 Li Fu
Li Fu Shouchao Lai1
Shouchao Lai1 Zhuo Zhou
Zhuo Zhou Lei Cheng
Lei Cheng- 1Key Laboratory of Development and Application of Rural Renewable Energy, Ministry of Agriculture and Rural Affairs, Biogas Institute of Ministry of Agriculture and Rural Affairs, Chengdu, China
- 2Research Institute of Petroleum Exploration and Development, Beijing, China
Coalbed water is a semi-open system connecting underground coalbeds with the external environment. Microorganisms in coalbed water play an important role in coal biogasification and the carbon cycle. The community assemblages of microorganisms in such a dynamic system are not well understood. Here, we used high-throughput sequencing and metagenomic analysis to investigate microbial community structure and identify the potential functional microorganisms involved in methane metabolism in coalbed water in the Erlian Basin, a preferred low-rank coal bed methane (CBM) exploration and research area in China. The results showed that there were differences in the responses of bacteria and archaea to seasonal variation. Bacterial community structure was affected by seasonal variation but archaea was not. Methane oxidation metabolism dominated by Methylomonas and methanogenesis metabolism dominated by Methanobacterium may exist simultaneously in coalbed water.
1. Introduction
The world’s total natural gas reserves include both conventional and non-conventional sources. Coal bed methane (CBM) is an unconventional natural gas produced during coal formation. CBM is a hydrocarbon gas that is adsorbed on the surface of coal matrix particles and dissociated in coal pores or dissolved in the water of coal seams (Moore, 2012). Methane in coal is derived from both biogenic and thermogenic processes (Strapoc et al., 2011), and approximately 20% of the world’s natural gas is biogenic (Wang et al., 2016). The origin of methane depends upon the maturity of coal. Generally, deep and highly matured coal is associated with thermogenic methane, whereas shallow and less mature coal is associated with biogenic methane. Coal reservoirs of moderate depth and maturity have a mixture of biogenic and thermogenic methane.
The biogasification of coal is a mixed fermentation process involving many different microorganisms (Faiz and Hendry, 2006; Park and Liang, 2016). Bacteria related to Firmicutes, Actinobacteria, and Bacteroidetes and all subgroups of Proteobacteria seem to be widespread in CBM (Beckmann et al., 2011a; Colosimo et al., 2016). These taxonomic groups are known for their versatile metabolic activity and hydrocarbon degrading capabilities. In the presence of microorganisms, coal (complex polymers) is degraded into a series of intermediates such as polycyclic aromatic hydrocarbons (PAHs), heterocyclic compounds, alkyl phenols, aromatic amines, alkyl aromatics, long-chain fatty acids, and aliphatic hydrocarbons (Park and Liang, 2016; Wang et al., 2018). These are intermediates to the formation of acetate and H2, which undergo methanogenesis (Conrad, 2020). Methanogens commonly detected in coal seams environments can be divided into three types according to their methanogenic pathways. Hydrogenotrophic methanogens like Methanobacterium consume H2 to reduce CO2 to generate CH4 (Rosewarne et al., 2013). Acetotrophic methanogens like Methanosarcina and Methanosaeta convert acetate into CH4 and CO2 (Krüger et al., 2008; Beckmann et al., 2011b). Methylotrophic methanogens like Methanolobus use methanol, methylamine, and other methyl substances to produce CH4 (Doerfert et al., 2009; Guo et al., 2012b). Unexpectedly, Mayumi et al. characterized a strain of Methermicoccus shengliensis that can produce methane from the dozens of methoxylated aromatic compounds found in a variety of coal types (Mayumi et al., 2016). This portends methane production from coal by a single methoxydotrophic methanogen. Methane in these environments can be utilized by methanotrophs as electron donors and energy sources (Smith and Wrighton, 2019). Methanotrophic microorganisms play roles in mitigating methane emissions. However, the function of these microorganisms is often neglected in studies of CBM.
The Erlian Basin investigated in this study has CBM geological characteristics similar to the Powder River basin in the United States (Ayers, 2002; Sun et al., 2017), and is the preferred low-rank CBM exploration and research area in China (Chen and Pan, 2019). The resource quantity of low-rank CBM in the Erlian Basin is approximately 2.48 × 1012 m3 (Sun et al., 2018). In order to effectively develop low-rank CBM in China, a simulation experiment was conducted to test the microbial community and clarify the methanogenic pathway and biological gas recovery potential in the coal seams of the Jiergalangtu depression, Erlian Basin. The results showed that both aceticlastic and hydrotrophic methanogenesis were detected in the coal and coalbed water (Sun et al., 2018). Previous studies have explored the methane production potential (Lai et al., 2020) and influencing factors (Chen et al., 2018) of coalbed organisms in the Erlian Basin. However, the above studies were based on simulation experiments, and cannot reflect the remodeling of microbial community structure that occurs through real environmental changes.
Coalbed water is a semi-open system connecting the underground coalbed with the external environment, and the microorganisms in coalbed water respond directly to various environmental changes. Their response mechanism is of great importance to both microbial mechanism research and production applications. In recent years, the application of high-throughput sequencing (Guo et al., 2012a; Wei et al., 2014; Beckmann et al., 2019) and metagenomics (Robbins et al., 2016) have gradually revealed the biogenic CBM microbial community. Metagenomics allows to assess the presence and genomic potential of an organism, from which hypotheses about its physiology can be derived. Therefore, in this study, coalbed water samples were collected from the Erlian Basin during different seasons. The seasonal variation in the microbial community and the functional microorganisms involved in methane metabolism were investigated using high-throughput sequencing and metagenomic analysis.
2. Materials and methods
2.1. Coalbed water sample collection
Twenty-three original samples of coalbed water were collected from the Erlian Basin (Supplementary Figure S1) in May, August, and October 2018, corresponding to the local spring, summer, and autumn. Meteorological conditions such as precipitation and temperature vary greatly between seasons in this region. In May, August, and October 2018, the total monthly precipitation was 6.6, 30.3, and 0.7 mm, and the mean temperature was 18.2, 23.1, and 4.4°C, respectively (Supplementary Table S2). The formation water samples were gathered from a depth of 400 meters. Water samples dedicated for chemical composition analysis were kept on ice. The basic physical and chemical properties of coalbed water were determined by the unconventional experimental center of China National Offshore Oil Corporation Energy Development Co., LTD. The pH of the samples ranged from 7.53 to 8.79, which was slightly alkaline. Nitrite ions were detected in only two samples (JM10O and JM13O). Nitrate and sulfate concentrations were also detected at low levels. Sodium concentration was detected at a high level based on the Chinese classification of mine water (Supplementary Table S1). Water samples dedicated for microbial analysis were stored and transported in ambient temperature. Water samples were transported by road, and the average transportation time was 3 days.
2.2. DNA extraction
For original coalbed water samples, 500 ml water sample was required per sample for high speed centrifugation (5 min at 14,000 g at 4°C). For the sample of the culture experiment, 2 ml of culture at the end of the experiment was required for high speed centrifugation (5 min at 14,000 g at 4°C). After the supernatant was discarded, the precipitate was resuspended with 20 ml phosphate buffer, and the suspension was transferred to a new, enzyme-free spiral tube for further centrifugation. The precipitate obtained after final centrifugation was stored at −80°C until DNA extraction. DNA was extracted using a Water DNA Isolation Kit (Foregene).
2.3. High-throughput sequencing of the 16S rRNA genes of bacteria and archaea
High-throughput sequencing was performed on the extracted DNA samples by Beijing Nuohe Zhiyuan Co. Ltd. DNA concentration and purity were determined by ultramicro-spectrophotometer, and the quality of all DNA samples met the sequencing requirements. The V3-V4 region of the bacterial 16S rRNA genes was amplified using the bacteria-specific primers 341F_CCTAYGGGRBGCASCAG/806R_GGACTACNNGGGTATCTAAT. The V4-V5 region of archaea 16S rRNA genes was amplified using the archaea-specific primers Arch519F_CAGCCGCCGCGGTAA/Arch915R_GTGCTCCCCCGCCAATTCCT. Illumina HiSeq 2,500 was used for pair-end sequencing after the amplification products were recovered by gel cutting.
2.4. High-throughput data analysis
QIIME software was used to conduct quality control, chimeric removal, re-sampling, OTU (Operational Taxonomic Unit) clustering, selection of representative sequences, species annotation, and singleton removal for sequencing data from the sequencing unloading machine. Because of limited primer specificity, a small number of archaea sequences were found in the amplified fragments of bacterial primers, and bacterial sequences were also amplified by archaea primers. After the mismatched OTUs were deleted by Excel, the structure of the microbial community was analyzed. For original coalbed water samples, a total of 673,216 high-quality sequences were obtained using high-throughput sequencing, among which 502,724 were bacterial sequences and 170,492 were archaeal sequences (Supplementary Table S3). A total of 1,009 archaeal OTUs were obtained based on a 97% sequence similarity and the average number of OTUs of the samples was 44. The number of OTUs in samples JM3A (104) and JM4-1A (102) was higher than that of other samples. A total of 20,082 bacterial OTUs were obtained, and the average number of OTUs per sample was 873. The Shannon index indicated high bacterial diversity (3.57 ± 0.51; mean ± SD). Coverage for bacteria and archaea ranged from 0.96 to 0.98, indicating that sequencing depth was sufficient.
2.5. Metagenomics sequencing and analysis
The genomic DNA was randomly sheared into short fragments. The obtained fragments were end repaired, A-tailed and further ligated with Illumina adapter. The fragments with adapters were PCR amplified, size selected, and purified. The library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. Quantified libraries will be pooled and sequenced on Illumina platforms, according to effective library concentration and data amount required. The raw reads were dereplicated and quality-trimmed using Trimmomatic (Bolger et al., 2014). High-quality metagenomic sequences were de novo assembled using SPAdes (v.3.12.0) with the default k-mer size (Nurk et al., 2017). The assembled scaffolds were binned using MetaBAT (v.2.12.1) based on tetranucleotide frequency, GC content, and coverage (Kang et al., 2019). Partial and near-complete genomes were recovered after binning. The completeness, contamination, and strain heterogeneity of MAGs were evaluated using CheckM (v.1.0.11) based on lineage-specific marker sets (Parks et al., 2015). The taxonomic assignment of the different bins was conducted with the GTDB-Tk package (v.1.3.0; Chaumeil et al., 2019), and bins were translated by Prodigal using the “-p meta” parameters (Hyatt et al., 2010). For each predicted coding sequence (CDS), protein function was annotated using the KEGG server (BlastKOALA) and eggNOG-mapper (Kanehisa et al., 2016).
2.6. Methanogenic culture experiment
Water sample JM13M was selected for methanogenic simulation culture. After shaking well, the 180 ml water sample was added to the 300 ml sterilized and dried serum bottle. The pH was adjusted to neutral. Na2S·9H2O (1.0 mM), resazurin (0.0005 g L−1), trace elements (2.0 ml/L), and vitamins (V284/VB1/VB12; 4.0 ml/L) were supplemented. Vitamin and trace element solutions were prepared as described previously (Lu and Lu, 2012). The headspace gas was flushed with N2 for 30 min to remove oxygen. Fourteen methanogenic precursors were selected for methanogenic simulation culture:H2/CO2 (50 ml/25 ml), sodium formate (20 mM), dimethyl sulfide (20 mM), dimethylamine (20 mM), methanol (20 mM), sodium acetate (20 mM), sodium propionate (20 mM), sodium butyrate (20 mM), ethanol (20 mM), 1-propanol (20 mM), 2-propanol (20 mM), 1-butanol (20 mM), 2-butanol (20 mM), and Choline hydroxide (20 mM). No precursor was added to the control. Three parallel were set for each treatment and incubated at 35°C in the dark.
2.7. CH4 detection
The CH4 content in the headspace of all culture bottles was measured using gas chromatograph 7820A (Agilent Technologies, United States) with Porapak Q capillary columns (3 m) at 65°C. High-purity hydrogen (99.999%) was used as the mobile phase. The column flow rate was 27 ml/min. TCD (thermal detector) at 130°C, the inlet temperature was 105°C, the reference flow was 30 ml/min, and the tail blow flow was 2 ml/min. The gas detection time was 1.5 min, and the injection volume was 0.2 ml. The relative proportions of N2, CH4, and CO2 in the sample gas were calculated using the corrected area normalization method for the peak diagram. All samples were tested at 5-day intervals.
2.8. Data availability
The datasets generated for this study have been deposited under NCBI. For original coalbed water samples, 16S rRNA genes of archaea: PRJNA681683. 16S rRNA genes of bacteria: PRJNA681635. For the sample of the culture experiment, 16S rRNA genes of archaea and bacteria: PRJNA923302. Metagenomic raw data: PRJNA682244 (Supplementary Table S9).
3. Results and discussion
3.1. Response of microbial community structure to seasonal variation
A total of 24 bacterial phyla, including the candidate divisions, were detected from coalbed water samples (Supplementary Figure S2). Proteobacteria was the most abundant phylum, and the average relative abundance of Proteobacteria in the 23 samples was 77.3% (range 25.6–93.0%). Other bacteria were mainly affiliated with the phyla Bacteroidetes (10.1%) and Firmicutes (6.7%; Supplementary Figure S2). Bacteria with a relative abundance of more than 5% (genus level) in each sample were selected, as shown in Figure 1A. The dominant Gammaproteobacteria genera were Methylomonas (15.1%), Methylobacter (2.8%), Thioalkalimicrobium (2.1%), Arenimonas (2.0%) Methylomicrobium (0.6%), Aeromonas (0.4%), Rheinheimera (0.3%), and Shewanella (0.3%). Epsilonproteobacteria included Arcobacter (5.2%), Sulfuricurvum (2.9%), Sulfurimonas (1.5%), and Sulfurospirillum (1.3%). Betaproteobacteria included Methylotenera (3.8%), Hydrogenophaga (1.8%), Acidovorax (1.1%), Malikia (1.1%), and Thiobacillus (0.6%). Alphaproteobacteria included Porphyrobacter (2.5%), Rhizobium (1.3%), Roseovarius (0.7%), Hyphomonas (0.6%), and Hoeflea (0.4%). Bacteroidetes included Flavobacterium (1.8%), Algoriphagus (1.3%), and Barnesiella (0.5%). In Firmicutes, only Acetobacterium (1.1%) showed slightly higher relative abundance.
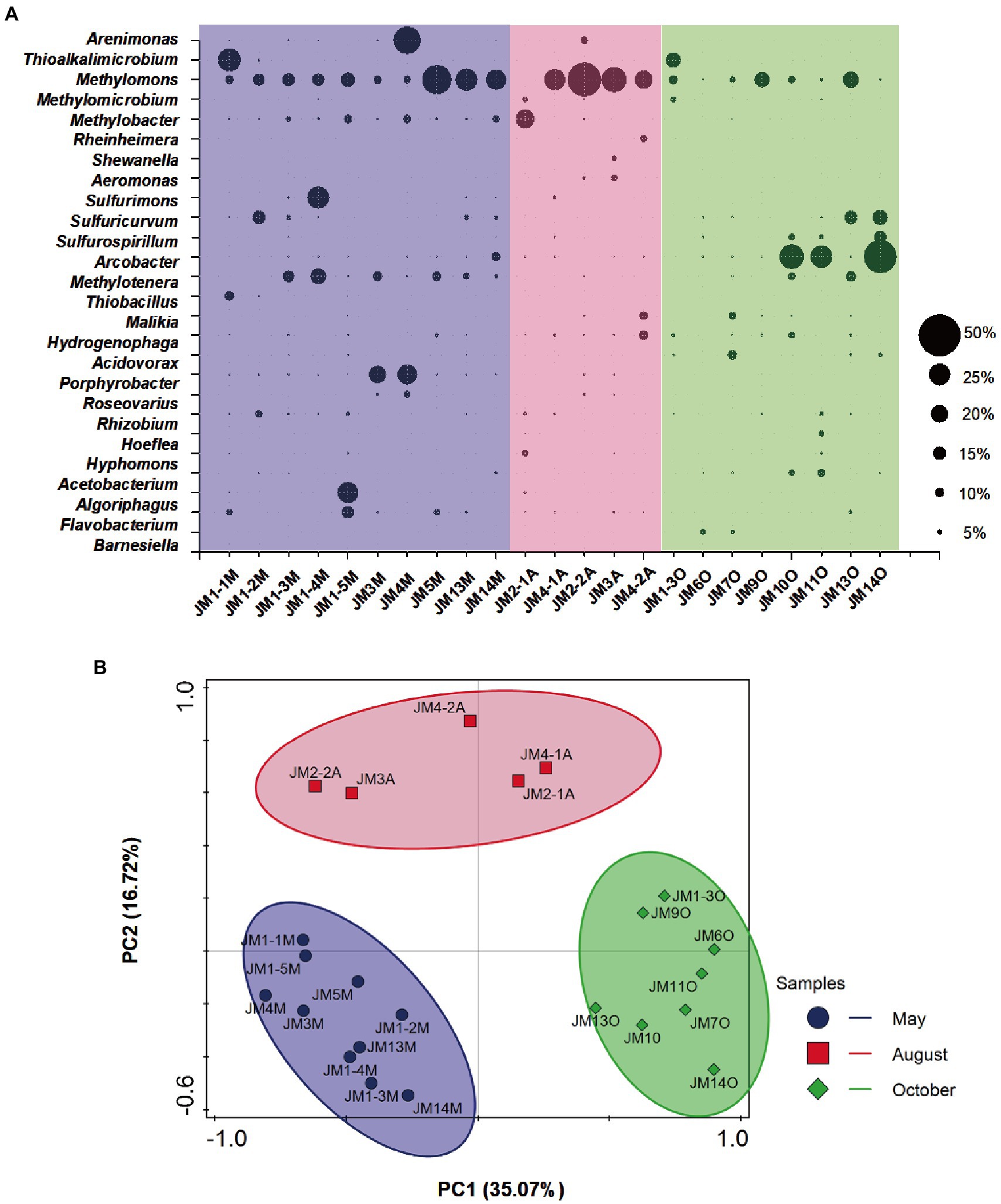
Figure 1. (A) Responses of bacterial community structure to seasonal variation in coalbed water based on 16S rRNA gene. The suffix letter of each sample name represents the month in which the sample was taken (M: May; A: August; O: October). The size of the bubble represents the relative abundance of the genus in each sample. (B) Principal components analysis (PCA) of bacterial 16S rRNA gene retrieved from coalbed water. Different symbols and colors represent different sampling months (purple circle: May; red square: August; green diamond: October).
OTUs with a relative abundance of >5% included primarily methanotrophs, methylotrophs, acetogenic bacteria, chemoautotrophic bacteria, and fermentative bacteria. The aerobic methanotrophs were Methylomonas and Methylobacter, and Methylomonas dominated the coalbed water with the highest relative abundance (up to 40%; Figure 1A). Methylotrophic bacteria included Methylotenera, whose substrate spectrum is narrow and could only metabolize methylamine, methanol, and other methyl compounds (Kalyuzhnaya et al., 2012; Mustakhimov et al., 2013). Acetobacterium can generate acetic acid using H2/CO2, short-chained alcohols, and various carbohydrates (sugars; Kremp et al., 2018; Westphal et al., 2018). Chemoautotrophic bacteria included Thiobacillus, Sulfurospirillum, Sulfurimonas, and Sulfuricurvum. Thiobacillus (Kellermann and Griebler, 2009), Sulfurospirillum (Stolz et al., 1999; Luijten et al., 2003; Hubert and Voordouw, 2007), and Sulfurimonas (Inagaki et al., 2003; Sievert et al., 2008) were the non-strict chemoautotrophic bacteria, which utilize reductive sulfide, hydrogen, and organic matter (glucose, volatile fatty acid, amino acid, etc.) as electron donors, nitrate or sulfate as electron acceptors, and carbon dioxide as a carbon source. Sulfuricurvum, however, is a strictly chemoautotrophic bacteria, and can only use hydrogen and reductive sulfides as an electron acceptor.
PCA results showed that the bacterial community structure of coalbed water samples in the Erlian Basin was clustered according to sampling months. There was no overlap or crossover between the bacterial communities in the coalbed water during the three sampling periods (Figure 1B). This indicated that the community structure of bacteria in coalbed water in this block differed among seasons. A large number of aerobic bacteria (Methylomonas, Methylobacter, Methylotenera, Sulfurospirillum, Acidovorax, and Thioalkalimicrobium) were detected in the coalbed water, which may be related to atmospheric precipitation. Atmospheric precipitation leads to surface runoff into coal seams and brings in oxygen, nutrients, and electron receptors. However, the periodicity of rainfall leads to an unsustainable supply of oxygen, while the metabolic activity of aerobic microorganisms causes periodic changes in oxygen concentration, resulting in co-existence of aerobic and anaerobic bacteria in coalbed water (Bates et al., 2011). The meteorological data from the National Meteorological Science Data Center1 show that there were significant differences in rainfall and mean temperature among the sampling sites during different seasons (Supplementary Table S2). However, due to the lack of reliable measured data, we are still unable to attribute bacterial community changes.
At the phylum level, archaea were dominated by Euryarchaeota (relative abundance 74.6–92.3%). Only some of the archaea in samples JM2 and JM9 were distributed in Nanoarchaeota (relative abundance 8.1–8.6%) and Thaumarchaeota (relative abundance 5.2–6.9%). At the genus level, the dominant archaea were hydrogenotrophic methanogens Methanobacterium, and the average relative abundance of the 23 samples was 61.6% (range 6.6–98.2%; Figure 2A). In addition, the dominant archaea in sample JM4A were hydrogenotrophic methanogens Methanocalculus (relative abundance 62.9%), and the dominant archaea in JM11O, JM1_2M, and JM14M were hydrogenotrophic methanogens Methanocorpusculum, with relative abundances of 87.7, 77.0, and 87.9%, respectively. The methylotrophic Methanolobus (Doerfert et al., 2009; relative abundance 36.9%) in JM9O was also relatively high.
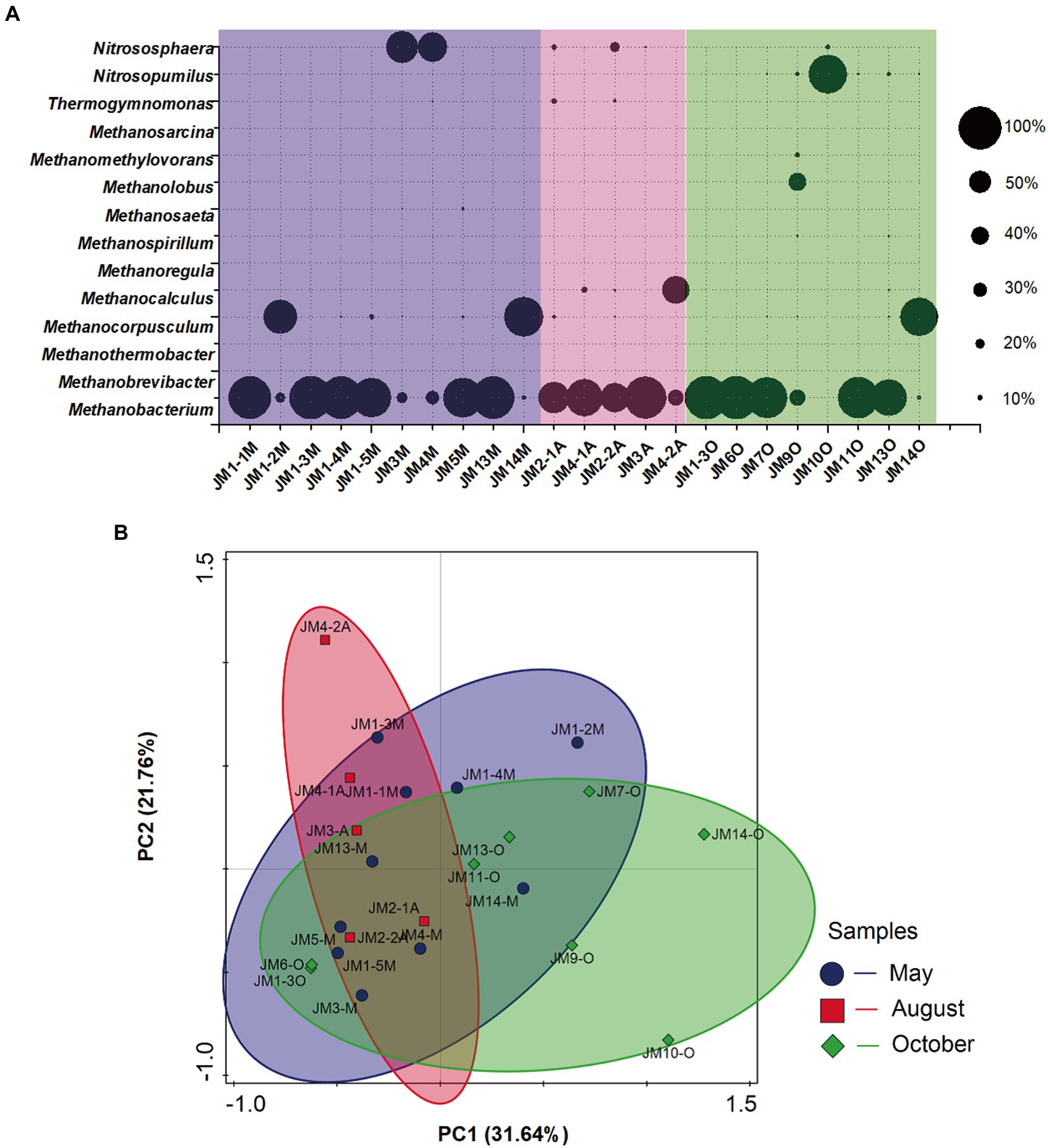
Figure 2. (A) Responses of archaeal community structure to seasonal variation in coalbed water based on 16S rRNA sequencing. The suffix letter of each sample name represents the month in which the sample was taken (M: May; A: August; O: October). The size of the bubble represents the relative abundance of the genus in each sample. (B) Principal component analysis (PCA) of archaeal 16S rRNA gene retrieved from coalbed water. Different symbols and colors represent different sampling months (purple circle: May; red square: August; green diamond: October).
PCA showed that the archaea communities of the 23 coalbed water samples were partially overlapping and were dispersed in different areas in the PCA diagram, with no obvious clustering (Figure 2B). This indicated that the community structure of archaea in coalbed water in this block was relatively stable. The archaea community structure is affected by many environmental factors. All methanogens are strict anaerobes, not documented to use O2 as an electron acceptor during energy-conserving metabolism (Horne and Lessner, 2013). Nutrient availability is one of the key factors determining the abundance of methanogens (Zhou et al., 2015). Salinity (Waldron et al., 2007; Zhang et al., 2020) and temperature (Lu et al., 2015) are also primary environmental factors regulating methanogenic community assemblage. The different responses of archaea and bacteria to seasonal variation in coalbed water reflect their distinct ecological niches.
3.2. Methane production potential of microorganisms In coalbed water
In order to verify the methane production potential of microorganisms in coalbed water and assess its methanogenic pathway, coalbed water sample JM13M was selected to study methanogenic potential using different precursor compounds. H2/CO2 and formate (Figure 3A), methyl compounds (methanol, dimethylamine, dimethyl sulfide, choline; Figure 3C), and short-chain fatty alcohols (propanol, isopropyl alcohol, butanol, and isobutanol; Figure 3B) may be used by microorganisms in JM13M to produce CH4, while short-chain fatty acids cannot be used (Figure 3D). Both the lag phase and the specific growth rate of methane production were different under different substrate treatment (Supplementary Table S4). The dominant archaea were the hydrogenotrophic methanogens Methanobacterium (relative abundance 2.9–91.7%) and Methanocorpusculum (relative abundance 4.9–91.6%), and the methylotrophic Methanolobus (relative abundance 1.2–32.9%; Figure 3E). The methanogenic activity of coalbed water was confirmed by the simulation culture (Figure 3). The bacterial community mainly consisted of Desulfomicrobium and Desulfovibrio (Figure 3F), which might be related to the addition of sodium sulfide.
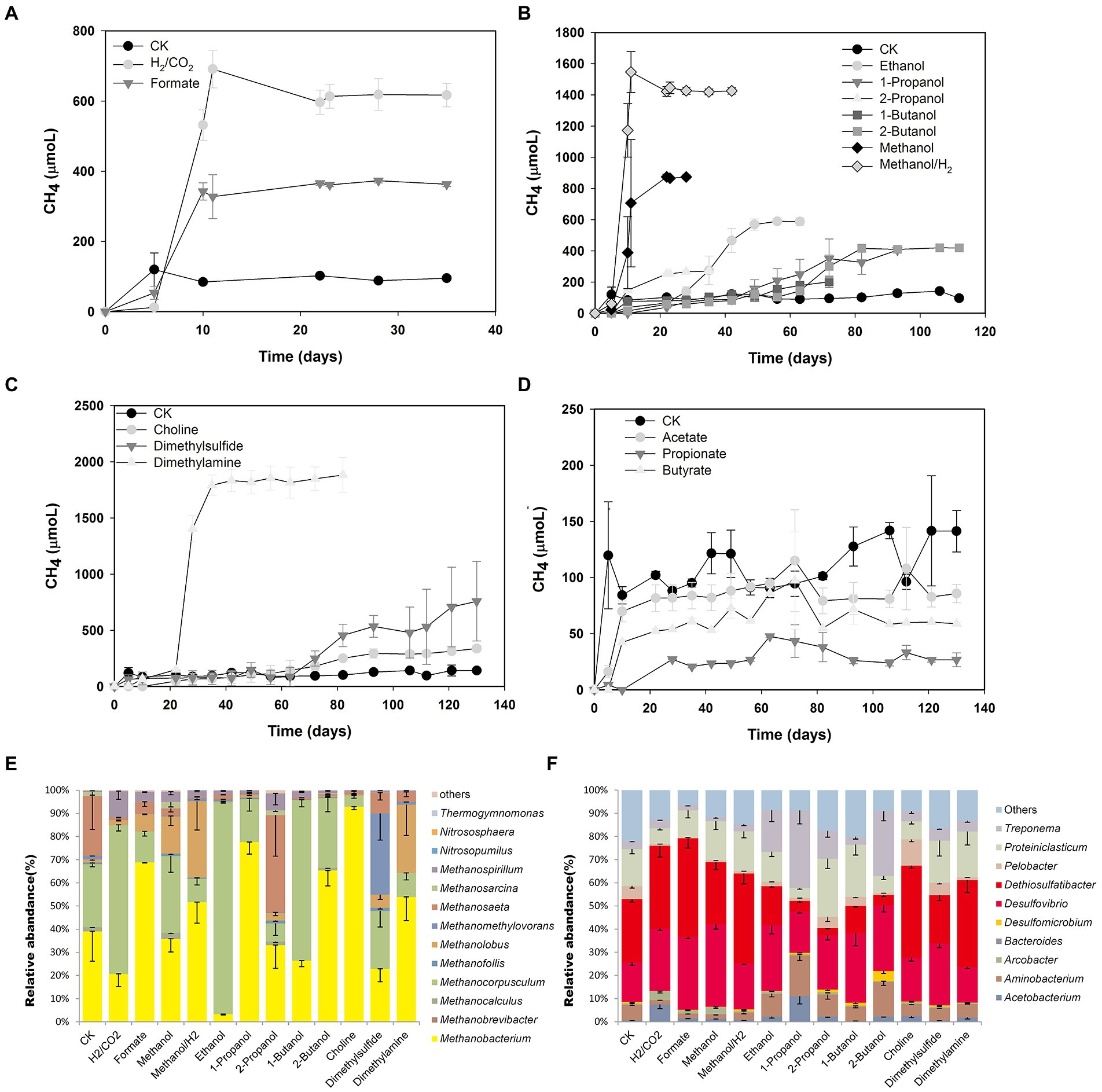
Figure 3. Methane production potential and microbial community structure of different methane precursors. Methane production from H2/CO2 and formate (A), short-chain fatty alcohols (B), methyl compounds (C), and short-chain fatty acids (D). The relative abundances of archaeal (E) and bacterial (F) 16S rRNA genes.
We also collected published and available coalbed archaea data from different basins, including Qinshui Basin (Guo et al., 2017; Yang et al., 2018), Hailar Basin, Jingmen-Dangyang Basin (Wei et al., 2014), Ordos Basin (Guo et al., 2012a), and Powder river Basin (Huang et al., 2017; Davis et al., 2018), and combined them with the Erlian Basin data for analysis (Figure 4). We found that most of these basins were dominated by hydrogenotrophic methanogens Methanobacterium (except the Powder River basin and JM-DY Basin, which were dominated by acetotrophic methanogens). Methanobacterium is widely distributed in anaerobic habitats such as marine and freshwater sediments, soils, animal gastrointestinal tracts, anaerobic sewage digesters, and geothermal habitats (Liu, 2010). Methanobacterium are typically autotrophic and use H2 and CO2, while some also use formate as a substrate for methanogenesis (Liu, 2010). Formate dehydrogenases that supply reductant from formate oxidation and that yield intracellular CO2 to allow for methanogenesis to proceed under otherwise dissolved inorganic carbon (DIC) limited conditions (Fones et al., 2021). Recent studies have shown that Methanobacterium are also able to engage in direct interspecific electron transfer (Zheng et al., 2020), which facilitates the formation of a more efficient connection between Methanobacterium and syntrophic bacteria. These properties allow Methanobacterium to thrive in a variety of methane-producing environments, including coalbed water. Methanocorpusculum was also dominant in some coalbed water samples, and was the most prominent genus in a coal bed of the Illinois Basin (Strapoc et al., 2008) and in shale in northern Michigan (Waldron et al., 2007). Methanocorpusculum belong to the order Methanomicrobiales within the archaeal phylum Euryarchaeota. Similar to Methanobacterium, Methanocorpusculum use either H2/CO2 or formate as substrate for methanogenesis. Compared to other strains of Methanomicrobiales, Methanocorpusculum labreanum was the only strain showing evidence of genome downsizing (Browne et al., 2017), which leads to niche specialization (Lee and Marx, 2012). However, M. labreanum’s putative specialization remains unclear.
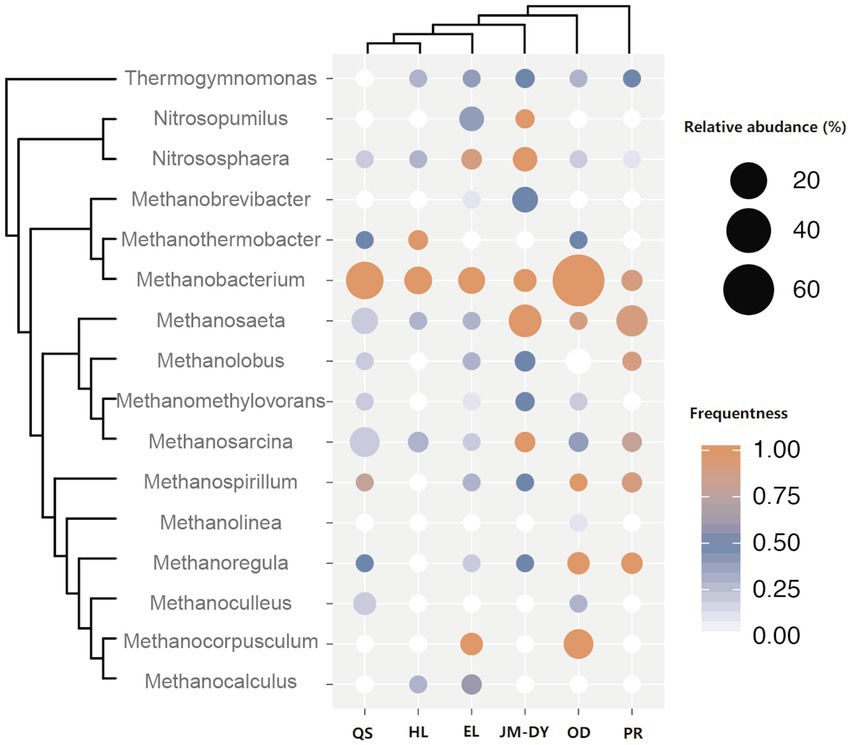
Figure 4. Comparative analysis of archaea community structure in coal seams in different basins (QS: Qinshui Basin; HL: Hailar Basin; EL: Erlian Basin; JM-DY: Jingmen-Dangyang Basin; OD: Ordos Basin; PB: Powder river Basin). The size of the bubble represents the relative abundance of the genus in each sample. The change in bubble color represents the frequency of the genus detected.
3.3. Aerobic methane-oxidizing bacteria in coalbed water presented by metagenomics
The previous high-throughput sequencing results revealed high relative abundance of aerobic methane-oxidizing bacteria in coalbed water. This implied that the methane oxidation reaction was very active. However, the mechanisms by which aerobic methane-oxidizing bacteria act in coalbed water warranted further study. For this, three coalbed water samples (JM1-2M, JM5M, and JM13M) were selected for metagenomic sequencing (Supplementary Table S5). After sequencing and quality control of the sequences, taxonomic classification was performed using two different tools, Kraken2 and MetaPhlAn2. Overall, bacteria (relative abundance >1%) accounted for 60.0–76.9% of the genera identified by Kraken2 and 96.7–97.4% of the genera identified by MetaPhlAn2 (Supplementary Figure S3). However, the relative abundance of archaea in metagenomic data was too low (<0.02%), as shown in the figure.
Kraken2 and MetaPhlAn2 produced similar mock community composition profiles. According to Kraken2, Methylomonas (19.0–51.7%) occurred in a higher proportion in bacteria (Supplementary Figure S3B), and the same result was found using MetaPhlAn2 (51.8–87.1%; Supplementary Figure S3A). Such a high abundance of Methylomonas was also consistent with our high-throughput sequencing results. In addition, Methylotenera, Pseudomonas, and Thiomonas occupied a certain proportion in some samples. Among them, Pseudomonas was particularly noteworthy due to its unique value to research and application to PAH and coal biodegradation (Foght and Westlake, 1988; Tang et al., 2011; Poblete-Castro et al., 2012).
Metagenomic data were used to assemble methane-oxidizing bacteria genomes. Seven high-quality genome bins (MAGs) were obtained (Supplementary Table S6), and after dereplication with dRep with an ANI cutoff of 97%, five cluster MAGs were obtained for subsequent analysis (Supplementary Table S7). Phylogenetic trees constructed with the genomes of methane-oxidizing bacteria (Supplementary Table S8) showed three clusters affiliated with Methylomonas (Supplementary Figure S4).
These three cluster MAGs contained complete metabolic pathways for aerobic methane oxidation. In the first step of methane metabolism, methane is captured and converted to methanol by granular methane monooxygenase (pMMO), an enzyme complex that uses oxygen to oxidize the C-H bonds in methane. In the second step of methane oxidation, methanol is catalyzed by methanol dehydrogenase (MDH) to form formaldehyde. There are two known MDH enzymes, the canonical and long-studied MxaF type and a novel type, XoxF (Chistoserdova et al., 2009). These three cluster MAGs encode both types of methanol dehydrogenases. In the next step, formaldehyde is a key metabolite. Formaldehyde can be converted to formate via the tetrahydromethanopterin (H4MPT) pathway or the methylene-tetrahydrofolate (methylene-H4F) pathway. Another part of formaldehyde can be metabolized through the RuMP cycle and the serine cycle. All genes coding for these pathways were detected in our MAGs (Figure 5). Succinate dehydrogenase (complex II, sdhABCD) was found in the MAGs, together with genes encoding cytochrome c oxidase. The proton motive force generated by the respiratory chain can be used by ATPase (complex V). Nitrogenase (NifDHK), dissimilatory nitrite reductase (NiRk), and nitric oxide reductase (NorB) were also found in these MAGs. This means that these genomes may have the ability to fix nitrogen and convert nitrite into ammonium through denitrification (Figure 5). Denitrification may support methane oxidation in environments with low or below-detectable oxygen concentrations (Padilla et al., 2017; Smith et al., 2018; Smith and Wrighton, 2019).
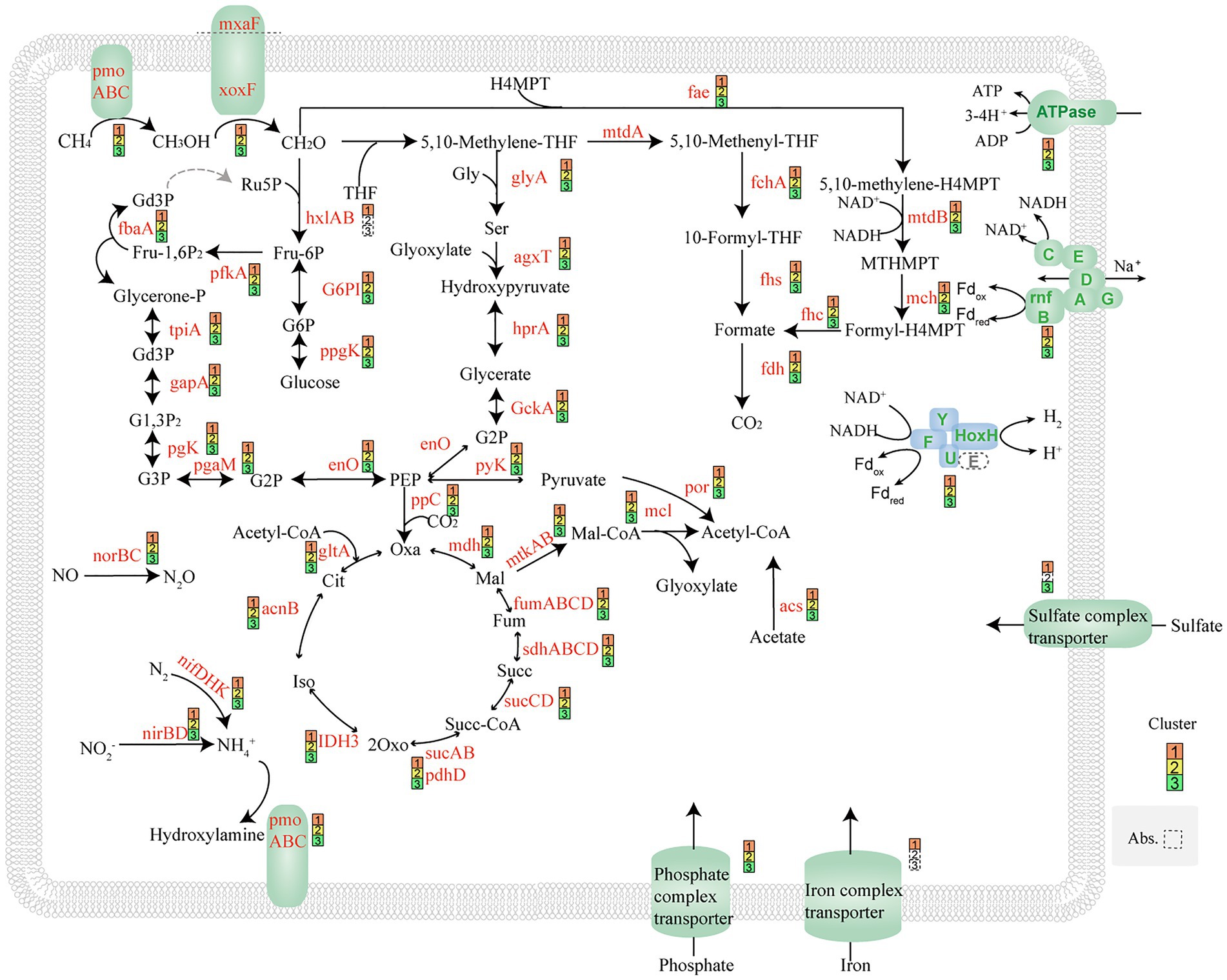
Figure 5. Proposed pathway by MAGs of Methylomonas based on metagenomics analyses. All MAGs use pmo complex to conduct the oxidation of methane to methanol. XoxF and mxaF dehydrogenase, which can catalyze methanol to formaldehyde, were found. In this study, MAGs have complete RuMP cycles, serine cycles, EMP, and TCA cycle pathways. NifDHK nitrogenase, NiRk dissimilatory nitrite reductase, NorB nitric oxide reductase, and HoxHY hydrogenase were also found in MAGs.
4. Conclusion
Microbial gas recovery is one of the effective means to improve the efficiency of low-rank CBM recovery. Low-rank CBM accumulation and coal reservoir characteristics are controlled by not only geological factors such as structure, coal rank, and hydrology, but also microbial factors. In this study, seasonal variation in microbial communities and the functional microorganisms involved in methane metabolism in Erlian Basin were investigated. There were differences in the responses of bacteria and archaea to seasonal variation. The bacterial community structure in coalbed water was affected by seasonal variation. The archaea community structure did not change with seasonal variation. Methylomonas may play an important role in methane oxidation metabolism in coalbed water. Methanobacterium, the dominant hydrogenotrophic methanogens, may play a dominant role in methanogenic metabolism. Further research should focus on the driving factors that cause seasonal variation of bacterial community structure. This will provide some theoretical basis for the regulation of methane metabolism of low-rank CBM in this basin.
Data availability statement
The original contributions presented in the study are publicly available. These data can be found in GenBank, 16S rRNA genes of archaea: PRJNA681683, 16S rRNA genes of bacteria: PRJNA681635, and Metagenomic raw data: PRJNA682244 and PRJNA923302 (Supplementary Table S8).
Author contributions
LC and ZC conceived the research. SL performed the sample collection, in situ simulated incubation, and molecular analysis. ZZ performed the Metagenomics analysis. LF wrote the manuscript. LF and LC edited the manuscript. All authors reviewed and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key R&D Program of China (2016ZX05041001-003), Agricultural Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2016-BIOMA), and the Central Public-interest Scientific Institution Basal Research Fund (No. 1610012020006-03104).
Acknowledgments
We thank Shanghai OE Biotech Inc. (Shanghai, China) for high-throughput sequencing services and bioinformatics support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1114201/full#supplementary-material
Footnotes
References
Ayers, W. B. (2002). Coalbed gas systems, resources, and production and a review of contrasting cases from the San Juan and Powder River basins. AAPG Bull. 86, 1853–1890. doi: 10.1306/61eeddaa-173e-11d7-8645000102c1865d
Bates, B. L., McIntosh, J. C., Lohse, K. A., and Brooks, P. D. (2011). Influence of groundwater flowpaths, residence times and nutrients on the extent of microbial methanogenesis in coal beds: Powder River Basin, USA. Chem. Geol. 284, 45–61. doi: 10.1016/j.chemgeo.2011.02.004
Beckmann, S., Krueger, M., Engelen, B., Gorbushina, A. A., and Cypionka, H. (2011a). Role of bacteria, archaea and fungi involved in methane release in abandoned coal mines. Geomicrobiol J. 28, 347–358. doi: 10.1080/01490451.2010.503258
Beckmann, S., Lueders, T., Krueger, M., von Netzer, F., Engelen, B., and Cypionka, H. (2011b). Acetogens and Acetoclastic Methanosarcinales govern methane formation in abandoned coal mines. Appl. Environ. Microbiol. 77, 3749–3756. doi: 10.1128/aem.02818-10
Beckmann, S., Luk, A. W. S., Gutierrez-Zamora, M.-L., Chong, N. H. H., Thomas, T., Lee, M., et al. (2019). Long-term succession in a coal seam microbiome during in situ biostimulation of coalbed-methane generation. ISME J. 13, 632–650. doi: 10.1038/s41396-018-0296-5
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Browne, P., Tamaki, H., Kyrpides, N., Woyke, T., Goodwin, L., Imachi, H., et al. (2017). Genomic composition and dynamics among Methanomicrobiales predict adaptation to contrasting environments. ISME J. 11, 87–99. doi: 10.1038/ismej.2016.104
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen, S., and Pan, Z. (2019). Current status, challenges, and policy suggestions for coalbed methane industry development in China: a review. Energy Sci. Eng. 7, 1059–1074. doi: 10.1002/ese3.358
Chen, H., Qin, Y., Deng, Z., Geng, M., Li, G., Sang, G., et al. (2018). Factors influencing the biogenic gas production of low rank coal beds in the Jiergalangtu sag, Erlian Basin. Nat. Gas Ind. 38, 27–32. doi: 10.1016/j.ngib.2019.01.001
Chistoserdova, L., Kalyuzhnaya, M. G., and Lidstrom, M. E. (2009). The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 63, 477–499. doi: 10.1146/annurev.micro.091208.073600
Colosimo, F., Thomas, R., Lloyd, J. R., Taylor, K. G., Boothman, C., Smith, A. D., et al. (2016). Biogenic methane in shale gas and coal bed methane: a review of current knowledge and gaps. Int. J. Coal Geol. 165, 106–120. doi: 10.1016/j.coal.2016.08.011
Conrad, R. (2020). Importance of hydrogenotrophic, aceticlastic and methylotrophic methanogenesis for methane production in terrestrial, aquatic and other anoxic environments: a mini review. Pedosphere 30, 25–39. doi: 10.1016/s1002-0160(18)60052-9
Davis, K. J., Lu, S., Barnhart, E. P., Parker, A. E., Fields, M. W., and Gerlach, R. (2018). Type and amount of organic amendments affect enhanced biogenic methane production from coal and microbial community structure. Fuel 211, 600–608. doi: 10.1016/j.fuel.2017.09.074
Doerfert, S. N., Reichlen, M., Iyer, P., Wang, M., and Ferry, J. G. (2009). Methanolobus zinderi sp nov., a methylotrophic methanogen isolated from a deep subsurface coal seam. Int. J. Syst. Evol. Microbiol. 59, 1064–1069. doi: 10.1099/ijs.0.003772-0
Faiz, M., and Hendry, P. (2006). Significance of microbial activity in Australian coal bed methane reservoirs - a review. Bull. Can. Petrol. Geol. 54, 261–272. doi: 10.2113/gscpgbull.54.3.261
Foght, J. M., and Westlake, D. W. S. (1988). Degradation of polycyclic aromatic hydrocarbons and aromatic heterocycles by a pseudomonas species. Can. J. Microbiol. 34, 1135–1141. doi: 10.1139/m88-200
Fones, E. M., Colman, D. R., Kraus, E. A., Stepanauskas, R., Templeton, A. S., Spear, J. R., et al. (2021). Diversification of methanogens into hyperalkaline serpentinizing environments through adaptations to minimize oxidant limitation. ISME J. 15, 1121–1135. doi: 10.1038/s41396-020-00838-1
Guo, H., Liu, R., Yu, Z., Zhang, H., Yun, J., Li, Y., et al. (2012a). Pyrosequencing reveals the dominance of methylotrophic methanogenesis in a coal bed methane reservoir associated with eastern Ordos Basin in China. Int. J. Coal Geol. 93, 56–61. doi: 10.1016/j.coal.2012.01.014
Guo, H., Yu, Z., Liu, R., Zhang, H., Zhong, Q., and Xiong, Z. (2012b). Methylotrophic methanogenesis governs the biogenic coal bed methane formation in eastern Ordos Basin, China. Appl. Microbiol. Biotechnol. 96, 1587–1597. doi: 10.1007/s00253-012-3889-3
Guo, H., Zhang, J., Han, Q., Huang, Z., Urynowicz, M. A., and Wang, F. (2017). Important role of fungi in the production of secondary biogenic coalbed methane in China's southern Qinshui Basin. Energy Fuel 31, 7197–7207. doi: 10.1021/acs.energyfuels.7b00925
Horne, A. J., and Lessner, D. J. (2013). Assessment of the oxidant tolerance of Methanosarcina acetivorans. FEMS Microbiol. Lett. 343, 13–19. doi: 10.1111/1574-6968.12115
Huang, Z., Sednek, C., Urynowicz, M. A., Guo, H., Wang, Q., Fallgren, P., et al. (2017). Low carbon renewable natural gas production from coalbeds and implications for carbon capture and storage. Nat. Commun. 8:568. doi: 10.1038/s41467-017-00611-7
Hubert, C., and Voordouw, G. (2007). Oil field souring control by nitrate-reducing Sulfurospirillum spp. that outcompete sulfate-reducing bacteria for organic electron donors. Appl. Environ. Microbiol. 73, 2644–2652. doi: 10.1128/aem.02332-06
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Inagaki, F., Takai, K., Hideki, K. I., Nealson, K. H., and Horikishi, K. (2003). Sulfurimonas autotrophica gen. Nov., sp nov., a novel sulfur-oxidizing epsilon-proteobacterium isolated from hydrothermal sediments in the mid-Okinawa trough. Int. J. Syst. Evol. Microbiol. 53, 1801–1805. doi: 10.1099/ijs.0.02682-0
Kalyuzhnaya, M. G., Beck, D. A. C., Vorobev, A., Smalley, N., Kunkel, D. D., Lidstrom, M. E., et al. (2012). Novel methylotrophic isolates from lake sediment, description of Methylotenera versatilis sp nov and emended description of the genus Methylotenera. Int. J. Syst. Evol. Microbiol. 62, 106–111. doi: 10.1099/ijs.0.029165-0
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/peerj.7359
Kellermann, C., and Griebler, C. (2009). Thiobacillus thiophilus sp nov., a chemolithoautotrophic, thiosulfate-oxidizing bacterium isolated from contaminated aquifer sediments. Int. J. Syst. Evol. Microbiol. 59, 583–588. doi: 10.1099/ijs.0.002808-0
Kremp, F., Poehlein, A., Daniel, R., and Mueller, V. (2018). Methanol metabolism in the acetogenic bacterium Acetobacterium woodii. Environ. Microbiol. 20, 4369–4384. doi: 10.1111/1462-2920.14356
Krüger, M., Beckmann, S., Engelen, B., Thielemann, T., Cramer, B., Schippers, A., et al. (2008). Microbial methane formation from hard coal and timber in an abandoned coal mine. Geomicrobiol J. 25, 315–321. doi: 10.1080/01490450802258402
Lai, S., Fang, X., Tu, B., Chen, H., Deng, Z., Li, G., et al. (2020). Methanogenic potential and community structure of coalbed-methane production water microbiome. Acta Microbiol Sin. 60, 727–738. doi: 10.13343/j.cnki.wsxb.20190291
Lee, M.-C., and Marx, C. J. (2012). Repeated, selection-driven genome reduction of accessory genes in experimental populations. PLoS Genet. 8:e1002651. doi: 10.1371/journal.pgen.1002651
Lu, Y., Fu, L., Lu, Y., Hugenholtz, F., and Ma, K. (2015). Effect of temperature on the structure and activity of a methanogenic archaeal community during rice straw decomposition. Soil Biol. Biochem. 81, 17–27. doi: 10.1016/j.soilbio.2014.10.031
Lu, Z., and Lu, Y. (2012). Methanocella conradii sp nov., a thermophilic, obligate Hydrogenotrophic methanogen, isolated from Chinese Rice field soil. PLoS One 7:e35279. doi: 10.1371/journal.pone.0035279
Luijten, M., de Weert, J., Smidt, H., Boschker, H. T. S., de Vos, W. M., Schraa, G., et al. (2003). Description of Sulfurospirillum halorespirans sp nov., an anaerobic, tetrachloroethene-respiring bacterium, and transfer of Dehalospirillum multivorans to the genus Sulfurospirillum as Sulfurospirillum multivorans comb. nov. Int. J. Syst. Evol. Microbiol. 53, 787–793. doi: 10.1099/ijs.0.02417-0
Mayumi, D., Mochimaru, H., Tamaki, H., Yamamoto, K., Yoshioka, H., Suzuki, Y., et al. (2016). Methane production from coal by a single methanogen. Science 354, 222–225. doi: 10.1126/science.aaf8821
Moore, T. A. (2012). Coalbed methane: a review. Int. J. Coal Geol. 101, 36–81. doi: 10.1016/j.coal.2012.05.011
Mustakhimov, I., Kalyuzhnaya, M. G., Lidstrom, M. E., and Chistoserdova, L. (2013). Insights into denitrification in Methylotenera mobilis from denitrification pathway and methanol metabolism mutants. J. Bacteriol. 195, 2207–2211. doi: 10.1128/jb.00069-13
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Padilla, C. C., Bertagnolli, A. D., Bristow, L. A., Sarode, N., Glass, J. B., Thamdrup, B., et al. (2017). Metagenomic binning recovers a transcriptionally active Gammaproteobacterium linking Methanotrophy to partial denitrification in an anoxic oxygen minimum zone. Front. Mar. Sci. 4:23. doi: 10.3389/fmars.2017.00023
Park, S. Y., and Liang, Y. (2016). Biogenic methane production from coal: a review on recent research and development on microbially enhanced coalbed methane (MECBM). Fuel 166, 258–267. doi: 10.1016/j.fuel.2015.10.121
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Poblete-Castro, I., Becker, J., Dohnt, K., dos Santos, V. M., and Wittmann, C. (2012). Industrial biotechnology of pseudomonas putida and related species. Appl. Microbiol. Biotechnol. 93, 2279–2290. doi: 10.1007/s00253-012-3928-0
Robbins, S. J., Evans, P. N., Parks, D. H., Golding, S. D., and Tyson, G. W. (2016). Genome-centric analysis of microbial populations enriched by hydraulic fracture fluid additives in a coal bed methane production well. Front. Microbiol. 7:731. doi: 10.3389/fmicb.2016.00731
Rosewarne, C.P., Greenfield, P., Li, D., Nai, T.-D., Midgley, D.J., and Hendry, P. (2013). Draft genome sequence of Methanobacterium sp. Maddingley, reconstructed from metagenomic sequencing of a methanogenic microbial consortium enriched from coal-seam gas formation water. Genome Announc. 1:e00082-12. doi: 10.1128/genomeA.00082-12
Sievert, S. M., Scott, K. A., Klotz, M. G., Chain, P. S. G., Hauser, L. J., Hemp, J., et al. (2008). Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl. Environ. Microbiol. 74, 1145–1156. doi: 10.1128/aem.01844-07
Smith, G. J., Angle, J. C., Solden, L. M., Borton, M. A., Morin, T. H., Daly, R. A., et al. (2018). Members of the genus Methylobacter are inferred to account for the majority of aerobic methane oxidation in Oxic soils from a freshwater wetland. MBio 9:e00815-18. doi: 10.1128/mBio.00815-18
Smith, G. J., and Wrighton, K. C. (2019). Metagenomic approaches unearth Methanotroph phylogenetic and metabolic diversity. Curr. Issues Mol. Biol. 33, 57–84. doi: 10.21775/cimb.033.057
Stolz, J. F., Ellis, D. J., Blum, J. S., Ahmann, D., Lovley, D. R., and Oremland, R. S. (1999). Sulfurospirillum barnesii sp nov and Sulfurospirillum arsenophilum sp nov., new members of the Sulfurospirillum clade of the epsilon Proteobacteria. Int. J. Syst. Bacteriol. 49 Pt 3, 1177–1180. doi: 10.1099/00207713-49-3-1177
Strapoc, D., Mastalerz, M., Dawson, K., Macalady, J., Callaghan, A. V., Wawrik, B., et al. (2011). “Biogeochemistry of microbial coal-bed methane,” in Annual review of earth and planetary sciences. eds. R. Jeanloz and K. H. Freeman, vol. 39, 617–656.
Strapoc, D., Picardal, F. W., Turich, C., Schaperdoth, I., Macalady, J. L., Lipp, J. S., et al. (2008). Methane-producing microbial community in a coal bed of the Illinois Basin. Appl. Environ. Microbiol. 74, 2424–2432. doi: 10.1128/aem.02341-07
Sun, B., Li, J., Cheng, L., Yang, Q., Tian, W., Li, X., et al. (2018). The feasibility of biological gas recovery in low-rank coal:a case study of Jiergalangtu depression in Erlian Basin. Acta Pet. Sin. 39:1272-1278,1291. doi: 10.7623/syxb201811007
Sun, F., Li, W., Sun, Q., Sun, B., Tian, W., Chen, Y., et al. (2017). Low-rank coalbed methane exploration in Jiergalangtu sag, Erlian Basin. Acta Petrolei Sinica 38, 485–492. doi: 10.7623/syxb201705001
Tang, H., Yu, H., Li, Q., Wang, X., Gai, Z., Yin, G., et al. (2011). Genome sequence of pseudomonas putida strain B6-2, a Superdegrader of polycyclic aromatic hydrocarbons and dioxin-like compounds. J. Bacteriol. 193:6789-+. doi: 10.1128/jb.06201-11
Waldron, P. J., Petsch, S. T., Martini, A. M., and Nuslein, K. (2007). Salinity constraints on subsurface archaeal diversity and methanogenesis in sedimentary rock rich in organic matter. Appl. Environ. Microbiol. 73, 4171–4179. doi: 10.1128/aem.02810-06
Wang, H., Ma, F., Tong, X., Liu, Z., Zhang, X., Wu, Z., et al. (2016). Assessment of global unconventional oil and gas resources. Pet. Explor. Dev. 43, 925–940. doi: 10.1016/s1876-3804(16)30111-2
Wang, A., Shao, P., Lan, F., and Jin, H. (2018). Organic chemicals in coal available to microbes to produce biogenic coalbed methane: a review of current knowledge. J. Nat. Gas Sci. Eng. 60, 40–48. doi: 10.1016/j.jngse.2018.09.025
Wei, M., Yu, Z., Jiang, Z., and Zhang, H. (2014). Microbial diversity and biogenic methane potential of a thermogenic-gas coal mine. Int. J. Coal Geol. 134-135, 96–107. doi: 10.1016/j.coal.2014.09.008
Westphal, L., Wiechmann, A., Baker, J., Minton, N. P., and Mueller, V. (2018). The Rnf complex is an energy-coupled transhydrogenase essential to reversibly link cellular NADH and ferredoxin pools in the Acetogen Acetobacterium woodii. J. Bacteriol. 200:e00357-18. doi: 10.1128/jb.00357-18
Yang, X., Chen, Y., Wu, R., Nie, Z., Han, Z., Tan, K., et al. (2018). Potential of biogenic methane for pilot-scale fermentation ex situ with lump anthracite and the changes of methanogenic consortia. J. Ind. Microbiol. Biotechnol. 45, 229–237. doi: 10.1007/s10295-018-2023-7
Zhang, C.-J., Chen, Y.-L., Pan, J., Wang, Y.-M., and Li, M. (2020). Spatial and seasonal variation of methanogenic community in a river-bay system in South China. Appl. Microbiol. Biotechnol. 104, 4593–4603. doi: 10.1007/s00253-020-10613-z
Zheng, S., Liu, F., Wang, B., Zhang, Y., and Lovley, D. R. (2020). Methanobacterium capable of direct interspecies electron transfer. Environ. Sci. Technol. 54, 15347–15354. doi: 10.1021/acs.est.0c05525
Zhou, Z., Chen, J., Cao, H., Han, P., and Gu, J.-D. (2015). Analysis of methane-producing and metabolizing archaeal and bacterial communities in sediments of the northern South China Sea and coastal Mai Po nature reserve revealed by PCR amplification of mcrA and pmoA genes. Front. Microbiol. 5:789. doi: 10.3389/fmicb.2014.00789
Keywords: methanogens, methanotrophs, coalbed water, Erlian Basin, metagenomic
Citation: Fu L, Lai S, Zhou Z, Chen Z and Cheng L (2023) Seasonal variation of microbial community and methane metabolism in coalbed water in the Erlian Basin, China. Front. Microbiol. 14:1114201. doi: 10.3389/fmicb.2023.1114201
Edited by:
Zhe Lyu, North Carolina State University, United StatesReviewed by:
Sina Schorn, Max Planck Society, GermanyZhongjun Jia, Institute of Soil Science (CAS), China
Copyright © 2023 Fu, Lai, Zhou, Chen and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhong Chen, ✉ Y2hlbnpoZW5ob25nQHBldHJvY2hpbmEuY29tLmNu; Lei Cheng, ✉ Y2hlbmdsZWlAY2Fhcy5jbg==