 Lixia Li1Xuedong Yang1,2Bingli Tong3Deng Wang4Xiu Tian1
Lixia Li1Xuedong Yang1,2Bingli Tong3Deng Wang4Xiu Tian1 Jiming Liu1*Jingzhong Chen1Xuefeng Xiao1Shu Wang1
Jiming Liu1*Jingzhong Chen1Xuefeng Xiao1Shu Wang1- 1Forest Ecology Research Center, College of Forestry, Guizhou University, Guiyang, Guihzou, China
- 2Guizhou Extension Station of Grassland Technology, Guiyang, Guizhou, China
- 3School of Life Sciences, Guizhou Normal University, Guiyang, Guizhou, China
- 4College of Urban and Rural Construction, Shaoyang University, Shaoyang, China
Introduction: Rhizobacterial communities and their metabolites can affect plant growth, development, and stress resistance, as well as the biosynthesis and accumulation of bioactive compounds in medicinal plants. This relationship has been well-characterized in many medicinal herbs, although much less commonly in medicinal trees.
Methods: Here, we analyzed the composition and structure of Cinnamomum migao rhizobacterial communities across nine growing regions in Yunnan, Guizhou and Guangxi, China, as well as differences in soil properties and fruit bioactive compounds.
Results: Results showed that the C. migao rhizobacterial communities exhibited high species richness, but location-specific differences in structure. Site-specific differences in soil properties and bioactive compounds were also observed. Furthermore, rhizobacterial community compositions were correlated with both soil properties and fruit bioactive compounds, metabolism-related functions were most common in C. migao rhizobacteria.
Discussion: Several bacterial genera, including Acidothermus, Acidibacter, Bryobacter, Candidatus_Solibacter, and Acidimicrobiales, potentially promote the biosynthesis and accumulation of 1,8-cineole, cypressene, limonene, and α-terpineol, Nitrospira and Alphaproteobacteria may play an inhibitory role. Finally, our results emphasized the critical role that soil pH and nitrogen levels play in driving rhizobacterial community structure, and specific functional bacteria can also counteract with soil properties, Acidibacter and Nitrospira can affect soil pH and nitrogen effectiveness. Overall, this study provides additional insight into the complex correlation of rhizosphere microorganisms with bioactive ingredients and soil properties of medicinal plants.
1. Introduction
Rhizosphere microorganisms are often considered “the second plant genome” because these microbes are so crucial for plant nutrition, growth, development, yield, secondary metabolite biosynthesis, and defense (Berendsen et al., 2012; Chaparro et al., 2012; Philippot et al., 2013; Qu et al., 2020; Wigginton and Amador, 2020). Both the community structure and diversity of rhizomicroorganisms are impacted by soil environmental conditions, including soil physicochemical properties, plant root exudates, and accumulated humus (Delgado-Baquerizo et al., 2018). Of these, soil physicochemical properties appear to be the primary drivers of rhizomicrobial community structure, likely because these properties pose the greatest constraints on microbial growth (Dunbar et al., 2002; de Carvalho et al., 2016). Because of this, soil microbial communities are highly variable among geographic locations and microclimates.
Bacteria are the most abundant rhizosphere denizens, particularly in nutrient-rich soils, and are vitally important for maintaining microecological stability (Marschner et al., 2001; Yang et al., 2019). Not only do rhizosphere bacteria and their metabolites affect the growth, development, and stress resistance of plants, but they can also impact plant metabolism and synthesis of bioactive compounds (Huang et al., 2018; Zhang et al., 2020). Rhizosphere bacteria can also regulate plant gene expression through the inducer effect. Specifically, microbes release small molecules into the rhizosphere and when plants recognize these chemical signals, it results in changes to both metabolism and gene expression, and the production and accumulation of plant secondary metabolites (Vives-Peris et al., 2020). This process is particularly important, from a human health perspective, in medicinal plants, where microbial induction leads to the bioaccumulation of medicinal bioactive compounds and other compounds of interest (Onrubia et al., 2013; Xie et al., 2019). However, the study of the relationship between medicinal plant metabolism and rhizosphere microorganisms is in its infancy.
While the majority of medicinal plant research is focused on roots and rhizomes, seeds are the second most important sources of medicinal and therapeutic compounds (Chinese Pharmacopoeia Commission, 2020). Additionally, the majority of medicinal plant research is focused on herbs and lianas of Asteraceae, Lamiaceae, Araliaceae, Apiaceae, Ranunculaceae, and Campanulaceae (Manero et al., 2012; Arora et al., 2016; Zeng et al., 2018; Xie et al., 2019; Li et al., 2021; Zhang et al., 2022), with some research on trees in Taxaceae (Taxus), Nyssaceae (Camptotheca acuminata), and Meliaceae (Dysoxylum binectariferum; Onrubia et al., 2013; Solaiman and Anawar, 2014). Although the subject of much less research, up to 20% of medicinal and therapeutic compounds are derived from trees (Chinese Pharmacopoeia Commission, 2020). There is a paucity of research regarding the correlation between rhizomicrobial community dynamics and changes in the bioactive constituents of medicinal plants, although some generalizations might be made. For example, medicinal plants with connected roots or rhizomes may experience more direct and intense microbial induction effects, while aerial plant parts, such as leaves, fruits, or flowers, may be less affected by rhizosphere activities. Additionally, herbs and some lianas may have much shorter lifespans than shrubs or trees, and the time of colonization of the recruited bacteria is inconsistent,potentially altering their relationship with and response to rhizomicrobes. Therefore, we may speculate that the magnitude of importance of either the direct or indirect effects of rhizomicrobes on the accumulation of bioactive compounds in medicinal plants is likely to be tissue-, contact-, time-, and environment-dependent, and highly variable across species, landscapes, and geographies.
Cinnamomum migao H. W. Li, endemic to southwestern China, bears fruit with high medicinal value. Specifically, C. migao fruit has been used in the treatment of coronary heart disease, angina pectoris, and asthma, among other conditions (Chen et al., 2021). However, due to high demand, the supply of C. migao fruit is experiencing a shortage. Therefore, understanding the ecological adaptability of this species and finding reliable methods to improve the yield and quality of C. migao fruit is of considerable importance. In this study, (a) we sought to characterize the relationships between soil chemical properties, rhizomicrobial community structure, and C. migao fruit bioactive compounds across several growing regions. (b) By understanding how soil chemistry interact with microorganisms, (c) and how microbial properties impact the accumulation of bioactive compounds in medicinal plants, growers can implement methods to increase the yield and quality of medicinal plant materials.
2. Materials and methods
2.1. Materials
Cinnamomum migao H. W. Li belongs to the family Lauraceae (Cinnamomum), which is an aiphyllium. It is a small population species, concentrated in the narrow area at the junction of Guizhou, Guangxi and Yunnan in China, its interpopulation gene differentiation coefficient (Gst) is 0.2636, the gene flow at the population level (Nm = 1.3968) > 1, and the gene flow between the populations of C. migao is confirmed to be large and the genetic differentiation is small (Li et al., 2018). The materials of C. migao in this experiment are wild-type.
2.2. Collection of soil and fruit samples
Rhizosphere soil and fruit samples were collected in October, 2018, from 5 healthy, wild C. migao trees (diameter 32–38 cm) at each of 9 sampling sites in Guizhou Province, Guangxi Province, and Yunnan Province, China. During the sampling process, the basic information such as sampling location, longitude, latitude and altitude were indicated. Information of the sampling sites are shown in Figure 1 and Table 1.
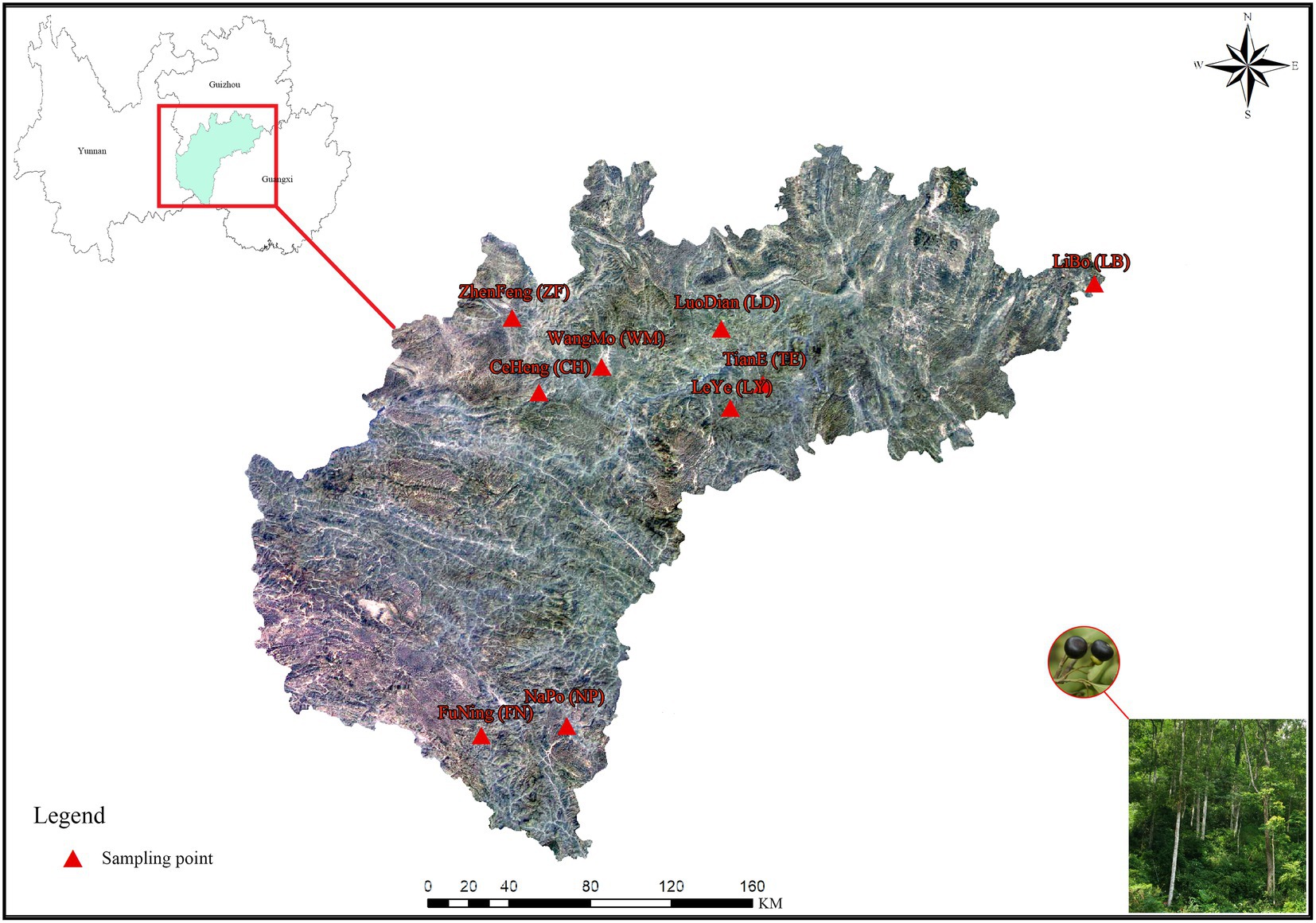
Figure 1. Map of the Cinnamomum migao sample collection sites. The map comes from the standard map service system of the Ministry of natural resources, PRC (http://bzdt.ch.mnr.gov.cn/), which is authorized for free. The picture in the right corner was taken by the first author during field investigation. The nine C. migao sampling points at the junction of Guizhou Province, Guangxi Province, and Yunnan Province, China.
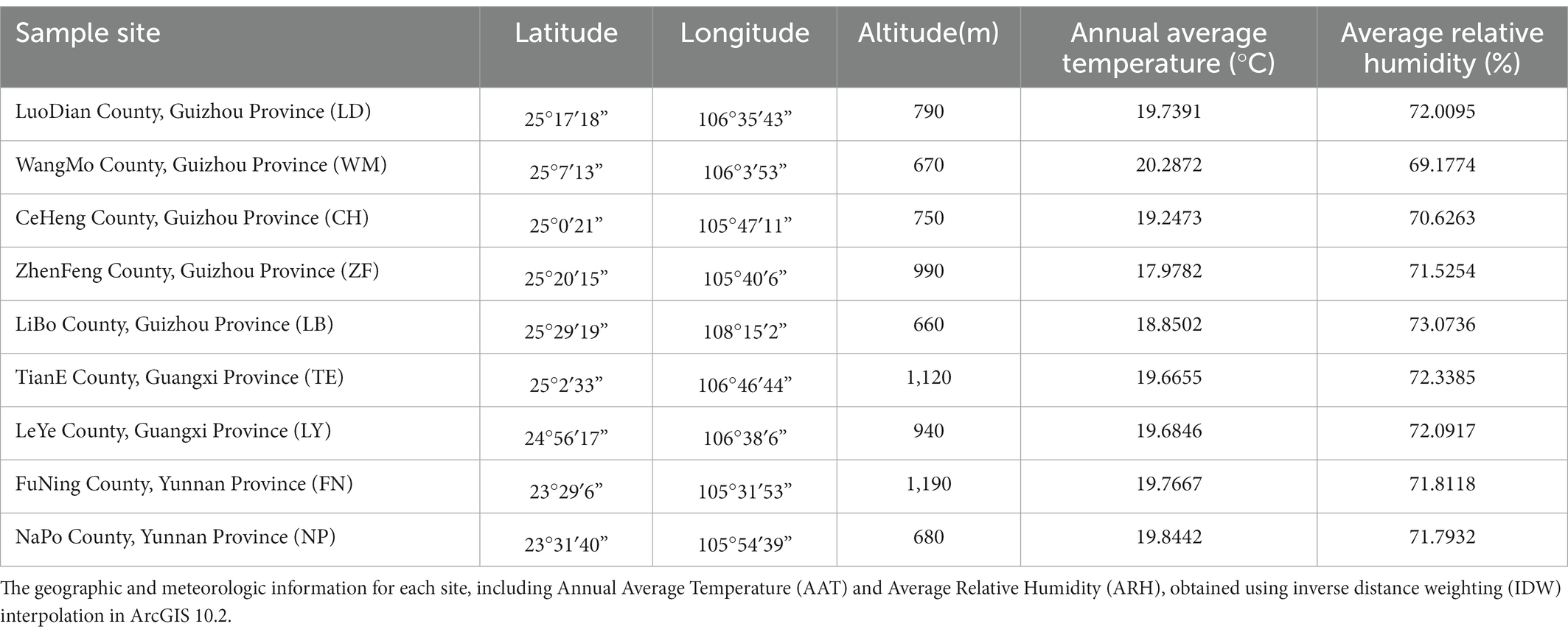
Table 1. Geographic and meteorologic information of each collection site.
To collect soil samples, after removing the humus from the soil surface, samples of healthy plant roots were collected at a depth of 0–20 cm along the base of each tree by using a drill. Soil samples were packed into the sterilized plastic bags respectively, and sampling records (time, place, number, etc.) were made on. Then they were quickly stored in ice boxes and brought back to the laboratory. Samples were processed as described by Beckers, the remaining soil was first manually removed by shaking the roots until approximately 0–2 mm soil was left attached to the roots, and then rhizosphere soils (100 g) were collected by shaking on a platform (20 min, 120 rpm; Beckers et al., 2017). The rhizosphere soil samples from 5 trees per site were mixed in equal proportions to form a single conglomerate sample (500 g; Shen et al., 2013). Homogenized soil samples were divided into two sub-samples using a 2-mm sieve: one subsample was dried at 45°C in a blast drying oven to a constant weight for determination of soil chemical properties; the other sample was stored in a − 80°C ultra-low temperature freezer for extraction of microbial DNA.
The C. migao fruits were harvested when it turns from green to brown or black (October). The collection of fruit and soil samples shall be consistent, and the corresponding fruits are taken separately and brought back to the laboratory. Fruits were dried at 45°C in a blast drying oven to a constant weight. The dried fruits was crushed into powder with multifunctional crusher, screened with 40-mesh sieve, and storaged at 4°C for determination of bioactive compounds (Sun et al., 2022).
2.3. Determination of soil chemical properties and fruit bioactive compounds
Soil chemical properties were determined according to the methods of Bao (2000). The potentiometric method was used to measure pH, water-soil ratio of 2.5:1(w/v); Organic matter (OM), Potassium dichromate oxidation volumetric method; Total nitrogen (TN), Alkaline hydrolysis diffusion method + semi micro Kjeldahl method; Total phosphorus (TP), Heating digestion method + Vanadium molybdate blue colorimetric method; Total potassium (TK), Heating digestion method + flame photometric method; Alkali hydrolyzed nitrogen (AN), Alkaline hydrolysis diffusion method + semi micro Kjeldahl method; Available phosphorus (AP), Sodium bicarbonate injection + Vanadium molybdate blue colorimetric method; Available potassium (AK), Ammonium acetate extraction + flame photometric method. The measured indicators of each soil sample (1–2 g) were measured five times in parallel, and the results were averaged.
Gas chromatography (GC) was used to quantify the bioactive compounds in C. migao fruit, including 1,8-Cineole, sabinene, limonene, and α-terpineol (Chen et al., 2021). The powder of C. migao samples was precisely weighed (1.00 g). Chromatographic conditions: the column was HP-5 capillary column (30 m × 320 mm, 0.25 μm); Carrier gas: high-purity nitrogen N2; Flow rate: 1 ml⋅min−1; Injection volume: 1 μl; Split ratio: 10:1, injector temperature: 200°C; temperature-rising program: the initial oven temperature was 50°C, and maintained for 3 min, raised the temperature to 85°C at 5°C⋅min−1 and held for 2 min, raised the temperature to 90°C at 5°C⋅min−1 and held for 2 min, raised the temperature to 160°C at 5°C⋅min−1 and held for 0 min, raised the temperature to 220°C at 20°C⋅min−1 and held for 0 min. Detector (FID) temperature: 235°C (Dai et al., 2013).
Analysis of variance (ANOVA) and Tukey T-test were employed to detect statistically significant differences (p < 0.05, p < 0.01) in these properties and compounds between samples using SPSS 22.0, Microsoft Excel 2010 and Origin 9.0 system software were used to analyze and map the data of soil chemical properties and fruit bioactive ingredients.
2.4. Extraction and sequencing of bacterial genome
The bacterial genomic DNA was extracted from of rhizosphere soil samples using the FastDNA® Spin Kit, the method was carried out according to the kit instructions. Added up to 0.5 g soil to Lysing Matrix E tube. Secured tubes in FastPrep Instrument and process for 30 s at speed 5.5. Centrifuged Lysing Matrix E tube at 14,000 ×g for 30 s. Transferred supernatant to a clean tube. Added 250 μl PPS and mixed by shaking the tube by hand 10 times. Centrifuged at 14,000 ×g for 5 min to pellet precipitate. Transferred supernatant to a clean 15 ml tube. Added 1 ml Binding Matrix Suspension to the supernatant. Placed on a rotator or inverted by hand for 2 min to allow binding of DNA to matrix. Placed tube in a rack for 3 min to allow settling of silica matrix. Discarded 500 μl of supernatant. Resuspend Binding Matrix in the remaining amount of supernatant. Transferred approximately 600 μl of the mixture to a SPINTМ Filter and Centrifuged at 14,000xg for 1 min. Emptied the catch tube and added the remaining supernatant to SPINTМ Filter and spun again. Added 500 uL SEWS-M to the SPINTМFilter and Centrifuged at 14,000 ×g for 1 min. Decanted flow-through and replaced SPINTМ Filter in catch tube. Centrifuged at 14,000 ×g for 2 min to “dry” the matrix of residual SEWS-M wash solution. Removed SPINTМ Filter and placed in fresh kit-supplied catch tube. Air dried the SPINTМ Filter for 5 min at room temperature. Added 50 μl DES and gently stirred matrix on filter membrane with a pipette tip or vortex/finger flipped to resuspend the silica for efficient elution of the DNA. Centrifuged at 14,000 ×g for 1 min to transfer eluted DNA to catch tube. DNA was application-ready.
DNA quality was assessed by 1% agarose gel electrophoresis (5 V/cm, 20 min). The V4-V5 region of the bacterial 16S rDNA was used for high-throughput sequencing. Prior to PCR amplification, the 338F forward primer was combined with 0.8 μl ACTCCTACGGGAGGCAGCAG (5 μmol/l) forward primer adapter, and the 806R reverse primer was combined with 0.8 μl GGACTACHVGGGTWTCTAAT (5 μmol/l) reverse primer adapter. PCR amplification was carried out in a 20-μL reaction system: 5× Buffer (4 μl), 2.5 mmol/l dNTPs (2 μl), TransStart Fastpfu DNA Polymerase (0.4 μl), template DNA (10 ng), plus ddH2O to 20 μl. The PCR reaction was carried out according to the following parameters: pre-denaturation at 95°C for 5 min, denaturation at 95°C for 3 min, annealing at 55°C for 3 s, 29 cycles; extension at 72°C for 10 min, hold at 10°C (ABI GeneAmp 9,700). The PCR products were sequenced reagents on an Illumina HiSeq platform. Data processing and biodiversity analysis were performed on the I-Sanger bioinformatics analysis cloud platform.1
2.5. Sequence data processing
After the paired-end (PE) sequence data were obtained by HiSeq sequencing, the PE reads were spliced into one coding sequence according to the overlap relationship between PE reads. Low-quality reads and chimera sequences were filtered out, and effective sequences were distinguished according to the barcode and primer sequences (Caporaso et al., 2010). Clean reads were used to perform taxonomic and operational taxonomic unit (OTU) cluster analysis. The QIIME platform was used to cluster sequences with a similarity of ≥0.97 into OTUs, and the longest sequence of each OTU was selected as the representative sequence. The representative OUT sequences were aligned using the Silva database2 to obtain taxonomic information for each OTU. Species composition and redundancy analysis (RDA) were performed on the I-Sanger bioinformatics analysis cloud platform. The Mothur v.1.30.1 index analysis package3 was used to calculate alpha diversity indices, including Shannon-Wiener, Simpson, ACE, and Chao1.
2.6. Bacterial ecological functional analysis
PICRUSt software was used for functional prediction of 16S amplicon sequencing results. First, PICRUSt was used to normalize OTU abundance. Then, the Greengene ID of each OTU was used to obtain COG (clusters of orthologs groups) family and KEGG (Kyoto encyclopedia of genes and genomes) Ortholog (KO) information. The eggNOG database was used to obtain the functional abundance spectrum based on the COG database. The KEGG database was used to obtain pathway, KO, and EC information, as well as the abundance of each functional category. Finally, PICRUSt was used to obtain three levels of metabolic pathway information and pathway abundance.
3. Results
3.1. Cinnamomum Migao rhizobacterial community composition across regions
3.1.1. Rhizobacterial alpha diversity
A total of 319,173 high-quality rhizobacterial sequences were obtained from samples spanning nine C. migao growing regions. From these, 2,112 distinct OTUs were obtained, with an average sequence length of 434 bp (Table 2). The coverage exceeded 99.00% across all nine growing regions, indicating that the results were valid and realistic. Standardized by the minimum number of sequences (27,740), both the Shannon and Simpson indices indicated that the WM site had the highest bacterial diversity while the LB site had the lowest. Specifically, the Shannon index indicated WM > CH > LY > FN > TE > LD > NP > ZF > LB and the Simpson index indicated LB > ZF > LD > TE > FN > NP > LY > CH > WM. According to the Chao1 and ACE indices, the WM and CM sites had the highest bacterial diversity, while the NP site had the lowest.
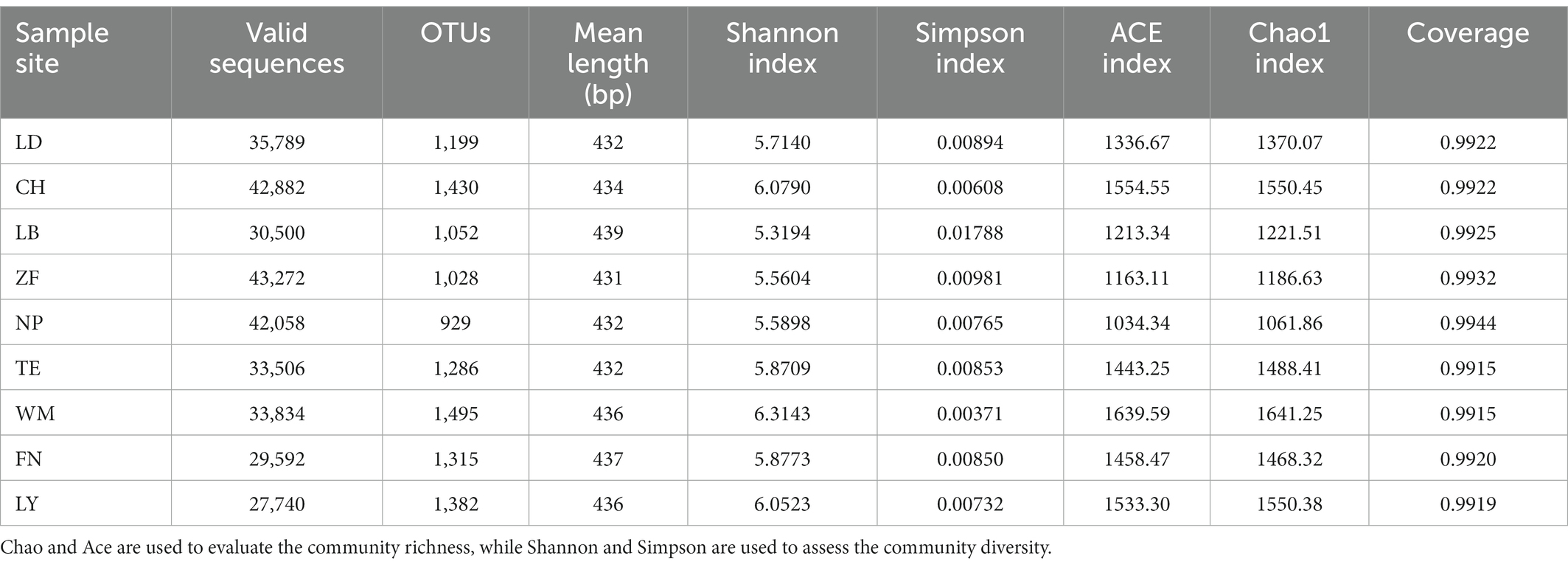
Table 2. Sequence statistics and rhizobacterial community alpha diversity indices obtained from samples spanning nine Cinnamomum migao growing regions.
3.1.2. Rhizobacterial community composition
We detected 29 phyla, 63 classes, 129 orders, 251 families, and 387 genera of bacteria across all soil samples. As shown in Figure 2A, the rhizobacteria were most commonly represented by the phyla Proteobacteria (34.81%), Acidobacteria (21.85%), Actinobacteria (16.71%), Chloroflexi (10.16%), Verrucomicrobia (3.48%), Nitrospirae (2.87%), Planctomycetes (2.38%), Firmicutes (2.38%), Gemmatimonadetes (1.68%), and Bacteroidetes (1.64%; Average value). The relative abundance of the top four bacterial phyla (Proteobacteria, Acidobacteria, Actinobacteria, and Chloroflexi) was all greater than 10%, which was significantly higher than any other bacterial phyla. Additionally, site-specific differences in bacterial community composition were also observed. Proteobacteria was the dominant phylum across all nine sample sites, which order was LB (43.78%), LY (40.13%), TE (39.91%), LD (36.47%), ZF (33.35%), FN (31.65%), NP (30.69%), CH (29.69%) and WM (27.57%), followed by Acidobacteria at all sites except LB and LY, which order was TE (30.05%), NP (27.27%), LD (25.42%), WM (24.09%), CH (23.87%), ZF (23.34%) and FN (20.32%), and Nitrospirae (12.77%) in LB samples. As shown in Figure 2B, the rhizobacteria were most commonly represented by the genera Acidobacteria (unidentified; 8.39%), Variibacter (5.40%), Acidobacteriaceae subgroup 1 (unidentified; 4.68%), Acidothermus (4.59%), Alphaproteobacteria (3.99%), and Candidatus-Solibacter (3.18%; Average value). Again, site-specific differences in bacterial community composition were also observed. The dominant genus at both ZF (11.86%) and NP (8.70%) was Acidothermus, followed by Acidobacteria (unidentified), Variibacter, Alphaproteobacteria, and Acidobacteriaceae subgroup 1 (unidentified). The dominant genus at LB was Nitrospira (12.77%), followed by Xanthobacteraceae (unidentified; 9.02%), H16 (5.06%), and Variibacter (4.93%). The dominant genera at FN were Acidobacteria (unidentified; 10.25%), Xanthobacteraceae (unidentified; 5.72%), Nitrospira (5.57%), and DA101 soil group (unidentified; 5.16%). The dominant genus at TE (9.34%), LD (8.08%), WM (8.63%), CH (15.39%), and LY (5.57%) was Acidobacteria (unidentified), followed by Alphaproteobacteria, and Variibacter.

Figure 2. Rhizobacterial community structure across nine sample sites. Phylum-level (A) and Genus-level (B). The color of the column represents the different genera, and the length of the column represents the proportion size of the genus. The species with <0.01% abundance were classified together as “others.” “norank” represent “unidentified” bacterial genus.
As shown in Figure 3, Site-specific differences in the abundance of rhizobacteria community of C. migao were observed obviously at genus level. The rhizobacteria of the nine samples can be divided into two categories: LD and TE and CH first clustered together, and then clustered with NP and ZF as one class, and FN, LY and WM and LB as the other category; Among them, the rhizobacterial community of C. migao is most similar between LD and TE, and between ZF and NP.
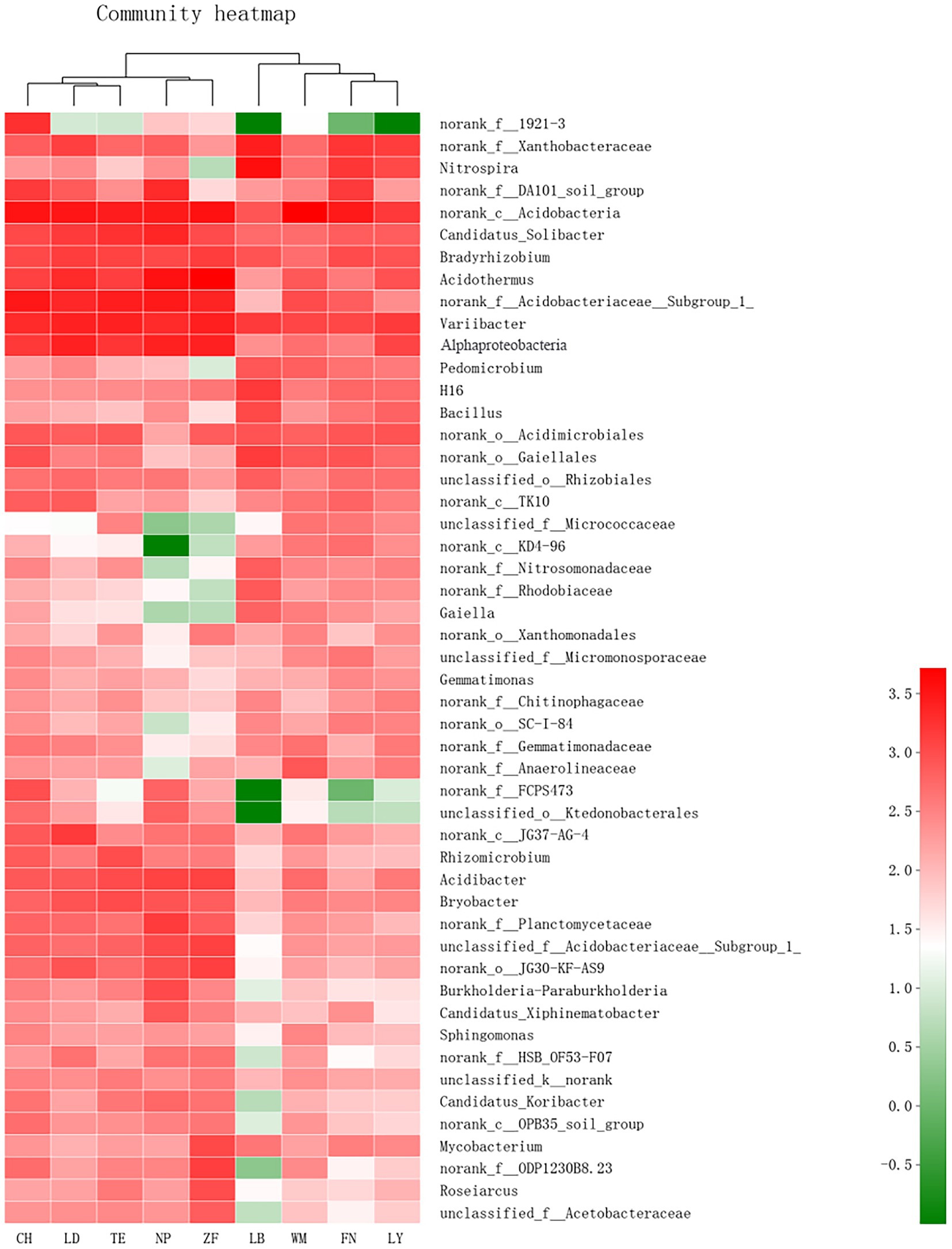
Figure 3. Heatmap of Bacteria in Rhizosphere Soil of C. migao. Heatmap of the top 50 most abundant genera in bacterial communities detected across nine sample sites. Dendrograms for hierarchical cluster analysis and sample locations were shown at the top.
3.1.3. Rhizobacterial ecological functions
The rhizobacterial unigenes across nine samples sites were classified into 24 gene function families based on the COG database (Figure 4). The eight most common functional gene families were energy production and conversion, amino acid transport and metabolism, carbohydrate transport and metabolism, transcription, cell wall/membrane/envelope biogenesis, general function prediction, function unknown, and signal transduction mechanisms, together representing 57.64–63.49% of all gene functions. At the primary functional later, KEGG analysis found six categories of biological functions across all samples, including Cellular Processes, Environmental Information Processing, Genetic Information Processing, Human Diseases, Metabolism, and Organismal Systems. Among these, Metabolism, Environmental Information Processing, and Genetic Information Processing were most common, accounting for 51.32–52.71%, 12.32–14.55%, and 14.81–15.77% of all gene functions, respectively. At the secondary functional layer, 41 sub-functions were identified across all samples, including Amino Acid Metabolism, Carbohydrate Metabolism, Cell Motility, Cellular Processes and Signaling, Energy Metabolism, Lipid Metabolism, Membrane Transport, Poorly Characterized, and Replication and Repair, among others.
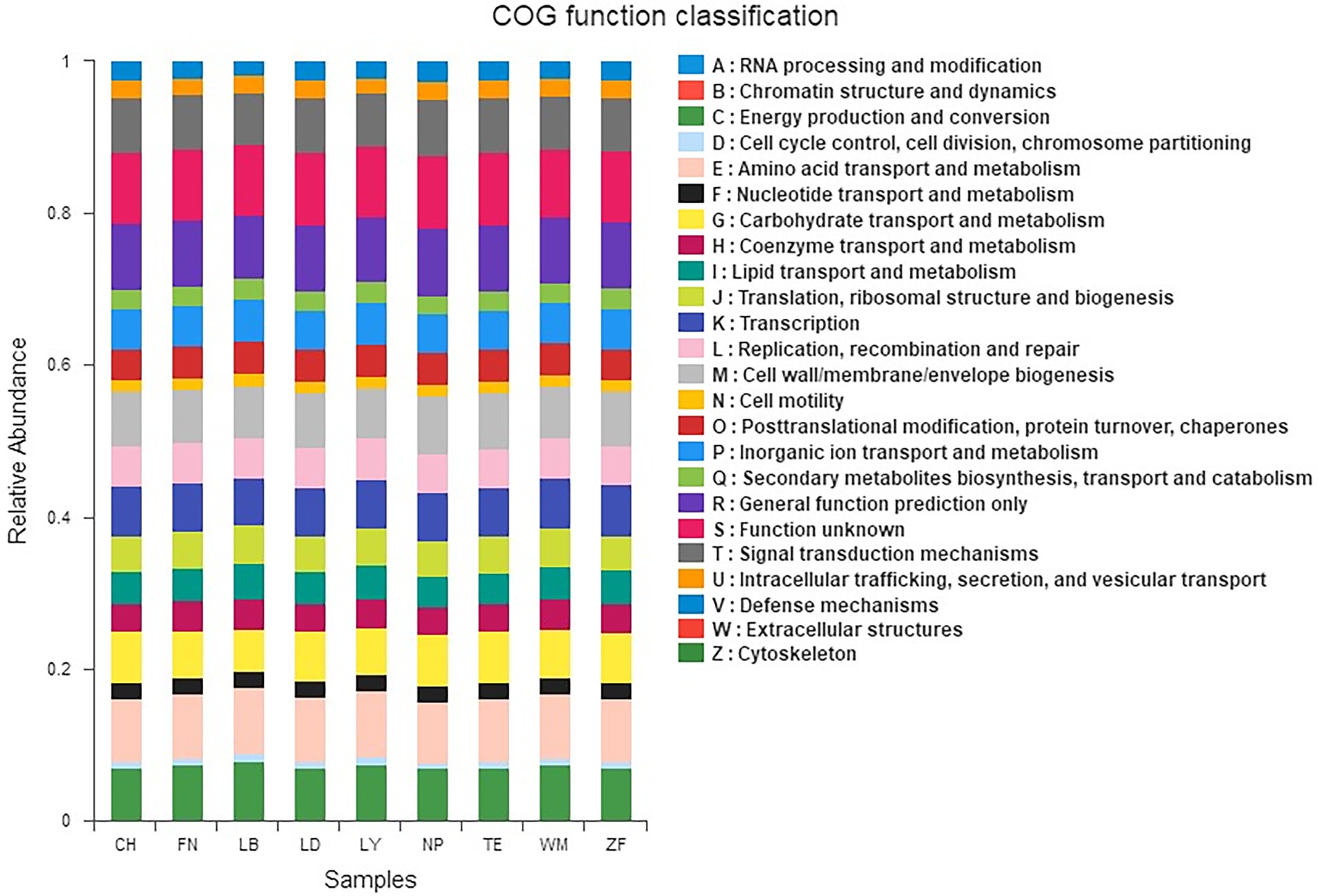
Figure 4. COG (clusters of orthologs groups) functional classification of the rhizobacterial communities across nine sample sites. The x-axis represents samples and the y-axis represents relative abundance presented as a percentage.
3.2. Cinnamomum Migao soil properties and bioactive compounds across regions
The rhizosphere soil chemical properties were analyzed across all nine sample sites (Figure 5). Overall, the soil across all sites was found to be more or less acidic, with significant differences between sites. Across sites, significant differences were found in OM, AN, TP, TK, and AK, with small differences in AP, and no significant difference in TN. Correlation analysis between soil chemical factors showed that soil pH were positively correlated with the 7 soil nutrient indicators, especially with AK, AP and OM (P < 0.01); TK with TP and TP with OM were negatively related (P < 0.01; Table 3). Overall, both the pH and nutrient contents of LB and LY were higher than the other sites, with ZF having the lowest pH and nutrient content. Across all sites, the AP content tended to be low while the AK and AN contents tended to be high. According to the “Nutrient Classification Standard of the Second Soil Census of China,” across sites the AP content would be classified as grade 4 (5–10 mg/kg) or grade 5 (3–5 mg/kg), the AK content would be classified as grade 1 (>200 mg/kg), and the AN content would be classified as grade 1 (>150 mg/kg).
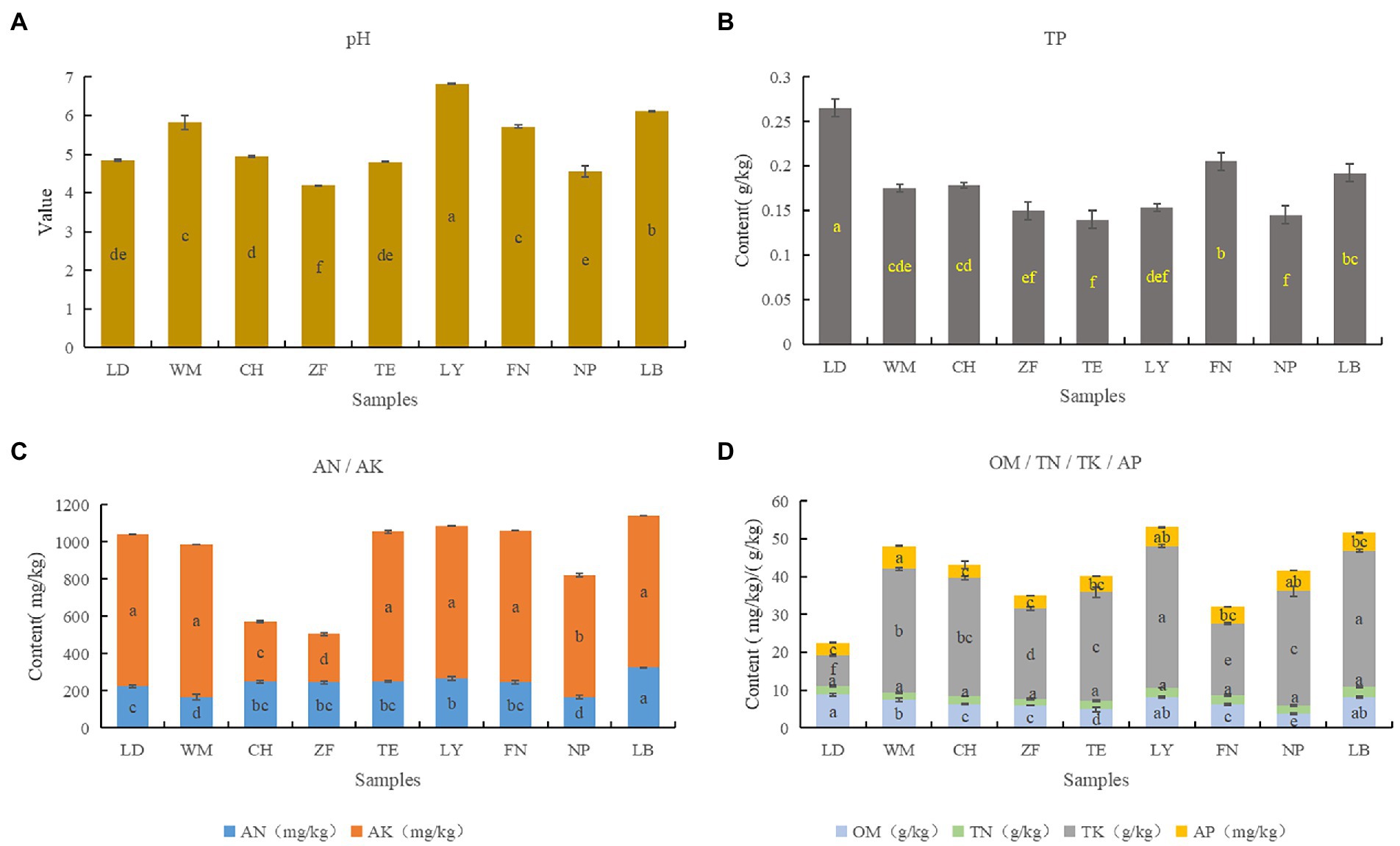
Figure 5. Nutrient and chemical properties of C. migao rhizosphere soils from nine sites. The ANOVA analysis and Turkey were used to compare the soil chemical properties across nine sample sites. Different letters indicate significant differences (p < 0.05; n = 5). The color of the column represents the different soil chemical properties, and the length of the column represents the content of the soil chemical property. (A) PH, (B) the content of total phosphorus (TP), (C) contents of alkali hydrolyzed nitrogen (AN) and total potassium (TK), (D) contents of organic matter (OM), total nitrogen (TN), available phosphorus (AP), and available potassium (AK).
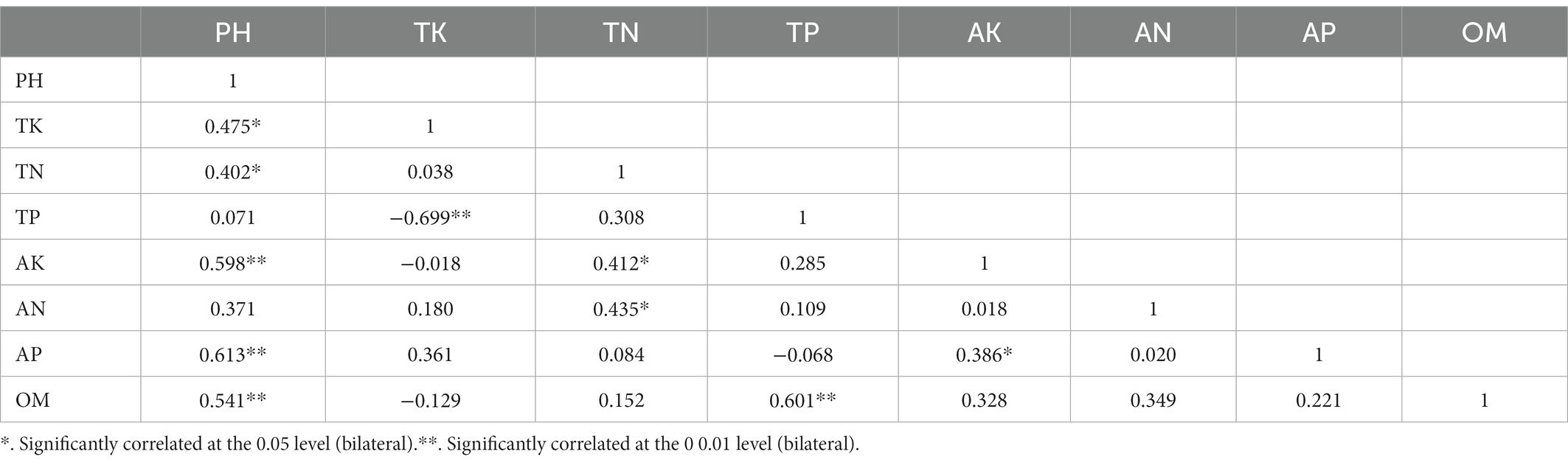
Table 3. Correlation of chemical Properties of Rhizosphere Soil of C. migao.
The content of several medicinally important bioactive compounds of C. migao fruit, including 1,8-cineole, sabinene, limonene, and α-terpineol, were determined by GC (Figure 6). The most plentiful compound was 1,8-cineole, followed by α-terpineol, sabinene, and limonene. Fruit from LY contained the highest content of 1,8-cineole, α-terpineol, and limonene, while fruit from FN contained the lowest content of all compounds. The differences in the content of bioactive compounds of C. migao fruit from different regions were considerable, suggesting that differences in environmental and soil characteristics translate into vast differences in bioactive compound accumulation.
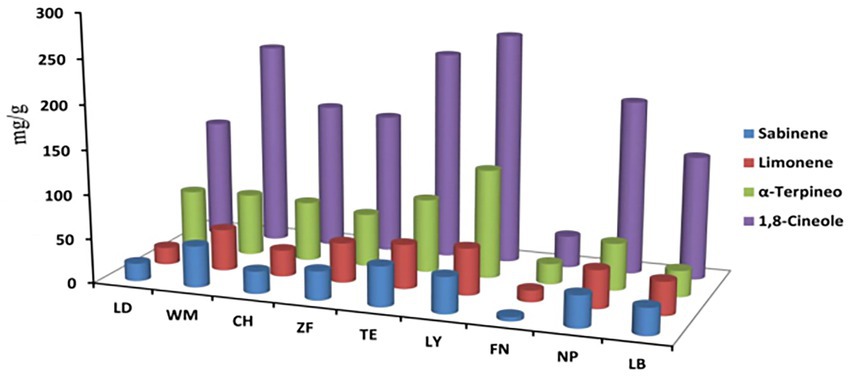
Figure 6. Content of medicinally important bioactive compounds of C. migao fruit across nine sites. The ANOVA analysis and Turkey were used to compare the soil chemical properties across nine sample sites. Different letters indicate significant differences (p < 0.05; n = 5). Four bioactive compounds, 1,8-cineole (blue), sabinene (orange), limonene (grey), and α-terpineol (yellow), the length of the column represents the content of the bioactive compound.
3.3. Correlations between soil chemical properties, rhizomicrobial community structure, and Cinnamomum migao fruit bioactive compounds
RDA was used to examine the relationships between the top 20 bacterial genera and soil chemical and nutritional properties (Figure 7A), with the horizontal axis as the first ranking axis (57.57% contribution) and the vertical axis as the second ranking axis (17.97% contribution). The bacterial genera Nitrospira, Xanthobacteraceae (unidentified), H16, and Gaiellales (unidentified) were positively correlated with all eight soil chemical indicators. Rhizobiales (unidentified) was positively correlated with AN, but not TN. Bryobacter and Acidobacteria (unidentified) were positively correlated with AP, but not TP. Bradyrhizobium was negatively correlated with seven soil chemical indicators. Most of the other bacterial genera, such as Acidibacter, Acidobacteriaceae subgroup 1 (unidentified), Candidatus-Solibacter, Variibacter, Acidothermus, Acidimicrobiales, and Alphaproteobacteria, were negatively correlated with the eight soil chemical indicators to different degrees. Additionally, pH, AN, and TN were strongly correlated, and together had the greatest influence on bacterial community composition at LB, LY, and FN.
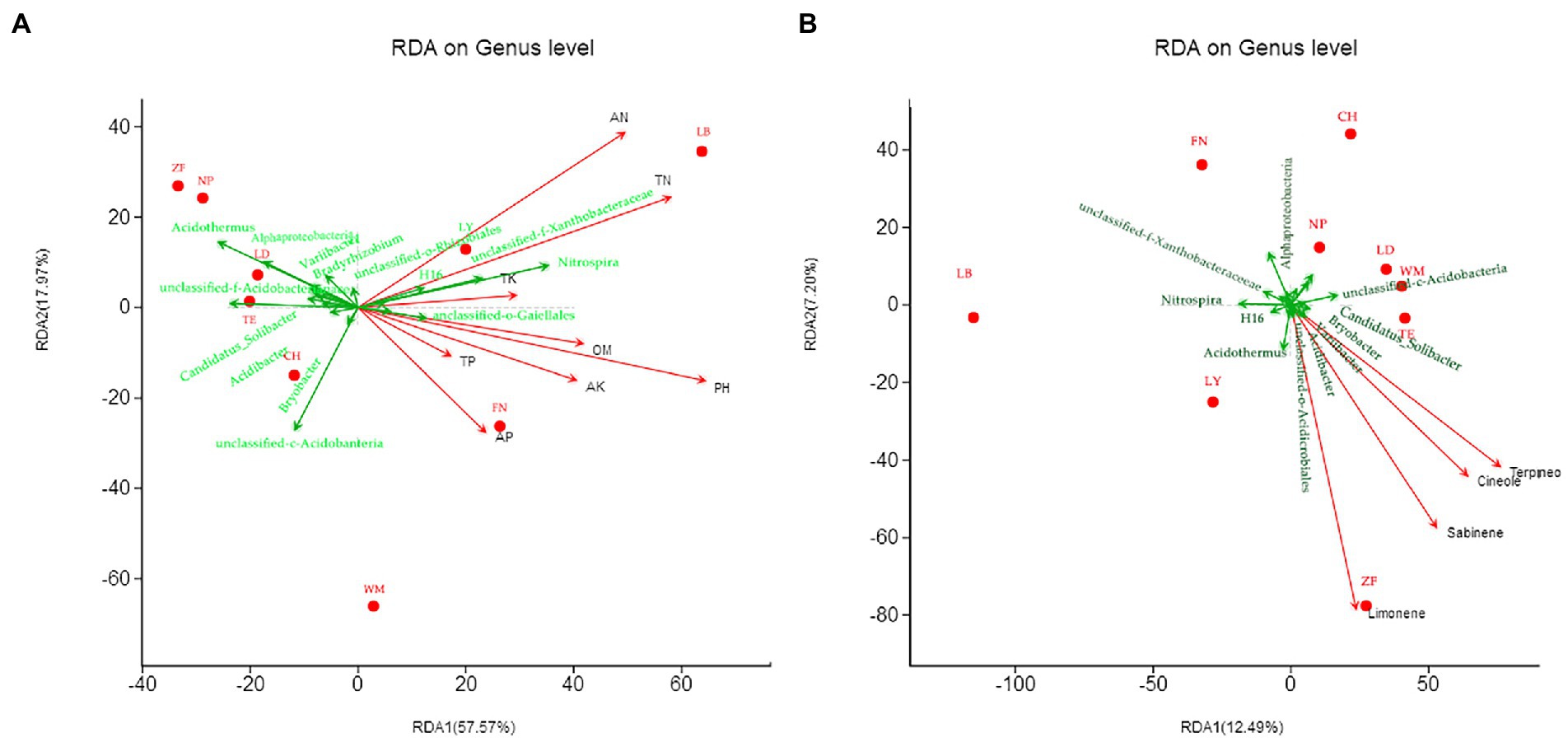
Figure 7. Canonical correspondence analysis (RDA) of rhizobacterial communities with soil chemical factors (A) and C. migao fruit bioactive compounds (B) across nine sites. The red dots represent different sample sites, the green arrows represent the top 20 abundance genera, and the red arrows represent soil chemistry and active ingredients, arrow length represents the degree of influence.
RDA was also used to examine the relationships between the top 20 bacterial genera and the content of C. migao fruit bioactive compounds (Figure 7B), with the horizontal axis as the first ranking axis (12.49% contribution) and the vertical axis as the second ranking axis (7.20% contribution). The bacterial genera Acidothermus, Acidibacter, Bryobacter, Candidatus_Solibacter, and Acidimicrobiales (unidentified) were positively correlated with all four bioactive compounds, particularly, Acidothermus and limonene have a strong positive correlation. Acidobacteria (unidentified) was positively correlated with all bioactive compounds except limonene. Nitrospira, Alphaproteobacteria and Xanthobacteraceae (unidentified) were negatively correlated with all four bioactive compounds.
4. Discussion
4.1. Variability of soil properties, bacterial communities, and bioactive compounds
Analysis of the bacteria present in the rhizosphere of C. migao from different regions revealed rich and varied communities. The dominant genera of rhizosphere bacteria from different habitats were basically the same, although there are some differences in abundance. Overall, Proteobacteria (34.81%), Acidobacteria (21.85%), Actinobacteria (16.71%), and Chloroflexi (10.16%) were the dominant bacterial phyla across all field sites; the defined bacterial genera, including Variibacter (5.40%), Acidothermus (4.59%), Alphaproteobacteria (3.99%), Candidatus_Solibacter (3.18%), Bradyrhizobium (3.05%), Nitrospira (2.86%), Acidibacter (1.91%) and Bryobacter (1.88%) were found to be the dominant genera. In comparing with previous studies, there are some species with similar dominant bacterial genera to C. migao, such as: natural mixed forests, apple trees, Abies Fabri, Robinia Pseudoacacia, and Cunninghamia Lanceolata (Jiang et al., 2017; Yang, 2019; Zhang, 2019; Nie et al., 2020; Xiong et al., 2021). However, some species dominant-phylum are as similar as 90% to C. migao, the specific bacterial genera differ from our results, such as: Taxus, which is an arboreal medicinal plant (Hao et al., 2016). Additionally, some plants have different genotypes from C. migao, and their dominant taxa are distinct (Li et al., 2021). Studies have shown that plants with different genotypes may differ in their morphological characteristics, ecological habits, and physiological traits. There may also be differences in the characteristics of the rhizomicroorganisms they recruit. For example, (a) species with different ecological habits have more discrepancy’s in their colonizing environments, e.g., rice vs. maize, and their rhizomicrobes also differ significantly (Li et al., 2014; Nevita et al., 2018). (b) Plants with different phenotypes have disparate environmental effects, e.g., variances in the depth, number and size of plant roots makes distinct effects on soil amelioration, ultimately leads to differences in enriched rhizobacteria. (c) Plants with different physiological characteristics may differ in their metabolites, and the secretion and release of specific metabolites may inhibit some genera that are not adapted to the type and concentration of the products, while attract those who use the products as a nutritional feedstocks and structural materials. It is worth mentioning that legumes actively recruit rhizobia for forming a mutually beneficial symbiosis (Glyan Ko, 2015).
After comprehensive analysis, we found that plants with similar dominant genera to C. migao were mostly derived from forestland (C. migao’s samples also from forests), and these dominant genera reflected the bacterial structure characteristics in specific ecological environment, which differed from the rhizobacterial compositions of crops or herbs in farmland (Zhang et al., 2022). This may be a commonality, as arboreal (forestland) recruited bacteria which have longer colonization periods and more stable ecotope than herbs (farmland). Besides, because of the abundant diversities of bacteria and the more complex mechanisms of interaction between arbors and microorganisms, there are still a large number of undefined bacterial taxa in forests (Du et al., 2017; Zhang et al., 2022), more than half taxa are undefined in C. migao rhizobacteria, and the defined are basically rare. Researches on the composition and function of sylvatic microorganisms should be strengthened in the future.
Analysis of soil chemical and nutritional properties indicated that there were significant differences in pH, TK, TP, and OM between sites, while differences in AN, AK, and AP differed only slightly, and TN did not differ at all between sites. Both the pH and nutrient content of soils from LB and LY were higher, while ZF soils had the lowest pH and nutrient content. Furthermore, the contents of four medicinally important bioactive compounds (1,8-cineole, sabinene, limonene, and α-terpineol) from C. migao fruit were different significantly between sites, with fruit from LY, WM, and TE containing the highest content of bioactive compounds, the minimum was ZF. This may be related to G × E (genotype × environment) interactions, Studies have confirmed that different genotypes have genetic traits for variable chemical compositions. For example, the active constituents of Taxus (Hao et al., 2016), Cornus officinalis (Sun et al., 2022), Astragalus mongholicus (Li et al., 2021), and Baphicacanthus cusia (Zeng et al., 2018) differ from C. migao. Besides, widespread species in order to adapt to different environments, leading to the creation of stably inheritable variation that results in multiple genotypes of the same species. The chemical components and contents of these genotypes may differ, e.g., the polyphenols and antioxidants contents of Sorghum were higher in two genotypes and simultaneously did not differ significantly between some genotypes (Aruna et al., 2020). Based on the small genetic differentiation among populations of C. migao (Li et al., 2018), we speculate that environmental heterogeneity are more likely to be responsible for the differences in active ingredients and dominance of homogeneous genera in C. migao. This is substantiated by the reports of others (Li et al., 2016), indicating that differences in environmental conditions, soil properties, and microorganisms can result in substantial differences in the quality of medicinal plant materials.
4.2. Effects of soil properties on rhizomicroorganisms
The structures and diversities of microbial community are meaningful indicators for evaluating ecosystem stability (Chapin et al., 2011; Romaniuk et al., 2011; Wigginton and Amador, 2020). Several environmental factors affect soil microbial community structure and diversity, including primarily climatic, phytic, and soil-specific factors (Marschner et al., 2001; Yang et al., 2019; Wang et al., 2021). Of these, soil properties have been found to be the primary driver of microbial community structure (Philippot et al., 2013), particularly soil pH (Scola et al., 2018). Our results are in agreement with these previous studies, indicating that pH is a key driver of rhizobacterial community structure (Jiang et al., 2017). Differences in response patterns of bacteria-specific to pH gradients were observed, for example, Nitrospira, Gaiellales (unidentified), Xanthobacteraceae (unidentified), and H16 were positively correlated with pH, while Acidothermus, Variibacter, Bradyrhizobium, Acidibacter, Acidobacterianace (unidentified), Alphaproteobacteria and Candidatus-Solibacter were negatively correlated with pH. This dual response is consistent with other studies (Jones et al., 2009; Dimitriu and Grayston, 2010; Rousk et al., 2010). The acidic soil environment of C. migao may provide suitable conditions for acidophiles to colonize and enrich, such as, Acidothermus, Alphaproteobacteria, Acidibacter, and Acidobacterianace (unidentified). Additionally, Nitrospira and Xanthobacteraceae (unidentified) were found to have a close positive correlation with TN and AN, nitrogen levels are also controlling factors for C. migao rhizobacteria.
In fact, there is an interaction, soil physicochemical factors not only affect microorganisms, but also microorganisms can react to soil properties. For example, Acidibacter as an acidophilic ferric reducing gammaproteobacterium can causing the pH to increase when it catalyzed the reductive dissolution of the ferric iron mineral schwertmannite (Falagán and Johnson, 2014); Nitrospira plays pivotal role in nitrification as an aerobic chemolithoautotrophic nitrite-oxidizing bacterium, metabolic versatility enables Nitrospira to colonize a broad range of habitats and to sustain shifts in environmental conditions such as changing oxygen concentrations and promoting nitrogen-cycling (Daims et al., 2015; Daims and Wagner, 2018), this is verified by the higher Nitrospira abundance and AN contents in LB samples. It’s showed that the enrichment of acidophiles and Nitrospira may be primarily responsible for the acidity and nitrogen-rich in C. migao rhizosphere soil. The further description is that C-, N- and P-cycles in soil are subject to the allocation of microbial community structures and functions, the growth and activity of endemic rhizomicroorganisms can promote the transformation and storage of effective nutrients in rhizosphere soil, which stimulate the uptake of nutrients by plant roots.
In addition, we found connection between soil pH, soil nutritional properties, and rhizobacterial diversity. On the one hand, pH and soil nutrients are positively correlated, especially with AK, AP and OM, pH can affect the effectiveness of soil nutrients, and it may be a key factor in regulating soil nutrients and promoting plant nutrient uptake. On the other hand, there are also positive correlation among pH, nutrients and rhizobacterial diversity. For example, soils with very low pHs tended to have lower nutritional content and, consequentially, lower rhizobacterial diversity, as was the case for ZF. Contrarily, soils with higher pH tended to have higher nutritional content and rhizobacterial diversity, as was the case for LY. This supported that soils with higher nutrient content tend to have higher microbial diversity (Maestre et al., 2015; Gebhardt et al., 2017), but other samples (WM, CH, LB, etc.) do not follow this universal rule. So other factors affecting rhizomicrobial diversity should be taken into account.
We comprehensively analyzed the geoclimatic information across all sample sites, the results showed that although a geoclimatic analysis of the nine sample sites indicated a significant elevation gradient, no significant correlations were found between elevation and soil properties, rhizobacterial diversity. This is consistent with reports indicating that bacterial community distribution may not follow the same elevational distribution patterns exhibited by plants or animals (Shen et al., 2013), but contrary to other reports indicating that bacterial communities do differ by elevation, despite perhaps indirectly (Fierer et al., 2011). Alternatively, temperature may be an important factor in the differentiation of the rhizobacterial diversity, for example, the low rhizobacterial diversity of ZF may be related to its relatively lower mean annual temperature (17.98°C), and the higher bacterial diversity of WM may be related to its relatively higher mean annual temperature (20.29°C). Soil properties explained 75.54% (RDA1 57.57%, RDA2 17.97%) of the variation in rhizobacterial community structure. This explanation rate was close to apple trees of around Bohai Gulf, with a cumulative explanation rate of 78.88% (RDA1 50.43%, RDA2 28.45%) for RDA (rhizobacteria × soil properties; Jiang et al., 2017), and both major contributing factors included pH and AN; Betula albosinensis was 57.43% (RDA1 42.65%, RDA2 14.72%), and its dominating factors showed SOC > C:N > TN > pH (Du et al., 2017). This discrepancy may be related to the different soil factors selected. The explanation rate of 75.54% indicates that soil chemistry is the dominant influence of rhizobacteria in C. migao, which may be connected with the existence of long-term, direct × indirect interactions with rhizomicroorganisms in a combination of multiple ways. The related percentage of variation unexplained by factors studied here (24.46%) suggests that there are other environmental or biological factors influencing C. migao rhizobacterial community structure. Future studies should examine the effect of other climatic, geographical, soil physiochemical properties (e.g., soil enzymatic activity, aeration, and water permeability), and associated plant communities on C. migao rhizobacterial community structure and diversity.
4.3. Effects of soil rhizobacteria on medicinal plants
Plant growth-promoting rhizobacteria (PGPR) colonize plant roots and stimulate plant growth, stress tolerance, and defense (Kloepper and Schroth, 1978). The widely reported PGPR in C. migao include Bradyrhizobium and Bacillus (Podile and Kishore, 2006; Lugtenberg and Kamilova, 2009; Haney et al., 2015; Xie et al., 2019), which have disease-preventing and growth-promoting effect on C. migao. They promote plant growth by enhancing nutrient acquisition, niche competition, the production of antimicrobial compounds, or inducing systemic resistance to prevent pathogen infection (Zamioudis et al., 2013; Pieterse et al., 2014, 2016; Chowdhury et al., 2015). Recent studies have demonstrated that not only do rhizobacteria and their metabolites affect the growth and stress resistance of plants, but they can also impact plant accumulation and synthesis of medicinal bioactive compounds (Köberl et al., 2013; Huang et al., 2018; Zhang et al., 2020). There are three primary mechanisms by which rhizomicroorganisms affect the accumulation of bioactive secondary metabolites in medicinal plants. First, microorganisms activate secondary metabolite signaling pathways in plants, resulting in the upregulation of secondary metabolite biosynthesis (Trivedi et al., 2020). Second, microorganisms themselves produce bioactive secondary metabolites which stimulate the upregulation of secondary metabolite biosynthesis in plants (Schmidt et al., 2014). Third, microorganisms improve the stress resistance of medicinal plants, which is often mediated by the biosynthesis of secondary metabolites (Taghinasab and Jabaji, 2020). Relevant representative studies, Piriformospora indica and Azotobacter chroococcum can significantly enhance the artemisinin content of Arteuaisia annua (Arora et al., 2016). Stenotrophomonas maltophilia and Pseudomonas fluorescens can significantly enhance the hypericin and hyperforin content of Hypericum perforatum (Manero et al., 2012). Pseudomonas can regulate gene expression in Taxus cells by releasing the phytotoxin-coronatine, which may be responsible for the resultant increase in taxane content (Onrubia et al., 2013). Burkholderia sp. increases the synthesis of indigo in Baphicacanthus cusia (Zeng et al., 2018). Bacillus pumilus enhances glycyrrhizic acid content by increasing the expression of key enzymes (Xie et al., 2019).
Here, we found that several bacteria, including Acidothermus, Acidibacter, Bryobacter, Variibacter, Candidatus_Solibacter, and Acidimicrobiales (unidentified) were positively correlated with the content of bioactive compounds in C. migao. Interestingly, Acidobacteria (unidentified) was positively correlated with all compounds except limonene, and Nitrospira, Alphaproteobacteria, and Xanthobacteraceae (unidentified) were negatively correlated with all four bioactive compounds. Another important finding is that the functional classification of C. migao rhizobacteria tended to be homogenous across regions, with metabolism-related functions being the most common. Therefore, we speculated that C. migao may selectively recruit specific microorganisms in rhizosphere that could metabolize certain substances to effect C. migao. Based on the data obtained in this study, the enrichment of bacterial genera Acidothermus, Acidibacter, Bryobacter, Variibacter, Candidatus_Solibacter, and Acidimicrobiales potentially promote the biosynthesis and accumulation of 1,8-cineole, cypressene, limonene, and α-terpineol in C. migao, while Nitrospira and Alphaproteobacteria may be repressive for the bioactive ingredients. However, the mechanism by which these rhizobacteria alter the accumulation of bioactive compounds remains to be identified.
Among the enriched taxa in C. migao rhizosphere. Acidothermus was widely known as the Acidothermus celulolyticus endoglucanase (E1; Larry et al., 2008). It has been recorded to colonize the rhizosphere of Lettuce (Cipriano et al., 2016), and found to be associated with the oxidation of sulfur, nitrogen and iron (Wang et al., 2022). This oxidation alters the uptake of sulfur, nitrogen, and iron by plants, affecting the synthesis of plant metabolites, in this study, Acidothermus may be a potentially beneficial bacteria used to promote the accumulation of bioactive compounds (especially limonene) of C. migao. Acidibacter, Bryobacter, Variibacterr, and Candidatus_Solibacter may have a slight stimulating effect on the accumulation of bioactive ingredients in the fruit of C. migao. Acidibacter colonized in many plant species, and associated with soil nutrient and iron cycles, plant blights, and soil pollution controls (Falagán and Johnson, 2014; Liu et al., 2016; Jiao et al., 2018; Huang et al., 2022), it may has a variety of potential functions. Bryobacter has been reported as beneficial bacteria in studies such as leguminous plants and Marchantia, where it plays roles in plant-growth promotion, complex exudate degradation, nitrogen fixation, methylotrophs, and disease-suppressive (Xiao et al., 2017; Luis et al., 2018). Variibacterr has been documented in soil contamination and nutrient cycle studies (Jiao et al., 2018; Wu et al., 2020), which may be a microbial community with degradation potential, although it has decreased abundance in tobacco-peanut intercropping soils with Burkholderia (Gao et al., 2019), it may also have biocontrol roles as Burkholderia. The abundance of Candidatus_Solibacter were increased in healthy sesame rhizosphere soils with Bryobacter and Bradyrhizobium, they were beneficial to improving the soil microecology and alleviating continuous cropping obstacle (Wang et al., 2017). Alphaproteobacteria is a diverse class of organisms within the phylum Proteobacteria, its members inhabit diverse environmental niches and have many important biological roles, they participate in a variety of metabolic strategies, including nitrogen fixation, photosynthesis, ammonia oxidation, and methylotrophy (Williams et al., 2007; Andreote et al., 2009; Collier, 2016), in this study, Alphaproteobacteria may suppresses the accumulation of C. migao bioactive ingredients by participating in a particular metabolisms. Nitrospira was widely reported as an aerobic autotrophic nitrite-oxidizing bacteria that plays a key role in plant nitrification and promotes efficient conversion and storage of nitrogen (Daims et al., 2015; Daims and Wagner, 2018); additionally, Nitrospira has also been reported to be associated with verticillium and fusarium wilts of cotton and watermelon (Zhu et al., 2019), the complexity of Nitrospira functions makes it possible to both as beneficial bacteria to stimulate plant absorption of effective nutrients, as well as a pathogen to induce systemic resistance of plants, Nitrospira may inhibit the accumulation of medicinal bioactive ingredients of C. migao. Overall, the enriched strains and their metabolites may promote or inhibit the growth and quality of C. migao through a variety of pathways. We suggest that the rhizosphere of wild-type-C. migao are enriched with a large number of potentially valuable bacteria. It provided a reference for the next step of screening beneficial bacteria to promote the growth and quality of C. migao. Meanwhile, the function of specific microbial strains should be validated, and the mechanisms of specific strains on bioactive ingredients in C. migao should be elucidated in the future.
The effective magnitude of rhizomicroorganisms on bioactive compounds is likely to be species-, tissue-, and contact-dependent. For example, in herbs with roots as medicinal parts, 57 OTUs are positively (0.9 ≥ R2 ≥ 0.51) or negatively (−0.85 ≤ R2 ≤ −0.52) correlated with the bioactive ingredient in Astragalus mongholicus (Li et al., 2021); 13 rhizobacteria genera are significantly (P < 0.05) associated with bioactive ingredient in Scutellaria baicalensis (Jiang, 2021). In herbs with the whole plant as medicinal parts, the correlation of bioactive ingredients of different medicinal parts with rhizobacteria is shown as aboveground < underground (R2) in Rumex japonicus (Ni, 2019). The relationship between three medicinal licorices endophytic bacteria and the bioactive ingredients showed that the cumulative interpretation rate was 22.33% (RDA1 12.55%, RDA2 9.78%; Dang et al., 2020). Combined with our results, soil properties explained 75.54% of the variation in rhizobacterial community structure, whereas rhizobacteria explained 19.69% of medicinal components. (a) We speculated that the above phenomena may be related to the effect of rhizobacteria on the medicinal ingredients in roots or rhizomes of herbs could rely on dual influences of direct and indirect, while fruit trees could basically depend on indirect effects. (b) The bioactive ingredients of fruits and rhizobacteria are not the dominant influencing factors for each other despite they somewhat correlated. The remaining 80.31% of unexplained factors may be related to other unmeasured indicators, such as: temperature, light, moisture, endophytes, other rhizomicroorganisms, and rare genera (relative abundance < 1%). Although the interpretation of interaction between arbor rhizobacteria and fruit medicinal ingredients is limited, it cannot be ignored, which can provide a possibility for understanding the complex ecological characteristics of C. migao and developing C. migao medicinal resources.
In summary, this study revealed the complex correlation of rhizobacteria with bioactive ingredients and soil properties, which provides us with an opportunity to improve and enhance the quality of C. migao from a microbial perspective.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
BT, XY, DW, XT, and XX: conceptualization. LL, BT, and XY: methodology. BT and JC: validation. LL and BT: formal analysis. BT, DW, and JC: investigation. LL: data curation and writing—original draft preparation. LL and XY: writing—review and editing. JL and SW: visualization. JL: supervision. XY and JL: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
The Guizhou Provincial Key Technology R&D Program of China (Qiankehezhicheng[2021]yiban143); National Natural Science Foundation of China (NSFC, 32171511); National Natural Science Foundation of China (NSFC, 31800335).
Acknowledgments
We would like to thank the Guizhou Provincial Key Technology R&D Program of China (Qiankehezhicheng[2021]yiban143) for support. We were also supported by awards from the National Natural Science Foundation of China (NSFC, grant nos. 31800335 and 32171511) to LL.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
OM, organic matter; TN, total nitrogen; AN, alkali hydrolyzed nitrogen; TP, total phosphorus; AP, available phosphorus; TK, total potassium, and available potassium (AK).
Footnotes
References
Andreote, F. D., Carneiro, R. T., Salles, J. F., Marcon, J., Labate, C. A., Azevedo, J. L., et al. (2009). Culture-independent assessment of rhizobiales-related alphaproteobacteria and the diversity of methylobacterium in the rhizosphere and rhizoplane of transgenic eucalyptus. Microb. Ecol. 57, 82–93. doi: 10.1007/s00248-008-9405-8
Arora, M., Saxena, P., Choudhary, D. K., Abdin, M. Z., and Varma, A. (2016). Dual symbiosis between Piriformospora indica and Azotobacter chroococcum enhances the artemisinin content in Arteuaisia annua L. World J. Microbiol. Biotech 32, 1–10. doi: 10.1007/s11274-015-1972-5
Aruna, C. R., Ratnavathi, C. V., Suguna, M., Ranga, B., Praveen Kumar, P., Annapurna, A., et al. (2020). Genetic variability and gxe interactions for total polyphenol content and antioxidant activity in white and red sorghums (sorghum bicolor). Plant Breed. 139, 119–130. doi: 10.1111/pbr.12756
Beckers, B., op de Beeck, M., Weyens, N., Boerjan, W., and Vangronsveld, J. (2017). Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome. 5:25. doi: 10.1186/s40168-017-0241-2
Berendsen, R. L., Pieterse, C., and Bakker, P. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., and Costello, E. K. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chaparro, J. M., Sheflin, A. M., Mantes, D. K., and Vivanco, J. M. (2012). Manipulating the soil microbiome to increase soil health and plant fertility. Biol. Fertil. Soils 48, 489–499. doi: 10.1007/s00374-012-0691-4
Chapin, F. S., Matson, P. A., and Mooney, H. A. (2011). Principles of terrestrial ecosystem ecology. Berlin, Germany: Springer.
Chen, J. Z., Huang, X. L., Tong, B. L., Wang, D., Liu, J. M., Liao, X. F., et al. (2021). Effects of rhizosphere fungi on the chemical composition of fruits of the medicinal plant Cinnamomum migao endemic to southwestern China. BMC Microbiol. 21:206. doi: 10.1186/s12866-021-02216-z
Chinese Pharmacopoeia Commission. (2020). Chinese pharmacopoeia, Beijing: China Medical Science and Technology Press.
Chowdhury, S. P., Hartmann, A., Gao, X., and Borriss, R. (2015). Biocontrol mechanism by root-associated bacillus amyloliquefaciens FZB42. Front. Microbiol. 6:780. doi: 10.3389/fmicb.2015.00780
Cipriano, M. A. P., Lupatini, M., Lopes-Santos, L., da Silva, M. J., Roesch, L. F. W., Destéfano, S. A. L., et al. (2016). Lettuce and rhizosphere microbiome responses to growth promoting pseudomonas species under field conditions. FEMS Microbiol. Ecol. 92:197. doi: 10.1093/femsec/fiw197
Collier, J. (2016). Cell cycle control in Alphaproteobacteria. Curr. Opin. Microbiol. 30, 107–113. doi: 10.1016/j.mib.2016.01.010
Dai, W., Xiao, S. P., Guo, K., Wang, J. Y., and Zhao, R. H. (2013). GC determined the content of 1,8-cineolein in seedling medicine Cinnamomum migao. Modern Chinese Medicine. 9, 1673–4890. doi: 10.13313/j.issn
Daims, H., Lebedeva, E. V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., et al. (2015). Complete nitrification by Nitrospira bacteria. Nature 528, 504–509. doi: 10.1038/nature16461
Daims, H., and Wagner, M. (2018). Nitrospira. Trends Microbiol. 26, 462–463. doi: 10.1016/j.tim.2018.02.001
Dang, H., Zhang, T., Li, G., Mu, Y., and Zhuang, L. (2020). Root-associated endophytic bacterial community composition and structure of three medicinal licorices and their changes with the growing year. BMC Microbiol. 20:291. doi: 10.1186/s12866-020-01977-3
de Carvalho, T. S., da Conceicao-Jesus, E., Barlow, J., Gardner, T. A., Soaves, I. C., Tiedje, J. M., et al. (2016). Land use intensification in the humid tropics increased both alpha and beta diversity of soil bacteria. Ecology 97, 2760–2771. doi: 10.1002/ecy.1513
Delgado-Baquerizo, M., Reith, F., Dennis, P. G., Llamonts, K., Powell, J. R., Young, A., et al. (2018). Ecological drivers of soil microbial diversity and soil biological networks in the southern hemisphere. Ecology 99, 583–596. doi: 10.1002/ecy.2137
Dimitriu, P. A., and Grayston, S. J. (2010). Relationship between soil properties and patterns of bacterial β-diversity across reclaimed and natural boreal forest soils. Microb. Ecol. 59, 563–573. doi: 10.1007/s00248-009-9590-0
Du, C., Geng, Z. C., Wang, Q., Zhang, T., He, W. X., Hou, L., et al. (2017). Variations in bacterial and fungal communities through soil depth profiles in a Betula albosinensis forest. J. Microbiol. 55, 684–693. doi: 10.1007/s12275-017-6466-8
Dunbar, J., Barns, S. M., Ticknor, L. O., and Kuske, C. R. (2002). Empirical and theoretical bacterial diversity in four Arizona soils. Appl. Environ. Microbiol. 68, 3035–3045. doi: 10.1128/AEM.68.6.3035-3045.2002
Falagán, C., and Johnson, B. D. (2014). Acidibacter ferrireducens gen. Nov., sp. nov.: an acidophilic ferric iron-reducing gammaproteobacterium. Extremophiles 18, 1067–1073. doi: 10.1007/s00792-014-0684-3
Fierer, N., McCain, C. M., Meir, P., Zimmermann, M., Rapp, J. M., Silman, M. R., et al. (2011). Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92, 797–804. doi: 10.1890/10-1170.1
Gao, L., Liu, X. M., Du, Y. M., Zong, H., and Shen, G. M. (2019). Effects of tobacco–peanut relay intercropping on soil bacteria community structure. Ann. Microbiol. 69, 1531–1536. doi: 10.1007/s13213-019-01537-9
Gebhardt, M., Fehmi, J. S., Rasmussen, C., and Gallery, R. E. (2017). Soil amendments alter plant biomass and soil microbial activity in a semi-desert grassland. Plant Soil 419, 53–70. doi: 10.1007/s11104-017-3327-5
Glyan Ko, A. K. (2015). Signaling systems of rhizobia (rhizobiaceae) and leguminous plants (fabaceae) upon the formation of a legume-rhizobium symbiosis (review). Applied Biochemistry & Microbiology. 51, 494–504. doi: 10.1134/S0003683815050063
Haney, C. H., Samuel, B. S., Bush, J., and Ausubel, F. M. (2015). Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nature Plants. 1:15051. doi: 10.1038/nplants.2015.51
Hao, D. C., Song, S. M., Mu, J., Hu, W. L., and Xiao, P. G. (2016). Unearthing microbial diversity of Taxus rhizosphere via MiSeq high-throughput amplicon sequencing and isolate characterization. Sci. Rep. 6:22006. doi: 10.1038/srep22006
Huang, Y. H., Liu, Y., Geng, J., Lü, H., Zhao, H. M., and Xiang, L. (2022). Maize root-associated niches determine the response variation in bacterial community assembly and function to phthalate pollution. J. Hazard. Mater. 429:128280. doi: 10.1016/j.jhazmat.2022.128280
Huang, W., Long, C., and Lam, E. (2018). Roles of plant-associated microbiota in traditional herbal medicine. Trends Plant Sci. 23, 559–562. doi: 10.1016/j.tplants.2018.05.003
Jiang, L. C. (2021). Study on key soil factors of active component accumulation of Scutellaria baicalensis. Yanan: Yan’ an University.
Jiang, J., Song, Z., Yang, X., Mao, Z., Nie, X., Guo, H., et al. (2017). Microbial community analysis of apple rhizosphere around bohai gulf. Sci. Rep. 7:8918. doi: 10.1038/s41598-017-08398-9
Jiao, S., Chen, W., Wang, J. L., Du, N. N., Li, Q. P., and Wei, G. H. (2018). Soil microbiomes with distinct assemblies through vertical soil profiles drive the cycling of multiple nutrients in reforested ecosystems. Microbiome 6:146. doi: 10.1186/s40168-018-0526-0
Jones, R. T., Robeson, M. S., Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. Int. Society Microb. Ecol. J. 3, 442–453. doi: 10.1038/ismej.2008.127
Kloepper, J. W., and Schroth, M. N. (1978). Plant growth promoting rhizobacteria on radishes. In: Proceedings of VIth International Conference on Plant Pathogenic Bacteria, Angres, France. 2, 879–882.
Köberl, M., Schmidt, R., Ramadan, E. M., Rudolf, B., and Gabriele, B. (2013). The microbiome of medicinal plants: diversity and importance for plant growth, quality and health. Front. Microbiol. 4:400. doi: 10.3389/fmicb.2013.00400
Larry, I. I., Dai, Z., Decker, S. R., Brunecky, R., Adney, W. S., and Ding, S. Y. (2008). Heterologous expression of glycosyl hydrolases in planta: a new departure for biofuels. Trends Biotechnol. 26, 413–424. doi: 10.1016/j.tibtech.2008.05.002
Li, L. X., Liu, J. M., Huang, X. L., Luo, C., Xiong, X., Liu, J. J., et al. (2018). Genetic diversity of Cinnamomum migao populations using ISSR markers. J. Northwest A&F University 46, 08–15. doi: 10.13207/j.cnki.jnwafu.2018.07.003
Li, Y. M., Liu, Y., Zhang, H., Yang, Y., Wei, G. H., and Li, Z. F. (2021). The composition of root-associated bacteria and fungi of Astragalus mongholicus and their relationship with the bioactive ingredients. Front. Microbiol. 12, 642–730. doi: 10.3389/fmicb.2021.642730
Li, X. Z., Rui, J. P., Mao, Y. J., Yannarell, A., and Mackie, R. (2014). Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol. Biochem. 68, 392–401. doi: 10.1016/j.soilbio.2013.10.017
Li, D. F., Voigt, T. B., and Kent, A. D. (2016). Plant and soil effects on bacterial communities associated with Miscanthus×giganteus rhizosphere and rhizomes. Glob. Change Biol. Bioenergy. 8, 183–193. doi: 10.1111/gcbb.12252
Liu, X., Zhang, S., Jiang, Q., Bai, Y., Shen, G., Li, S., et al. (2016). Using community analysis to explore bacterial indicators for disease suppression of tobacco bacterial wilt. Sci. Rep. 6:36773. doi: 10.1038/srep36773
Lugtenberg, B., and Kamilova, F. (2009). Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 63, 541–556. doi: 10.1146/annurev.micro.62.081307.162918
Luis, D. A., Mariana, P., Hugo, R. B., Ana, E. D., John, L. B., and Mario, A. A. (2018). Marchantia liverworts as a proxy to plants’ basal microbiomes. Sci. Rep. 8:12712. doi: 10.1038/s41598-018-31168-0
Maestre, F. T., Delgado-Baquerizo, M., Jeffries, T., Eldridge, D. J., and Singh, B. K. (2015). Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proceedings of the National Academy of Sciences of the United States of America (PNAS). 51, 15684–15689.
Manero, F. J. G., Algar, E., Gomez, M. S. M., Sierra, M. D. S., and Solano, B. R. (2012). Elicitation of secondary metabolism in Hypericum perforatum by rhizosphere bacteria and derived elicitors in seedlings and shoot cultures. Pharm. Biol. 50, 1201–1209. doi: 10.3109/13880209.2012.664150
Marschner, P., Yang, C. H., Lieberei, R., and Crowley, D. E. (2001). Soil and plant specific effects on bacterial community composition in the rhizosphere. Soil Biol. Biochem. 33, 1437–1445. doi: 10.1016/S0038-0717(01)00052-9
Nevita, T., Sharma, G. D., and Pandey, P. (2018). Differences in rice rhizosphere bacterial community structure by application of lignocellulolytic plant-probiotic bacteria with rapid composting traits. Ecol. Eng. 120, 209–221. doi: 10.1016/j.ecoleng.2018.06.007
Ni, Y. B. (2019). Comparative study on soil microbes and medicinal components in rhizosphere of Rumex japonicas Houtt under different soil conditions. Changchun: Jilin University.
Nie, H., Qin, T., Yan, D., Lv, X., and Liu, F. (2020). How do tree species characteristics affect the bacterial community structure of subtropical natural mixed forests? Sci. Total Environ. 764:144633. doi: 10.1016/j.scitotenv.2020.144633
Onrubia, M., Moyano, E., Bonfill, M., Cusidó, R. M., Goossens, A., and Palazóna, J. (2013). Coronatine, a more powerful elicitor for inducing taxane biosynthesis in taxes media cell cultures than methyl jasmonate. Plant Physiol. 170, 211–219. doi: 10.1016/j.jplph.2012.09.004
Philippot, L., Raaijmakers, J. M., Lemanceau, P., and van der Putten, W. H. (2013). Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. doi: 10.1038/nrmicro3109
Pieterse, C. M., de Jonge, R., and Berendsen, R. L. (2016). The soil-borne supremacy. Trends Plant Sci. 21, 171–173. doi: 10.1016/j.tplants.2016.01.018
Pieterse, C. M., Zamioudis, C., Berendsen, R. L., Weller, D. M., Van Wees, S. C., and Bakker, P. A. (2014). Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 52, 347–375. doi: 10.1146/annurev-phyto-082712-102340
Podile, A. R., and Kishore, G. K. (2006). “Plant growth promoting rhizobacteria” in Plant-associated bacteria. ed. S. S. Gnanamanickam (Amsterdam: Springer), 195–230.
Qu, Q., Zhang, Z. Y., Peijnenburg, W. J. G. M., Liu, W. Y., Lu, T., Hu, B. L., et al. (2020). Rhizosphere microbiome assembly and its impact on plant growth. J. Agric. Food Chem. 68, 5024–5038. doi: 10.1021/acs.jafc.0c00073
Romaniuk, R., Giuffre, L., Costantini, A., and Nannipieri, P. (2011). Assessment of soil microbial diversity measurements as indicators of soil functioning in organic and conventional horticulture systems. Ecol. Indic. 11, 1345–1353. doi: 10.1016/j.ecolind.2011.02.008
Rousk, J., Baath, E., Brookes, P. C., Lauber, C. L., Lozupone, C., Caporaso, J. G., et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. Int. Society Microb. Ecol. 4, 1340–1351. doi: 10.1038/ismej.2010.58
Schmidt, R., Koberl, M., Mostafa, A., Ramadan, E. M., Monschein, M., Jensen, K. B., et al. (2014). Effects of bacterial inoculants on the indigenous microbiome and secondary metabolites of chamomile plants. Front. Microbio. 5:64. doi: 10.3389/fmicb.2014.00064
Scola, V., Ramond, J., Frossard, A., Zablocki, O., Adriaenssens, E. M., Johnson, R. M., et al. (2018). Namib desert soil microbial community diversity, assembly, and function along a natural xeric gradient. Microb. Ecol. 75, 193–203. doi: 10.1007/s00248-017-1009-8
Shen, C. C., Xiong, J. B., Zhang, H. Y., Feng, Y. Z., Lin, X. G., Li, X. Y., et al. (2013). Soil ph drives the spatial distribution of bacterial communities along elevation on changbai mountain. Soil Biol. Biochem. 57, 204–211. doi: 10.1016/j.soilbio.2012.07.013
Solaiman, Z. M., and Anawar, H. M. (2014). Rhizosphere microbes interactions in medicinal plants. Plant-Growth-Promoting Rhizobacteria (PGPR) and Medicinal Plants. 42, 19–41. doi: 10.1007/978-3-319-13401-7
Sun, H. Q., Han, B. K., Yang, X. L., He, C. F., Zhao, K., Wang, T., et al. (2022). Correlates of rhizosphere soil properties, fungal community composition, and active secondary metabolites in cornus officinalis in different regions of China. J. Soil Sci. Plant Nutr. doi: 10.1007/s42729-022-01062-w
Taghinasab, M., and Jabaji, S. (2020). Cannabis microbiome and the role of endophytes in modulating the production of secondary metabolites: An overview. Microorganisms 8:355. doi: 10.3390/microorganisms8030355
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T. M., and Singh, B. K. (2020). Plant-microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621. doi: 10.1038/s41579-020-0412-1
Vives-Peris, V., de Ollas, C., Gomez-Cadenas, A., and Perez-Clemente, R. M. (2020). Root exudates: from plant to rhizosphere and beyond. Plant Cell Rep. 39, 3–17. doi: 10.1007/s00299-019-02447-5
Wang, W. Q., Cheng, X. Y., Song, Y. Y., Wang, H. M., Wu, M. X. J., Ma, L. Y., et al. (2022). Elevated antimony concentration stimulates rare taxa of potential autotrophic bacteria in the Xikuangshan groundwater. Science of The Total Environment 864:161105. doi: 10.1016/j.scitotenv.2022.161105
Wang, R. Q., Xiao, Y. P., Lv, F. J., Hu, L. Y., Wei, L. G., Yuan, Z. Q., et al. (2017). Bacterial community structure and functional potential of rhizosphere soils as influenced by nitrogen addition and bacterial wilt disease under continuous sesame cropping. Appl. Soil Ecol. 125, 117–127. doi: 10.1016/j.apsoil.2017.12.014
Wang, S., Zuo, X., Awada, T., and Medima-Roldán, E.,Cheng, H. (2021). Changes of soil bacterial and fungal community structure along a natural aridity gradient in desert grassland ecosystems, Inner Mongolia. Catena. 205,:105470. doi: 10.1016/j.catena.2021.105470
Williams, K. P., Sobral, B. W., and Dickerman, A. W. (2007). A robust species tree for the Alphaproteobacteria. J. Bacteriol. 189, 4578–4586. doi: 10.1128/JB.00269-07
Wu, Y., Wu, J., Tan, H., Song, Q., and Liu, X. (2020). Distributions of chlorinated paraffins and the effects on soil microbial community structure in a production plant brownfield site. Environ. Pollut. 262:114328. doi: 10.1016/j.envpol.2020.114328
Xiao, X., Fan, M., Wang, E., Chen, W., and Wei, G. (2017). Interactions of plant growth-promoting rhizobacteria and soil factors in two leguminous plants. Applied Microbiol. Biotechnol. 101, 8485–8497. doi: 10.1007/s00253-017-8550-8
Xie, Z. C., Chu, Y. K., Zhang, W. J., Lang, D. Y., and Zhang, X. H. (2019). Bacillus pumilus alleviates drought stress and increases metabolite accumulation in Glycyrrhiza uralensis Fisch. Environ. Exp. Bot. 158, 99–106. doi: 10.1016/j.envexpbot.2018.11.021
Xiong, W. J., Zhu, H., Li, J. B., Xie, T., Wu, Y. H., and Li, X. Z. (2021). Spatiotemporal distribution patterns and drivers of bacterial communities in the rhizosphere and bulk soil under an Abies fabri forest on Gongga Mountain. Chin J Appl Environ Biol 27, 1130–1137. doi: 10.19675/j.cnki.1006-687x.2021.02037
Yang, K. (2019). Effects of phosphorus addition on soil carbon, nitrogen and phosphorus stoichiometry and microbial diversity in Cunninghamia Lanceolata plantations. Changsha: Central South University of Forestry and Technology.
Yang, Y. Y., Raphael, A., Rossel, V., Li, S., Bissett, A., Lee, J., et al. (2019). Soil bacterial abundance and diversity better explained and predicted with spectro-transfer functions. Soil Biol. Biochem. 129, 29–38. doi: 10.1016/j.soilbio.2018.11.005
Zamioudis, C., Mastranesti, P., Dhonukshe, P., Blilou, I., and Pieterse, C. M. (2013). Unravelling root developmental programs initiated by beneficial pseudomonas spp. bacteria. Plant Physiol. 162, 304–318. doi: 10.1104/pp.112.212597
Zeng, M. J., Zhong, Y. J., Cai, S. J., and Diao, Y. (2018). Deciphering the bacterial composition in the rhizosphere of Baphicacanthus cusia (NeeS) Bremek. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-34177-1
Zhang, X. (2019). Stoichiometric characteristics and soil microbial diversity of Robinia pseudoacacia sprout forests. Shandong: Shandong Agricultural University.
Zhang, J. Y., Liu, P., Nie, B. T., Liu, X. X., Zhang, Z., He, R. L., et al. (2022). Effects of genotype and ecological environment on the community structure and function of symbiotic bacteria in rhizosphere of ginseng. BMC Microbiolog. 22:235. doi: 10.1186/s12866-022-02649-0
Zhang, Y., Zheng, L., Zheng, Y., Xue, S., and Li, C. (2020). Insight into the assembly of root-associated microbiome in the medicinal plant Polygonum cuspidatum. Ind. Crop. Prod. 145:112163. doi: 10.1016/j.indcrop.2020.112163
Keywords: Cinnamomum migao, soil rhizobacteria, soil chemical properties, medicinal bioactive ingredients, redundancy analysis
Citation: Li L, Yang X, Tong B, Wang D, Tian X, Liu J, Chen J, Xiao X and Wang S (2023) Rhizobacterial compositions and their relationships with soil properties and medicinal bioactive ingredients in Cinnamomum migao. Front. Microbiol. 14:1078886. doi: 10.3389/fmicb.2023.1078886
Edited by:
Xiancan Zhu, Anhui Normal University, ChinaReviewed by:
Ni Luh Suriani, Udayana University, IndonesiaArchna Suman, Indian Agricultural Research Institute (ICAR), India
Copyright © 2023 Li, Yang, Tong, Wang, Tian, Liu, Chen, Xiao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiming Liu, ✉ a2Fyc3QwNjIzQDE2My5jb20=