 Min Wang1,2†
Min Wang1,2† Xiao-Yang Zhi
Xiao-Yang Zhi- 1Key Laboratory of Microbial Diversity in Southwest China of Ministry of Education, School of Life Sciences, Yunnan Institute of Microbiology, Yunnan University, Kunming, China
- 2Zhaotong Health Vocational College, Zhaotong, China
As the canonical model organism to dissect bacterial morphological development, Streptomyces species has attracted much attention from the microbiological society. However, the evolution of development-related genes in Streptomyces remains elusive. Here, we evaluated the distribution of development-related genes, thus indicating that the majority of these genes were ubiquitous in Streptomyces genomes. Furthermore, the phylogenetic topologies of related strict orthologous genes were compared to the species tree of Streptomyces from both concatenation and single-gene tree analyses. Meanwhile, the reconciled gene tree and normalization based on the number of parsimony-informative sites were also employed to reduce the impact of phylogenetic conflicts, which was induced by uncertainty in single-gene tree inference based merely on the sequence and the bias in the amount of phylogenetic information caused by variable numbers of parsimony-informative sites. We found that the development-related genes had higher congruence to the species tree than other strict orthologous genes. Considering that the development-related genes could also be tracked back to the common ancestor of Streptomyces, these results suggest that morphological development follows the same pattern as species divergence.
Introduction
Streptomyces are Gram-positive and filamentous bacteria belonging to the phylum Actinobacteria. They have complex multicellular life cycles involving the transformation of a vegetative mycelium into the reproductive spores that are essential for their propagation (Kämpfer, 2015). Moreover, it is also the largest genus of prokaryotes, comprising 685 species with a validly published name (827 species including synonyms, data from LPSN)1 (Parte et al., 2020). The flourishing of species diversity is undoubtedly the result of its broad ecological adaptability (Andam et al., 2016; Li et al., 2019) and is also linked to its irreplaceable role in producing commercially and medically important bioactive substances (Anderson and Wellington, 2001). As we know, morphological differentiation and secondary metabolism are two physiologically coupled biological processes that strongly underpin the environmental adaptability of Streptomyces species (Sun et al., 2017; Xu et al., 2019). They have attracted the most interest and scrutiny of microbiological society.
The life cycle of Streptomyces species begins with the germination of a spore, followed by the formation of vegetative mycelia, including tip extension and the initiation of new branches (Flärdh and Buttner, 2009; van Dissel et al., 2014). Then, in response to nutrient depletion and other signals, the morphological differentiation and the production of secondary metabolites is initiated (Chater, 2006). Most of the genes involved in this intricate developmental process, e.g., bld genes related to the deficiency of aerial mycelium (Willey et al., 1991), whi genes related to spore formation (Soliveri et al., 2000), ram genes related to the hydrophobic protein synthesis (Capstick et al., 2007), ssg genes associated with the sporulation-specific cell division (Kormanec and Sevcikova, 2002), were considerably widespread in Streptomyces. Intriguingly, these two coupled biological processes were governed by complex regulatory networks (Bibb, 2005; Ohnishi et al., 2005). However, distinct from morphological differentiation, the secondary metabolism exhibits a high level of species specificity (Liu et al., 2013). Only some genes involved in regulation and signal transduction might be conserved among Streptomyces species (Bibb, 2005; Rodríguez et al., 2013).
Comparative genomics indicated that morphological development and its regulation in Streptomyces was probably achieved through the progressive acquisition of the laterally transferred DNA (Chater and Chandra, 2006). But most development-related genes may have been assembled in ancestors of Streptomyces species, at least before Streptomyces species branched off from the actinobacterial phylogenetic tree (Chandra and Chater, 2014). In this respect, Streptomyces differs from cyanobacteria, which also have the capacity for multicellular differentiation and a more complex early evolutionary history on the filamentous morphology (Sanchez-Baracaldo et al., 2005; Schirrmeister et al., 2011; Hammerschmidt et al., 2021). After that, the pan-genome analysis reaffirmed that many genes involved in stress response and morphological development were commonly expressed in Streptomyces species (Kim et al., 2015). Since 2013, fueled by rapid advances in high-throughput sequencing, the expansion of genome data of Streptomyces, especially the non-model streptomycetes (Lee et al., 2020), will aid in better comprehending the evolution of Streptomyces development. The relationship between gene phylogenies of specific core biosynthetic gene clusters and species phylogeny has been uncovered separately in a limited number of species (Joynt and Seipke, 2018; Waglechner et al., 2019; Navarro-Muñoz et al., 2020; Creamer et al., 2021). Nevertheless, the phylogenies of morphological development-related genes and their relationships with the phylogeny of species in Streptomyces remain elusive.
In this study, to address the above question, we performed an in-depth comparative phylogenetic investigation based on a large genome dataset of Streptomyces. The distributions of 65 development-related genes supported the previous conclusion that these genes were progressively assembled during the early evolution of Streptomyces, and some genes had been equipped in the genome of the common ancestor. Remarkably, the phylogenetic comparisons revealed that development-related genes had a higher degree of topological congruences to the species tree for both concatenated and single-gene trees. Whether by reconciling single-gene trees with the species tree using amalgamated likelihood estimation or by normalizing the topological distance of single-gene trees with a linear regression model, the development-related genes are always the gene sets that are considerably concordant with the species tree. These results demonstrated that the evolution of morphological development of Streptomyces kept in step with the speciation of this largest taxonomic taxon over the past 380 million years (McDonald and Currie, 2017).
Materials and methods
Genome and gene datasets preparation
The genome data (≥5 Mb) of type strains of 403 validly published Streptomyces species were collected from NCBI Assembly (344 genomes), GCM Type (43 genomes), and JGI IMG (16 genomes) databases (Supplementary Table 1). To reduce the redundancy of sequence data, the genome-pairwise average nucleotide identities (ANIs) were calculated by FastANI (Jain et al., 2018). And, if the genomes of two species shared an ANI value greater than 95%, which is the threshold for demarcating prokaryotic species (Chun et al., 2018), the genome of the species whose taxonomic name that had the priority of publication was retained (Li et al., 2022). Therefore, 388 genomes constituted a basic genome dataset (BGD) and were annotated using Prokka (version 1.12) with default parameters (Seemann, 2014). It needs to be stated that all subsequent phylogenetic analyses are based on the protein sequence. Firstly, 40 universal marker genes were extracted from BGD by SpecI (Mende et al., 2013), and the resultant gene datasets were denoted as UGs. Genomes with less than 38 extracted universal markers were further excluded from the BGD (Supplementary Table 2). Secondly, to collect the translated products of development-related genes in BGD, the reference protein sequences encoded by 65 development-related genes were retrieved from UniProt2 (Table 1) and used as queries to search against BGD by blastp (Altschul et al., 1990) with an e-value cut-off of 1 × 10–5. In each genome, all hits with identity ≥40% and coverage ≥50% were kept to constitute the original datasets for 65 development-related genes (denoted as DGs).
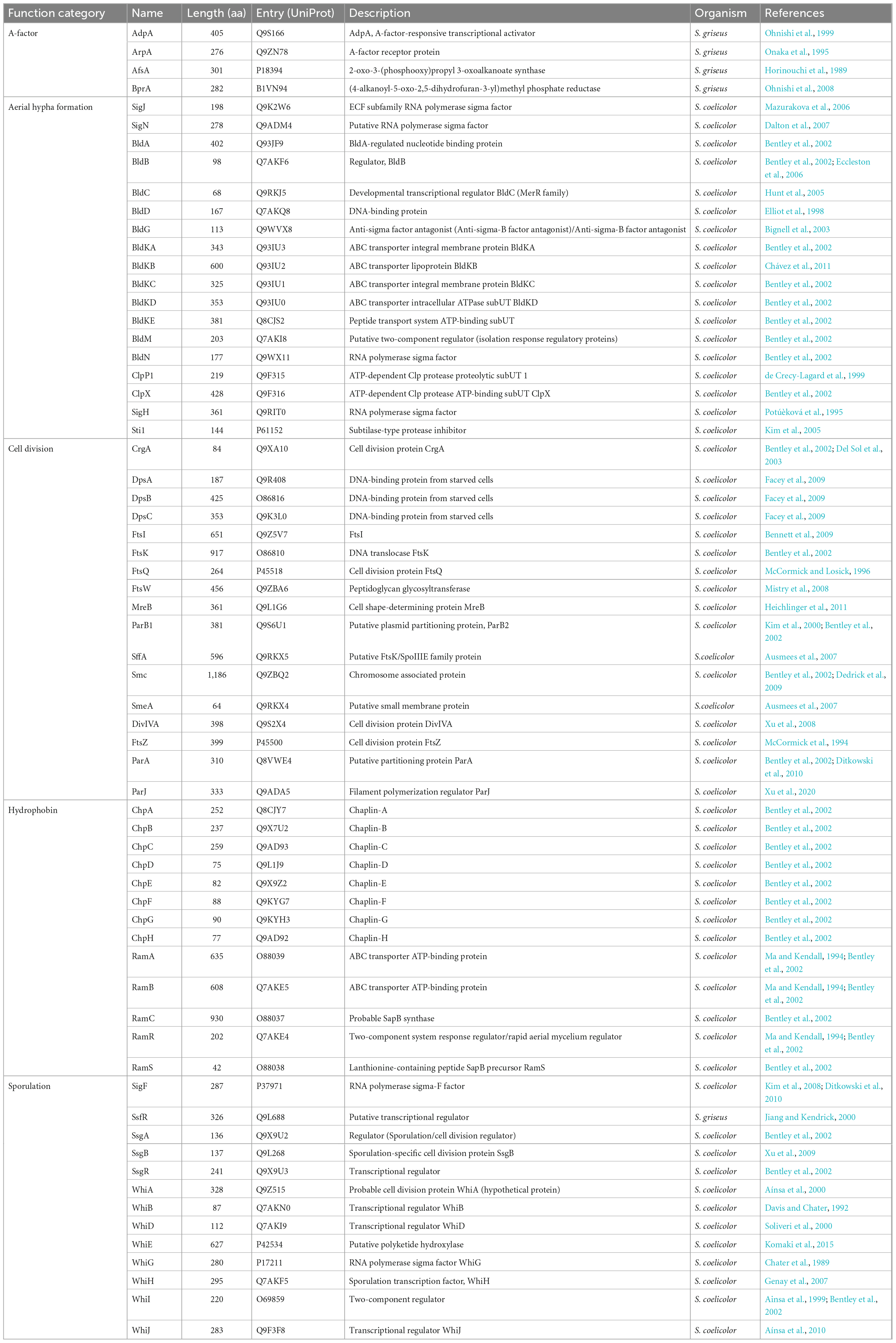
Table 1. The detailed information of development-related proteins in the genus Streptomyces.
Random genome datasets generation
Because of the large number of genomes in the basic genome dataset, the current orthology inference method cannot obtain sufficient strict orthogroups [theoretically refer to a gene family including genes that are orthologous to each other and do not involve pairs of inparalogs (Boeckmann et al., 2011); technically refer to a gene family including genes that are not only homologous to each other but also single copy in the source genomes]. Therefore, we used a random resampling strategy similar to previous works (Canback et al., 2002; Zhi et al., 2014) to decrease the number of genomes and to cover as much species diversity presented in BGD as possible. Briefly, a reference species tree (denoted as RST) was first reconstructed based on a concatenated alignment of UGs, using IQ-tree (version 2.1.2) (Nguyen et al., 2015). Due to the asymmetry of RST in the number of species on the deep branches, it was re-rooted to enable the numbers of species contained in the two deepest branches to be close. And then, based on re-rooted RST, all species were divided into groups based on different clades (Supplementary Table 3). The corresponding genomes were randomly selected in terms of the species constitution of various clades in RST. Finally, random sampling was repeated to generate 100 random genome datasets (denoted as RGDs, Supplementary Table 4).
Phylogenetic tree inference
All RGDs were individually subjected to orthology inference using OrthoFinder (version 2.5.2) (Emms and Kelly, 2015, 2019). The orthogroups with precisely one gene from each genome were regarded as the strict orthogroups (sOGs) and were aligned separately by ClustalO (version 1.2.3) (Sievers et al., 2011). Then, all alignments of sOGs were concatenated, and the ambiguous sites were trimmed by trimAl (version 1.4) (Capella-Gutierrez et al., 2009) with the “automated” option. A species tree based on supermatrix (denoted as ST1) was inferred using IQ-tree, based on the best model with the following options: “–m TEST –B 1000 –T AUTO.” To alleviate the bias induced by a single method, an additional species tree based on supertree (denoted as ST2) under the multispecies coalescent model was also built using ASTRAL (version 5.7.4) (Mirarab et al., 2014). The single-gene maximum likelihood (ML) trees used in the supertree approach were inferred using IQ-tree with options: “-m TEST –score-diff ALL –B 1000 –wbtl –T AUTO.”
Besides ST1 and ST2, other two concatenated ML trees were reconstructed based on UGs and DGs, respectively. Because the universal marker gene had at most one sequence in each genome, UGs can be treated like sOGs, and their concatenated tree (denoted as UT) could be built using the same method. However, unlike UG, the development-related gene sequences belonging to one RGD were first extracted from the original DG (based on BGD) and then subjected to the orthology assessment by UPhO (Ballesteros and Hormiga, 2016). To a DG family, its subfamilies that formed the largest monophyletic subtrees in the tree of the whole family were recognized as strict orthogroups and used to build the concatenated ML tree (denoted as DT). In summary, for each RGD, two species trees (ST1 and ST2), two concatenated ML trees (UT and DT), and a batch of single-gene trees (genes in sOGs and DGs) were reconstructed as well.
Phylogenetic topology comparison
The Robinson-Foulds (RF) distance (Briand et al., 2020; Hayati and Chindelevitch, 2020) was employed to quantify the difference in phylogenetic topologies by using the Python package ete3 (version 3.1.2). The species trees were compared with concatenated ML trees and single-gene trees. However, because multichotomous branching profoundly affects the RF distance calculation, the single-gene trees with multichotomous branches involving ≥50% of species were excluded from the topological comparison. To examine the difference in phylogenetic topologies among various functional groups, the Clusters of Orthologous Genes (COG) annotation was performed on all sOGs using the online version of eggNOG (version 5.0) (Huerta-Cepas et al., 2019). The single-gene trees of sOGs could be divided into subgroups according to the COG categories (Galperin et al., 2021), and their RF distances to species trees (ST1 and ST2) were analyzed separately. The whole data analysis procedure in this work is illustrated schematically in Figure 1.
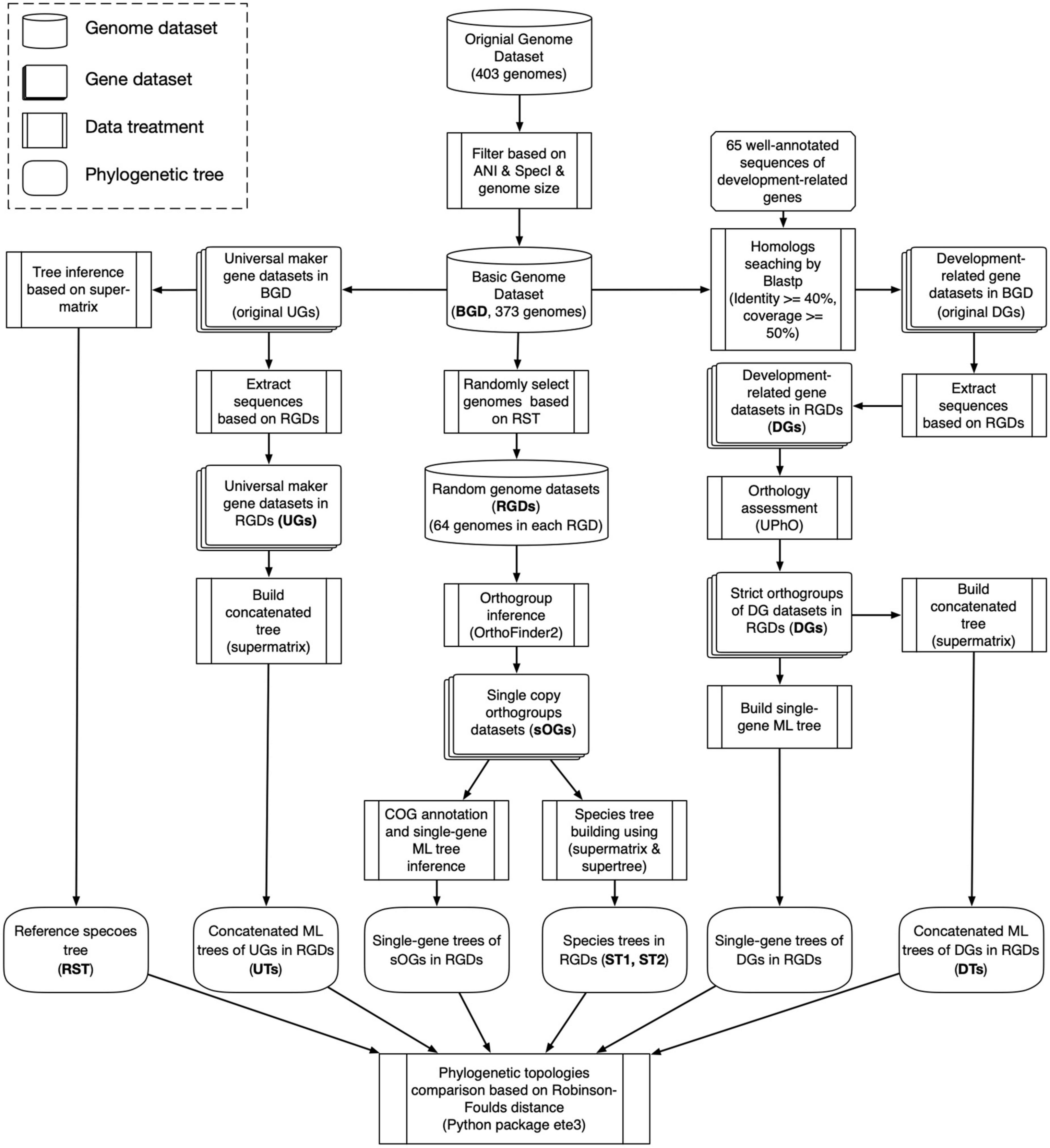
Figure 1. Flowchart of data analysis in this study.
The underpinning evolutionary processes causing phylogenetic conflict are diverse (Schrempf and Szöllősi, 2020). Besides biological factors like gene duplication and loss, and horizontal gene transfer, a wide variety of analytical factors, e.g., the tree reconstruction based on sequence alone (Szöllősi et al., 2013) and the limited amount of phylogenetic information (Steel, 2005), can lead to overestimating the phylogenetic conflict. Here, to reduce the discordance between gene trees inferred based on sequence alone, single-gene trees were reconciled using amalgamated likelihood estimation (ALE version 0.4) (Szöllősi et al., 2013, 2015). Briefly, the ultrafast bootstrap tree distributions generated by IQ-tree were converted into ALE objects using ALEobserve. And then, each ALE object was reconciled using ALEml_undated with the species tree.
Additionally, to reduce the gene tree incongruence due to differences in the number of parsimony-informative sites (PIS), the RF distance between the species tree and single-gene trees were normalized using a linear model between the PIS numbers and the actual RF distances. All RF distance values were sorted by corresponding PIS numbers ascendingly and divided into equal-sized bins (each bin contained 1,000 RF values). In each bin, the top 5% of RF values (the 5th percentile of RF values) representing “good” single-gene trees were selected for regression analysis with the relevant PIS numbers. A linear model was used to fit a line (RF = alog10NPIS+b, NPIS: the number of PIS) using least squares fitting. Then, all original RF values were normalized using the formula
Results
The morphological development predated the species divergence of Streptomyces
Based on 40 universal markers, a reference species tree (RST) of 373 Streptomyces genomes (basic genome datasets, BGD) was reconstructed. According to the evolutionary relationships reflected in RST, 373 Streptomyces species were segregated into eight major clades labeled as C1–C8 (Supplementary Figure 1). The species numbers of these clades were 1 (C1), 2 (C2), 18 (C3), 159 (C4), 36 (C5), 56 (C6), 15 (C7), and 86 (C8), respectively (Supplementary Table 3). Namely, the two deepest internal branches of the re-rooted RST possessed four descendant clades: C1–C4 (180 species) and C5–C8 (193 species). Parts of Streptomyces genomes were randomly selected according to the proportion of species number in eight clades, and their genomes constituted a small random genome dataset (RGD) containing 64 species (Supplementary Table 4). It’s worth noting that the number of species in each major clade is varied; hence the probabilities of 373 species being selected are also different. The highest proportion of overlapped species between RGDs was less than 37.2%. And each species was re-sampled an average of 17.3 times (standard deviation, sd: 6.2).
In terms of the distribution of homologous sequences, as expected, 44 of 65 development-related genes were identified over 90% of genomes in BGD (Figure 2, upper panel). Those genes ought to play crucial roles in the development of Streptomyces. For instance, AdpA, an essential early regulator of the development (Horinouchi, 2007), and MreB, involved in the spore cell wall assembly (Heichlinger et al., 2011) could be identified in all Streptomyces genomes. Nevertheless, nearly a third of the development-related genes showed varying degrees of loss. Most of them were either unnecessary for morphological development or could be replaced by alternative genes. For example, the narrow phylogenetic distribution of the SapB biosynthesis gene cluster, including the ramCSAB operon and ramR (a response regulator gene that controls the activation of the convergently transcribed ramCSAB operon), might relate to a SapB-independent pathway that is mediated by the chaplins (Nguyen et al., 2002; O’Connor et al., 2002). These results are consistent with the previous report (Chandra and Chater, 2014; Kim et al., 2015). However, the distribution of homologous sequences cannot reflect evolutionary events such as horizontal gene transfer and gene duplication. In other words, a gene of late origin can exhibit a wider phylogenetic distribution through HGT. Therefore, we evaluated the orthologies of these DGs and further investigated the distribution of orthologous DGs.
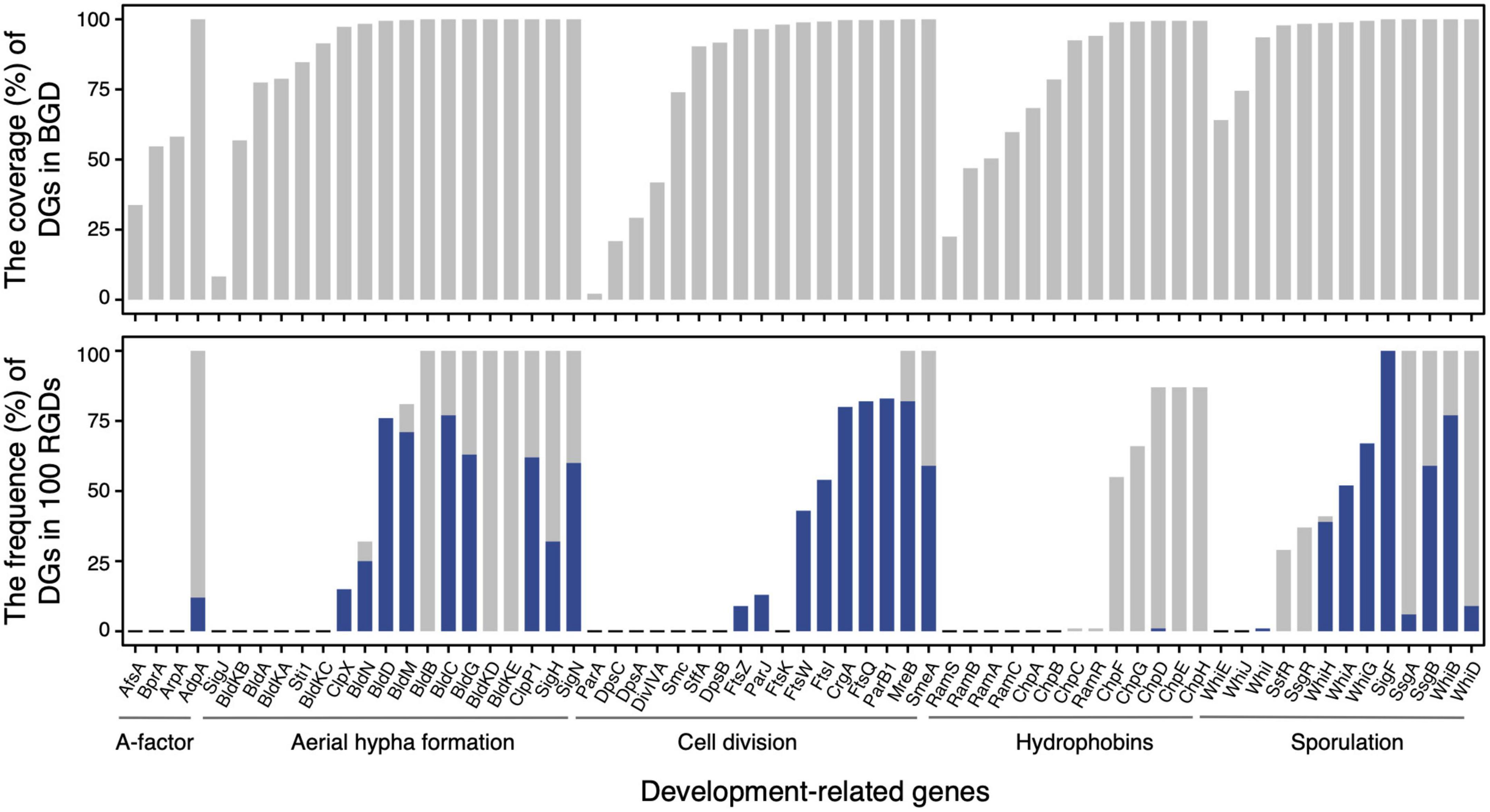
Figure 2. The distributions of development-related genes in the basic genome dataset (upper panel) and 100 random genome datasets (lower panel). In the lower panel, the frequencies of genome dataset (DG) as core gene family (having homolog in each genome) and strict orthogroup (subfamily assessed by UPhO) are visualized as gray and blue bars, respectively.
However, because it is difficult to obtain a reliable and stable phylogenetic result for the large sequence dataset, evaluating the orthology of DG directly based on BGD is not feasible. Therefore, the assessment of orthology was carried out based on RGDs in this work. With the reduction of the BGD to an RGD comprising 64 genomes, the development-related protein sequences only associated with focal RGD were extracted from the original DGs. In focal RGD, the development-related protein family distributed in all 64 genomes (number of sequences ≥64) was regarded as a core protein family and subjected to the assessment of orthology. The subfamilies of the core protein family corresponding to strict orthogroups were finally retained. Due to the high similarity between some query sequences (e.g., the similarities among SigH, SigF, and SigN were more than 70%), the identical strict orthogroup might be extracted from different DGs. In this case, only the strict orthogroup shared the highest similarity to the corresponding reference was extracted for further investigation. In addition, five protein families (ClpP1, SigN, SigF, WhiD, and SmeA) could be decomposed into two subfamilies as strict orthogroups and involved in 33 RGDs. Particularly, this situation occurs mainly in two protein families: ClpP1 (19 RGDs) and SigN (10 RGDs). These 33 RGDs included one and only one development-related gene that has this situation except for RGD064 and RGD090. All these strict orthogroups were retained for subsequent analysis.
The distributions of orthologous DGs in BGD were exhibited through the frequencies of DGs as core protein families (gray bars) and as strict orthogroups (blue bars) in 100 RGDs (Figure 2, lower panel). A total of 29 DGs involved in aerial hyphae and mature spore formation, cell division, and hydrophobin synthesis were recognized as core protein families in more than 50 % of RGDs. And 17 of 29 could extract strict orthogroups. These protein families from which strict orthogroups can be extracted tend to be more conserved than others. Notably, three protein families (BldB, BldKE, and BldKD) failed to extract the strict orthogroups, due to the high identity between sequences (resulting in a multifurcating tree). In addition, even though the strict orthogroups could be retrieved, the trimmed alignments were so short and conservative that the comparable trees of BldC, BldM, SsgB, WhiB, CrgA, and SmeA were unavailable. Besides the genome incompleteness and the species specificity of certain proteins, the progressive acquisition of these genes in the early ancestors of Streptomyces might also account for the differences in protein family distribution. Based on the larger genome datasets and relatively comprehensive protein collection, these results demonstrated that development-related genes had already been equipped in the genome of the common ancestor of Streptomyces and underwent a vertical evolutionary history, confirming that the origin of development should occur before the flourishing of its species diversity.
The concatenated DGs phylogeny has a higher degree of congruence with the species tree
In each RGD, two species trees based on supermatrix (ST1) and supertree (ST2) approaches and two concatenated ML trees, DT (based on DGs) and UT (based on UGs), were reconstructed. And a batch of single-gene trees was also inferred for strict orthogroups (sOGs) (496 60.8 trees) and DGs (10 2.6 trees). The Robinson-Foulds (RF) distance was implemented to evaluate the topological differences. Noticeably, when RST was compared to ST1, ST2, DT, and UT; it was required to be pruned to fit the different comparison objects. As shown in Figure 3A, the RF distances from UT to RST (mean sd: 0.36 0.1) were not significantly different from the distances from ST1 to RST (0.37 0.08) (Student’s t-test, p = 0.167). Compared to RST, ST1 differed in both the underlying gene dataset and the number of species involved; but UT only differed in the number of species involved. Therefore, it indicated that the random resampling strategy could achieve the goal of maintaining topological stability when reducing the number of species involved. Nevertheless, in addition to the underlying gene dataset and the number of species involved, the species tree-building approach of ST2 was also different. That might cause a significant difference between the distances from ST2 to RST and that from UT to RST (t-test, p = 0.048). The RF distances from DT to RST were significantly higher than that from ST1, ST2, and UT to RST (t-test, all p-values < 10–5).
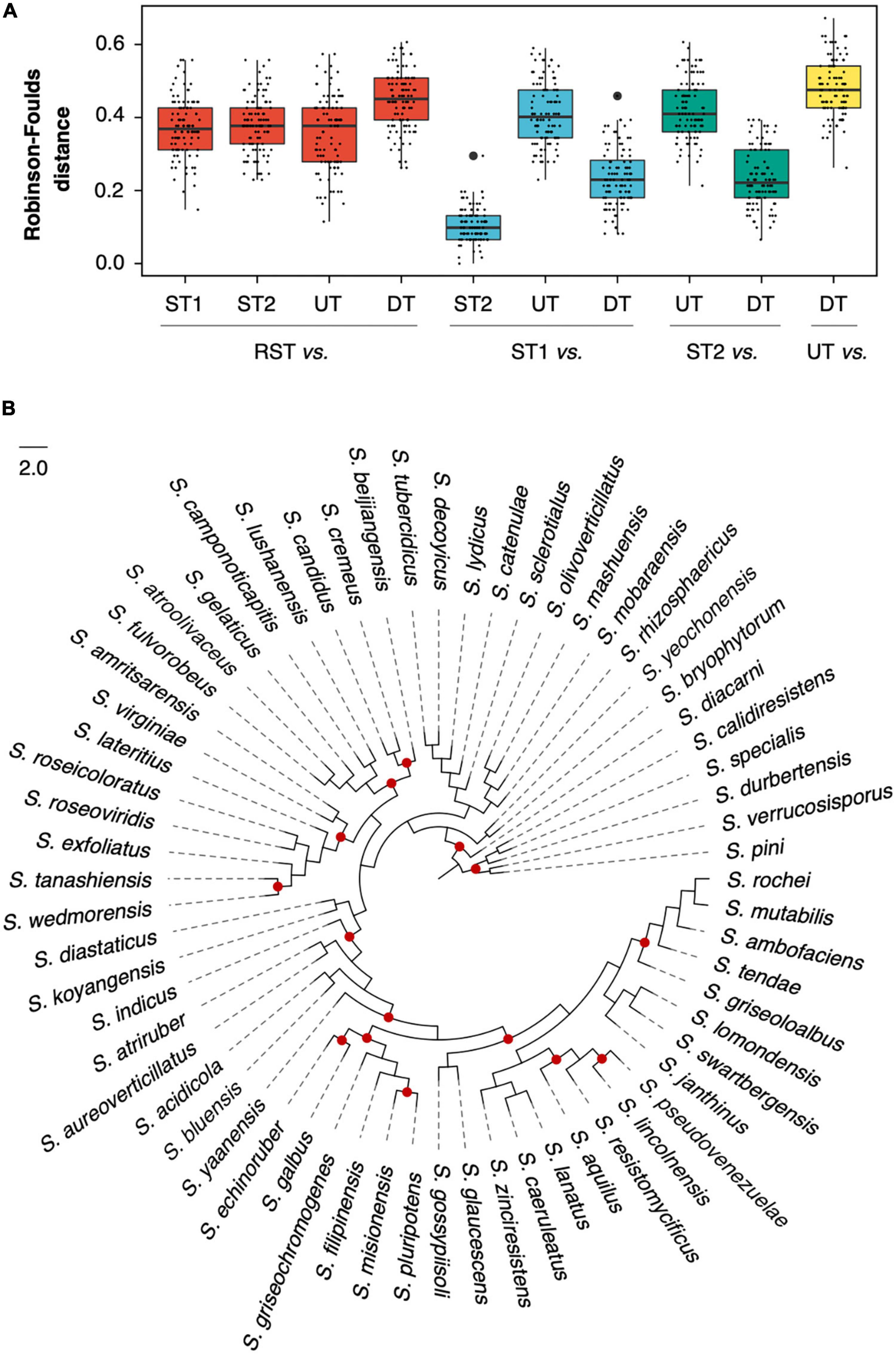
Figure 3. Topological comparison among species trees and concatenated maximum likelihood (ML) trees. (A) Robinson-Foulds distance between two compared trees in one of 100 random genome datasets (RGDs); (B) the species tree of RGD008 based on the supermatrix approach. The red dots represent where the phylogenetic conflict between DT and ST1 occurred.
When ST1 was used as a reference, the RF distances from DT to ST1 decreased dramatically (from 0.45 0.08 to 0.23 0.08) and were even significantly lower than that from UT to ST1 (t-test, p < 10–5). For example, Figure 3B illustrates the degree of congruence between DT and ST1, which were generated from dataset RGD008 (RF = 0.23). Given the negligible difference between ST1 and ST2 (their average RF distance was only 0.10 0.05), the distances of DT decreased similarly when ST2 was used as a reference. For further understanding of the distances from DT to ST1, sOGs were divided according to COG functional categories and then used to infer concatenated ML trees for each COG category. The average RF distances from COG categories to ST1 ranged from 0.11 (COG Others, including orthogroups that could not be assigned to COGs or whose COG category appeared once in 100 RGDs) to 0.33 (COG P) (Supplementary Table 5). Interestingly, DT had a lower RF distance to ST1 than half of the COG categories. Concerning concatenation analysis, the phylogenies of concatenated DGs had a higher degree of congruence with the specie trees of the genus Streptomyces.
The single-gene phylogenies of DGs have no significant difference from that of sOGs on phylogenetic conflict
The concatenation approach integrating phylogenetic information from multiple gene loci suppresses the phylogenetic heterogeneity between different genes (Pease et al., 2016; Wu et al., 2018; Jiang et al., 2020). To investigate the phylogenetic conflicts of single genes with the species tree, all single-gene trees in different gene datasets were individually compared with the species tree (Figure 4 and Supplementary Figure 2, detailed data see Supplementary Table 5). For each gene dataset, the mean value of RF distances from single-gene trees to the species tree was calculated and used to measure the topological difference between the whole gene dataset and the species tree. The distribution of mean values of RF distances from sOGs to ST1 demonstrated that the single-gene tree had a higher degree of incongruence to the species tree than the concatenated tree, as expected.
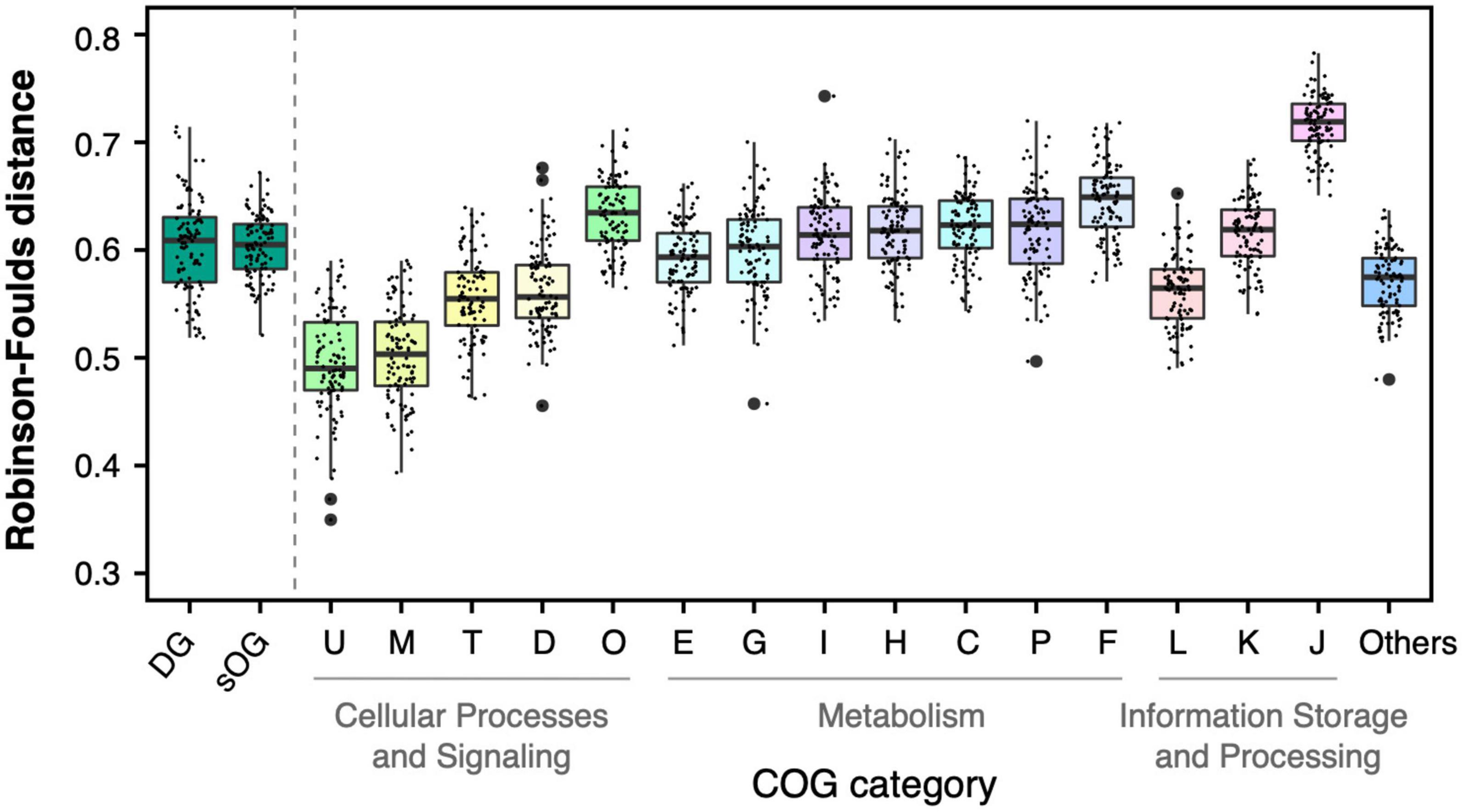
Figure 4. Topological comparison between single-gene trees and species tree based on supermatrix (ST1). Each single data point represented a mean value of Robison-Foulds distances between single-gene trees in the corresponding dataset [e.g., genome dataset (DG)] to ST1 in one of 100 random genome datasets (RGDs).
The mean RF distances from DGs to ST1 (0.60 0.04) were not significantly different from the mean RF distances from sOGs to ST1 (0.60 0.03) (t-test, p = 0.9). And it is consistent with the results of concatenation analysis, DGs had lower RF distance to ST1 than parts of COG categories (e.g., COGs F and J). sOGs belonging to different COG categories showed different degrees of topological incongruence with species trees. For instance, COG M (cell wall/membrane/envelope biogenesis) and COG U (intracellular trafficking, secretion, and vesicular transport) had relatively lower mean RF distances to the species tree. And, at the higher level of functional classification, the single-gene trees of COGs related to cellular processes and signaling were closer to the species tree than those of other COGs involved in metabolism and information storage and processing. Astonishingly, COG J (translation, ribosomal structure, and biogenesis), including all ribosomal proteins usually used in phylogenomic analysis exhibited the highest degree of incongruence with the species tree. However, the concatenated tree of COG J had only 0.26 RF distance on average to ST1 (Supplementary Table 5). This suggested that the concatenation would exert a complex effect on species tree reconstruction.
The phylogenetic inference of a single gene based on sequence alone often lacks enough information to confidently support one gene tree topology. This may be one of the major reasons why the single gene has a higher degree of topological incongruence than the concatenated alignment. By reconciling the gene tree with a putative species tree based on the joint likelihood, its incongruence with the species tree was substantially reduced (Figure 5 and Supplementary Figure 3). However, the mean RF distances of reconciled gene trees are still higher than those of the corresponding concatenation trees. Interestingly, although the RF distances were decreased after the gene trees were reconciled, the magnitudes of the RF distance reduction of various gene datasets relative to sOGs were discrepant. Generally, gene datasets whose single-gene trees were closer to the species tree, such as COGs M and U, had more magnitudes of RF distance reduction. In contrast, gene datasets whose single-gene trees were more distant from the species tree, such as COG J, had fewer magnitudes of RF distance reduction.
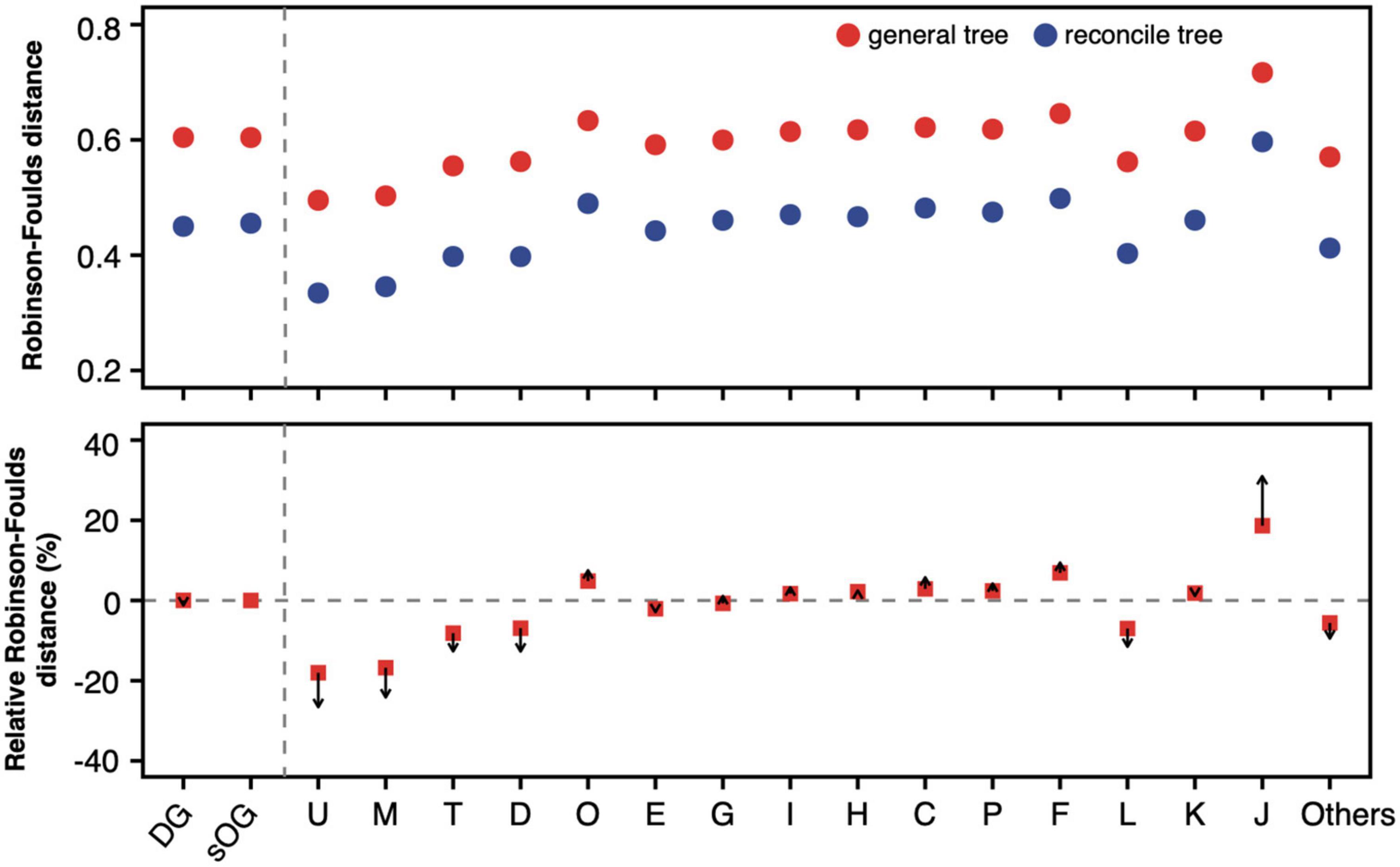
Figure 5. Robinson-Foulds distance of general gene tree and reconciled gene tree to supermatrix (ST1). For a gene dataset, 100 mean values of Robinson-Foulds (RF) distances from 100 random genome datasets (RGDs) were averaged and shown in the (upper panel). Red solid circles represent the average RF distance of general gene trees to ST1, and solid blue circles represent the average RF distance of reconciled gene trees to ST1. The relative RF distance was calculated as formula, , where tJ is general gene tree of the gene in Clusters of Orthologous Genes (COG) J. In the (lower panel), arrows start from the relative RF distance of general gene trees and point to the relative RF distance of reconciled gene trees.
DGs exhibited the highest topological concordance with the species tree based on the normalized RF distance
As we know, the number of information sites used for phylogenetics profoundly impacted the topology of the resultant tree. Therefore, the limited number of parsimony-informative sites might contribute to the topological incongruence between the single gene tree and the species tree. Meanwhile, we noted that the numbers of parsimony-informative sites of proteins seem to be negatively correlated to their RF distances (Pearson’s r = -0.51, p < 0.01, Supplementary Table 5). Therefore, to determine the correlation between the number of parsimony-informative sites and the topological incongruence, all single genes in different gene datasets were divided into bins according to the number of parsimony-informative sites. The RF distance values in each bin were sorted ascendingly, and the top 5% of RF values representing ones with “good” single-gene phylogeny (similar to species tree) were used for the regression analysis (Figure 6 and Supplementary Figure 4).
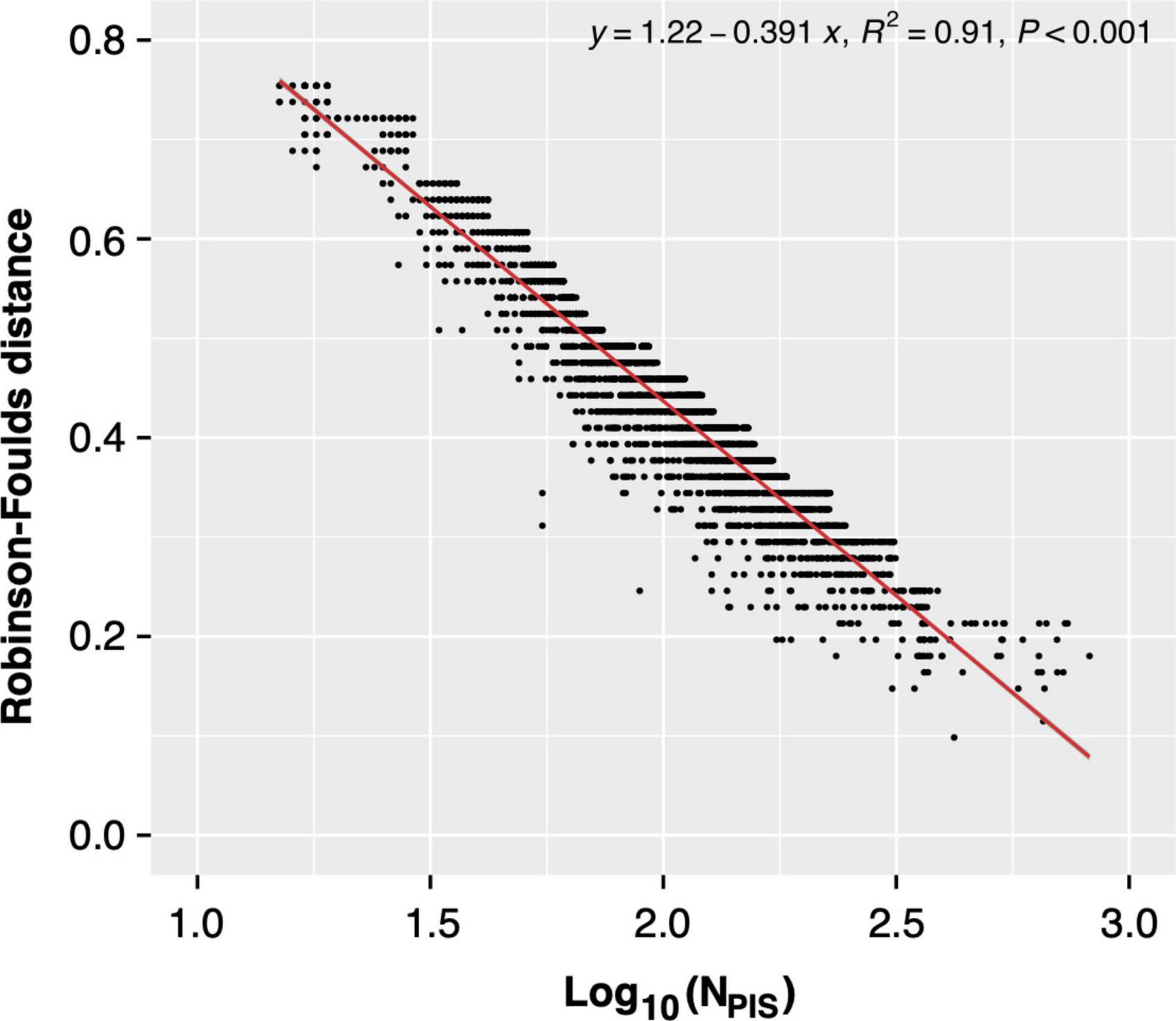
Figure 6. Regression analysis between Robinson-Foulds (RF) distance [between the single-gene tree and supermatrix (ST1)] and the number of parsimony-informative sites.
The actual RF distances were normalized based on the linear regression model (Figure 7 and Supplementary Figure 5). However, the normalized RF distance scale changed, which made the direct comparison to the original RF distance inapplicable. So, just as reconciled gene tree comparison, relative RF distance was used to present the variation of RF distance caused by the normalization based on the number of parsimony-informative sites. The relative mean RF distance from single-gene trees of COG J to ST1 decreased from 18.6 to 1.2%; on the contrary, the relative mean RF distance from single-gene trees of COG L to ST1 increased from –6.9 to 6.0%. Generally, the regression model decreased the RF distances of genes with more parsimony-informative sites (such as COGs U, M, E, G, C, and L). In contrast, the RF distances of genes with a lower number of parsimony-informative sites were increased (such as COGs J, K, and DGs). Remarkably, the RF distances of DGs had dramatically changed relative to sOGs same as COG J (decreased from <0.1 to –13.2%), and became the lowest ones. Taken together, these results indicated that development-related genes exhibited more congruent single-gene phylogenies to the species tree versus strict orthogroups, especially when technical factors like the limitation of parsimony-informative sites are excluded.
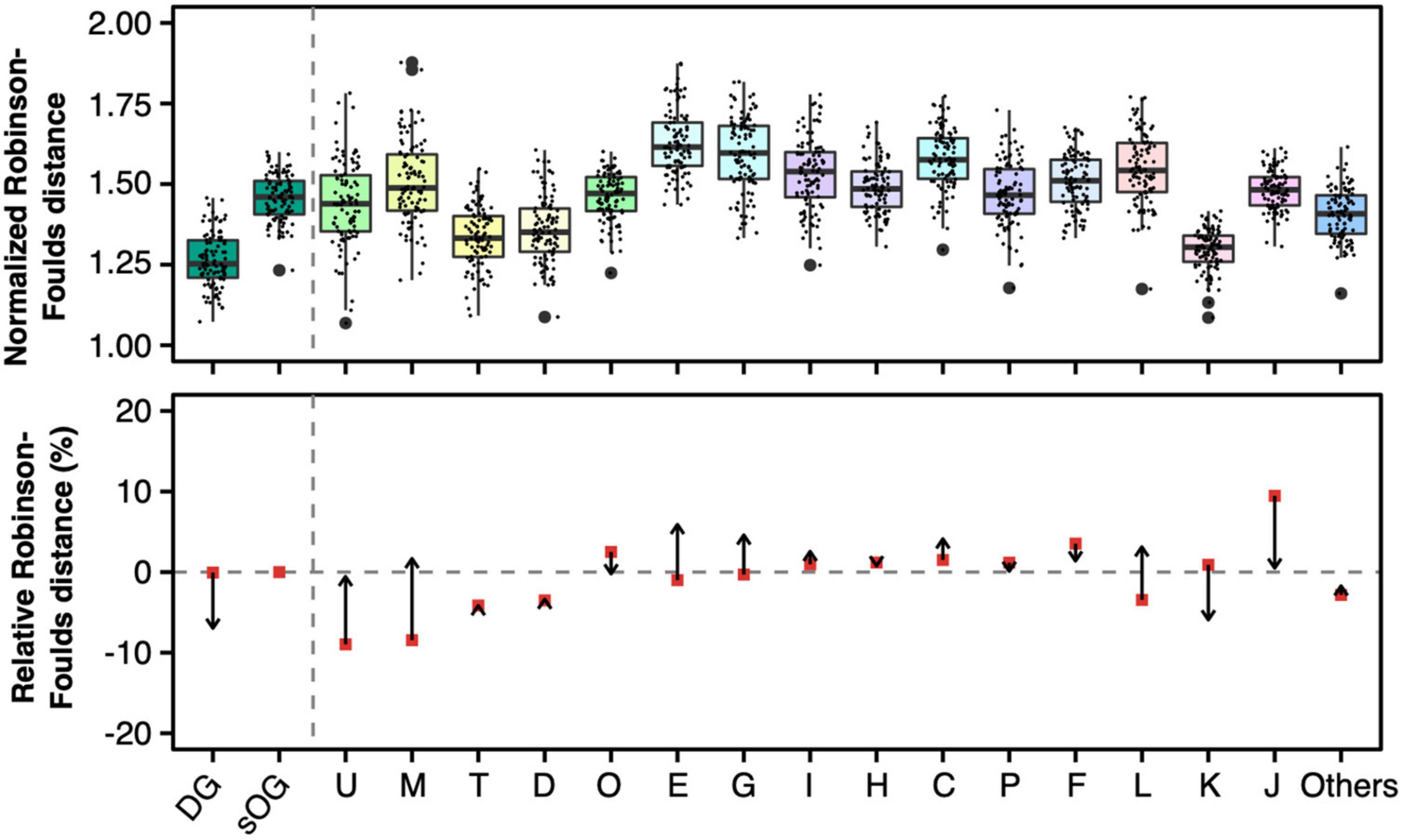
Figure 7. Normalized Robison-Foulds distances (upper panel) and relative normalized Robison-Foulds distances (lower panel) between single-gene trees and supermatrix (ST1). Like the lower panel of Figure 4, the arrows start from the relative Robison-Foulds (RF) distances of general gene trees and point to the relative RF distances after normalization based on the number of parsimony-informative sites.
Discussion
The development of Streptomyces, including both morphological and physiological differentiation, was controlled by the same complex regulatory system and endowed streptomycetes with the ability to cope with complex living environments. Based on a large genome dataset, we found that development-related genes present differential distribution patterns in Streptomyces genomes. Parts of development-related genes had been equipped in the genome of the common ancestor of Streptomyces bacteria. Although it remains unknown whether these are sufficient to support the morphological development of the common ancestor of Streptomyces, the origin of these genes can be traced back to the early stages of Streptomyces speciation or even earlier. The phylogenetic distribution of homologous genes is an essential tool for studying the origin of genes and relevant biological functions, relying on the assumption that ancient genes often have a broader phylogenetic distribution. However, the natural evolutionary process is not entirely immune to confounding factors, such as horizontal gene transfer, which inevitably complicate the evolution of the gene family. In this study, the orthologies of development-related genes were also considered besides their phylogenetic distribution.
Furthermore, we compared the phylogenetic topologies of related genes to the species tree on both concatenation and single-gene levels. We found that the evolution of development-related genes had higher congruence to the species tree. Especially when the analytical factors, such as tree reconstruction based on sequence alone and the limited amount of phylogenetic information, were excluded, the development-related genes exhibited the lowest degree of topological incongruence with the species tree. This is consistent with the above conclusion that parts of development-related genes have been assembled in the genome of the comment ancestor of Streptomyces. Consequently, the subsequent evolution of these genes should be synchronized with the history of species differentiation. Or in other words, the development-related genes documented the evolutionary history of Streptomyces. Meanwhile, it must be noted that the phylogenies of some development-related genes originating earlier in Streptomyces were relatively complex. These genes were either not conservative enough, or their genuine orthologies were obscured by other evolutionary events like lateral gene transfer, gene duplication, and loss.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: doi: 10.5061/dryad.00000005d.
Author contributions
MW and C-JL: initial drafting, data analysis, figures generation, and discussion. ZZ and P-PL: raw data processing and discussion. L-LY: discussion and draft correction. X-YZ: project coordination, discussion, and draft correction. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (Grant Number: 32060003); Program for Excellent Young Talents, Yunnan University; and grants from the Major Science and Technology Projects of Yunnan Province (Digitalization, development and application of biotic resource, 202002AA100007).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1102250/full#supplementary-material
Footnotes
References
Aínsa, J. A., Bird, N., Ryding, N. J., Findlay, K. C., and Chater, K. F. (2010). The complex whiJ locus mediates environmentally sensitive repression of development of Streptomyces coelicolor A3(2). Antonie Van Leeuwenhoek 98, 225–236. doi: 10.1007/s10482-010-9443-3
Ainsa, J. A., Parry, H. D., and Chater, K. F. (1999). A response regulator-like protein that functions at an intermediate stage of sporulation in Streptomyces coelicolor A3(2). Mol. Microbiol. 34, 607–619. doi: 10.1046/j.1365-2958.1999.01630.x
Aínsa, J. A., Ryding, N. J., Hartley, N., Findlay, K. C., Bruton, C. J., and Chater, K. F. (2000). WhiA, a protein of unknown function conserved among gram-positive bacteria, is essential for sporulation in Streptomyces coelicolor A3(2). J. Bacteriol. 182, 5470–5478. doi: 10.1128/JB.182.19.5470-5478.2000
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andam, C. P., Doroghazi, J. R., Campbell, A. N., Kelly, P. J., Choudoir, M. J., and Buckley, D. H. (2016). A latitudinal diversity gradient in terrestrial bacteria of the genus Streptomyces. mBio 7, e2200–e2215. doi: 10.1128/mBio.02200-15
Anderson, A. S., and Wellington, E. M. (2001). The taxonomy of Streptomyces and related genera. Int J. Syst. Evol. Microbiol. 51, 797–814. doi: 10.1099/00207713-51-3-797
Ausmees, N., Wahlstedt, H., Bagchi, S., Elliot, M. A., Buttner, M. J., and Flärdh, K. (2007). SmeA, a small membrane protein with multiple functions in Streptomyces sporulation including targeting of a SpoIIIE/FtsK-like protein to cell division septa. Mol. Microbiol. 65, 1458–1473. doi: 10.1111/j.1365-2958.2007.05877.x
Ballesteros, J. A., and Hormiga, G. (2016). A New orthology assessment method for phylogenomic data: Unrooted phylogenetic orthology. Mol. Biol. Evol. 33, 2117–2134. doi: 10.1093/molbev/msw069
Bennett, J. A., Yarnall, J., Cadwallader, A. B., Kuennen, R., Bidey, P., Stadelmaier, B., et al. (2009). Medium-dependent phenotypes of Streptomyces coelicolor with mutations in ftsI or ftsW. J. Bacteriol. 191, 661–664. doi: 10.1128/JB.01048-08
Bentley, S. D., Chater, K. F., Cerdeño-Tárraga, A.-M., Challis, G. L., Thomson, N. R., James, K. D., et al. (2002). Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147. doi: 10.1038/417141a
Bibb, M. J. (2005). Regulation of secondary metabolism in streptomycetes. Curr. Opin. Microbiol. 8, 208–215. doi: 10.1016/j.mib.2005.02.016
Bignell, D. R. D., Lau, L. H., Colvin, K. R., and Leskiw, B. K. (2003). The putative anti-anti-sigma factor BldG is post-translationally modified by phosphorylation in Streptomyces coelicolor. FEMS Microbiol. Lett. 225, 93–99. doi: 10.1016/S0378-1097(03)00504-4
Boeckmann, B., Robinson-Rechavi, M., Xenarios, I., and Dessimoz, C. (2011). Conceptual framework and pilot study to benchmark phylogenomic databases based on reference gene trees. Brief Bioinform. 12, 423–435. doi: 10.1093/bib/bbr034
Briand, S., Dessimoz, C., El-Mabrouk, N., Lafond, M., and Lobinska, G. (2020). A generalized robinson-foulds distance for labeled trees. BMC Genomics 21:779. doi: 10.1186/s12864-020-07011-0
Canback, B., Andersson, S. G. E., and Kurland, C. G. (2002). The global phylogeny of glycolytic enzymes. Proc. Natl. Acad. Sci. U.S.A. 99, 6097–6102. doi: 10.1073/pnas.082112499
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Capstick, D. S., Willey, J. M., Buttner, M. J., and Elliot, M. A. (2007). SapB and the chaplins: Connections between morphogenetic proteins in Streptomyces coelicolor. Mol. Microbiol. 64, 602–613. doi: 10.1111/j.1365-2958.2007.05674.x
Chandra, G., and Chater, K. F. (2014). Developmental biology of Streptomyces from the perspective of 100 actinobacterial genome sequences. FEMS Microbiol. Rev. 38, 345–379. doi: 10.1111/1574-6976.12047
Chater, K. F. (2006). Streptomyces inside-out: A new perspective on the bacteria that provide us with antibiotics. Phil. Trans. R. Soc. B 361, 761–768. doi: 10.1098/rstb.2005.1758
Chater, K. F., and Chandra, G. (2006). The evolution of development in Streptomyces analysed by genome comparisons. FEMS Microbiol. Rev. 30, 651–672. doi: 10.1111/j.1574-6976.2006.00033.x
Chater, K. F., Bruton, C. J., Plaskitt, K. A., Buttner, M. J., Méndez, C., and Helmann, J. D. (1989). The developmental fate of S. coelicolor hyphae depends upon a gene product homologous with the motility σ factor of B. subtilis. Cell 59, 133–143. doi: 10.1016/0092-8674(89)90876-3
Chávez, A., Forero, A., Sánchez, M., Rodríguez-Sanoja, R., Mendoza-Hernández, G., Servín-Gonzalez, L., et al. (2011). Interaction of SCO2127 with BldKB and its possible connection to carbon catabolite regulation of morphological differentiation in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 89, 799–806. doi: 10.1007/s00253-010-2905-8
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Creamer, K. E., Kudo, Y., Moore, B. S., and Jensen, P. R. (2021). Phylogenetic analysis of the salinipostin γ-butyrolactone gene cluster uncovers new potential for bacterial signalling-molecule diversity. Microbial. Genomics 7:000568. doi: 10.1099/mgen.0.000568
Dalton, K. A., Thibessard, A., Hunter, J. I. B., and Kelemen, G. H. (2007). A novel compartment, the ?subapical stem? of the aerial hyphae, is the location of a sigN-dependent, developmentally distinct transcription in Streptomyces coelicolor. Mol. Microbiol. 64, 719–737. doi: 10.1111/j.1365-2958.2007.05684.x
Davis, N. K., and Chater, K. F. (1992). The Streptomyces coelicolor whiB gene encodes a small transcription factor-like protein dispensable for growth but essential for sporulation. Mol. Gen. Genet. 232, 351–358. doi: 10.1007/BF00266237
de Crecy-Lagard, V., Servant-Moisson, P., Viala, J., Grandvalet, C., and Mazodier, P. (1999). Alteration of the synthesis of the Clp ATP-dependent protease affects morphological and physiological differentiation in Streptomyces. Mol. Microbiol. 32, 505–517. doi: 10.1046/j.1365-2958.1999.01364.x
Dedrick, R. M., Wildschutte, H., and McCormick, J. R. (2009). Genetic interactions of smc, ftsK, and parB genes in Streptomyces coelicolor and their developmental genome segregation phenotypes. J. Bacteriol. 191, 320–332. doi: 10.1128/JB.00858-08
Del Sol, R., Pitman, A., Herron, P., and Dyson, P. (2003). The product of a developmental gene, crgA, that coordinates reproductive growth in Streptomyces belongs to a novel family of small actinomycete-specific proteins. J. Bacteriol. 185, 6678–6685. doi: 10.1128/JB.185.22.6678-6685.2003
Ditkowski, B., Troæ, P., Ginda, K., Donczew, M., Chater, K. F., Zakrzewska-Czerwiñska, J., et al. (2010). The actinobacterial signature protein ParJ (SCO1662) regulates ParA polymerization and affects chromosome segregation and cell division during Streptomyces sporulation: ParJ – regulator of ParA polymerization. Mol. Microbiol. 78, 1403–1415. doi: 10.1111/j.1365-2958.2010.07409.x
Eccleston, M., Willems, A., Beveridge, A., and Nodwell, J. R. (2006). Critical residues and novel effects of overexpression of the Streptomyces coelicolor developmental protein BldB: Evidence for a critical interacting partner. J. Bacteriol. 188, 8189–8195. doi: 10.1128/JB.01119-06
Elliot, M., Damji, F., Passantino, R., Chater, K., and Leskiw, B. (1998). The bldD gene of Streptomyces coelicolor A3(2): A regulatory gene involved in morphogenesis and antibiotic production. J. Bacteriol. 180, 1549–1555. doi: 10.1128/JB.180.6.1549-1555.1998
Emms, D. M., and Kelly, S. (2015). OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Facey, P. D., Hitchings, M. D., Saavedra-Garcia, P., Fernandez-Martinez, L., Dyson, P. J., and Del Sol, R. (2009). Streptomyces coelicolor Dps-like proteins: Differential dual roles in response to stress during vegetative growth and in nucleoid condensation during reproductive cell division. Mol. Microbiol. 73, 1186–1202. doi: 10.1111/j.1365-2958.2009.06848.x
Flärdh, K., and Buttner, M. J. (2009). Streptomyces morphogenetics: Dissecting differentiation in a filamentous bacterium. Nat. Rev. Microbiol. 7, 36–49. doi: 10.1038/nrmicro1968
Galperin, M. Y., Wolf, Y. I., Makarova, K. S., Vera Alvarez, R., Landsman, D., and Koonin, E. V. (2021). COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281. doi: 10.1093/nar/gkaa1018
Genay, M., Decaris, B., and Dary, A. (2007). Implication of stringent response in the increase of mutability of the whiG and whiH genes during Streptomyces coelicolor development. Mutat. Res. 624, 49–60. doi: 10.1016/j.mrfmmm.2007.03.016
Hammerschmidt, K., Landan, G., Domingues Kümmel Tria, F., Alcorta, J., and Dagan, T. (2021). The order of trait emergence in the evolution of cyanobacterial multicellularity. Genome Biol. Evol. 13:evaa249. doi: 10.1093/gbe/evaa249
Hayati, M., and Chindelevitch, L. (2020). Computing the distribution of the Robinson-Foulds distance. Comput. Biol. Chem. 87:107284. doi: 10.1016/j.compbiolchem.2020.107284
Heichlinger, A., Ammelburg, M., Kleinschnitz, E.-M., Latus, A., Maldener, I., Flärdh, K., et al. (2011). The MreB-Like Protein Mbl of Streptomyces coelicolor A3(2) depends on MreB for proper localization and contributes to spore wall synthesis. J. Bacteriol. 193, 1533–1542. doi: 10.1128/JB.01100-10
Horinouchi, S. (2007). Mining and polishing of the treasure trove in the bacterial genus streptomyces. Biosci. Biotechnol. Biochem. 71, 283–299. doi: 10.1271/bbb.60627
Horinouchi, S., Suzuki, H., Nishiyama, M., and Beppu, T. (1989). Nucleotide sequence and transcriptional analysis of the Streptomyces griseus gene (afsA) responsible for A-factor biosynthesis. J. Bacteriol. 171, 1206–1210. doi: 10.1128/jb.171.2.1206-1210.1989
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Hunt, A. C., Servín-González, L., Kelemen, G. H., and Buttner, M. J. (2005). The bldC developmental locus of Streptomyces coelicolor encodes a member of a family of small DNA-binding proteins related to the DNA-binding domains of the MerR family. J. Bacteriol. 187, 716–728. doi: 10.1128/JB.187.2.716-728.2005
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jiang, H., and Kendrick, K. E. (2000). Characterization of ssfR and ssgA, Two genes involved in sporulation of Streptomyces griseus. J. Bacteriol. 182, 5521–5529. doi: 10.1128/JB.182.19.5521-5529.2000
Jiang, X., Edwards, S. V., and Liu, L. (2020). the multispecies coalescent model outperforms concatenation across diverse phylogenomic data sets. Syst. Biol. 69, 795–812. doi: 10.1093/sysbio/syaa008
Joynt, R., and Seipke, R. F. (2018). A phylogenetic and evolutionary analysis of antimycin biosynthesis. Microbiology 164, 28–39. doi: 10.1099/mic.0.000572
Kämpfer, P. (2015). “Streptomyces,” in Bergey’s manual of systematics of archaea and bacteria, eds W. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, et al. (New York, NY: John Wiley & Sons). doi: 10.1002/9781118960608.gbm00191
Kim, D.-W., Chater, K., Lee, K.-J., and Hesketh, A. (2005). Changes in the extracellular proteome caused by the absence of the bldA gene product, a developmentally significant tRNA, reveal a new target for the pleiotropic regulator AdpA in Streptomyces coelicolor. J. Bacteriol. 187, 2957–2966. doi: 10.1128/JB.187.9.2957-2966.2005
Kim, E. S., Song, J. Y., Kim, D. W., Chater, K. F., and Lee, K. J. (2008). A possible extended family of regulators of sigma factor activity in Streptomyces coelicolor. J. Bacteriol. 190, 7559–7566. doi: 10.1128/JB.00470-08
Kim, H.-J., Calcutt, M. J., Schmidt, F. J., and Chater, K. F. (2000). Partitioning of the linear chromosome during sporulation of Streptomyces coelicolor A3(2) involves an oriC -Linked parAB locus. J. Bacteriol. 182, 1313–1320. doi: 10.1128/JB.182.5.1313-1320.2000
Kim, J.-N., Kim, Y., Jeong, Y., Roe, J.-H., Kim, B.-G., and Cho, B.-K. (2015). Comparative genomics reveals the core and accessory genomes of Streptomyces Species. J. Microbiol. Biotechnol. 25, 1599–1605. doi: 10.4014/jmb.1504.04008
Komaki, H., Ichikawa, N., Hosoyama, A., Fujita, N., and Igarashi, Y. (2015). Draft genome sequence of marine-derived Streptomyces sp. TP-A0598, a producer of anti-MRSA antibiotic lydicamycins. Stand. Genomic Sci. 10:58. doi: 10.1186/s40793-015-0046-5
Kormanec, J., and Sevcikova, B. (2002). The stress-response sigma factor σH controls the expression of ssgB, a homologue of the sporulation-specific cell division gene ssgA, in Streptomyces coelicolor A3(2). Mol. Gen. Genomics 267, 536–543. doi: 10.1007/s00438-002-0687-0
Lee, N., Hwang, S., Kim, J., Cho, S., Palsson, B., and Cho, B.-K. (2020). Mini review: Genome mining approaches for the identification of secondary metabolite biosynthetic gene clusters in Streptomyces. Comput. Struct. Biotechnol. J. 18, 1548–1556. doi: 10.1016/j.csbj.2020.06.024
Li, C.-J., Wang, M., Zhang, Z., Zhi, X.-Y., and Yang, L.-L. (2022). Genome-based analyses reveal heterotypic synonyms of Streptomyces species and associated subspecies. Arch. Microbiol. 204:581. doi: 10.1007/s00203-022-03204-1
Li, Y., Pinto-Tomás, A. A., Rong, X., Cheng, K., Liu, M., and Huang, Y. (2019). Population genomics insights into adaptive evolution and ecological differentiation in Streptomycetes. Appl. Environ. Microbiol. 85, e2555–e2518. doi: 10.1128/AEM.02555-18
Liu, G., Chater, K. F., Chandra, G., Niu, G., and Tan, H. (2013). Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 77, 112–143. doi: 10.1128/MMBR.00054-12
Ma, H., and Kendall, K. (1994). Cloning and analysis of a gene cluster from Streptomyces coelicolor that causes accelerated aerial mycelium formation in Streptomyces lividans. J. Bacteriol. 176, 3800–3811. doi: 10.1128/jb.176.12.3800-3811.1994
Mazurakova, V., Sevcikova, B., Rezuchova, B., and Kormanec, J. (2006). Cascade of sigma factors in streptomycetes: Identification of a new extracytoplasmic function sigma factor σJ that is under the control of the stress-response sigma factor σH in Streptomyces coelicolor A3(2). Arch. Microbiol. 186, 435–446. doi: 10.1007/s00203-006-0158-9
McCormick, J. R., and Losick, R. (1996). Cell division gene ftsQ is required for efficient sporulation but not growth and viability in Streptomyces coelicolor A3(2). J. Bacteriol. 178, 5295–5301. doi: 10.1128/jb.178.17.5295-5301.1996
McCormick, J. R., Su, E. P., Driks, A., and Losick, R. (1994). Growth and viability of Streptomyces coelicolor mutant for the cell division gene ftsZ. Mol. Microbiol. 14, 243–254. doi: 10.1111/j.1365-2958.1994.tb01285.x
McDonald, B. R., and Currie, C. R. (2017). Lateral Gene transfer dynamics in the ancient bacterial genus Streptomyces. mBio 8, e644–e617. doi: 10.1128/mBio.00644-17
Mende, D. R., Sunagawa, S., Zeller, G., and Bork, P. (2013). Accurate and universal delineation of prokaryotic species. Nat. Methods 10, 881–884. doi: 10.1038/nmeth.2575
Mirarab, S., Reaz, R., Bayzid, M., Zimmermann, T., Swenson, M. S., and Warnow, T. (2014). ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 30, i541–i548. doi: 10.1093/bioinformatics/btu462
Mistry, B. V., Del Sol, R., Wright, C., Findlay, K., and Dyson, P. (2008). FtsW is a dispensable cell division protein required for Z-ring stabilization during sporulation septation in Streptomyces coelicolor. J. Bacteriol. 190, 5555–5566. doi: 10.1128/JB.00398-08
Navarro-Muñoz, J. C., Selem-Mojica, N., Mullowney, M. W., Kautsar, S. A., Tryon, J. H., Parkinson, E. I., et al. (2020). A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60–68. doi: 10.1038/s41589-019-0400-9
Nguyen, K. T., Willey, J. M., Nguyen, L. D., Nguyen, L. T., Viollier, P. H., and Thompson, C. J. (2002). A central regulator of morphological differentiation in the multicellular bacterium Streptomyces coelicolor. Mol. Microbiol. 46, 1223–1238. doi: 10.1046/j.1365-2958.2002.03255.x
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
O’Connor, T. J., Kanellis, P., and Nodwell, J. R. (2002). The ramC gene is required for morphogenesis in Streptomyces coelicolor and expressed in a cell type-specific manner under the direct control of RamR. Mol. Microbiol. 45, 45–57. doi: 10.1046/j.1365-2958.2002.03004.x
Ohnishi, Y., Ishikawa, J., Hara, H., Suzuki, H., Ikenoya, M., Ikeda, H., et al. (2008). Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 190, 4050–4060. doi: 10.1128/JB.00204-08
Ohnishi, Y., Kameyama, S., Onaka, H., and Horinouchi, S. (1999). The A-factor regulatory cascade leading to streptomycin biosynthesis in Streptomyces griseus?: Identification of a target gene of the A-factor receptor. Mol. Microbiol. 34, 102–111. doi: 10.1046/j.1365-2958.1999.01579.x
Ohnishi, Y., Yamazaki, H., Kato, J., Tomono, A., and Horinouchi, S. (2005). AdpA, a central transcriptional regulator in the A-factor regulatory cascade that leads to morphological development and secondary metabolism in Streptomyces griseus. Biosci. Biotechnol. Biochem. 69, 431–439. doi: 10.1271/bbb.69.431
Onaka, H., Ando, N., Nihira, T., Yamada, Y., Beppu, T., and Horinouchi, S. (1995). Cloning and characterization of the A-factor receptor gene from Streptomyces griseus. J. Bacteriol. 177, 6083–6092. doi: 10.1128/jb.177.21.6083-6092.1995
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Pease, J. B., Haak, D. C., Hahn, M. W., and Moyle, L. C. (2016). Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PLoS Biol. 14:e1002379. doi: 10.1371/journal.pbio.1002379
Potúèková, L., Kelemen, G. H., Findlay, K. C., Lonetto, M. A., Buttner, M. J., and Kormanec, J. (1995). A new RNA polymerase sigma factor,? F is required for the late stages of morphological differentiation in Streptomyces spp. Mol. Microbiol. 17, 37–48. doi: 10.1111/j.1365-2958.1995.mmi_17010037.x
Rodríguez, H., Rico, S., Díaz, M., and Santamaría, R. I. (2013). Two-component systems in Streptomyces: Key regulators of antibiotic complex pathways. Microb. Cell Fact. 12:127. doi: 10.1186/1475-2859-12-127
Sanchez-Baracaldo, P., Hayes, P. K., and Blank, C. E. (2005). Morphological and habitat evolution in the Cyanobacteria using a compartmentalization approach. Geobiology 3, 145–165. doi: 10.1111/j.1472-4669.2005.00050.x
Schirrmeister, B. E., Antonelli, A., and Bagheri, H. C. (2011). The origin of multicellularity in cyanobacteria. BMC Evol. Biol. 11:45. doi: 10.1186/1471-2148-11-45
Schrempf, D., and Szöllősi, G. (2020). “The sources of phylogenetic conflicts,” in phylogenetics in the genomic era, eds C. Scornavacca, F. Delsuc, and N. Galtier, 3.1:1–3.1:23. Available online at: https://hal.inria.fr/PGE
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Soliveri, J. A., Gomez, J., Bishai, W. R., and Chater, K. F. (2000). Multiple paralogous genes related to the Streptomyces coelicolor developmental regulatory gene whiB are present in Streptomyces and other actinomycetes. Microbiology 146, 333–343. doi: 10.1099/00221287-146-2-333
Steel, M. (2005). Should phylogenetic models be trying to ‘fit an elephant’? Trends Genet. 21, 307–309. doi: 10.1016/j.tig.2005.04.001
Sun, D., Liu, C., Zhu, J., and Liu, W. (2017). Connecting metabolic pathways: Sigma factors in Streptomyces spp. Front. Microbiol. 8:2546. doi: 10.3389/fmicb.2017.02546
Szöllősi, G. J., Davín, A. A., Tannier, E., Daubin, V., and Boussau, B. (2015). Genome-scale phylogenetic analysis finds extensive gene transfer among fungi. Phil. Trans. R. Soc. B 370:20140335. doi: 10.1098/rstb.2014.0335
Szöllősi, G. J., Rosikiewicz, W., Boussau, B., Tannier, E., and Daubin, V. (2013). Efficient exploration of the space of reconciled gene trees. Syst. Biol. 62, 901–912. doi: 10.1093/sysbio/syt054
van Dissel, D., Claessen, D., and van Wezel, G. P. (2014). Morphogenesis of Streptomyces in submerged cultures. Adv. Appl. Microbiol. 89, 1–45. doi: 10.1016/B978-0-12-800259-9.00001-9
Waglechner, N., McArthur, A. G., and Wright, G. D. (2019). Phylogenetic reconciliation reveals the natural history of glycopeptide antibiotic biosynthesis and resistance. Nat. Microbiol. 4, 1862–1871. doi: 10.1038/s41564-019-0531-5
Willey, J., Santamaria, R., Guijarro, J., Geistlich, M., and Losick, R. (1991). Extracellular complementation of a developmental mutation implicates a small sporulation protein in aerial mycelium formation by S. coelicolor. Cell 65, 641–650. doi: 10.1016/0092-8674(91)90096-H
Wu, M., Kostyun, J. L., Hahn, M. W., and Moyle, L. C. (2018). Dissecting the basis of novel trait evolution in a radiation with widespread phylogenetic discordance. Mol. Ecol. 27, 3301–3316. doi: 10.1111/mec.14780
Xu, H., Chater, K. F., Deng, Z., and Tao, M. (2008). A Cellulose synthase-like protein involved in hyphal tip growth and morphological differentiation in Streptomyces. J. Bacteriol. 190, 4971–4978. doi: 10.1128/JB.01849-07
Xu, L., Ye, K.-X., Dai, W.-H., Sun, C., Xu, L.-H., and Han, B.-N. (2019). Comparative genomic insights into secondary metabolism biosynthetic gene cluster distributions of marine Streptomyces. Mar. Drugs 17:498. doi: 10.3390/md17090498
Xu, M., Wang, W., Waglechner, N., Culp, E. J., Guitor, A. K., and Wright, G. D. (2020). GPAHex-A synthetic biology platform for Type IV–V glycopeptide antibiotic production and discovery. Nat. Commun. 11:5232. doi: 10.1038/s41467-020-19138-5
Xu, Q., Traag, B. A., Willemse, J., McMullan, D., Miller, M. D., Elsliger, M.-A., et al. (2009). Structural and functional characterizations of SsgB, a conserved activator of developmental cell division in morphologically complex actinomycetes. J. Biol. Chem. 284, 25268–25279. doi: 10.1074/jbc.M109.018564
Keywords: Streptomyces, morphological development, comparative phylogenetics, phylogenetic conflict, Robinson-Foulds distance
Citation: Wang M, Li C-J, Zhang Z, Li P-P, Yang L-L and Zhi X-Y (2023) The evolution of morphological development is congruent with the species phylogeny in the genus Streptomyces. Front. Microbiol. 14:1102250. doi: 10.3389/fmicb.2023.1102250
Received: 18 November 2022; Accepted: 14 March 2023;
Published: 29 March 2023.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Francisco Barona-Gomez, Leiden University, NetherlandsMichele Cloete, Agricultural Research Council-Vegetable and Ornamental Plants (ARC-VOPI), South Africa
Copyright © 2023 Wang, Li, Zhang, Li, Yang and Zhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Yang Zhi, eHl6aGlAeW51LmVkdS5jbg==
†These authors have contributed equally to this work