 Bo Liu
Bo Liu Ye-Song Ren
Ye-Song Ren Cheng-Yuan Su
Cheng-Yuan Su Yoshihisa Abe3
Yoshihisa Abe3 Dao-Hong Zhu
Dao-Hong Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 February 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1084839
Introduction: The genus Wolbachia provides a typical example of intracellular bacteria that infect the germline of arthropods and filarial nematodes worldwide. Their importance as biological regulators of invertebrates, so it is particularly important to study the evolution, divergence and host adaptation of these bacteria at the genome-wide level.
Methods: Here, we used publicly available Wolbachia genomes to reconstruct their evolutionary history and explore their adaptation under host selection.
Results: Our findings indicate that segmental and single-gene duplications, such as DNA methylase, bZIP transcription factor, heat shock protein 90, in single monophyletic Wolbachia lineages (including supergroups A and B) may be responsible for improving the ability to adapt to a broad host range in arthropod-infecting strains. In contrast to A strains, high genetic diversity and rapidly evolving gene families occur in B strains, which may promote the ability of supergroup B strains to adapt to new hosts and their large-scale spreading. In addition, we hypothesize that there might have been two independent horizontal transfer events of cif genes in two sublineages of supergroup A strains. Interestingly, during the independent evolution of supergroup A and B strains, the rapid evolution of cif genes in supergroup B strains resulted in the loss of their functional domain, reflected in a possible decrease in the proportion of induced cytoplasmic incompatibility (CI) strains.
Discussion: This present study highlights for reconstructing of evolutionary history, addressing host adaptation-related evolution and exploring the origin and divergence of CI genes in each Wolbachia supergroup. Our results thus not only provide a basis for further exploring the evolutionary history of Wolbachia adaptation under host selection but also reveal a new research direction for studying the molecular regulation of Wolbachia- induced cytoplasmic incompatibility.
Wolbachia belongs to the Anaplasmataceae in Rickettsiales, and its members are common intracellular symbionts of arthropods and nematodes (Wolbach and Hertig, 1924). Wolbachia species not only have a wide host range, including species of Culex (Werren et al., 2008), Aedes (Trpis et al., 1981), Drosophila (Teixeira et al., 2008; Klasson et al., 2009), parasitic wasps (Monnin et al., 2017) and a variety of lepidopteran pests (Delgado and Cook, 2009; Ju et al., 2020), but also exert various regulatory effects on their hosts. Not surprisingly given this high incidence and wide host range, the Wolbachia clade exhibits high genetic diversity (Zug and Hammerstein, 2015; Detcharoen et al., 2019; Kaur et al., 2021).
Wolbachia strains are distributed in several large clades referred to as ‘supergroups’ that have likely diverged over hundreds of millions of years (Bordenstein and Rosengaus, 2005; Lo et al., 2007; Glowska et al., 2015; Gerth and Bleidorn, 2016; Lefoulon et al., 2020). However, these large groups could in principle take on species status, which is a matter of ongoing debate (Shamayim et al., 2015; Gerth, 2016; Shamayim et al., 2016; Bleidorn and Gerth, 2018). Wolbachia classification is based on molecular data and loci that are regularly employed for strain discrimination at various levels, such as the 16S rRNA gene, five multilocus sequence typing loci (MLST) and the Wolbachia surface protein gene (wsp). A total of 14 Wolbachia supergroups (designated A–O) have been described in different host taxa. Most arthropod-associated Wolbachia strains are defined as belonging to supergroups A and B (Baldo et al., 2006; Lo et al., 2007), nematode-infecting strains are defined as belonging to supergroups C and D (Bandi et al., 1998). Supergroups E and F have been found in, arthropods (Czarnetzki and Tebbe, 2004; Panaram and Marshall, 2007) and nematodes (Fenn and Blaxter, 2004). Supergroup G is restricted to spiders (Rowley et al., 2004), supergroup H has been identified in association with dampwood termites (Bordenstein and Rosengaus, 2005), and supergoups M and N have been found in aphids (Augustinos et al., 2011; Wang et al., 2014).
In recent years, with the rapid development of technologies for DNA sequencing and extracting DNA from whole insect hosts, the whole-genome sequencing of Wolbachia has been realized (Darby et al., 2012). The wMel strain of Drosophila melanogaster was the first Wolbachia strain to have its full genome sequence published (Wu et al., 2004). The genome size of the wMel strain is approximately 1.27 Mb and contains a large number of repeated sequences and mobile elements, which is rare among intracellular species. In contrast, the wBm strain hosted by filarial nematodes contains no prophage and fewer repeat sequences (Foster et al., 2005). To date, many Wolbachia strain genomes have been released in the National Center for Biotechnology Information (NCBI) database, which provides data support for revealing the evolution of host adaptation and regulation of host interactions between strains and their hosts.
The predominant mode of Wolbachia transmission within a species occurs via the egg cytoplasm, resulting in vertical transmission. Due to this transmission pattern, Wolbachia exerts regulatory effects on host reproduction, the most common of which changing the sex ratio of the host population (Engelstaedter and Hurst, 2009). Wolbachia was first discovered in the reproductive tissues of Culex pipiens (Werren et al., 2008), in which the bacterium showed cytoplasmic incompatibility with its host (Yen and Barr, 1971). Wolbachia has since been found to have other reproductive regulatory functions, such as male killing (Stouthamer et al., 1999), feminization (Rousset et al., 1992) and parthenogenesis (Stouthamer et al., 1990), making it a hot topic of research. Wolbachia manipulates insect reproduction by enhancing its inheritance through the female germline. The most common mode of reproductive manipulation is the induction of cytoplasmic incompatibility (CI) (Yen and Barr, 1971; Hunter et al., 2003), in which eggs from uninfected females fail to develop when fertilized by sperm from Wolbachia-infected males, which results in embryonic lethality in crosses between infected males and uninfected females. Based on comparative and transgenic approaches, previous studies have shown that two differentially transcribed, codiverging genes in the eukaryotic association module of prophage WO from Wolbachia strain wMel recapitulate and enhance cytoplasmic incompatibility (Lindsey et al., 2018). Another study revealed that CI-like embryonic lethality could be recapitulated in Drosophila melanogaster males through the transgenic coexpression of homologous transgenes cifA and cifB, encoded by the wPip strain of Wolbachia, which naturally infects Culex mosquitoes (Beckmann et al., 2017). In previous research, the CI factors cifA (locus WD0631) always encoded directly upstream of cifB (locus WD0632) in the genome of wMel strain (LePage et al., 2017). In vitro functional analyses revealed that cifB encodes deubiquitylase activity, and cifA encodes a protein that binds cifB (Beckmann et al., 2017). Mutating the predicted catalytic residue in the deubiquitylating domain of cidB results in a loss of the CI-like function in transgenic flies (Beckmann et al., 2017). The presence of the two genes within prophage WO has implications for the transmission of these genes since temperate phage WO exhibits frequent lateral transfers between Wolbachia (Bordenstein and Wernegreen, 2004; Chafee et al., 2010). Whether the origin and evolution of these genes are important for their function remains an open question.
In the present research, we aim to reconstruct the evolutionary history, investigate the host adaptation-related evolution and explore the origin and divergence of CI genes in each Wolbachia supergroup based on the analysis of gene family expansion, genetic diversity and syntenic relationships in comparisons of 57 Wolbachia genomes. Our study not only provides guidance regarding the coevolution of intracellular symbionts and hosts but also generates new ideas about the origin and evolution of key genes involved in cytoplasmic incompatibility.
The genome sequences used in this study were downloaded from the National Center for Biotechnology Information (NCBI)1 up to May 2020. We filtered the genomes according to the following criteria: (1) we filtered out Wolbachia strains without available host information; (2) when the strains had the same name, we retained the more recently submitted genome version; and (3) the genome sequences of strains without predictive genes were filtered out. Finally, a total of 57 Wolbachia strain genomes were analyzed in this study. The NCBI accession numbers of all Wolbachia genome sequences are given in Supplementary Table S1.
Gene functional annotation was performed by aligning the corresponding protein sequences to the NCBI nonredundant (NR), Universal Protein (UniProt) (Wu et al., 2006), Evolutionary Genealogy of Genes: Nonsupervised Orthologous Groups (eggNOG) (Huerta-Cepas et al., 2017) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (Kanehisa et al., 2004) by using BLASTP v2.3.0+ with an E-value cut-off of 10−5. InterProScan v2.0 (Quevillon et al., 2005) was used to assign preliminary Gene Ontology (GO) terms, Pfam domains and IPR domains to each gene. The enrichment analysis of GO and KEGG pathways was performed using the online OmicShare platform2.
Orthologous and paralogous gene families of 57 Wolbachia strains, one Ehrlichia canis str. YZ-1 (PRJNA429059) and one Anaplasma marginale str. Florida (PRJNA16369) were assigned by OrthoFinder v0.4 (Emms and Kelly, 2015) with the parameters “-f –t 30.” The orthologous groups that contained only one gene in each strain were selected to construct the phylogenetic tree. The protein sequences of each orthologous group were independently aligned with MUSCLE v3.8.31 (Edgar, 2004) with the parameters “-maxiters 16” and then concatenated into one supersequence. The phylogenetic tree was constructed based on maximum likelihood (ML) using PhyML v3.0 (Guindon et al., 2010) with the best-fit model (HIVb+I + G + F) that was estimated by ProtTest3 (Darriba et al., 2011). Node support was estimated with 1,000 bootstrap replicates.
The nucleotide sequences of five housekeeping genes (gatB, coxA, hcpA, ftsZ, and fbpA) that were downloaded from the PubMLST database3 were aligned to the gene sets of all strains by using BLASTP v2.3.0 with the parameter setting of 10−20. Then, the maximum likelihood tree was constructed by using PhyML v3.0 with the parameters “-d aa –m LG –c 4.” Node support was estimated with 1,000 bootstrap replicates.
To infer the divergence times of different Wolbachia supergroups in the phylogeny, divergence time estimates were calculated using r8s v1.8.1 (Sanderson, 2003) with the parameters “-b -f” by fitting branch lengths of an ML tree using penalized likelihood and a smoothing parameter of 8, chosen as optimal by cross-validation. The two secondary calibration points obtained from Michael et al. (2016), where ~217 million years ago (Mya) was the split time of wAu and wNo.
To determine the change in orthologous group members of Wolbachia strains during evolution, the analysis of gene gains and losses was conducted using CAFÉ v3.0 (De Bie et al., 2006), in which orthologous group change was simulated using a stochastic birth and death model. The optimal lambda parameter was automatically determined independently. The orthologous groups with p values <0.05 were defined as rapidly evolving families in the CAFÉ results. We also used the z-test (p < 0.05) to identify the expanded orthologous group in each Wolbachia supergroup based on the gene numbers. We used OmicShare and WEGO v2.0 (Ye et al., 2018) to analyse the functional enrichment of expansive orthologous groups.
Genomic synteny fragments were identified with MCscanX (2012) (Wang et al., 2012), requiring at least five gene pairs per collinear block. Then, we used the duplicate_gene_classififier (2012) of MCscanX to identify duplicated genes and classified the origins of the genes into different types, including segmental, tandem, proximal and dispersed duplications. We employed the inter- and intrasyntentic gene pairs to calculate synonymous mutation (Ks) values by using KaKs_Calculator v2.0 (Wang et al., 2010) with the parameter “-c 1 -m MS.” The orthologous groups that contained only one gene for each strain were selected. Then, the nucleotide diversity of the single-copy genes within each Wolbachia supergroup was calculated by using DnaSP v6.12.03 (Rozas et al., 2017). The identity of genome-wide nucleotide sequences in each pair of strains was determined by using Mummer v3.23 package (Kurtz et al., 2004; delta-filter -i 75 -l 1,000 and show-coords –r –c -l).
The cifA (GeneID: 69724995 and GeneID: 61803217) and cifB (GeneID: 69724996 and GeneID: 61803216) gene sequences used in this study were downloaded from the National Center for Biotechnology Information (NCBI)4. To identify the cifA and cifB genes in each Wolbachia strain, we used the two cif gene sequences to align to the gene set of each strain by using BLASTN v2.3.0+ with the parameters word_size = 4 and Evalue = 10. Then, to avoid missing the cif genes of each strain, synteny analysis was performed between the prophage WO genome and each Wolbachia genome to find more cif genes. To identify the divergence times of the cif genes in Wolbachia strains, we used the cifA and cifB gene pairs within supergroups A and B, respectively, to calculate the synonymous (Ks) and nonsynonymous (Ka) mutation rates by using KaKs_Calculator v2.0 with the parameters “-c 1 -m MS.” The nucleotide diversity (π) and genetic distance of cif genes within supergroups A and B were calculated by using DnaSP v6.12.03. The motifs of the cifA and cifB gene sequences in each Wolbachia strain were analysed by using MEME5 with 10 motifs should MEME find. We used JCVI (Tang et al., 2015) to construct the local syntenic relationships of each gene in the supergroup A strain.
To assess the paleohistory of Wolbachia strains, we downloaded 57 available genomic sequences of Wolbachia, including sequences of supergroups A, B, C, D, E and F as well as the unclassified Wolbachia supergroup, and performed a comparative genomic investigation with Ehrlichia canis and Anaplasma marginale as outgroups (Supplementary Table S1). Among these genomes, a total of 60,932 genes were clustered into 2,381 orthologous groups containing 109 single-copy orthologues. The phylogenetic trees showed that 24 and 23 Wolbachia strains were clustered into supergroups A and B, respectively. Supergroups A and B shared a common ancestor (Figure 1A) corresponding to the topological structure based on housekeeping genes (Supplementary Figure S1A). The estimated divergence time analysis indicated that Wolbachia diverged from the two outgroup genera (Anaplasma and Ehrlichia) ~1799 million years ago (Mya). Arthropod-infecting Wolbachia supergroups A and B strains were reciprocally monophyletic and diverged from their common ancestor 217 Mya. supergroups C, D, F, and wCfeJ formed a monophyletic group, which corresponded to previously published results (Gerth et al., 2014; Michael et al., 2016). Arthropod-infecting Wolbachia strains diverged from nematode-infecting Wolbachia strains 278 Mya (Figure 1A).
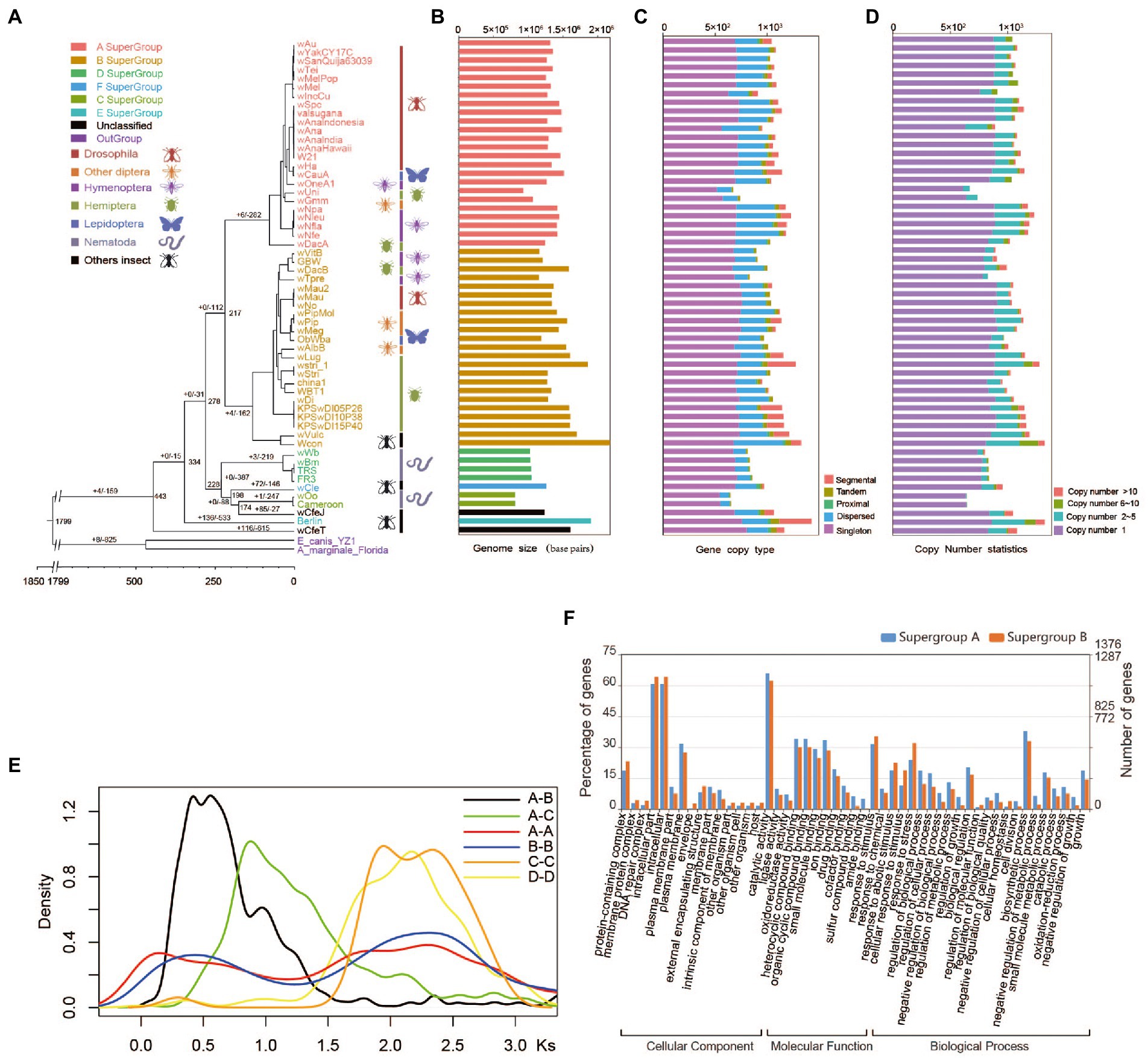
Figure 1. Evolution of Wolbachia genomes. (A) Paleohistory of Wolbachia with Ehrlichia canis and Anaplasma marginale as outgroups. The number at the nodes represents the divergence time of species. (B) Genome sizes of Wolbachia strains. (C) The different gene copy types in Wolbachia strains were identified and classified by using the software duplicate_gene_classififier. The gene duplication types were classified as segmental, tandem, proximal and dispersed duplications. Segmental indicates anchor/collinear genes in syntenic blocks; tandem indicates continuous repeat; proximal indicates in nearby chromosomal region but not adjacent; dispersed indicates other modes than segmental, tandem and proximal. (D) Numbers of gene copies. (E) Ks distribution of syntenic orthologues from four Wolbachia supergroups. The y-axis shows the ratio of gene pairs in the syntenic block. (F) Functional enrichment of all duplicated genes in supergroups A and B according to Gene Ontology (GO) classification.
The nature and relative importance of the molecular mechanisms and evolutionary forces underlying genome size variation have been the subject of intense research and debate (Petrov, 2001; Elliott and Gregory, 2015). A number of correlative associations between genome size and phenotypic traits suggest that natural selection and adaptive processes also shape genome size evolution (Cavalier-Smith, 1982; Andrews et al., 2009; Wright et al., 2014; Ellegren and Galtier, 2016). The present study showed distinct differences in genome size between Wolbachia supergroups, indicating that the average genome size of arthropod-infecting Wolbachia strains was 1.47 times larger than that of nematode-infecting Wolbachia strains (t test, p = 5.82E−06; Figure 1B). Previous studies have documented that the differential expansion, accumulation and removal of transposable element (TE) sequences are major determinants of genome size variation between Wolbachia strains wBm and wMel (Foster et al., 2005). However, we found that the larger genome size of arthropod-infecting Wolbachia strains, in which the gene numbers were significantly greater than those in nematode-infecting Wolbachia strains, was due not only to an increase in repeat sequences but also to gene duplications (Figures 1B–D; Supplementary Figure S1B). Based on gene copy number analysis, 684, 717, 692 and 547 genes (on average) were assigned to single-copy genes in supergroups A, B, C and D, respectively. Unexpectedly, it was found that approximately 33.9% of the total genes in arthropod-infecting Wolbachia strains were likely produced through small-scale gene duplication events (Figure 1C; Supplementary Figure S1C), dominated by genes showing 2–5 copies (Figure 1D; Supplementary Figure S1D), which was significantly greater than the number in supergroups C (16.9%) and D (15.6%). Among these duplicated genes in Wolbachia supergroup A, an average of 280 dispersed duplicated genes were found, which was similar to the number in supergroup B (242 genes) but significantly higher than that in supergroups C and D (85 and 118 genes, respectively).
To study the history of gene duplications, we identified the genes showing inter- and intraspecies homology between each supergroup and then calculated the synonymous mutation rates (Ks) of the syntenic fragments of orthologous pairs. Apparent Ks peaks were observed in all of the four supergroups (A, B, C and D) (Figure 1E), which have a complex history of duplication involving two small-scale gene duplications instead of a whole-genome duplication. According to a Ks value of less than 1, 34.1 and 59.1% segmentally duplicated gene pairs were identified within supergroups A and B, respectively, indicating that a recent duplication event occurred during the divergence of supergroup A and B strains. Single-gene duplication events with a peak of Ks = 1.75–3, dominated by dispersed gene duplications, were shared by the common ancestor of each supergroup. In contrast to the evolutionary history of supergroups A and B, no evidence of a recent gene duplication event was detected in supergroups C and D based on the Ks distribution (Figure 1E).
All of duplicated genes identified in supergroups A and B were significantly enriched in the following functional categories based on the GO and KEGG analyses: catalytic and hydrolase activity, nitrogen compound metabolism, transcription factor, response to stimulus and chemical (Figure 1F; Supplementary Figure S2), such as DNA methylase, bZIP transcription factor, heat shock protein 90, DNA mismatch repair protein MLH1, cysteine protease and so on. In addition, the enrichment of ABC transporters in supergroups A and B was mainly due to the duplication of ATP-binding cassette subfamily A/B/D/G genes (EC:7.6.2.2 and EC:7.6.2.4). The gene encoding cucurbitadienol synthase (EC:5.4.99.33), 5-phosphonooxy-L-lysine phospho-lyase (EC:4.2.3.134), vanillin aminotransferase (EC:2.6.1.119) and vanillin aminotransferase (EC:2.6.1.119) were also expansion in the supergroups A and B, in which the cucurbitadienol synthase was not present in the supergroups C and D. This demonstrated that extensive gene fractionation occurred during the evolutionary history of arthropod-infecting Wolbachia strain genomes, which promoted the retention of essential genes for survival and host adaptation.
In this study, 2,381 orthologous groups were found in 57 Wolbachia strains, in which the average number of orthologous groups in arthropod-infecting Wolbachia supergroups (A and B) was significantly higher (1.6 times) than that in nematode-infecting Wolbachia supergroups (C and D) (Figure 2A). Otherwise, the supergroup-specific orthologous groups displayed a similar variation pattern, in which the average number of orthologous groups in arthropod-infecting Wolbachia was 4.6 times higher than that in nematode-infecting groups (Figure 2A).
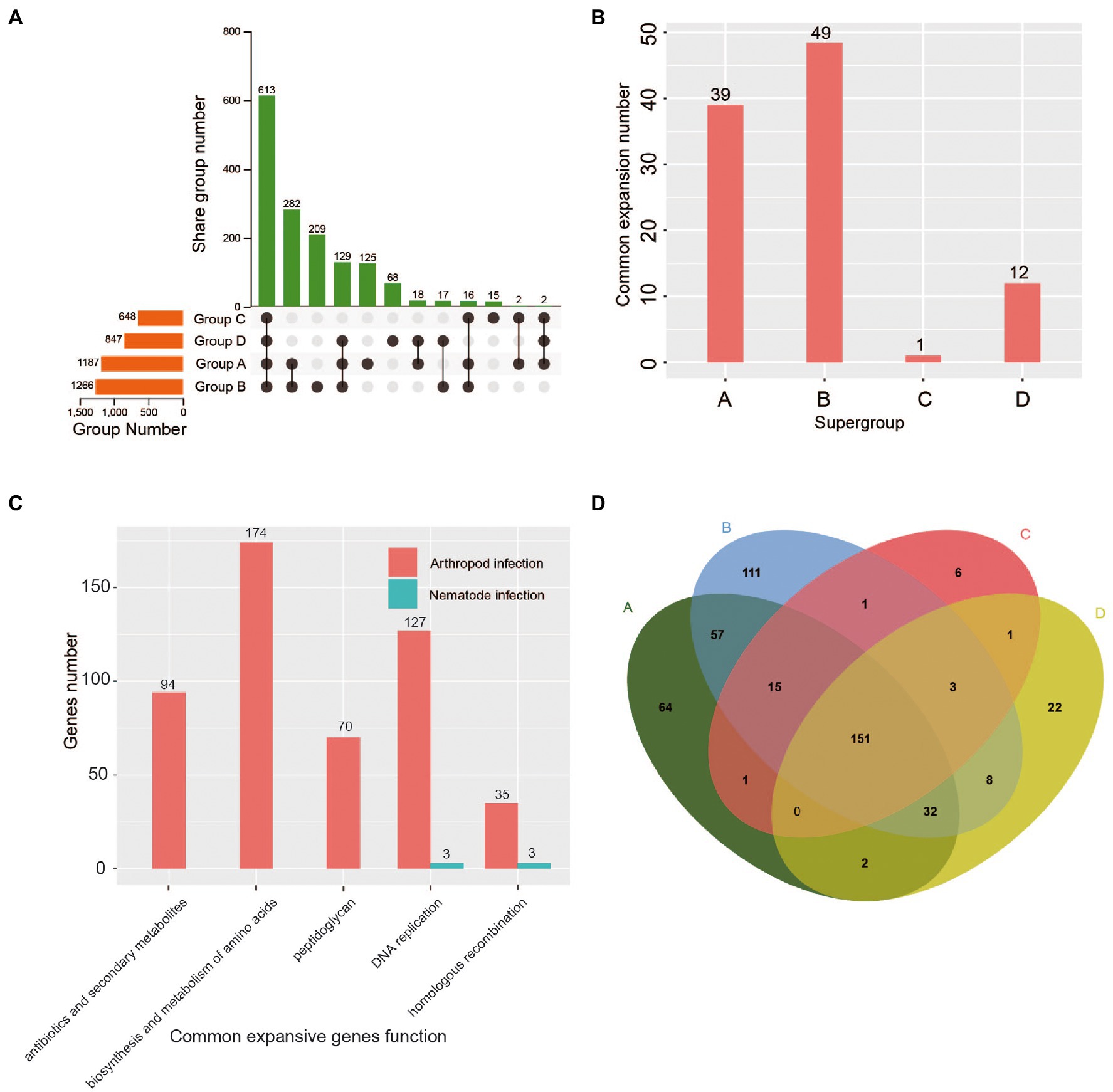
Figure 2. Comparison of gene families and functional enzyme expansions among the four Wolbachia supergroups (A, B, C, and D). (A) Unique and shared orthologous groups between and among the four supergroup genomes. (B) Number of coexpansive orthologous groups within each supergroup. (C) KEGG functional enrichment analysis of coexpanded genes in arthropod-infecting and nematode-infecting Wolbachia strains. (D) Venn diagram showing the number of unique and shared functional enzymes between and among the four supergroups, A, B, C and D.
Unexpectedly, the average number of expanded orthologous groups was significantly greater in arthropod-infecting Wolbachia groups than in nematode-infecting groups (Supplementary Figure S3). Among those expanded orthologous groups, the number of orthologous groups that underwent common expansion within arthropod-infecting Wolbachia strains was significantly higher than that in nematode-infecting Wolbachia strains (A/C p < 0.001, A/D p < 0.001, B/C p < 0.001, B/D p < 0.001; Figure 2B). In contrast to nematode-infecting Wolbachia, the function of common expansive genes in arthropod-infecting strains was primarily enriched in DNA replication, homologous recombination and the biosynthesis and metabolism of amino acids, peptidoglycan, antibiotics and secondary metabolites (Figure 2C).
The same conclusion was reached for functional enzymes, where an average of 350 kinds of enzymes were identified in the arthropod-infecting supergroup, which was significantly higher than the number in the nematode-infecting supergroup (Figure 2D). The number of supergroup-specific enzymes in arthropod-infecting Wolbachia strains was also significantly greater than that in nematode-infecting strains (A/C p < 0.001, A/D p < 0.001, B/C p < 0.001, B/D p < 0.001; Figure 2D).
The above evidence indicated a large amount of gene over retention, which was related to the synthesis and metabolism of amino acids and other important compounds and has previously been observed in the genomes of arthropod-infecting Wolbachia strains, improving the ability to adapt to a broad host range (Gerth et al., 2014). In contrast to arthropod-infecting Wolbachia, the more host-specific supergroups C and D have established long-lasting mutualistic relationships with their hosts, leading to a stable state of the genome that does not require large amounts of gene duplication.
According to previous studies on Wolbachia strains conducted in the last 20 years, supergroup A strains have infected approximately 162 arthropods, including members of 14 orders, 80 families and 126 genera (Figure 3A; Supplementary Table S2). In stark contrast, the supergroup B strains present a wider host range, infecting 185 arthropods of 19 orders, 100 families and 185 genera (Figure 3A; Supplementary Table S2). To investigate the adaptive evolution of the host range of Wolbachia strains, the genetic diversity within supergroups A and B was assessed in this study. In the whole-genome alignments used to analyse the ingroup sequence identity estimated from all Wolbachia strains, high conservation was detected in supergroup A, in which the sequence identity between the two strains was 97% on average, ranging from 94 to 99%. In contrast, sequence identity at the genome level between the two supergroup B strains presented significant variance compared with that in supergroup A (t test, p = 1.7E−24; Figure 3B). The further analysis of nucleotide diversity (π) among single-copy genes within supergroups A and B revealed a similar variation pattern, in which the π value of conserved genes within supergroup A strains was significantly lower than that within supergroup B strains (t test, p = 1.6E−6; Figure 3C). The results showed that the π values of genes between the supergroup A strains varied from 0.00187 to 0.04841 (0.01639 on average), whereas it varied from 0.01391 to 0.06408 (0.0342 on average) in the B strains.
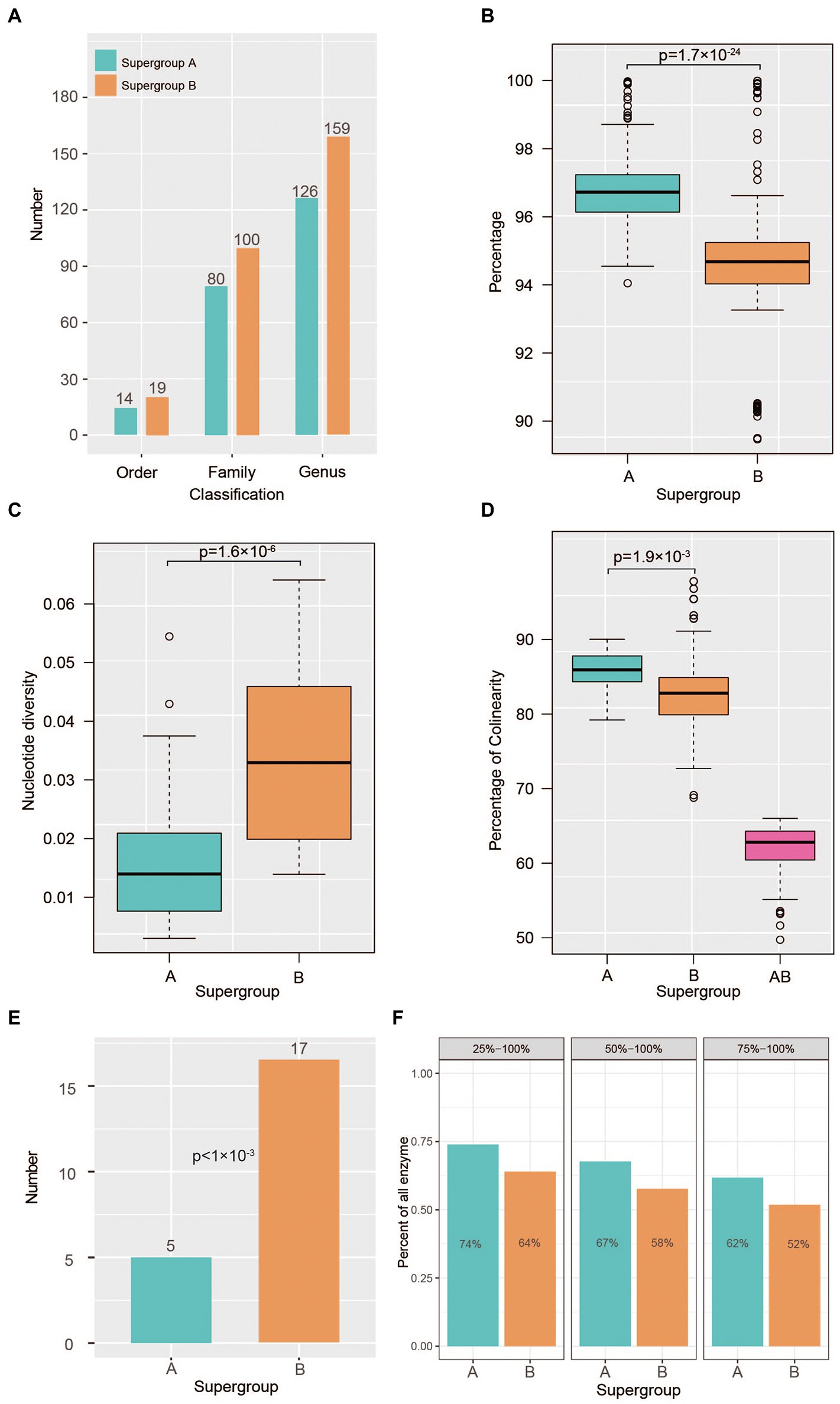
Figure 3. Rapid genomic evolution in Wolbachia supergroup B strains. (A) Statistics of host species numbers among supergroups A (green box) and B (orange box) based on studies in the last 20 years. (B) Analysis of whole-genome identity within supergroups A and B. (C) Nucleotide diversity (Pi) within supergroups A (green box) and B (orange box). (D) Percentage of syntenic genes of each pair of strains within supergroups A (green box) and B (orange box). And percentage of syntenic genes of each pair of strains between supergroups A and B (purple box) (E) Number of rapidly evolving gene families in the ancestors of supergroups A (green box) and B (orange box). Gene families with a p value <0.05 were defined as rapidly evolving families in the CAFÉ results. (F) Percentage of shared functional enzymes within supergroups A (green box) and B (orange box), where 25–100% indicates that more than 25% of Wolbachia strains in each supergroup shared the same functional enzyme; 50–100% indicates that more than 50% of Wolbachia strains in each supergroup shared the same functional enzyme; and 75–100% indicates that more than 75% of Wolbachia strains in each supergroup shared the same functional enzyme.
To investigate the evolutionary dynamics of genome structure, we performed a comparative genomic analysis among supergroup A and B strains using proteins as markers to identify syntenic genes. Within the supergroup A genomes, 85.9% of genes (median value; range 79.2 to 90.0%) showed syntenic relationships between any two strains, whereas 82.8% of genes (range 68.8 to 97.8%) showed syntenic relationships in B strains (t test, p = 0.0019; Figure 3D). By examining the syntenic relationships between the supergroup A and B strains, we found that the collinearity ratio between the two supergroups was low (Figure 3D), although it was significantly higher than that of nematode-infecting Wolbachia strains (Supplementary Figure S4), which was reasonable considering their phylogenetic distance.
According to the CAFÉ analysis, we found that the number of rapidly evolving gene families was significantly higher in the ancestors of supergroup A than in those of supergroup B (p < 0.001; Figure 3E). Based on the gene functional analysis, these rapidly evolving gene families were associated with multiple metabolism-related pathways (Supplementary Figure S5), such as acarbose and validamycin biosynthesis, biosynthesis of vancomycin group antibiotics, polyketide sugar unit biosynthesis. Furthermore, we analysed the number of enzymes shared among Wolbachia strains, and it was found that there were significantly more kinds of common enzymes in supergroup A strains than in supergroup B strains (Figure 3F), indicating that the rapid evolution of supergroup B strains has enabled them to retain more enzymes for adaptation to a broad host range.
To investigate the evolution of cif genes in each Wolbachia supergroup, a comparative genomic strategy was applied in this study. A literature review focusing on parasitic reproductive modulation by Wolbachia showed that a total of 28 CI-inducing Wolbachia strains have been identified (Table 1). Among these CI-inducing Wolbachia strains, 42.9% (12 of 28) belonged to supergroup A, which was nearly twice the percentage in supergroup B. Notably, based on the retention and deletion analysis of cif genes in the genomes of Wolbachia strains, approximately 86.3% (44 of 51) of strains contained both cif genes in supergroup A, which was significantly higher than the percentage in supergroup B (chi-squared test, p < 0.0001; Figure 4A), while none of the cif genes were detected in the other supergroups.
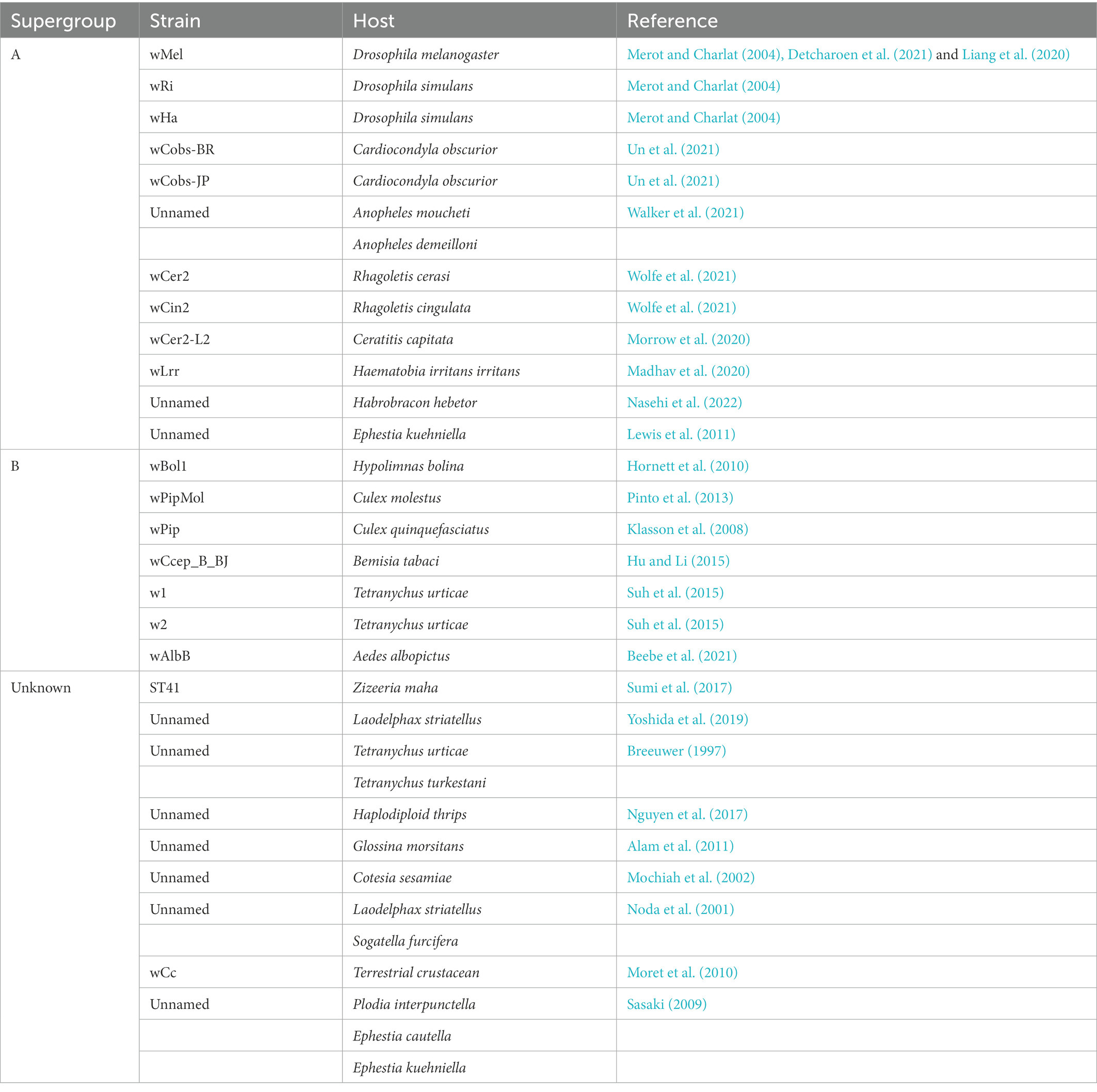
Table 1. Information of CI-inducing Wolbachia strains.

Figure 4. Evolution and diversity of Wolbachia cif genes in supergroups A and B. (A) Proportion of Wolbachia strains containing two cif genes in different supergoups. Supergroup A (green box), Supergroup B (orange box) and the other supergroups (red box) (B) Ks distribution of cif genes in Wolbachia supergroups A and B. A-CifA and A-CifB indicate the Ks distribution of cifA and cifB genes, respectively, between two strains in supergroup A. B-CifA and B-CifB indicate the Ks distribution of cifA and cifB genes, respectively, between two strains in supergroup B. (C) Maximum likelihood tree of cifB genes in supergroup A strains that contained both cif genes. The red circle represents the ancient cif gene insertion event. The red square represents the recent cif gene insertion event. (D) Multiple sequence alignment of intergenic sequences between two cif genes. (E) Motif composition of the cifB gene-encoding sequence. Each colour box of rectangle represents a different motif. The dials at the bottom indicate the length of the coding sequence. (F) Microsyntenic graph of ancient insertions with the same haplotype in Wolbachia genomes. The red and yellow lines represent the syntenic relationships of the cifA and cifB genes, respectively. The green and grey lines represent the syntenic relationships of phage WO and Wolbachia genes, respectively. The blue boxes indicate genes in each strain genome. (G) Microsyntenic graph of recent insertions with the same haplotype in Wolbachia genomes. The red and yellow lines represent the syntenic relationships of the cifA and cifB genes, respectively. The green and grey lines represent the syntenic relationships of phage WO and Wolbachia genes, respectively. The blue boxes indicate genes in each strain genome. (H) Comparison of the synonymous mutation rate (Ks) and nonsynonymous mutation rate (Ka) in Wolbachia supergroups A and B. ka.cifA. A indicates the Ka values of cifA gene between two strains in supergroup A. ka.cifB. B indicates the Ka values of cifB gene between two strains in supergroup B, ks.cifA. B indicates the Ks values of cifA gene between two strains in supergroup B, ks.cifB. A indicates the Ks values of cifB gene between two strains in supergroup A.
To study the history of cif gene origination, a total of 58 strains were used to calculate the synonymous mutation rates (Ks) of the cifA and cifB genes within each pair of Wolbachia strains (Figure 4B). Interestingly, two independent insertion events may have occurred in Wolbachia supergroup A, in which an ancient insertion event involved an independent clade (including wNfe, wNfla, wNleu and wNpa strains) and a recent insertion event involved another clade containing almost all supergroup A strains. The phylogenetic analysis of the cifB gene showed that there were two different clades in supergroup A, in which the cifB genes of four strains (wNfe, wNfla, wNleu, and wNpa) were more ancient (Figure 4C). Further analysis revealed that the physical distance between the cifA and cifB genes was 53 bp during the ancient insertion event, and the coding sequence (CDS) lengths of the cifB genes in the four strains were completely consistent (Supplementary Table S3). In contrast, the physical distance between the two cif genes was 75 bp during the recent insertion event, and the CDS length of the cifB genes was twice that in the ancient insertion event (Supplementary Table S3). The sequence similarity analysis of the intergenic region between the cifA and cifB genes showed that the intergenic region sequences involved in each insertion event shared high identity, suggesting that there were distinct haplotypes in the two insertion events (Figure 4D). The cifB gene sequences showed a distinct motif composition between the two insertion events (Figure 4E; Supplementary Figure S6). Further analysis showed that the two insertion segments were located in different genome regions in the two lineages of Wolbachia strains based on the analysis of microsynteny (Supplementary Figure S7). However, the fragments that contained both cifA and cifB were inserted at the same location in the Nomada-associated Wolbachia strains (Figure 4F), and the same pattern was found in the Wolbachia strains with the recent insertion (Figure 4G). The results showed that Wolbachia strains with the ancestral insertion were only identified in host insects of the genus Nomada within Hymenoptera, while the recent insertion was detected in insects belonging to Diptera, Lepidoptera, and Coleoptera (Supplementary Table S3).
Furthermore, more than 52.9% (18 of 34) of supergroup B strains may have lost the cifA and cifB genes, whereas the corresponding proportion among supergroup A strains was only 13.7% (7 of 51). In contrast to supergroup B strains, the retained cif genes of supergroup A strains were highly conserved and displayed lower mean nucleotide diversity (π = 0.10371 and 0.08647 in cifA and cifB, respectively). However, the cif genes of supergroup B strains showed a markedly higher evolutionary rate (π = 0.11429 and 0.1455 in cifA and cifB, respectively) than those in supergroup A strains. It is noteworthy that despite the conservation of cif gene order, the functional domains of these genes in supergroup B strains showed extensive divergence and differences, in which most important domains were lost (Supplementary Figure S8). In addition, both the synonymous (Ks) and nonsynonymous (Ka) mutation rates of cif genes in supergroup B strains were significantly higher than those in supergroup A strains (Figure 4H). This result suggested that during the independent evolution of supergroup A and B strains, the rapid evolution of cif genes in supergroup B strains resulted in the loss of their function, reflected in a decrease in the proportion of induced CI strains.
Here, we present a phylogenetic hypothesis for Wolbachia supergroups A, B, C and D based on the analysis of whole genome single copy gene and five housekeeping genes. Our findings indicate that the Wolbachia genomes have a complex evolutionary history, including ancient duplication events (Figure 1E) in the ancestor of the four supergroup (A, B, C and D) and a recent duplication event that were occurred in the ancestor of supergroup A and B. These recent duplication events generated abundant overretentive genes related to functions including the synthesis/metabolism of important compounds and the response to stimuli and chemicals, which are important for the diversity of gene functions and adaptation to changing environments. In addition, we found that the genes related to growth and development (Pratt, 1998; Sánchez-Romero et al., 2015) were significant expansion both in supergroup A and B, such as ATPase family associated with various cellular activities, DNA methylase, heat shock protein 90, DNA mismatch repair protein MLH1, cysteine protease and so on, which were perhaps significantly increase the gene repertoires and the genome complexity and could provide a greater chance for natural selection to generate a novel function (Long et al., 2003; Zhang, 2003; Conant and Wolfe, 2008; Lynch et al., 2008; Lipinski et al., 2011; Gao et al., 2017). So, we speculate that extensive gene fractionation occurred during the evolutionary history of arthropod-infecting Wolbachia strain genomes, which promoted the retention of genes that are essential for survival and host adaptation. In contrast, nematode-infecting Wolbachia strains have established long-lasting mutualistic relationships with their specific hosts (Darby et al., 2012; Godel et al., 2012), leading to a stable state of the genome that may lead to the loss of large numbers of genes (Gerth et al., 2014) or to form species-specific novel genes (Weyandt et al., 2022), as recently reported in another Wolbachia study.
Even under perfect transmission fidelity, Wolbachia would have limited chances of spreading. In addition, deleterious fitness effects and imperfect transmission impose further restrictions on the spread of Wolbachia within a population (Sanaei et al., 2021). Consequently, without the induction of a phenotype driving its the spread of Wolbachia, the bacteria may easily be lost from a new host species (Sanaei et al., 2021). In this study, we identified multiple gene duplication events (Figure 1E) in the ancestor of Wolbachia A and B strains, which resulted in many gene redundancies in those genomes. Following duplication, the effect of purifying selection on any one duplicated gene is relaxed (Cheng et al., 2018), permitting the loss or differentiation of duplicated genes and regulatory elements (Conant and Wolfe, 2008). Further analysis revealed clear differences in nucleotide diversity, genomic structural mutations, rapidly evolving gene families and functional gene diversity within each Wolbachia B strain. This high rate variability may be not due to Wolbachia but rather due to peculiar genetic selection in its hosts. From an evolutionary viewpoint, these genetic variations can all be explained as adaptations enhancing bacterial fitness through the fitness of the infected host, which is straightforward in the case of direct positive effects, such as protection against pathogens or nutrient provision (Sanaei et al., 2021). Overall, the random genetic drift of Wolbachia strains may promote their adaptability to widespread hosts and may provide direct fitness benefits to their hosts. Therefore, we hypothesize that supergroup B strains responded to host selection via rapid genomic and genic evolution, a high degree of instability, and recurrent rearrangements and recombination events (Lo et al., 2007) to adapt to new hosts and achieve large-scale spreading after the divergence of supergroups A and B.
Based on comparative and transgenic approaches, two differentially genes (cifA nad cifB) of prophage WO from Wolbachia strain wMel recapitulate and enhance cytoplasmic incompatibility (Lindsey et al., 2018). In this study, based on the comparative genomic strategy between Wolbachia supergroups A and B, we found that two distinct haplotypes of supergroup A strains were detectable based on the analysis of cifA, cifB and intergenic sequences, suggesting that there may have been two independent horizontal gene transfer events involving prophage WO. The lineage consisted of wNfe, wNfla, wNleu and wNpa strains with the same haplotype and same insertion position, in which the inserted fragment from the prophage genome may have appeared in the common ancestor of this lineage. This also suggested that the cif genes were not present in the last common ancestor of supergroup A strains but rather that they were acquired independently by Nomada-associated Wolbachia. In contrast, the Wolbachia strains of a more recently diverged lineage presented another identical haplotype, and they showed an almost identical insertion location, suggesting that the insertion event may have occurred in the ancestor of the lineage, possibly before the divergence of each strain. Regrettably, complete sequences were not available for the Wolbachia strains in other clades, which made it inconvenient to identify the location of the insertion fragment of prophage WO. We believe that the publication of more complete sequences of Wolbachia strains will be helpful to systematically study the origin and evolution of cif genes in different supergroups.
In this study, we aimed to reconstruct the evolutionary history, address host adaptation-related evolution and explore the origin and divergence of CI genes in each Wolbachia supergroup. Our results thus not only provide a basis for further exploring the evolutionary history of Wolbachia adaptation under host selection but also reveal a new research direction for studying the molecular regulation of Wolbachia-induced cytoplasmic incompatibility.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Y-SR, BL, and C-YS were responsible for comparative genome analysis. BL and Y-SR draft the paper. D-HZ, BL, and YA coordinated the project. All authors contributed to the article and approved the submitted version.
The work was funded by the National Key Research and Development Program of China (Grant No. 2018YFE0127100), Shenzhen Science and Technology Program (Grant No. KQTD20180411143628272 and JCYJ20190813144407666) and science technology innovation and industrial development of Shenzhen Dapeng New District (Grant No. KJYF202001-03).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1084839/full#supplementary-material
Alam, U., Medlock, J., Brelsfoard, C., Pais, R., Lohs, C., Balmand, S., et al. (2011). Wolbachia symbiont infections induce strong cytoplasmic incompatibility in the tsetse fly Glossina morsitans. PLoS Pathog. 7:e1002415. doi: 10.1371/journal.ppat.1002415
Andrews, C. B., Mackenzie, S. A., and Gregory, T. R. (2009). Genome size and wing parameters in passerine birds. Proc. R. Soc. B Biol. Sci. 276, 55–61. doi: 10.1098/rspb.2008.1012
Augustinos, A. A., Santos-Garcia, D., Dionyssopoulou, E., Moreira, M., and Bourtzis, K. (2011). Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS One 6:e28695. doi: 10.1371/journal.pone.0028695
Baldo, L., Bordenstein, S., Wernegreen, J., and Werren, J. (2006). Widespread recombination throughout Wolbachia genomes. Mol. Biol. Evol. 23, 437–449. doi: 10.1093/molbev/msj049
Bandi, C., Anderson, T., Genchi, C., and Blaxter, M. (1998). Phylogeny of Wolbachia in filarial nematodes. Proc. R. Soc. B Biol. Sci. 265, 2407–2413. doi: 10.1098/rspb.1998.0591
Beckmann, J. F., Ronau, J. A., and Hochstrasser, M. (2017). A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nat. Microbiol. 2:5. doi: 10.1038/nmicrobiol.2017.7
Beebe, N. W., Pagendam, D., Trewin, B. J., Boomer, A., Bradford, M., Ford, A., et al. (2021). Releasing incompatible males drives strong suppression across populations of wild and Wolbachia-carrying Aedes aegypti in Australia. Proc. Natl. Acad. Sci. U. S. A. 118:118. doi: 10.1073/pnas.2106828118
Bleidorn, C., and Gerth, M. (2018). A critical re-evaluation of multilocus sequence typing (MLST) efforts in Wolbachia. FEMS Microbiol. Ecol. 94:1. doi: 10.1093/femsec/fix163
Bordenstein, S., and Rosengaus, R. (2005). Discovery of a novel Wolbachia supergroup in isoptera. Curr. Microbiol. 51, 393–398. doi: 10.1007/s00284-005-0084-0
Bordenstein, S. R., and Wernegreen, J. J. (2004). Bacteriophage flux in endosymbionts (Wolbachia): infection frequency, lateral transfer, and recombination rates. Mol. Biol. Evol. 21, 1981–1991. doi: 10.1093/molbev/msh211
Breeuwer, J. (1997). Wolbachia and cytoplasmic incompatibility in the spider mites Tetranychus urticae and T-turkestani. Heredity 79, 41–47. doi: 10.1038/hdy.1997.121
Cavalier-Smith, T. (1982). Skeletal DNA and the evolution of genome size. Annu. Rev. Biophys. 11, 273–302.
Chafee, M. E., Funk, D. J., Harrison, R. G., and Bordenstein, S. R. (2010). Lateral phage transfer in obligate intracellular bacteria (Wolbachia): verification from natural populations. Mol. Biol. Evol. 27, 501–505. doi: 10.1093/molbev/msp275
Cheng, F., Wu, J., Cai, X., Liang, J., Freeling, M., and Wang, X. (2018). Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4, 258–268. doi: 10.1038/s41477-018-0136-7
Conant, G. C., and Wolfe, K. H. (2008). Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 9, 938–950. doi: 10.1038/nrg2482
Czarnetzki, A., and Tebbe, C. (2004). Detection and phylogenetic analysis of Wolbachia in collembola. Environ. Microbiol. 6, 35–44. doi: 10.1046/j.1462-2920.2003.00537.x
Darby, A. C., Armstrong, S. D., Bah, G. S., Kaur, G., Hughes, M. A., Kay, S. M., et al. (2012). Analysis of gene expression from the Wolbachia genome of a filarial nematode supports both metabolic and defensive roles within the symbiosis. Genome Res. 22, 2467–2477. doi: 10.1101/gr.138420.112
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
De Bie, T., Cristianini, N., Demuth, J., and Hahn, M. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Delgado, A. M., and Cook, J. M. (2009). Effects of a sex-ratio distorting endosymbiont on mtDNA variation in a global insect pest. BMC Evol. Biol. 9:49. doi: 10.1186/1471-2148-9-49
Detcharoen, M., Arthofer, W., Schlick-Steiner, B. C., and Steiner, F. M. (2019). Wolbachia megadiversity: 99% of these microorganismic manipulators unknown. FEMS Microbiol. Ecol. 95:151. doi: 10.1093/femsec/fiz151
Detcharoen, M., Schilling, M. P., Arthofer, W., Schlick-Steiner, B. C., and Steiner, F. M. (2021). Differential gene expression in Drosophila melanogaster and D. nigrosparsa infected with the same Wolbachia strain. Sci. Rep. 11:11336. doi: 10.1038/s41598-021-90857-5
Edgar, R. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ellegren, H., and Galtier, N. (2016). Determinants of genetic diversity. Nat. Rev. Genet. 17, 422–433. doi: 10.1038/nrg.2016.58
Elliott, T. A., and Gregory, T. R. (2015). Do larger genomes contain more diverse transposable elements? BMC Evol. Biol. 15:69. doi: 10.1186/s12862-015-0339-8
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Engelstaedter, J., and Hurst, G. D. D. (2009). The ecology and evolution of microbes that manipulate host reproduction. Ann. Rev. Ecol. Evol. Syst. 40, 127–149. doi: 10.1146/annurev.ecolsys.110308.120206
Fenn, K., and Blaxter, M. (2004). Are filarial nematode Wolbachia obligate mutualist symbionts? Trends Ecol. Evol. 19, 163–166. doi: 10.1016/j.tree.2004.01.002
Foster, J., Ganatra, M., Kamal, L., Ware, J., Makarova, K., Lvanova, N., et al. (2005). The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 3, 599–614. doi: 10.1371/journal.pbio.0030121
Gao, Y., Zhao, H., Jin, Y., Xu, X., and Han, G. Z. (2017). Extent and evolution of gene duplication in DNA viruses. Virus Res. 240, 161–165. doi: 10.1016/j.virusres.2017.08.005
Gerth, M. (2016). Classification of Wolbachia (Alphaproteobacteria, Rickettsiales): no evidence for a distinct supergroup in cave spiders. Infect. Genet. Evol. 43, 378–380. doi: 10.1016/j.meegid.2016.05.034
Gerth, M., and Bleidorn, C. (2016). Comparative genomics provides a timeframe for Wolbachia evolution and exposes a recent biotin synthesis operon transfer. Nat. Microbiol. 2:16241. doi: 10.1038/nmicrobiol.2016.241
Gerth, M., Gansauge, M., Weigert, A., and Bleidorn, C. (2014). Phylogenomic analyses uncover origin and spread of the Wolbachia pandemic. Nature. Communications 5:5117. doi: 10.1038/ncomms6117
Glowska, E., Dragun-Damian, A., Dabert, M., and Gerth, M. (2015). New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infect. Genet. Evol. 30, 140–146. doi: 10.1016/j.meegid.2014.12.019
Godel, C., Kumar, S., Koutsovoulos, G., Ludin, P., Nilsson, D., Comandatore, F., et al. (2012). The genome of the heartworm, Dirofilaria immitis, reveals drug and vaccine targets. FASEB J. 26, 4650–4661. doi: 10.1096/fj.12-205096
Guindon, S., Dufayard, J., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hornett, E. A., Duplouy, A. M. R., Davies, N., Roderick, G. K., and Charlat, S. (2010). You can’t keep a good parasite down: evolution of a male-killer suppressor uncovers cytoplasmic incompatibility. Evolution 62, 1258–1263. doi: 10.1111/j.1558-5646.2008.00353.x
Hu, H. Y., and Li, Z. X. (2015). A novel Wolbachia strain from the rice moth Corcyra cephalonica induces reproductive incompatibility in the white fly Bemisia tabaci: sequence typing combined with phenotypic evidence. Environ. Microbiol. Rep. 7, 508–515. doi: 10.1111/1758-2229.12279
Huerta-Cepas, J., Forslund, K., Coelho, L., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Hunter, M. S., Perlman, S. J., and Kelly, S. E. (2003). A bacterial symbiont in the Bacteroidetes induces cytoplasmic incompatibility in the parasitoid wasp Encarsia pergandiella. Proc. R. Soc. B Biol. Sci. 270, 2185–2190. doi: 10.1098/rspb.2003.2475
Ju, J. F., Bing, X. L., Zhao, D. S., Guo, Y., Xi, Z. Y., Xi, Z. Y., et al. (2020). Wolbachia supplement biotin and riboflavin to enhance reproduction in planthoppers. ISME J. 14, 676–687. doi: 10.1038/s41396-019-0559-9
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, 277D–2280D. doi: 10.1093/nar/gkh063
Kaur, R., Shropshire, J. D., Cross, K. L., Leigh, B., and Bordenstein, S. R. (2021). Living in the endosymbiotic world of Wolbachia: a centennial review. Cell Host Microbe 29, 879–893. doi: 10.1016/j.chom.2021.03.006
Klasson, L., Walker, T., Sebaihia, M., Sanders, M., Quail, M. A., Lord, A., et al. (2008). Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol. Biol. Evol. 25, 1877–1887. doi: 10.1093/molbev/msn133
Klasson, L., Westberg, J., Sapountzis, P., Nasiund, K., Lutnaes, P., Darby, A., et al. (2009). The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc. Natl. Acad. Sci. U. S. A. 106, 5725–5730. doi: 10.1073/pnas.0810753106
Kurtz, S., Phillippy, A., Delcher, A., Smoot, M., Shumway, M., Antinescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lefoulon, E., Clark, T., Borveto, F., Perriat-Sanguinet, M., Moulia, C., Slatko, B. E., et al. (2020). Pseudoscorpion Wolbachia symbionts: diversity and evidence for a new supergroup S. BMC Microbiol. 20:188. doi: 10.1186/s12866-020-01863-y
LePage, D. P., Metcalf, J. A., Bordenstein, S. R., On, J., Perlmutter, J. I., Shropshire, J. D., et al. (2017). Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 543, 243–247. doi: 10.1038/nature21391
Lewis, Z., Champion de Crespigny, F. E., Sait, S. M., Tregenza, T., and Wedell, N. (2011). Wolbachia infection lowers fertile sperm transfer in a moth. Biol. Lett. 7, 187–189. doi: 10.1098/rsbl.2010.0605
Liang, X., Liu, J., Bian, G., and Xi, Z. (2020). Wolbachia inter-strain competition and inhibition of expression of cytoplasmic incompatibility in mosquito. Front. Microbiol. 11:1638. doi: 10.3389/fmicb.2020.01638
Lindsey, A. R. I., Rice, D., Bordenstein, S. R., Brooks, A. W., Bordenstein, S. R., and Newton, I. L. G. (2018). Evolutionary genetics of cytoplasmic incompatibility genes cifA and cifB in prophage WO of Wolbachia. Genome Biol. Evol. 10, 434–451. doi: 10.1093/gbe/evy012
Lipinski, K. J., Farslow, J. C., Fitzpatrick, K. A., Lynch, M., Katju, V., and Bergthorsson, U. (2011). High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr. Biol. 21, 306–310. doi: 10.1016/j.cub.2011.01.026
Lo, N., Paraskevopoulos, C., Bourtzis, K., O’Neill, S. L., Werren, J. H., Bordenstein, S. R., et al. (2007). Taxonomic status of the intracellular bacterium Wolbachia pipientis. Int. J. Syst. Evol. Microbiol. 57, 654–657. doi: 10.1099/ijs.0.64515-0
Long, M., Betrán, E., Thornton, K., and Wang, W. (2003). The origin of new genes: glimpses from the young and old. Nat. Rev. Genet. 4, 865–875. doi: 10.1038/nrg1204
Lynch, M., Sung, W., Morris, K., Coffey, N., Landry, C. R., Dopman, E. B., et al. (2008). A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. U. S. A. 105, 9272–9277. doi: 10.1073/pnas.0803466105
Madhav, M., Parry, R., Morgan, J., James, P., and Asgari, S. (2020). Wolbachia endosymbiont of the horn Fly (Haematobia irritans irritans): a supergroup A strain with multiple horizontally acquired cytoplasmic incompatibility genes. Appl. Environ. Microbiol. 86:6. doi: 10.1128/AEM.02589-19
Merot, H., and Charlat, S. (2004). Wolbachia infections in Drosophila melanogaster and D. simulans: polymorphism and levels of cytoplasmic incompatibility. Genetica 120, 51–59. doi: 10.1023/B:GENE.0000017629.31383.8f
Michael, G., and Bleidorn, C. (2016). Comparative genomics provides a timeframe for Wolbachia evolution and exposes a recent biotin synthesis operon transfer. Nature. Microbiology 2:16241. doi: 10.1038/nmicrobiol.2016.241
Mochiah, M. B., Ngi-Song, A. J., Overholt, W. A., and Stouthamer, R. (2002). Wolbachia infection in Cotesia sesamiae (Hymenoptera: Braconidae) causes cytoplasmic incompatibility: implications for biological control. Biol. Control 25, 74–80. doi: 10.1016/S1049-9644(02)00045-2
Monnin, D., Kremer, N., Desouhant, E., and Vavre, F. (2017). Impact of Wolbachia on oxidative stress sensitivity in the parasitic wasp Asobara japonica. PLoS One 12:e0175974. doi: 10.1371/journal.pone.0175974
Moret, Y., Juchault, P., and Rigaud, T. (2010). Wolbachia endosymbiont responsible for cytoplasmic incompatibility in a terrestrial crustacean: effects in natural and foreign hosts. Heredity 86, 325–332. doi: 10.1046/j.1365-2540.2001.00831.x
Morrow, J. L., Schneider, D., Klasson, L., Janitz, C., Miller, W. J., and Riegler, M. (2020). Parallel sequencing of Wolbachia wCer2 from donor and novel hosts reveals multiple incompatibility factors and genome stability after host transfers. Genome Biol. Evol. 12, 720–735. doi: 10.1093/gbe/evaa050
Nasehi, S. F., Fathipour, Y., Asgari, S., and Mehrabadi, M. (2022). Environmental temperature, but not male age, affects Wolbachia and prophage WO thereby modulating cytoplasmic incompatibility in the Parasitoid wasp, Habrobracon hebetor. Microb. Ecol. 83, 482–491. doi: 10.1007/s00248-021-01768-x
Nguyen, D. T., Morrow, J. L., Spooner-Hart, R. N., and Riegler, M. (2017). Independent cytoplasmic incompatibility induced by Cardinium and Wolbachia maintains endosymbiont co-infections in haplodiploid thrips populations. Evolution 71, 995–1008. doi: 10.1111/evo.13197
Noda, H., Koizumi, Y., Qiang, Z., and Deng, K. (2001). Infection density of Wolbachia and incompatibility level in two Planthopper species, Laodelphax striatellus and Sogatella furcifera. Insect Biochem. Mol. Biol. 31, 727–737. doi: 10.1016/S0965-1748(00)00180-6
Panaram, K., and Marshall, J. L. (2007). F supergroup Wolbachia in bush crickets: what do patterns of sequence variation reveal about this supergroup and horizontal transfer between nematodes and arthropods? Genetica 130, 53–60. doi: 10.1007/s10709-006-0020-7
Petrov, D. (2001). Evolution of genome size: new approaches to an old problem. Trends Genet. 17, 23–28. doi: 10.1016/S0168-9525(00)02157-0
Pinto, S. B., Stainton, K., Harris, S., Kambris, Z., Sutton, E., Bonsall, M. B., et al. (2013). Transcriptional regulation of Culex pipiens mosquitoes by Wolbachia influences cytoplasmic incompatibility. PLoS Pathog. 9:e1003647. doi: 10.1371/journal.ppat.1003647
Pratt, W. B. (1998). The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc. Soc. Exp. Biol. Med. 217, 420–434. doi: 10.3181/00379727-217-44252
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Lopez, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Rousset, F., Bouchon, D., Pintureau, B., Juchault, P., and Solignac, M. (1992). Wolbachia endosymbionts responsible for various alterations of sexuality in arthropods. Proc. R. Soc. B Biol. Sci. 250, 91–98. doi: 10.1098/rspb.1992.0135
Rowley, S., Raven, R., and McGraw, E. (2004). Wolbachia pipientis in Australian spiders. Curr. Microbiol. 49, 208–214. doi: 10.1007/s00284-004-4346-z
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sanaei, E., Charlat, S., and Engelstdter, J. (2021). Wolbachia host shifts: routes, mechanisms, constraints and evolutionary consequences. Biol. Rev. 96, 433–453. doi: 10.1111/brv.12663
Sánchez-Romero, M. A., Cota, I., and Casadesús, J. (2015). DNA methylation in bacteria: from the methyl group to the methylome. Curr. Opin. Microbiol. 25, 9–16. doi: 10.1016/j.mib.2015.03.004
Sanderson, M. (2003). r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
Sasaki, T. (2009). Wolbachia infections and cytoplasmic incompatibility in the almond moth and the Mediterranean flour moth. J. Am. Mosq. Control Assoc. 16, 739–744. doi: 10.2108/zsj.16.739
Shamayim, T., Puebla, R., Luis, E., Ernesto, O., Arturo, V., Rosenblueth, M., et al. (2015). Species in Wolbachia? Proposal for the designation of ‘Candidatus Wolbachia bourtzisii’, ‘Candidatus Wolbachia onchocercicola’, ‘Candidatus Wolbachia blaxteri’, ‘Candidatus Wolbachia brugii’, ‘Candidatus Wolbachia taylori’, ‘Candidatus Wolbachia collemboli. Syst. Appl. Microbiol. 38, 390–399. doi: 10.1016/j.syapm.2015.05.005
Shamayim, T., Puebla, R., Luis, E., Ernesto, O., Arturo, V., Rosenblueth, M., et al. (2016). A response to Lindsey et al. ‘Wolbachia pipientis should not be split into multiple species: a response to Ramirez-Puebla et al. Syst. Appl. Microbiol. 39, 223–225. doi: 10.1016/j.syapm.2016.03.004
Stouthamer, R., Breeuwer, J., and Hurst, G. (1999). Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu. Rev. Microbiol. 53, 71–102. doi: 10.1146/annurev.micro.53.1.71
Stouthamer, R., Luck, R. F., and Hamilton, W. D. (1990). Antibiotics cause parthenogenetic Trichogramma (Hymenoptera/Trichogrammatidae) to revert to sex. Proc. Natl. Acad. Sci. U. S. A. 87, 2424–2427. doi: 10.1073/pnas.87.7.2424
Suh, E., Sim, C., Park, J. J., and Cho, K. (2015). Inter-population variation for Wolbachia induced reproductive incompatibility in the haplodiploid mite Tetranychus urticae. Exp. Appl. Acarol. 65, 55–71. doi: 10.1007/s10493-014-9846-3
Sumi, T., Miura, K., and Miyatake, T. (2017). Wolbachia density changes seasonally amongst populations of the pale grass blue butterfly, Zizeeria maha (Lepidoptera: Lycaenidae). PLoS One 12:e0175373. doi: 10.1371/journal.pone.0175373
Teixeira, L., Ferreira, A., and Ashburner, M. (2008). The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 6, 2753–2763. doi: 10.1371/journal.pbio.1000002
Trpis, M., Perrone, J. B., Reissig, M., and Parker, K. L. (1981). Control of cytoplasmic incompatibility in the Aedes scutellaris complex. Incompatible crosses become compatible by treatment of larvae with heat or antibiotics. J. Hered. 72, 313–317. doi: 10.1093/oxfordjournals.jhered.a109513
Un, C., Schultner, E., Manzano-Marín, A., Flórez, L. V., Seifert, B., Heinze, J., et al. (2021). Cytoplasmic incompatibility between old and New World populations of a tramp ant. Evolution 75, 1775–1791. doi: 10.1111/evo.14261
Walker, T., Quek, S., Jeffries, C. L., Janvier, B. B., and Hughes, G. (2021). Stable high-density and maternally inherited Wolbachia infections in Anopheles moucheti and Anopheles demeilloni mosquitoes. Curr. Biol. 31, 2310–2320.e5. doi: 10.1016/j.cub.2021.03.056
Wang, Z., Su, X. M., Wen, J., Jiang, L. Y., and Qiao, G. X. (2014). Widespread infection and diverse infection patterns of Wolbachia in Chinese aphids. Insect Sci. 21, 313–325. doi: 10.1111/1744-7917.12102
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Weyandt, N., Aghdam, S. A., and Brown, A. M. V. (2022). Discovery of early-branching Wolbachia reveals functional enrichment on horizontally transferred genes. Front. Microbiol. 13:867392. doi: 10.3389/fmicb.2022.867392
Wolbach, S. B., and Hertig, M. Studies on rickettsia-like microrganisms in insects. J. Med. Res. 44, 329–374.
Wolfe, T. M., Bruzzese, D. J., Klasson, L., Corretto, E., and Schuler, H. (2021). Comparative genome sequencing reveals insights into the dynamics of Wolbachia in native and invasive cherry fruit flies. Mol. Ecol. 30, 6259–6272. doi: 10.1111/mec.15923
Wright, N. A., Gregory, T. R., and Witt, C. C. (2014). Metabolic ‘engines’ of flight drive genome size reduction in birds. Proc. R. Soc. B Biol. Sci. 281:20132780. doi: 10.1098/rspb.2013.2780
Wu, C. H., Apweiler, R., Bairoch, A., Natale, D. A., Barker, W. C., Boeckmann, B., et al. (2006). The universal protein resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 34, D187–D191. doi: 10.1093/nar/gkj161
Wu, M., Sun, L., Vamathevan, J., Riegler, M., Deboy, R., Brownlie, J. C., et al. (2004). Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2, E69–E341. doi: 10.1371/journal.pbio.0020069
Ye, J., Zhang, Y., Cui, H., Liu, J., Wu, Y., Cheng, Y., et al. (2018). WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 46, W71–W75. doi: 10.1093/nar/gky400
Yen, J. H., and Barr, A. R. (1971). New hypothesis of the cause of cytoplasmic incompatibility in Culex pipiens L. Nature 232, 657–658. doi: 10.1038/232657a0
Yoshida, K., Sanada-Morimura, S., Huang, S.-H., and Tokuda, M. (2019). Influences of two coexisting endosymbionts, CI-inducing Wolbachia and male-killing Spiroplasma, on the performance of their host Laodelphax striatellus (Hemiptera: Delphacidae). Ecol. Evol. 9, 8214–8224. doi: 10.1002/ece3.5392
Zhang, J. (2003). Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298. doi: 10.1016/S0169-5347(03)00033-8
Keywords: Wolbachia, host adaptation, cytoplasmic incompatibility, evolution, genomics
Citation: Liu B, Ren Y-S, Su C-Y, Abe Y and Zhu D-H (2023) Pangenomic analysis of Wolbachia provides insight into the evolution of host adaptation and cytoplasmic incompatibility factor genes. Front. Microbiol. 14:1084839. doi: 10.3389/fmicb.2023.1084839
Edited by:
Andrew Paul Jackson, University of Liverpool, United KingdomReviewed by:
Beatriz Sabater-Munoz, Polytechnic University of Valencia, SpainCopyright © 2023 Liu, Ren, Su, Abe and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dao-Hong Zhu, ✉ ZGFvaG9uZ3podUB5ZWFoLm5ldA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.