13C (‰) values of CO2-C, MBC, DOC, and MASOC were measured using an elemental analyzer-isotope ratio mass spectrometer (Thermo Fisher Scientific, Waltham, MA, United States).
The activities of β-glucosidase (GC), N-acetyl-β-glucosaminidase (NAG), and alkaline phosphatase (AKP) were quantified by measuring the rate at which the substrates were degraded using p-nitrophenyl-β-D-glucoside, p-nitrophenyl-N-acetyl-β-D-glucosaminide and p-nitrophenyl phosphate, respectively (Geisseler and Horwath, 2009). The specific enzyme activities were calculated as enzyme activities divided by the amount of MBC.
The soil microbial DNA was extracted using a FastDNA Spin Kit (MP Biomedicals, Santa Ana, CA, United States). The DNA concentrations and quality were measured using a NanoDrop spectrophotometer and 1% agarose gel electrophoresis, respectively. Qualified DNA was randomly fragmented to <500 bp by sonication with an S220 Focused-ultrasonicator (Covaris LLC, Woburn, MA, United States) for library construction. Sequencing was performed on an Illumina HiSeq 2,500 platform (Illumina, Inc., San Diego, CA, United States) using a 2 × 150 paired-end (PE) configuration. Quality control was conducted in Trimmomatic to remove low quality reads (quality value <30 or the presence of undetermined bases) and short reads (length < 50 bp) (Bolger et al., 2014). Clean reads were then de novo assembled into contigs by MEGAHIT (K-mer parameters: k-min 35, k-max 95, and k-step 20) (Li et al., 2015). The contigs >500 bp in length were selected for subsequent analysis. The open reading frames (ORFs) for the assembled contigs were predicted using Prodigal (Hyatt et al., 2010). The CD-HIT version 4.6 was used to construct a non-redundant gene catalogue with 95% identity and 90% coverage (Fu et al., 2012), and the gene counts and abundances in each sample were evaluated using Bowtie2 (Langmead and Salzberg, 2012) and SAMtools (Li et al., 2009). The assembled unigenes were compared with the NR database of NCBI using DIAMOND (blastp, evalue ≤1e-5) (Buchfink et al., 2015), and the LCA algorithm was used for taxonomic annotation. The functions of predicted protein sequences were annotated against the KEGG database using GhostKOALA (Kanehisa et al., 2016).
The theoretical partitioning of microbial life history strategies was proposed by Malik et al. (2020), and they used microbial functions as traits and assigned Y-A-S strategies to dominant populations in the community (Malik and Bouskill, 2022). The Y-strategists were defined as microbes that maximize their growth yield by enhancing central metabolism (e.g., C, N, and P metabolism) and biosynthesis (Malik et al., 2020; Li C. N. et al., 2022). The A-strategists were defined as microbes that invested more in functional traits that are associated with the complex substrates degradation, extracellular enzymes production, cell motility, transporters and siderophores (Malik et al., 2020; Shao et al., 2021). The S-strategists were defined as microbes that invested more in microbial traits that related to biomolecular damage repair, maintenance of cellular integrity, and osmolyte production (Malik et al., 2020; Malik and Bouskill, 2022). Next, the top 20 most abundant taxa at genus level with significant differences (p < 0.05) using an independent t-test between high- and low-salinity soils, were classified based on the Y-A-S strategies framework. The top 30 abundant functional categories at KEGG level 3 that differed significantly (p < 0.05) between high- and low-salinity soils were classified as Y-A-S strategies. In addition, specific genes were allocated to the Y-A-S strategies as described above. The abundance of microbial genes assigned to the Y-A-S strategies was transformed by a Z-score and visualized in heatmaps by the “pheatmap” package in R (v.4.1.1).
The amounts of 13C-C pools (i.e., 13C-CO2, 13C-DOC, and 13C-MASOC) derived from glucose were estimated using the following equations (Arcand et al., 2017):
where F is the fraction (CO2-C, DOC, or MASOC) derived from the glucose, and δ13Csample and δ13Ccontrol are the corresponding δ13C (‰) value in the sample and control soil (without glucose), respectively. δ13Cglucose is the δ13C (‰) value of the 13C-labeled glucose. Cpools (μg g−1) is the amount of CO2-C, DOC, and MASOC in the soil samples amended with glucose, and 13C-Cpools is the amount of 13C-CO2, 13C-DOC, and 13C-MASOC derived from glucose.
The glucose-derived MBC (13C-MBC) and CUE were calculated as follows (Kallenbach et al., 2016; Fang et al., 2020):
where δ13MBC is the δ13C (‰) value of the MBC; δ13CFum and δ13CNFum are the δ13C (‰) values of fumigated and non-fumigated extracts, respectively, and CFum and CNFum represent the DOC concentrations (μg g−1) of the fumigated and non-fumigated extracts, respectively. δ13MBCsample and δ13MBCcontrol are the corresponding δ13C (‰) value in the sample and control soil (without glucose), respectively. 13C-MBC and 13C-CO2 are the MBC and CO2 derived from glucose, respectively.
A one-way analysis of variance (ANOVA) with the least significant difference (LSD) test was performed in SPSS 19.0 (IBM, Inc., Armonk, NY, United States). Bar plots and boxplots were drawn in Origin2017 (OriginLab, Northampton, MA, United States). The relationships of microbial CUE and Y-A-S strategies were outlined using Pearson’s correlation analysis in the “corrplot” package in R, and significant differences between CUE and Y-A-S strategies were indicated by p < 0.05. The differences in genes between treatments with or without nutrient additions in low-salinity soil were analyzed using STAMP 2.1.3 (Parks et al., 2014).
The raw data of metagenomic sequencing were deposited in the NCBI SRA database under accession number PRJNA891869.
The contents of 13C-MASOC and 13C-MBC were higher in low-salinity soil than in high-salinity soil. The addition or non-addition of nutrients showed no significant variation on the contents of 13C-MASOC, 13C-MBC, and 13C-DOC in high-salinity soil. Whereas, the addition of nutrients decreased 13C-MASOC and 13C-MBC by 24.5 and 42.5% in low-salinity soil, respectively (Figure 1).
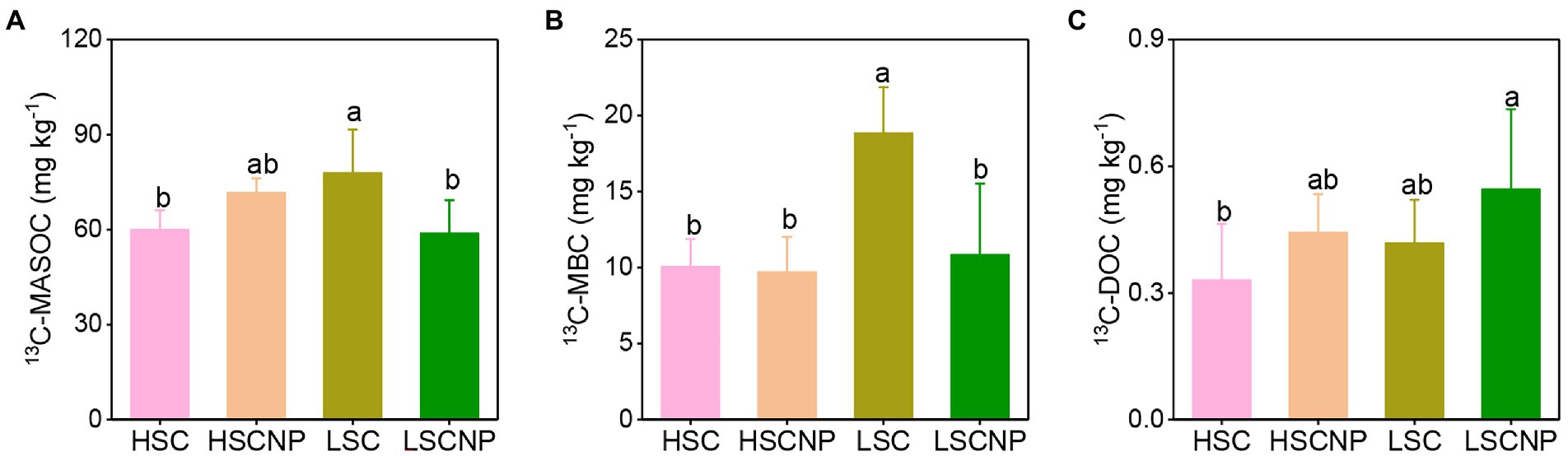
Figure 1. 13C-Glucose-derived carbon in the pools of mineral-associated soil organic carbon (MASOC) (A), microbial biomass carbon (MBC) (B), and dissolved organic carbon (DOC) (C) after 56 days of incubation. Different letters indicate significant differences (p < 0.05). HSC, high-salinity soil added with 13C-glucose; HSCNP, high-salinity soil added with 13C-glucose and NP nutrients; LSC, low-salinity soil added with 13C-glucose; LSCNP, low-salinity soil added with 13C-glucose and NP nutrients.
The soil specific enzyme activity indicated the effectiveness of the substrate and the strategy of the microorganisms to acquire nutrients. In this study, the activity of AKP per unit of MBC was significantly higher in high-salinity than in low-salinity soil. The addition of nutrients resulted in a significant decrease in the specific activities of NAG and AKP in the early stage of incubation and a significant increase in the specific activities of GC and NAG during the later stage of incubation in low-salinity soil (Supplementary Figure S1).
The microbial CUE was the highest (9.8%) in low-salinity soil without nutrients amended, which was significantly higher than that in high-salinity soil (7.8 and 7.3%). Moreover, the input of nutrients could not effectively increase the microbial CUE in high-salinity soil (Figure 2A).

Figure 2. Soil microbial carbon use efficiency (CUE) after 7 days of incubation (A). The sum of normalized read numbers of the top 20 abundant genera involved in the growth yield (Y-strategy), resource acquisition (A-strategy), and stress tolerance (S-strategy) (B). The reads numbers of specific genes that were assigned to the Y-strategy (C), A-strategy (D), S-strategy (E). Different letters indicate significant differences (p < 0.05), *p < 0.05, **p < 0.01. ***p < 0.001. HSC, high-salinity soil added with 13C-glucose; HSCNP, high-salinity soil added with 13C-glucose and NP nutrients; LSC, low-salinity soil added with 13C-glucose; LSCNP, low-salinity soil added with 13C-glucose and NP nutrients.
The metagenome sequencing generated a total of 198.36 GB of raw data, and 189.89 GB of clean data were obtained after removing low-quality reads. 5,614,187 unique genes were identified, and 3,281,396 genes were phylogenetically classified into the bacteria (99.36%), micro-eukaryotes (0.04%), and archaea (0.60%). After excluding unassigned genes, 40.56% of genes presented a genus-level classification. The top 20 dominant genera (accounting for 27.23 ~ 45.55% of the total reads) were selected for in-depth analysis. We classified and summarized these top 20 genera according to the Y-A-S strategies (Supplementary Tables S1). The results showed that the A- and S-strategy microorganisms were dominant in high-salinity soil, while the abundance of Y-strategy microbes was the highest in low-salinity soil, followed by the A-strategy, with a small proportion of S-strategy microbes. The addition of nutrients resulted in a reduction in the abundance of Y-strategy microbes in low-salinity soil (p < 0.05) (Figure 2B). In particular, the genera Sphingomonas, Gemmatirosa, Steroidobacter, and Agromyces, which were involved in the Y-strategy, were prominently present in greater abundance in the low-salinity soil than in the high-salinity soil (p < 0.001) (Figure 2C). The abundance of seven genera, including Nocardioides, Erythrobacter, Sphingosinicella, Lysobacter, Pontibacter, Arthrobacter, and Gramella, that were associated with the A-strategy were found in remarkably higher levels in the high-salinity soil than in the low-salinity soil (p < 0.01) (Figure 2D). Streptomyces, Salinimicrobium, Enhygromyxa, Haliangium, Marinobacter, Altererythrobacter, and Halogeometricum assigned to the S-strategy were extremely significantly increased in the high-salinity soil (p < 0.001) (Figure 2E). Unexpectedly, the abundances of Lysobacter, Enhygromyxa, Haliangium, and Marinobacter that were involved in the A- and S-strategies increased with the input of nutrients to the low-salinity soil.
The top 30 abundant functional categories at KEGG level 3 with significant differences (p < 0.05) between high- and low-salinity soils were summarized in Supplementary Tables S2. Typically, the abundance of microbial functions involved in the growth yield was higher in low-salinity soil, but the addition of nutrients was detrimental to the increase of functions related to the Y-strategy in low-salinity soil, while the abundance of microbial functions involved in the A- and S-strategies were notably increased in high-salinity soils compared with low-salinity soils (p < 0.05) (Figure 3A). In particular, the functional categories related to amino acid metabolism (alanine, aspartate, and glutamate and arginine), carbohydrate metabolism (starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism, pentose phosphate pathway, glyoxylate and dicarboxylate metabolism, carbon fixation pathways in prokaryotes) and cell processes (cell cycle-Caulobacter, pyrimidine metabolism and RNA polymerase) were especially reduced under high salt stress (Figure 3B). Instead, the microbial functions of ABC transporters and flagellar assembly were increased in high-salinity soil (Figure 3C). The microbial functions associated with S-strategy, such as homologous recombination, pantothenate and CoA biosynthesis, peptidoglycan biosynthesis, ubiquinone and other terpenoid-quinone biosynthesis, biofilm formation and base excision repair, were also significantly increased under high salt stress (Figure 3D). Furthermore, the input of nutrients to low-salinity soil remarkably increased the function of bacterial chemotaxis that were assigned to A-strategy, and decreased the functions of two-component system, pyrimidine metabolism, and arginine biosynthesis that were linked with the Y-strategy (Supplementary Figure S2).
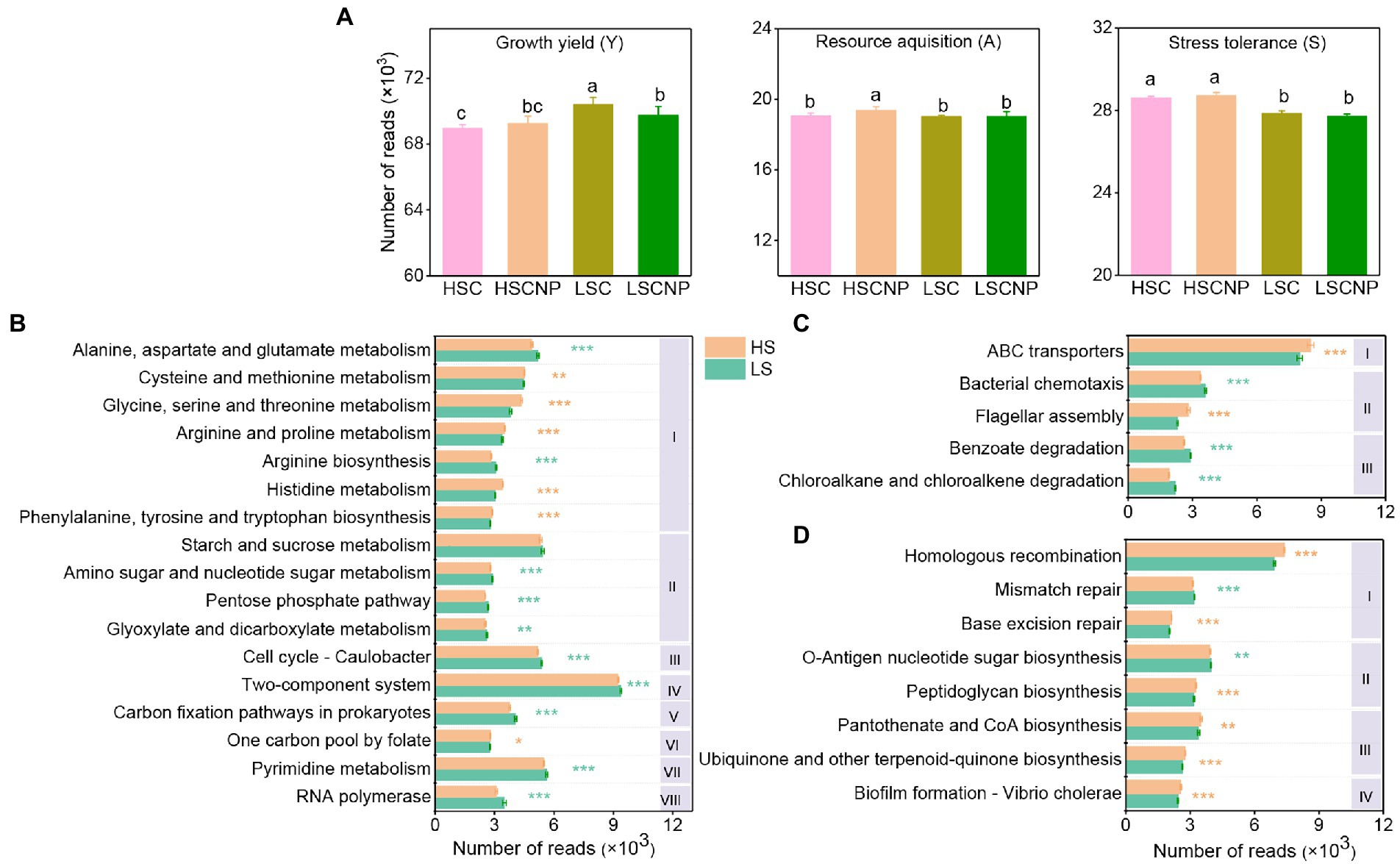
Figure 3. The reads numbers of functions related to the growth yield (Y-strategy), resource acquisition (A-strategy), and stress tolerance (S-strategy) according to KEGG annotations (A). Specific KEGG functional categories assigned to the Y-strategy (B), A-strategy (C), and S-strategy (D). The Roman numerals I, II, III, IV, V, VI, VII, and VII in subpanel (B) represent amino acid metabolism, carbohydrate metabolism, cell growth and death, signal transduction, energy metabolism, metabolism of cofactors and vitamins, nucleotide metabolism and transcription at the KEGG level 2, respectively. The Roman numerals I, II, and III in subpanel (C) represent membrane transport, cell motility and xenobiotics biodegradation and metabolism at the KEGG level 2, respectively. The Roman numerals I, II, III, and IV in subpanel (D) represent replication and repair, glycan biosynthesis and metabolism, metabolism of cofactors and vitamins and cellular community – prokaryotes, respectively. Orange asterisks indicate a higher abundance of functions in high-salinity soil, whereas green asterisks indicate a higher abundance of functions in low-salinity soil. Different letters indicate significant differences (p < 0.05). *p < 0.05, **p < 0.01, ***p < 0.001. HSC, high-salinity soil added with 13C-glucose; HSCNP, high-salinity soil added with 13C-glucose and NP nutrients; LSC, low-salinity soil added with 13C-glucose; LSCNP, low-salinity soil added with 13C-glucose and NP nutrients.
In the present study, a total of 198 genes that encoded the functions involved in the Y-A-S strategies were detected. Most of them (116) were assigned to growth yield, and the abundance of these genes decreased with increasing salinity. A total of 54 and 28 genes that were assigned to resource acquisition and stress tolerance, respectively, were more abundant in high-salinity soil (Figure 4A and Supplementary Data S1). To ensure that the heatmap was clear and concise, genes of the same gene cluster were combined and displayed together. More details are shown in Supplementary Data S1. The genes that encoded functions in C (i.e., cbbL, rbcS, nifJ, aclB, porA/B/C/D, ppc, korA/B/D, malE/F/K/Q/Z, abfA, manB, pmm-pgm, xylA/B/F/G/H, cbh, and pel), N (i.e., nifA/D/H/K, nasA/B, napA/B, nrfA, asnA/B, aspA, arcC, GLT1, ncd2, tdcB, and tnaA), and P (i.e., phoA/B/D/N/P/R, appA, ppk1, ugpA/B/C/E and gcd) metabolism contributed to the increase in Y-strategy in low-salinity soil (Figure 4B). The genes that encoded flagellar assembly (flg, flh, and fli gene clusters) and siderophore synthesis (entA/C and ccmA/B/C/D genes) represented A-strategy under high salt stress (Figure 4C). The S-strategy related genes that encoded osmolyte production, such as ectA/B/C/D, gbsA/B, opuA/B/C/D, otsA/B, treS, gdh, GDH2, and proA/B/C, and genes that encoded compatible solute transport (kdpA/B/C) and polysaccharide biosynthesis transport (exoP) were strikingly higher (p < 0.001) in high-salinity soils than in low-salinity soils (Figure 4D). Moreover, the four genes that encoded C (rbcS), N (nirK), and P (ugpE) cycle and osmolyte production (opuB/D) decreased (p < 0.05) in the nutrient-added treatment of the low-salinity soil (Supplementary Figure S2). In addition, the abundance of genes that were assigned to Y-strategy, especially those involved in C and N cycle, were significantly (p < 0.05) positively correlated with microbial CUE. Whereas the genes encoding flagellar assembly and siderophore synthesis assigned to A-strategy were strongly negatively correlated with microbial CUE (Supplementary Figure S3).
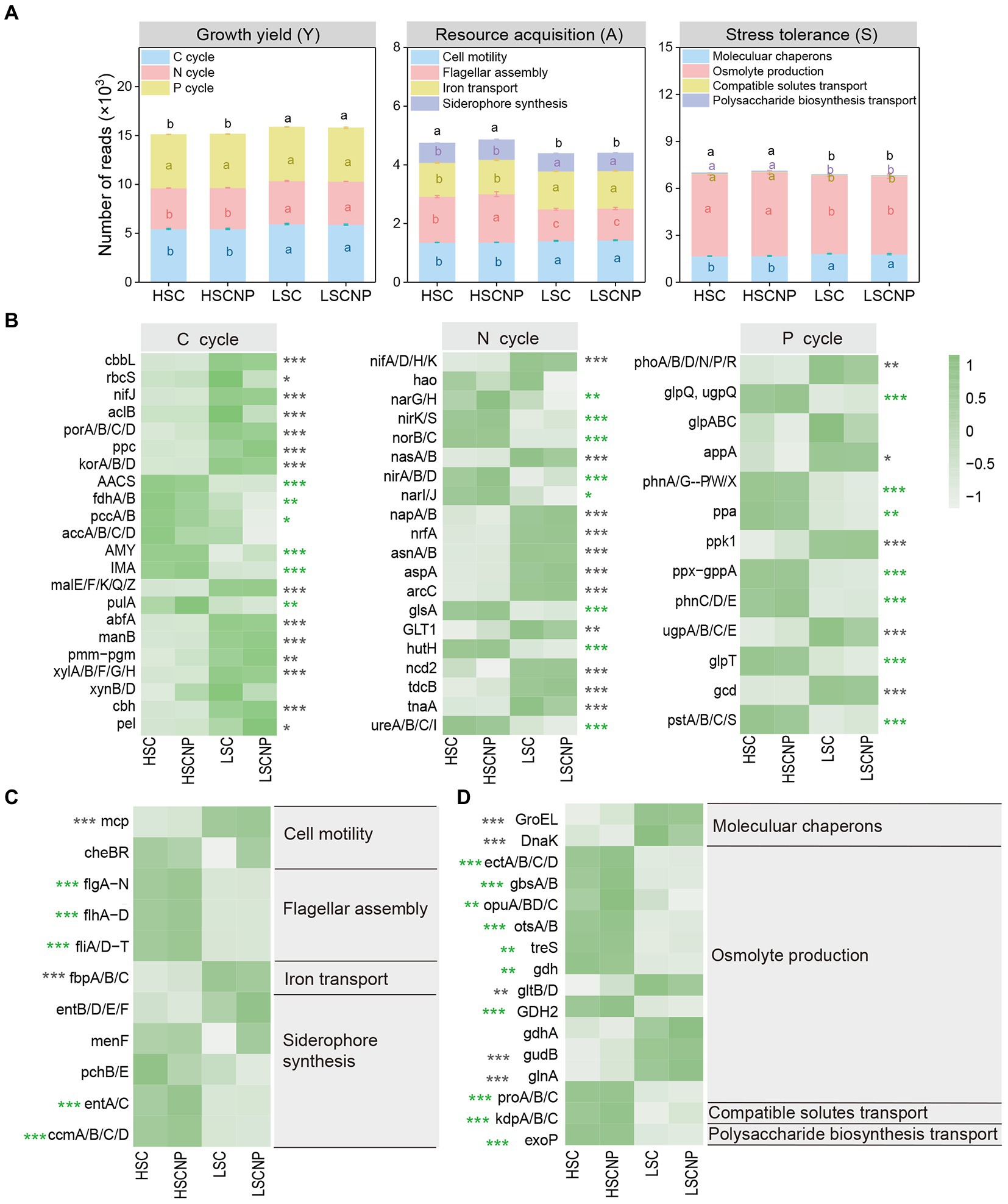
Figure 4. The normalized read numbers of genes assigned to growth yield (Y-strategy), resource acquisition (A-strategy), and stress tolerance (S-strategy) (A). The heatmaps showed specific genes that corresponded to the Y-strategy (B), A-strategy (C), and S-strategy (D), respectively. Green asterisks indicate a higher abundance of genes in high-salinity soil, whereas gray asterisks indicate a higher abundance of genes in low-salinity soil. To ensure that the heatmap was clear and concise, genes of the same gene cluster were combined and displayed together. More details are shown in Supplementary Data S1. Different letters indicate significant differences (p < 0.05). *p < 0.05, **p < 0.01, ***p < 0.001. HSC, high-salinity soil added with 13C-glucose; HSCNP, high-salinity soil added with 13C-glucose and NP nutrients; LSC, low-salinity soil added with 13C-glucose; LSCNP low-salinity soil added with 13C-glucose and NP nutrients.
The input of balanced nutrients did not exert a significant increase in MASOC and MBC in high-salinity soil, and the same trend was observed for microbial CUE (Figures 1, 2A), indicating that salt stress is detrimental to microbial growth and turnover, and the formation of microbial-derived SOC under high salt stress was independent from nutrient stoichiometry. A higher microbial CUE indicates that a greater proportion of substrate C was assimilated and converted toward microbial biomass synthesis, which potentially increased the generation and sequestration of stable SOC (Fang et al., 2020). In high-salinity soil, microorganisms sacrifice their growth to resist stress (Dong et al., 2022), which results in the decrease of microbial CUE and mineral-associated SOC content. Although previous results have shown that stoichiometry influenced the microbial CUE and SOC storage in soil that did not exhibit stress conditions (Mo et al., 2021), this study suggested that in saline soil, salt stress exerted stronger effects on CUE and MASOC, and the effects of stoichiometry were negligible, which is inconsistent with our hypothesis. These results were further confirmed by the trade-off of microbial life history strategies in high-salinity soil that involves stress tolerance combined with resource acquisition strategies.
Soil salinity altered the CUE due to its direct effect on microbial biomass, but it also indirectly affected the CUE by selecting different microbial life history strategies, which ultimately influenced the microbial-derived SOC formation. In high-salinity habitats, microbial taxa, functions, and genes that involved in S-strategy were prominently enriched. For example, several genera that increased (Figure 2), such as Streptomyces, Salinimicrobium, Enhygromyxa, Haliangium, Marinobacter, Altererythrobacter, and Halogeometricum, have been previously reported to be halotolerant microbes (Fudou et al., 2002; Cui et al., 2010; Gemperlein et al., 2018; Bachran et al., 2019; Etesami and Glick, 2020; Li et al., 2021). Under salt scenarios, these microbes have specific functional traits and genes to improve their resistance to salinity and could be divided into the following three categories (Figures 3, 4): (i) The microorganisms were capable of repairing biomolecular damage and maintaining cellular integrity, such as homologous recombination, base excision repair, and biofilm formation; (ii) the microorganisms induced the biosynthesis of extracellular polysaccharides, such as peptidoglycan biosynthesis and the exoP genes that are encoded; and iii) they produced osmolytes to maintain an osmotic balance by encoding the ectA/B/C/D, gbsA/B, opuA/BD, otsA/B, treS, gdh, GDH2, proA/B/C, and kdpA/B/C genes. Specifically, the ability to repair biomolecular damage was an essential metabolic trait under high-salinity stress, and similar results were found in other studies conducted in a saline environment (Wilson et al., 2004; Xu et al., 2022). The exoP gene is a polysaccharide biosynthetic transport protein that encodes biofilm formation. Biofilm formation and extracellular polysaccharide biosynthesis facilitate the concentration of resources to maintain microbial metabolic activity, which is consistent with previous studies on drought (Malik and Bouskill, 2022) and cold (Feng et al., 2021) stress. The production of osmolytes could maintain the cellular osmotic equilibrium and help to tolerate scarce resources, which is a very expensive energy consumption metabolic trait, and comes at the expense of reducing growth yield (Malik et al., 2020; Li C. N. et al., 2022). It takes a large amount of energy (30–110 ATP) to synthesize osmotic substances, which is much greater than those required to synthesize cell walls (30 ATP) (Chowdhury et al., 2011). This resulted in a significant metabolic burden on the microbes and reduces their energy for microbial growth. Some osmolytes are rich in C and N and can serve as energy sources after the stress has diminished (Malik and Bouskill, 2022). Ectoine, trehalose, glycine betaine, proline, and glutamate are the most common osmolytes that have been previously described (Malik et al., 2020; Malik and Bouskill, 2022). In addition, K+ uptake and transport and Na+/H+ antiporters are other metabolic methods to regulate osmotic pressure. Due to K+ is less toxic to the microbial community and metabolic function than Na+, microorganisms usually select the accumulation of K+ to reach an osmotic balance in a high-salinity environment (Rath and Rousk, 2015). All the microbial metabolic functions described that involved in the S-strategy are energy (C) expensive and result in a lower microbial CUE and microbial-derived SOC formation (Dong et al., 2022).
Strikingly, the A-strategy was also dominant in high-salinity soil, independent of nutrient input. The production of extracellular enzymes is a common way to increase resource capture. The specific enzyme activity per unit of MBC provides more sensitive indications for the microbial metabolic status and stability in extracellular enzymes activity, independently of changes in the MBC contents (Raiesi and Beheshti, 2014). Our research showed that salt stress results in a limitation of resources, and microorganisms produce more phosphatases and acquire P from soil organic matter to meet their growth under high salt stress (Supplementary Figure S1). Some specific genera, such as Nocardioides, Erythrobacter, Sphingosinicella, Lysobacter, Pontibacter, Arthrobacter, and Gramella, that involved in A-strategy were detected in high salt conditions (Figure 2). These taxa invest in the degradation of complex resources and secretion of hydrolytic enzymes to improve the availability of nutrients. For example, Nocardioides was reported to degrade xylan (Li et al., 2021). Erythrobacter is halophilic and can hydrolyze epoxides (Woo et al., 2007; Wang et al., 2021). Sphingosinicella, Lysobacter, and Pontibacter can produce hydrolytic enzymes, chitin-degrading enzymes, and glycoside hydrolase, respectively (Kato et al., 2009; Zhou et al., 2016; Fu et al., 2022). Arthrobacter and Gramella have ability to degrade recalcitrant polysaccharides, solubilize phosphate, and also obtain nutrients by cell motility (Bauer et al., 2006; Chen et al., 2006; Fan et al., 2014; Kabisch et al., 2014; Chen et al., 2019). Furthermore, resource acquisition markers, such as the microbial genes that encode ABC transporters (ccmA/B/C/D), flagellar assembly (flg, flh, and fli gene clusters), and siderophore biosynthesis (entA/C), were also widely found in high-salinity soils (Figure 4). The ABC transporters transport a variety of substrates using energy from ATP (Badri et al., 2009), and a greater investment in transporters contributed to the uptake of nutrient substrates by microorganisms under higher salt stress. Flagellar assembly was an indicator of the microbial capabilities of resource discovery (Malik et al., 2020). Previous studies confirmed that the flg, flh, and fli gene clusters that encode flagellar assembly are an expensive energy- consuming process and are intrinsically linked to biofilm formation (Wilkinson et al., 2011), which inevitably occurs at the expense of other physiological processes, such as growth yield. The genes entA/C that encode siderophore biosynthesis provide an increase in iron availability for microbial populations in highly saline soil. Iron plays an important role in biological systems under salt stress, such as those that catalyze enzymatic processes related to oxygen, hydrogen, and N, and are involved in DNA and RNA synthesis and repair processes (Ferreira et al., 2019).
Therefore, microbial physiology and the key pathways indicate that stress tolerance and resource acquisition are dominant life history strategies under high salt stress. Such strategies lead to resource reallocation as the microbes divert investments away from microbial growth yield and increase their investments in microbial survival and resource capture, thereby decreasing the efficiency of cellular growth and impacting the soil microbial-derived SOC formation.
Compared with high-salinity soil, microbial CUE and MASOC were higher in low-salinity soil, demonstrating that soil microorganisms have a high growth yield strategy in the absence of salt stress, which is proved by the strongly positively correlations between microbial CUE and Y- strategy (Supplementary Figure S3). Specifically, the abundance of some microbial populations, such as Sphingomonas, Gemmatirosa, Steroidobacter, and Agromyces, increased when the salt stress was somewhat alleviated (Figure 2). These genera generally play a role in C and N metabolism, and thus, were assigned to the Y-strategy. Sphingomonas can metabolize various C sources, some species participate in N fixation and denitrification (Yang et al., 2016), and some strains produce salicylic acid to improve plant growth under salt stress (Etesami and Glick, 2020). Li W. J. et al. (2022) reported that species of Gemmatirosa were symbiotic flora and proliferated with the increase in the availability of water and organic matter. Steroidobacter possesses a strong ability to biodegrade sulfonamides, which are N-containing organic compounds (Zhang et al., 2021). As a member of the Actinobacteria, Agromyces was reported to be capable of N fixation (Marcos et al., 2019) and can decompose cellulolytic compounds by producing the β-glucosidase gene (Nevins et al., 2018). In addition, microbes can reduce the costs in stress tolerance and resource acquisition and increase their investment in central metabolism and biosynthesis, such as amino acid metabolism, carbohydrate metabolism, and cell processes, to maximize their growth yield in low-salinity habitats. For example, the increase in abundance of genes involved in the Calvin cycle, the reductive tricarboxylic acid cycle, starch catabolism, cellulose and hemicellulose catabolism, and pectin catabolism that related to Y-strategy are favored microbial C formation (Figure 4 and Supplementary Data S1). The Calvin cycle and reductive tricarboxylic acid cycle were the common C fixation pathways in farmland ecosystems (Huang et al., 2022). The higher abundance of the cbbL gene that is responsible for the Calvin cycle in low-salinity soil indicated a greater ability to fix CO2 than in high-salinity soil. Similar results in a previous study reported that salt stress inhibited the expression of activities of the Calvin cycle enzymes (Yu et al., 2011). Several genes, including nifJ, aclB, porA/B/C/D, ppc, and korA/B/D, that encode the reductive tricarboxylic acid cycle participate in the CO2 fixation metabolic pathway, which is favored to maximize the microbial growth yield owing to the low energy consumption of the reductive tricarboxylic acid cycle (Huang et al., 2022). In addition, starch, cellulose, hemicellulose, and pectin are easily taken up and metabolized by microorganisms (Chen et al., 2014), which thereby promotes their biomass growth.
However, the input of nutrients had negative effects on the microbial CUE and MASOC formation in low-salinity soil (Figure 1). The reduction in microbial CUE with the addition of nutrients suggested that a greater proportion of C was mineralized and released as the form of CO2 by microbes, while less C was converted to microbial biomass (Mo et al., 2021). The addition of N and P sources affected specific activities of GC, NAG, and AKP in low-salinity soil (Supplementary Figure S1). This probably occurred due to the N and P substrates that added were directly available for microbial growth, and there was no need to secrete additional extracellular enzymes during the early stage of incubation, which thereby decreased the activities of NAG and AKP. In contrast, during the later stage of incubation, the availability of substrates was reduced, and microorganisms needed to produce more GC and NAG to break down complex compounds into smaller substrates for uptake. Furthermore, the differences in life history traits also affected the microbial competition for C and nutrient flows (Mo et al., 2021). Our study showed that, compared with the lack of addition of nutrients, the metabolic traits and markers, such as Lysobacter, bacterial chemotaxis, and the flgM gene, that associated with the A-strategy were depleted in low-salinity soil that contained added nutrients. Lysobacter can produce a variety of lytic enzymes to degrade chitin, glucans, and proteins (Puopolo et al., 2018). Chemotaxis is a foraging strategy, which is primarily characterized by the use of flagella to swim and be motile, which enhances the bacterial uptake of nutrients and energy (Keegstra et al., 2022). The flgM gene, which encodes the flagellar assembly, has an important role in bacterial swimming and the formation of biofilms by regulating flagellar synthesis and flagellum numbers (Wilkinson et al., 2011). Taken together, the soil microbial community invested more C in the A-strategy and primarily through the secretion of extracellular enzymes and bacterial chemotaxis to enhance their resource capture ability, thereby influencing microbial-derived SOC formation.
Overall, the tradeoff of microbial life history strategies in the soils of different salinity affected the microbial CUE and microbial-derived SOC formation. Microbes invested more energy and resources in S- and A- strategies at the expense of growth yield in high-salinity habitats. Specifically, microorganisms increased their metabolic traits that are involved in biomolecular damage repair, extracellular polysaccharide biosynthesis, and osmolyte production and transport to improve their resistance to salinity. They also increased the synthesis of siderophores and flagellar assembly to acquire nutrient resources. In contrast, microbes primarily invested in a Y-strategy with a small proportion in A-strategy in low-salinity soil. The adjustment of stoichiometry had negligible effects on the promotion of microbial CUE and microbial-derived SOC formation in high-salinity soil. Moreover, the alteration of soil stoichiometry by nutrient addition increased the microbial metabolic traits in the shift to A-strategy in low-salinity soil, which was also detrimental to microbial-derived SOC formation. Therefore, for saline soil, the most important thing is to alleviate salt stress rather than regulate stoichiometry, so as to shift the microbial life strategy from S- and A- strategies to the Y-strategy, thereby increasing the microbial CUE and enhancing microbial-derived SOC formation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
JZ, QN, and LC jointly developed the overall approach. QN and LC carried out the experimental work, did metagenomic sequencing data analysis, and manuscript preparation. FL, GZ, CZ, DM, and JZ revised the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
This work was jointly supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA24020104 and XDA28020203), the National Key Research and Development Program of China (2022YFD1500203 and 2022YFD1500401), the National Natural Science Foundation of China (42177332), the China Agriculture Research System (CARS-03-15 and CARS-52), and the Jiangsu Provincial Postdoctoral Science Foundation (2021Z241).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1141436/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Soil specific enzyme activities per unit of microbial biomass (mg product g−1 MBC) in day 7 (A-C) and day 56 (D-F). Different letters indicate significant differences (P < 0.05). HSC, high-salinity soil added with 13C-glucose; HSCNP, high-salinity soil added with 13C-glucose and NP nutrients; LSC, low-salinity soil added with 13C-glucose; LSCNP low-salinity soil added with 13C-glucose and NP nutrients.
SUPPLEMENTARY FIGURE S2 | Genes with significant differences between treatments with or without nutrient additions in low salinity soil. LSC, low-salinity soil added with 13C-glucose; LSCNP low-salinity soil added with 13C-glucose and NP nutrients.
SUPPLEMENTARY FIGURE S3 | Pearson correlations of microbial CUE and Y-A-S strategies (A) and the relationships between Y-A-S strategies based on related genes classification and microbial CUE (B). * mean P < 0.05. Y-strategy, growth yield; A-strategy, resource acquisition; S-strategy, stress tolerance.
Arcand, M. M., Levy-Booth, D. J., and Helgason, B. L. (2017). Resource legacies of organic and conventional management differentiate soil microbial carbon use. Front. Microbiol. 8:8. doi: 10.3389/fmicb.2017.02293
Auwal, M., Singh, B. P., Chen, Z. Y., Kumar, A., Pan, S. T., Luo, Y., et al. (2021). Nutrients addition regulates temperature sensitivity of maize straw mineralization. J. Soils Sediments 21, 2778–2790. doi: 10.1007/s11368-021-02960-9
Bachran, M., Kluge, S., Lopez-Fernandez, M., and Cherkouk, A. (2019). Microbial diversity in an arid, naturally saline environment. Microb. Ecol. 78, 494–505. doi: 10.1007/s00248-018-1301-2
Badri, D. V., Quintana, N., El Kassis, E. G., Kim, H. K., Choi, Y. H., Sugiyama, A., et al. (2009). An ABC transporter mutation alters root exudation of phytochemicals that provoke an overhaul of natural soil microbiota. Plant Physiol. 151, 2006–2017. doi: 10.1104/pp.109.147462
Bauer, M., Kube, M., Teeling, H., Richter, M., Lombardot, T., Allers, E., et al. (2006). Whole genome analysis of the marine Bacteroidetes 'Gramella forsetii' reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 8, 2201–2213. doi: 10.1111/j.1462-2920.2006.01152.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Chen, L. J., Jiang, Y. J., Liang, C., Luo, Y., Xu, Q. S., Han, C., et al. (2019). Competitive interaction with keystone taxa induced negative priming under biochar amendments. Microbiome 7:18. doi: 10.1186/s40168-019-0693-7
Chen, Z. Y., Kumar, A., Brookes, P. C., Kuzyakov, Y., Luo, Y., and Xu, J. M. (2022). Three source-partitioning of CO2 fluxes based on a dual-isotope approach to investigate interactions between soil organic carbon, glucose and straw. Sci. Total Environ. :811. doi: 10.1016/j.scitotenv.2021.152163
Chen, Y. P., Rekha, P. D., Arun, A. B., Shen, F. T., Lai, W. A., and Young, C. C. (2006). Phosphate solubilizing bacteria from subtropical soil and their tricalcium phosphate solubilizing abilities. Appl. Soil Ecol. 34, 33–41. doi: 10.1016/j.apsoil.2005.12.002
Chen, L., Zhang, J. B., Zhao, B. Z., Yan, P., Zhou, G. X., and Xin, X. L. (2014). Effects of straw amendment and moisture on microbial communities in Chinese fluvo-aquic soil. J. Soils Sediments 14, 1829–1840. doi: 10.1007/s11368-014-0924-2
Chowdhury, N., Marschner, P., and Burns, R. G. (2011). Soil microbial activity and community composition: impact of changes in matric and osmotic potential. Soil Biol. Biochem. 43, 1229–1236. doi: 10.1016/j.soilbio.2011.02.012
Cleveland, C. C., and Liptzin, D. (2007). C: N: P stoichiometry in soil: is there a "Redfield ratio" for the microbial biomass? Biogeochemistry 85, 235–252. doi: 10.1007/s10533-007-9132-0
Cui, H. L., Yang, X., Gao, X., Li, X. Y., Xu, X. W., Zhou, Y. G., et al. (2010). Halogeometricum rufum sp. nov., a halophilic archaeon from a marine solar saltern, and emended description of the genus Halogeometricum. Int. J. Syst. Evol. Microbiol. 60, 2613–2617. doi: 10.1099/ijs.0.019463-0
Dong, Y., Chen, R. R., Petropoulos, E., Yao, T. Y., Yu, B. Q., Lin, X. G., et al. (2022). Microbial carbon use efficiency in coastal soils along a salinity gradient revealed by ecoenzymatic stoichiometry. J. Geophys. Res.-Biogeosci. 127:e2022JG006800. doi: 10.1029/2022JG006800
Etesami, H., and Glick, B. R. (2020). Halotolerant plant growth-promoting bacteria: prospects for alleviating salinity stress in plants. Environ. Exp. Bot. 178:104124. doi: 10.1016/j.envexpbot.2020.104124
Fan, F. L., Yin, C., Tang, Y. J., Li, Z. J., Song, A., Wakelin, S. A., et al. (2014). Probing potential microbial coupling of carbon and nitrogen cycling during decomposition of maize residue by 13C-DNA-SIP. Soil Biol. Biochem. 70, 12–21. doi: 10.1016/j.soilbio.2013.12.002
Fang, Y. Y., Singh, B. P., Collins, D., Armstrong, R., Van Zwieten, L., and Tavakkoli, E. (2020). Nutrient stoichiometry and labile carbon content of organic amendments control microbial biomass and carbon-use efficiency in a poorly structured sodic-subsoil. Biol. Fertil. Soils 56, 219–233. doi: 10.1007/s00374-019-01413-3
Feng, J., Zeng, X. M., Zhang, Q., Zhou, X. Q., Liu, Y. R., and Huang, Q. (2021). Soil microbial trait-based strategies drive metabolic efficiency along an altitude gradient. ISME J. 1:71. doi: 10.1038/s43705-021-00076-2
Ferreira, M. J., Silva, H., and Cunha, A. (2019). Siderophore-producing rhizobacteria as a promising tool for empowering plants to cope with iron limitation in saline soils: a review. Pedosphere 29, 409–420. doi: 10.1016/S1002-0160(19)60810-6
Fu, Y., Luo, Y., Tang, C., Li, Y., Guggenberger, G., and Xu, J. (2022). Succession of the soil bacterial community as resource utilization shifts from plant residues to rhizodeposits. Soil Biol. Biochem. 173:108785. doi: 10.1016/j.soilbio.2022.108785
Fu, L. M., Niu, B. F., Zhu, Z. W., Wu, S. T., and Li, W. Z. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Fudou, R., Jojima, Y., Iizuka, T., and Yamanaka, S. (2002). Haliangium ochraceum gen. Nov., sp nov and Haliangium tepidum sp nov.: novel moderately halophilic myxobacteria isolated from coastal saline environments. J. Gen. Appl. Microbiol. 48, 109–115. doi: 10.2323/jgam.48.109
Geisseler, D., and Horwath, W. R. (2009). Relationship between carbon and nitrogen availability and extracellular enzyme activities in soil. Pedobiologia 53, 87–98. doi: 10.1016/j.pedobi.2009.06.002
Gemperlein, K., Zaburannyi, N., Garcia, R., La Clair, J. J., and Muller, R. (2018). Metabolic and biosynthetic diversity in marine myxobacteria. Mar. Drugs 16:314. doi: 10.3390/md16090314
Georgiou, K., Jackson, R. B., Vinduskova, O., Abramoff, R. Z., Ahlstrom, A., Feng, W., et al. (2022). Global stocks and capacity of mineral-associated soil organic carbon. Nat. Commun. 13:3797. doi: 10.1038/s41467-022-31540-9
Huang, Q., Huang, Y., Wang, B., Dippold, M. A., Li, H., Li, N., et al. (2022). Metabolic pathways of CO2 fixing microorganisms determined C-fixation rates in grassland soils along the precipitation gradient. Soil Biol. Biochem. 172:108764. doi: 10.1016/j.soilbio.2022.108764
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Kabisch, A., Otto, A., Konig, S., Becher, D., Albrecht, D., Schuler, M., et al. (2014). Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes 'Gramella forsetii' KT0803. ISME J. 8, 1492–1502. doi: 10.1038/ismej.2014.4
Kallenbach, C. M., Frey, S. D., and Grandy, A. S. (2016). Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls. Nat. Commun. 7:10. doi: 10.1038/ncomms13630
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kato, H., Tsuji, K., and Harada, K. (2009). Microbial degradation of cyclic peptides produced by bacteria. J. Antibiot. 62, 181–190. doi: 10.1038/ja.2009.8
Keegstra, J. M., Carrara, F., and Stocker, R. (2022). The ecological roles of bacterial chemotaxis. Nat. Rev. Microbiol. 20, 491–504. doi: 10.1038/s41579-022-00709-w
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, C. N., Liao, H. J., Xu, L., Wang, C. T., He, N. P., Wang, J. M., et al. (2022). The adjustment of life history strategies drives the ecological adaptations of soil microbiota to aridity. Mol. Ecol. 31, 2920–2934. doi: 10.1111/mec.16445
Li, W. J., Li, Y., Lv, J., He, X. M., Wang, J. L., Teng, D. X., et al. (2022). Rhizosphere effect alters the soil microbiome composition and C, N transformation in an arid ecosystem. Appl. Soil Ecol. 170:104296. doi: 10.1016/j.apsoil.2021.104296
Li, D. H., Liu, C. M., Luo, R. B., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, H. Y., Luo, N. Y., Ji, C. L., Li, J., Zhang, L., Xiao, L., et al. (2021). Liquid organic fertilizer amendment alters rhizosphere microbial community structure and co-occurrence patterns and improves sunflower yield under salinity-alkalinity stress. Microb. Ecol. 84, 423–438. doi: 10.1007/s00248-021-01870-0
Liang, C., Schimel, J. P., and Jastrow, J. D. (2017). The importance of anabolism in microbial control over soil carbon storage. Nat. Microbiol. 2:2. doi: 10.1038/nmicrobiol.2017.105
Liu, F. D., Mo, X., Kong, W. J., and Song, Y. (2020). Soil bacterial diversity, structure, and function of Suaeda salsa in rhizosphere and non-rhizosphere soils in various habitats in the Yellow River Delta, China. Sci. Total Environ. :140144:740. doi: 10.1016/j.scitotenv.2020.140144
Malik, A. A., and Bouskill, N. J. (2022). Drought impacts on microbial trait distribution and feedback to soil carbon cycling. Funct. Ecol. 36, 1442–1456. doi: 10.1111/1365-2435.14010
Malik, A. A., Martiny, J. B. H., Brodie, E. L., Martiny, A. C., Treseder, K. K., and Allison, S. D. (2020). Defining trait-based microbial strategies with consequences for soil carbon cycling under climate change. ISME J. 14, 1–9. doi: 10.1038/s41396-019-0510-0
Marcos, M. S., Bertiller, M. B., and Olivera, N. L. (2019). Microbial community composition and network analyses in arid soils of the Patagonian Monte under grazing disturbance reveal an important response of the community to soil particle size. Appl. Soil Ecol. 138, 223–232. doi: 10.1016/j.apsoil.2019.03.001
Mo, F., Zhang, Y. Y., Liu, Y., and Liao, Y. C. (2021). Microbial carbon-use efficiency and straw-induced priming effect within soil aggregates are regulated by tillage history and balanced nutrient supply. Biol. Fertil. Soils 57, 409–420. doi: 10.1007/s00374-021-01540-w
Nevins, C. J., Nakatsu, C., and Armstrong, S. (2018). Characterization of microbial community response to cover crop residue decomposition. Soil Biol. Biochem. 127, 39–49. doi: 10.1016/j.soilbio.2018.09.015
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Puopolo, G., Tomada, S., and Pertot, I. (2018). The impact of the omics era on the knowledge and use of Lysobacter species to control phytopathogenic micro-organisms. J. Appl. Microbiol. 124, 15–27. doi: 10.1111/jam.13607
Raiesi, F., and Beheshti, A. (2014). Soil specific enzyme activity shows more clearly soil responses to paddy rice cultivation than absolute enzyme activity in primary forests of Northwest Iran. Appl. Soil Ecol. 75, 63–70. doi: 10.1016/j.apsoil.2013.10.012
Rath, K. M., and Rousk, J. (2015). Salt effects on the soil microbial decomposer community and their role in organic carbon cycling: a review. Soil Biol. Biochem. 81, 108–123. doi: 10.1016/j.soilbio.2014.11.001
Shao, P. S., Lynch, L., Xie, H. T., Bao, X. L., and Liang, C. (2021). Tradeoffs among microbial life history strategies influence the fate of microbial residues in subtropical forest soils. Soil Biol. Biochem. 153:108112. doi: 10.1016/j.soilbio.2020.108112
Simpson, A. J., Simpson, M. J., Smith, E., and Kelleher, B. P. (2007). Microbially derived inputs to soil organic matter: are current estimates too low? Environ. Sci. Technol. 41, 8070–8076. doi: 10.1021/es071217x
Sokol, N. W., and Bradford, M. A. (2019). Microbial formation of stable soil carbon is more efficient from belowground than aboveground input. Nat. Geosci. 12, 46–53. doi: 10.1038/s41561-018-0258-6
Vance, E. D., Brookes, P. C., and Jenkinson, D. S. (1987). An extraction method for measuring soil microbial biomass-C. Soil Biol. Biochem. 19, 703–707. doi: 10.1016/0038-0717(87)90052-6
Wang, M., Wang, L. F., Shi, H. D., Liu, Y. B., and Chen, S. B. (2021). Soil bacteria, genes, and metabolites stimulated during sulfur cycling and cadmium mobilization under sodium sulfate stress. Environ. Res. 201:111599. doi: 10.1016/j.envres.2021.111599
Wilkinson, D. A., Chacko, S. J., Venien-Bryan, C., Wadhams, G. H., and Armitage, J. P. (2011). Regulation of flagellum number by fliA and flgM and role in biofilm formation by Rhodobacter sphaeroides. J. Bacteriol. 193, 4010–4014. doi: 10.1128/JB.00349-11
Wilson, C., Caton, T. M., Buchheim, J. A., Buchheim, M. A., Schneegurt, M. A., and Miller, R. V. (2004). DNA-repair potential of Halomonas spp. from the salt plains microbial observatory of Oklahoma. Microb. Ecol. 48, 541–549. doi: 10.1007/s00248-004-0243-z
Woo, J. H., Hwang, Y. O., Kang, S. G., Lee, H. S., Cho, J. C., and Kim, S. J. (2007). Cloning and characterization of three epoxide hydrolases from a marine bacterium, Erythrobacter litoralis HTCC2594. Appl. Microbiol. Biotechnol. 76, 365–375. doi: 10.1007/s00253-007-1011-z
Xia, J. B., Ren, J. Y., Zhang, S. Y., Wang, Y. H., and Fang, Y. (2019). Forest and grass composite patterns improve the soil quality in the coastal saline-alkali land of the Yellow River Delta, China. Geoderma 349, 25–35. doi: 10.1016/j.geoderma.2019.04.032
Xie, J. X., Li, Y., Zhai, C. X., Li, C. H., and Lan, Z. D. (2009). CO2 absorption by alkaline soils and its implication to the global carbon cycle. Environ. Geol. 56, 953–961. doi: 10.1007/s00254-008-1197-0
Xu, Y., You, G. X., Zhang, M. R., Peng, D. Y., Jiang, Z. W., Qi, S. T., et al. (2022). Antibiotic resistance genes alternation in soils modified with neutral and alkaline salts: interplay of salinity stress and response strategies of microbes. Sci. Total Environ. 809:152246. doi: 10.1016/j.scitotenv.2021.152246
Yang, J. S. (2008). Development and prospect of the research on salt-affected soils in China (in Chinese). Acta Pedol. Sin. 5, 837–845.
Yang, H., Hu, J. X., Long, X. H., Liu, Z. P., and Rengel, Z. (2016). Salinity altered root distribution and increased diversity of bacterial communities in the rhizosphere soil of Jerusalem artichoke. Sci. Rep. 6:20687. doi: 10.1038/srep20687
Yang, C., Lv, D. T., Jiang, S. Y., Lin, H., Sun, J. Q., Li, K. J., et al. (2021). Soil salinity regulation of soil microbial carbon metabolic function in the Yellow River Delta, China. Sci. Total Environ. 790:148258. doi: 10.1016/j.scitotenv.2021.148258
Yu, J. J., Chen, S. X., Zhao, Q., Wang, T., Yang, C. P., Diaz, C., et al. (2011). Physiological and proteomic analysis of salinity tolerance in Puccinellia tenuiflora. J. Proteome Res. 10, 3852–3870. doi: 10.1021/pr101102p
Zhang, G. X., Zhao, Z. H., Yin, X. A., and Zhu, Y. E. (2021). Impacts of biochars on bacterial community shifts and biodegradation of antibiotics in an agricultural soil during short-term incubation. Sci. Total Environ. 771:144751. doi: 10.1016/j.scitotenv.2020.144751
Zhao, Q. Q., Bai, J. H., Gao, Y. C., Zhao, H. X., Zhang, G. L., and Cui, B. S. (2020). Shifts in the soil bacterial community along a salinity gradient in the Yellow River Delta. Land Degrad. Dev. 31, 2255–2267. doi: 10.1002/ldr.3594
Zhou, J. P., Liu, Y., Lu, Q., Zhang, R., Wu, Q., Li, C. Y., et al. (2016). Characterization of a glycoside hydrolase family 27 alpha-galactosidase from Pontibacter reveals its novel salt-protease tolerance and transglycosylation activity. J. Agric. Food Chem. 64, 2315–2324. doi: 10.1021/acs.jafc.6b00255
Keywords: life history strategy, metagenome, metabolic traits, coastal saline soil, soil organic carbon, microbial carbon use efficiency
Citation: Ning Q, Chen L, Li F, Zhou GX, Zhang CZ, Ma DH and Zhang JB (2023) Tradeoffs of microbial life history strategies drive the turnover of microbial-derived organic carbon in coastal saline soils. Front. Microbiol. 14:1141436. doi: 10.3389/fmicb.2023.1141436
Edited by:
Kim Yrjälä, Zhejiang Agriculture and Forestry University, ChinaReviewed by:
Artur Banach, The John Paul II Catholic University of Lublin, PolandCopyright © 2023 Ning, Chen, Li, Zhou, Zhang, Ma and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiabao Zhang, amJ6aGFuZ0Bpc3Nhcy5hYy5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.