 Cristina Lara-Ochoa1,2*
Cristina Lara-Ochoa1,2* Alejandro Huerta-Saquero1,3
Alejandro Huerta-Saquero1,3 Abraham Medrano-López1
Abraham Medrano-López1 Wanyin Deng4B. Brett Finlay4Ygnacio Martínez-Laguna5
Wanyin Deng4B. Brett Finlay4Ygnacio Martínez-Laguna5 José L. Puente1*
José L. Puente1*- 1Departamento de Microbiología Molecular, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
- 2Centro de Detección Biomolecular, Benemérita Universidad Autónoma de Puebla, Puebla, Mexico
- 3Departamento de Bionanotecnología, Centro de Nanociencias y Nanotecnología, Universidad Nacional Autónoma de México, Ensenada, Mexico
- 4Michael Smith Laboratories, Department of Microbiology and Immunology, and Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC, Canada
- 5Vicerrectoría de Investigación y Estudios de Posgrado, Benemérita Universidad Autónoma de Puebla, Puebla, Mexico
Introduction: Enteropathogenic Escherichia coli (EPEC), enterohemorrhagic E. coli (EHEC) and Citrobacter rodentium (CR) belong to a group of pathogens that share the ability to form “attaching and effacing” (A/E) lesions on the intestinal epithelia. A pathogenicity island known as the locus of enterocyte effacement (LEE) contains the genes required for A/E lesion formation. The specific regulation of LEE genes relies on three LEE-encoded regulators: Ler activates the expression of the LEE operons by antagonizing the silencing effect mediated by the global regulator H-NS, GrlA activates ler expression and GrlR represses the expression of the LEE by interacting with GrlA. However, despite the existing knowledge of LEE regulation, the interplay between GrlR and GrlA and their independent roles in gene regulation in A/E pathogens are still not fully understood.
Methods: To further explore the role that GrlR and GrlA in the regulation of the LEE, we used different EPEC regulatory mutants and cat transcriptional fusions, and performed protein secretion and expression assays, western blotting and native polyacrylamide gel electrophoresis.
Results and discussion: We showed that the transcriptional activity of LEE operons increased under LEE-repressing growth conditions in the absence of GrlR. Interestingly, GrlR overexpression exerted a strong repression effect over LEE genes in wild-type EPEC and, unexpectedly, even in the absence of H-NS, suggesting that GrlR plays an alternative repressor role. Moreover, GrlR repressed the expression of LEE promoters in a non-EPEC background. Experiments with single and double mutants showed that GrlR and H-NS negatively regulate the expression of LEE operons at two cooperative yet independent levels. In addition to the notion that GrlR acts as a repressor by inactivating GrlA through protein-protein interactions, here we showed that a DNA-binding defective GrlA mutant that still interacts with GrlR prevented GrlR-mediated repression, suggesting that GrlA has a dual role as a positive regulator by antagonizing GrlR’s alternative repressor role. In line with the importance of the GrlR-GrlA complex in modulating LEE gene expression, we showed that GrlR and GrlA are expressed and interact under both inducing and repressing conditions. Further studies will be required to determine whether the GrlR alternative repressor function depends on its interaction with DNA, RNA, or another protein. These findings provide insight into an alternative regulatory pathway that GrlR employs to function as a negative regulator of LEE genes.
Introduction
Enteropathogenic Escherichia coli (EPEC) is one of the main etiological agents of severe diarrhea in children under 2 years of age, predominantly in developing countries (Pearson et al., 2016). EPEC, enterohemorrhagic E. coli (EHEC) and Citrobacter rodentium belong to a group of pathogens that possess the ability to induce a unique histopathological lesion known as attaching and effacing (A/E) (Croxen and Finlay, 2010). The localized destruction of the microvilli of intestinal epithelial cells followed by rearrangements of the cytoskeleton beneath the site of bacterial adherence, leading to the formation of actin-rich cup-like structures that favor an intimate interaction between the bacterium and the host cell, are hallmarks of this lesion (Spears et al., 2006; Frankel and Phillips, 2008).
Most genes required for A/E lesion formation are located within a pathogenicity island known as the locus of enterocyte effacement (LEE) (Gaytan et al., 2016). The LEE region contains five polycistronic operons (LEE1-LEE5), two bicistronic operons (espG-rorf1 and grlRA) and four transcriptional units (etgA, cesF, map, and escD). LEE1 to LEE3 encode the structural components of a type III secretion system (T3SS) responsible for translocating effector proteins into the enterocyte. The LEE4 operon codes for translocator proteins (EspA, B, D), and LEE5 encodes proteins involved in the intimate attachment (intimin and Tir). Genes encoding effector proteins, chaperones and transcriptional regulators are distributed within and outside the LEE (Pearson et al., 2016; Serapio-Palacios and Finlay, 2020). The cooperative action of the EPEC translocated effector proteins leads to cytoskeleton rearrangements, increased cell permeability, decreased absorption of ions and nutrients, alterations of tight junctions and modulation of the inflammatory response in the host intestinal cells and thus diarrheal disease (Guttman and Finlay, 2008; Croxen et al., 2013).
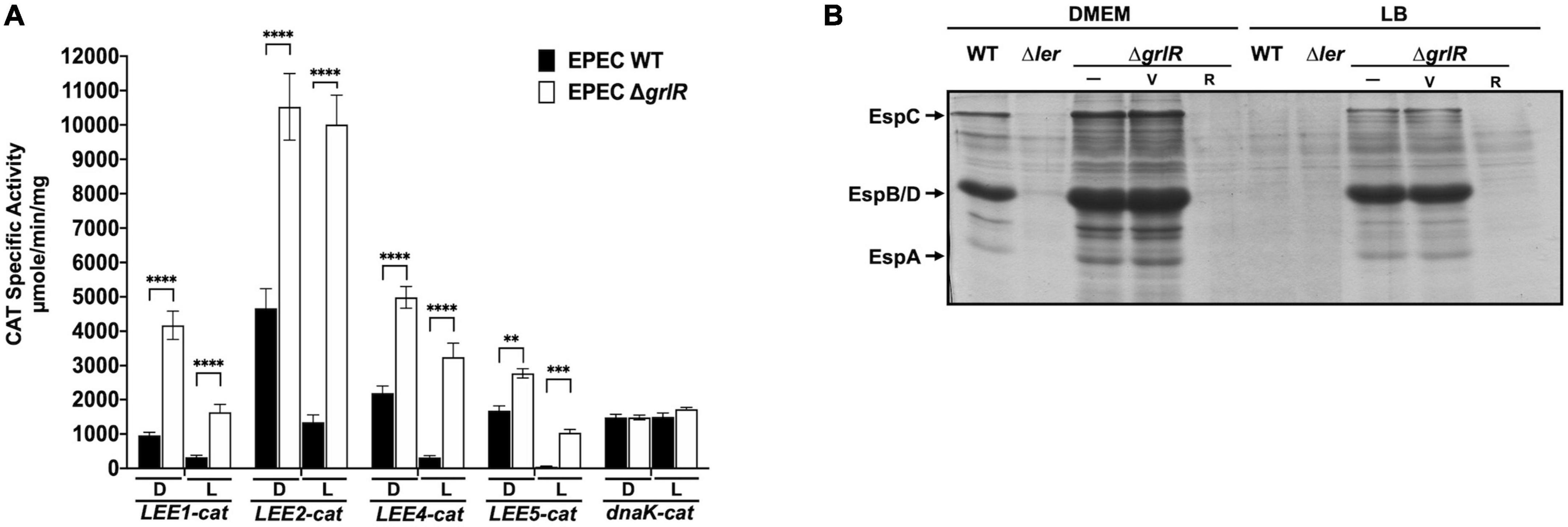
Figure 1. The absence of GrlR derepresses the expression of locus of enterocyte effacement (LEE) operons under repressing conditions. (A) The transcriptional activity of the LEE1-cat, LEE2-cat, LEE4-cat, LEE5-cat, and dnaK-cat fusions was analyzed in WT EPEC (black bars) and its ΔgrlR isogenic mutant (white bars), grown in 50 ml DMEM (D) or LB medium (L) with shaking at 37°C. Specific chloramphenicol acetyltransferase (CAT) activity was determined from samples collected at an OD600 of 1. Values are an average of three independent experiments performed in duplicate. Error bars indicate standard deviations. Statistically different values are indicated (**p-value < 0.01; ***p-value < 0.001; ****p-value < 0.0001). (B) Profile of secreted proteins of EPEC WT, Δler and ΔgrlR (carrying the empty vector pMPM-T3 or its derivative pT3GrlR) grown under the same conditions as in panel (A). Secreted proteins were concentrated from culture supernatants by precipitation with trichloroacetic acid (TCA) and separated by 12% SDS-PAGE. V: pMPM-T3, R: pT3GrlR.
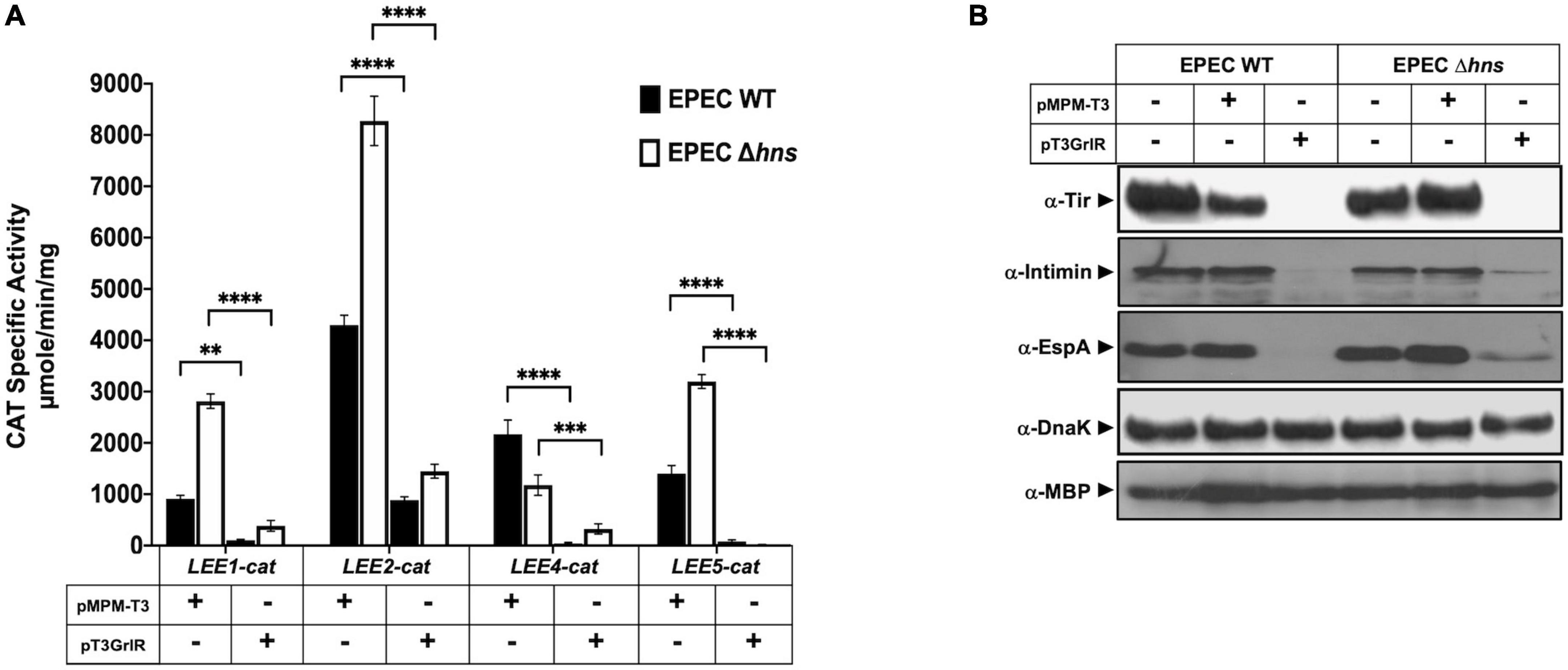
Figure 2. GrlR represses the expression of locus of enterocyte effacement (LEE) genes in the absence of H-NS. (A) Expression of the LEE1-cat, LEE2-cat, LEE4-cat, and LEE5-cat fusions was analyzed in WT enteropathogenic Escherichia coli (EPEC) (black bars) and its Δhns isogenic mutant (white bars) carrying the empty vector pMPM-T3 (V) or its derivative pT3GrlR (R), grown in DMEM with shaking at 37°C. Specific chloramphenicol acetyltransferase (CAT) activity was determined using samples collected from cultures grown in 50 ml Dulbecco’s modified Eagle’s medium (DMEM) or Lysogeny Broth (LB) at an OD600 of 1. Values are an average of three independent experiments performed in duplicate. Error bars indicate standard deviations. Statistically different values are indicated (**p-value < 0.01; ***p-value < 0.001; ****p-value < 0.0001). (B) Total extracts were prepared from the same culture samples and separated by 12% SDS-PAGE. The expression of Tir, intimin and EspA was analyzed by western blotting using polyclonal anti-intimin and anti-EspA and monoclonal anti-Tir antibodies. As controls for protein loading, maltose binding protein (MBP) and DnaK were also detected using monoclonal anti-DnaK and polyclonal anti-MBP antibodies.
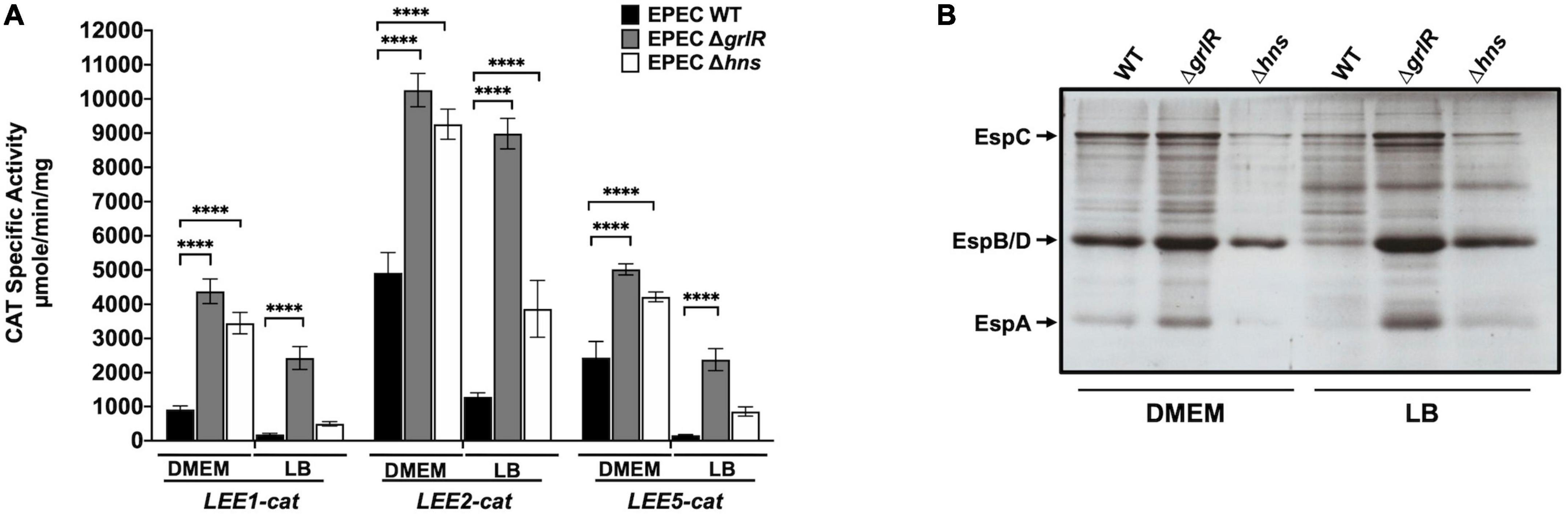
Figure 3. GrlR is the main repressor of locus of enterocyte effacement (LEE) gene expression under repressing conditions. (A) Expression of the LEE1-cat, LEE2-cat and LEE5-cat fusions was analyzed in WT EPEC (black bars) and its ΔgrlR (gray bars) and Δhns (white bars) isogenic mutants, grown in 50 ml of Dulbecco’s modified Eagle’s medium (DMEM) or Lysogeny Broth (LB) with shaking at 37°C. Specific chloramphenicol acetyltransferase (CAT) activity was determined from samples collected from cultures at an OD600 of 1. Values are an average of three independent experiments performed in duplicate. Error bars indicate standard deviations. Statistically different values are indicated (****p-value < 0.0001). (B) Secreted proteins of the same cultures were concentrated from supernatants by precipitation with trichloroacetic acid (TCA), separated by 12% SDS-PAGE and stained with Coomassie brilliant blue.
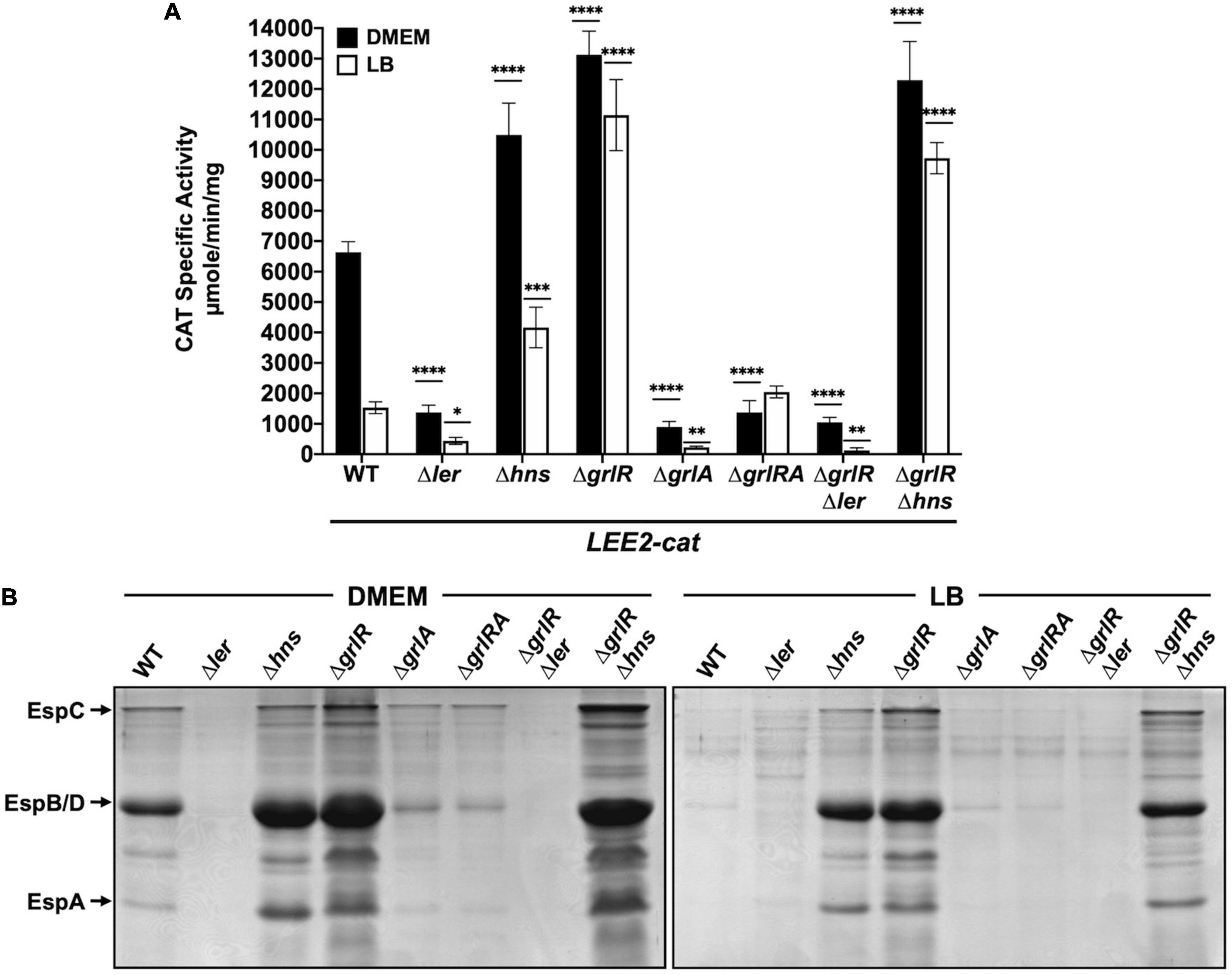
Figure 4. GrlR and H-NS repress locus of enterocyte effacement (LEE) gene expression by an indirect cooperative mechanism. (A) chloramphenicol acetyltransferase (CAT) activity of the LEE2-cat fusion was determined in WT enteropathogenic Escherichia coli (EPEC) and its Δler, Δhns, ΔgrlR, ΔgrlA, ΔgrlRA, ΔgrlRΔler, and ΔgrlRΔhns derivative mutants grown in 50 ml of Dulbecco’s modified Eagle’s medium (DMEM) (black bars) or Lysogeny Broth (LB) (white bars) with shaking at 37°C. Specific CAT activity was determined from samples collected at an OD600 of 1. Values are an average of three independent experiments performed in duplicate. Error bars indicate standard deviations. Statistically different values are indicated (*p-value < 0.1; **p-value < 0.01; ***p-value < 0.001; ****p-value < 0.0001). (B) Secreted proteins of the same cultures were concentrated from supernatants by precipitation with TCA, separated by 12% SDS-PAGE and stained with Coomassie brilliant blue.
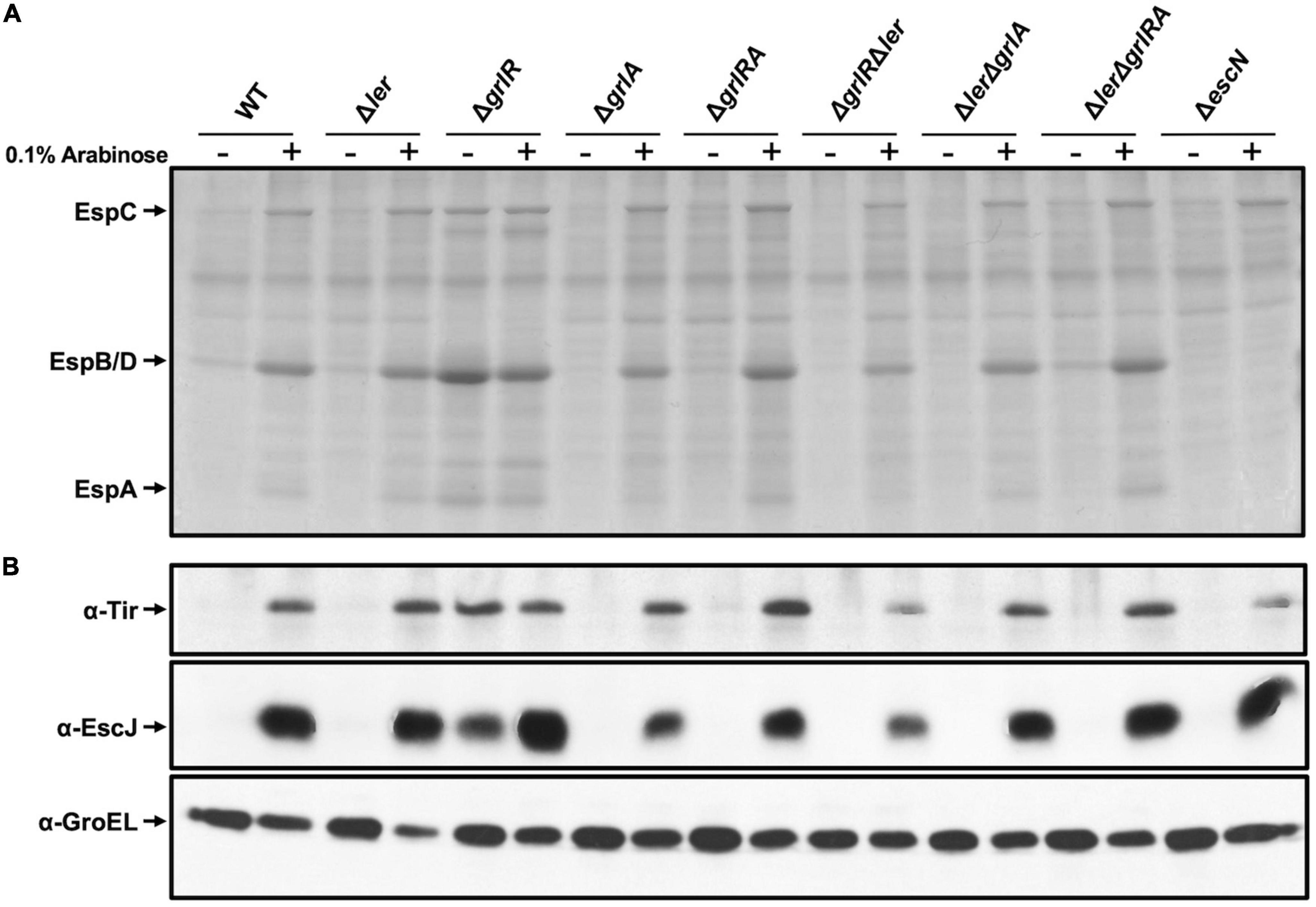
Figure 5. Protein secretion profile in the presence of a dominant negative H-NS mutant. (A) Secreted proteins from WT enteropathogenic Escherichia coli (EPEC) and its Δler, ΔgrlR, Δhns, ΔgrlA, ΔgrlRA, ΔgrlRΔler, ΔlerΔgrlA, ΔlerΔgrlRA, and ΔescN derivative mutants, carrying plasmid pT6-HNS/G113D expressing H-NSG113D, grown in 50 ml Lysogeny Broth (LB) with (+) and without (–) 0.1% arabinose at 37°C with shaking, were concentrated from culture supernatants by precipitation with trichloroacetic acid (TCA), separated by 12% SDS-PAGE and stained with Coomassie brilliant blue. (B) Total extracts were prepared from the corresponding bacterial pellets and separated by 12% SDS-PAGE. Tir and EscJ expression was analyzed by western blotting using an anti-Tir monoclonal antibody and anti-EscJ polyclonal antibodies. As a control for protein loading, GroEL expression was also analyzed using an anti-GroEL monoclonal antibody.
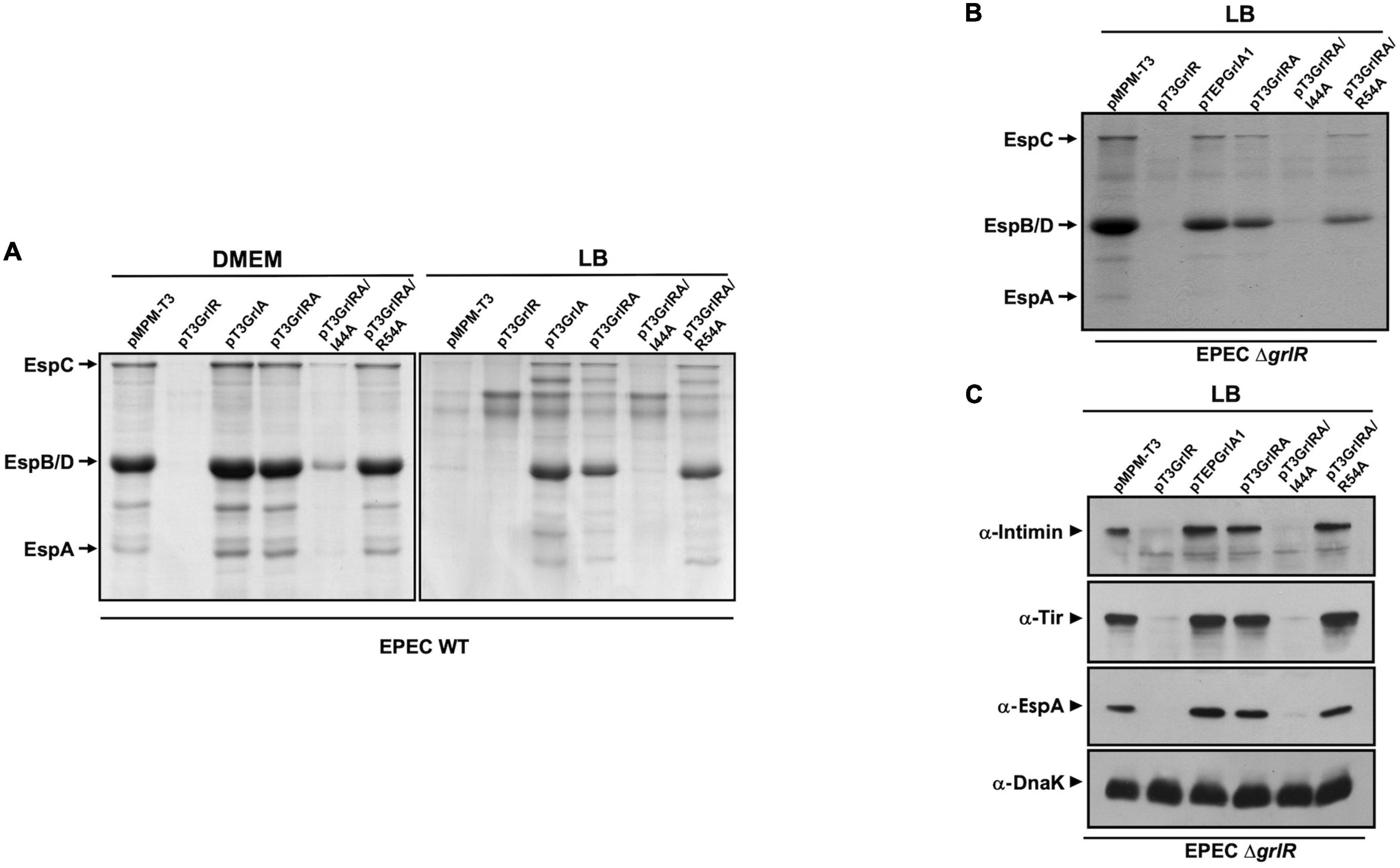
Figure 6. GrlA antagonizes GrlR-mediated repression. (A) Profile of secreted proteins from WT enteropathogenic Escherichia coli (EPEC) carrying the empty vector pMPM-T3 or its derivatives pT3GrlR, pTEPGrlA1, and pT3GrlRA, pT3GrlRA/I44A and pT3GrlRA/R54A grown in 50 ml Dulbecco’s modified Eagle’s medium (DMEM) or Lysogeny Broth (LB) to an OD600 of 1. Secreted proteins were concentrated from culture supernatants by precipitation with trichloroacetic acid (TCA), separated by 12% SDS-PAGE and stained with Coomassie brilliant blue. (B) Secreted proteins from EPEC ΔgrlR carrying the same plasmids as in panel (A) grown in 50 ml of LB with shaking at 37°C. (C) Total extracts were prepared from bacterial samples of the same cultures described in panel (B). Proteins were separated by 12% SDS-PAGE and expression of intimin, Tir and EspA was analyzed by western blotting using anti-intimin, anti-Tir and anti-EspA antibodies. As a control for protein loading, DnaK expression was also analyzed using anti-DnaK antibodies.
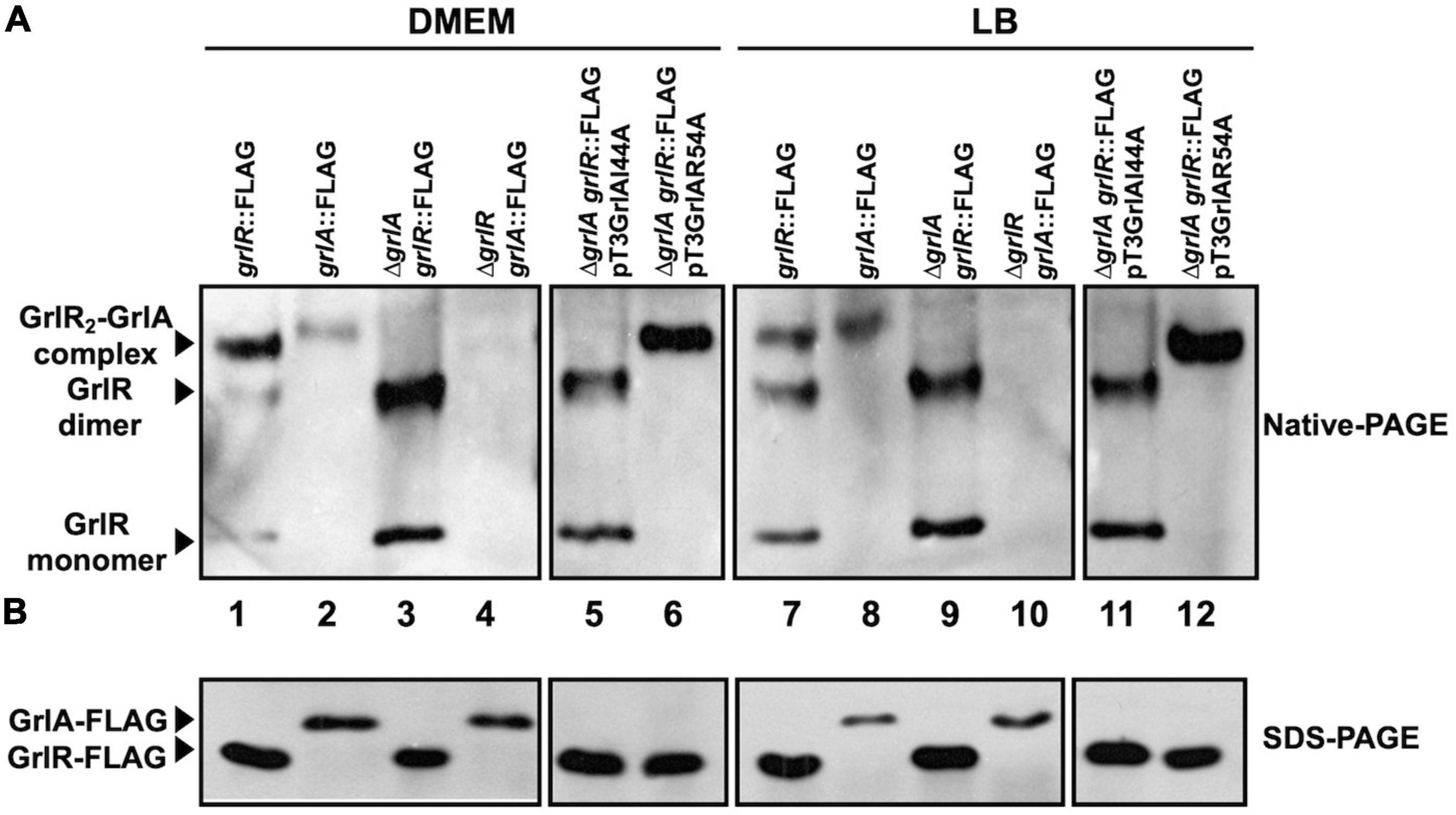
Figure 7. Analysis of GrlR2-GrlA complex formation by native gel electrophoresis. (A) GrlR2-GrlA protein complex formation in the enteropathogenic Escherichia coli (EPEC) strains grlR:3xFLAG, grlA:3xFLAG, ΔgrlR_grlA:3xFLAG, grlR:3xFLAG_ΔgrlA, as well as in the grlR:3xFLAG_ΔgrlA strain carrying plasmids pTEPGrlA1/I44A or pTEPGrlA1/R54A, was analyzed from samples taken from cultures grown in Dulbecco’s modified Eagle’s medium (DMEM) or Lysogeny Broth (LB) at an OD600 of 1. Total extracts were obtained from bacterial pellets and separated by 12% native PAGE. The GrlR2-GrlA and GrlR-GrlR complexes and the GrlR monomer (indicated by arrows) were identified by western blotting using anti-FLAG monoclonal antibodies. (B) The same cell extracts were also separated by 12% SDS-PAGE and transferred to a PVDF membrane for western blotting to confirm GrlR-3xFLAG and GrlA-3xFLAG expression in the different strains using anti-FLAG antibodies.
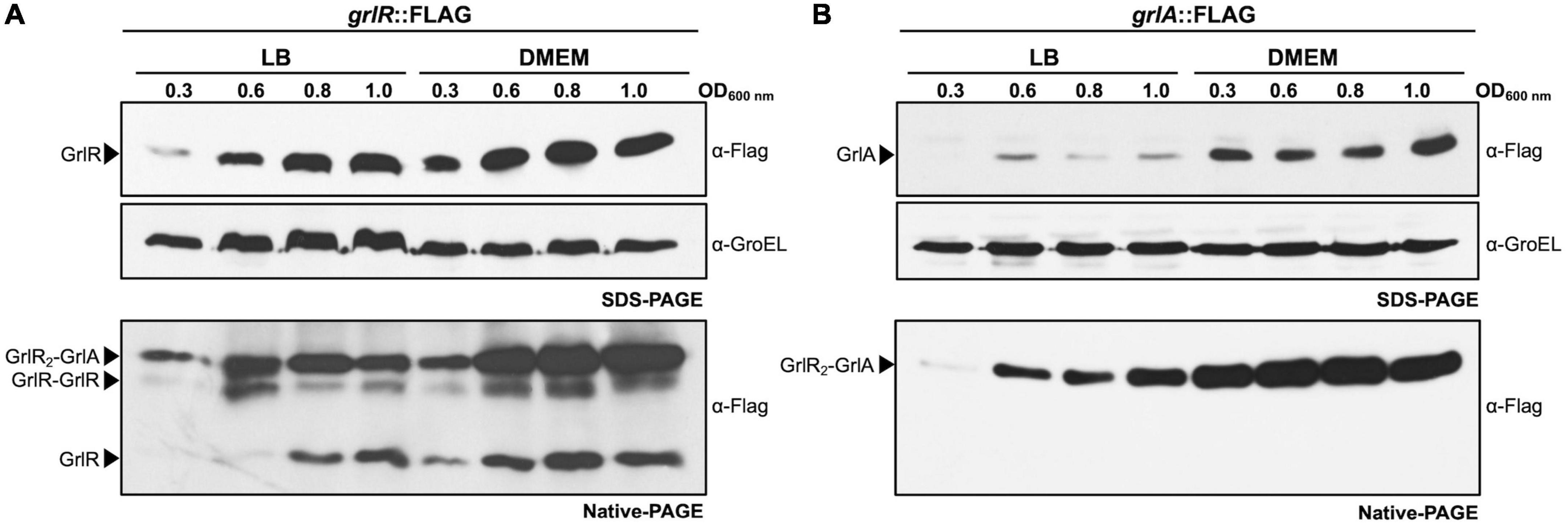
Figure 8. GrlR and GrlA are expressed under both repressing and inducing conditions and interact to form the GrlR2-GrlA heterotrimer. The enteropathogenic Escherichia coli (EPEC) grlR:3xFLAG (A) and grlA:3xFLAG (B) strains were grown in 50 ml of Dulbecco’s modified Eagle’s medium (DMEM) or Lysogeny Broth (LB) with shaking at 37°C. Total cell extracts were collected at OD600 of 0.3, 0.6, 0.8, and 1.0 and separated by 12% native PAGE. GrlR2-GrlA and GrlR-GrlR complexes and GrlR monomer are indicated by arrows and were identified by western blotting using anti-FLAG monoclonal antibodies. The same cell extracts were also separated by 12% SDS-PAGE to evaluate the expression of GrlR-FLAG and GrlA-FLAG by western blotting using anti-FLAG antibodies. As a control for protein loading, GroEL expression was also analyzed using anti-GroEL antibodies.
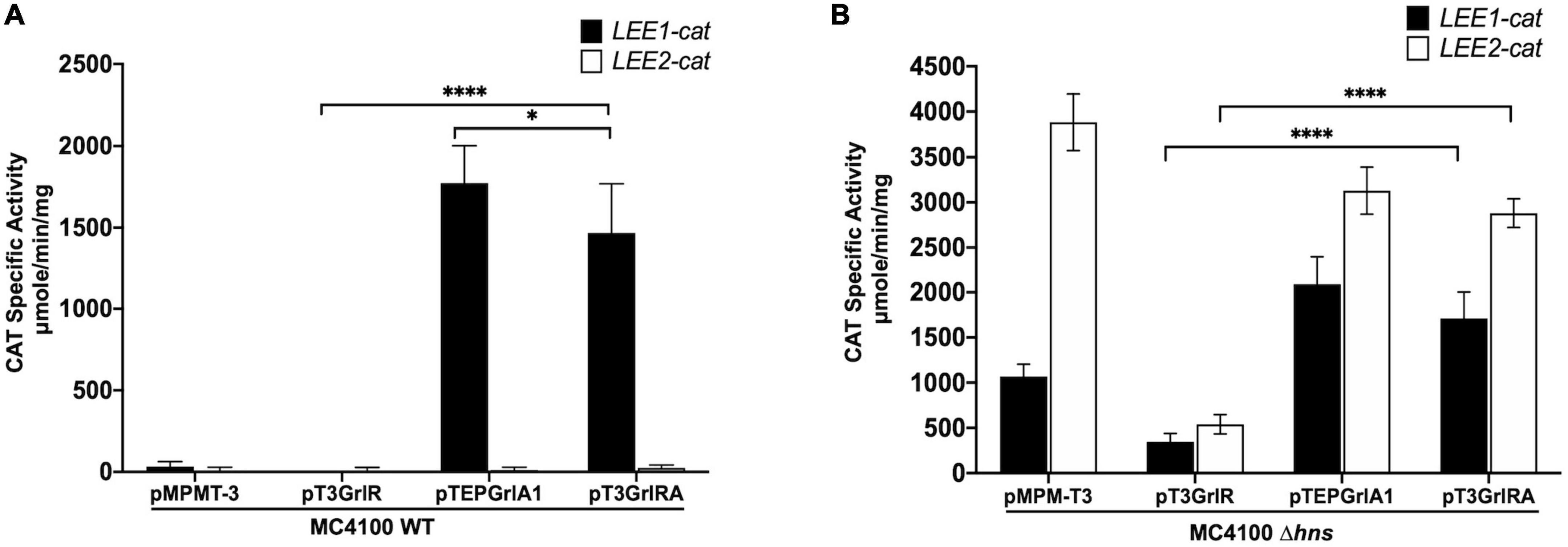
Figure 9. GrlR represses the expression of LEE1 and LEE2 operons in the absence of H-NS, GrlA and Ler. Expression of LEE1-cat (black bars) and LEE2-cat (white bars) fusions was analyzed in panel (A) WT Escherichia coli MC4100 and (B) MC4100 Δhns (JPMC1) strains, carrying the empty vector pMPMT-3 or its derivatives pT3GrlR, pTEPGrlA1 and pT3GrlRA grown in 50 ml of Dulbecco’s modified Eagle’s medium (DMEM) with shaking at 37°C. Specific chloramphenicol acetyltransferase (CAT) activity was determined from samples collected from cultures grown in DMEM at an OD600 of 1. Values are an average of 3 independent experiments performed in duplicate. Error bars indicate standard deviations. Statistically different values are indicated (*p-value < 0.1; ****p-value < 0.0001).
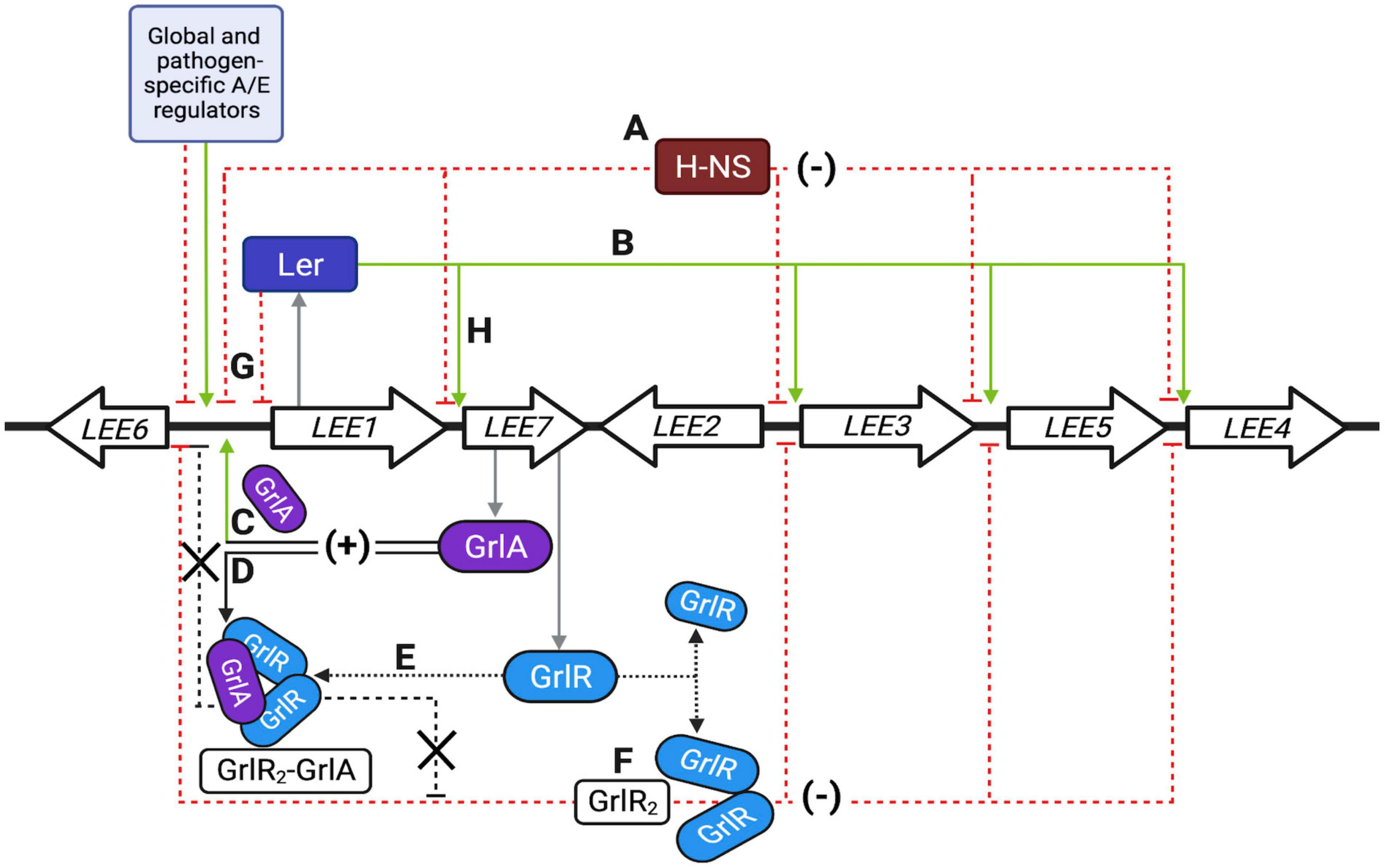
Figure 10. Schematic representation of the locus of enterocyte effacement (LEE) regulatory network. (A–H) Represent the primary regulatory circuits controlling LEE gene expression, as described in detail in the discussion. Green and red dashed lines indicate positive and negative regulatory pathways; black arrows indicate protein interactions. Thick arrows show a simplified representation of the seven operons previously reviewed in detail (Gaytan et al., 2016). Single genes are not illustrated. The first gene of the polycistronic LEE1 operon encodes Ler, while the bicistronic LEE7 operon codes for GrlR and GrlA. The light blue box represents several other global, ancestral and pathotype-specific regulators that modulate LEE gene expression, mainly by acting, but not exclusively, on Ler expression. Other colleagues have extensively and elegantly reviewed the regulatory elements known to modulate the regulation of the LEE (Furniss and Clements, 2018; Platenkamp and Mellies, 2018; Turner et al., 2019; Serapio-Palacios and Finlay, 2020). Created with BioRender.com.
In A/E bacteria, the LEE pathogenicity island is a distinctive example of a genetic element whose expression is controlled by a complex network of global (ancestral) and horizontally acquired regulatory proteins in response to a wide variety of environmental factors (Furniss and Clements, 2018; Platenkamp and Mellies, 2018). One of the main regulatory mechanisms involved in controlling LEE gene expression is the xenogeneic silencing exerted by H-NS (Bustamante et al., 2001). As described in different enterobacteria, H-NS specifically silences gene transcription by binding to AT-rich DNA sequences of exogenous origin to maintain cell integrity but also contributes to the acquisition of beneficial sequences (Navarre, 2016). Interestingly, within the LEE, the first gene of the LEE1 operon encodes Ler (LEE-encoded regulator), a protein belonging to the H-NS family of nucleoid-associated proteins, which is the central positive regulator controlling the expression of LEE genes, and genes located outside the LEE, by counteracting the silencing effect exerted by H-NS on the LEE operons (Elliott et al., 2000; Sperandio et al., 2000; Bustamante et al., 2001; Sanchez-SanMartin et al., 2001; Haack et al., 2003; Barba et al., 2005; Bingle et al., 2014). Due to its essential role, modulation of LEE1 operon expression is key for activating or repressing all the LEE genes and non-LEE co-regulated genes; thus, its regulation is complex and multifactorial (Furniss and Clements, 2018; Platenkamp and Mellies, 2018; Turner et al., 2019).
The LEE encodes two additional regulatory proteins: GrlA (Global regulator of LEE-activator) and GrlR (Global regulator of LEE-repressor). GrlA shares homology with a small number of predicted uncharacterized proteins in different bacterial species and with CaiF, an activator of genes involved in carnitine utilization (Deng et al., 2004; Jimenez et al., 2010). A predicted helix-turn-helix (HTH) motif is found at the N-terminus of these proteins, where most of the conservation is observed. GrlA binds to the ler promoter to positively regulate its expression (Huang and Syu, 2008; Jimenez et al., 2010; Bustamante et al., 2011; Padavannil et al., 2013), while Ler also induces the grlRA operon establishing a positive regulatory loop that enhances LEE gene expression under inducing conditions (Barba et al., 2005). Point mutations at the HTH motif affect the activation function of GrlA (Jimenez et al., 2010; Islam et al., 2011; Padavannil et al., 2013). Moreover, GrlA is present in the cell in a membrane-associated inactive state that responds to mechanical stimuli (Alsharif et al., 2015; Sirisaengtaksin et al., 2020).
In contrast, GrlR acts as a repressor by forming a dimeric structure of antiparallel beta-barrel subunits with a molecular mass of 29 kDa, which exerts its function as a negative regulator through the interaction with the HTH motif of GrlA, forming a complex with a molecular mass of 47.3 kDa that prevents the activation of the ler promoter (Deng et al., 2004; Lio and Syu, 2004; Iyoda and Watanabe, 2005; Jobichen et al., 2007; Jimenez et al., 2010; Padavannil et al., 2013). Orthologs of GrlR are found in species of the genera Proteus, Morganella, Serratia, Klebsiella and Salmonella, among others, sharing between 30 to 42% identity; however, these GrlR orthologs are hypothetical proteins with no assigned function yet. The ClpXP complex, an AAA + protease, positively controls LEE gene expression in EHEC through direct regulation of GrlR levels during the stationary phase of growth (Iyoda and Watanabe, 2005). Moreover, Hfq, an RNA chaperone, together with the small RNAs MgrR, RyhB and McaS, negatively modulate LEE gene expression in EPEC at the post-transcriptional level by destabilizing the grlRA mRNA and, consequently, the expression of GrlR and GrlA (Hansen and Kaper, 2009; Bhatt et al., 2017; Sudo et al., 2022). Furthermore, GrlA and GrlR also regulate the expression of genes located outside the LEE, such as the hemolysin and flagellar genes in EHEC (Iyoda et al., 2006; Saitoh et al., 2008), as well as some non-LEE encoded effector genes in EPEC (Garcia-Angulo et al., 2012), indicating that during the evolution of A/E organisms other genes were incorporated into the Ler-GrlRA regulatory network to coordinate other functions that enhanced the pathogenic capabilities of these bacteria.
Despite the current knowledge, the mechanisms underlying the interplay between GrlA and GrlR in regulating LEE and non-LEE genes under different environmental conditions are still poorly understood. In this work, we show that in addition to the notion that GrlR functions as a repressor by forming a complex with GrlA, it can also act as a repressor of LEE genes independently of this interaction, while GrlA has a dual role as a positive regulator by also antagonizing GrlR through protein-protein interactions. Our data further illustrate that LEE gene expression is negatively regulated at two levels mediated by global (H-NS) and EPEC-specific (GrlR) regulators.
Materials and methods
Bacterial strains, plasmids, and culture conditions
The bacterial strains and plasmids used in this study are listed in Supplementary Table 1. Bacteria were routinely cultured in Lysogeny Broth (LB) or in Dulbecco’s modified Eagle’s medium (DMEM) containing glucose [0.45% (wt/vol)] and L-glutamine (584 mg/l), but not sodium pyruvate (Gibco-BRL Life Technologies, Waltham, MA, USA), supplemented with 1% LB. When necessary, antibiotics were added at the following concentrations: kanamycin (Km) 30 μg ml–1, ampicillin (Ap) 100 μg ml–1, tetracycline (Tc) 12 μg ml–1, streptomycin (Sm) 100 μg ml–1 and chloramphenicol (Cm) 25 μg ml–1. To induce the expression of the LEE virulence genes, bacteria were grown either in 50 ml DMEM under shaken or static + 5% CO2 conditions at 37°C, while shaken LB broth was used as the non-inducing or repressing condition (Martinez-Laguna et al., 1999; Bustamante et al., 2001, 2011).
DNA manipulations
Standard genetic and molecular techniques were applied as described previously (Sambrook and Russell, 2001). Restriction enzymes were obtained from Thermo Scientific, Waltham, MA, USA and used according to the manufacturer’s instructions. PCR reactions were performed in 50 μl using Platinum Taq DNA polymerase (Invitrogen, Waltham, MA, USA). The oligonucleotides used for PCR amplification were synthesized at the Oligonucleotide Synthesis Facility of the Instituto de Biotecnología/UNAM, Cuernavaca, México, and are listed in Supplementary Table 2.
Construction of plasmids
To construct the pT3GrlR plasmid, a DNA sequence including the RBS and the coding region of grlR was amplified by PCR using the GREPKE-F/GREPX-R oligonucleotide pair. The product obtained was digested with KpnI-XhoI enzymes and ligated into vector pMPM-T3 (Mayer, 1995), digested with the same enzymes. To generate plasmids pT3GrlRA, pT3GrlRA/I44A, and pT3GrlRA/R54A fragments were amplified by PCR with the oligonucleotides XHINTERGRLAF/HIGRLAR (Supplementary Table 2), using plasmids pTEPGrlA1, pTEPGrlA1/I44A, and pTEPGrlA1/R54A as templates (Jimenez et al., 2010), respectively. The resulting products were digested with XhoI-HindIII and cloned into the pT3GrlR plasmid digested with the same enzymes.
The pDnaK-CAT plasmid was constructed by PCR amplifying a fragment comprising from position −394 to + 127 of the transcription start site of the dnaK gene, using the DnaKF/DnaKR oligonucleotides. The product was digested with BamHI-HindIII enzymes and cloned into the pKK232-8 vector (Pharmacia Biotech) (Brosius, 1984), previously digested with the same enzymes.
Chromosomal DNA from the EPEC E2348/69 strain was used as the PCR template. All plasmids generated were verified by DNA sequencing and evaluated for their ability to complement the corresponding mutants through a profile of secreted proteins or western blotting against virulence proteins.
Construction of mutants and strains expressing FLAG-tagged proteins
The grlR and grlA mutants were produced by generating chromosomal in-frame deletions of grlR codons 6 to 118 or grlA codons 6 to 132 by the sacB gene-based allelic exchange method as described previously (Edwards et al., 1998). Deletion of the grlRA operon spans from codon 6 of grlR to codon 132 of grlA. Suicide plasmids were generated by cloning PCR-amplified fragments containing the described deletions flanked by approximately 800 to 1000 bp on each side into XbaI/SacI-digested plasmid pRE112. The resulting suicide plasmids pRE112DgrlREP, pRE112DgrlAEP, and pRE112DgrlRAEP, respectively (Supplementary Table 1), were conjugated into strains WT EPEC E2348/69, Δhns (JPEP36) and Δler to generate single and double mutants (Supplementary Table 1).
To generate the chromosomally 3xFLAG-tagged strains, a modification of the λRed recombinase system was used as described previously (Datsenko and Wanner, 2000; Uzzau et al., 2001). The PCR fragment to tag the native grlR gene was generated using oligonucleotides grlR-FLAGH1P1 and grlR-FLAGH2P2 and the grlA gene using oligonucleotides grlA-FLAGH1P1 and grlA-FLAGH2P2, and plasmid pSUB11 DNA as template. The resulting PCR products were electroporated into WT EPEC or its ΔgrlR or ΔgrlA mutants to generate the EPEC grlR:3xFLAG, grlA:3xFLAG, grlR:3xFLAG ΔgrlA and ΔgrlR grlA:3xFLAG (Supplementary Table 1).
Single and double mutants were verified by PCR amplification and DNA sequencing. The tagged strains contain the tag in their native gene chromosomal locations; thus, are expressed from the grlRA operon promoter.
CAT assay
Chloramphenicol acetyltransferase (CAT) activity, derived from the expression of the transcriptional fusions to the cat reporter gene, was determined as follows (Martinez-Laguna et al., 1999). The strains containing the transcriptional fusions were grown in 5 ml of LB supplemented with antibiotics and incubated overnight at 37°C. The next day, culture pellets were adjusted to an OD600 = 1.0 with 1X phosphate buffered saline (PBS) (10 mM Na2HPO4, 2 mM KH2PO4, 137 mM NaCl and 2.7 mM KCl) and used to inoculate 50 ml of LB or DMEM supplemented with antibiotics with one ml of each suspension. Cultures were incubated at 37°C under shaking or static plus 5% CO2 growth conditions, and 1 ml samples were collected when the cultures reached an OD600 = 0.8 and 1.0. The cell pellet was obtained by centrifugation at 14,000 rpm for 2 minutes and washed with 1 ml of TDTT buffer (Tris-Hcl 50 mM pH 7.8 and dithiothreitol 30 μM), centrifuged again and resuspended in 0.5 ml of the same buffer. Lysis was achieved by sonication for 5 min with pulses of 10 s per minute and 5 s rest. Soluble extracts were separated from cell debris by centrifugation at 12,000 rpm for 20 min at 4°C, and 5 μl aliquots of each extract were added in duplicate to a 96-well microtiter plate followed by the addition of 200 μl of the reaction mix containing 1 mM DTNB [5,58-dithio-bis(2-nitrobenzoic acid)] (Research Organics), 0.1 mM Tris-Hcl pH 7.8, 0.1 mM acetyl-CoA (Pharmacia LKB Biotech Inc., Alameda, CA, USA), and 0.1 mM chloramphenicol (Sigma Chemical Co., Saint Louis, MO, USA). Changes in absorbance at 410 nm were recorded every 5 s for 5 min with a CERES 900C automatic microplate reader and the KC3 program set to kinetic mode. A CAT standard curve (from 0 to 2500 U/ml) was used to interpolate the activities of each sample. The protein concentration of each extract was determined by adding 10 μl of each extract in duplicate and 200 μl of the reaction mixture (25 ml of solution A + 500 μl of solution B) of the “BCA Protein Assay Kit” (Pierce) to a 96-well plate. Then incubated at 37°C for 30 min; the absorbance was read at a wavelength of 562 nm using an automatic microplate reader CERES 900 C and the KC3 program. The CAT-specific activity was determined by dividing the CAT activity by the protein concentration of each extract. The data are expressed as μmol/min/mg of protein and are the results from at least three independent assays done in duplicate. The empty vector pKK232-8 did not show measurable levels of CAT activity in any of the strains used in the study (data not shown).
Protein secretion assay
EPEC secreted proteins were analyzed as described previously (Deng et al., 2004). Briefly, bacterial cultures in LB and DMEM were incubated at 37°C under shaking or static conditions. Triplicate 1.5 ml samples were collected per culture when the cultures reached an OD600 of 1.0 and were subjected to centrifugation at 17,900 x g for 5 min in Eppendorf tubes. Bacterial pellets were saved when convenient, and 1.3 ml of each supernatant was separated into fresh tubes and 160 μl of 100% trichloroacetic acid (TCA) was added per tube. Proteins were allowed to precipitate at 4°C overnight. Subsequently, the proteins were concentrated by centrifugation at 20,000 × g for 30 min and resuspended in 10 μl of 1× Laemmli loading buffer and the triplicate samples were mixed in the same tube, boiled for 5 min and resolved by 12% SDS-polyacrylamide gel electrophoresis (PAGE).
Western blotting
Bacteria were grown in DMEM or LB under shaking or static conditions until they reached an OD600 of 1. Total extracts were obtained from 3 ml samples of the bacterial cultures. Cells were resuspended in 500 μl of urea solution (8 M) and lysed by sonication. Aliquots of total extracts were mixed with Laemmli buffer, boiled for 5 min and analyzed by SDS-PAGE (12% acrylamide) and, subsequently, transferred to 0.45-μm-pores-size PVDF membrane (Millipore) using a semi-dry transfer chamber (BioRad, Hercules, CA, USA).
The membrane was blocked in 10% non-fat milk and incubated with one of the following primary antibodies: a 1:10,000 dilution of monoclonal anti-Tir, a 1:7,500 dilution of polyclonal anti-intimin (kindly provided by J.A. Giron), a 1:20,000 dilution of polyclonal anti-EspA (kindly provided by J. Kaper), a 1:10,000 dilution of polyclonal anti-EscJ (kindly provided by Dr. Bertha González-Pedrajo), a 1:5,000 dilution of monoclonal anti-FLAG M2 (Sigma-Aldrich, Saint Louis, MO, USA), a 1:15,000 dilution of polyclonal anti-maltose binding protein (MBP) (New England Biolabs, Ipswich, MA, USA), a 1:10,000 dilution of polyclonal anti-DnaK (Invitrogen, Waltham, MA, USA) and a 1:50,000 dilution of monoclonal anti-GroEL (Sigma-Aldrich, Saint Louis, MO, USA). The membrane was washed with 1x phosphate buffer saline (PBS) containing 0.05% Tween 20 and incubated with a 1:10,000 dilution of secondary anti-rabbit or anti-mouse antibodies coupled to horseradish peroxidase (Thermo Scientific, Waltham, MA, USA). The membrane was developed with the commercial kit “Western Lightning Chemiluminescence Reagent Plus” (Perkin Elmer, Waltham, MA, USA) according to the manufacturer’s instructions.
Native gel electrophoresis
EPEC E2348/69 grlR:3xFLAG, grlA:3xFLAG, ΔgrlR grlA:3xFLAG, and grlR:3xFLAG ΔgrlA strains were transformed with pT3GrlA1/I44A or pT3GrlA1/R54A and grown overnight in 5 ml of LB medium supplemented with antibiotics. The next day, 0.5 ml of these cultures were inoculated into 20 ml DMEM or LB medium supplemented with antibiotics and allowed to grow at 37°C with shaking. When the cultures reached an OD600 of 1.0, 5 ml samples were taken and the cells were concentrated by centrifugation at 18,000 xg at 4°C, resuspended in 500 μl PBS and lysed by sonication. Samples were taken at an OD600 of 0.3, 0.6, 0.8, and 1.0 from 50 ml cultures when needed.
Total extracts were resuspended in a non-denaturing buffer (50% glycerol, 25 mM Tris-Hcl pH 6.8 and 0.05% bromophenol Blue) and separated by 12% native PAGE at 4°C. The gel was transferred to a 0.45-μm-pore-size PVDF membrane (Millipore) and processed for western blotting.
Simultaneously, aliquots obtained from the same assays were processed for 12% SDS-PAGE and western blotting.
Statistical analysis
Data analyses were performed by two-way ANOVA followed by Tukey’s multiple comparisons posttest using GraphPad Prism version 8.4.3 (471) for Mac OS. P-values < 0.05 were considered statistically significant.
Results
GrlR negatively regulates LEE gene expression under repression conditions
GrlR represses LEE gene expression mainly under non-permissive conditions such as growth in LB (Deng et al., 2004; Lio and Syu, 2004; Iyoda and Watanabe, 2005). To further explore this feature, we evaluated the expression of the LEE1, LEE2, LEE4, LEE5, and dnaK promoters fused to the cat reporter gene in WT EPEC E2348/69 and its ΔgrlR non-polar deletion mutant cultured under LEE inducing (DMEM/37°C) and repressing (LB/37°C) growth conditions. Under inducing conditions, the CAT reporter activity of all LEE fusions increased between 1.5- to 4-fold in EPEC ΔgrlR compared to the WT strain (Figure 1A); however, under repression conditions, the fold increase was higher, between 5 and 13.5 (Figure 1A). In contrast, dnaK-cat expression, used as a LEE unrelated control, was similar in both strains and growth conditions (Figure 1A).
The analysis of the T3SS-dependent protein secretion profile of WT, ΔgrlR and Δler EPEC strains showed that, under repressing conditions (LB), the ΔgrlR mutant abundantly secreted proteins into the medium, while the WT strain did not secrete detectable levels of LEE-encoded proteins (Figure 1B). This phenotype reversed by complementing the mutant strain with a plasmid containing the grlR gene (pT3GrlR) (Figure 1B). Growth under inducing conditions (DMEM) allowed secretion of LEE-encoded proteins by the WT strain and higher secretion levels by the ΔgrlR strain. Moreover, complementing the ΔgrlR mutant with the pT3GrlR plasmid suppressed protein secretion, even under inducing conditions (DMEM), indicating that GrlR overexpression can override the presence of GrlA under T3S permissive conditions (Figure 1B). As expected, the Δler mutant did not secrete under both growth conditions.
These results highlight the negative regulatory effect that GrlR exerts on the LEE genes in agreement with previous reports (Deng et al., 2004; Lio and Syu, 2004; Iyoda and Watanabe, 2005).
GrlR represses the expression of LEE operons in the absence of H-NS
H-NS globally represses LEE-gene expression, while Ler acts as an antagonist to overcome this repression; thus, in the absence of H-NS, the role of Ler is dispensable (Friedberg et al., 1999; Leh et al., 2017; Shin, 2017). Moreover, GrlR expression from a plasmid strongly represses the expression of the LEE genes in WT EPEC even under conditions permissive to LEE expression (Deng et al., 2004; Lio and Syu, 2004; Jobichen et al., 2007). Evidence showing that GrlR and GrlA establish protein-protein interactions led to propose that GrlR negatively regulates LEE gene expression by preventing GrlA from binding to and activating the ler promoter (Creasey et al., 2003; Jobichen et al., 2007; Jimenez et al., 2010; Padavannil et al., 2013).
Based on such evidence, we hypothesized that overexpression of GrlR in EPEC lacking H-NS, which expresses LEE genes constitutively even under non-permissive growth conditions in a Ler- and GrlA-independent manner, would not affect LEE gene expression. Therefore, we analyzed the transcriptional activity of the LEE1-, LEE2-, LEE4-, and LEE5-cat fusions and the expression of LEE-encoded proteins in WT and Δhns EPEC strains carrying or not plasmid pT3GrlR (Supplementary Table 1). Consistent with previous reports for other A/E pathogens, overexpression of GrlR in WT EPEC repressed the transcriptional activity of all LEE fusions tested (Figure 2A), as well as the expression of Tir, intimin and EspA, as shown by western blot (Figure 2B), and of EspC (Supplementary Figure 1A), an autotransporter encoded outside the LEE and whose expression is also regulated by Ler (Elliott et al., 2000; Li et al., 2004; Vidal and Navarro-Garcia, 2008). Unexpectedly, GrlR also significantly repressed the transcriptional activity of the LEE promoters in the EPEC Δhns mutant (Figures 2A, B). Under these conditions, all strains similarly expressed proteins DnaK and MBP, used as loading controls, indicating that repression did not result from an unspecific pleiotropic effect in EPEC physiology due to GrlR overexpression.
To address this concept, we evaluated the effect of GrlR expressed from a plasmid under growth conditions where GrlA is not the primary activator of Ler expression. When EPEC grows in static DMEM cultures under 5% CO2 atmosphere at 37°C, the EAF plasmid-encoded regulator PerC is the main activator of LEE1 promoter expression and therefore of ler (Bustamante et al., 2011). Thus, we analyzed the secreted protein profile and intimin and EspA expression in WT EPEC and the Δhns mutant harboring plasmid pT3GrlR or the empty vector pMPM-T3, grown in shaken and static + 5% CO2 DMEM. As shown, GrlR expressed from pT3GrlR repressed protein secretion (Supplementary Figure 1A) and expression of EspA and Intimin in both strains and conditions, but not that of DnaK (Supplementary Figure 1B), confirming the results shown in Figure 1B.
These data suggested that GrlR can repress LEE gene expression through an alternative pathway, probably independent of its interaction with GrlA.
The global regulator H-NS and the EPEC specific regulator GrlR act cooperatively, but at different levels, to repress the expression of LEE genes
Ler and GrlA establish a positive regulatory feedback loop that counteracts the repression exerted by H-NS (Barba et al., 2005; Jimenez et al., 2010). According to the results described above, GrlR and H-NS act cooperatively to repress the expression of LEE genes. To further explore this observation, we evaluated the expression of the LEE1-cat, LEE2-cat, and LEE5-cat transcriptional fusions in EPEC WT and Δhns grown under inducing and repressing conditions and also analyzed the secretion profile of these strains from samples of their culture supernatants. Under inducing conditions, the activity of the LEE2-cat and LEE5-cat fusions increased about 2-fold in the absence of either of the two repressors, whereas that of the LEE1-cat fusion was 4.7-fold and 3.7-fold in the ΔgrlR and Δhns mutants, respectively (Figure 3A). Under repressing conditions, the derepression observed for the LEE1, LEE2, and LEE5 promoters operons was about 13-, 7-, and 14-fold, respectively, in the absence of GrlR; whereas in the Δhns mutant it was 2-, 3-, and 5-fold, respectively, relative to the activity displayed in the WT strain (Figure 3A). In agreement with these results, while the WT strain poorly secreted LEE-encoded proteins under repressing conditions (LB), the ΔgrlR and Δhns mutants showed a clear increase in protein secretion (Figure 3B).
To delve deeper into the role of both H-NS and GrlR in the negative regulation of the LEE genes, we analyzed the transcriptional activity of the LEE2-cat fusion and the protein secretion profile in WT EPEC and its Δler, Δhns, ΔgrlR, ΔgrlA, ΔgrlRA, ΔlerΔgrlR, and ΔgrlRΔhns isogenic mutants grown under inducing and repressing conditions (Figure 4). As expected, compared to the WT strain, the Δler and ΔgrlA strains significantly downregulated LEE2-cat fusion expression and protein secretion, while the ΔgrlR mutant expressed higher levels of LEE2-cat activity (Figures 4A, B). Interestingly, the ΔgrlRA double mutant displayed a phenotype similar to the ΔgrlA single mutant, confirming the important role of GrlA in ler activation and, indirectly, in derepression of Ler-dependent promoters that in the absence of GrlR remained repressed by H-NS (Figures 4A, B). In the ΔgrlRΔhns double mutant, the transcriptional activity of the LEE2-cat fusion showed expression levels similar to those observed in the ΔgrlR and Δhns single mutants under inducing conditions, while under repressing conditions, its expression was closer to the activity observed in the ΔgrlR strain (Figure 4A). These results coincided with the secreted protein profile since, in DMEM and LB, the ΔgrlR and Δhns single mutants and the double ΔgrlRΔhns mutant showed increased secretion in contrast to the WT strain (Figure 4B). Finally, the phenotype of the ΔgrlRΔler mutant confirmed that Ler is essential to antagonizing the repression exerted by H-NS regardless of the growth condition (Figures 4A, B).
The inclusion of the ΔlerΔhns double and the ΔlerΔhnsΔgrlRA quadruple mutant in this assay was cumbersome due to the poor growth shown by these strains. To overcome this limitation, we expressed ectopically H-NSG113D, a dominant negative version of H-NS that carries a mutation affecting its DNA binding capacity without altering the oligomerization domain (Ueguchi et al., 1996), to inhibit H-NS activity. WT EPEC and its mutant derivatives Δler, Δhns, ΔgrlR, ΔgrlA, ΔgrlRA, ΔlerΔgrlR,ΔlerΔgrlA, ΔlerΔgrlRA and ΔescN carrying plasmid pT6-HNS/G113D, expressing H-NSG113D under the control of an arabinose inducible promoter (Bustamante et al., 2008; Jimenez et al., 2010), were grown under repressing conditions with and without arabinose. We collected samples from these cultures to analyze the secreted proteins by PAGE and the expression of Tir and EscJ in total cell extracts by western blotting. In the absence of arabinose, we did not observed T3S of EspA, EspB and EspD nor Tir and EscJ expression in the WT or any strain carrying the Δler or ΔgrlA deletions (Figures 5A, B). In contrast, upon induction of H-NSG113D, de-repression of T3S was evident even in the absence of Ler and/or GrlA, while the ΔgrlR mutant showed protein secretion and Tir and EscJ expression both without or with H-NSG113D induction (Figures 5A, B). The ΔescN strain, a mutant defective in T3S, was used as a control that did not secrete virulence proteins even upon H-NSG113D induction, except for EspC, a non-T3 secreted protein, whose gene is also repressed by H-NS and derepressed by Ler (Figure 5A). In contrast, Tir and EscJ were observed in total cell extracts of this strain when H-NSG113D was induced (Figure 5B).
Taken together, these results indicate that GrlR and H-NS negatively regulate the expression of LEE operons at two independent levels that are indirectly cooperative (see Section “Discussion”).
GrlA counteracts the repressor effect of GrlR through protein-protein interactions
We previously observed that under inducing conditions, the negative effect of GrlR is counteracted when co-expressed with GrlA in C. rodentium (Deng et al., 2004). A possible explanation of this phenotype is that the co-expression of both proteins prevents the titration effect that overexpressing only GrlR from a plasmid has on the native levels of GrlA. However, there is also the possibility that under these conditions, the interaction between GrlR and GrlA also plays a role in preventing the GrlR-mediated independent repression described above (Figure 2 and Supplementary Figure 1).
To evaluate this possibility, we transformed plasmids pT3GrlRA, pT3GrlR, pTEPGrlA1 and the empty vector pMPM-T3 (Supplementary Table 1) into WT EPEC and grew the resulting strains under inducing and repressing conditions to analyze the secretion profile. As shown in Figure 2, GrlR overexpression repressed the LEE genes and inhibited T3S even under inducing conditions, while overexpression of GrlA overcame the repressing effect of growth in LB (Figure 6A). In turn, as observed in C. rodentium (Deng et al., 2004), GrlR-GrlA co-expression prevented the negative effect of GrlR, even under repressing conditions, but to a lesser extent (Figure 6A).
To further explore this notion, we took advantage of the phenotype of two GrlA mutants, GrlA/I44A and GrlA/R54A, that cannot activate the LEE1 operon because they no longer bind to the ler regulatory region due to a single amino acid change in the HTH domain. Additionally, the GrlA/I44A mutant cannot interact with GrlR (Jimenez et al., 2010; Padavannil et al., 2013). The genes encoding these mutants were cloned into pT3GrlR to co-express them with WT grlR. WT EPEC containing the resulting plasmids pT3GrlRA/I44A or pT3GrlRA/R54A were then grown under inducing and repressing conditions to examine their T3S profile. Under inducing conditions, T3S in the presence of pT3GrlRA/I44A was similar to the secretion profile seen with pT3GrlR, as in both cases protein secretion was significantly reduced (Figure 6A). In contrast, in the presence of GrlR-GrlA/R54A, T3S was unaltered (Figure 6A), suggesting that GrlA/R54A probably prevented GrlR-mediated repression by interacting with it and not because it was over inducing Ler expression. Moreover, in LB medium, where the WT strain does not secrete T3S substrates, we observed a clear de-repression of virulence protein secretion in the presence of GrlR-GrlA/R54A, but not with GrlR-GrlA/I44A, suggesting that GrlA/R54A titrated away both the endogenous GrlR expressed from the chromosomal gene and that expressed from the plasmid (Figure 6A). To confirm the above observations, we performed a similar experiment with the EPEC ΔgrlR strain but grown under repressing conditions (LB medium). Co-expression of GrlR with GrlA/I44A did not prevent GrlR from complementing the repression phenotype and the effect was similar to that observed for the strain with pT3GrlR (Figure 6B); however, this was not the case when GrlR was co-expressed with GrlA/R54A, as protein secretion was still observed (Figure 6B). Western blotting to detect intimin, Tir and EspA in total cell extracts of the same strains, showed similar results (Figure 6C).
These results are consistent with the notion that GrlA, in addition to its activating effect on the ler promoter as a DNA binding protein, could also counteract the alternative GrlR repression effect by interacting with it.
GrlR and GrlA are expressed and interact under inducing and repressing conditions
The results described above further indicated that GrlR has a critical role as a repressor under non-permissive growth conditions for LEE expression, thus implying that EPEC expresses it in both LB and DMEM. To analyze GrlR and GrlA expression and their interaction in more detail, we grew the EPEC grlR:3xFLAG, grlA:3xFLAG, ΔgrlA-grlR:3xFLAG and ΔgrlR-grlA:3xFLAG strains, as well as the ΔgrlA-grlR:3xFLAG strain carrying the plasmids encoding the GrlA/I44A and GrlA/R54A mutants (Supplementary Table 1), under inducing and repressing conditions and analyzed total cell extracts by native PAGE and western blotting from samples taken from each culture. As shown in Figure 7, the formation of the GrlR-GrlA complex occurred under both growth conditions (Figure 7A, lanes 1, 2, 7, and 8, upper band). The EPEC grlR:3xFLAG strain also showed the GrlR dimer and monomer (Figure 7A, lanes 1 and 7), while the banding pattern of the ΔgrlA-grlR:3xFLAG strain, where the upper band is not present due to the absence of GrlA (Figure 7A, lines 3 and 9), confirmed the nature of the second and third bands. The EPEC grlA:3xFLAG strain only showed a band corresponding to the GrlR-GrlA complex (Figure 7A, lines 2 and 8), while in the absence of GrlR, GrlA-3xFLAG did not enter the gel (Figure 7A, lines 4 and 10), likely due to its isoelectric point (theoretical pI: 9.71), which changes when in complex with GrlR. Moreover, the expression of the plasmid-encoded GrlA/I44A and GrlA/R54A mutants in the ΔgrlA-grlR:3xFLAG strain further confirmed these observations as the GrlA/I44A mutant, which does not interact with GrlR, did not form the GrlR2-GrlA complex (Figure 7A, lines 5 and 11) seen with the GrlA/R54A mutant (Figure 7A, lines 6 and 12). Interestingly, the GrlA/R54A mutant drove GrlR to mainly form the complex with GrlA since the dimeric and monomeric forms were not visible. In part, this may be why overexpression of GrlA in the EPEC WT strain overrode the repression in LB (Figure 6A). SDS-PAGE and anti-FLAG western blot with the same cell extracts showed the presence of the tagged proteins (Figure 7B).
Native PAGE analysis also suggested, based on the banding pattern shown by GrlR and GrlA (as discussed above), that GrlR and GrlA are expressed and interact, although not to the same extent, independently of the growth conditions. To analyze this observation in more detail, we took samples from LB and DMEM cultures of the EPEC grlR:3xFLAG and grlA:3xFLAG strains at different growth stages and analyzed them by SDS-PAGE and native PAGE followed by western blot. The qualitative analysis showed that GrlR and GrlA were expressed at moderately higher levels in DMEM than in LB; however, although enriched in DMEM, heterotrimers formed under both growth conditions independently of the expression levels of these proteins (Figures 8A, B, upper panels).
These data allowed us to hypothesize that heterotrimer formation plays a bidirectional role, on the one hand, preventing the activation of ler by reducing the levels of free GrlA and, on the other, antagonizing the independent repressor function of GrlR. A future more quantitative analysis of the relative concentrations of GrlA under both growth conditions, as well as the effect of these conditions on the ability of GrlA to activate the ler promoter and on the kinetics of GrlR-GrlA interaction, will allow a better understanding of the mechanism that regulates the expression of LEE genes in response to environmental cues.
Together, these results further supported the notion that GrlA counteracts the repressor effect of GrlR by interacting with it and showed that the GrlR2-GrlA heterotrimeric complex, the GrlR-GrlR dimer and the GrlR monomer are present in EPEC under both growth conditions.
GrlR represses the expression of LEE promoters in a non-EPEC background
To further explore the notion that GrlR can repress the constitutive expression of LEE promoters independently of its interaction with GrlA, we assessed the activity of the LEE1 and LEE2 promoters in a background lacking not only H-NS, but also GrlA and Ler. We then transformed the empty vector pMPM-T3 or plasmids pT3GrlR, pTEPGrlA1 o pT3GrlRA into the Δhns mutant of the LEE-negative lab strain E. coli MC4100 (JPMC1) containing the LEE1-cat or LEE2-cat fusion. The WT strain was the control, where the LEE1-cat fusion, but not the LEE2-cat fusion, was active in the presence of pTEPGrlA1 (Figure 9A), as previously shown (Bustamante et al., 2011). Interestingly, GrlR also repressed the constitutive expression of the LEE1-cat and LEE2-cat fusions in the Δhns background but not when co-expressed with GrlA (Figure 9B). This result supported the proposal that GrlR also negatively regulates LEE gene expression independently of its interaction with GrlA. Also, GrlA had a dual role as a positive regulator by, in addition to its function as a ler activator, preventing GrlR’s negative effect, most likely by trapping it in the heterotrimer. Defining the underlying mechanism by which GrlR represses the constitutive expression of the LEE promoters will require further investigation.
Discussion
In this study, we further investigated the role of GrlR as a repressor and the function of its interaction with GrlA in modulating LEE gene expression. Our results revealed a novel feature of its mechanism of action as GrlR also seems to act as a repressor independently of its interaction with GrlA; however, it remains to be seen if GrlR exerts this function by directly interacting with Ler-dependent promoters or with Ler-derived transcripts recognizing a common motif or indirectly by interacting with or modulating the function of a conserved element that is also present in E. coli K12.
A summary of the events leading to the control of LEE gene expression in response to the growth conditions is shown in Figure 10 and described below. GrlR and H-NS act independently to negatively regulate the LEE at two different levels, establishing an indirect cooperative repressive effect on the expression of LEE operons when EPEC is grown under repressing conditions (Figures 10A, F, red dotted lines). In the WT strain, H-NS silences Ler-dependent promoters (Figure 10A), while GrlR inactivates GrlA by forming GrlR2-GrlA heterotrimers previously shown to prevent GrlA binding to the ler promoter (Jimenez et al., 2010; Padavannil et al., 2013; Figure 10F). When GrlR is not present, free GrlA efficiently activates ler expression (Figure 10C), leading to Ler-dependent elimination of H-NS-mediated repression of LEE promoters (Figure 10B), thus resembling the phenotype shown by the double ΔgrlRΔhns mutant; however, mutants lacking grlR but also ler or grlA, cannot activate LEE gene expression because H-NS is still present and there is no Ler to overcome this repression.
In addition to identifying the role of GrlR as a negative regulator independent of its interaction with GrlA, our findings show that GrlA plays a dual role in the positive regulation of LEE genes by binding to the ler regulatory region and activating its expression (Jimenez et al., 2010; Islam et al., 2011), and antagonizing the independent repressor activity of GrlR through the formation of the GrlR2-GrlA heterotrimer (Figures 10C, D, respectively); thus establishing an indirect coherent-positive feedforward loop (Shoval and Alon, 2010).
How the growth conditions determine whether EPEC induces or represses the LEE genes when both proteins are present and interact needs to be clearly understood; however, current knowledge and the results described here open up interesting possibilities for future investigation. For example, GrlR, in addition to reciprocally antagonizing GrlA through their interaction, may also interact with other proteins to modulate the transcriptional activity of LEE genes or directly with DNA promoter regions or the RNA transcripts derived from Ler-regulated genes. Along with our results, reports showing that GrlA activity in EHEC is mechanoresponsive and modulated by its membrane-bound state (Sirisaengtaksin et al., 2020), that Hfq differentially regulates GrlR and GrlA synthesis (Hansen and Kaper, 2009; Bhatt et al., 2017; Sudo et al., 2022) and that ClpXP posttranslationally regulates GrlR at the stationary phase (Iyoda and Watanabe, 2005), illustrate that there is more to learn about the mechanism of action of the GrlR and GrlA duet. How the growth conditions impact the GrlR and GrlA ratio in the cell, which seems to have a profound effect on the function and interaction between GrlR and GrlA, and thus in LEE gene expression, warrants future investigation.
The presence of the GrlR2-GrlA complex, GrlR dimer and its monomer under both growth conditions, which is consistent with previous observations indicating that the basal activity of the grlRA promoter is higher than most of the LEE promoters under non-inducing conditions (Yerushalmi et al., 2014), indicate that certain levels of free GrlA are also present at any given moment during growth. Thus, the growth conditions must also play a role in determining GrlR and GrlA competency to repress or activate, respectively, when the complex is disrupted, either due to its natural dissociation or by the influence of additional environmental factors. GrlR can displace GrlA from the ler promoter (Padavannil et al., 2013) and here we showed that overexpression of GrlR or GrlA drives the other protein to predominantly form the GrlR2-GrlA complex (Figures 10D, E). In this complex, they reciprocally antagonize each other and, consequently, free GrlR or GrlA would act without competition with the other protein to either strongly repress (Figure 10F) or activate (Figure 10C) LEE gene expression, respectively. Thus, this dynamic behavior is likely modulating the activation or repression state of the LEE, whose expression has, unsurprisingly, been shown to be bimodal and render a fitness advantage to the cell (Leh et al., 2017; Ronin et al., 2017).
The acquisition of a pathogenicity island imposes on bacteria the need to adopt and adapt pre-existent and horizontally acquired regulatory mechanisms to prevent the deleterious expression of recently incorporated genes (Navarre, 2016). Although LEE gene expression is modulated by a complex assortment of global regulatory proteins, its specific activation relies on the LEE-encoded regulators Ler and GrlA. In contrast, its negative control is mediated at two levels by a global regulator (H-NS) and an EPEC-specific regulator (GrlR). In this context, self-regulation of gene expression mediated by GrlR and GrlA may have provided a fitness advantage during the acquisition of the LEE, where GrlR down-regulated its uncontrolled expression preventing detrimental effects on bacterial fitness, while GrlA counteracted this self-encoded sentinel function. It is tempting to speculate that the incorporation of pre-existing or ancestral regulatory mechanisms, such as the repression exerted by H-NS to finetune LEE gene expression further, led to the incorporation of additional regulatory proteins, such as Ler, as well as other regulators that are known to modulate Ler expression in response to various environmental, stress or metabolic signals (Furniss and Clements, 2018; Platenkamp and Mellies, 2018; Turner et al., 2019).
In line with the diversification and opposing effects of GrlR and GrlA functions, a ΔgrlR mutant showed reduced levels of flagellin (FliC) expression and motility, while overexpression of GrlA had the opposite effect suggesting that GrlA may be acting directly as a repressor on flagellar genes (Iyoda et al., 2006; Kitagawa et al., 2011). Iyoda’s work also indicated that GrlA represses the expression of flhD, which encodes a master regulator of flagellar gene expression; consistently, GrlA binds to the flhDC regulatory region, while GrlR outcompetes this interaction (Padavannil et al., 2013), further supporting the notion that GrlA can also act as a repressor. Although details of the underlying mechanism remain undescribed, in the context of our work, the previously cited papers support the notion that the GrlR2:GrlA interaction plays a reciprocal modulatory role in GrlR and GrlA activities.
Overall, LEE regulation is a fine example of the integration of ancestral and horizontally acquired regulators that now comprise a complex and coordinated set of network motifs (Shoval and Alon, 2010), orchestrating a consensus response to a myriad of signals concurring to regulate the expression of the LEE genes. For example, Ler negatively regulates its own expression, establishing a checkpoint that prevents the overexpression of the LEE (Figure 10G; Berdichevsky et al., 2005), while Ler and GrlA represent a positive feedback loop (Figures 10C, H; Barba et al., 2005); however, depending on the growth conditions, activation of the grlRA operon by Ler may also lead to an indirect coherent-negative feedforward loop where GrlR inactivates GrlA through the formation of heterotrimers and independently represses the ler and other LEE promoters (Figures 10E, F; Jimenez et al., 2010; Padavannil et al., 2013). In contrast, GrlA controls an indirect coherent-positive feedforward loop by activating ler expression and antagonizing GrlR (Figures 10C, D; Jimenez et al., 2010; Islam et al., 2011; Padavannil et al., 2013; this work). Deciphering the unknown nuances of this intertwined regulatory network and the implications for virulence gene regulation during the transit of the pathogen from the environment to the host or within the host during the colonization process is an exciting challenge for future research.
Data availability statement
The original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
CL-O and JP conceived the project, designed the experiments, and wrote the original draft of the manuscript with input and edits from all authors. CL-O, AH-S, WD, and AM-L performed the experiments. CL-O, AH-S, AM-L, WD, BF, YM-L, and JP performed data analysis. BF, YM-L, and JP provided funding. All authors approved the submitted version.
Funding
This work was supported by Consejo Nacional de Ciencia y Tecnología (60796, 239659, and FC-2015-2/950) and Dirección General de Personal Académico (PAPIIT IN224107 and IN218322) to JP and Programa institucional del fortalecimiento a grupos de investigación de la VIEP/BUAP and a CIHR Foundation grant to BF.
Acknowledgments
We are particularly thankful to A. Vázquez for mutant construction, B. González-Pedrajo for the generous donation of anti-Tir, -EspA and -Intimin antibodies, F. Santana for technical assistance, R. Jiménez for sharing constructs and J. A. Ibarra for valuable comments on the manuscript. We also thank the Oligonucleotide Synthesis Facility of the Instituto de Biotecnología. CL-O and AM-L were supported by fellowships from CONACyT (165332 and 173678, respectively).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1063368/full#supplementary-material
References
Alsharif, G., Ahmad, S., Islam, M. S., Shah, R., Busby, S. J., and Krachler, A. M. (2015). Host attachment and fluid shear are integrated into a mechanical signal regulating virulence in Escherichia coli O157:H7. Proc. Natl. Acad. Sci. U.S.A. 112, 5503–5508. doi: 10.1073/pnas.1422986112
Barba, J., Bustamante, V. H., Flores-Valdez, M. A., Deng, W., Finlay, B. B., and Puente, J. L. (2005). A positive regulatory loop controls expression of the locus of enterocyte effacement-encoded regulators Ler and GrlA. J. Bacteriol. 187, 7918–7930.
Berdichevsky, T., Friedberg, D., Nadler, C., Rokney, A., Oppenheim, A., and Rosenshine, I. (2005). Ler is a negative autoregulator of the LEE1 operon in enteropathogenic Escherichia coli. J. Bacteriol. 187, 349–357. doi: 10.1128/JB.187.1.349-357.2005
Bhatt, S., Egan, M., Ramirez, J., Xander, C., Jenkins, V., Muche, S., et al. (2017). Hfq and three Hfq-dependent small regulatory RNAs-MgrR, RyhB and McaS-coregulate the locus of enterocyte effacement in enteropathogenic Escherichia coli. Pathog Dis. 75:ftw113. doi: 10.1093/femspd/ftw113
Bingle, L. E., Constantinidou, C., Shaw, R. K., Islam, M. S., Patel, M., Snyder, L. A., et al. (2014). Microarray analysis of the Ler regulon in enteropathogenic and enterohaemorrhagic Escherichia coli strains. PLoS One 9:e80160. doi: 10.1371/journal.pone.0080160
Bustamante, V. H., Martinez, L. C., Santana, F. J., Knodler, L. A., Steele-Mortimer, O., and Puente, J. L. (2008). HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc. Natl. Acad. Sci. U.S.A. 105, 14591–14596. doi: 10.1073/pnas.0801205105
Bustamante, V. H., Santana, F. J., Calva, E., and Puente, J. L. (2001). Transcriptional regulation of type III secretion genes in enteropathogenic Escherichia coli: Ler antagonizes H-NS-dependent repression. Mol. Microbiol. 39, 664–678. doi: 10.1046/j.1365-2958.2001.02209.x
Bustamante, V. H., Villalba, M. I., Garcia-Angulo, V. A., Vazquez, A., Martinez, L. C., Jimenez, R., et al. (2011). PerC and GrlA independently regulate Ler expression in enteropathogenic Escherichia coli. Mol. Microbiol. 82, 398–415. doi: 10.1111/j.1365-2958.2011.07819.x
Creasey, E. A., Delahay, R. M., Daniell, S. J., and Frankel, G. (2003). Yeast two-hybrid system survey of interactions between LEE-encoded proteins of enteropathogenic Escherichia coli. Microbiology 149(Pt 8), 2093–2106. doi: 10.1099/mic.0.26355-0
Croxen, M. A., and Finlay, B. B. (2010). Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8, 26–38.
Croxen, M. A., Law, R. J., Scholz, R., Keeney, K. M., Wlodarska, M., and Finlay, B. B. (2013). Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 26, 822–880. doi: 10.1128/CMR.00022-13
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Deng, W., Puente, J. L., Gruenheid, S., Li, Y., Vallance, B. A., Vazquez, A., et al. (2004). Dissecting virulence: Systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U.S.A. 101, 3597–3602.
Edwards, R. A., Keller, L. H., and Schifferli, D. M. (1998). Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207, 149–157. doi: 10.1016/s0378-1119(97)00619-7
Elliott, S. J., Sperandio, V., Giron, J. A., Shin, S., Mellies, J. L., Wainwright, L., et al. (2000). The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 68, 6115–6126. doi: 10.1128/IAI.68.11.6115-6126.2000
Frankel, G., and Phillips, A. D. (2008). Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: Getting off the pedestal. Cell Microbiol. 10, 549–556. doi: 10.1111/j.1462-5822.2007.01103.x
Friedberg, D., Umanski, T., Fang, Y., and Rosenshine, I. (1999). Hierarchy in the expression of the locus of enterocyte effacement genes of enteropathogenic Escherichia coli. Mol. Microbiol. 34, 941–952. doi: 10.1046/j.1365-2958.1999.01655.x
Furniss, R. C. D., and Clements, A. (2018). Regulation of the locus of enterocyte effacement in attaching and effacing pathogens. J. Bacteriol. 200:e00336-17. doi: 10.1128/JB.00336-17
Garcia-Angulo, V. A., Martinez-Santos, V. I., Villasenor, T., Santana, F. J., Huerta-Saquero, A., Martinez, L. C., et al. (2012). A distinct regulatory sequence is essential for the expression of a subset of nle genes in attaching and effacing Escherichia coli. J. Bacteriol. 194, 5589–5603. doi: 10.1128/JB.00190-12
Gaytan, M. O., Martinez-Santos, V. I., Soto, E., and Gonzalez-Pedrajo, B. (2016). Type three secretion system in attaching and effacing pathogens. Front. Cell Infect. Microbiol. 6:129. doi: 10.3389/fcimb.2016.00129
Guttman, J. A., and Finlay, B. B. (2008). Subcellular alterations that lead to diarrhea during bacterial pathogenesis. Trends Microbiol. 16, 535–542. doi: 10.1016/j.tim.2008.08.004
Haack, K. R., Robinson, C. L., Miller, K. J., Fowlkes, J. W., and Mellies, J. L. (2003). Interaction of Ler at the LEE5 (tir) operon of enteropathogenic Escherichia coli. Infect. Immun. 71, 384–392. doi: 10.1128/IAI.71.1.384-392.2003
Hansen, A. M., and Kaper, J. B. (2009). Hfq affects the expression of the LEE pathogenicity island in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 73, 446–465. doi: 10.1111/j.1365-2958.2009.06781.x
Huang, L. H., and Syu, W. J. (2008). GrlA of enterohemorrhagic Escherichia coli O157:H7 activates LEE1 by binding to the promoter region. J. Microbiol. Immunol. Infect. 41, 9–16.
Islam, M. S., Bingle, L. E., Pallen, M. J., and Busby, S. J. (2011). Organization of the LEE1 operon regulatory region of enterohaemorrhagic Escherichia coli O157:H7 and activation by GrlA. Mol. Microbiol. 79, 468–483. doi: 10.1111/j.1365-2958.2010.07460.x
Iyoda, S., and Watanabe, H. (2005). ClpXP protease controls expression of the type III protein secretion system through regulation of RpoS and GrlR levels in enterohemorrhagic Escherichia coli. J. Bacteriol. 187, 4086–4094. doi: 10.1128/JB.187.12.4086-4094.2005
Iyoda, S., Koizumi, N., Satou, H., Lu, Y., Saitoh, T., Ohnishi, M., et al. (2006). The GrlR-GrlA regulatory system coordinately controls the expression of flagellar and LEE-encoded type III protein secretion systems in enterohemorrhagic Escherichia coli. J. Bacteriol. 188, 5682–5692. doi: 10.1128/JB.00352-06
Jimenez, R., Cruz-Migoni, S. B., Huerta-Saquero, A., Bustamante, V. H., and Puente, J. L. (2010). Molecular characterization of GrlA, a specific positive regulator of ler expression in enteropathogenic Escherichia coli. J. Bacteriol. 192, 4627–4642. doi: 10.1128/JB.00307-10
Jobichen, C., Li, M., Yerushalmi, G., Tan, Y. W., Mok, Y. K., Rosenshine, I., et al. (2007). Structure of GrlR and the implication of its EDED motif in mediating the regulation of type III secretion system in EHEC. PLoS Pathog. 3:e69. doi: 10.1371/journal.ppat.0030069
Kitagawa, R., Takaya, A., and Yamamoto, T. (2011). Dual regulatory pathways of flagellar gene expression by ClpXP protease in enterohaemorrhagic Escherichia coli. Microbiology 157(Pt 11), 3094–3103. doi: 10.1099/mic.0.051151-0
Leh, H., Khodr, A., Bouger, M. C., Sclavi, B., Rimsky, S., and Bury-Mone, S. (2017). Bacterial-chromatin structural proteins regulate the bimodal expression of the locus of enterocyte effacement (LEE) pathogenicity island in enteropathogenic Escherichia coli. mBio 8:e00773-17. doi: 10.1128/mBio.00773-17
Li, M., Rosenshine, I., Tung, S. L., Wang, X. H., Friedberg, D., Hew, C. L., et al. (2004). Comparative proteomic analysis of extracellular proteins of enterohemorrhagic and enteropathogenic Escherichia coli strains and their ihf and ler mutants. Appl. Environ. Microbiol. 70, 5274–5282. doi: 10.1128/AEM.70.9.5274-5282.2004
Lio, J. C., and Syu, W. J. (2004). Identification of a negative regulator for the pathogenicity island of enterohemorrhagic Escherichia coli O157:H7. J. Biomed. Sci. 11, 855–863. doi: 10.1007/BF02254371
Martinez-Laguna, Y., Calva, E., and Puente, J. L. (1999). Autoactivation and environmental regulation of bfpT expression, the gene coding for the transcriptional activator of bfpA in enteropathogenic Escherichia coli. Mol. Microbiol. 33, 153–166. doi: 10.1046/j.1365-2958.1999.01460.x
Mayer, M. P. (1995). A new set of useful cloning and expression vectors derived from pBlueScript. Gene 163, 41–46. doi: 10.1016/0378-1119(95)00389-n
Navarre, W. W. (2016). The impact of gene silencing on horizontal gene transfer and bacterial evolution. Adv. Microb. Physiol. 69, 157–186. doi: 10.1016/bs.ampbs.2016.07.004
Padavannil, A., Jobichen, C., Mills, E., Velazquez-Campoy, A., Li, M., Leung, K. Y., et al. (2013). Structure of GrlR-GrlA complex that prevents GrlA activation of virulence genes. Nat. Commun. 4:2546. doi: 10.1038/ncomms3546
Pearson, J. S., Giogha, C., Wong Fok Lung, T., and Hartland, E. L. (2016). The genetics of enteropathogenic Escherichia coli virulence. Annu. Rev. Genet. 50, 493–513. doi: 10.1146/annurev-genet-120215-035138
Platenkamp, A., and Mellies, J. L. (2018). Environment controls LEE regulation in enteropathogenic Escherichia coli. Front. Microbiol. 9:1694. doi: 10.3389/fmicb.2018.01694
Ronin, I., Katsowich, N., Rosenshine, I., and Balaban, N. Q. (2017). A long-term epigenetic memory switch controls bacterial virulence bimodality. Elife 6:e19599. doi: 10.7554/eLife.19599
Saitoh, T., Iyoda, S., Yamamoto, S., Lu, Y., Shimuta, K., Ohnishi, M., et al. (2008). Transcription of the ehx enterohemolysin gene is positively regulated by GrlA, a global regulator encoded within the locus of enterocyte effacement in enterohemorrhagic Escherichia coli. J. Bacteriol. 190, 4822–4830. doi: 10.1128/JB.00231-08
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd Edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Sanchez-SanMartin, C., Bustamante, V. H., Calva, E., and Puente, J. L. (2001). Transcriptional regulation of the orf19 gene and the tir-cesT-eae operon of enteropathogenic Escherichia coli. J. Bacteriol. 183, 2823–2833. doi: 10.1128/JB.183.9.2823-2833.2001
Serapio-Palacios, A., and Finlay, B. B. (2020). Dynamics of expression, secretion and translocation of type III effectors during enteropathogenic Escherichia coli infection. Curr. Opin. Microbiol. 54, 67–76. doi: 10.1016/j.mib.2019.12.001
Shin, M. (2017). The mechanism underlying Ler-mediated alleviation of gene repression by H-NS. Biochem. Biophys. Res. Commun. 483, 392–396. doi: 10.1016/j.bbrc.2016.12.132
Shoval, O., and Alon, U. (2010). SnapShot: Network motifs. Cell 143, 326–e321. doi: 10.1016/j.cell.2010.09.050
Sirisaengtaksin, N., Odem, M. A., Bosserman, R. E., Flores, E. M., and Krachler, A. M. (2020). The E. coli transcription factor GrlA is regulated by subcellular compartmentalization and activated in response to mechanical stimuli. Proc. Natl. Acad. Sci. U.S.A. 117, 9519–9528. doi: 10.1073/pnas.1917500117
Spears, K. J., Roe, A. J., and Gally, D. L. (2006). A comparison of enteropathogenic and enterohaemorrhagic Escherichia coli pathogenesis. FEMS Microbiol. Lett. 255, 187–202. doi: 10.1111/j.1574-6968.2006.00119.x
Sperandio, V., Mellies, J. L., Delahay, R. M., Frankel, G., Crawford, J. A., Nguyen, W., et al. (2000). Activation of enteropathogenic Escherichia coli (EPEC) LEE2 and LEE3 operons by Ler. Mol. Microbiol. 38, 781–793. doi: 10.1046/j.1365-2958.2000.02168.x
Sudo, N., Lee, K., Sekine, Y., Ohnishi, M., and Iyoda, S. (2022). RNA-binding protein Hfq downregulates locus of enterocyte effacement-encoded regulators independent of small regulatory RNA in enterohemorrhagic Escherichia coli. Mol. Microbiol. 117, 86–101. doi: 10.1111/mmi.14799
Turner, N. C. A., Connolly, J. P. R., and Roe, A. J. (2019). Control freaks-signals and cues governing the regulation of virulence in attaching and effacing pathogens. Biochem. Soc. Trans. 47, 229–238. doi: 10.1042/BST20180546
Ueguchi, C., Suzuki, T., Yoshida, T., Tanaka, K., and Mizuno, T. (1996). Systematic mutational analysis revealing the functional domain organization of Escherichia coli nucleoid protein H-NS. J. Mol. Biol. 263, 149–162. doi: 10.1006/jmbi.1996.0566
Uzzau, S., Figueroa-Bossi, N., Rubino, S., and Bossi, L. (2001). Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. U.S.A. 98, 15264–15269.
Vidal, J. E., and Navarro-Garcia, F. (2008). EspC translocation into epithelial cells by enteropathogenic Escherichia coli requires a concerted participation of type V and III secretion systems. Cell Microbiol. 10, 1975–1986. doi: 10.1111/j.1462-5822.2008.01181.x
Keywords: EPEC, type III secretion, LEE regulation, GrlR, GrlA, transcription, A/E pathogens, CAT reporter assay
Citation: Lara-Ochoa C, Huerta-Saquero A, Medrano-López A, Deng W, Finlay BB, Martínez-Laguna Y and Puente JL (2023) GrlR, a negative regulator in enteropathogenic E. coli, also represses the expression of LEE virulence genes independently of its interaction with its cognate partner GrlA. Front. Microbiol. 14:1063368. doi: 10.3389/fmicb.2023.1063368
Received: 07 October 2022; Accepted: 23 January 2023;
Published: 16 February 2023.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Bindu Subhadra, Long Island University, United StatesRuojun Wang, Princeton University, United States
Ilan Rosenshine, The Hebrew University of Jerusalem, Israel
Copyright © 2023 Lara-Ochoa, Huerta-Saquero, Medrano-López, Deng, Finlay, Martínez-Laguna and Puente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Lara-Ochoa,  Y3Jpc3RpbmEubGFyYW9jaG9hQGNvcnJlby5idWFwLm14; José L. Puente, am9zZS5wdWVudGVAaWJ0LnVuYW0ubXg=
Y3Jpc3RpbmEubGFyYW9jaG9hQGNvcnJlby5idWFwLm14; José L. Puente, am9zZS5wdWVudGVAaWJ0LnVuYW0ubXg=