 Loic Deblais1†
Loic Deblais1† Hyein Jang2†
Hyein Jang2† Mike Kauffman1
Mike Kauffman1 Jayanthi Gangiredla2Marianne Sawyer2Saritha Basa2Jelmer W. Poelstra3
Jayanthi Gangiredla2Marianne Sawyer2Saritha Basa2Jelmer W. Poelstra3 Uma S. Babu2
Uma S. Babu2 Lisa M. Harrison2
Lisa M. Harrison2 Kelli L. Hiett2
Kelli L. Hiett2 Kannan V. Balan2
Kannan V. Balan2 Gireesh Rajashekara1*
Gireesh Rajashekara1*- 1Department of Animal Sciences, Center for Food Animal Health, The Ohio Agricultural Research and Development Center, The Ohio State University, Wooster, OH, United States
- 2Center for Food Safety and Applied Nutrition (CFSAN), Office of Applied Research and Safety Assessment (OARSA), U.S. Food and Drug Administration, Laurel, MD, United States
- 3Molecular and Cellular Imaging Center, The Ohio Agricultural Research and Development Center, The Ohio State University, Wooster, OH, United States
Introduction: With more public interest in consuming locally grown produce, small specialty crop farms (SSCF) are a viable and growing segment of the food production chain in the United States.
Methods: The goal of this study was to investigate the genomic diversity of Campylobacter isolated from dairy manure (n = 69) collected from 10 SSCF in Northeast Ohio between 2018 and 2020.
Results: A total of 56 C. jejuni and 13 C. coli isolates were sequenced. Multi-locus sequence typing (MLST) identified 22 sequence types (STs), with ST-922 (18%) and ST-61 (13%) predominant in C. jejuni and ST-829 (62%) and ST-1068 (38%) predominant in C. coli. Interestingly, isolates with similar genomic and gene contents were detected within and between SSCF over time, suggesting that Campylobacter could be transmitted between farms and may persist in a given SSCF over time. Virulence-associated genes (n = 35) involved in the uptake and utilization of potassium and organic compounds (succinate, gluconate, oxoglutarate, and malate) were detected only in the C. jejuni isolates, while 45 genes associated with increased resistance to environmental stresses (capsule production, cell envelope integrity, and iron uptake) were detected only in the C. coli isolates. Campylobacter coli isolates were also sub-divided into two distinct clusters based on the presence of unique prophages (n = 21) or IncQ conjugative plasmid/type-IV secretion system genes (n = 15). Campylobacter coli isolates harbored genes associated with resistance to streptomycin (aadE-Cc; 54%) and quinolone (gyrA-T86I; 77%), while C. jejuni had resistance genes for kanamycin (aph3’-IIIa; 20%). Both species harbored resistance genes associated with β-lactam (especially, blaOXA-193; up to 100%) and tetracycline (tetO; up to 59%).
Discussion/Conclusion: Our study demonstrated that Campylobacter genome plasticity associated with conjugative transfer might provide resistance to certain antimicrobials and viral infections via the acquisition of protein-encoding genes involved in mechanisms such as ribosomal protection and capsule modification.
Introduction
Campylobacter is a leading cause of bacterial foodborne gastroenteritis worldwide and is a major public health problem (Moffatt et al., 2021; Mortensen et al., 2021). It has been estimated that 2.4 million people are affected by Campylobacter in the United States annually, causing a $1.3 billion deficit in medical care (Zhou et al., 2022). Campylobacteriosis is associated with abdominal pain, fever, and bloody diarrhea, but can also lead to Guillain-Barre syndrome, Miller Fisher syndrome, and reactive arthritis (Kassem et al., 2016; Scallan Walter et al., 2020). Thermophilic Campylobacter (especially, C. jejuni) are the leading cause of campylobacteriosis cases (90%; Kassem et al., 2016), and can be frequently detected in livestock and livestock-based products (i.e., beef and poultry; Buzby et al., 1997; An et al., 2018; Halpin et al., 2018; Thépault et al., 2018; Scharff, 2020; Heimesaat et al., 2021).
Small specialty crop farms (SSCF) are a viable and growing segment of the food production chain in the United States (valued at $4.7 billion in the Midwest in 2012; Midwest Climate and Specialty Crops, n.d.). With society looking more towards locally grown produce as their source of fresh food, the farmers provide a direct and local connection between producer and consumer. However, these farms frequently practice mixed farming (animal and vegetable farming; Kim, 2016). Further, biological amendments produced by these farms are applied as a natural fertilizer for vegetable production. As a consequence, this practice may increase the risk that crops generated on SSCF are contaminated with foodborne pathogens present in livestock feces (Sharma and Reynnells, 2016; Ramos et al., 2019; Nutrition, 2021). In addition, antibiotics may be used to treat the animals, creating a risk for emergence of potential antimicrobial resistance (AMR) organisms and genes being transferred to the soil when manure is applied. To date, very little is known about the prevalence of foodborne pathogens in this specific agricultural niche and the impact of SSCF agricultural practices on these pathogens in terms of food safety and public health risks. Since 2016, our team has been working with SSCF in Northeast Ohio to understand agricultural practices used by SSCF and assess the impact of biological amendments on public health and food safety in Ohio (Hailu et al., 2021). We demonstrated that manure (dairy and poultry) collected from these farms between 2016 and 2020 harbored thermophilic bacteria such as Campylobacter spp. (8%), Listeria monocytogenes (7.9%), Escherichia coli O157 (1.8%), and Salmonella spp. (1.5%; Hailu et al., 2021). The majority of the Campylobacter isolates (57.3%) from this study possessed multiple drug resistance genes (especially, blaOXA-61, tetO, and aadE) based on conventional PCR analysis. Thus, our previous study, as well as studies conducted by other groups, have highlighted the potential public health and food safety risks associated with the use of biological amendments in SSCF (Ramos et al., 2019, 2021; Black et al., 2021; Hailu et al., 2021).
This study presents whole genome sequencing (WGS) analyses of Campylobacter spp. isolated from diary manure from a longitudinal study performed between 2018 and 2020 in 10 SSCF of Northeast Ohio. A total of 69 thermophilic Campylobacter isolates were recovered from the manure samples. Specifically, the goal of this study was to investigate the genomic diversity of Campylobacter isolated from dairy manure. We focused on the variable-genome, -resistome, and -virulome to determine how their predicted metabolic capabilities might contribute to their fitness in a specific niche. Correlation analyses between the whole genome sequence and metadata (farm location, sample collection, sequence type, and clonal complex) were performed to better understand the spatiotemporal dynamics of thermophilic Campylobacter in SSCF of Northeast Ohio.
Materials and methods
Farm selection and sample collection
A total of 10 small specialty crop farms (SSCF) in Northeast Ohio (United States) were selected for this study, based on their availability for longitudinal sampling throughout the study period (2018–2020; Hailu et al., 2021). Farms were asked to continue performing management activities, which followed normal and customary practices of producers utilizing limited mechanization. The spatial distribution of the sample collection sites is provided in Figure 1. Each farm cluster (FC; n = 5) was created based on the geographic proximity of the farms with each other. The dairy manure was obtained from open dairy heifers housed on a bedded pack during the winter and raised on pasture in the summer. Dairy manure was not treated by the farmers. Dairy manure samples were collected monthly across the 10 SSCF between 2018 and 2020 (n = 140 total samples). Some samples were missing due to events surrounding the COVID-19 pandemic. Samples were collected aseptically into Nasco Whirl-Pak™ bags (Fisher Scientific, Waltham, MA, United States) and stored on ice until processed immediately in the lab.

Figure 1. Multi-dimensional scaling of 10 SSCF positive for thermophilic Campylobacter. Dimensions 1 and 2 represent distance between farms in meters. Each dot represents a farm selected for the study. #, farm ID; FC, farm cluster.
Isolation of thermophilic Campylobacter from manure
Manure samples (25 g) were resuspended into 225 mL of phosphate buffered saline (PBS; Fisher Scientific, Waltham, MA, United States; 1:10 ratio), homogenized, serial diluted (10-fold), plated on RAPID’ Campylobacter (BioRad, Hercules, CA, United States), and then incubated at 42°C in microaerobic conditions (5% O2, 10% CO2, and 85% N2) for 48 h. Resuspended manure samples (1 mL) were also enriched for 48 h at 42°C in 9 mL of Preston enrichment broth containing Campylobacter growth supplements (product No. CM067, Lysed Horse Blood product No. SR0048, Preston Campylobacter Selective Supplement product No. SR0117 and Campylobacter Growth Supplement product No. SR0232; Oxoid Ltd., Cambridge, United Kingdom; 1:10 ratio), plated on RAPID’ Campylobacter, and incubated under microaerobic condition at 42°C for 48 h. Colonies growing on RAPID’ Campylobacter plates were sub-cultured onto a fresh modified charcoal cefoperazone deoxycholate agar (mCCDA; Fisher Scientific, Waltham, MA, United States) plated and incubated at 42°C for 48 h under microaerobic conditions. A Campylobacter genus-specific colony-PCR was performed on all the Campylobacter-suspected colonies, as described below. Isolates confirmed to be Campylobacter were frozen at −80°C in Brucella broth supplemented with 30% glycerol (v/v).
Campylobacter genus-specific colony PCR
Colony-PCR was performed on all isolates suspected to be Campylobacter using genus-specific primers (MD16S F: ATCTAATGGCTTAACCATTAAAC; C1228R: GGACGGTAACTAG TTTAGTATT; Final concentration = 0.11 μM; product size = 857 bp; Denis et al., 1999). Briefly, colonies were resuspended in 100 μL sterile DNase-free water and boiled at 95°C for 10 min. The product was centrifuged for 10 min at 4,000 ×g and 2 μL of the lysate was used for the colony-PCR (25 μL final volume; 25 cycles). The following program was used: one cycle of 10 min at 95°C, 35 cycles of 30s at 95°C, 1.5 min at 59°C, 1 min at 72°C, and a final extension step of 10 min at 72°C. PCR products were visualized using 2% agarose gels. Isolates confirmed as Campylobacter by PCR were selected for whole genome sequencing.
DNA extraction and whole genome sequencing and assembly
Genomic DNA from pure Campylobacter isolates (n = 69) was extracted using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, United States). The DNA was quantified using a Qubit dsDNA BR assay kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA, United States) and Qubit 3.0 fluorometer (Invitrogen). The extracted DNA samples were diluted in molecular biology grade nuclease-free deionized water (Thermo Fisher Scientific) to a final concentration of 0.2 ng/μL for whole genome sequencing (WGS). Genomic libraries were constructed using the Nextera XT DNA sample preparation kit as described in the manufacturer’s protocol (Illumina, San Diego, CA, United States) and sequenced on the MiSeq platform using 500 cycles of paired-end reads (Illumina). Quality control of the FASTQ datasets (raw reads) obtained from each sequence run was assessed using FastQC (Andrews, 2010). Low-quality reads < Q20 were trimmed and adaptor sequences were removed using the default parameter in Trimmomatic (Bolger et al., 2014). Trimmomatic modifications were conducted with a sliding window trimming option where the number of bases to average across was set to 4 and the average quality required was set to 20. The cleaned reads were subsequently de novo assembled using SKESA (Strategic K-mer Extension for Scrupulous Assemblies; Souvorov et al., 2018). Assembly qualities were also evaluated by QUAST (Quality Assessment Tool; Gurevich et al., 2013; Mikheenko et al., 2018). The analytical tools for FastQC, Trimmomatic, SKESA, and QUAST are available on the GalaxyTrakr platform (https://galaxytrakr.org, accessed 27 May 2022) which is an open-source bioinformatics platform maintained by the Center for Food Safety and Applied Nutrition (CFSAN) at the U.S. Food and Drug Administration (Gangiredla et al., 2021).
Multi-locus sequence typing: Traditional and core genome MLST
Sequence types (STs) of Campylobacter genomes investigated in this study were initially analyzed using the traditional seven-loci Multi-locus sequence typing (MLST) scheme based on the following housekeeping genes (aspA, glnA, gltA, glyA, pgm, tkt, and uncA; Dingle et al., 2001). Briefly, the genome assemblies were scanned against the PubMLST schemes where the ST of each genome was assigned by comparing the alleles of the seven genes in the MLST open database (Jolley and Maiden, 2010; Seemann, 2022b).
To cover the expanded resolution of the MLST concept, a core genome MLST (cgMLST) profiling against the PubMLST database (Jolley et al., 2018) was performed using a modified Seemann’s MLST tool which used contig files to scan against the Oxford C. jejuni and C. coli cgMLST scheme (Mulder et al., 2020). The cgMLST scheme describes the genetic variation among the strains by utilizing the allele sequences of 1,343 loci which were defined from genome sequences of 2,472 representative United Kingdom campylobacteriosis isolates including C. jejuni and C. coli (Cody et al., 2017). The traditional MLST tool was used on the GalaxyTrakr platform (accessed 28 April 2022) while the cgMLST tool was accessed (28 April and 16 December 2022) from an open-source platform1 to query the genomes against the current C. jejuni and C. coli scheme. The closest ST match for these isolates were assigned using the cgMLST scheme on the PubMLST Campylobacter jejuni/coli typing database2 (Cody et al., 2017; Jolley et al., 2018).
Taxonomic classification and annotation
To identify the isolates at the genus and species level, the sequence data were analyzed by Kraken 2, which is a k-mer-based taxonomic sequence classifier using k-mers of 35 bp (Wood and Salzberg, 2014; Wood et al., 2019). The Kraken 2 algorithm assigns taxonomic labels by matching each k-mer within a query sequence to the lowest common ancestor (LCA) of the pre-computed database of genomes containing the given k-mer (Wood et al., 2019). In addition, the genomes were annotated by uploading the assemblies of FASTA datasets through the National Center for Biotechnology Information (NCBI) prokaryotic genome annotation pipeline (PGAP) with its best-placed reference protein set GeneMarkS+ application (Haft et al., 2018; Li et al., 2021). Assembled genomes were also annotated using RAST (Aziz et al., 2008; Overbeek et al., 2014) to provide an overall picture of the gene content across the Campylobacter isolates.
Identification of antimicrobial resistance and virulence genes based on WGS
The predicted antimicrobial resistance (AMR) genes were identified using the National Antimicrobial Resistance Monitoring System (NARMS) Campylobacter workflow on GalaxyTrakr which uses BLAST techniques against the NCBI’s comprehensive AMR gene database (Feldgarden et al., 2019a,b, 2021). The AMRFinderPlus tool accessed from the GalaxyTrakr platform was used to detect acquired AMR genes in bacterial proteins or assembled nucleotide sequences, along with point mutations that were cross-referenced with a core set of AMR elements and the expanded subset of reference genes related to biocide, stress response, and virulence factors (Feldgarden et al., 2021). The AMRFinderPlus tool was designed to utilize the Reference Gene Catalog database of NCBI Pathogen Detection, which consists of 6,428 genes (5,588 AMR genes, 210 stress response genes, and 630 virulence genes), 627 hidden Mark models (HMMs), and 682 point-mutations (Feldgarden et al., 2021). Virulence gene profiles of Campylobacter isolates were determined by BLASTN comparison against the virulence factor database (VFDB) with the threshold of 80% identity and 80% sequence coverage (Chen et al., 2016; Liu et al., 2019) by using an ABRicate tool which performed mass screening of contigs for antimicrobial resistance and virulence genes (Seemann, 2022a).
Phylogenetic analysis based on single nucleotide polymorphism
To understand relatedness among the strains, the Campylobacter genome assemblies were analyzed using the CFSAN SNP Pipeline (Davis et al., 2015) which is accessible on the GalaxyTrakr platform. The single nucleotide polymorphism (SNP) pipeline performs the alignment of mapped reads of sequences to a reference genome, which creates high-quality SNP matrices for sequences and determines phylogenetic relatedness among the strains (Davis et al., 2015). The SNP analysis was conducted against the reference genome chosen by the SNP pipeline, and a reference strain was chosen based on the best assembly metrics from the quast report, such as the one with the longest N50. For this analysis, sample #112962 (Biosample accession # SAMN29955749) was selected as a reference to build the tree. The phylogenetic tree was constructed using the Neighbor-Joining method (Saitou and Nei, 1987). The evolutionary analyses were employed using MEGA7 software (Kumar et al., 2016, p. 7), with evolutionary distances computed using the Maximum Composite Likelihood method (Tamura et al., 2004).
Plasmid annotation
Assembled genomes were scanned for plasmids using PlasmidFinder v. 2.1.6 (Carattoli et al., 2014), which detects plasmids via replicons only, and MOB-Suite v. 3.1.0 (Robertson and Nash, 2018), which detects plasmids with a database containing replicons, relaxases, and complete plasmid sequences. PlasmidFinder was run using the plasmidfinder.py script, after downloading the database with the download-db.sh script (on 16 July 2022), with a minimum coverage threshold of 0.60 (the default) and an identity threshold of 0.95 (the default for the webserver, whereas the default for the Python script is 0.90). MOB-Suite was run via the “mob_recon” command, after running “mob_init” to download the databases (on 18 November 2022) with default parameters.
Statistical analyses
Genomic data were synchronized with the metadata (e.g., collection time point and location, and source) and analyzed using JMP Pro 16 software (SAS Institute Inc., Cary, NC, United States). The analyses described below focus essentially on the variable genomes (protein-encoded genes not consistently detected within all the C. jejuni or C. coli genomes) and not the core genome (protein-encoded genes consistently detected within all the C. jejuni or C. coli genomes). Multi-dimensional scaling analysis combined with K-means clustering were used to create a two-dimensional plot displaying the farm geographic distribution and associated clusters based on the GPS coordinates of the SSCF. Hierarchical clustering analysis was conducted to identify similar variable gene profiles among the 69 C. jejuni and C. coli isolates. Principal component (PCA) and agglomerative hierarchical clustering (HCA) analyses were used to study the distribution of the Campylobacter isolates based on their functional genomic profiles and cgMLST. Multi-correspondence analysis (MCA) combined with a Chi2 test were used to determine whether the prevalence of certain genes was influenced by the Campylobacter species, SSCF, and sample collection time point (e.g., year and month of collection). MCA and multivariate analyses were used to identify co-occurrence of specific genes for a given Campylobacter species. In addition, an association study (also called produce market analysis) was conducted to determine whether the prevalence of specific genes could be linked with specific geographic or temporal parameters associated with the isolates. A minimal confidence level of 95% and lift of 2 were used to identify associations of statistical importance.
Results
Thermophilic Campylobacter are frequently detected in dairy manure from Northeast Ohio
Out of the 140 dairy manure samples collected from the 10 SSCF between 2018 and 2020, 49% (n = 69) were positive for thermophilic Campylobacter (Table 1). Dairy manure collected in 2020 and 2019 harbored the highest prevalence (88% and 69%, respectively) compared to 2018 (18%; p < 0.001). On the other hand, dairy manure collected in 2020 harbored the lowest abundance of Campylobacter (2.92 [IC95%: 2.61–3.22]) log CFU/g compared to 2019 and 2018 (3.89 [IC95%: 3.64–4.13] and 3.28 [IC95%: 2.99–3.57] log CFU/g, respectively Table 1; p < 0.008). Equivalent Campylobacter prevalence (60%–100%) and load (2.76 [IC95%: 2.31–3.21] to 4.21 [IC95%: 3.82–4.60] log CFU/g) in the dairy manure were detected between farms and seasons (p > 0.01).

Table 1. Prevalence of thermophilic Campylobacter spp. in dairy manure from SSCF of Northeast Ohio.
Whole genome sequence of Campylobacter isolates
The whole genome analyses revealed 56 C. jejuni (genome length = 1,683,112 bp [IC95%: 1,664,247–1,701,977]) and 13 C. coli (genome length = 1,691,610 bp [IC95%: 1,680,103-1,703,117]) isolates (Supplementary Table 1). All associated metadata and genome quality statistics are included in Supplementary Table 2. Campylobacter jejuni was recovered across all 3 years of sampling, predominantly in 2019 (n = 33/56; Table 2). Most of the C. coli isolates were recovered in 2020 (n = 12/13; Table 2). Isolates were collected in the winter, spring, and summer seasons (n = 16–19 C. jejuni and 3–6 C. coli isolates per season), but rarely in the fall (n = 3/56 C. jejuni isolates and no C. coli isolates).
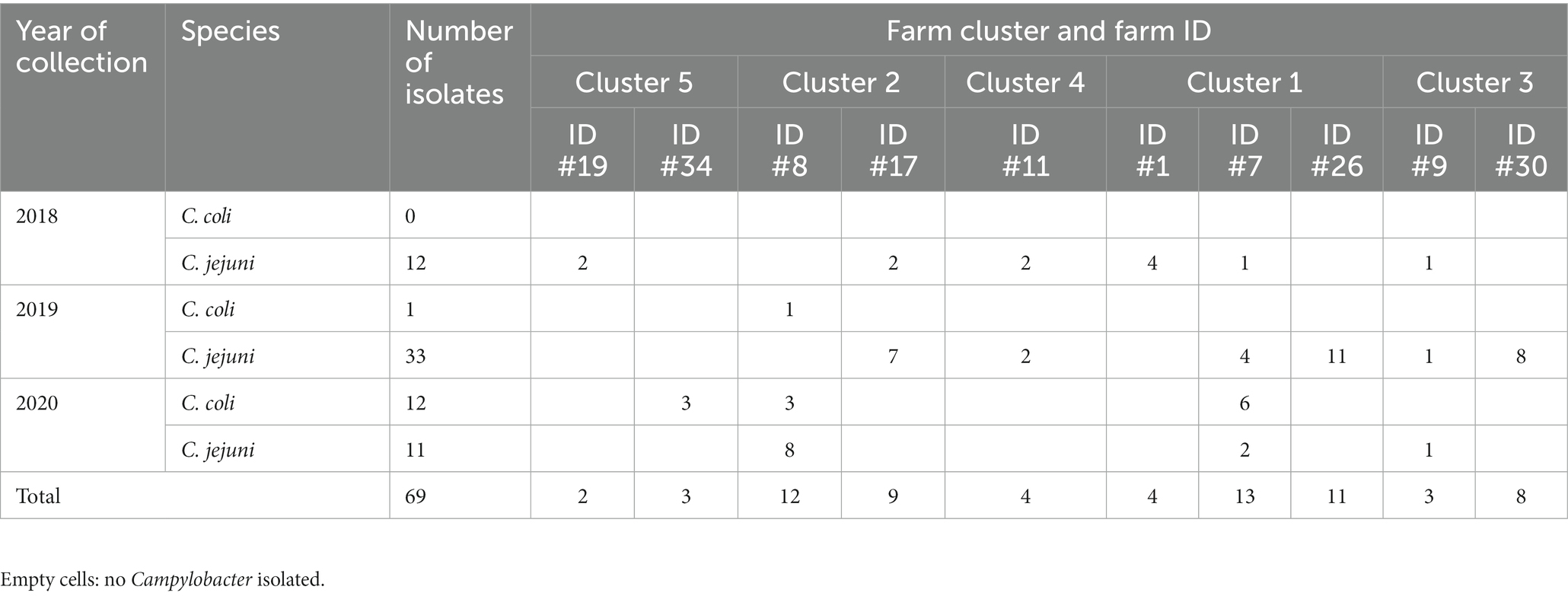
Table 2. Distribution of the sequenced thermophilic Campylobacter isolates based on farm ID, farm clusters and years of collection.
MLST analysis of the Campylobacter isolates
A total of 20 STs were identified among the 56 C. jejuni isolates, with ST-922 (n = 10) and ST-61 (n = 7) being the most predominant (Table 3). These 20 STs belonged to 10 clonal complexes (CCs), with CC-21 (n = 26) being the most predominant. The geographic location of the farms affected the type of ST and CC detected. The majority of the ST-61 (n = 6/7) were detected in farm #30 in 2019; ST-922 (n = 7/10) were detected in farm #26 in 2019 and ST-829 (n = 5/8) were detected in farm #7 in 2020. The spatiotemporal distribution of the CC and ST was studied across the 10 farms over 3 years. Similar CCs were detected in FC #1 (farm #1, 7 and 26) and FC #2 (farm #8 and 1 7; Supplementary Figure 1A), which is in concordance with the geographic proximity of both FC (Figure 1). However, FC #3, #4, and #5 harbored different CCs compared to FC #1 and #2 (Supplementary Figure 1A), which is also in agreement with the distant geographic location of the FCs between each other (Figure 1). Two STs (ST-829 [n = 8] and ST-1068 [n = 5]) were detected among the 13 C. coli isolates and belonged to CC-828. The number of STs and CCs (C. jejuni and C. coli combined) were equivalent across collection years (n = 9–12 and 5–7, respectively), but each year a different ST/CC profile was observed (Supplementary Figure 1B). No differences in ST and CC profiles were detected between seasons.
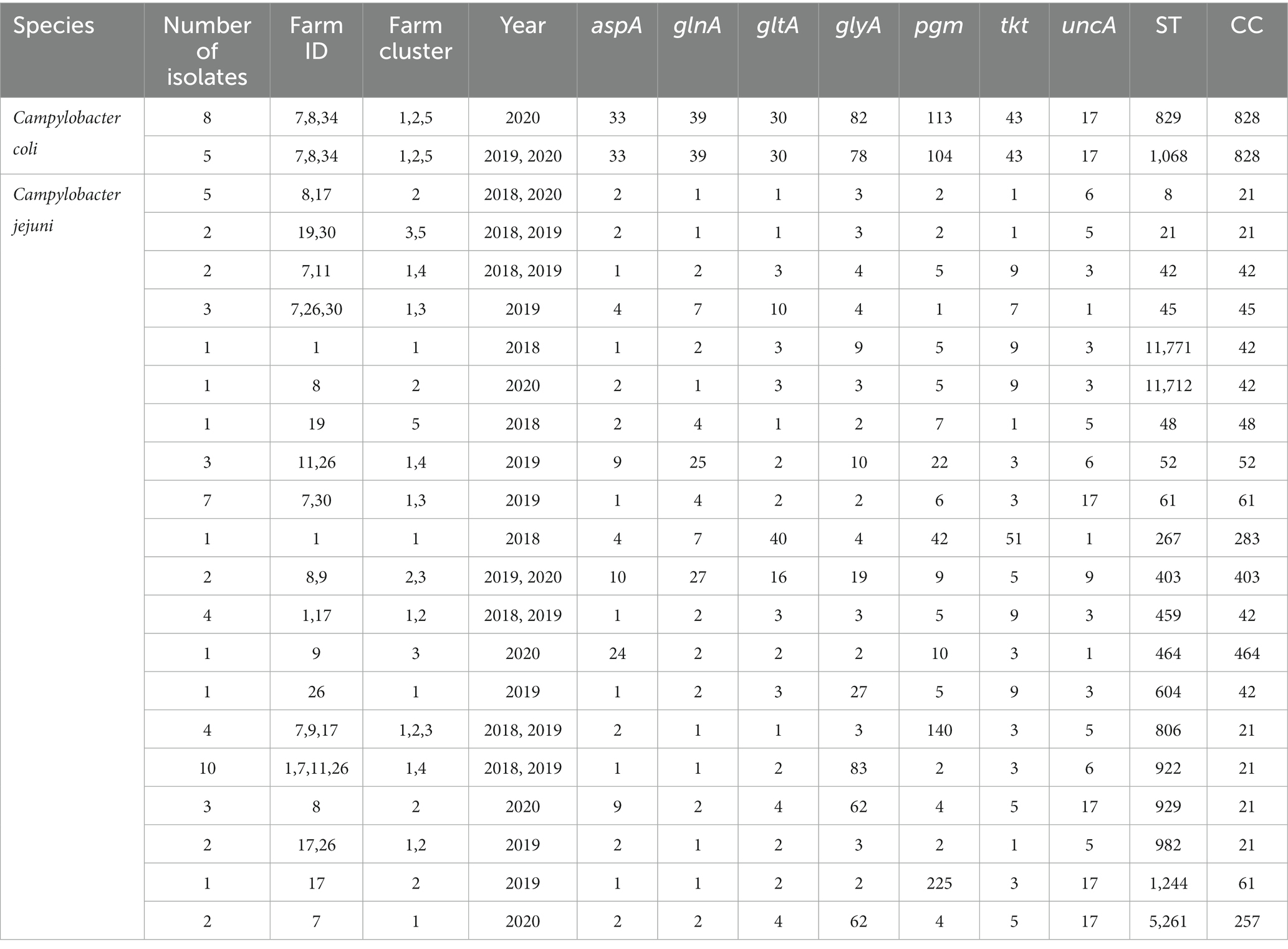
Table 3. Multi-locus sequence typing of thermophilic Campylobacter isolates using the traditional seven gene schema.
Potential associations between the spatial and temporal distributions of the Campylobacter isolates and their genomic profile
Distinct SNP signatures were detected between different Campylobacter species, as well as, between isolates of the same species (Figure 2). As expected, C. jejuni isolates segregated from the C. coli isolates. A total of six major C. jejuni clusters were observed based on the SNP profiles. The composition of these clusters was associated to some extent with the geographic location, and less with the time of collection (Figure 2). Most of the C. jejuni isolates collected from farm #30 (n = 6/8) clustered together and displayed identical SNP profiles (cluster 2). The majority of C. jejuni isolates collected from FC #1 (n = 16/21; farm #1, #7, and #26; Figure 1) clustered away from the other C. jejuni isolates (n = 31; cluster 4; Figure 2). Interestingly, FC #1 was mainly composed of C. jejuni isolated between the summers of 2018 and 2019, and harbored high SNP profile similarity with a C. jejuni isolated in early 2018 from farm #11. Farm #11 belongs to FC #4 which is in close proximity to FC #1 (Figure 1). High SNP profile similarities were also observed between the C. jejuni isolates collected between FC #1 and FC #2 (farm #8 and #17; cluster 3 and 5; Figure 2). A similar trend was observed with C. coli isolates between FC#1, FC#2 and FC#5 (C. coli cluster; Figure 2).
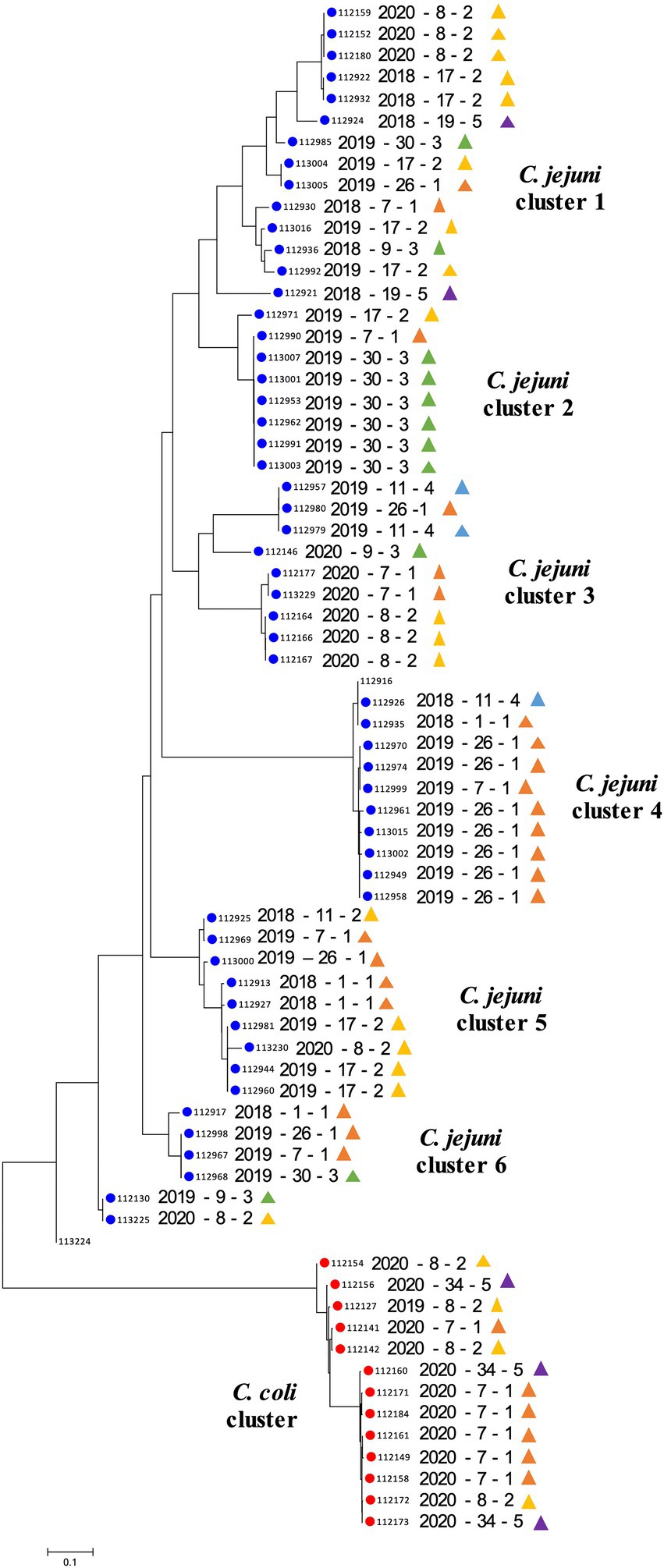
Figure 2. Phylogenetic diversity based on single nucleotide polymorphisms (SNPs) analysis. The phylogenetic tree was constructed using the Neighbor-Joining method. The optimal tree with the sum of branch length = 2.14 is shown. The tree was generated to scale with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The analysis involved 69 genomes. There was a total of 2,748 positions in the final dataset. The scale bar represents a 0.1 base substitution per site. Each row indicates a specific Campylobacter isolate (year of collection—farm ID—farm cluster). Blue and red circles indicate Campylobacter jejuni and Campylobacter coli, respectively. Colored triangles are associated with the farm cluster number; orange, yellow, green, blue and purple triangles indicate farm clusters 1 to 5, respectively (Figure 1).
The 69 Campylobacter isolates were also analyzed using core genome MLST (cgMLST). A total of 869/1,343 alleles were detected across the 69 isolates (Supplementary Figure 2). Overall, a distinct separation between C. jejuni and C. coli was observed. For the C. coli isolates, ST-829 isolates (n = 8) clustered away from the ST-1068 isolates (n = 5). Interestingly, C. jejuni CC-21 isolates were subdivided into four clusters; all C. jejuni ST-922 CC-21 isolates (n = 10) clustered away from the second (i.e., C. jejuni CC-21 ST-8 [n = 5]), third (i.e., C. jejuni CC-21 ST-929 [n = 3]) and fourth cluster (i.e., C. jejuni CC-21 ST-806 [n = 4] and ST-21 [n = 1]). The allelic profile of C. jejuni ST-922 CC-21 isolates was also closely similar to C. jejuni CC-403, CC-463 and CC-52. Similarly, other C. jejuni CC-21 (i.e., ST-806 and ST-982) were closely related to C. jejuni CC-48 and CC-61. Campylobacter jejuni ST-5261 CC-257 were closely related to of C. jejuni ST-929 CC-21 cluster, which itself displayed similarities with C. jejuni CC-42 isolates. Only C. jejuni ST-8 CC-21 and ST-45 CC-45 isolates stood apart from other C. jejuni isolates.
Campylobacter jejuni and Campylobacter coli isolates possess distinct gene content profiles
After annotating the genomes using RAST (general gene content profile), VFDB (virulome annotation) and NDARO (resistome annotation), a total of 1,784 annotated genes were detected across the 69 Campylobacter isolates. Among them, 35.7% (n = 637/1,784) of annotated genes were not always detected in the 69 Campylobacter isolates studied (referred to as variable genes). As observed with the SNP analysis (Figure 2), C. coli isolates displayed a different gene content profile compared to the C. jejuni isolates. Campylobacter jejuni isolates possessed several unique virulence-associated genes involved in the uptake of potassium, carbon utilization (e.g., succinate, gluconate, oxoglutarate and malate), and chemotaxis (n = 35; Supplementary Table 3). Similarly, C. coli isolates also had unique virulence genes associated with capsule production, cell envelope integrity, and membrane transporters, increasing resistance to environmental stresses (Supplementary Table 3). Campylobacter coli isolates also possessed unique sets of genes coding for enzymes (e.g., histidine kinase, acylamide amidohydrolase, methylcitrate dehydratase and synthase, adenine-specific DNA methyltransferase, adenylylsulfate kinase, L-carnitine dehydratase, and methionine synthase), transporters (e.g., creD, yihN, and yihY) essential for the utilization of specific energy sources, and proteins involved in iron availability and/or acquisition (e.g., ycsG, hemerythrin, hemolysin, and peroxide stress regulator/ferric uptake regulation protein; Supplementary Table 3).
Functional genomic analyses of Campylobacter jejuni demonstrated associations between the geographic location and sequence type of the isolates
A total of 401 protein-encoding genes were identified as part of the C. jejuni variable genome (all variable genes identified across the 56 C. jejuni genomes). Specific gene content profiles (n = 12 major clusters; Table 4; Supplementary Table 4) were identified. We observed that the gene content profiles were closely associated with ST of the isolates (Figure 3). Additional details about the variable gene profile distribution observed for each C. jejuni isolate are presented in Supplementary Figure 3A.
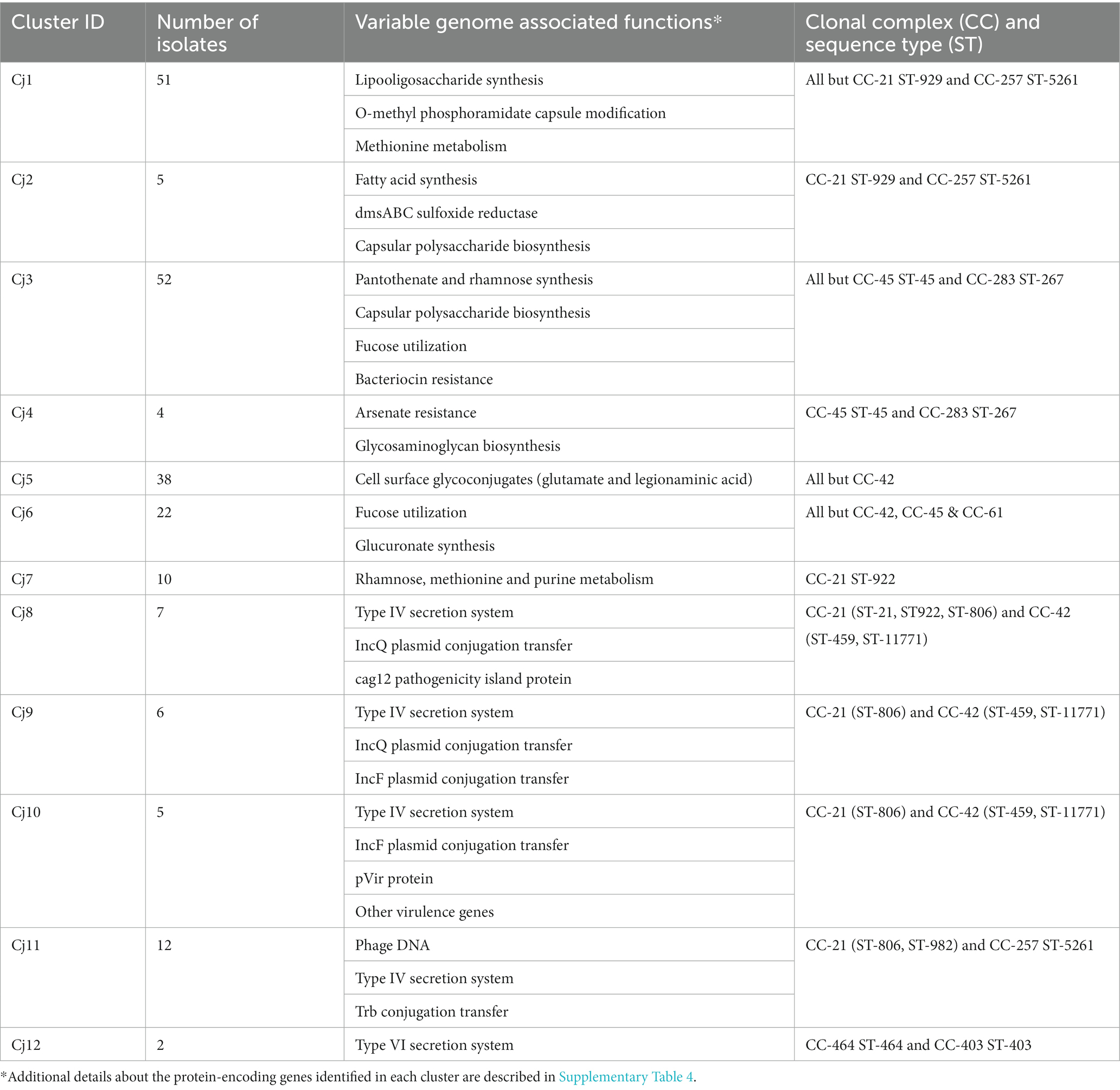
Table 4. Variable genome clusters in Campylobacter jejuni isolates.
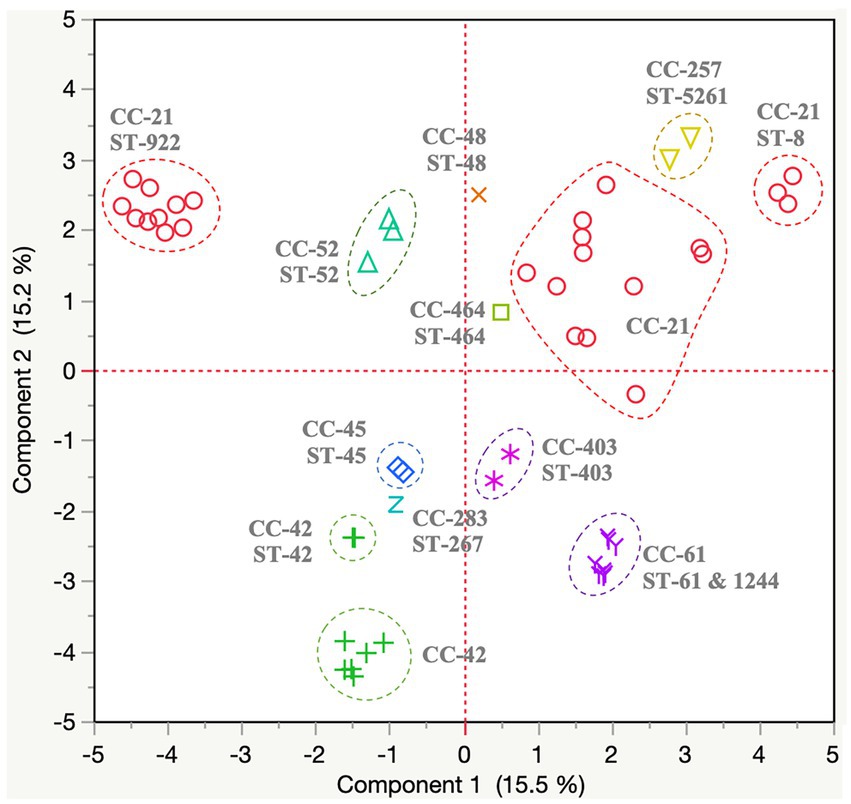
Figure 3. Variable genome distribution of Campylobacter jejuni (n = 56) isolated from dairy manure in small specialty crops farms in Northeast Ohio between 2018 and 2020. A total of 401 protein-encoding genes were identified as part of the C. jejuni variable genome and were used to create this distance matrix plot. Dotted circles indicate specific clonal complex (CC)-sequence type (ST) clusters. The color of each dot within the plot represents a different CC. The principal component plot is defined by component 1 and 2, which explains 15.5% and 15.2%, respectively of variations based on the variable-genome (n = 401 protein encoding genes) of the 56 C. jejuni isolates.
Most C. jejuni isolates (n = 51/56 isolates) possessed genes associated with lipooligosaccharide synthesis, O-methyl phosphoramidate capsule modification and methionine metabolism (n = 12 genes; Cluster Cj1 in Table 4; Supplementary Table 4). On the other hand, five isolates missing the genes from cluster Cj1 possessed genes involved in fatty acid synthesis, dmsABC sulfoxide reductase and capsular polysaccharide biosynthesis (n = 8 genes; Cluster Cj2 in Table 4; Supplementary Table 4). Additionally, most isolates (n = 52/56 isolates) possessed genes associated with pantothenate and rhamnose synthesis, capsular polysaccharide biosynthesis, fucose utilization, and bacteriocin resistance (n = 12; Cluster Cj3 in Table 4; Supplementary Table 4). However, the four isolates missing the genes from cluster Cj3 possessed other genes involved in arsenate resistance and glycosaminoglycan biosynthesis (n = 5 genes; Cluster Cj4 in Table 4; Supplementary Table 4).
A fraction of the isolates (n = 38/56 isolates) possessed genes associated with cell surface glycoconjugates (glutamate and legionaminic acid; n = 8 genes; Cluster Cj5 in Table 4; Supplementary Table 4). Among these isolates, 22 C. jejuni (especially from FC #2) also harbored genes for the utilization of fucose and glucuronate synthesis (n = 6 genes; Cluster Cj6 in Table 4; Supplementary Table 4).
Several isolates from FC #1 (n = 10/22 isolates; especially ST-922 CC-21 from farm #26) harbored genes associated with rhamnose, methionine, and purine metabolism (n = 10 genes; Cluster Cj7 in Table 4; Supplementary Table 4). The majority of these isolates also possessed type IV secretion systems (T4SS) protein-encoding genes (virB2,3,5–9, and virD4; n = 10 genes), IncQ plasmid conjugative transfer-related genes (traC, traE, traG and traR; n = 4 genes), and the gene encoding cag pathogenicity island protein (cag12; Cluster Cj8 in Table 4; Supplementary Table 4). A similar T4SS/conjugation profile was observed in C. jejuni isolates from FC #2 (n = 6/17 isolates, especially farm #17). These isolates also harbored genes associated with IncF plasmid conjugative transfer pilus assembly (n = 11 genes; traB,C,E,H,K,L,N,T,U,V, and W; Cluster Cj9 in Table 4; Supplementary Table 4). Further, 5 of these isolates also possessed additional protein-encoding genes associated with T4SS (virB4,7) and conjugative transfer (traG), hypothetical pVir protein (n = 10 genes, pVir0004,7,8,9,12,15,19,20,29,42), and other virulence-associated genes (n = 15; Cluster Cj10 in Table 4; Supplementary Table 4).
Another group of isolates (n = 12 isolates) harbored genes associated with conjugative transfer (n = 6; trbB,D,E,F,I,L) and T4SS (virB1), as well as a high abundance of bacteriophage DNA (n = 29 genes) especially from the Escherichia coli phage protein families Gp (phage lambda) and Mup (phage Mu; Cluster Cj11 in Table 4; Supplementary Table 4). Three isolates harbored type VI secretion system genes (T6SS; n = 6 genes; hcp, impB,C,G,I,K; Cluster Cj12 in Table 4; Supplementary Table 4).
Eighty-one protein encoding genes belonging to the variable genome were not clustered based on the CC or ST of the C. jejuni isolates. A majority of the protein-encoding genes (n = 27) were labeled as possible, putative, uncharacterized or hypothetical proteins (Supplementary Table 4).
Functional genomic profile analysis of Campylobacter coli demonstrated associations between the geographic location and sequence type of the isolates
A total of 102 protein-encoding genes were identified as part of the C. coli variable genome. Specific gene content profiles (n = 2 major clusters) were identified based on the ST associated with the isolates (Table 5; Supplementary Table 5). Additional details about the variable gene profile distribution observed for each C. coli isolate are presented in Supplementary Figure 3B. Interestingly, the major difference between these two clusters is the presence/absence of genes involved in the release and uptake of DNA and protein. All C. coli ST-829 CC-828 (n = 8 isolates) possessed phage DNA (n = 24 genes, especially from Gp and Mup families) and genes encoding enzymes associated with hexose utilization (glucose, abequose, fucose, mannose, glycerate, glucoronate and rhamnose; n = 14 genes; cluster Cc1 in Table 5; Supplementary Table 5). On the other hand, C. coli ST-1068 CC-828 (n = 5 isolates) possessed genes associated with conjugation transfer (n = 4 genes; traC,E,G, and Q), T4SS (n = 10 genes; virB2,4–11 and virD4), capsule modification (n = 5 genes), and cell wall synthesis (n = 2 genes; cluster Cc2 in Table 5; Supplementary Table 5).
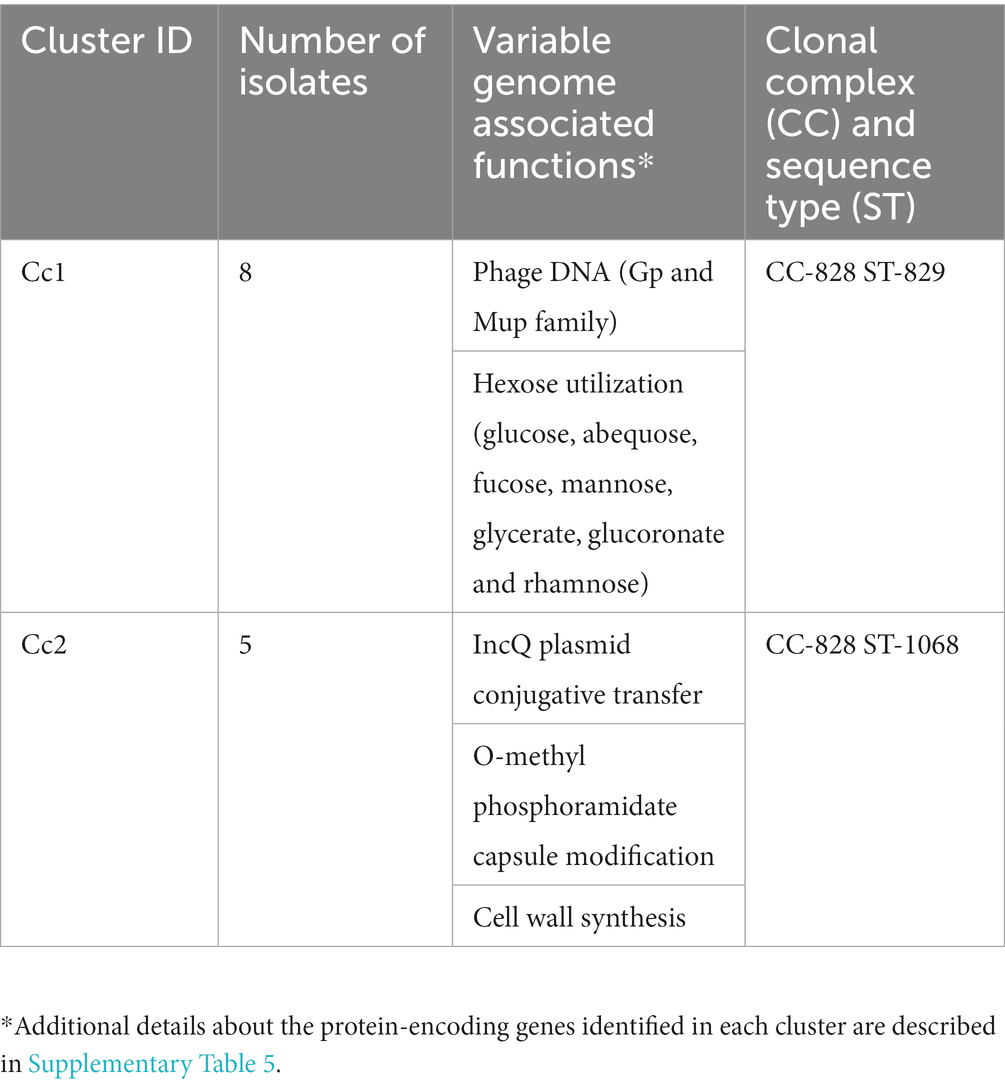
Table 5. Variable genome clusters in Campylobacter coli isolates.
Antimicrobial resistance profile of Campylobacter jejuni and Campylobacter coli isolates
Using the National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS) database, a total of 11 antimicrobial resistance genes (ARGs) were detected across the 69 Campylobacter isolates (Table 6; Figure 4). Three ARGs (blaOXA-193, gyrA_T86I, and tetO) were detected in both C. jejuni and C. coli isolates (Table 6). Campylobacter jejuni isolates were characterized with beta-lactamase (blaOXA-449, blaOXA-461, blaOXA-603, and blaOXA-61), macrolide (50S_L22_A103V), and aminoglycoside ARGs (rpsL_K88R and aph3’IIIa; Figure 4A), while C. coli isolates were characterized with an aminoglycoside ARG (aadE-Cc; Figure 4B). Interestingly, all C. coli isolates also harbored genes encoding proteins associated with resistance to arsenic (arsenate reductase [EC 1.20.4.4] thioredoxin-coupled and arsenical-resistance protein ACR3). All isolates with aadE-Cc also harbored tetO and gyrA_T86I (r2 = −0.85; p = 0.0002), and all isolates with tetO also had gyrA_T86I (r2 = 1; p < 0.0001) in C. jejuni. No ARG co-occurrence was detected in C. coli. A total of 14 ARG profiles were observed across the 69 isolates (Supplementary Table 6). Most of the isolates possessed genes conferring resistance to at least two antibiotics (n = 43/69 isolates). The co-occurrence of genes involved in the resistance to streptomycin (aadE-Cc) and beta-lactam (blaOXA-193) or to tetracycline (tetO), quinolone (gyrA_T86I), and beta-lactam (blaOXA-193) were predominant in C. coli (n = 7/13 and 5/13 isolates, respectively; Supplementary Table 7). Similarly, for most C. jejuni, the resistance to tetracycline (tetO) and beta-lactam (blaOXA-193, −461 or −449; n = 16/56 isolates), or to kanamycin (aph[3’IIIa]), beta-lactam (blaOXA-193), and tetracycline (tetO) were predominant (n = 8/56 isolates; Supplementary Table 7). Interestingly, gyrA_T86I and tetO were more likely detected in the C. coli isolates collected in the winter (60% confidence level and lift of 2), while aadE-CC was more likely detected in the C. coli isolates collected in the summer (57% confidence level and lift of 2.1). A similar trend was detected with blaOXA-193 in the summer with C. jejuni (40% confidence level and lift of 1.2).
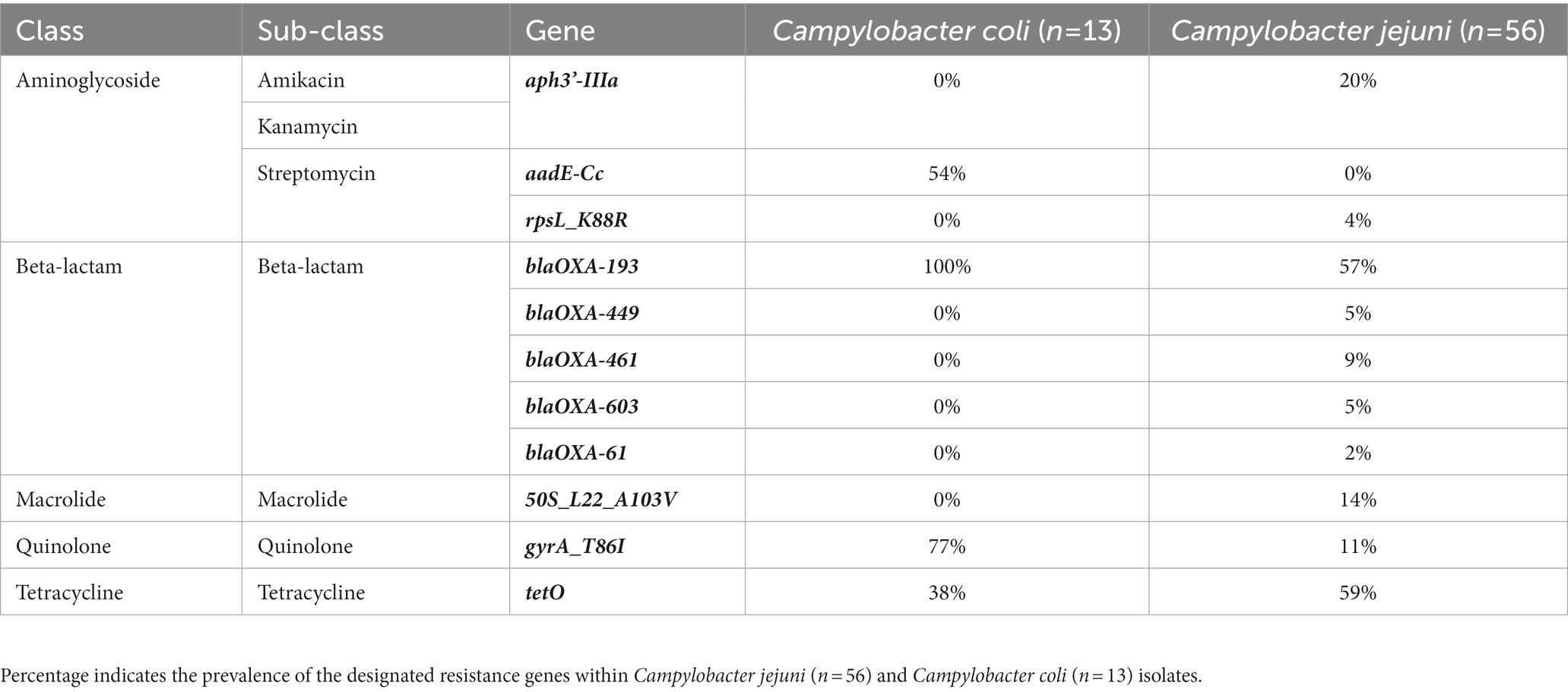
Table 6. Antimicrobial resistance genes in thermophilic Campylobacter.
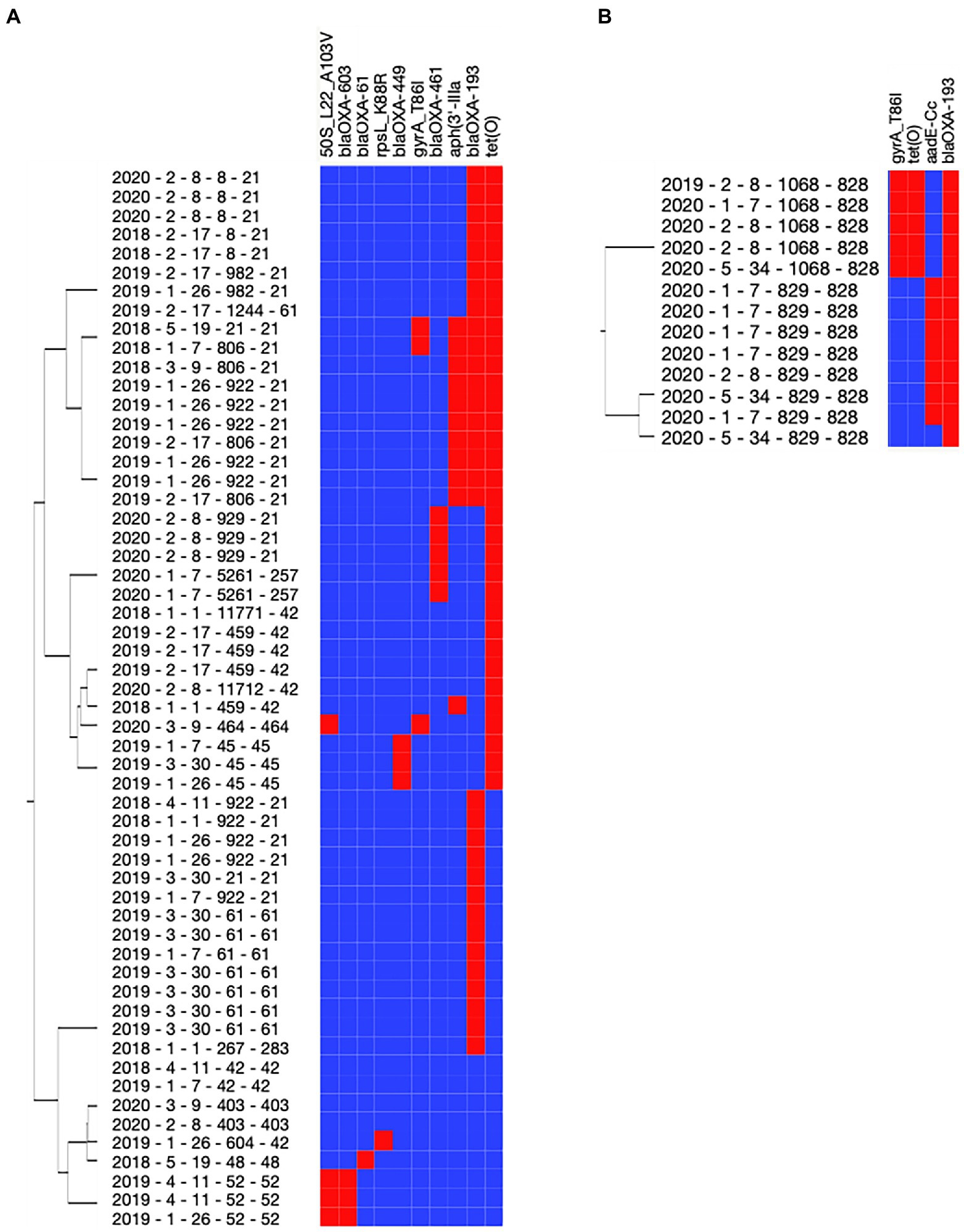
Figure 4. Antimicrobial resistance gene content of Campylobacter spp. isolated from dairy manure in small specialty crops farms in Northeast Ohio from 2018 to 2020. Resistome of Campylobacter jejuni (A) and Campylobacter coli (B). Eleven genes were detected across the 69 Campylobacter isolates using the NARMS database. The tree associated with the Y-axis displays the similarities of resistome profile among the isolates. Red and blue cells represent genes detected or missing (Y-axis) in the designated Campylobacter isolate (X-axis), respectively. Metadata associated with the Campylobacter spp. isolates are presented in the following order: year of collection (2018–2020), farm cluster (1–5), farm ID (#1–#34), sequence type (ST), and clonal complex (CC).
Co-occurrence of genes from the variable genomes of Campylobacter isolates
The co-occurrence of genes from the variable genome was investigated in both C. jejuni (n = 401 genes) and C. coli (n = 102 genes). Overall, the C. coli variable genome was divided into three major clusters with co-occurring genes (Figure 5A; p < 0.01). The first co-occurring cluster named “conjugation/T4SS” (r2 > 0.8; n = 47 genes; Supplementary Table 8) was composed of genes involved in conjugative transfer and T4SS (n = 17 genes), capsule associated genes (n = 5), cag12 (encoding cag pathogenicity island protein) and other virulence genes (e.g., rfbA, rfbB, and rhmA). Campylobacter coli isolates possessing the tetO gene, conferring tetracycline resistance, also possessed genes associated with conjugation/T4SS (r2 = 1; p < 0.001). A similar observation was made regarding C. jejuni (r2 > 0.43; p < 0.01). Interestingly, C. coli isolates possessing these conjugation/T4SS associated genes were less likely to possess prophages (n = 22 genes), and virulence genes (e.g., ceuB, fabG, glxK, rfbC, and rfbF; r2 < −0.91; p < 0.00 l n = 47 genes; Figure 5A; Supplementary Table 8). The opposite trend between the co-occurrence of prophage and conjugative/T4SS clusters was observed in C. jejuni (r2 = 0.69; p < 0.01). Both C. coli and C. jejuni isolates possessing genes implicated in O-methyl phosphoramindate capsule modification (hddA, hddC, and hddD) were also more likely to possess prophages (r2 = 0.78 in C. coli and r2 = 0.37 in C. jejuni; p < 0.01). Campylobacter coli isolates possessing genes associated with arsenical resistance (n = 3 genes) were likely to have genes involved in streptomycin resistance (n = 1 gene) and other genes (e.g., yrrC, yraQ family and ydeQ/yrkL/ywrO family). Unlike C. coli, the C. jejuni variable genome displayed less pronounced and indistinct gene co-occurrence profiles (Figure 5B).
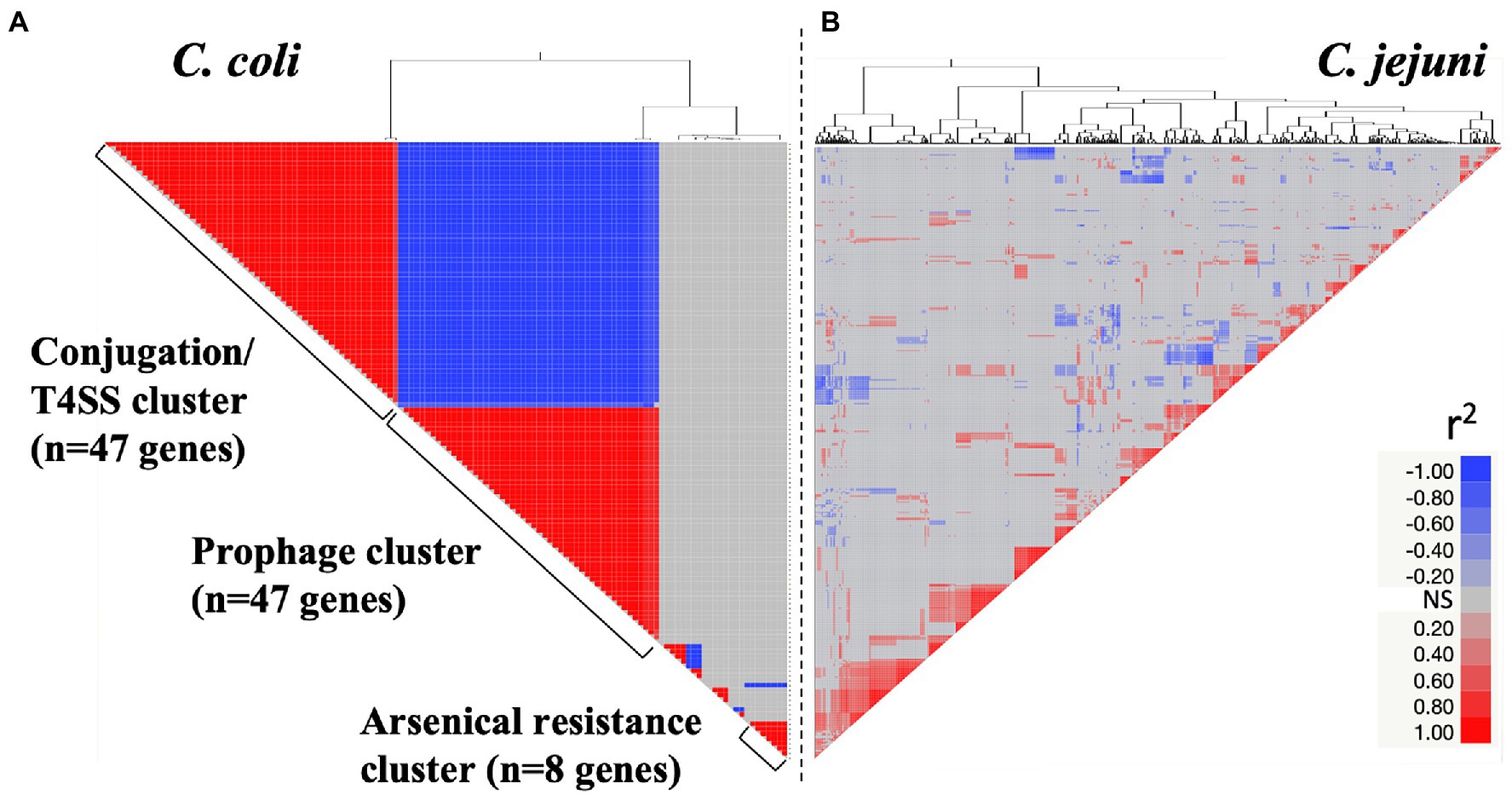
Figure 5. Co-occurrence of genes from the variable genome for both Campylobacter coli (A; n = 102 genes) and Campylobacter jejuni (B; n = 401 genes). X- and Y-axes display the gene co-occurrence profile (r2 value). Red and blue cells represent positive and negative correlations between genes of the variable genome (P < 0.01), respectively. Gray cells indicate non-significant correlations (NS; P > 0.01). Details about the gene co-occurrence profile for C. coli are displayed in Supplementary Table 8.
Plasmid composition
While no plasmids were detected by PlasmidFinder, MOB-Suite identified a total of 223 plasmids across 69 assembled Campylobacter genomes. Only five plasmids were detected in C. coli (on average 0.54 plasmids per assembly), whereas 216 plasmids were detected in C. jejuni (on average 3.86 plasmids per assembly). Multiple plasmids were detected in each farm. Most plasmids were highly similar and grouped into a total of eight clusters (an approximation of Operational Taxonomic Units for plasmids) and only 18 unique reference plasmids were the nearest neighbors to each detected plasmid (Supplementary Figure 4; Supplementary Table 9). However, it is important to mention that sequencing was conducted using a protocol designed to extract genomic DNA and not plasmid DNA. By consequence, results presented in this section might underestimate the plasmid composition in the Campylobacter isolates. Overall, among the 18 plasmids, 10 of them harbored ARG associated with the resistance to tetracyclines and aminoglycosides, four of them harbored genes associated with the T6SS and two other plasmids harbored genes associated with the T4SS. The prevalence of certain plasmids was closely associated with the ST-CC profile of the isolates.
Discussion
This study further demonstrates that dairy manure is a potential reservoir of thermophilic Campylobacter and a possible mode of transmission of Campylobacter to the human food chain (An et al., 2018; Sheng et al., 2019; Szott and Friese, 2021). In this study, whole genome sequencing (WGS) was conducted to identify the Campylobacter isolates obtained from the dairy manure used in SSCF as well as to better understand the diversity of genomic traits in the isolates. WGS demonstrated that C. jejuni (n = 56/69) was more predominant in dairy manure compared to C. coli (n = 13/69), which mirrors previous findings (Ocejo et al., 2019; Hansson et al., 2020). Overall, Campylobacter was recurrently detected in the SSCF between 2018 and 2020. Distinct genomic profiles were observed between C. coli and C. jejuni isolates, independent of the location and time of collection. Furthermore, the variable genomes of the Campylobacter isolates were closely associated with their ST and CC profile. Based on SNP profiling and MLST data, Campylobacter isolates collected from the same SSCF were more likely to belong to the same CC and harbor high genomic content similarity compared to isolates from other SSCF; Nevertheless, genomic profile similarities were also detected to some extent between SSCF. Thereby, our data suggest that Campylobacter may persist for an extended period of time within a cattle herd of a SSCF and could be transmitted between SSCF. We hypothesize that the dissemination of Campylobacter within or between the SSCFs could be due to sharing of agricultural equipment or fields. However, our current data does not allow determination as to whether (1) the infected cattle shed Campylobacter for an extended period, (2) Campylobacter persist in the SSCF by re-infecting cattle at different times over the course of the year, (3) the same Campylobacter isolates are re-introduced into the farms over time through interactions with the ecosystem surrounding the farm, or (4) how many cattle within the herd are infected with Campylobacter.
Distinct differences in functional genomics (gene content, virulome, and resistome) were observed between and within Campylobacter species and were also influenced by the spatial distribution of the SSCF where the isolates were collected. Several pathways associated with the capsule modification/biosynthesis, sugar synthesis/utilization, conjugative transfer, T4SS, and prophage diversity were associated with specific CC-ST profiles (Tables 4, 5). The main variations in gene content detected between isolates were related to gene transfer such as conjugative transfer (IncQ/IncF) and T4SS, which are key bacterial mechanisms involved in the virulence of Campylobacter and in the survival in specific environmental niches (Bacon et al., 2000; Huddleston, 2014; Deblais et al., 2018; Marasini et al., 2020; Panzenhagen et al., 2021). Campylobacter coli (n = 5/13) and C. jejuni (n = 22/59) isolates recovered in this study possessed several IncQ/IncF (traB,C,E,G,H,K,L,N,Q,R,T,U,V, and W) and T4SS (virB2-11 and virD4) protein-encoding genes. The presence of these genes was previously reported to be rare in C. jejuni and C. fetus subsp. venerealis isolates worldwide (Bacon et al., 2000; Panzenhagen et al., 2021; Silva et al., 2021). A mutation in virB11 was also shown to reduce C. jejuni 81–176 adhesion and invasion by 6- and 11-fold, respectively compared to wild-type (Bacon et al., 2000). Conjugation/T4SS-associated genes are frequently detected in Campylobacter isolated from livestock and are transmitted by plasmids also carrying ARG (Marasini et al., 2018, 2020; Ocejo et al., 2019). Similar trends were observed in our study. The resistance to tetracycline (tetO) and amikacin/kanamycin (aph3’-IIIa) in both C. jejuni and C. coli were highly correlated with the presence of IncQ-IncF/T4SS genes (r2 > 0.5; p < 0.001). Similarly, the gene encoding the Cag12 pathogenicity island protein was also highly correlated with the genes mentioned above (r2 > 0.8; p < 0.001). Overall, our study highlights that thermophilic Campylobacter recovered from SSCF carry plasmid-encoded proteins essential for its virulence (Bacon et al., 2000; Marasini et al., 2018, 2020). Based on these observations, DNA extraction using protocols allowing for the isolation of both genomic and plasmid DNA will be used for future studies to enhance the resolution of the genomic interpretations and better understand horizontal transfer dynamics. Preliminary data showed that plasmids associated with tetracycline resistance, the T4SS, and the T6SS were predominant, especially in C. jejuni isolates. Similar plasmids (e.g., plasmid accession number CP023447) were previously reported in C. jejuni isolated from poultry meat in Brazil (de Fátima Rauber Würfel et al., 2020). However, it is important to mention that the DNA extraction method used in this study limited the in-depth analysis and resolution of the plasmid composition in the Campylobacter isolates.
Interestingly, the presence of genes involved in DNA/protein transfer was negatively correlated with the prevalence of prophages in C. coli genomes, while the opposite trend was observed in C. jejuni. Bacteriophages/prophages have been linked to the acquisition of novel host survival strategies, and virulence and antimicrobial resistance genes (Kondo et al., 2021; Torres-Barceló, 2018; Pratama and van Elsas, 2019). Similarly, there were strong, negative correlations of bacteriophages/prophages with certain protein-encoded genes associated with O-methyl phosphoramidate modification of the capsule (gamma-glutamyl-CDP-amidate hydrolase, L-glutamine kinase, methyltransferase [EC 2.1.1.-], and phosphoglutamine cytidylyltransferase [EC 2.7.7.-]), and the utilization of UDP-activated sugars (UDP-glucose, UDP-galactopyranose and UDP-glucuronate, and L-glutamine kinase) in both C. coli and C. jejuni. Other genes implicated in O-methyl phosphoramidate capsule modification (hddA, hddC, and hddD) displayed the opposite trend, which follow previously published data concerning the role of capsule polysaccharides in phage sensitivity (Cai et al., 2019). A previous study demonstrated that the modification of the capsule polysaccharides modulates the phage infectivity in C. jejuni (Sørensen et al., 2012). Further, it was hypothesized that the level of UDP-activated sugars may be associated with the cell wall integrity due to phage infection (Ankrah et al., 2014). Interestingly, similar trends concerning the composition of conjugation/T4SS genes and prophages were observed with Salmonella enterica subsp. enterica serotype Heidelberg isolates collected from poultry farms in the Midwest (Deblais et al., 2018). Thus, our data suggests the importance of conjugation/T4SS genes and prophages in the genome plasticity as well as the emergence of antibiotic resistance, survival, and virulence abilities in thermophilic Campylobacter.
The prevalence of ARGs for aminoglycoside, beta-lactam, quinolone, and tetracycline was predominant (>29%) in Campylobacter isolates in this study. Similar trends were observed in published studies (Ocejo et al., 2019; Cobo-Díaz et al., 2021; Hailu et al., 2021; Hull et al., 2021). As previously described (Ocejo et al., 2019; Mouftah et al., 2021), blaOXA-193 was the most predominant ARG to beta-lactam compared to other beta-lactamase encoding genes (blaOXA-449, -461, -603, and 61; <10%). However, some other studies have reported blaOXA-61 being predominant in Campylobacter (Cobo-Díaz et al., 2021; Hull et al., 2021). Such discrepancies could be associated with annotation errors, which may lead to wrongly identifying bla-OXA-193 as bla-OXA-61 (Feldgarden et al., 2019b). It was previously shown that human contact with cattle could be linked to the transmission of tetracycline resistant C. jejuni (ST-464, ST-459, and ST-982; Cha et al., 2016, 2017). However, in the current study, resistance to tetracycline was broadly distributed across 12 other STs, especially in CC-21 (n = 25/56) which is the most predominant CC identified similar to a previously published study (An et al., 2018). CC-828 was the most prevalent clonal complex detected among the C. coli isolates (n = 13/13), which agrees with previous studies performed on ruminants (especially sheep) from Nigeria, Scotland, and Spain (Sproston et al., 2011; Ngulukun et al., 2016; Ocejo et al., 2019). The predominance of CC-21 and CC-828 in the dairy manure collected in SSCF from Northeast Ohio, United States is expected given they are known host generalist clonal complexes (Ocejo et al., 2021). Other STs (ST-829) were previously detected in chicken farms and slaughter houses, suggesting that horizontal transmission of Campylobacter may occur between livestock species (Frosth et al., 2020). ST-8 was previously isolated from sheep and cattle and was associated with abortion (Sahin et al., 2008; Wu et al., 2014; Tang et al., 2017).
Data obtained in this study showed that C. jejuni isolated from dairy manure harbored genes associated with legionaminic acid, glutamate/fucose, and pantothenic acid (vitamin B5) metabolism. It was previously reported that vitamin B5 biosynthesis enhances C. jejuni colonization of cattle and was associated with human infections (Sheppard et al., 2013; Buchanan et al., 2017). Further, the competition for vitamin B5 between Campylobacter and the host may affect the host immune system and increase susceptibility to other pathogens (Yoshii et al., 2019). Legionaminic acid is involved in the modification of bacterial flagellin which is key for the persistence of Campylobacter in poultry and host interactions (Howard et al., 2009; Stephenson et al., 2014; Ardissone et al., 2020; Vieira et al., 2021); however, its role in the persistence of Campylobacter in cattle and dairy manure remains unknown. The utilization of fucose in nutrient-limited conditions has been reported to provide a competitive advantage to C. jejuni by enhancing cell adhesions and biofilm production (Day et al., 2009; Muraoka and Zhang, 2011; Stahl et al., 2011; Dwivedi et al., 2016). Similar observations were reported regarding the vital role of glutamate in C. jejuni metabolism and as a chemoattractant (Lübke et al., 2018). However, it is important to mention that most of these studies have been performed in poultry.
Conclusion
In conclusion, our study demonstrated that several thermophilic Campylobacter STs isolated from dairy manure in SSCF from Northeast Ohio were previously associated with human campylobacteriosis cases, and thus, signals public health risks associated with the presence of dairy cattle in these mixed farming systems. Furthermore, the thermophilic Campylobacter isolates studied frequently harbored ARGs related to quinoline, beta-lactam, tetracycline, and streptomycin resistance. Our data also highlights potential risks associated with the long-term persistence of thermophilic Campylobacter within the same SSCF farm over time. Similarly, our findings suggest potential interactions between farms leading to the dissemination of thermophilic Campylobacter isolates between farms of proximity. Therefore, an emphasis on the understanding of SSCF practices and potential interactions between SSCF will help in identifying factor(s) that influence the persistence and dissemination of Campylobacter within and between SSCF, and thus, develop new guidelines to mitigate Campylobacter burden in SSCF.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
LD, MK, HJ, KB, UB, LH, KH, and GR: experimental designing. MK: collection and processing of the samples. MK, HJ, MS, and SB: extraction of DNA and preparation of samples for sequencing. LD, MK, GR, HJ, KB, JP, and JG: analyzing and interpretation of sequencing results. LD, GR, HJ, JG, and KB: writing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by state and federal funds appropriated to the Ohio Agricultural Research and Development Center, The Ohio State University. Funds for sequencing was made available by the Produce Safety Research Consortium, CFSAN, FDA. MS and SB are Fellows supported by Goldbelt C6, LLC.
Acknowledgments
We thank Katie Dodson for technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1074548/full#supplementary-material
Footnotes
References
An, J.-U., Ho, H., Kim, J., Kim, W.-H., Kim, J., Lee, S., et al. (2018). Dairy Cattle, a Potential Reservoir of Human Campylobacteriosis: Epidemiological and Molecular Characterization of Campylobacter jejuni From Cattle Farms. Front. Microbiol. 9:3136. doi: 10.3389/fmicb.2018.03136
Andrews, S. (2010). Babraham Bioinformatics—FastQC A Quality Control tool for High Throughput Sequence Data. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed 9 June 2022).
Ankrah, N. Y. D., May, A. L., Middleton, J. L., Jones, D. R., Hadden, M. K., Gooding, J. R., et al. (2014). Phage infection of an environmentally relevant marine bacterium alters host metabolism and lysate composition. ISME J. 8, 1089–1100. doi: 10.1038/ismej.2013.216
Ardissone, S., Kint, N., and Viollier, P. H. (2020). Specificity in glycosylation of multiple flagellins by the modular and cell cycle regulated glycosyltransferase FlmG. Elife 9:e60488. doi: 10.7554/eLife.60488
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bacon, D. J., Alm, R. A., Burr, D. H., Hu, L., Kopecko, D. J., Ewing, C. P., et al. (2000). Involvement of a Plasmid in Virulence of Campylobacter jejuni 81-176. Infect. Immun. 68, 4384–4390. doi: 10.1128/IAI.68.8.4384-4390.2000
Black, Z., Balta, I., Black, L., Naughton, P. J., Dooley, J. S. G., and Corcionivoschi, N. (2021). The Fate of Foodborne Pathogens in Manure Treated Soil. Front. Microbiol. 12:781357. doi: 10.3389/fmicb.2021.781357
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchanan, C. J., Webb, A. L., Mutschall, S. K., Kruczkiewicz, P., Barker, D. O. R., Hetman, B. M., et al. (2017). A Genome-Wide Association Study to Identify Diagnostic Markers for Human Pathogenic Campylobacter jejuni Strains. Front. Microbiol. 8:1224. doi: 10.3389/fmicb.2017.01224
Buzby, J. C., Allos, B. M., and Roberts, T. (1997). The economic burden of Campylobacter-associated Guillain-Barré syndrome. J Infect Dis 176, S192–S197. doi: 10.1086/513785
Cai, R., Wang, G., Le, S., Wu, M., Cheng, M., Guo, Z., et al. (2019). Three Capsular Polysaccharide Synthesis-Related Glucosyltransferases, GT-1, GT-2 and WcaJ, Are Associated With Virulence and Phage Sensitivity of Klebsiella pneumoniae. Front. Microbiol. 10:1189. doi: 10.3389/fmicb.2019.01189
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Cha, W., Mosci, R., Wengert, S. L., Singh, P., Newton, D. W., Salimnia, H., et al. (2016). Antimicrobial Susceptibility Profiles of Human Campylobacter jejuni Isolates and Association with Phylogenetic Lineages. Front. Microbiol. 7:589. doi: 10.3389/fmicb.2016.00589
Cha, W., Mosci, R. E., Wengert, S. L., Venegas Vargas, C., Rust, S. R., Bartlett, P. C., et al. (2017). Comparing the Genetic Diversity and Antimicrobial Resistance Profiles of Campylobacter jejuni Recovered from Cattle and Humans. Front. Microbiol. 8:818. doi: 10.3389/fmicb.2017.00818
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Cobo-Díaz, J. F., González del Río, P., and Álvarez-Ordóñez, A. (2021). Whole Resistome Analysis in Campylobacter jejuni and C. coli Genomes Available in Public Repositories. Front. Microbiol. 12:662144. doi: 10.3389/fmicb.2021.662144
Cody, A. J., Bray, J. E., Jolley, K. A., McCarthy, N. D., and Maiden, M. C. J. (2017). Core Genome Multilocus Sequence Typing Scheme for Stable, Comparative Analyses of Campylobacter jejuni and C. coli Human Disease Isolates. J. Clin. Microbiol. 55, 2086–2097. doi: 10.1128/JCM.00080-17
Davis, S., Pettengill, J. B., Luo, Y., Payne, J., Shpuntoff, A., Rand, H., et al. (2015). CFSAN SNP Pipeline: an automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput. Sci. 1:e20. doi: 10.7717/peerj-cs.20
Day, C. J., Tiralongo, J., Hartnell, R. D., Logue, C.-A., Wilson, J. C., von Itzstein, M., et al. (2009). Differential Carbohydrate Recognition by Campylobacter jejuni Strain 11168: Influences of Temperature and Growth Conditions. PLoS One 4:e4927. doi: 10.1371/journal.pone.0004927
de Fátima Rauber Würfel, S., Jorge, S., de Oliveira, N. R., Kremer, F. S., Sanchez, C. D., Campos, V. F., et al. (2020). Campylobacter jejuni isolated from poultry meat in Brazil: in silico analysis and genomic features of two strains with different phenotypes of antimicrobial susceptibility. Mol. Biol. Rep. 47, 671–681. doi: 10.1007/s11033-019-05174-y
Deblais, L., Lorentz, B., Scaria, J., Nagaraja, K. V., Nisar, M., Lauer, D., et al. (2018). Comparative Genomic Studies of Salmonella Heidelberg Isolated From Chicken- and Turkey-Associated Farm Environmental Samples. Front. Microbiol. 9:1841. doi: 10.3389/fmicb.2018.01841
Denis, M., Soumet, C., Rivoal, K., Ermel, G., Blivet, D., Salvat, G., et al. (1999). Development of a m-PCR assay for simultaneous identification of Campylobacter jejuni and C. coli. Lett. Appl. Microbiol. 29, 406–410. doi: 10.1046/j.1472-765X.1999.00658.x
Dingle, K. E., Colles, F. M., Wareing, D. R. A., Ure, R., Fox, A. J., Bolton, F. E., et al. (2001). Multilocus Sequence Typing System for Campylobacter jejuni. J. Clin. Microbiol. 39, 14–23. doi: 10.1128/JCM.39.1.14-23.2001
Dwivedi, R., Nothaft, H., Garber, J., Xin Kin, L., Stahl, M., Flint, A., et al. (2016). L-fucose influences chemotaxis and biofilm formation in Campylobacter jejuni. Mol. Microbiol. 101, 575–589. doi: 10.1111/mmi.13409
Feldgarden, M., Brover, V., Gonzalez-Escalona, N., Frye, J. G., Haendiges, J., Haft, D. H., et al. (2021). AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 11:12728. doi: 10.1038/s41598-021-91456-0
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019b). Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 63:e00483. doi: 10.1128/AAC.00483-19
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019a). Using the NCBI AMRFinder Tool to Determine Antimicrobial Resistance Genotype-Phenotype Correlations Within a Collection of NARMS Isolates. bioRxiv [Preprint], 550707.
Frosth, S., Karlsson-Lindsjö, O., Niazi, A., Fernström, L.-L., and Hansson, I. (2020). Identification of Transmission Routes of Campylobacter and On-Farm Measures to Reduce Campylobacter in Chicken. Pathogens 9:363. doi: 10.3390/pathogens9050363
Gangiredla, J., Rand, H., Benisatto, D., Payne, J., Strittmatter, C., Sanders, J., et al. (2021). GalaxyTrakr: a distributed analysis tool for public health whole genome sequence data accessible to non-bioinformaticians. BMC Genomics 22:114. doi: 10.1186/s12864-021-07405-8
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haft, D. H., DiCuccio, M., Badretdin, A., Brover, V., Chetvernin, V., O’Neill, K., et al. (2018). RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 46, D851–D860. doi: 10.1093/nar/gkx1068
Hailu, W., Helmy, Y. A., Carney-Knisely, G., Kauffman, M., Fraga, D., and Rajashekara, G. (2021). Prevalence and Antimicrobial Resistance Profiles of Foodborne Pathogens Isolated from Dairy Cattle and Poultry Manure Amended Farms in Northeastern Ohio, the United States. Antibiotics 10:1450. doi: 10.3390/antibiotics10121450
Halpin, A. L., Gu, W., Wise, M. E., Sejvar, J. J., Hoekstra, R. M., and Mahon, B. E. (2018). Post-Campylobacter Guillain Barré Syndrome in the USA: secondary analysis of surveillance data collected during the 2009–2010 novel Influenza A (H1N1) vaccination campaign. Epidemiol. Infect. 146, 1740–1745. doi: 10.1017/S0950268818001802
Hansson, I., Olsson Engvall, E., Ferrari, S., Harbom, B., and Lahti, E. (2020). Detection of Campylobacter species in different types of samples from dairy farms. Vet. Rec. 186:605. doi: 10.1136/vr.105610
Heimesaat, M. M., Backert, S., Alter, T., and Bereswill, S. (2021). “Human Campylobacteriosis—A Serious Infectious Threat in a One Health Perspective” in Fighting Campylobacter Infections: Towards a One Health Approach Current Topics in Microbiology and Immunology. ed. S. Backert (Cham: Springer International Publishing), 1–23.
Howard, S. L., Jagannathan, A., Soo, E. C., Hui, J. P. M., Aubry, A. J., Ahmed, I., et al. (2009). Campylobacter jejuni Glycosylation Island Important in Cell Charge, Legionaminic Acid Biosynthesis, and Colonization of Chickens. Infect. Immun. 77, 2544–2556. doi: 10.1128/IAI.01425-08
Huddleston, J. R. (2014). Horizontal gene transfer in the human gastrointestinal tract: potential spread of antibiotic resistance genes. Infect Drug Resist 7, 167–176. doi: 10.2147/IDR.S48820
Hull, D. M., Harrell, E., Vliet, A. H. M.Van, Correa, M., and Thakur, S. (2021). Antimicrobial resistance and interspecies gene transfer in Campylobacter coli and Campylobacter jejuni isolated from food animals, poultry processing, and retail meat in North Carolina, 2018–2019. PLoS One 16:e0246571. doi: 10.1371/journal.pone.0246571
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org/ website and their applications. Wellcome Open Res 3:124. doi: 10.12688/wellcomeopenres.14826.1
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Kassem, I., Kehinde, O., Helmy, Y., Piña Mimbela, R., Kumar, A., Chandrashekhar, K., et al. (2016). “Campylobacter in Poultry: The Conundrums of Highly Adaptable and Ubiquitous Foodborne Pathogens,” in foodborne diseases, 1st edn. eds. J. M. Soon, L. Manning, C. A. Wallace (Boca Raton, FL: CRC press). 79–112.
Kim, H.-J. (2016). Opportunities and Challenges of Alternative Specialty Crops: The Global Picture. HortScience 51, 1316–1319. doi: 10.21273/HORTSCI10659-16
Kondo, K., Kawano, M., and Sugai, M. (2021). Distribution of Antimicrobial Resistance and Virulence Genes within the Prophage-Associated Regions in Nosocomial Pathogens. mSphere 6, e00452–e00421. doi: 10.1128/mSphere.00452-21
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, W., O’Neill, K. R., Haft, D. H., DiCuccio, M., Chetvernin, V., Badretdin, A., et al. (2021). RefSeq: expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 49, D1020–D1028. doi: 10.1093/nar/gkaa1105
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Lübke, A.-L., Minatelli, S., Riedel, T., Lugert, R., Schober, I., Spröer, C., et al. (2018). The transducer-like protein Tlp12 of Campylobacter jejuni is involved in glutamate and pyruvate chemotaxis. BMC Microbiol. 18:111. doi: 10.1186/s12866-018-1254-0
Marasini, D., Karki, A. B., Bryant, J. M., Sheaff, R. J., and Fakhr, M. K. (2020). Molecular characterization of megaplasmids encoding the type VI secretion system in Campylobacter jejuni isolated from chicken livers and gizzards. Sci. Rep. 10:12514. doi: 10.1038/s41598-020-69155-z
Marasini, D., Karki, A. B., Buchheim, M. A., and Fakhr, M. K. (2018). Phylogenetic Relatedness Among Plasmids Harbored by Campylobacter jejuni and Campylobacter coli Isolated From Retail Meats. Front. Microbiol. 9:2167. doi: 10.3389/fmicb.2018.02167
Midwest Climate and Specialty Crops (n.d.). Available at: https://store.extension.iastate.edu/product/Midwest-Climate-and-Specialty-Crops (Accessed 10 May 2020).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D., and Gurevich, A. (2018). Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150. doi: 10.1093/bioinformatics/bty266
Moffatt, C. R. M., Kennedy, K. J., Selvey, L., and Kirk, M. D. (2021). Campylobacter-associated hospitalisations in an Australian provincial setting. BMC Infect. Dis. 21:10. doi: 10.1186/s12879-020-05694-0
Mortensen, N., Jonasson, S. A., Lavesson, I. V., Emberland, K. E., Litleskare, S., Wensaas, K.-A., et al. (2021). Characteristics of hospitalized patients during a large waterborne outbreak of Campylobacter jejuni in Norway. PLoS One 16:e0248464. doi: 10.1371/journal.pone.0248464
Mouftah, S. F., Pascoe, B., Calland, J. K., Mourkas, E., Tonkin, N., Lefèvre, C., et al. (2021). Local accessory gene sharing drives lineage-specific acquisition of antimicrobial resistance in Egyptian Campylobacter spp. Biorxiv [Preprint]. 2021.09.24.461243.
Mulder, A. C., Franz, E., de Rijk, S., Versluis, M. A. J., Coipan, C., Buij, R., et al. (2020). Tracing the animal sources of surface water contamination with Campylobacter jejuni and Campylobacter coli. Water Res. 187:116421. doi: 10.1016/j.watres.2020.116421
Muraoka, W. T., and Zhang, Q. (2011). Phenotypic and Genotypic Evidence for l-Fucose Utilization by Campylobacter jejuni. J. Bacteriol. 193, 1065–1075. doi: 10.1128/JB.01252-10
Ngulukun, S., Oboegbulem, S., and Klein, G. (2016). Multilocus sequence typing of Campylobacter jejuni and Campylobacter coli isolates from poultry, cattle and humans in Nigeria. J. Appl. Microbiol. 121, 561–568. doi: 10.1111/jam.13185
Nutrition (2021). Risk Assessment of Foodborne Illness Associated With Pathogens From Produce Grown in Fields Amended With Untreated Biological Soil Amendments of Animal Origin. FDA. Available at: https://www.fda.gov/food/cfsan-risk-safety-assessments/risk-assessment-foodborne-illness-associated-pathogens-produce-grown-fields-amended-untreated (Accessed 8 March 2022).
Ocejo, M., Oporto, B., and Hurtado, A. (2019). Occurrence of Campylobacter jejuni and Campylobacter coli in Cattle and Sheep in Northern Spain and Changes in Antimicrobial Resistance in Two Studies 10-years Apart. Pathogens 8:98. doi: 10.3390/pathogens8030098
Ocejo, M., Oporto, B., Lavín, J. L., and Hurtado, A. (2021). Whole genome-based characterisation of antimicrobial resistance and genetic diversity in Campylobacter jejuni and Campylobacter coli from ruminants. Sci. Rep. 11:8998. doi: 10.1038/s41598-021-88318-0
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Panzenhagen, P., Portes, A. B., Dos Santos, A. M. P., Duque, S. D. S., and Conte Junior, C. A. (2021). The Distribution of Campylobacter jejuni Virulence Genes in Genomes Worldwide Derived from the NCBI Pathogen Detection Database. Genes 12:1538. doi: 10.3390/genes12101538
Pratama, A. A., and van Elsas, J. D. (2019). Gene mobility in microbiomes of the mycosphere and mycorrhizosphere –role of plasmids and bacteriophages. FEMS Microbiol. Ecol. 95:fiz053. doi: 10.1093/femsec/fiz053
Ramos, T. D. M., Jay-Russell, M. T., Millner, P. D., Baron, J. N., Stover, J., Pagliari, P., et al. (2021). Survival and Persistence of Foodborne Pathogens in Manure-Amended Soils and Prevalence on Fresh Produce in Certified Organic Farms: A Multi-Regional Baseline Analysis. Front. Sustain. Food Syst :67:5. doi: 10.3389/fsufs.2021.674767
Ramos, T. M., Jay-Russell, M. T., Millner, P. D., Shade, J., Misiewicz, T., Sorge, U. S., et al. (2019). Assessment of Biological Soil Amendments of Animal Origin Use, Research Needs, and Extension Opportunities in Organic Production. Front. Sustain. Food Syst. 3:73. doi: 10.3389/fsufs.2019.00073
Robertson, J., and Nash, J. H. E. Y. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microbial Genomics 4:e000206. doi: 10.1099/mgen.0.000206
Sahin, O., Plummer, P. J., Jordan, D. M., Sulaj, K., Pereira, S., Robbe-Austerman, S., et al. (2008). Emergence of a tetracycline-resistant Campylobacter jejuni clone associated with outbreaks of ovine abortion in the United States. J. Clin. Microbiol. 46, 1663–1671. doi: 10.1128/JCM.00031-08
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Scallan Walter, E. J., Crim, S. M., Bruce, B. B., and Griffin, P. M. (2020). Incidence of Campylobacter-Associated Guillain-Barré Syndrome Estimated from Health Insurance Data. Foodborne Pathog. Dis. 17, 23–28. doi: 10.1089/fpd.2019.2652
Scharff, R. L. (2020). Food Attribution and Economic Cost Estimates for Meat- and Poultry-Related Illnesses. J. Food Prot. 83, 959–967. doi: 10.4315/JFP-19-548
Seemann, T. (2022a). ABRicate. Available at: https://github.com/tseemann/abricate (Accessed 9 June 2022).
Seemann, T. (2022b). mlst. Available at: https://github.com/tseemann/mlst (Accessed 9 June 2022).
Sharma, M., and Reynnells, R. (2016). Importance of Soil Amendments: Survival of Bacterial Pathogens in Manure and Compost Used as Organic Fertilizers. Microbiol Spectr 4, 1–13. doi: 10.1128/microbiolspec.PFS-0010-2015
Sheng, L., Shen, X., Benedict, C., Su, Y., Tsai, H.-C., Schacht, E., et al. (2019). Microbial Safety of Dairy Manure Fertilizer Application in Raspberry Production. Front. Microbiol. 10, 1–10. doi: 10.3389/fmicb.2019.02276
Sheppard, S. K., Didelot, X., Meric, G., Torralbo, A., Jolley, K. A., Kelly, D. J., et al. (2013). Genome-wide association study identifies vitamin B5 biosynthesis as a host specificity factor in Campylobacter. Proc. Natl. Acad. Sci. U. S. A. 110, 11923–11927. doi: 10.1073/pnas.1305559110
Silva, M. F., Pereira, A. L., Fraqueza, M. J., Pereira, G., Mateus, L., Lopes-da-Costa, L., et al. (2021). Genomic and Phenotypic Characterization of Campylobacter fetus subsp. venerealis Strains. Microorganisms 9:340. doi: 10.3390/microorganisms9020340
Sørensen, M., van Alphen, L., Frodor, C., Crowley, S., Christensen, B., Szymanski, C., et al. (2012). Phase Variable Expression of Capsular Polysaccharide Modifications Allows Campylobacter jejuni to Avoid Bacteriophage Infection in Chickens. Front. Cell. Infect. Microbiol. 2, 1–11. doi: 10.3389/fcimb.2012.00011
Souvorov, A., Agarwala, R., and Lipman, D. J. (2018). SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 19:153. doi: 10.1186/s13059-018-1540-z
Sproston, E. L., Ogden, I. D., MacRae, M., Dallas, J. F., Sheppard, S. K., Cody, A. J., et al. (2011). Temporal Variation and Host Association in the Campylobacter Population in a Longitudinal Ruminant Farm Study. Appl. Environ. Microbiol. 77, 6579–6586. doi: 10.1128/AEM.00428-11
Stahl, M., Friis, L. M., Nothaft, H., Liu, X., Li, J., Szymanski, C. M., et al. (2011). l-Fucose utilization provides Campylobacter jejuni with a competitive advantage. Proc. Natl. Acad. Sci. 108, 7194–7199. doi: 10.1073/pnas.1014125108
Stephenson, H. N., Mills, D. C., Jones, H., Milioris, E., Copland, A., Dorrell, N., et al. (2014). Pseudaminic Acid on Campylobacter jejuni Flagella Modulates Dendritic Cell IL-10 Expression via Siglec-10 Receptor: A Novel Flagellin-Host Interaction. J Infect Dis 210, 1487–1498. doi: 10.1093/infdis/jiu287
Szott, V., and Friese, A. (2021). “Emission Sources of Campylobacter from Agricultural Farms, Impact on Environmental Contamination and Intervention Strategies” in Fighting Campylobacter Infections: Towards a One Health Approach Current Topics in Microbiology and Immunology. ed. S. Backert (Cham: Springer International Publishing), 103–125.
Tamura, K., Nei, M., and Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Tang, Y., Meinersmann, R. J., Sahin, O., Wu, Z., Dai, L., Carlson, J., et al. (2017). Wide but Variable Distribution of a Hypervirulent Campylobacter jejuni Clone in Beef and Dairy Cattle in the United States. Appl. Environ. Microbiol. 83, e01425–e01417. doi: 10.1128/AEM.01425-17
Thépault, A., Poezevara, T., Quesne, S., Rose, V., Chemaly, M., and Rivoal, K. (2018). Prevalence of Thermophilic Campylobacter in Cattle Production at Slaughterhouse Level in France and Link Between C. jejuni Bovine Strains and Campylobacteriosis. Front. Microbiol. 9:471. doi: 10.3389/fmicb.2018.00471
Torres-Barceló, C. (2018). The disparate effects of bacteriophages on antibiotic-resistant bacteria. Emerg Microbes Infect 7, 168–112. doi: 10.1038/s41426-018-0169-z
Vieira, A. Z., Raittz, R. T., and Faoro, H. (2021). Origin and evolution of nonulosonic acid synthases and their relationship with bacterial pathogenicity revealed by a large-scale phylogenetic analysis. Microb Genom 7:000563. doi: 10.1099/mgen.0.000563
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Wu, Z., Sippy, R., Sahin, O., Plummer, P., Vidal, A., Newell, D., et al. (2014). Genetic Diversity and Antimicrobial Susceptibility of Campylobacter jejuni Isolates Associated with Sheep Abortion in the United States and Great Britain. J. Clin. Microbiol. 52, 1853–1861. doi: 10.1128/JCM.00355-14
Yoshii, K., Hosomi, K., Sawane, K., and Kunisawa, J. (2019). Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front. Nutr. 6, 1–12. Available at:. doi: 10.3389/fnut.2019.00048
Keywords: Campylobacter jejuni, Campylobacter coli, whole genome sequencing, dairy manure, small specialty crop farm, resistome, virulome, prophage
Citation: Deblais L, Jang H, Kauffman M, Gangiredla J, Sawyer M, Basa S, Poelstra JW, Babu US, Harrison LM, Hiett KL, Balan KV and Rajashekara G (2023) Whole genome characterization of thermophilic Campylobacter species isolated from dairy manure in small specialty crop farms of Northeast Ohio. Front. Microbiol. 14:1074548. doi: 10.3389/fmicb.2023.1074548
Edited by:
Christophe Bordi, Aix Marseille Université, FranceReviewed by:
Kinga Anna Wieczorek, National Veterinary Research Institute (NVRI), PolandPatrick Jon Biggs, Massey University, New Zealand
Hiroshi Asakura, National Institute of Health Sciences (NIHS), Japan
Copyright © 2023 Deblais, Jang, Kauffman, Gangiredla, Sawyer, Basa, Poelstra, Babu, Harrison, Hiett, Balan and Rajashekara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gireesh Rajashekara, cmFqYXNoZWthcmEuMkBvc3UuZWR1
†These authors have contributed equally to this work