
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 21 March 2023
Sec. Microbiological Chemistry and Geomicrobiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1131000
This article is part of the Research TopicSoil-Microbial InteractionsView all 6 articles
 Qian Zhang
Qian Zhang Shuxia Lyu*
Shuxia Lyu*Microbial consortia play a key role in human health, bioenergy, and food manufacturing due to their strong stability, robustness and versatility. One of the microbial consortia consisting of Ketogulonicigenium vulgare and Bacillus megaterium for the production of the vitamin C precursor, 2-keto-L-gulonic acid (2-KLG), has been widely used for large-scale industrial production. To further investigate the cell–cell communication in microbial consortia, a microbial consortium consisting of Ketogulonicigenium vulgare and Bacillus pumilus was constructed and the differences in protein expression at different fermentation time points (18 h and 40 h) were analyzed by iTRAQ-based proteomics. The results indicated that B. pumilus was subjected to acid shocks in the coculture fermentation system and responded to it. In addition, the quorum sensing system existed in the coculture fermentation system, and B. pumilus could secrete quorum-quenching lactonase (YtnP) to inhibit the signaling pathway of K. vulgare. This study offers valuable guidance for further studies of synthetic microbial consortia.
In natural environments, microorganisms normally exist as communities of multiple species, capable of performing a variety and complexity of tasks and different members of a consortium assuming different responsibilities, increasing a higher overall productivity and allowing a more complex behavior than that of a monoculture (Brune and Bayer, 2012; Wang et al., 2016). With the development of synthetic communities, microbial consortia have become increasingly important in industrial applications (Jia et al., 2016). Therefore, there is an increasing focus on the development of synthetic consortia in industry. In general, several types of microbial interactions, i.e., food competition, mutualism, amensalism, synergism, predation, and parasitism could be involved in a mixed culture fermentation process (Sabra et al., 2010). However, microbial consortia in industry are complex, as the exchange of biomolecules (e.g., proteins, nucleic acids and metabolites) and information signals with the surroundings have to be taken into consideration, and the relationships among the strains are often too diverse to analyze (Wang et al., 2016; Ma et al., 2019; Roell et al., 2019).
Here, an example is described of using microbial consortia in industry for the production of vitamin C (L-ascorbic acid) to reduce costs and increase product quality. Microbial consortia of Ketogulonicigenium vulgare and Bacillus spp. have been widely used in a two-step vitamin C fermentation process and have achieved great economic benefits (Zhang and Lyu, 2022). K. vulgare is responsible for producing 2-keto-L-gulonic acid (2-KLG, the precursor of vitamin C) from L-sorbose via L-sorbosone but with an extremely low yield. Bacillus strains (e.g., Bacillus megaterium, Bacillus endophyticus, Bacillus subtilis, Bacillus pumilus), as helper strains, are cocultured to significantly stimulate the growth of K. vulgare and 2-KLG yields (Yakushi et al., 2020; Zeng et al., 2020; Fang et al., 2021; Yang et al., 2021; Zhang et al., 2022). The relationship between K. vulgare and the helper strain has been one of the hot topics focused on in the field of vitamin C fermentation.
In recent years, the interaction and communication between K. vulgare and the helper strain have been well explained with the development and application of omics technology (e.g., proteomics, metabolomics, and genomics) (Ma et al., 2011; Zhou et al., 2011; Jia et al., 2015; Ma et al., 2019; Zhang and Lyu, 2022). Most of the relevant studies have illustrated that the interaction between the two bacteria is a synergistic combination of mutualism and antagonism (Zhou et al., 2011). The helper strain provides key elements necessary to promote the growth and 2-KLG production of K. vulgare, whereas K. vulgare may accelerate the sporulation of the helper strain (Ma et al., 2011). In addition, Zhang et al. (2015) used Monod-type equations to describe the interactions between the two bacteria, which showed that K. vulgare was a predator and that the helper strain was prey. Moreover, it has been reported that L-sorbose, the substrate for 2-KLG production, not only promotes the growth of K. vulgare but also inhibits the growth of the helper strain, and the helper strain that is L-sorbose-tolerant enhances 2-KLG productivity (Mandlaa et al., 2018). Although it seems that the growth of the helper strain is inhibited in the coculture fermentation system, the cell–cell communication and the specific biomolecules that inhibit the growth of the helper strain are still not clear.
In this paper, our aim was to investigate the cell–cell communication and the reasons for inhibiting the growth of the helper strain in the coculture fermentation system. We therefore hypothesized that K. vulgare produced signals in the environment and that the oxidization products, including 2-KLG, could inhibit the growth of the helper strain. To test these hypotheses, iTRAQ-based proteomics analysis of an artificial consortium of K. vulgare and B. pumilus was performed, and the results used to derive novel information for further understanding of the synthetic consortium in Vc microbial fermentation and hopefully shed light on other complex microbial consortia.
Ketogulonicigenium vulgare 25B-1 (K. vulgare 25B-1) and Bacillus pumilus SY-A9 (B. pumilus SY-A9) were kindly provided by Northeast Pharmaceutical Group Co. Ltd. (Shenyang, China). These strains were stored at −80°C in glycerol.
Three media were used. Isolation medium: 20 g/L L-sorbose (autoclaved separately), 10 g/L peptone, 1 g/L urea, 5 g/L corn-steep liquor, 3 g/L yeast extract, 3 g/L beef extract, 1 g/L KH2PO4, 0.4 g/L MgSO4, 1 g/L CaCO3, 20 g/L agar powder, pH 6.7. The seed medium was the same as the isolation medium without the addition of agar powder. Fermentation medium: 80 g/L L-sorbose (autoclaved separately), 12 g/L urea, 10 g/L corn-steep liquor, 0.2 g/L MgSO4, 1 g/L KH2PO4, 5 g/L CaCO3, pH 6.7. These media were autoclaved at 121°C for 15 min before use.
First, approximately 1,000 colonies of K. vulgare were collected from the isolation medium plate (K. vulgare had been incubated for 48 h at 28°C) and suspended in 2 mL of sterile water. Second, one colony of B. pumilus with a diameter of 1 mm from the isolation medium plate (B. pumilus had been incubated for 24 h at 28°C) was mixed with 2 mL of sterile water. Then, 1 mL of B. pumilus solution and 2 mL of K. vulgare solution was mixed and the mixture was inoculated into a 20-mL seed culture medium in a 250-mL flask. After shaking (170 rpm) for 16 h at 28°C, the seeds were obtained, and K. vulgare colony number reached 1 × 109 CFU/mL and B. pumilus colony number reached 1 × 106 CFU/mL. Subsequently, 2 mL of seed was inoculated into a 20-mL fermentation medium in a 250-mL flask and the flask was cultivated on a shaker with 170 rpm at 28°C.
The fermentation broths at 18 h and 40 h were centrifuged at 13,000 × g for 20 min to obtain 0.5 g of wet bacteria for protein extraction. Each sample was mixed with 300 μl of breaking buffer (consisting of 50 mM Na3PO4, 1 mM phenylmethanesulfonyl fluoride (PMSF), 1 mM ethylenediaminetetraacetic acid (EDTA) and 5% glycerol) and complete protease inhibitor cocktail (4,693,116,001; Roche) on ice, followed by centrifugation at 4°C and 15,000 × g for 15 min. The supernatant containing intracellular proteins was further quantified for protein concentration using the Bradford assay. 200 μg protein was mixed with 5 mL of 1 M dithiothreitol (DTT) at 37°C for 1 h and alkylated with 20 μL of 1 M indole acetic acid (IAA) at room temperature for 1 h in the dark. Then, trypsin digestion (protein:trypsin ratio of 20:1) was performed for 24 h at 37°C. Using an iTRAQ Reagent-8Plex Multiplex Kit (AB SCIEX), peptides from the four samples were labeled with the iTRAQ tags as follows: K. vulgare + B. pumilus18 (tags 113 and 114) and K. vulgare + B. pumilus40 (tags 115 and 116), respectively.
The peptides were further fractionated using a C18 column (4.6 mm × 250 mm, 300 Å) with a high-performance liquid chromatography (HPLC) system (Agilent Technologies, United States) at a flow rate of 0.2 mL/min. The peptides were suspended in mobile phase A (100% H2O, pH 10.0, consisting of 20 mM NH4Ac) and eluted at a flow rate of 0.2 mL/min with the following gradient: 5 to 35% mobile phase B (90% acetonitrile (ACN), pH 10.0, consisting of 20 mM NH4Ac) for 30 min, 35 to 90% mobile phase B for 5 min, 90% mobile phase B for 10 min. The elution peak was monitored at 214 nm and one component per minute was collected and then lyophilized.
The lyophilized peptides were resuspended in 5 μl of 0.5% formic acid (FA), transferred to a C18 trap column (100 μm × 2 cm, 3 μm), and then separated using an Easy nLC 1,000 system with a C18 column (75 μm × 15 cm, 3 μm) at a flow rate of 200 nl/min. The mobile phases were solvent A (0.1% FA) and solvent B (0.1% FA in ACN). The elution gradient was as follows: 0–1 min, 1–5% solvent B; 1–6 min, 5–10% solvent B; 6–85 min, 10–30% solvent B; 85–87 min, 30–40% solvent B; 87–90 min, 40–90% solvent B; 90–100 min, 90% solvent B; 100–101 min, 90–1% solvent B; 101–120 min, 1% solvent B. The end of the nanoliter liquid-phase separation was connected directly to the Q-Exactive mass spectrometer (Thermo Fisher Scientific, United States) with a set of the following parameters: polarity, positive ion mode; MS scan range, 300–1,800 m/z; MS/MS scan resolution, 17,500; normalized collision energy, 30.
The acquired raw mass spectrometry data were processed and identified using the Sequest server and Proteome Discoverer (PD) (Thermo Fisher Scientific, United States) software. The spectra extracted using the PD software were searched against the Uniprot-Ketogulonicigenium+vulgare&Uniprot-Organism%3Abacillus+ subtilis database using the Sequest server with the following identification parameters: max missed cleavages, 2; fixed modifications, carbamidomethyl (C), iTRAQ8plex (N-term), iTRAQ8plex (K), iTRAQ8plex (Y); variable modifications, oxidation (M), deamidated (N, Q); peptide mass tolerance, 10 ppm; fragment mass tolerance, 0.02 Da; enzyme, trypsin. Proteins were quantified based on the PD software. In this paper, proteins with two unique peptides, p-values <0.05, fold changes ≥1.2 or ≤ 0.83, were identified as differentially expressed proteins (DEPs) between the different B. pumilus strains, whereas p-values <0.05, fold changes ≥1.1 or ≤ 0.91 were identified as DEPs between the different K. vulgare strains. In other words, in B. pumilus, the fold changes of the upregulated DEPs were more than 1.2, while those of the downregulated DEPs were less than 0.83. In K. vulgare, the fold changes of the upregulated DEPs were more than 1.1, while those of the downregulated DEPs were less than 0.91. For the convenience of expression, B. pumilus18 (B18) vs. B. pumilus40 (B40) and K. vulgare18 (K18) vs. K. vulgare40 (K40) were used to compare the proteome of the former with that of the latter to screen DEPs.
Hierarchical cluster analysis of DEPs was performed using R package. Clusters of orthologous genes (COG) annotations of DEPs were carried out using NCBI Blast.1 The biological pathway levels of DEPs were determined using the KEGG database.2 The functional interaction between the DEPs was performed using String3 and Cytoscape (3.8.2).
The sample was collected from the coculture fermentation system at 18 h and 40 h of incubation. Total RNA from each sample was extracted with SteadyPure Universal RNA Extraction Kit (AG21017, Accurate Biotechnology, Hunan, Co., Ltd., China). The synthesis of the first-strand cDNA was performed by an Evo M-MLV RT Permix for quantitative PCR (qPCR) (AG11706, Accurate Biotechnology, Hunan, Co., Ltd., China) after measuring the RNA quality and quantity by a BioDrop Spectrophotometer (Biochrom, England). Specific primers of selected DEPs were designed by NCBI Primer-BLAST (Supplementary Table S1), and 16S rRNA was used as the reference gene. The related gene expression level was determined on a CFX96 real-time PCR system (Bio-Rad, United States) with a total 10 μl volume of reaction mixture containing 0.8 μl template DNA, 0.4 μl forward primer (10 μM), 0.4 μl reverse primers (10 μM), 5 μl SYBR Green Premix Pro Taq HS qPCR Kit (AG11701, Accurate Biotechnology, Hunan, Co., Ltd., China), and 3.4 μl sterile purified water. Relative gene expression was calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001), followed by statistical analysis with GraphPad Prism 9 (GraphPad Software).
In total, 1,202 peptides and 584 proteins of B. pumilus SY-A9 and 5,475 peptides and 1,134 proteins of K. vulgare 25B-1 were obtained via iTRAQ labeling and 2D LC-MS/MS analysis in this study. Compared with the control group (samples at 40 h fermentation broth), 82 DEPs (38 upregulated and 44 downregulated) of B. pumilus SY-A9 and 96 DEPs (47 upregulated and 49 downregulated) of K. vulgare 25B-1 were identified in the 18 h fermentation broth group (Figure 1).
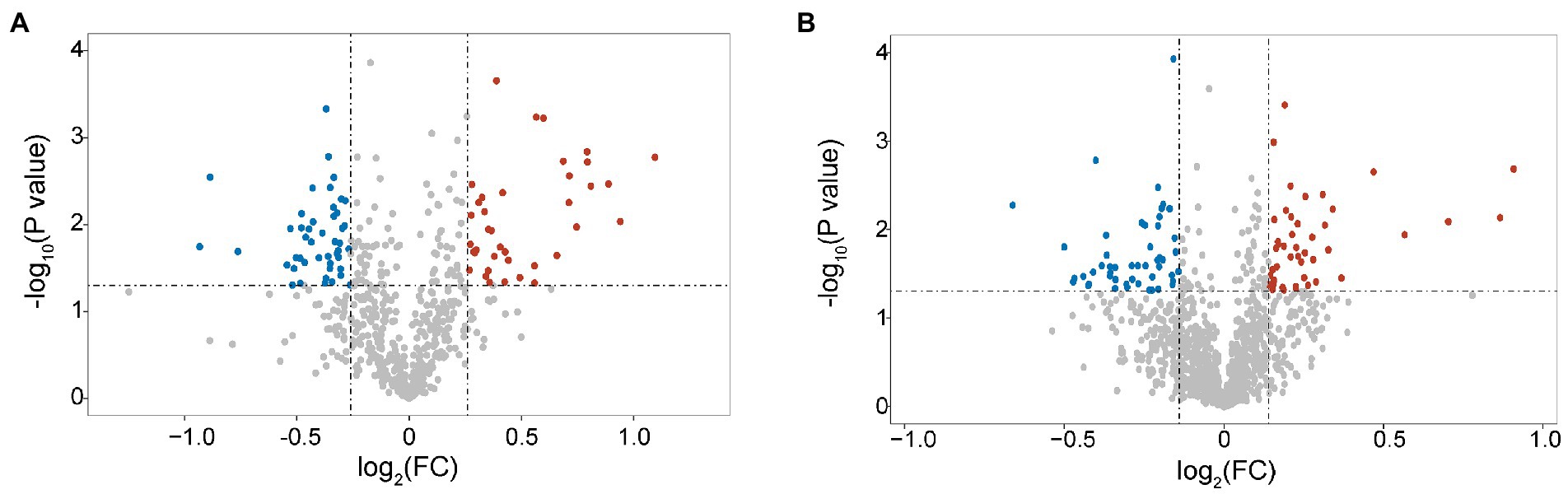
Figure 1. Volcano plot of differentially expressed proteins (DEPs) in B. pumilus (B18 vs. B40) (A) and K. vulgare (K18 vs. K40) (B). Each point represents a protein, and the red and blue areas represent upregulated and downregulated proteins (in B. pumilus, FC ≥ 1.2 or FC ≤ 0.83 and p < 0.05; in K. vulgare FC ≥ 1.1 or FC ≤ 0.91 and p < 0.05), respectively.
All acquired DEPs of B. pumilus SY-A9 and K. vulgare 25B-1 was subjected to COG functional annotation, and 25 categories were assigned with A-Z representation (Figure 2).
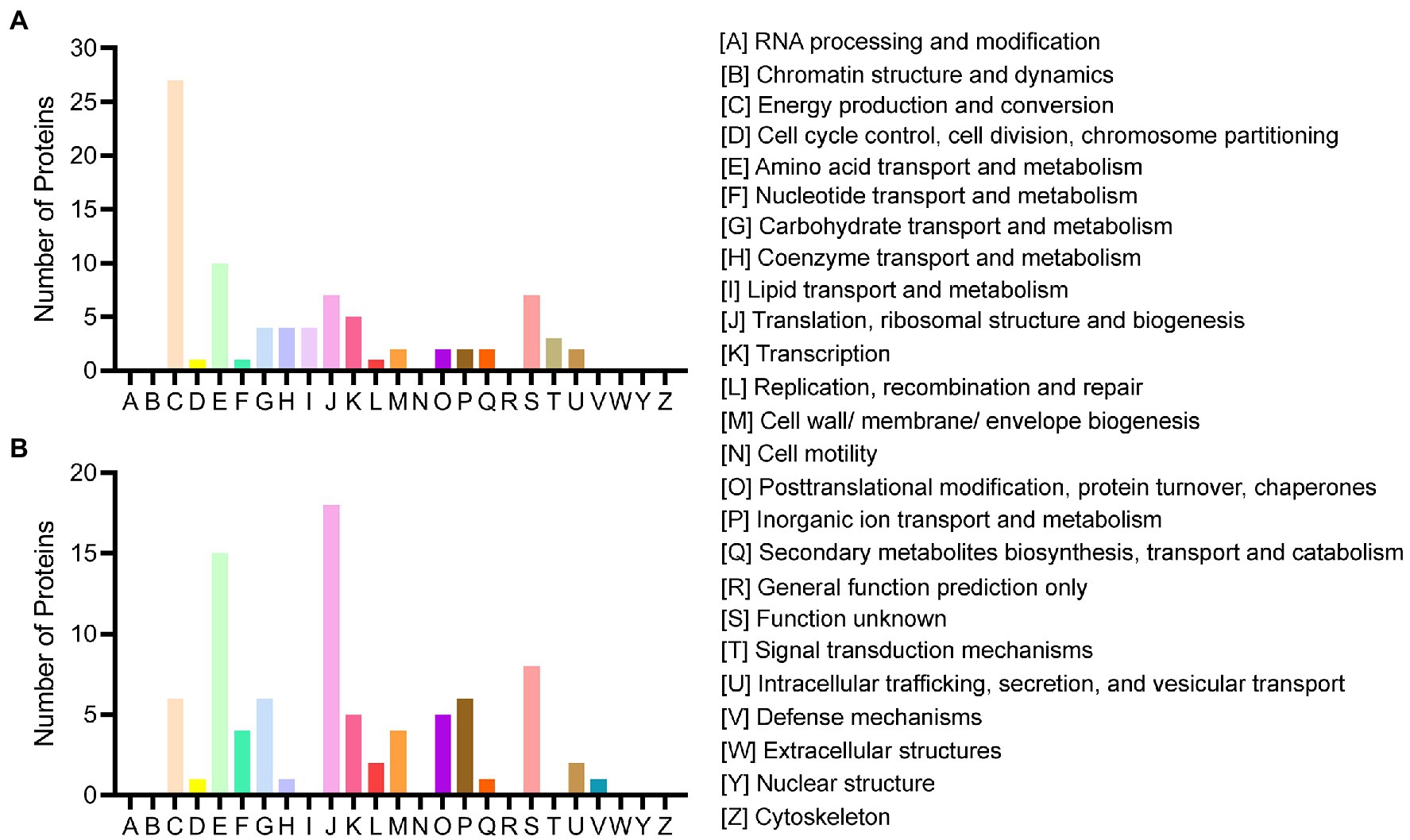
Figure 2. COG functional classification of the DEPs in B. pumilus (B18 vs. B40) (A) and K. vulgare (K18 vs. K40) (B).
Among all the DEPs of B. pumilus SY-A9, five were annotated with multiple functions, and three were not annotated. The five main functional categories were as follows: [C] energy production and conversion (27, 32.14%), [E] amino acid transport and metabolism (10, 11.90%), [J] translation, ribosomal structure and biogenesis (7, 8.33%), [S] function unknown (7, 8.33%), and [K] transcription (5, 5.95%). The detailed functional annotation of the up- and downregulated DEPs of B. pumilus SY-A9 was shown in Supplementary Table S2. The obtained DEPs were classified into four main categories, including “information storage and processing,” “cellular processes and signaling,” “metabolism,” and “poorly characterized.” There were 3, 5, 29, and 1 of the upregulated DEPs involved in “information storage and processing,” “cellular processes and signaling,” “metabolism,” and “poorly characterized,” respectively, and 1 had no annotation function. There were 10, 5, 25, and 6 of the downregulated DEPs involved in “information storage and processing,” “cellular processes and signaling,” “metabolism,” and “poorly characterized,” respectively, and 2 had no annotation function.
Among all the DEPs of K. vulgare 25B-1, three were annotated with multiple functions, and 14 were not annotated. The three main functional categories were as follows: [J] translation, ribosomal structure and biogenesis (18, 21.18%), [E] amino acid transport and metabolism (15, 17.65%), and [S] function unknown (8, 9.41%). The detailed functional annotation of the up- and downregulated DEPs of K. vulgare 25B-1 was shown in Supplementary Table S3. There were 17, 3, 17, and 3 of the upregulated DEPs involved in “information storage and processing,” “cellular processes and signaling,” “metabolism,” and “poorly characterized,” respectively, and 9 had no annotation function. There were 8, 10, 22, and 5 of the downregulated DEPs involved in “information storage and processing,” “cellular processes and signaling,” “metabolism,” and “poorly characterized,” respectively, and 5 had no annotation function.
Seven significantly enriched KEGG pathways (p < 0.05) were obtained based on the DEPs of B. pumilus (B18 vs. B40) (Figure 3A), including biosynthesis of secondary metabolites, microbial metabolism in diverse environments, carbon metabolism, citrate cycle (TCA cycle), pyruvate metabolism, propanoate metabolism, and C5-branched dibasic acid metabolism. Nine significantly enriched KEGG pathways (p < 0.05) were acquired based on the DEPs of K. vulgare (K18 vs. K40) (Figure 3B), including biosynthesis of secondary metabolites, ribosome, biosynthesis of amino acids, microbial metabolism in diverse environments, glycolysis/gluconeogenesis, C5-branched dibasic acid metabolism, nitrogen metabolism, butanoate metabolism, and valine, leucine and isoleucine biosynthesis.
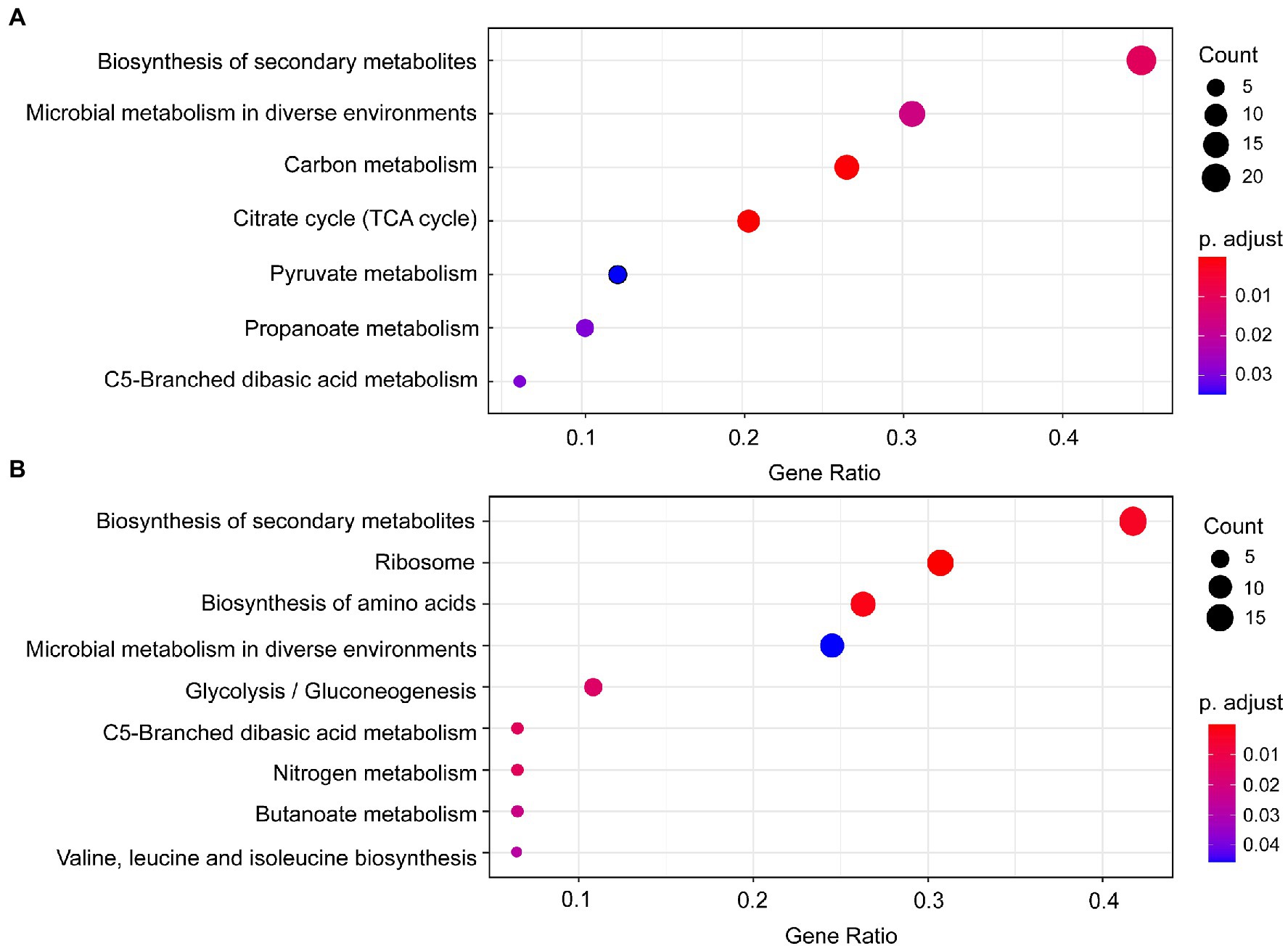
Figure 3. KEGG pathway enrichment of the DEPs in B. pumilus (B18 vs. B40) (A) and K. vulgare (K18 vs. K40) (B). A redder color represents a greater p-value, and a larger circle represents a greater number of proteins.
Protein–protein interaction (PPI) network analysis (Figure 4A) showed that the number of downregulated proteins in B. pumilus SY-A9 was higher than that of the upregulated proteins, and the core proteins were mostly related to energy production and conversion.
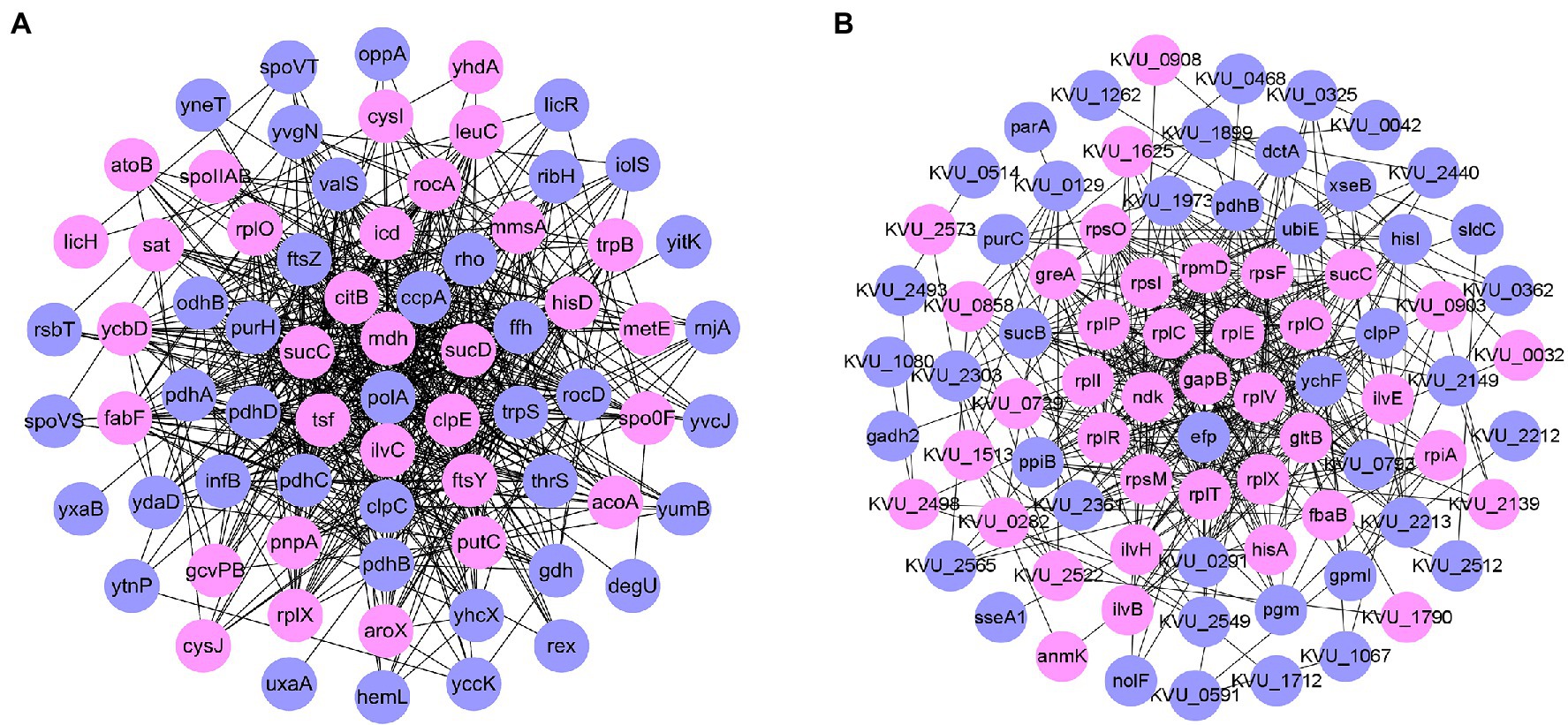
Figure 4. Network analysis of proteins based on differentially expressed proteins (DEPs) in B. pumilus (B18 vs. B40) (A) and K. vulgare (K18 vs. K40) (B). Network analysis revealing the protein–protein interactions among upregulated proteins (pink circle) and downregulated proteins (blue circle).
PPI network analysis (Figure 4B) showed that, the number of downregulated proteins in K. vulgare 25B-1 was higher than that of the upregulated proteins, and the core proteins were mostly related to translation, ribosomal structure and biogenesis.
To test the correspondence between transcript levels and protein expression levels, the transcript levels of 9 DEPs were measured by qPCR, and the result are shown in Table 1. The nine genes represent the predominant metabolic pathways of energy production and conversion, carbohydrate transport and metabolism, post-translational modification, cell wall biogenesis and inorganic ion transport of B. pumilus and K. vulgare, respectively, which showed similar trends in terms of changes in their transcript levels and protein expression levels.
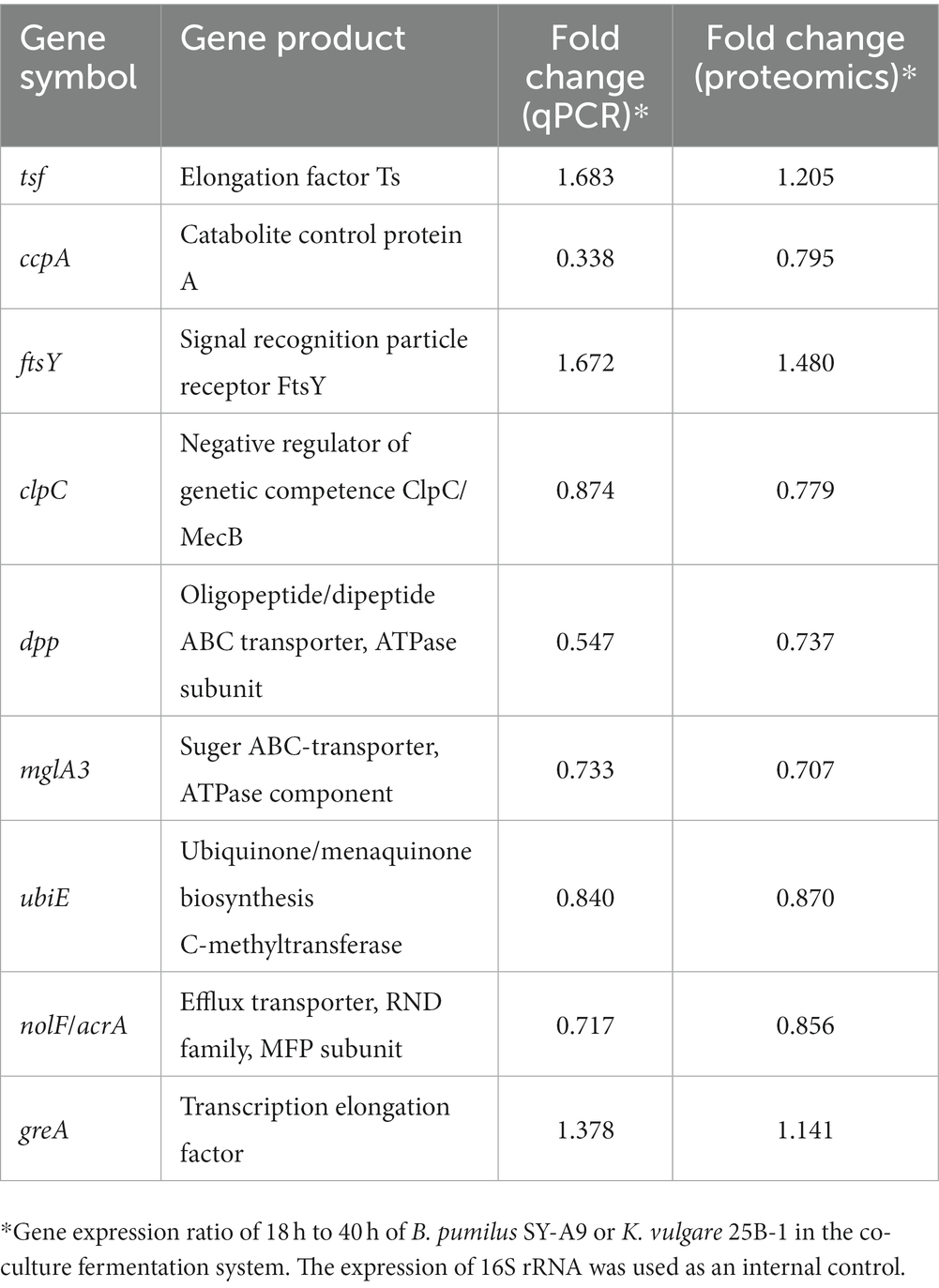
Table 1. Transcriptional complementation of the proteomic output by qPCR assays.
To explore the reasons for inhibiting the growth of B. pumilus, iTRAQ-based proteomic analysis was used in a coculture fermentation system, and a total of 178 DEPs were obtained in the B18 vs. B40 and K18 vs. K40.
Twelve DEPs of B. pumilus, including M5P1Y9, G4EPA3, A0A0C5CAF1, Q3EJU2, M4KMP7, W7R788, L8ASG9, A0A0C5CB21, A0A0K6MRG1, A0A0C5CMU8, Q9KEE6, and A0A132BIK5, were predicted to be involved in the response to acid stress. Here, the acid resistance mechanisms of B. pumilus including the general stress response, pH homeostasis maintenance, metabolic rearrangements and alkali production, and the secondary oxidative stress response are discussed. In Gram-positive bacteria, the alternative sigma factor σB, as the key sigma factor, plays a vital role in regulating the expression of approximately 150 genes in response to significant changes in the surroundings and appear to control, at least in part, the general stress response (Desriac et al., 2013; Rothstein et al., 2017). In general, σB is activated by several stress conditions, e.g., heat shock, acid shock, NaCl or H2O2 exposure and ethanol shock and the activity is controlled by the regulator of Sigma B (RsbRST) complex (Yang et al., 1996; Chaturongakul and Boor, 2004; Lee et al., 2004; Ramesh et al., 2021). In the RsbRST-complex, serine/threonine-protein kinase (RsbT, M5P1Y9) acts as a switch that primarily receives stress signals to regulate the activity of σB (Ramesh et al., 2021). In this study, RsbT was downregulated in B18 vs. B40, indicating that B. pumilus could be under environmental stresses such as osmotic stress, oxidative stress, and acid stress in the coculture fermentation system at 40 h. Moreover, the expression of proteins regulated by σB was downregulated, such as general stress protein 30 (YxaB, G4EPA3), general stress protein 39 (YdaD, A0A0C5CAF1), glucose 1-dehydrogenase (Gdh, Q3EJU2), and uncharacterized oxidoreductase (YccK, M4KMP4). The negative regulator of genetic competence ClpC/MecB (ClpC, L8ASG9) is reported to be required for various processes, including the stress response, sporulation and competence in B. subtilis (Singh et al., 2015), whereas ATP-dependent Clp protease ATP-binding subunit (ClpE, W7R788) acts synergistically with ClpC (Nair et al., 1999). Therefore, the downregulation of ClpC and ClpE also indicated that B. pumilus was under oxidative stress and acid stress in the coculture fermentation system.
Under acidic conditions, bacteria activate enzymes such as FoF1-ATPase, proton transporter, and amino-acid decarboxylase systems to maintain intracellular pH homeostasis (Mols and Abee, 2011). In aerobic bacteria, active H+ transport is integrated with electron transport in the respiratory chain, while anaerobic bacteria use ATP hydrolysis for H+-ATPase molecules (Gandhi and Chikindas, 2007). B. pumilus, as a facultative anaerobic bacterium, may use both ATP synthesis and hydrolysis to maintain its pH homeostasis. The NADH dehydrogenase-like protein (YumB, M4KYK9) involved in electron transport in the respiratory chain was downregulated, and the nucleotide-binding protein (YvcJ, A0A0C5C8R2) that displays ATPase activity was also downregulated. The downregulation of these two DEPs suggested that B. pumilus could use both ATP synthesis and hydrolysis to regulate and thereby maintain intracellular pH homeostasis.
Under acidic conditions, alkali production is observed, such as urea, arginine and ammonia (Desriac et al., 2013). Urea and arginine are rapidly hydrolyzed to ammonia, which then combines with H+ to produce NH4+ to raise the internal pH (Lemos and Burne, 2008). Moreover, it has been reported that up regulation of the acid-inducible glycine decarboxylase system (GcvPA, GcvPB, and GcvT) can produce ammonia and assist in counteracting cytoplasmic acidification (Okamura-Ikeda et al., 1993). The expression of the enzyme catalyzing pyruvate to acetoin was induced upon exposure to acid shocks (Kitko et al., 2009; Mols and Abee, 2011). Here, acetoin: 2,6-dichlorophenolindophenol oxidoreductase subunit alpha (AcoA, A0A0K6MRG1), which cleaves acetoin into acetate and acetaldehyde, was upregulated. The probable glycine dehydrogenase (GcvPB, A0A0C5CMU8) was also upregulated. The upregulation of AcoA and GcvPB suggested that B. pumilus was under acid shocks at 18 h in the coculture fermentation system and could produce ammonia in response to acid stress.
Gram-positive bacteria exposed to inorganic and organic acids under aerobic conditions have been reported to show a major oxidative response (Desriac et al., 2013), and the products of oxygen include superoxide, hydroxyl radicals, and hydrogen peroxide. To survive the toxic effects of reactive oxygen species (ROS), the secondary oxidative stress response upon exposure to acid shocks is indicated by the induction of oxidative stress-associated genes, such as thioredoxins, catalases, and superoxide dismutase (Boylan et al., 2008; Mols et al., 2010). In this paper, upregulation of catalase-peroxidase (KatG, Q9KEE6) indicated that B. pumilus induced KatG to scavenge intracellular H2O2 in response to oxidative stress and acid shocks. In addition, the redox-sensing transcriptional repressor (Rex, A0A0132BIK5), which controls the respiratory pathway by regulating transcription in response to changes in the cellular NADH/NAD+ radox state, was downregulated (Park et al., 2018). The downregulation of Rex indicated that B. pumilus was under acid stress and could resist acid shock.
2-KLG, the acidic and oxidation product of K. vulgare, has been reported to cause oxidative stress, osmotic stress, and DNA damage in Gluconobacter oxydans (Fang et al., 2021). In this paper, it was found that B. pumilus might be under acid shocks from K. vulgare. In addition, based on proteomics analysis, 8 DEPs of K. vulgare, including E3F3W9, E3EYA2, A0A1B1VM42, F9Y8N8, A0A1B1VND4, E3EZ83, E3EZI0, and A0A1B1VNF7, were associated with quorum sensing (QS) and virulence. The LysR-type transcriptional regulator (KVU_1790, E3F3W9) was upregulated and regulated a diverse set of genes involved in virulence, metabolism, QS and motility (Maddocks and Oyston, 2008). The GntR-type transcriptional regulators (KVU_2493, E3EYA2; KVU_0514, A0A1B1VM42) were downregulated and are capable of regulating a variety of biological processes, including metabolic pathways, morphogenesis, the cell envelope stress response and antibiotic production (Lemmens et al., 2019). The ATP-dependent Clp protease proteolytic subunit (ClpP, A0A1B1VND4) was upregulated, which degrades or hydrolyzes misfolded or damaged proteins in response to environmental stress and regulates the production of virulence factors (Michel et al., 2006; Cohn et al., 2007). The upregulation of ClpP indicated that K. vulgare might be under environmental stress in the coculture fermentation system and produce virulence factors in response to it. Interestingly, involved in multidrug efflux proteins, ABC-type multidrug transport system ATPase component-like protein (KVU_2149, E3EZI0), efflux transporter, RND family, MFP subunit (AcrA, A0A1B1VNF7), and oligopeptide/dipeptide (Opp/Dpp) ABC transporter, ATPase subunit (KVU_1262, E3EZ83) were downregulated. In Gram-negative bacteria, the multidrug efflux mechanism is mediated by transport proteins that are resistant to toxic compounds, and these transporters are associated with intrinsic and acquired antimicrobial resistance (Moreira et al., 2004). In addition, KVU_1262 is reported to be involved in the transport of amino acids, virulence factors, and QS factors (Masukagami et al., 2018). The CRISPR-associated protein (KVU_1368, F9Y8N8) correlated with QS (Hoyland-Kroghsbo et al., 2017) was upregulated, implying the presence of a QS system in the coculture fermentation system at 18 h.
The QS system is known as a cell–cell communication mechanism that is mediated by cell density and involved in the production, release, and detection of extracellular signal molecules (autoinducers, AI) (Hoyland-Kroghsbo et al., 2017). When the threshold concentration of AI is reached and detected by the group, a population-wide alteration in gene expression will occur in response to it. QS is a mechanism that controls virulence factor secretion, competence, biofilm formation, bioluminescence, and sporulation (Bassler and Losick, 2006). In general, N-acylhomoserine lactones (AHLs) are one of the AIs used by Gram-negative bacteria (Kaufmann et al., 2005). However, the probable quorum-quenching lactonase (YtnP, D4G066) of B. pumilus was downregulated, which was reported to hydrolyze AI to inhibit the signaling pathway, biofilm formation, and production of virulence factors (Sun et al., 2021). In vitamin C fermentation, Wang et al. (2019) established the programmed cell death module based on the AHLs-LuxR QS system in G. oxydans to reduce the competition between G. oxydans and K. vulgare, but not mentioned the QS system of K. vulgare. Although the QS system in other Gram-negative bacteria is well defined, such as Pseudomonas aeruginosa, Vibrio fischeri, and Vibrio harveyi, it is not clear what kind of autoinducers are produced by K. vulgare. Moreover, it is not known whether K. vulgare produces virulence factors, and the relationship between virulence factors and autoinducers in the QS system of K. vulgare has rarely been mentioned.
In our previous study, we found that K. vulgare contains an N-acyl-L-homoserine lactone synthetase-like protein (KVU_2075) and LuxR (KVU_2076), indicating that K. vulgare could use the AHLs-LuxR QS system to regulate its own cell density (Zhang and Lyu, 2022). Here, to further investigate the availability of QS in the coculture fermentation system, the expression of genes KVU_2075 (encoded N-acyl-L-homoserine lactone synthetase-like protein), KVU_2076 (encoded LuxR family transcriptional regulator), and ytnP (encoded probable quorum-quenching lactonase) were measured in monoculture and coculture fermentation systems at 6, 18, 40, and 72 h, respectively (Supplementary Figure S1). The expression of genes KVU_2075 and KVU_2076 of K. vulgare in the coculture fermentation system was higher than that in the monoculture fermentation system at 6 h and 18 h. The high expression of KVU_2075 implies a high cell density of K. vulgare in the coculture fermentation system (Subramoni and Vittorio, 2009). However, at 40 h and 72 h, the expression of K. vulgare genes KVU_2075 and KVU_2076 in the coculture fermentation system was significantly lower than that in the monoculture fermentation system. At 6 h and 40 h, no expression levels of the ytnP gene of B. pumilus were detected in either monoculture or coculture fermentation systems, whereas at 18 h and 72 h, ytnP expression levels were much higher in the coculture fermentation system than in the monoculture fermentation system. The lower expression of KVU_2075 and KVU_2076 and the higher expression of ytnP suggested that B. pumilus could secrete YtnP to hydrolyze the AHLs produced by K. vulgare to interfere with the normal signaling pathway of K. vulgare in the coculture fermentation system.
In this paper, we analyzed the reasons for inhibiting the growth of B. pumilus by iTRAQ-based proteomics analysis. In conclusion, B. pumilus might be under the acid shock and quorum sensing system of K. vulgare and the relationship between the two bacteria could be antagonistic (Figure 5). In addition, the synthetic consortium used in this study was screened from soil. Thus, the study of the interactions between K. vulgare and B. pumilus not only provides a better insight into the interactions between synthetic consortia in vitamin C microbial fermentation, but also further contributes to the understanding of soil-microbe interactions.
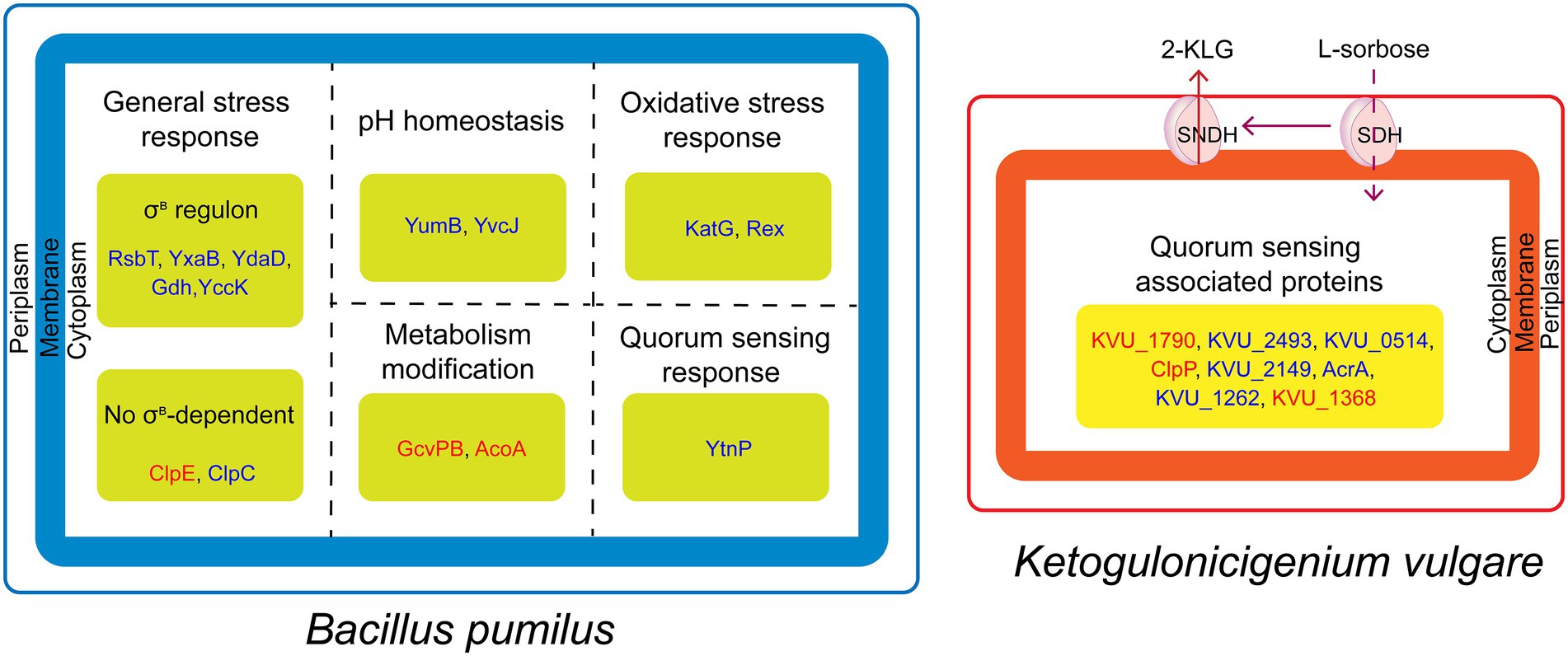
Figure 5. Schematic representation of antagonistic mechanism between B. pumilus and K. vulgare. K. vulgare produce 2-KLG and quorum sensing associated virulence factors against B. pumilus, whereas, B. pumilus produce proteins in response to acid stress and quorum sensing. The red and blue fonts represent upregulated and downregulated proteins, respectively.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
SL contributed to conception and design of the study. QZ performed the statistical analysis and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by the National Science Foundation of China (Grant number: 31370077) and Liaoning Province Science Research Fund (Grant number: LSNJC201915). Shenyang Agricultural University Postgraduate Innovation Cultivation Project (Grant number: 2022YCXB11).
The authors thank Yunhe Zhang and Beijing Bangfei Bioscience Co., Ltd. for their excellent technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1131000/full#supplementary-material
Bassler, B. L., and Losick, R. (2006). Bacterially speaking. Cells 125, 237–246. doi: 10.1016/j.cell.2006.04.001
Boylan, J. A., Lawrence, K. A., Downey, J. S., and Gherardini, F. C. (2008). Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Mol. Microbiol. 68, 786–799. doi: 10.1111/j.1365-2958.2008.06204.x
Brune, K. D., and Bayer, T. S. (2012). Engineering microbial consortia to enhance biomining and bioremediation. Front. Microbiol. 3:203. doi: 10.3389/fmicb.2012.00203
Chaturongakul, S., and Boor, K. J. (2004). RsbT and RsbV contribute to σB-dependent survival under environmental, energy, and intracellular stress conditions in listeria monocytogenes. J. Bacteriol. 70, 5349–5356. doi: 10.1128/aem.70.9.5349-5356.2004
Cohn, M. T., Ingmer, H., Mulholland, F., Jorgensen, K., Wells, J. M., and Brondsted, L. (2007). Contribution of conserved ATP-dependent proteases of Campylobacter jejuni to stress tolerance and virulence. Appl. Environ. Microbiol. 73, 7803–7813. doi: 10.1128/aem.00698-07
Desriac, N., Broussolle, V., Postollec, F., Mathot, A. G., Sohier, D., Coroller, L., et al. (2013). Bacillus cereus cell response upon exposure to acid environment: Toward the identification of potential biomarkers. Front. Microbiol. 4:284. doi: 10.3389/fmicb.2013.00284
Fang, J., Wan, H., Zeng, W. Z., Li, J. H., Chen, J., and Zhou, J. W. (2021). Transcriptome analysis of Gluconobacter oxydans WSH-003 exposed to elevated 2-keto-L-gulonic acid reveals the response to osmotic and oxidative stress. Appl. Biochem. Biotechnol. 193, 128–141. doi: 10.1007/s12010-020-03405-8
Gandhi, M., and Chikindas, M. L. (2007). Listeria: A foodborne pathogen that knows how to survive. Int. J. Food Microbiol. 113, 1–15. doi: 10.1016/j.ijfoodmicro.2006.07.008
Hoyland-Kroghsbo, N. M., Paczkowski, J., Mukherjee, S., Broniewski, J., Westra, E., Bondy-Denomy, J., et al. (2017). Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci. U. S. A. 114, 131–135. doi: 10.1073/pnas.1617415113
Jia, N., Ding, M. Z., Gao, F., and Yuan, Y. J. (2016). Comparative genomics analysis of the companion mechanisms of Bacillus thuringiensis Bc601 and Bacillus endophyticus Hbe603 in bacterial consortium. Sci. Rep. 6:28794. doi: 10.1038/srep28794
Jia, N., Du, J., Ding, M. Z., Gao, F., and Yuan, Y. J. (2015). Genome sequence of Bacillus endophyticus and analysis of its companion mechanism in the Ketogulonicigenium vulgare-Bacillus strain consortium. PLoS One 10:e0135104. doi: 10.1371/journal.pone.0135104
Kaufmann, G. F., Sartorio, R., Lee, S. H., Rogers, C. J., Meijler, M. M., Moss, J. A., et al. (2005). Revisiting quorum sensing: Discovery of additional chemical and biological functions for 3-oxo-N-acylhomoserine lactones. Proc. Natl. Acad. Sci. U. S. A. 102, 309–314. doi: 10.1073/pnas.0408639102
Kitko, R. D., Cleeton, R. L., Armentrout, E. I., Lee, G. E., Noguchi, K., Berkmen, M. B., et al. (2009). Cytoplasmic acidification and the benzoate transcriptome in Bacillus subtilis. PLoS One 4:e8255. doi: 10.1371/journal.pone.0008255
Lee, E. J., Cho, Y. H., Kim, H. S., Ahn, B. E., and Roe, J. H. (2004). Regulation of σB by an anti- and an anti-anti-sigma factor in Streptomyces coelicolor in response to osmotic stress. J. Bacteriol. 186, 8490–8498. doi: 10.1128/jb.186.24.8490-8498.2004
Lemmens, L., Tilleman, L., De Koning, E., Valegard, K., Lindas, A. C., Van Nieuwerburgh, F., et al. (2019). YtrASa, a GntR-family transcription factor, represses two genetic loci encoding membrane proteins in Sulfolobus acidocaldarius. Front. Microbiol. 10:2084. doi: 10.3389/fmicb.2019.02084
Lemos, J. A., and Burne, R. A. (2008). A model of efficiency: Stress tolerance by Streptococcus mutans. Microbiology 154, 3247–3255. doi: 10.1099/mic.0.2008/023770-0
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Ma, Q., Bi, Y. H., Wang, E. X., Zhai, B. B., Dong, X. T., Qiao, B., et al. (2019). Integrated proteomic and metabolomic analysis of a reconstructed three-species microbial consortium for one-step fermentation of 2-keto-L-gulonic acid, the precursor of vitamin C. J. Ind. Microbiol. Biotechnol. 46, 21–31. doi: 10.1007/s10295-018-2096-3
Ma, Q., Zhou, J., Zhang, W. W., Meng, X. X., Sun, J. W., and Yuan, Y. J. (2011). Integrated proteomic and metabolomic analysis of an artificial microbial community for two-step production of vitamin C. PLoS One 6:e26108. doi: 10.1371/journal.pone.0026108
Maddocks, S. E., and Oyston, P. C. F. (2008). Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154, 3609–3623. doi: 10.1099/mic.0.2008/022772-0
Mandlaa, S. Z. Y., Wang, R. G., Han, X. D., Xu, H., and Yang, W. C. (2018). Enhanced 2-keto-L-gulonic acid production by applying L-sorbose-tolerant helper strain in the coculture system. AMB Express 8:30. doi: 10.1186/s13568-018-0562-y
Masukagami, Y., Nijagal, B., Tseng, C. W., Dayalan, S., Tivendale, K. A., Markham, P. F., et al. (2018). Metabolite profiling of Mycoplasma gallisepticum mutants, combined with bioinformatic analysis, can reveal the likely functions of virulence-associated genes. Vet. Microbiol. 223, 160–167. doi: 10.1016/j.vetmic.2018.08.001
Michel, A., Agerer, F., Hauck, C. R., Herrmann, M., Ullrich, J., Hacker, J., et al. (2006). Global regulatory impact of ClpP protease of Staphylococcus aureus on regulons involved in virulence, oxidative stress response, autolysis, and DNA repair. J. Bacteriol. 188, 5783–5796. doi: 10.1128/jb.00074-06
Mols, M., and Abee, T. (2011). Bacillus cereus responses to acid stress. Environ. Microbiol. 13, 2835–2843. doi: 10.1111/j.1462-2920.2011.02490.x
Mols, M., Van Kranenburg, R., Van Melis, C. C. J., Moezelaar, R., and Abee, T. (2010). Analysis of acid-stressed Bacillus cereus reveals a major oxidative response and inactivation-associated radical formation. Environ. Microbiol. 12, 873–885. doi: 10.1111/j.1462-2920.2009.02132.x
Moreira, M. A. S., Souza, E. C., and Moraes, C. A. (2004). Multidrug efflux system in gram-negative bacteria. Braz. J. Microbiol. 35, 19–28. doi: 10.1590/s1517-83822004000100003
Nair, S., Frehel, C., Nguyen, L., Escuyer, V., and Berche, P. (1999). ClpE, a novel member of the HSP100 family, is involved in cell division and virulence of Listeria monocytogenes. Mol. Microbiol. 31, 185–196. doi: 10.1046/j.1365-2958.1999.01159.x
Okamura-Ikeda, K., Ohmura, Y., Fujiwara, K., and Motokawa, Y. (1993). Cloning and nucleotide sequence of the GCV operon encoding the Escherichia coli glycine-cleavage system. Eur. J. Biochem. 216, 539–548. doi: 10.1111/j.1432-1033.1993.tb18172.x
Park, Y. W., Jang, Y. Y., Joo, H. K., and Lee, J. Y. (2018). Structural analysis of redox-sensing transcriptional repressor rex from Thermotoga maritima. Sci. Rep. 8:13244. doi: 10.1038/s41598-018-31676-z
Ramesh, M., Nitharwal, R. G., Behra, P. R. K., Fredrik, P. B. M., Dasgupta, S., and Kirsebom, L. A. (2021). Intracellular localization of the mycobacterial stressosome complex. Sci. Rep. 11:10060. doi: 10.1038/s41598-021-89069-8
Roell, G. W., Zha, J., Carr, R. R., Koffas, M. A., Fong, S. S., and Tang, Y. J. J. (2019). Engineering microbial consortia by division of labor. Microb. Cell Factories 18:35. doi: 10.1186/s12934-019-1083-3
Rothstein, D. M., Lazinski, D., Osburne, M. S., and Sonenshein, A. L. (2017). A mutation in the Bacillus subtilis rsbU gene that limits RNA synthesis during sporulation. J. Bacteriol. 199, 212–217. doi: 10.1128/JB.00212-17
Sabra, W., Dietz, D., Tjahjasari, D., and Zeng, A. P. (2010). Biosystems analysis and engineering of microbial consortia for industrial biotechnology. Eng. Life Sci. 10, 407–421. doi: 10.1002/elsc.201000111
Singh, L. K., Dhasmana, N., Sajid, A., Kumar, P., Bhaduri, A., Bharadwaj, M., et al. (2015). clpC operon regulates cell architecture and sporulation in Bacillus anthracis. Environ. Microbiol. 17, 855–865. doi: 10.1111/1462-2920.12548
Subramoni, S., and Vittorio, V. (2009). LuxR-family 'solos': Bachelor sensors/regulators of signalling molecules. Microbiology 155, 1377–85–1385. doi: 10.1099/mic.0.026849-0
Sun, X. H., Hill, P., Liu, J., Qian, J., Ma, Y. T., and Zhou, S. F. (2021). Marine-source quorum quenching enzyme YtnP to improve hygiene quality in dental units. Mar. Drugs 19:225. doi: 10.3390/md19040225
Wang, E. X., Ding, M. Z., Ma, Q., Dong, X. T., and Yuan, Y. J. (2016). Reorganization of a synthetic microbial consortium for one-step vitamin C fermentation. Microb. Cell Factories 15:21. doi: 10.1186/s12934-016-0418-6
Wang, E. X., Liu, Y., Ma, Q., Dong, X. T., Ding, M. Z., and Yuan, Y. J. (2019). Synthetic cell-cell communication in a three-species consortium for one-step vitamin C fermentation. Biotechnol. Lett. 41, 951–961. doi: 10.1007/s10529-019-02705-2
Yakushi, T., Takahashi, R., Matsutani, M., Kataoka, N., Hours, R. A., Ano, Y., et al. (2020). The membrane-bound sorbosone dehydrogenase of Gluconacetobacter liquefaciens is a pyrroloquinoline quinone-dependent enzyme. Enzym. Microb. Technol. 137:109511. doi: 10.1016/j.enzmictec.2020.109511
Yang, X., Kang, C. M., Brody, M. S., and Price, C. M. (1996). Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to active a bacterial transcription factor. Genes Dev. 10, 2265–2275. doi: 10.1101/gad.10.18.2265
Yang, W. C., Sun, H., Dong, D., Ma, S., Mandlaa, W. Z. X., and Xu, H. (2021). Enhanced 2-keto-L-gulonic acid production by a mixed culture of Ketogulonicigenium vulgare and bacillus megaterium using three-stage temperature control strategy. Braz. J. Microbiol. 52, 257–265. doi: 10.1007/s42770-020-00396-w
Zeng, W. Z., Wang, P. P., Li, N., Li, J. H., Chen, J., and Zhou, J. W. (2020). Production of 2-keto-L-gulonic acid by metabolically engineered Escherichia coli. Bioresour. Technol. 318:124069. doi: 10.1016/j.biortech.2020.124069
Zhang, Q., Lin, Y., Zhang, H. H., Shen, G. Z., and Lyu, S. X. (2022). Siderophores of Bacillus pumilus promote 2-keto-L-gulonic acid production in a vitamin C microbial fermentation system. J. Basic Microbiol. 62, 833–842. doi: 10.1002/jobm.202200237
Zhang, Q., and Lyu, S. X. (2022). Microbial interactions in a vitamin C industrial fermentation system: Novel insights and perspectives. Appl. Environ. Microb. 88:e0121222. doi: 10.1128/aem.01212-22
Zhang, Z. X., Sun, J. W., and Yuan, J. Q. (2015). Investigating the interaction between Gluconobacter oxydans and Bacillus megaterium for 2-keto-L-gulonic acid biosynthesis in the two-step vitamin C fermentation. J. Shanghai Jiaotong Univ. 20, 281–285. doi: 10.1007/s12204-015-1622-y
Keywords: microbial consortium, quorum sensing, proteomics, communication, vitamin C
Citation: Zhang Q and Lyu S (2023) iTRAQ-based proteomics analysis of Bacillus pumilus responses to acid stress and quorum sensing in a vitamin C fermentation system. Front. Microbiol. 14:1131000. doi: 10.3389/fmicb.2023.1131000
Edited by:
Ashton Keith Cowan, Rhodes University, South AfricaReviewed by:
Tianwen Wang, Anhui Polytechnic University, ChinaCopyright © 2023 Zhang and Lyu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuxia Lyu, bHVzaHV4aWFAc3lhdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.