 Jannick Van Cauwenberghe
Jannick Van Cauwenberghe Rosa I. Santamaría
Rosa I. Santamaría Patricia Bustos
Patricia Bustos Víctor González
Víctor González
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 September 2022
Sec. Phage Biology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.990394
This article is part of the Research Topic Rhizobiaceae mediated HGT: facts, mechanisms, and evolutionary consequences View all 6 articles
This study describes novel single-stranded DNA phages isolated from common bean agriculture soils by infection of the nitrogen-fixing symbiotic bacteria Rhizobium etli and R. phaseoli. A total of 29 phages analyzed have 4.3–6 kb genomes in size and GC 59–60%. They belong to different clades unrelated to other Microviridae subfamilies. Three-dimensional models of the major capsid protein (MCP) showed a conserved β-barrel structural “jelly-roll” fold. A variable-length loop in the MCPs distinguished three Rhizobium microvirus groups. Microviridae subfamilies were consistent with viral clusters determined by the protein-sharing network. All viral clusters, except for Bullavirinae, included mostly microviruses identified in metagenomes from distinct ecosystems. Two Rhizobium microvirus clusters, chaparroviruses, and chicoviruses, were included within large viral unknown clusters with microvirus genomes identified in diverse metagenomes. A third Rhizobium microvirus cluster belonged to the subfamily Amoyvirinae. Phylogenetic analysis of the MCP confirms the divergence of the Rhizobium microviruses into separate clades. The phylogeny of the bacterial hosts matches the microvirus MCP phylogeny, suggesting a coevolutionary history between the phages and their bacterial host. This study provided essential biological information on cultivated microvirus for understanding the evolution and ecological diversification of the Microviridae family in diverse microbial ecosystems.
Viruses are the most abundant, ubiquitous, and diverse biological entities on earth (Breitbart and Rohwer, 2005; Suttle, 2007; Dion et al., 2020). These obligatory pathogens infect all known taxa of organisms, but most viruses are specialized to infect bacteria. These viruses are called bacteriophages or phages and play a key role in the evolution and ecology of bacteria. Phages shape bacterial community structure through the lysis of diverse bacterial genera, species, or even strains, on which they are specialized because of coevolution (Poullain et al., 2008; Parsons et al., 2012; Weitz et al., 2013; Morella et al., 2018; Wang et al., 2019). Phages can also transduce bacterial genes by erroneously packing bacterial DNA in the viral capsid or when prophages are excised inaccurately from the host genome (Pedulla et al., 2003; Canchaya et al., 2004; Touchon et al., 2017). Prophages are phages integrated into the host genome, which can excise and induce lysis after multiple host generations or remain stranded as cryptic prophages when excision genes experience loss-of-function mutations (Casjens, 2003; Wang et al., 2010; Ramisetty and Sudhakari, 2019).
Single-stranded (ss) DNA viruses are less thoroughly studied and are a minor fraction of the phages found in databases, which abound in double-stranded (ds) DNA viruses (16.2% as of 2022; Roux et al., 2016a, 2016b). However, they appear to be prevalent in aquatic ecosystems and the human gut microbiome (López-Bueno et al., 2009; Shkoporov et al., 2019). The most common ssDNA viruses are Microviridae, composed of a small (~25 nm) capsid with icosahedral symmetry and a 4,000–6,500 bp genome (Maclean and Hall, 1962; Roux et al., 2012; Doore and Fane, 2016). Bullavirinae and Gokushovirinae are the two Microviridae subfamilies recognized by the International Committee on Taxonomy of Viruses (ICTV) based on structural and genomic differences (King et al., 2011). Other subfamilies suggested are Aravirinae (Quaiser et al., 2015), Pichovirinae (Roux et al., 2012), and Alpavirinae (Krupovic and Forterre, 2011). Most known Bullavirinae have been isolated infecting Enterobacteria and have been the subject of early research on phage biology. Gokushovirinae and suggested subfamilies are known to infect diverse bacterial taxa (Roux et al., 2012; Labonté and Suttle, 2013; Quaiser et al., 2015; Zheng et al., 2018).
Microviridae were long believed to be exclusively lytic, with the only exemption being the temperate Alpavirinae (Krupovic and Forterre, 2011). Until recently, some studies discovered prophages belonging to Gokushovirinae (Kirchberger and Ochman, 2020) and Bullavirinae (Kirchberger et al., 2021) through the analysis of host genomes. These findings demonstrate the limited understanding of the diversity and prevalence of Microviridae prophages. Microviridae have been detected in human and animal microbiomes and aquatic systems (Roux et al., 2012; Kirchberger and Ochman, 2020). In contrast, the diversity of phages infecting soil and rhizosphere-dwelling bacteria is poorly described (Zablocki et al., 2016; Pratama and van Elsas, 2018). Rhizobia is a particular group of bacteria adapted to the rhizosphere (Nautiyal, 1997; Wheatley et al., 2020). They engage in a mutualistic association with legumes, forming nodules on the roots and fixing atmospheric nitrogen in exchange for photosynthates. Rhizobia are infected by various families of Caudovirales (Werquin et al., 1988; Santamaría et al., 2022). In our recent study, we report that the Microviridae family likely includes a significant fraction of phage communities infecting rhizobia associated with common beans (Van Cauwenberghe et al., 2021).
Here, we aim to investigate the infection properties of Rhizobium microviruses and the structural features of the capsid surface. Moreover, we want to determine the phylogenetic relationship among Microviridae associated with rhizobia and the coevolutionary history of these phages with their hosts.
Three phages, RHEph17, X92, and X94, originating from common bean agriculture soils in Mexico, were isolated by infection enrichment according to Santamaría et al. (2014) using R. etli N741, R. etli GR14, and R. phaseoli GR75 as host, respectively. Pure phages were obtained after plaquing three times in double-layer plates. Next, the phage genomic DNA was extracted following the next protocol: phages were propagated in 2 × 6 ml of PY broth, adding 0.1 ml of phage stock solution (PFUs 109) to the host strain at OD600 at 0.1, and incubated overnight.
Next, the lysate was added to 10% v/v chloroform to remove cell debris and centrifuged to recover 10 ml of the supernatant. Next, the lysate was treated with 0.1 ml of DNAse (10 mg/ml; Roche Diagnostics) and RNase (10 mg/ml; MP Biomedicals) to remove the DNA and RNA of bacteria. Phages were precipitated with 12% w/v PEG8000 and 1 M NaCl overnight at 4°C, then centrifuged (30 min at 10,000 rpm). Finally, concentrated phage was treated with the DNA Isolation Kit for Cells and Tissues (Roche Life Sciences, CA, United States) following the procedure for DNA purification for gram-negative bacteria, adjusting the volumes used to 0.5 ml of a lysis solution with 10 μl of proteinase K, 20 μl RNase and 0.24 ml precipitation solution. DNA was precipitated with 0.7 volumes of isopropanol and 1 ml of 70% ethanol and resuspended in 0.1 ml water. Phage genome mobility and integrity were evaluated by agarose gel electrophoresis.
Genome sequencing was performed with pair-end libraries made with the Nextera Kit and sequenced in an Illumina NextSeq 500 sequencer at Unidad Universitaria de Secuenciación Masiva de DNA [UUSMD]-Universidad Nacional Autónoma de México [UNAM]. Readings of 135 bp were trimmed with TrimGalore1 and assembled using Spades v. 3.13.1, Velvet v. 1.2.10, and Phred/Phrap/Consed v. 23.0 (Zerbino and Birney, 2008; Bankevich et al., 2012). The assembled sequence demonstrated that the samples are small circular genomes of about 4.3–6.3 kb, according to their mobility in gel electrophoresis.
In addition to the three microviruses isolated in this work, we included 26 previously reported microviruses (Van Cauwenberghe et al., 2021; Supplementary Table S1). The current study describes the features of the 29 microviruses in this collection. These belong to three genomic clusters, defined by the average nucleotide identity based on MUMmer (ANIm; Pritchard et al., 2016) above 80%, provisionally referred to as F02, F08, and F40 (Van Cauwenberghe et al., 2021). The second set of microvirus genomes was obtained by downloading 2,147 genomes of Microviridae from GenBank. They consist of 1,605 microviruses identified in metagenomes, and 542 represent the genome sequence of isolated virions (Supplementary Table S2). Moreover, 25 recently published microvirus genomes were identified in metagenomic fecal samples of flying foxes (Lopez et al., 2022). Finally, they were incorporated into a local database to run the vConTACT v2 viral clustering method (Supplementary Table S3).
Rhizobium strains were grown in peptone-yeast extract broth (PY added with 7 mM CaCl2 and 20 μg/ml nalidixic acid) in an incubator at 30°C with shaking at 200 rpm. Phage titer was estimated by mixing 200 μl of the corresponding host strain in the exponential growth phase and 100 μl of 10-fold phage dilutions with top agar (soft medium with 0.4% agar melted at 42°C) and poured onto plates with 1.5% agar. Double-layer plates were allowed to cool and were subsequently incubated at 30°C overnight.
Host-range determination was carried out with the spot-test technique range in double-layer plates (Hyman and Abedon, 2009). First, bacterial lawns were made by mixing 300 μl of strain with a soft medium and pouring it over PY-agar plates. Next, 10 μl of the phage solution from stocks were dropped onto the lawns and incubated at 30°C. After incubation, spots were registered as complete lysis (transparent plaque), partial lysis (translucent plaque), or resistance to lysis when no plaque was observed. R (v. 3.6.1) programming was used to perform box plots and two-sample T-test to assess the statistical significance of the host-range infection rates.
Infection kinetics analyses were performed in 96-well microplates in a BioTek Synergy 2 Multi-Mode Microplate Reader (Agilent, United States) using the R. phaseoli N2.5 strain as the host. Infections were tested at Multiplicities of Infection (MOI) of 1–0.00006. The change in optical density was tracked in time by measuring optical density every 30 min.
The one-step growth curve of phage TM23, TM24, and Y67 was determined using the R. etli N2.5 strain. Bacterial culture and phage were mixed in 1 ml medium at MOI of 0.01 (1 × 108 bacterial cells and 1 × 106 phages) and incubated for 30 min at 30°C to allow phage adsorption. Subsequently, the unabsorbed phage was removed by centrifugation, discarding the supernatant. Pellets with infected cells were diluted in 1 ml of fresh medium, transferred to a 250 ml flask with 100 ml of fresh PY broth, and incubated at 30°C. Samples were taken every 30 or 60 min for 8.5 h, and 10-fold dilutions were performed for plaquing. The experiment was performed in triplicate, and PFUs (plaque-forming units) were registered and plotted using ggplot2 (v. 3.3.6) in R (v. 3.6.1). Burst size was calculated by the difference between the average PFUs at the maximum virus release stage and the average PFUs at the eclipse period and by dividing that number by the number of infected cells (total phages minus unabsorbed phages).
Phage samples were propagated in a 250 ml flask by adding 1 ml of phage solution from the stock (109 PFUs/ml) in 100 ml of PY medium containing the corresponding host strain at an OD of 0.1 and incubated overnight. The lysate was then centrifuged (10 min at 10,000 rpm), passed through a 0.22-μm membrane filter, and subject to precipitation using 12% w/v PEG8000 and 1 M NaCl overnight at 4°C. Then, the solution was centrifuged (30 min at 10,000 rpm), and the pellet was resuspended in 5 ml 10 mM MgSO4. One volume of chloroform was added to remove PEG, and the mixture re-centrifuged. Subsequently, the aqueous phase was recovered, and phages were concentrated using an Amicon Ultra-15 100 K filter (Merck Millipore) to a volume of 0.2 ml. The phage titer was about 1 × 1010 PFUs/ml at the end of the purification. A droplet of these viral particles was negatively stained with 1% uranyl acetate on a copper grid covered with evaporated carbon and Formvar film (E.M.S. FF200-Cu). Electron microscopy was performed in a transmission electron microscope Libra 120 (Zeiss, Germany) coupled with a Multiscan Camera (GATAN Inc., United States) at the Unidad de Microscopía Electrónica, UNAM.
The amino acid sequences of the MCPs of TM23, TM24, Y67, phiX174 (Bullavirinae), and SpV4 (Gokushovirinae) were used to obtain the three-dimensional models (3D) with AlphaFold-2.1.0 in ColabFold server using the MMseq2 algorithm (Jumper et al., 2021; Mirdita et al., 2021). The unrelaxed rank 1 models were visually inspected with the RSCB PDB 3D Viewer Mol* (Sehnal et al., 2021) and then were aligned in pairs to superimpose the 3D models using the matchmaker tool in UCSF Chimera X v. 1.2 software (Pettersen et al., 2021). The quality of the models was assessed first by the local Distance Difference Test (lDDT) method (Mariani et al., 2013) and second by the superposition of pairs of predicted structures, including the predicted 3D MCP models with the experimental structure of MCP- phiX174 (RCSB PDB Protein Data Bank, ID 2BPA; PDB DOI: 10.2210/pdb2BPA/pdb). High lDDT scores (>80) were assigned for the MCPs TM23, TM24, and Y67 at the equivalent structural regions in the phiX174 structure. The root mean square deviation (RMSD), which represents the dissimilarity of the predicted protein structures, was calculated by measuring the average distance between atoms of the proteins when superimposed. Lower RMSD values indicate a more significant similarity of predicted protein structures. The similarity between MCP 3D models was determined by calculating the distance matrix with the R package “dist” using the method “maximum.” The dendrogram was built with the R package “hclust” using the method “complete.” An additional comparison included the 3D model of the MCP of phage HK97 (Caudovirales) as a control.
Average nucleotide identity (ANI) among phages described here and representative phages of different Microviridae subfamilies was calculated with pyani v.0.2.9 using the ANIm MUMmer method (Pritchard et al., 2016). In addition, average amino acid identity (AAI) was calculated with the script aai.rb from the enveomics collection (Rodriguez-R and Konstantinidis, 2016) and plotted with ggplot2 function heatmap 2. Gene-sharing networks were created using vConTACT v2 (Jang et al., 2019). First, the vConTACT v2 tool clusters similar proteins into protein clusters (PCs) using the Markov cluster algorithm (MCL). Then, viral clusters (VCs) were calculated according to the maximum probabilities of sharing PCs (edges) between the genomes (nodes) to produce a bipartite network. The VCs were defined using ClusterONE, with default parameters (MCL inflation: 2; penalty value: 2; edge weight: 10). The networks generated by vConTACT v2 were visualized using Cytoscape v3.8.2.2
Phylogenetic trees of Microviridae were constructed using the major capsid protein (MCP) identified in Microviridae isolated here and previously(Van Cauwenberghe et al., 2021), in previously suggested Microviridae subfamilies, and detected using BLASTp (e-value <10−6, % identity >70%, query cover >30%). An alignment of MCP was created using MUSCLE (Edgar, 2004). A maximum-likelihood phylogenetic tree was constructed with IQTREE with 1,000 bootstrap replicates, using the GTR+F+R6 model selected using the Bayesian Information Criterion (BIC; Nguyen et al., 2015).
Genome phylogeny of R. etli and R. phaseoli strains was based on concatenated 3,602 core proteins obtained with the Bacterial Pan Genome Analysis Tool (BPGA; Chaudhari et al., 2016). A protein super alignment was performed using MUSCLE (Edgar, 2004), and gaps were processed with TrimAI (Capella-Gutiérrez et al., 2009). The phylogenetic tree was constructed with IQTREE with 1,000 bootstrap replicates with the JTT+F+R3 model.
We used ParaFit (Legendre et al., 2002) in the ape package in R (Paradis and Schliep, 2019) to test for significant similarities between bacterial phylogenies (based on 16S nucleotide sequences) and the MCP phylogenies of both the 29 lytic Microviridae used in this study and MCPs located in bacterial genomes identified by BlastP at the species level or available on the NCBI RefSeq database.
Each tree was built using IQTREE using the LG+I+G4+F substitution model for phages, and the TIM3+I+G4+F substitution model for bacteria, with 1,000 bootstrap replicates. Models were selected using the Bayesian Information Criterion (BIC) implemented on the IQTREE web server (Nguyen et al., 2015). Patristic distances of the phylogenies were calculated using cophenetic.phylo from the ape package in R.
To investigate the infection features and host range of Rhizobium microviruses, we chose one representative phage per each of the already-described ANIm Microviridae clusters: F02, F08, and F40 (Van Cauwenberghe et al., 2021). Infection kinetics at different MOIs was performed using the R. etli strain N2.5 host (Figure 1A; Supplementary Figure S1). Although the three phages differ in the genome sequence, they are equally competent to lyse the N2.5 host. The phage Y67 (belonging to ANIm cluster F40) was very efficient to lyse cultures of N2.5, even at lower MOIs in comparison with TM24 (F08) and TM23 (F02). Despite slight differences in the infection kinetics of TM23 and TM24, they both eliminate the bacterial cells at the lowest MOI (0.00006) after 6 h of incubation. Moreover, the titer of phages after 9 h of incubation with N2.5 strain was moderately high for Y67 and TM23 (9 × 108 and 7 × 108 PFUs/ml, respectively) and low for TM24 (9 × 107 PFUs/ml; Figure 1B).
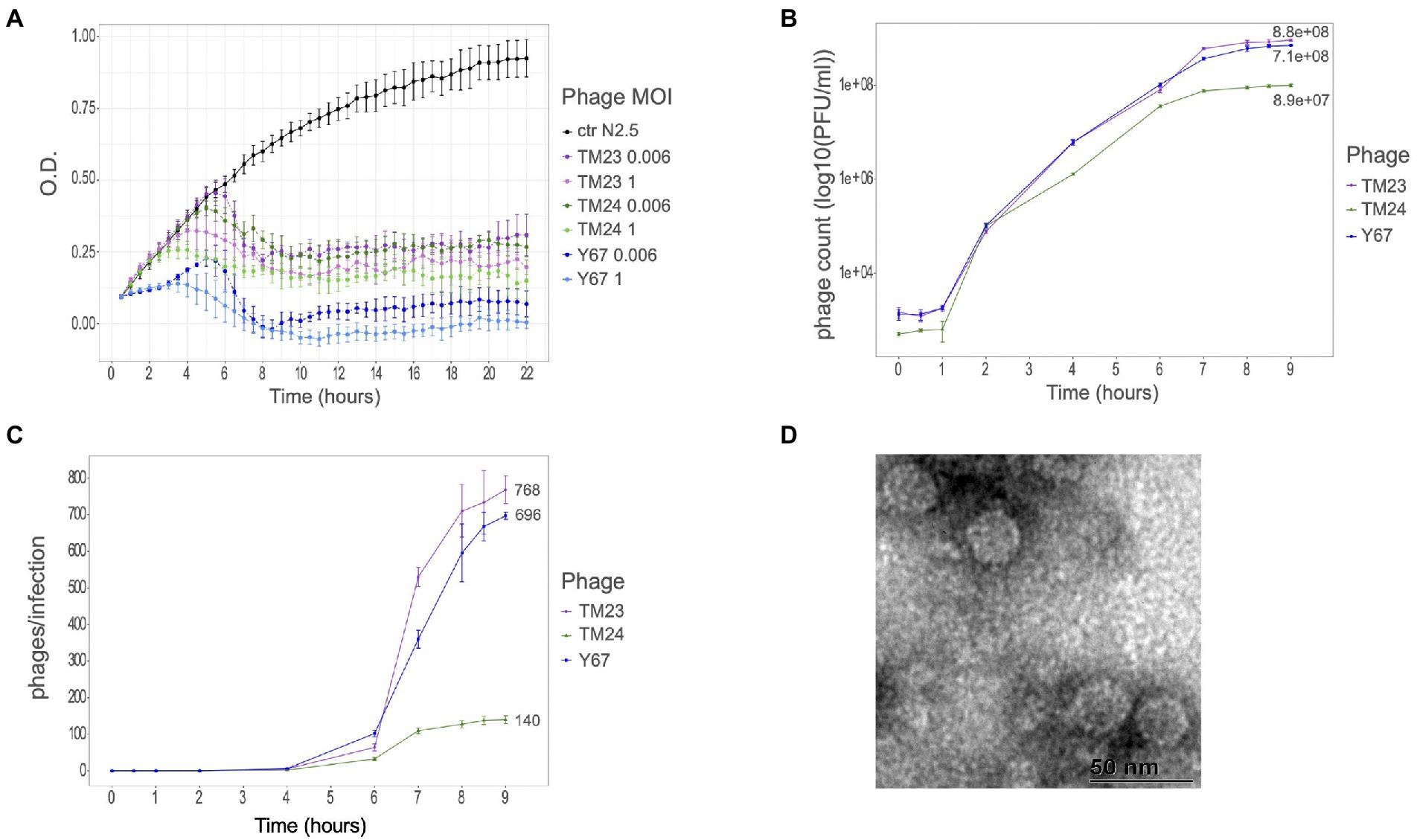
Figure 1. Infectious properties and morphology of Rhizobium microvirus. (A) Infection kinetics in PY-broth culture of phages TM23 (representing ANIm cluster F02), TM24 (F08), and Y67 (F40) at two different MOIs (1 and 0.006) on R. etli N2.5. (B) Phage titers after 9 h of infection on N2.5. (C) Two-step growth curve of the three phages at MOI 0.01. Colors code indicating the respective strains is at the right of the panels. (D) Electron micrograph of TM23 at 50 nm.
To assess the infection features of Rhizobium microvirus, we did the one-step-growth kinetics for the three phages TM23, TM24, and Y67 (Figure 1B). The latent phase took about 4 h to yield the first virions in all the three phages; after 9 h, the virion production stopped in TM23 and Y67, but phage TM24 developed slow kinetics with a very short exponential phase (Figure 1C). The estimated burst size of TM23 and Y67 was 768 and 669 virions per cell, respectively. In contrast, TM24 had the smallest burst size of 140 virions per cell.
To look for species-specific infection preferences of these three phages, we compare their range of infection by spot-assays over a collection of 10 R. etli strains and 11 R. phaseoli strains whose complete genome is known and devoid of Microviridae-related sequences. Spot phenotypes appear as evident lysis (transparent), medium lysis (translucid), and resistant (no phenotype). TM23 and TM24 infection yielded transparent spots in 12 and 10 out of 21 strains (proportion 0.6 of R. etli over R. phaseoli) of both species without any observable species-specificity (Figure 2A). These spot phenotypes did not associate with the phylogenetic separation of the strains into the species R. etli and R. phaseoli. In contrast, the Y67 phage showed marked preference and limited host range for R. phaseoli strains (Figure 2A). The proportion of infection (clear spots) in 63 microviruses of the collection previously reported (Van Cauwenberghe et al., 2021) showed a significant trend to infect R. etli over R. phaseoli (two-samples t-test p = 2 × 10−6; Figures 2B,C). Moreover, the three Rhizobium microvirus tested produced partially lytic spots (translucid) in some strains, indicating differences in the efficiency of infection, an issue that deserves further experimentation.
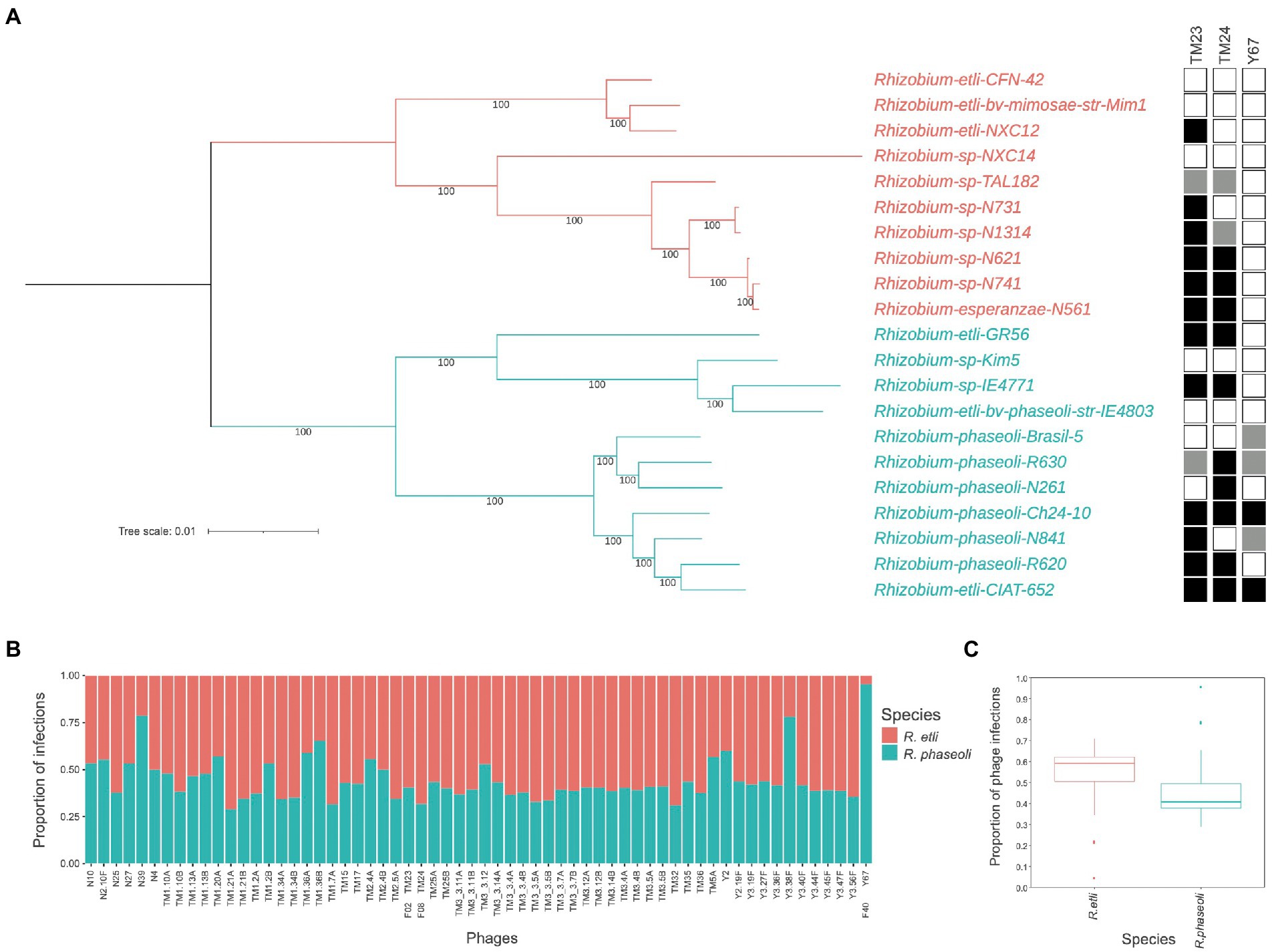
Figure 2. Host infection range of Rhizobium microvirus. (A) Maximum likelihood unrooted tree with the common core genome of R. etli and R. phaseoli strains and the infection by TM23, TM24, and Y67 in spot-assays. Black squares indicate complete lysis; gray, partial lysis; and empty squares, absence of lysis. (B) The proportion of infection of 63 microviruses on R. etli and R. phaseoli strains. (C) Box plots of the proportion of phage infections on the two Rhizobium species with the median and the quartile distribution. Data on infection for B and C were extracted from the phage-bacteria infection matrix reported in (Van Cauwenberghe et al., 2021, “Dataset 1”).
Using uranyl acetate, we observed the morphology of Rhizobium TM23 virions by negative staining electron microscopy. TM23 likely has an icosahedral appearance of about 25 nm in diameter (Figures 1D–F). The surface features of TM23 seemingly display mushroom-like protrusions like those observed in SpV4 (McKenna et al., 1992; Chipman et al., 1998). The protrusions have been associated with an inserted loop within the MCP protein (Roux et al., 2012).
To search for structural differences in the surface of TM24, TM23, and Y67 phages, we made 3D models of the MCP proteins with AlphaFold2 and compared them with the MCP 3D models of SpV4 and phiX174, and the solved crystal structure of MCP-phiX174 (McKenna et al., 1992). MCP-TM24 and MCP-TM23 have a similar length of 556 and 554 amino acids, respectively, while the MCP-Y67 is minor with 425 amino acids in length. The 3D models made with AlphaFold2 predicted a core of eight antiparallel β-sheets and a variable number of α-helix and loops, consistent with the MCP of SpV4 and phiX174, both 3D model and solved structure (Figure 3). In addition, low IDDT scores were observed between amino acid positions 200–300 (Supplementary Figure S2), a region that corresponds to the large loop followed in the 3D models of MCP-TM23, TM24, and SpV4 but not in the MCP-phiX174 (3D model and structure), where the loop is short, and in MCP-Y67 which lacks a loop in this region, which may be part of the reason why its host range differs from other phages (Supplementary Figure S3).
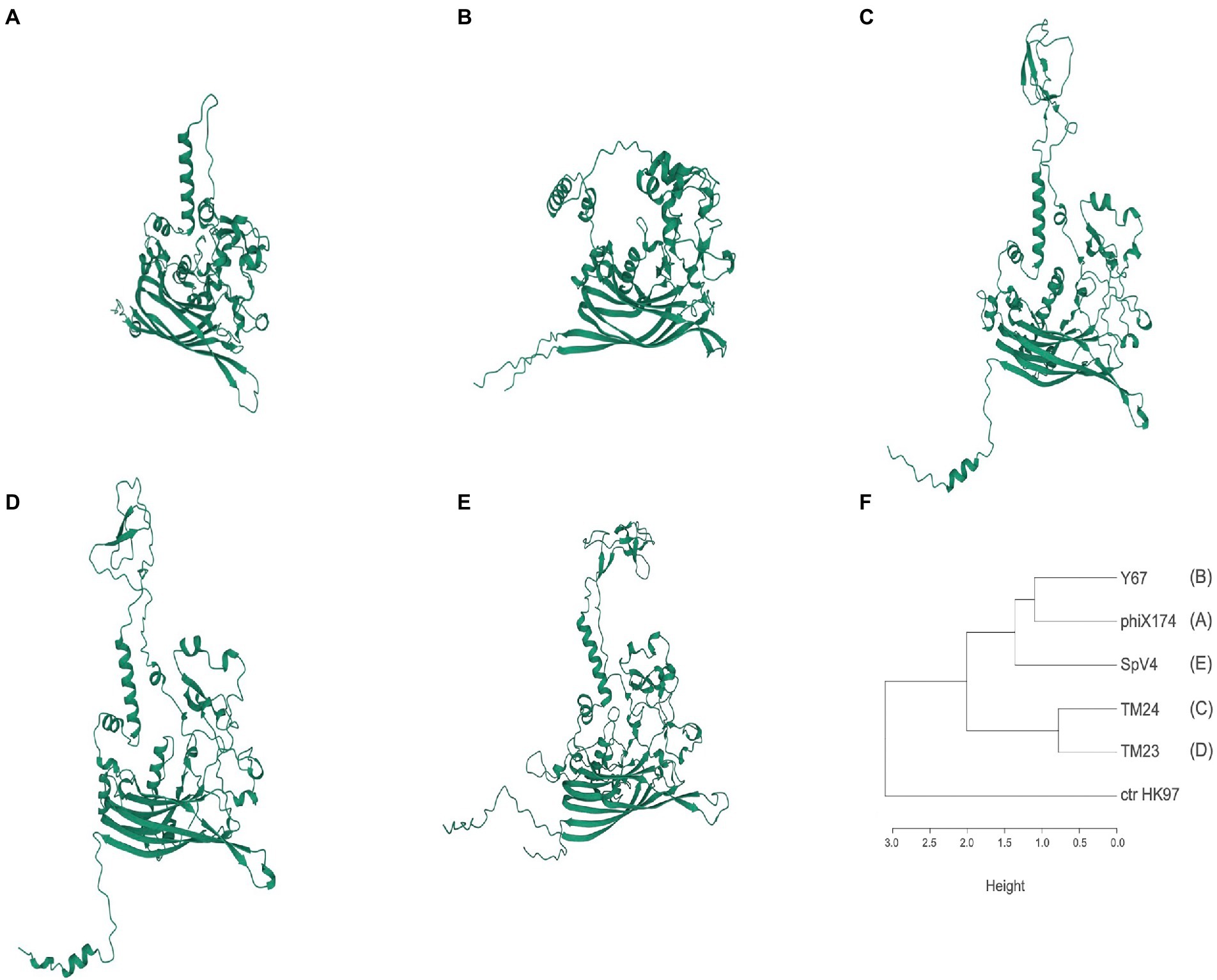
Figure 3. Predicted three-dimensional structure models of MCPs. (A) PhiX174; (B) Y67 (representing cluster ANIm F40). (C) TM24 (F08); (D) TM23 (F02); (E) SpV4 (Spiroplasma, Goshukovirus); (F) MCP similarity according to the RMSD values from the superposition between pairs of models (Supplementary Table S4; Supplementary Movies S1–S3). Models were performed with the alphaFold 2.1 program (Jumper et al., 2021).
The superposition of the 3D models assessed by RMSD (Root Median Standard Deviation) showed that MCP-Y67 has a lower RMSD (11.8 A) than MCP-TM23 and TM24 (16.8 and 18.9 A, respectively) when compared with MCP-phiX174 3D model (Supplementary Movie S1–S3). Similarly, the RMSDs between MCP-TM23 and MCP-TM24 3D models are more similar than those with MCP-Y67 (Supplementary Table S4). Although MCP-Y67 is more like the SpV4 model in the core of β-sheets, the large loop made them different. The dendrogram in Figure 3F shows the similarities between the models according to the RMSD. The comparison between the crystallographic structure of the MCP of phiX174 and the AlphaFold2 model obtained from the same protein, indicates RMSD values of 1,267 angstroms. Equivalent RMSD values were observed between the 3D models of the MCPs of microviruses TM23, TM24, and Y67 compared to either the crystallographic structure of MCP-phiX174 or its AlphaFold2 model (Supplementary Table S4).
To look for identities below the ANI threshold of 70% that we used in previous ANIm and ANIb assessments, we made whole genome amino acid comparisons (AAI; Supplementary Figures S4, S5). The three comparative methods detected the largest Microviridae cluster, designated as F02. It consisted of 24 Rhizobium Microviridae genomes and the Microviridae-sp-isolate-ctcf-4 (MH616837.2) that was identified in metagenomes of animal samples, and it was the most divergent genome in the F02 cluster (52% AAI; Lopez et al., 2022). F02 comprises genomes of 6 kb and GC content of about 58–59% related by nucleotide identities of 95% on average and AAI above 60%. The F02 cluster contains four subgroups: (1) The main group (n = 20), (2) a group that includes solely Argentinian phages (n = 2; AAI ≥ 98%; ANIb = 93%, when compared to other F02 members), (3) the novel phage X92 (AAI = 80%, ANIb = 74%), and (4) RHEph17 (AAI > 98%, ANIb = 93%).
ANIb and AAI comparisons revealed two small clusters consistent with the previous F08 and F40 ANIm clusters. They consist of three microviruses of 6.2 Kb (GC 59%) and two microviruses of 4.7–4.8 kb (GC 57–58%), respectively. In addition, by AAI comparisons, phages (microvirus sp. 1712115-248 and 166) isolated from a sewage oxidation pond and two phages (microvirus Tbat2-88 and 91) isolated from feces samples of the flying fox bat were included in the F08 cluster at AAI 60% (Lopez et al., 2022). The three clusters described here showed little to no ANIb or AAI relationship with genomes of previously described Microviridae subfamilies.
To uncover the genomic relationships of Rhizobium microviruses with the other Microviridae subfamilies, we used a network method based on scoring the protein-sharing families between genomes (Jang et al., 2019). The dataset of 2,176 microvirus genomes was included in the vConTACT v2 database (see “Materials and Methods” section). Most of the microvirus genomes (73.5%) in such a database come from metagenomic samples of the gut and feces of humans and other mammals and tortoises (Supplementary Table S3; Orton et al., 2020; Lopez et al., 2022). A small subset of the microvirus genomes included in the database has been determined from viruses isolated in vivo by infecting a few bacterial species (Supplementary Table S3). The microviruses are distributed in the vConTACT v2 network (Figure 4; Supplementary Figure S6) in 16 viral clusters (VCs) corresponding to Microviridae subfamilies; one is the Bullavirinae, which group the phages related to phiX174 and was found in two separated unrelated VCs in the network. Other Microviridae subfamilies also appear distributed in two or three VCs. For instance, Gokushovirinae and Alpavirinae are contained in two and three VCs. Although unexpected, multiple VC clusters in the same subfamily reflect the wide diversity of microviruses within the already defined taxons.
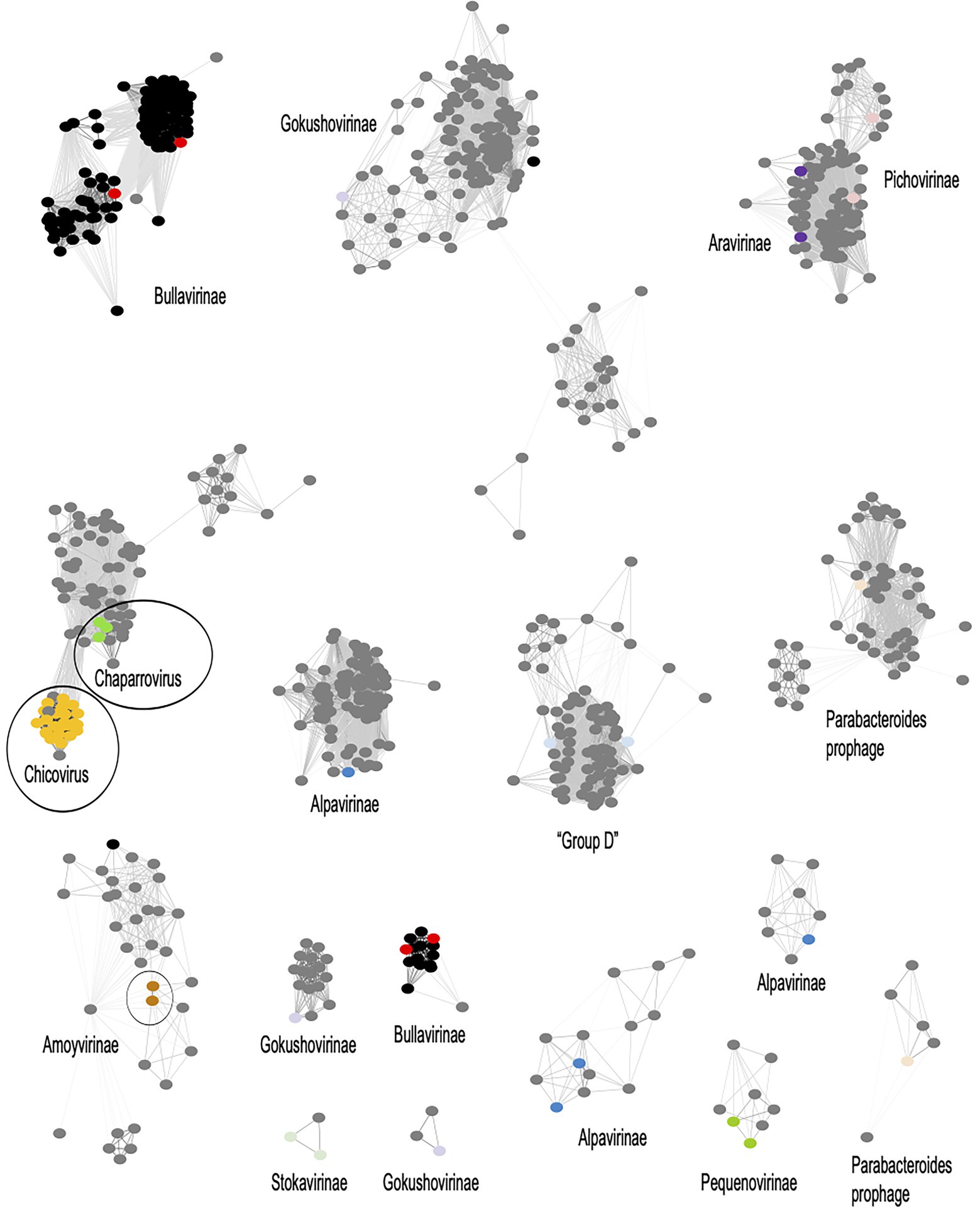
Figure 4. Viral clusters (VCs) of Microviridae phages. The network performed with Cytoscape v3.8.2 (https://cytoscape.org/) represents the genomic relationships of Microviridae as were inferred from vConTACT-v2. Nodes represent genomes, and the edges connecting nodes are the scores of protein shared clusters, including similarity measures and ICTV inferences. Black dots are microviruses cultivated isolates; grey dots are microviruses identified in metagenomic samples. Microviridae subfamilies are named in the corresponding VC, and a color dot within the cluster identifies the subfamily in the MCP phylogenetic tree in Figure 5. Encircled are the Rhizobium microviruses within a VC supercluster.
The F02 and F08 Rhizobium microviruses were embedded into a VC supercluster of Microviridae genomes identified in metagenomic samples (encircled in Figure 4). They are undescribed taxons of Microviridae that we propose to name chicoviruses (F02) and chaparroviruses, respectively (F08; see below).
The Rhizobium microviruses N39 and Y67 of the F40 cluster were included in the vConTACT-2 network within the Amoyvirinae subfamily even though they are far related to vB Cib; an isolate that infects the marine α-proteobacteria Citromicrobium (Zheng et al., 2018), to a prophage in the genome of Novospinghobium NBRC 16725 from sewage sludge, and with metagenomic samples of marine origin (SOG00694).
To investigate the genetic relatedness between Rhizobium microviruses and Microviridae subfamilies that either have been recognized by the ICTV or have been proposed in previous studies, we constructed a maximum-likelihood (ML) phylogenetic tree based on amino acid sequences of the major capsid protein (MCP; Figure 5). First, MCPs were obtained from microviruses belonging to known subfamilies and metagenomes and isolated microviruses from the GenBank database, and MCPs in bacterial genomes (as part of intact or degenerated prophages) were available on GenBank using BLASTp searches (Supplementary Table S5). MCPs are highly divergent in amino acid sequence, indicating an extensive evolutionary history. The MCP phylogeny yields four major clades. One clade (I) leads to Bullavirinae; the second (II), represented by Amoyvirinae; a third (III), that contains the Gokushovirinae subfamilies and most of the proposed subfamilies from metagenomic samples; and the fourth (IV), large and divergent major clade that includes unknown subfamilies distant from any recognized subfamily of Microviridae (Figure 5).
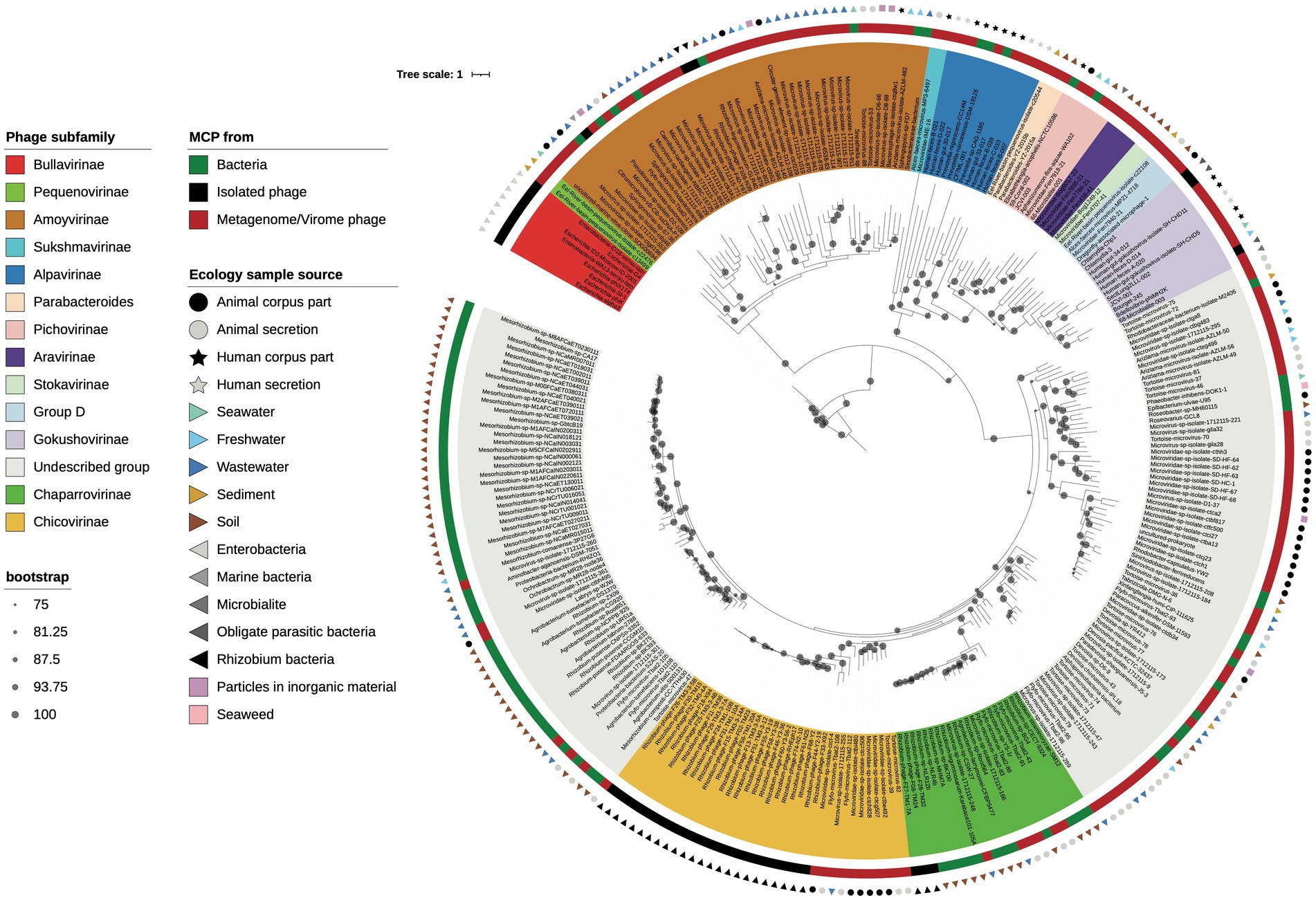
Figure 5. MCP phylogeny. Maximum likelihood phylogenetic tree based on the major capsid protein (MCP) sequences of the Rhizobium microviruses, related BlastP hits, and representative phages from different proposed Microviridae subfamilies. Bootstrap scores greater than 75% are indicated with a black dot. Phage names are highlighted with colors corresponding to subfamily identity (see color legend to the left).
Then, within the clade IV, the Rhizobium microviruses that belonged to the VCs of the chicoviruses (F02) and chaparroviruses (F08) were clustered with MCPs of phages detected in fecal samples of vertebrates (Orton et al., 2020; Lopez et al., 2022), and with putative prophages in hosts closely related to Rhizobium (Mesorhizobium, Agrobacterium, Ochrobactrum, and R. pusense; Figure 5).
Two Rhizobium microviruses (ANIm cluster F40) were grouped with the uncultured marine virus clone SOG00694 and various phages and prophages associated with Sphingomonadaceae and other proteobacteria (Figure 5), previously described as Amoyvirinae (Zheng et al., 2018).
Blast searches with the MCP sequences of the three Rhizobium Microviridae clusters showed similarities with phages detected in diverse ecological niches, including metagenome samples from animal-associated microbiomes (Orton et al., 2020; Tisza et al., 2021; Lopez et al., 2022), but also from a sewage oxidation pond (Kraberger et al., 2021). MCPs were also found in genomes of bacteria associated with soil and aquatic environments suggesting lysogenic interaction with microviruses (Figure 5). The ample genetic and ecological diversification of microviruses indicates that the phylogenetic history of microviruses may be discordant with the phylogeny of the bacterial host they infect. To investigate this possibility, we compared the topologies of the 16S phylogeny bacterial host with the MCP phylogenies using ParaFit (Legendre et al., 2002).
Moreover, most of these soil bacteria were closely related to Rhizobium, within the order of Hyphomicrobiales. In contrast, bacteria with more distantly related MCPs, like those from aquatic environments, were more distantly related to Rhizobium.
The 16S phylogeny of the MCP-housing bacteria, for which we could obtain species identity and/or were available in the RefSeq database, was significantly similar to the MCP phylogeny (i.e., co-phylogeny ParaFit: p = 3e-04) since genetically similar MCPs were shared among genetically similar bacterial hosts (Figure 6). Although the presence of MCP alone does not necessarily indicate the presence of microvirus prophages, the result of single-marker phylogenies suggested that microviruses have coevolved with their bacterial host.

Figure 6. Co-phylogeny between microviruses and their bacterial hosts. Phage major capsid protein (MCP) phylogeny was compared to the bacterial 16S sequence phylogeny with Parafit (Legendre et al., 2002). The lines between the two phylogenies connect phages with their corresponding hosts.
Knowledge of the abundance and diversity of the Microviridae family is expanding, mainly due to the resolution of metagenomic methods and powerful bioinformatics algorithms. Metagenomics has allowed the identification and classification of single-stranded DNA viruses in DNA obtained from various ecosystems (Kirchberger et al., 2022). In contrast, the isolation of viral particles of microviruses by bacterial infection has progressed slowly. Cultivated microviruses constitute 16% of the complete Microviridae genomes represented in GenBank. Furthermore, most known isolated microviruses are relatives of phiX174 (Bullavirinae), infecting Enterobacteria (Brentlinger et al., 2002). In this work, a comprehensive collection of cultivated microvirus isolates that infect the symbiotic bacterium Rhizobium was placed in a phylogenetic context revealing two novel lineages and one included within the already proposed subfamily Amoyvirinae (Zheng et al., 2018). The new lineages are unrelated to recognized ICTV Microviridae subfamilies (Bullavirinae and Gokushovirinae) and other proposed subfamilies (Krupovic and Forterre, 2011; Roux et al., 2012; Quaiser et al., 2015; Zheng et al., 2018). The biological properties of representative Rhizobium microviruses and the proposed taxonomic affiliation reported here make them a valuable resource for further experimental studies.
The diversity of bacteriophages infecting soil rhizobia, well-studied and economically significant bacteria, such as Rhizobium, remains underexplored. Until recently, Caudovirales were the only group of phages known to infect rhizobia (Werquin et al., 1988; Santamaría et al., 2014, 2022). Recently, a collection of 63 lytic ssDNA viruses was isolated from agricultural soils and showed abundances ranging from 0 to 60%, predominating over Caudovirales in one field (Van Cauwenberghe et al., 2021). The genome sequence of 29 microviruses, representative of the entire collection, shows the basic genetic structure of most microviruses with genes encoding for the capsid (mcp), the replication (rep), and the endolysin (lys). The genes encoding for the pilot protein and other auxiliary proteins for capsid assembly and replication, as in Bullavirinae and Gokushovirinae members, were undetected in our BlastP searches. Though the pilot protein has been involved in the superinfection exclusion of phages of Gokushovirinae, it has remote sequence similarity with sequences of microviruses of other subfamilies (Krupovic and Forterre, 2011; Kirchberger et al., 2021; Zucker et al., 2022).
We describe Rhizobium microviruses clustered in three distinct groups based on ANIb, AAI, protein-sharing networks, and MCP phylogeny. Two novel Microviridae clusters were included within an ample viral cluster of uncultivated microviruses of metagenomic origin (Figure 4). We refer to these as chaparroviruses and chicoviruses from the synonymous words chaparro- and chico-, meaning “small in size” in Spanish. They formed groups distinct from any previously described subfamilies of Microviridae. Chaparroviruses and chicoviruses are sister clades that share similar proteins yet form separate clusters in protein-sharing networks and show low nucleotide identity (ANIb <70%). Chaparroviruses share about 98% ANIm, and chicoviruses share 95% ANIm. This difference in cohesion is due to the presence of four subclusters within the chicoviruses, which partially reflect the different spatial origins of the phages. The phylogeny of the MCP protein confirms the relationships between chaparroviruses and chicoviruses and various undescribed clades, highlighting how little is known about Microviridae diversity.
The third group of Rhizobium microviruses, represented by two phages, is included within the Amoyvirinae subfamily, a recently described group of lytic phages and prophages infecting marine Sphingomonadaceae (Zheng et al., 2018). Amoyvirinae have notably smaller genomes than most Microviridae, about 4.3 kb vs. about 6 kb, yet those infecting rhizobia appear to have larger genomes of approximately 4.8 kb. The two Rhizobium Amoyvirinae share nearly 84% ANIm and form a coherent viral cluster within the protein-sharing network. In addition, these phages are far related to vB_Cib of Citromicrobium, another cultivated representant of Amoyvirinae of marine origin (Zheng et al., 2018).
The MCP structure differs among microvirus subfamilies (Roux et al., 2012; Quaiser et al., 2015). Three-dimensional models of the MCP of Rhizobium microviruses showed a jelly-roll fold, commonly found in capsids of microviruses and RNA and ssDNA virus of eukaryotes (Krupovic and Koonin, 2017). The 3D models of the MCP of the phages TM23 (chicovirus) and TM24 (chaparrovirus) are similar to each other and to the published MCP 3D models of Gokushovirus, Alphavirus, Pichovirus, Stokavirus, and Aravirus (Roux et al., 2012; Quaiser et al., 2015). They have a large variable loop at the same position in the structure of phiX174, which in contrast, has a small loop (McKenna et al., 1992). Although the model of MCP-Y67 (Amoyvirinae) is reminiscent of MCP-phiX174, its loop is reduced to a hypervariable region. The alignment of the amino acid sequence of the MCP-TM23 and MCP-Y67 showed a region from the proline 205 to glycine 310 in the first protein that is absent in the last (Supplementary Figure S3). Comparing the MCP models suggests that the hypervariable loop may have a role in the host recognition and diversification of the Microviridae.
A key element to understanding the diversification and adaptation of microviruses is the identification of their bacterial hosts and their range of infection. Nevertheless, most microviruses genomes come from metagenomes from various ecological niches, and there is limited knowledge about the bacterial species they infect. Rhizobium microviruses have a broad host range of infection of diverse strains of R. etli and R. phaseoli. Chicoviruses, the group more represented in the agriculture plot sampled, possibly represents an ongoing evolving lineage specializing in infecting Rhizobium. Few members of the currently known subfamilies have been identified in the Rhizobium Microviridae (e.g., Bullavirinae or Gokushovirinae). Few or none of the Gokushovirinae are presently known to infect hosts of the superphylum Proteobacteria, suggesting that different clades of Microviridae have specialized to infect particular taxa of bacteria or at least are more prevalent in these taxa. Chaparroviruses and chicoviruses infecting Rhizobium are closely related to various phages detected mainly in fecal viromes of vertebrates (Tisza et al., 2021; Lopez et al., 2022) whose gut microbiomes are noticeably richer in Proteobacteria (Sun et al., 2020; Kim et al., 2021). Proteobacteria are rarer in human gut microbiomes (Reiss et al., 2016; Senghor et al., 2018) and increased abundances indicate dysbiosis and disease (Morgan et al., 2012; Shin Na-Ri et al., 2015), while Bacteroidota, common hosts of other Microviridae (e.g., Alpavirinae and Pichovirinae), are more dominant (Reiss et al., 2016). Furthermore, MCPs closely related to chaparroviruses and chicoviruses are found as prophages in a small subset of genomes of host strains closely related to Rhizobium within the order of Hyphomicrobiales. In contrast, more distantly related MCPs are found in members of the corresponding order of Rhodobacterales (Figures 5,6).
The co-phylogeny of MCPs and 16S sequences suggests coevolutionary history between the Microviridae described here and their hosts. The observed coevolutionary pattern may be due to reciprocal adaptation and counter-adaption between the virus and its host or long-term prophage integration. Lysogeny of microviruses has been experimentally demonstrated for synthetic Gokushoviruses, which can integrate into the host genome and produce lytic plaques (Kirchberger and Ochman, 2020). Lysogenic microviruses have also been described in Alpavirinae (Krupovic and Forterre, 2011) and Bullavirinae (Kirchberger et al., 2021). The discovery of microvirus sequences (e.g., mcp) integrated into genomes of some species of bacteroidetes, proteobacteria, and enterobacteria support a lysogenic cycle. Nevertheless, the divergence of microvirus sequences is so great that it has been challenging to demonstrate the presence of complete microvirus prophages. Recent work using HMM profiles and recursive Blast has allowed the identification of Microviridae prophages in a vast collection of genomes, including some in Rhizobiaceae (Kirchberger et al., 2022).
Some MCPs related to Rhizobium Microviridae could be leftovers from degenerated prophages, also called cryptic prophages (Casjens, 2003). In the long term, footprints of the interaction between microviruses and their hosts remain in the genomes. Microviruses may have been initially integrated before the divergence of their respective hosts and got stranded due to a deleterious mutation (Wang et al., 2010; Bobay et al., 2014). However, prophages and cryptic prophages are expected to degenerate rapidly out of existence since they represent a metabolic cost (Campbell, 1998) unless they experience purifying selection due to a benefit they provide to the host (Bobay et al., 2014).
Capsid proteins are generally not retained and are increasingly rare in more degenerated prophages (Khan et al., 2020; Pattenden et al., 2022). Our observation of virion sequences clustering intertwined with prophages indicates relatively recent prophage integrations and continuous dynamic alternations between virulent and temperate lifestyles.
Recent efforts have been made to transform viral taxonomy from a morphology-based into a genome-based classification (Aiewsakun and Simmonds, 2018; Turner et al., 2021). One key aspect of this effort is setting out generalizable and clear delineations of various taxonomic levels (e.g., for Caudovirales in Turner et al., 2021). In analogy with such guidelines, we identify two new species within the Amoyvirinae and four new species in the chicovirus group. In a recent preprint, the same chaparroviruses, chicoviruses, and related prophages described here were classified as one subfamily named Occultatumvirinae (Zucker et al., 2022). Then, this would suggest that chaparroviruses and chicoviruses are two genera within this subfamily. Though both groups are connected in a protein-sharing network, they form distinct clusters and share little average nucleotide identity.
Moreover, various previously suggested subfamilies are either internally divided into multiple monophyletic clusters in the protein-sharing network or are more connected than chaparroviruses and chicoviruses. Turner and coauthors (Turner et al., 2021) proposed to define Caudovirales phage families as “cohesive and monophyletic groups in the … proteome-based clustering tools.” This definition does not apply to the current classification of Microviridae, indicating the need for a more precise delineation of taxonomic levels within this group. The taxonomy of Microviridae could be revised to match delineations set out for Caudovirales. One step in this direction would be to elevate Microviridae to the rank of order and its subfamilies to families (Kirchberger et al., 2022). However, the difference in genome sizes and evolutionary relevant genomic sizes might challenge the idea of a generalizable classification system.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
VG: conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, visualization, writing—original draft, writing—review and editing. RS: data curation, formal analysis, methodology, software, validation, visualization. PB: data curation, methodology, resources, software. JVC: conceptualization, data curation, formal analysis, investigation, methodology, resources, software, writing—original draft, writing—review and editing. All authors contributed to the article and approved the submitted version.
The work was supported by PAPIIT-UNAM 2017–2019 (IN209817 to VG). JVC received Postdoctoral Scholarship from DGAPA-UNAM (2016–2018); JVC was partially supported by National Science Foundation (NSF; DEB-1457508, IOS-1759048, to E.L. Simms).
We thank José Espíritu, Víctor del Moral, and Alfredo Hernández for their support in software installation. Thanks to Guadalupe Zavala (Unidad de Microscopia Electrónica – UNAM) for help in phage micrographs. Thanks are extended for the reviewers for helpful comments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.990394/full#supplementary-material
1. ^https://github.com/FelixKrueger/TrimGalore (Accessed August 24, 2022).
2. ^https://cytoscape.org/ (Accessed August 24, 2022).
Aiewsakun, P., and Simmonds, P. (2018). The genomic underpinnings of eukaryotic virus taxonomy: creating a sequence-based framework for family-level virus classification. Microbiome 6:38. doi: 10.1186/s40168-018-0422-7
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bobay, L. M., Touchon, M., and Rocha, E. P. (2014). Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. U. S. A. 111, 12127–12132. doi: 10.1073/pnas.1405336111
Breitbart, M., and Rohwer, F. (2005). Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 13, 278–284. doi: 10.1016/j.tim.2005.04.003
Brentlinger, K. L., Hafenstein, S., Novak, C. R., Fane, B. A., Borgon, R., McKenna, R., et al. (2002). Microviridae, a family divided: isolation, characterization, and genome sequence of φMH2K, a bacteriophage of the obligate intracellular parasitic bacterium Bdellovibrio bacteriovorus. J. Bacteriol. 184, 1089–1094. doi: 10.1128/jb.184.4.1089-1094.2002
Campbell, A. (1998). “Prophages and cryptic prophages,” in Bacterial Genomes Physical Structure and Analysis. eds. F. J. De Bruijn, J. R. Lupski, and G. M. Weinstock (Springer: Boston, MA), 23–29. doi: 10.1007/978-1-4615-6369-3_3
Canchaya, C., Fournous, G., and Brussow, H. (2004). The impact of prophages on bacterial chromosomes. Mol. Microbiol. 53, 9–18. doi: 10.1111/j.1365-2958.2004.04113.x
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Casjens, S. (2003). Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 49, 277–300. doi: 10.1046/j.1365-2958.2003.03580.x
Chaudhari, N., Gupta, V., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chipman, P. R., Agbandje-McKenna, M., Renaudin, J., Baker, T. S., and McKenna, R. (1998). Structural analysis of the spiroplasma virus, SpV4: implications for evolutionary variation to obtain host diversity among the Microviridae. Structure 6, 135–145. doi: 10.1016/s0969-2126(98)00016-1
Dion, M. B., Oechslin, F., and Moineau, S. (2020). Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138. doi: 10.1038/s41579-019-0311-5
Doore, S. M., and Fane, B. A. (2016). The Microviridae: diversity, assembly, and experimental evolution. Virology 491, 45–55. doi: 10.1016/j.virol.2016.01.020
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5:113. doi: 10.1186/1471-2105-5-113
Hyman, P., and Abedon, S. T. (2009). “Practical methods for determining phage growth parameters,” in Bacteriophages Methods and Protocols. Vol. 1. eds. M. R. J. Clokie and A. M. Kropinski, 175–202.
Jang, B. H., Bolduc, B., Zablocki, O., Kuhn, J. H., Roux, S., Adriaenssens, E. M., et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 37, 632–639. doi: 10.1038/s41587-019-0100-8
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Khan, A., Burmeister, A. R., and Wahl, L. M. (2020). Evolution along the parasitism-mutualism continuum determines the genetic repertoire of prophages. PLoS Comput. Biol. 16:e1008482. doi: 10.1371/journal.pcbi.1008482
Kim, P. S., Shin, N. R., Lee, J. B., Kim, M. S., Whon, T. W., Hyun, D. W., et al. (2021). Host habitat is the major determinant of the gut microbiome of fish. Microbiome 9:166. doi: 10.1186/s40168-021-01113-x
King, A. M. Q., Adams, M. J., Carstens, E. B., and Lefkowitz, E. J. (2011). Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. Cambridge, MA: Elsevier Academic Press.
Kirchberger, P. C., Martinez, Z. A., Luker, L. J., and Ochman, H. (2021). Defensive hypervariable regions confer superinfection exclusion in microviruses. Proc. Natl. Acad. Sci. U. S. A. 118:e2102786118. doi: 10.1073/pnas.2102786118/
Kirchberger, P. C., Martinez, Z. A., and Ochman, H. (2022). Organizing the global diversity of microviruses. MBio 13:e0058822. doi: 10.1128/mbio.00588-22
Kirchberger, P. C., and Ochman, H. (2020). Resurrection of a global, metagenomically defined gokushovirus. elife 9:e51599. doi: 10.7554/eLife.51599
Kraberger, S., Schreck, J., Galilee, C., and Varsani, A. (2021). Genome sequences of microviruses identified in a sample from a sewage treatment oxidation pond. Microbiol. Resour. Announc. 10, e00373–e02110. doi: 10.1128/mra.00373-21
Krupovic, M., and Forterre, P. (2011). Microviridae goes temperate: microvirus-related proviruses reside in the genomes of bacteroidetes. PLoS One 6:e19893. doi: 10.1371/journal.pone.0019893
Krupovic, M., and Koonin, E. v. (2017). Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. U. S. A. 114, E2401–E2410. doi: 10.1073/pnas.1621061114
Labonté, J. M., and Suttle, C. A. (2013). Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front. Microbiol. 4:404. doi: 10.3389/fmicb.2013.00404
Legendre, P., Desdevises, Y., and Bazin, E. (2002). A statistical test for host-parasite coevolution. Syst. Biol. 51, 217–234. doi: 10.1080/10635150252899734
Lopez, J. K. M., Aleamotu’, M., Kami, V., Stainton, D., Lund, M. C., Kraberger, S., et al. (2022). Genomes of bacteriophages belonging to the orders Caudovirales and Petitvirales identified in fecal samples from Pacific flying fox (Pteropus tonganus) from the Kingdom of Tonga. Microbiol. Resour. Announc. 11:2. doi: 10.1128/mra.00038-22
López-Bueno, A., Tamames, J., Velázquez, D., Moya, A., Quesada, A., and Alcamí, A. (2009). High diversity of the viral community from an Antarctic Lake. Science 326, 858–861. doi: 10.1126/science.1179287
Maclean, E. C., and Hall, C. E. (1962). Studies on bacteriophage φX174 and its DNA by electron microscopy. J. Mol. Biol. 4:173. doi: 10.1016/S0022-2836(62)80049-7
Mariani, V., Biasini, M., Barbato, A., and Schwede, T. (2013). IDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 29, 2722–2728. doi: 10.1093/bioinformatics/btt473
McKenna, R., Xia, D., Willingmann, P., Llag, L. L., Krishnaswamy, S., Rossmann, M. G., et al. (1992). Atomic structure of single-stranded DNA bacteriophage phiX 174 and its functional implications. Nature 355, 137–143. doi: 10.1038/355137a0
Mirdita, M., Steinegger, M., Breitwieser, F., Söding, J., and Levy Karin, E. (2021). Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 37, 3029–3031. doi: 10.1093/bioinformatics/btab184
Morella, N. M., Gomez, A. L., Wang, G., Leung, M. S., and Koskella, B. (2018). The impact of bacteriophages on phyllosphere bacterial abundance and composition. Mol. Ecol. 27, 2025–2038. doi: 10.1111/mec.14542
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., and Devaney, K. L., Ward, D. v., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. doi: 10.1186/gb-2012-13-9-r79
Na-Ri, S., Whon, T. W., and Bae, J.-W. (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Nautiyal, C. S. (1997). A method for selection and characterization of rhizosphere-competent bacteria of chickpea. Curr. Microbiol. 34, 12–17. doi: 10.1007/s002849900136
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Orton, J. P., Morales, M., Fontenele, R. S., Schmidlin, K., Kraberger, S., Leavitt, D. J., et al. (2020). Virus discovery in desert tortoise fecal samples: novel circular single-stranded DNA viruses. Viruses 12:143. doi: 10.3390/v12020143
Paradis, E., and Schliep, K. (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Parsons, R. J., Breitbart, M., Lomas, M. W., and Carlson, C. A. (2012). Ocean time-series reveals recurring seasonal patterns of virioplankton dynamics in the northwestern Sargasso Sea. ISME J. 6, 273–284. doi: 10.1038/ismej.2011.101
Pattenden, T., Eagles, C., and Wahl, L. M. (2022). Host life-history traits influence the distribution of prophages and the genes they carry. Philos. Trans. R. Soc. B: Biol. Sci. 377:20200465. doi: 10.1098/rstb.2020.0465
Pedulla, M. L., Ford, M. E., Houtz, J. M., Karthikeyan, T., Wadsworth, C., Lewis, J. A., et al. (2003). Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182. doi: 10.1016/s0092-8674(03)00233-2
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. doi: 10.1002/pro.3943
Poullain, V., Gandon, S., Brockhurst, M. A., Buckling, A., and Hochberg, M. E. (2008). The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 62, 1–11. doi: 10.1111/j.1558-5646.2007.00260.x
Pratama, A. A., and van Elsas, J. D. (2018). The “neglected” soil virome: potential role and impact. Trends Microbiol. 26, 649–662. doi: 10.1016/j.tim.2017.12.004
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Quaiser, A., Dufresne, A., Ballaud, F., Roux, S., Zivanovic, Y., Colombet, J., et al. (2015). Diversity and comparative genomics of Microviridae in sphagnum-dominated peatlands. Fronti. Microbiol. 6:375. doi: 10.3389/fmicb.2015.00375
Ramisetty, B. C. M., and Sudhakari, P. A. (2019). Bacterial “grounded” prophages: hotspots for genetic renovation and innovation. Front. Genet. 10:65. doi: 10.3389/fgene.2019.00065
Reiss, A., Jacobi, M., Rusch, K., and Schwiertz, A. (2016). Association of dietary type with fecal microbiota and short chain fatty acids in vegans and omnivores. J. Int. Soc. Microbiota 1:1. doi: 10.18143/JISM_v1i1.782
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1
Roux, S., Brum, J. R., Dutilh, B. E., Sunagawa, S., Duhaime, M. B., Loy, A., et al. (2016a). Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 537, 689–693. doi: 10.1038/nature19366
Roux, S., Krupovic, M., Poulet, A., Debroas, D., and Enault, F. (2012). Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS One 7:e40418. doi: 10.1371/journal.pone.0040418
Roux, S., Solonenko, N. E., Dang, V. T., Poulos, B. T., Schwenck, S. M., Goldsmith, D. B., et al. (2016b). Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 4:e2777. doi: 10.7717/peerj.2777
Santamaría, R. I., Bustos, P., Sepúlveda-Robles, O., Lozano, L., Rodríguez, C., Fernández, J. L., et al. (2014). Narrow-host-range bacteriophages that infect Rhizobium etli associate with distinct genomic types. Appl. Environ. Microbiol. 80, 446–454. doi: 10.1128/AEM.02256-13
Santamaría, R. I., Bustos, P., van Cauwenberghe, J., and González, V. (2022). Hidden diversity of double-stranded DNA phages in symbiotic Rhizobium species. Philos. Trans. R. Soc. B: Biol. Sci. 377:20200468. doi: 10.1098/rstb.2020.0468
Sehnal, D., Bittrich, S., Deshpande, M., Svobodová, R., Berka, K., Bazgier, V., et al. (2021). Mol∗Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 49, W431–W437. doi: 10.1093/nar/gkab314
Senghor, B., Sokhna, C., Ruimy, R., and Lagier, J. C. (2018). Gut microbiota diversity according to dietary habits and geographical provenance. Hum. Microbiome J. 7–8, 1–9. doi: 10.1016/j.humic.2018.01.001
Shkoporov, A. N., Clooney, A. G., Sutton, T. D. S., Ryan, F. J., Daly, K. M., Nolan, J. A., et al. (2019). The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe 26, 527–541.e5. doi: 10.1016/j.chom.2019.09.009
Sun, D. L., Gao, Y. Z., Ge, X. Y., Shi, Z. L., and Zhou, N. Y. (2020). Special features of bat microbiota differ from those of terrestrial mammals. Front. Microbiol. 11:1040. doi: 10.3389/fmicb.2020.01040
Suttle, C. A. (2007). Marine viruses - major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812. doi: 10.1038/nrmicro1750
Tisza, M. J., Buck, C. B., and Chang, Y. (2021). A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. U.S. A. 118:e2023202118. doi: 10.1073/pnas.2023202118
Touchon, M., Moura de Sousa, J. A., and Rocha, E. P. (2017). Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr. Opin. Microbiol. 38, 66–73. doi: 10.1016/j.mib.2017.04.010
Turner, D., Kropinski, A. M., and Adriaenssens, E. M. (2021). A roadmap for genome-based phage taxonomy. Viruses 13:13. doi: 10.3390/v13030506
Van Cauwenberghe, J., Santamaría, R. I., Bustos, P., Juárez, S., Ducci, M. A., Figueroa Fleming, T., et al. (2021). Spatial patterns in phage-Rhizobium coevolutionary interactions across regions of common bean domestication. ISME J. 15, 2092–2106. doi: 10.1038/s41396-021-00907-z
Wang, X., Kim, Y., Ma, Q., Hong, S. H., Pokusaeva, K., Sturino, J. M., et al. (2010). Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 1:147. doi: 10.1038/ncomms1146
Wang, X., Wei, Z., Yang, K., Wang, J., Jousset, A., Xu, Y., et al. (2019). Phage combination therapies for bacterial wilt disease in tomato. Nat. Biotechnol. 37, 1513–1520. doi: 10.1038/s41587-019-0328-3
Weitz, J. S., Poisot, T., Meyer, J. R., Flores, C. O., Valverde, S., Sullivan, M. B., et al. (2013). Phage-bacteria infection networks. Trends Microbiol. 21, 82–91. doi: 10.1016/j.tim.2012.11.003
Werquin, M., Ackermann, H. W., and Levesque, R. C. (1988). A study of 33 bacteriophages of rhizobium meliloti. Appl. Environ. Microbiol. 54, 188–196. doi: 10.1128/AEM.54.1.188-196.1988
Wheatley, R. M., Ford, B. L., Li, L. N., Aroney, S. T., Knights, H. E., Ledermann, R., et al. (2020). Lifestyle adaptations of Rhizobium from rhizosphere to symbiosis. Proc. Natl. Acad. Sci. U. S. A. 117, 23823–23834. doi: 10.1073/pnas.2009094117
Zablocki, O., Adriaenssens, E. M., and Cowan, D. (2016). Diversity and ecology of viruses in hyperarid desert soils. Appl. Environ. Microbiol. 82, 770–777. doi: 10.1128/AEM.02651-15
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zheng, Q., Chen, Q., Xu, Y., Suttle, C. A., and Jiao, N. (2018). A virus infecting marine photoheterotrophic alphaproteobacteria (Citromicrobium spp.) defines a new lineage of ssDNA viruses. Front. Microbiol. 9:1418. doi: 10.3389/fmicb.2018.01418
Keywords: Rhizobium, phages, Microviridae, MCP, viral clusters, coevolution
Citation: Van Cauwenberghe J, Santamaría RI, Bustos P and González V (2022) Novel lineages of single-stranded DNA phages that coevolved with the symbiotic bacteria Rhizobium. Front. Microbiol. 13:990394. doi: 10.3389/fmicb.2022.990394
Edited by:
Heejoon Myung, Hankuk University of Foreign Studies, South KoreaReviewed by:
Thomas Klose, Purdue University, United StatesCopyright © 2022 Van Cauwenberghe, Santamaría, Bustos and González. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jannick Van Cauwenberghe, anZhbmNhdUBiZXJrZWxleS5lZHU=; Víctor González, dmdvbnphbEBjY2cudW5hbS5teA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.