 Yong-Hong Liu†1,2
Yong-Hong Liu†1,2 Yi-Min Ma1Hong-Ou Tian1Bo Yang3Wen-Xiong Han4Wei-Hong Zhao1Hai-Liang Chai1Zhan-Sheng Zhang1Li-Feng Wang1Lei Chen5Yu Xing5Yu-Lin Ding1,2
Yi-Min Ma1Hong-Ou Tian1Bo Yang3Wen-Xiong Han4Wei-Hong Zhao1Hai-Liang Chai1Zhan-Sheng Zhang1Li-Feng Wang1Lei Chen5Yu Xing5Yu-Lin Ding1,2 Li Zhao†1,2*
Li Zhao†1,2*- 1College of Veterinary Medicine, Inner Mongolia Agricultural University, Hohhot, China
- 2Key Laboratory of Clinical Diagnosis and Treatment Technology in Animal Disease, Ministry of Agriculture and Rural Affairs, Hohhot, China
- 3Animal Disease Control Center of Ordos, Ordos City, China
- 4Inner Mongolia Saikexing Reproductive Biotechnology (Group) Co., Ltd., Hohhot, China
- 5Shanghai Origingene Bio-pharm Technology Co., Ltd., Shanghai, China
Melophagus ovinus (sheep ked) is one of the common ectoparasites in sheep. In addition to causing direct damage to the host through biting and sucking blood, sheep ked is a potential vector of helminths, protozoa, bacteria, and viruses. Sheep M. ovinus samples from three regions in Tibet were selected for DNA extraction. The 16S rDNA V3-V4 hypervariable region was amplified, after genomic DNA fragmentation, Illumina Hiseq libraries were constructed. The 16S rRNA sequencing and viral metagenomics sequencing were separately conducted on the Illumina Novaseq 6000 platform and molecular biology software and platforms were employed to analyze the sequencing data. Illumina PE250 sequencing results demonstrated that the dominant bacteria phylum in M. ovinus from Tibet, China was Proteobacteria, where 29 bacteria genera were annotated. The dominant bacterial genera were Bartonella, Wolbachia, and Arsenophonus; Bartonella chomelii, Wolbachia spp., and Arsenophonus spp. were the dominant bacterial species in M. ovinus from Tibet, China. We also detected Kluyvera intermedia, Corynebacterium maris DSM 45190, Planomicrobium okeanokoites, and Rhodococcus erythropolis, of which the relative abundance of Kluyvera intermedia was high. Illumina Hiseq sequencing results demonstrated that 4 virus orders were detected in M. ovinus from Tibet, China, and 3 samples were annotated into 29 families, 30 families, and 28 families of viruses, respectively. Virus families related to vertebrates and insects mainly included Mimiviridae, Marseilleviridae, Poxviridae, Ascoviridae, Iridoviridae, Baculoviridae, Hytrosaviridae, Nudiviridae, Polydnaviridae, Adomaviridae, Asfarviridae, Hepeviridae, Herpesviridae, and Retroviridae; at the species level, the relative abundance of Tupanvirus_soda_lake, Klosneuvirus_KNV1, and Indivirus_ILV1 was higher. African swine fever virus and many poxviruses from the family Poxviridae were detected, albeit their relative abundance was low. The dominant bacterial phylum of M. ovinus from Tibet, China was Proteobacteria, and the dominant bacterial genera were Bartonella, Wolbachia, and Arsenophonus, where 23 out of 29 annotated bacteria genera were first reported in M. ovinus. Kluyvera intermedia, Corynebacterium maris DSM 45190, Planomicrobium okeanokoites, and Rhodococcus erythropolis were detected for the first time. All DNA viruses detected in this study have been reported in M. ovinus for the first time.
Introduction
There is a type of wingless fly that has dense setae on the body surface and three pairs of legs with claws and belongs to the genus Melophagus and family Hippoboscidae (Diptera: Hippoboscoidea), which is a hematophagous ectoparasite of animals (Small, 2005). Its scientific name is Melophagus ovinus (M. ovinus) and the proper name is sheep ked (Ulicsni et al., 2016). M. ovinus is also known as ked or louse fly (Tang et al., 2018), and it is called by different folk names in different regions (such as uhkullancs [sheep kullancs] and kullancs) (Ulicsni et al., 2016). M. ovinus is commonly wrongly named as sheep tick (Small, 2005). Sheep is the permanent host of M. ovinus (Halos et al., 2004; Small, 2005; Kumsa et al., 2012; Liu et al., 2018, 2019; Zhao et al., 2018, 2020), but M. ovinus is also found in goats (Bequaert, 1942; Seyoum et al., 2015), rabbits (Small, 2005), dogs (Tetley, 1958), donkeys (Flores-Mendoza et al., 2021), European bison (Izdebska, 2001), red foxes (Lassnig et al., 1998), Tibetan sheep (Hao et al., 2020), Tibetan antelope (Liu et al., 2018; Zhao et al., 2018, 2020) and humans (Tetley, 1958). M. ovinus has a broad geographical distribution and is reported in Europe [e.g., Czechia (Chrudimský et al., 2012; Nováková et al., 2015; Rudolf et al., 2016), Hungary (Hornok et al., 2011), Slovakia (Hubálek et al., 1986), Croatia (Martinković et al., 2012), St Kilda (Gibson et al., 2010), France (Halos et al., 2004), Poland (Werszko et al., 2021), Russia (Litov et al., 2021)], Africa [e.g., Ethiopia (Kumsa et al., 2012, 2014; Seyoum et al., 2015; Hadgu et al., 2021), Algeria (Boucheikhchoukh et al., 2019), Kenya (Liu et al., 2016)], Oceania [e.g., Australia (Liu et al., 2016; Litov et al., 2021), New Zealand (Liu et al., 2016)], North America [e.g., United States (Halos et al., 2004; Kosoy et al., 2004, 2016)], South America [e.g., Peru (Flores-Mendoza et al., 2021)] and Asia [e.g., Turkey (Payzin, 1953), Mongolia, India, Japan (Liu et al., 2016)]. In recent years, M. ovinus has frequently appeared in China, Asia in Tibet (Chu et al., 2011), Gansu (Duan et al., 2017, 2020; Tang et al., 2018; Zhao et al., 2020), Xinjiang (Liu et al., 2016, 2017, 2019; Zhao et al., 2018, 2020), Sichuan (Hao et al., 2020), and Qinghai (Zhang et al., 2021).
Melophagus ovinus causes damage to the host via biting and blood-sucking in two aspects. On the hand, it causes restlessness, anemia, weight loss, wool loss, skin irritation and pruritus, skin damage, and reduced skin value in the host, which further causes inflammation, secondary microbial infections, cutaneous myiasis (Small, 2005), and scabies (No Author List., 2000), and decreases meat, milk, and fur yields in the host (Small, 2005). More importantly on the other hand, M. ovinus can carry or spread pathogens such as Rickettsia spp. (Hornok et al., 2011), R. raoultii, R. slovaca (Liu et al., 2016), R. melophagi (Arkwright et al., 1919; Cowdry, 1923; Hertig and Wolbach, 1924; Kligler and Aschner, 1931; Rudolf et al., 2016), Bartonella spp. (Halos et al., 2004; Reeves et al., 2006; Kumsa et al., 2014; Duan et al., 2020; Werszko et al., 2021), Ba. melophagi (Halos et al., 2004; Kosoy et al., 2004, 2016; Bemis and Kania, 2007; Kumsa et al., 2014; Rudolf et al., 2016; Liu et al., 2018; Boucheikhchoukh et al., 2019; Flores-Mendoza et al., 2021), Ba. melophagi variants variants, Ba. chomelii (Duan et al., 2020), Ba. schoenbuchensis (Halos et al., 2004), Wolbachia sp. (Duan et al., 2017, 2020; Liu et al., 2018; Boucheikhchoukh et al., 2019), Trypanosoma spp. (Werszko et al., 2021), Tr. melophagium (Hoare, 1923, 1972; Gibson et al., 2010; Martinković et al., 2012), Tr. theodori (Small, 2005), Anaplasma ovis (Zaugg and Coan, 1986; Hornok et al., 2011; Zhao et al., 2018; Zhang et al., 2021), An. phagocytophilum, An. bovis (Zhang et al., 2021), Acinetobacter spp. (Kumsa et al., 2012; Duan et al., 2017), Ac. lwoffii (Kumsa et al., 2012), Borrelia burgdorferi sensu lato (Werszko et al., 2021), Bo. garinii, Bo. spirochetes belonging to Bo. valaisiana-related group (Chu et al., 2011), Theileria Luwenshuni (Hao et al., 2020), Th. ovis (Zhao et al., 2020; Zhang et al., 2021), Anaplasmataceae family (Boucheikhchoukh et al., 2019), Arsenophonus spp. (Duan et al., 2017, 2020), Ar. melophagi (Husnik et al., 2020), Candidatus Ar. melophagi (Nováková et al., 2015), Coxiella burnetii (Pavilanis et al., 1958), Candidatus Sodalis melophagi (Chrudimský et al., 2012; Nováková et al., 2015; Boyd et al., 2016), Streptococcus hyointestinalis, Lactobacillus amylovorus, L. johnsonii, L. intestinalis, L. salivarius, L. murinus, L. agilis, Escherichia coli, Enterococcus cecorum, Megasphaera elsdenii, Helicobacter ganmani, Clostridium leptum, Clostridium sp. Culture 27, Actinobacillus minor, Aureococcus anophagefferens, Dorea ormicigenerans, Lachnospiraceae bacterium A2, Eubacterium coprostanoligenes, Blautia coccoides (Duan et al., 2020), Pseudomonas aeruginosa, Shewanella spp., Sh. algae, Staphylococcus spp., St. xylosus, Enterobacter spp., Halomonas spp., Bacillus spp. (Duan et al., 2017; Bezerra-Santos and Otranto, 2020). There are very few reports on M. ovinus-related viruses, which are all RNA viruses, such as blue-tongue virus (Luedke et al., 1965), Border disease virus (Liu et al., 2019), dengue virus (Setién et al., 2017), Khandagaity Melophagus iflfla-like virus, Bayan-Khairhan-Ula Melophagus solemo-like virus, Ulaatai Melophagus solemo-like virus, Aksy-Durug Melophagus sigmavirus, Berke-Baary Melophagus reo-like virus (Litov et al., 2021). In addition, M. ovinus from different regional sources were tested as negative for several specific pathogens, including those that were reportedly positive for M. ovinus in other regions (Dungal, 1946; Payzin, 1953; Nelder et al., 2008; Rudolf et al., 2016; Duan et al., 2017; Boucheikhchoukh et al., 2019; Flores-Mendoza et al., 2021; Werszko et al., 2021; Zhang et al., 2021). It is well-known that microorganisms in ectoparasites are unstable and that the microbiome composition and abundance are affected by the geographical origin and blood-feeding behavior. Therefore, there is a need for a comprehensive analysis of the diversity of microorganisms carried by M. ovinus from different regions.
For a long period, Hippoboscid flies (Diptera: Hippoboscidae) have been ignored by the scientific community, and its role as a vector of pathogens for humans and domestic animals has been scantly investigated; as a result, there is a lack of scientific knowledge on its biology, epidemiology as well as vector competence (Bezerra-Santos and Otranto, 2020). Examining the pathogens carried by M. ovinus in livestock or wild animals can help analyze the role of M. ovinus in epidemiology. High-throughput sequencing is expedient and efficient for the analysis of the microbial community structure of M. ovinus. However, high-throughput sequencing has been conducted only on microbial population and RNA virome in M. ovinus from Gansu province in China (Duan et al., 2017, 2020) as well as the Republic of Tuva in Russia (Litov et al., 2021) so far. However, to date, no study has employed modern metaviromics techniques to reveal the DNA virome of M. ovinus, and no study has conducted specific tests, such as PCR, to detect DNA viruses in M. ovinus. In this study, the Illumina Novaseq 6000 platform was employed to sequence M. ovinus from Tibet, China so as to determine the microbial community structure and diversity and analyze the DNA virome of M. ovinus in order to provide a foundation for predicting emerging pathogens, evaluating potential risks, preventing insect-borne diseases, and developing biological control tools.
Materials and methods
Melophagus ovinus collection
Melophagus ovinus (Figure 1) was collected from Garze County in Rikaze, Tibet (4029 m above sea level; 28°91′N, 89°60′E) and Lhari County in Nagqu, Tibet (4,498 m above sea level; 30°63′N, 93°24′E) from April to May 2020. and from Nyingchi, Tibet (3,008 m above sea level; 29°57′N, 94°48′E) in July 2019. The hosts were all sheep, and the samples were subjected to identification using a combination of morphological and molecular biology methods. The sample was stored at −80°C. For the convenience of description, the abbreviations used to represent each sample are SK1, SK2, and SK3.

Figure 1. Photomicrographs of Melophagus ovinus (Left: Ventral view of the female; Right: Ventral view of the male).
DNA extraction and high-throughput sequencing
Four M. ovinus (two of each gender) were collected from each sampling point and mixed as one sample. The body surfaces of the insects in every sample were cleaned thrice with 70% ethanol and sterile deionized water. Next, TaKaRa MiniBEST Universal Genomic DNA Extraction Kit Ver. 5.0 (Takara, Beijing, China, Code No. 9765) was used to extract total DNA from the samples according to the manufacturer’s protocol in a biosafety cabinet, and the extracted DNA was stored at −20°C.
One aliquot of each sample, which served as a DNA template, and specific primers with barcodes were used to amplify the 16S rDNA V3-V4 hypervariable region using the TransStart FastPfu DNA Polymerase (TransStart®, Beijing, China, Code No. AP221-02) kit. The universal primers were 338F: 5′-ACT CCT ACG GGA GGC AGC A-3′, 806R: 5′-GGA CTA CHV GGG TWT CTA AT-3′. The PCR reaction volume was 25 μL, and the annealing temperature was 55°C. PCR products were recovered using the AXYPREP DNA Gel Extraction Kit (AXYGEN, Suzhou, China, Code No. AP-GX-50G). After the samples were subjected to 2% agarose gel electrophoresis, Illumina PE250 libraries were constructed using the VAHTS® ssDNA Library Prep Kit (Illumina, San Diego, CA, United States, Code No. ND6201). Subsequently, 16S rRNA sequencing was conducted on the Illumina Novaseq 6000 platform (San Diego, CA, United States).
An additional portion of DNA from each sample was subjected to genomic DNA fragmentation using Covaris M220 Focused Ultrasonicator (Covaris, MA, United States, M220), and an Illumina Hiseq library was constructed using the TruSeq™ DNA Sample Prep Kit (Illumina, San Diego, CA, United States, catalog No. FC-121-2003). Then, bridge PCR was performed using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, San Diego, CA, United States, catalog No. PE-401-3001). Finally, viral metagenomic sequencing was performed using the Truseq SBS Kit v3-HS (Illumina, San Diego, CA, United States, catalog No. FC-401-3001) on the Illumina Novaseq 6000 platform (San Diego, CA, United States).
Sequencing was completed in collaboration with the Origin-gene biology Co., Ltd. (Shanghai, China).
Data analysis
The Illumina PE250 sequencing first obtained valid sequences from all samples according to the barcodes before paired-end reads from the original DNA fragments were merged using FLASH (version 1.2.11). UPARSE software package (Edgar, 2013) (version 7.0.1090)1 was employed for sequencing analysis. Sequences with ≥97% similarity were assigned to the same operational taxonomic units (OTUs), and RDP classifier (Wang et al., 2007) (version 2.2)2 Bayesian algorithm was used for the taxonomic analysis of the representatives OTU sequences from the phylum to species. For the diversity analysis of a single sample (alpha diversity), we calculated the number of unique OTUs, community richness marker Chao-the Chao1 estimator,3 community diversity marker Shannon-the Shannon index,4 Simpson-the Simpson index,5 and sequencing depth index coverage-the Good’s coverage6 for every sample. Inter-sample diversity analysis (beta analysis) was conducted on the Qiime platform (Caporaso et al., 2010) (version 1.9.0)7 and weighted and unweighted unifrac distances were calculated. Bray-Curtis and complete methods in the vegan package of R were applied for distance calculation and clustering analysis, respectively. R was used to calculate the number of common and unique OTUs in multiple samples, as depicted with Venn diagrams (Fouts et al., 2012).
Illumina Hiseq sequencing chromatogram data was converted to sequencing data using the Base Calling software. The Cutadapt8 software was used for quality cleavage and the quality control of source data. BWA9 was used for the alignment of high-quality data and host genome to eliminate host DNA contamination. The megahit (version 1.1.3)10 software was used for de novo assembly of the sequencing data. Contigs in the assembled results were applied for the ORF prediction to construct non-redundant gene sets. Next, SOAPaligner (version 2.21)11 was used to align high-quality reads of every sample and non-redundant gene sets (95% identity), and the abundant information of genes in corresponding samples was calculated. Then, the blast was used for blastp alignment of protein sequences of non-redundant genes and virus sequences. Finally, the optimal alignment results of every sequence were employed for species taxonomic annotation. BLASTP (BLAST Version 2.2.28 +)12 and MEGAN (Huson et al., 2011)13 were used to return the species abundance information from sequencing in the NCBI database.
Results
Illumina PE250
General statistics
Sequencing data statistics
After calibration and chimera removal of the sequencing results from 3 samples (SK1, SK2, and SK3), a total of 182,116 (54691, 53493, and 73932, respectively) optimized data sequences were obtained, where the length of this optimized data was 74,265,823 bp (22,156,008, 21,666,667, and 30,443,148 bp, respectively). The mean length of these optimized sequences was 407.79 bp (405.11, 405.04, and 411.77 bp, respectively), and 99.99% of the sequences were mapped to a 401–440-bp region. OTU selection and taxonomic assignments were employed to generate 174,533 (51,972, 51,609, and 70,952, respectively) high-quality, effective clean reads for analysis.
Alpha-diversity analysis
A similarity level of 97% was applied to calculate the alpha-diversity indices (Shannon, Simpson, Chao, ACE, and Good’s coverage). The results demonstrated that the SK3 sample had the highest Shannon value and the lowest Simpson value and that the SK1 sample had the highest Chao and Ace indices (Table 1). The Good’s coverage of the three samples exceeded 99.99%.
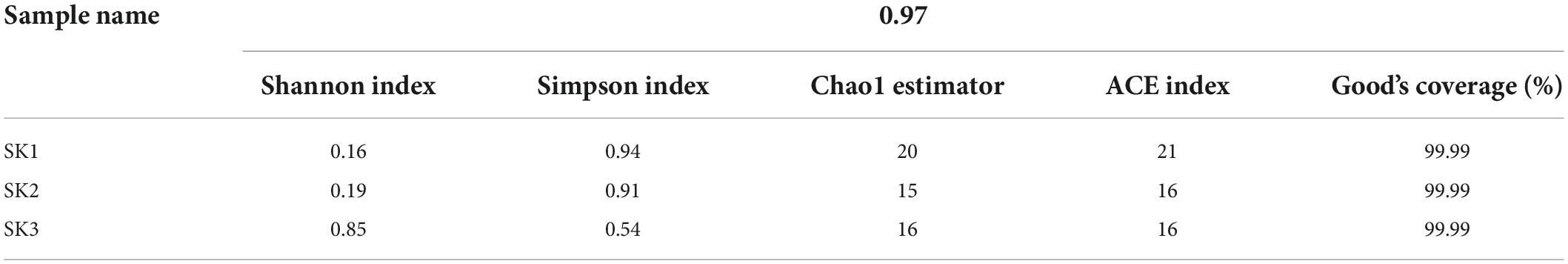
Table 1. Microbial richness and alpha-diversity indices of the samples.
Operational taxonomic unit cluster analysis
As shown in the Venn diagram in Figure 2, 19, 15, and 15 OTUs were obtained from SK1, SK2, and SK3 samples, respectively. Among these, nine OTUs showed high similarity between SK1 and SK2 samples, five OTUs showed high similarity between SK1 and SK3 samples, five OTUs showed high similarity between SK2 and SK3 samples, and three OTUs showed high similarity in all three samples. In all samples, most tags were classified, of which 97% of the tags were assigned to the genus level. Only a minority of tags were classified at the species level.
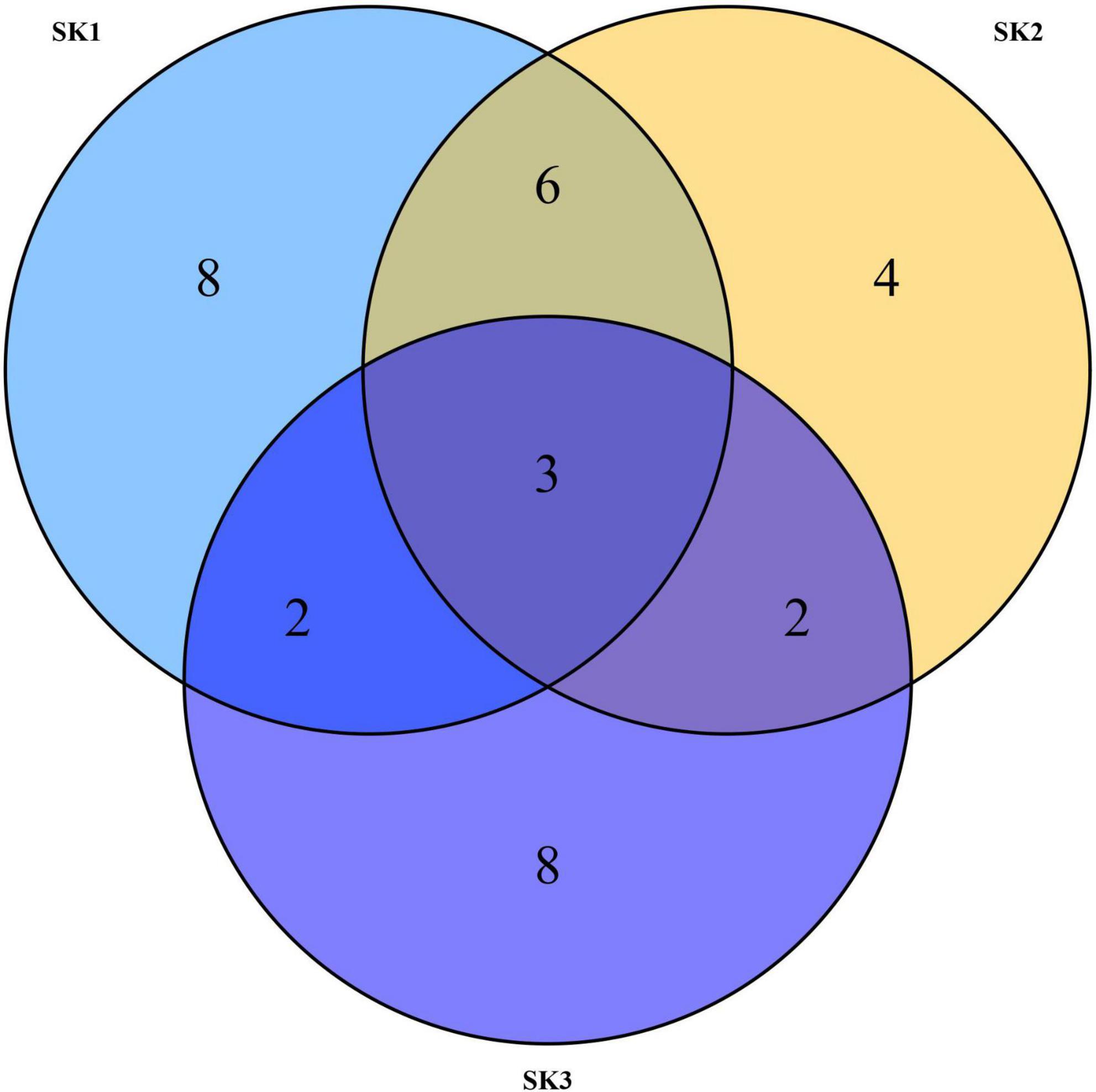
Figure 2. Venn diagrams of three Melophagus ovinus samples based on OTUs.
Microbial population
Microbial characteristics at the phyla level
Three phyla were annotated to SK1 and SK2 (Proteobacteria, Firmicutes, and Actinobacteria), and three phyla (Chloroflexi, Cyanobacteria, and Bacteria unclassified) were annotated to SK3. The abundance of Proteobacteria in the three samples was 98.96, 99.91, and 99.84%, respectively, showing a marked predominance. In addition, the relative abundances of Actinobacteria (0.42%), Firmicutes (0.61%) in SK1, and Actinobacteria (0.14%) in SK3 were also high.
Microbial characteristics at the genus level
A total of 29 bacterial genera were annotated in 3 samples, of which 16, 14, and 14 genera were present in SK1, SK2, and SK3 samples, and only 4 genera (Bartonella, Wolbachia, Arsenophonus, and Corynebacterium 1) were common among the 3 samples. Eighteen genera (Acinetobacter, Brachybacterium, Candidatus Pelagibacter, Cellulomonas, Glutamicibacter, JG30-KF-CM45 norank, Kluyvera, Micrococcaceae unclassified, Microcoleus, Micromonosporaceae unclassified, Nocardiopsis, Ornithinimicrobium, Propioniciclava, SAR116 clade norank, SAR86 clade norank, Salinicoccus, Staphylococcus, and ZD0405 norank) were present in only one of the three samples. Bartonella and Wolbachia were common dominant bacterial genera in three samples, and the relative abundance of Arsenophonus was relatively high in SK3 but low in SK1 and SK2. In SK1, the relative abundances of Corynebacterium 1, Jeotgalicoccus, Nocardiopsis, and Staphylococcus were high. In SK3, the relative abundances of Kluyvera and Rhodococcus were high, but the relative abundances of other genera were low (Table 2 and Figure 3 left).
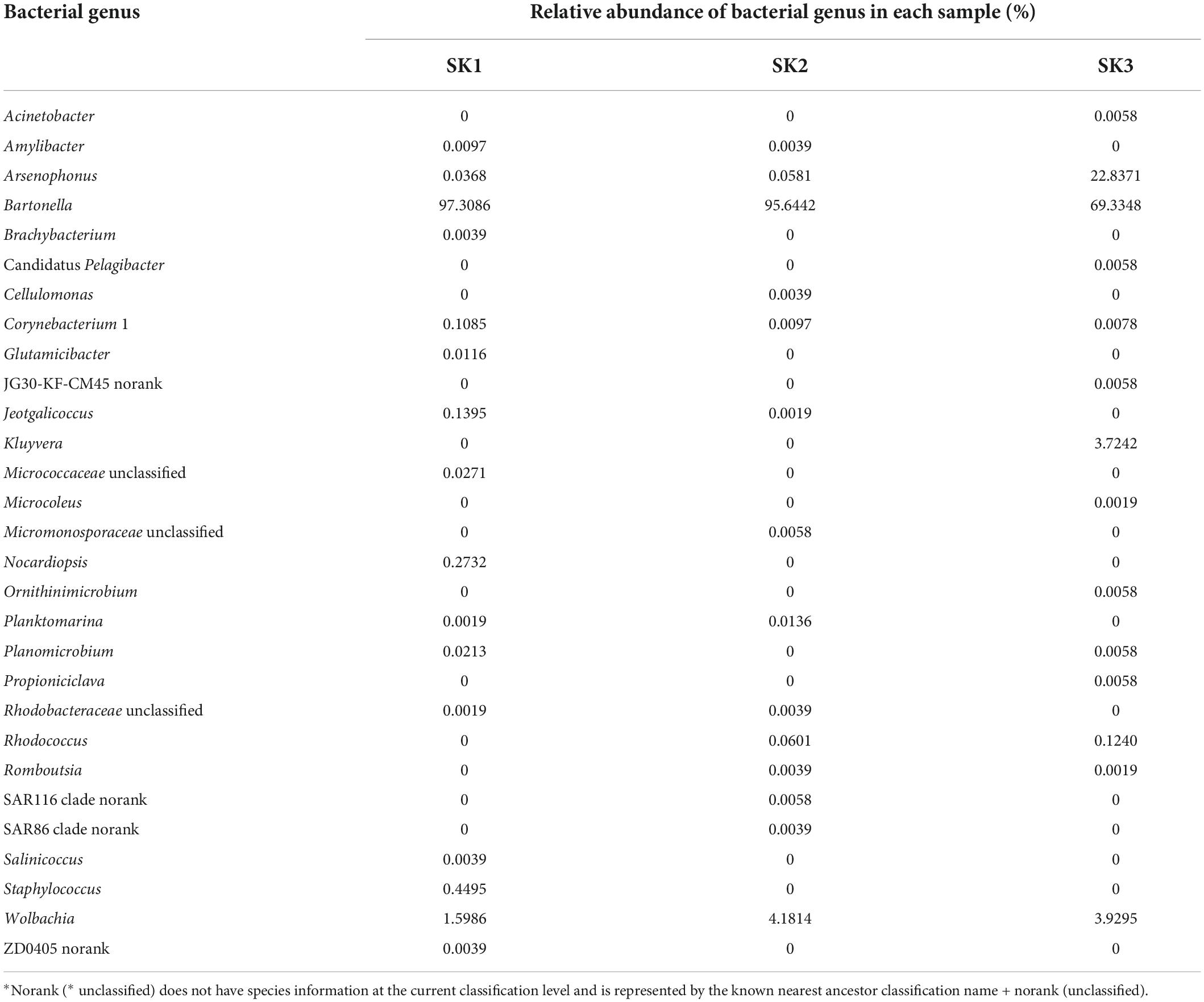
Table 2. The relative abundance of 29 bacterial genera in the 3 samples.
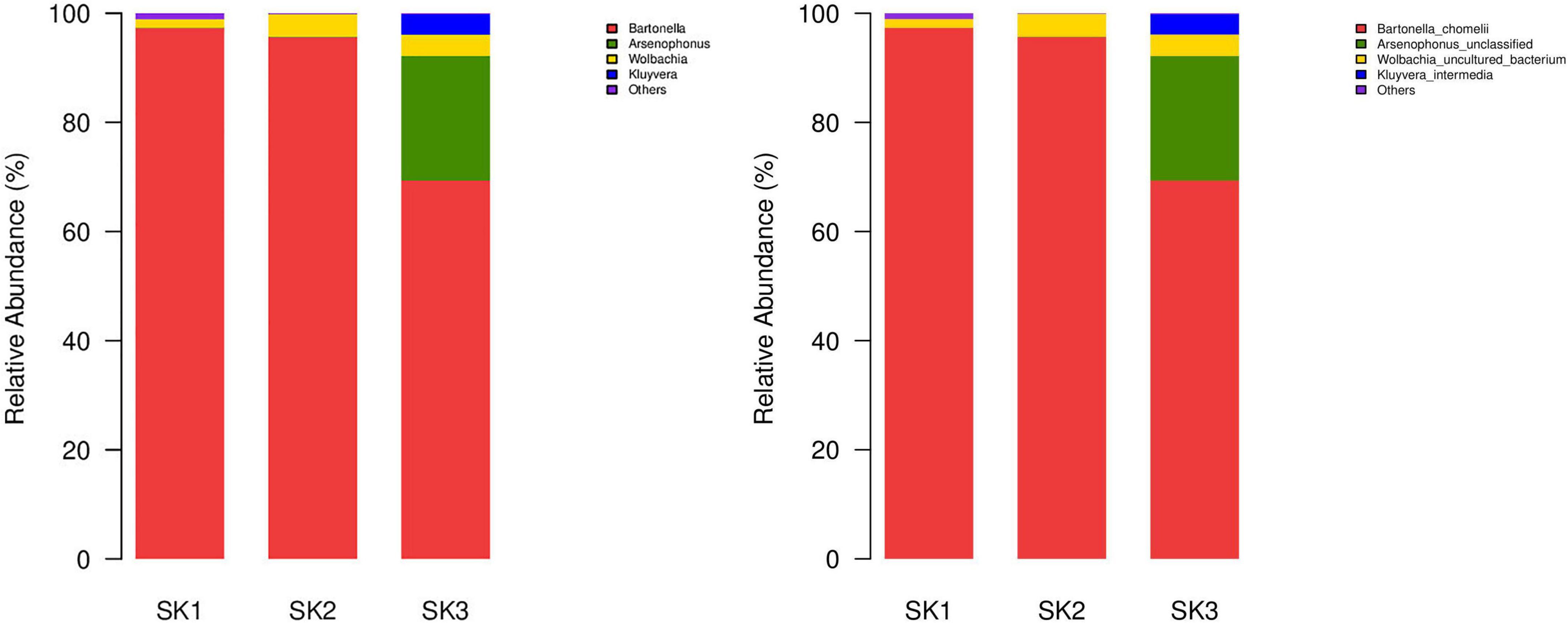
Figure 3. Microbial community bar plot of bacteria from three Melophagus ovinus samples (Left: Bacterial genus; Right: Bacterial species).
Microbial characteristics at the species level
At least seven bacterial species were identified in the three samples, of which the predominant bacteria Ba. chomelii and Wolbachia spp. were common to the three samples, and the relative abundances of Ba. chomelii and Wolbachia spp. in SK1, SK2, and SK3 were 97.31 and 1.60%, 95.64, and 4.18%, and 69.33 and 3.93%, respectively. Arsenophonus spp. was present in three samples, but only SK3 had a high relative abundance (SK1—0.04%, SK2—0.06%, SK3—22.84%) (Figure 3 Right). Kluyvera intermedia was only identified in SK3, and its relative abundance (3.72%) was also high. Corynebacterium maris DSM 45190 (0.09, 0.002%) were also identified in the SK1 and SK2 samples. Planomicrobium okeanokoites (0.02, 0.01%) were also identified in the SK2 and SK3 samples. Rhodococcus erythropolis (0.06, 0.12%) were also identified in the SK2 and SK3 samples, but their relative abundances were low.
Illumina hiseq sequencing results
A total of 162,989,660 reads were obtained from the three samples (SK1: 62474042, SK2: 42703260, and SK3: 57812358) on the Illumina Novaseq 6000 platform. After quality control, the raw data was 104,774,210 reads (37,614,416, 29,461,180, and 37,698,614, respectively) and GC% was 35.21, 33.45, and 33.17, respectively. After de novo assembly of the quality control data, the assembled genome sequences were obtained. After ORF prediction, 2,156,816 reads (5.7340%, 2,156,816/37,614,416), 1,377,322 reads (4.67504%, 1,377,322/29,461,180), and 2,156,990 reads (5.7217%, 2,156,990/37,698,614) were annotated as viruses, respectively.
Four identical virus orders were annotated in the three samples (except for Caudovirales, the virus gene prediction reads were mainly annotated as o__norank_d__Viruses, and a few reads were annotated as Herpesvirales and Ortervirales, Table 3). The 3 samples were, respectively, annotated into 29 families (corresponding to 4 aforementioned orders: 5 families, 17 families, 4 families, and 3 families), 30 families (corresponding to 4 aforementioned orders: 5 families, 18 families, 4 families, and 3 families), and 28 families (corresponding to 4 aforementioned orders: 5 families, 17 families, 3 families, and 3 families). In o__norank_d__Viruses, 3 samples were annotated into 16 common families (f__norank_d__Viruses, Mimiviridae, Phycodnaviridae, Poxviridae, Pithoviridae, Inoviridae, Baculoviridae, Polydnaviridae, Marseilleviridae, Iridoviridae, Nudiviridae, Adomaviridae, Geminiviridae, Fuselloviridae, Hytrosaviridae, Asfarviridae), Hepeviridae was found in SK1 and SK2, Ascoviridae was found in SK3, and Microviridae was found in SK2. Among o__norank_d__Viruses, the number of annotated reads of certain families in one of the three samples that accounted for >1% of annotated reads were in four identical families, namely f__norank_d__Viruses (22.10, 23.57, and 23.89%, respectively), Mimiviridae (13.47, 13.78, and 12.51%, respectively), Phycodnaviridae (9.17, 9.79, and 8.27%, respectively), and Poxviridae (1.55, 1.51, and 1.44%, respectively). Herpesviridae had the highest number of annotated reads in Herpesvirales in all three samples (1492, 860, and 414, respectively), and Retroviridae had the highest number of annotated reads in Ortervirales (5856, 6538, and 4462, respectively).
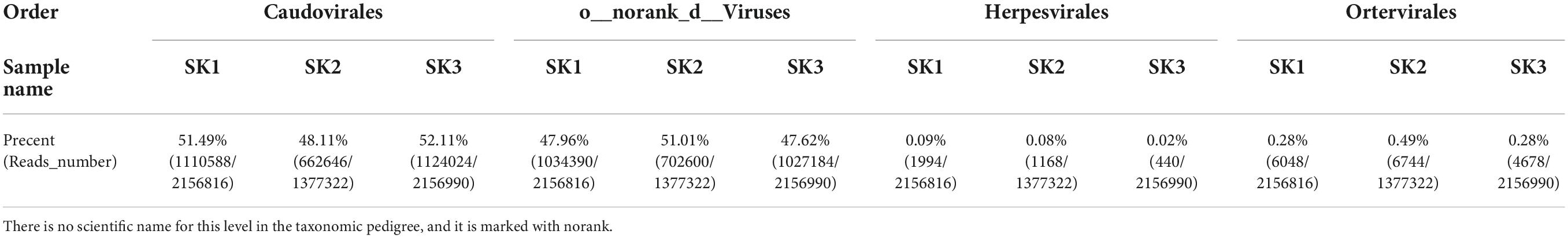
Table 3. Statistical table for the number of reads of three samples at the order level.
At the genus level, genera with species transcripts per million (TPM) abundance to the sample’s total abundance ≥1% (Table 4) include six genera in four families in Caudovirales and also included two genera from the f__norank_d__Viruses family (g__norank_d__Viruses and Pandoravirus), three genera from the Phycodnaviridae family (norank_f__Phycodnaviridae, Prasinovirus, Chlorovirus), and four genera from the Mimiviridae family (Tupanvirus, Klosneuvirus, Indivirus, norank_f__Mimiviridae) (Figure 4).
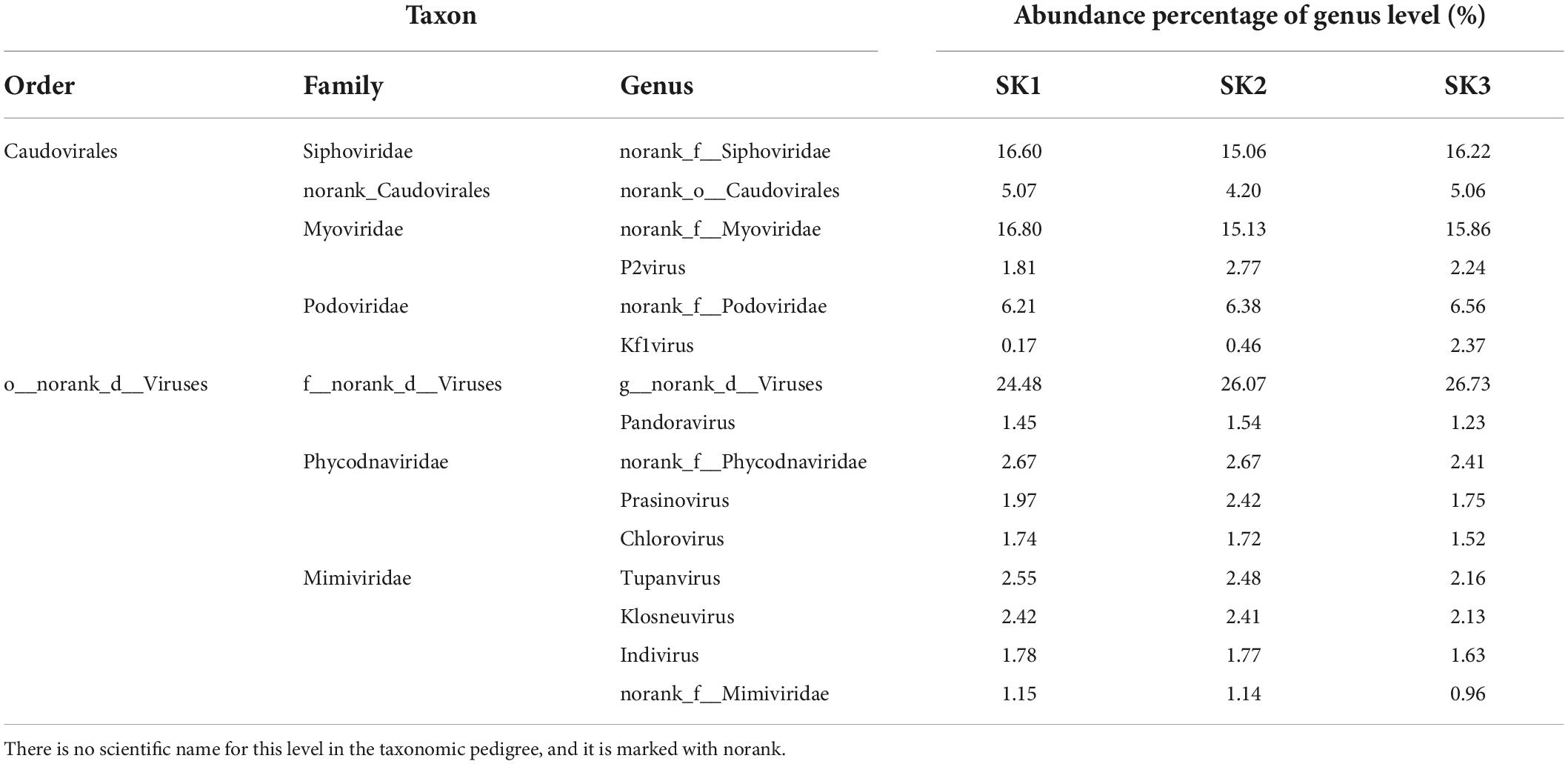
Table 4. Transcripts per million (TPM) abundance percentage of samples at the genus level.
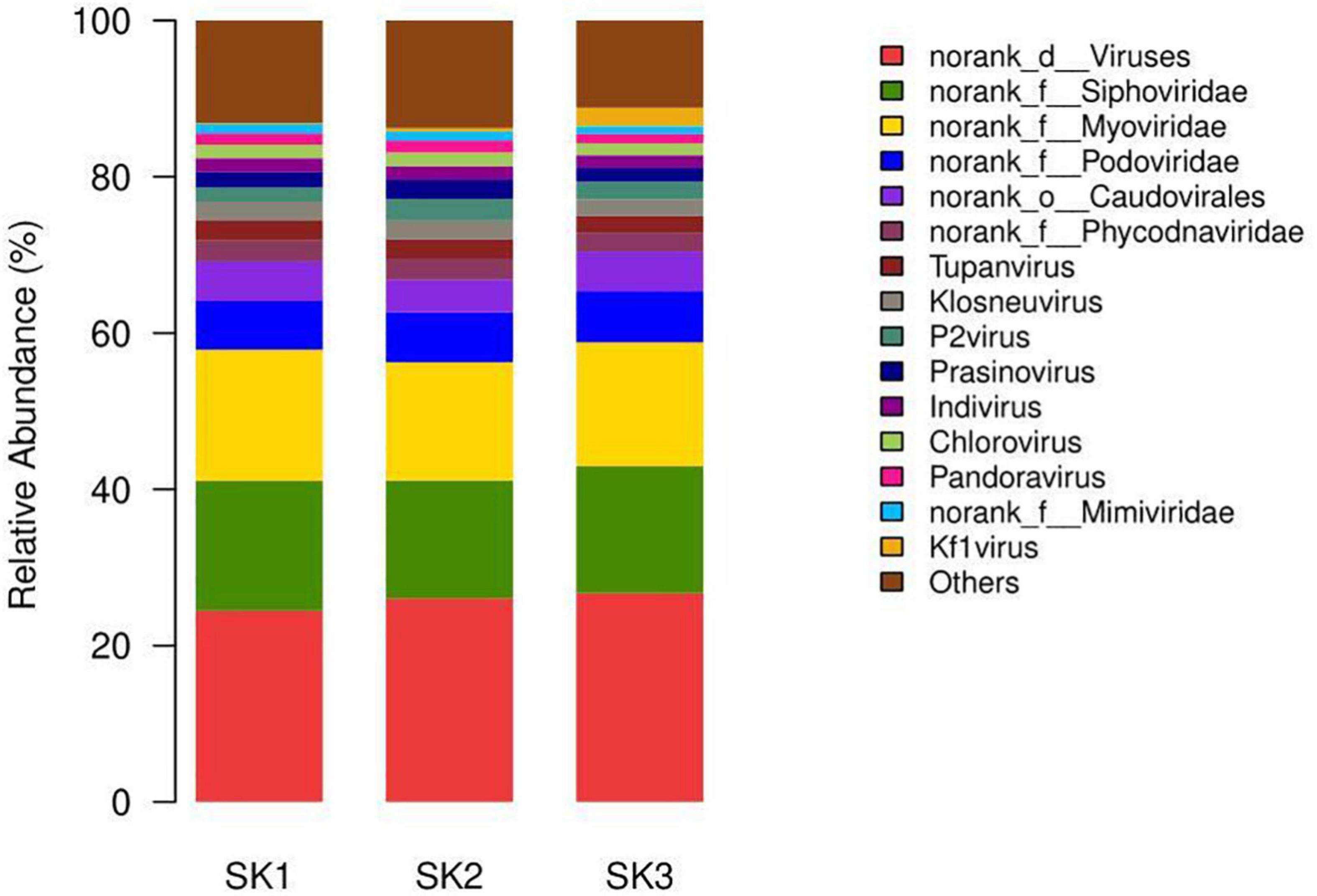
Figure 4. The microbial community bar plot of virus genus of the three Melophagus ovinus samples.
At the species level, those with TPM abundance ≥1% of the sample’s total abundance (Table 5) included 12 species, 5 genera, and 4 families in Caudovirales and 7 species in the g__norank_d__Viruses genus in the f__norank_d__Viruses family. In addition, it included three species and three genera from the Mimiviridae family: Tupanvirus (Tupanvirus_soda_lake), Klosneuvirus (Klosneuvirus_KNV1), and Indivirus (Indivirus_ILV1). Furthermore, more viruses were simultaneously detected in the three samples. Although the relative abundances were low, these pathogens could cause common and serious diseases to some livestock, such as the African swine fever virus and different Poxviruses in Poxviridae.

Table 5. Transcripts per million (TPM) abundance percentage of samples at the species level.
Discussion
From 1950 to 2018, 124 tick species, 103 tick-borne agents, and 29 tick-borne pathogens (Zhao et al., 2021) infecting humans have been reported in China. In the 33 years since 1982, 33 emerging tick-associated agents have been identified in mainland China, of which 20 have been confirmed to cause human disease (Fang et al., 2015). With regards to pathogen carriage and transmission, various species of mosquitoes and ixodid ticks have been actively studied (Litov et al., 2021). More than 213 species belonging to 21 genera (Liu et al., 2017; Werszko et al., 2021) in the family Hippoboscidae (Diptera) were reported globally, but less attention has been paid to these insects. Among them, M. ovinus has been the most studied species relatively. Undoubtedly, advances in molecular technologies and their applications have led to the discovery of novel agents. However, most studies on M. ovinus are limited to the detection of specific pathogens. High-throughput sequencing is expedient and efficient for the analysis of the microbial community structure of M. ovinus. However, high-throughput sequencing has only been conducted on microbial population and RNA virome in M. ovinus from the Gansu province in China (Duan et al., 2017, 2020) and the Republic of Tuva in Russia (Litov et al., 2021) so far. Several factors such as biogeography, the season of sample collection, and the number of samples may induce differences in pathogenic microorganisms carried by M. ovinus in different regions (Zhang et al., 2021). In this study, we employed high-throughput sequencing to analyze the microbial community structure and DNA viruses in M. ovinus from three regions in Tibet, China.
In 16S rRNA V3-V4 high-throughput sequencing, a greater Shannon index and a lower Simpson index indicate higher microbial community diversity of the sample. A higher Ace value indicates that the total number of species is higher (Duan et al., 2017, 2020). The sequencing results showed that the SK3 sample had the highest microflora diversity and the SK1 sample had the highest total number of species. The sequencing depth Good’s coverage of the three samples all exceeded 99.9%, indicating that the sequencing depth was sufficient to demonstrate microbial diversity of the samples and reflected the actual status of microorganism carriage in the samples. OTU cluster analysis revealed that some microbial populations were identical between the samples. On the basis of the aforementioned estimators, an analysis of the sequencing data revealed that Proteobacteria is a dominant bacterial phyla common in the three M. ovinus samples from Tibet, China, which is consistent with the M. ovinus results reported from Gansu province, China (Duan et al., 2017, 2020). Twenty-nine bacteria genera were annotated in M. ovinus from Tibet, China, which included six bacteria genera reported in the past M. ovinus studies (Bartonella, Wolbachia, Arsenophonus, Staphylococcus, Acinetobacter, and Salinicoccus). Among them, 23 bacteria genera (Amylobacter, Brachybacterium, Candidatus Pelagibacter, Cellulomonas, Corynebacterium 1, Glutamicibacter, JG30-KF-CM45 norank, Jeotgalicoccus, Kluyvera, Micrococcaceae unclassified, Microcoleus, Micromonosporaceae unclassified, Nocardiopsis, Ornithinimicrobium, Planktomarina, Planomicrobium, Propioniciclava, Rhodobacteraceae unclassified, Rhodococcus, Romboutsia, SAR116 clade norank, SAR86 clade norank, ZD0405 norank) were not previously reported in M. ovinus. Bartonella and Wolbachia were dominant bacterial genera common to the three samples. Arsenophonus was the dominant bacterial genus of SK3, where the dominant bacterial genera were also consistent with the results of M. ovinus reported in the Gansu province, China (Duan et al., 2017, 2020). The relative abundances of bacterial genera Corynebacterium 1, Jeotgalicoccus, Kluyvera, Nocardiopsis, and Staphylococcus in M. ovinus from Tibet, China were also high, particularly that of Kluyvera. At the species level, the reported Ba. chomelii, Wolbachia spp., and Arsenophonus spp. were identified. In addition, K. intermedia, Corynebacterium maris DSM 45190, Planomicrobium okeanokoites, and Rhodococcus erythropolis were discovered for the first time. As per the literature (Liu et al., 2016) and our previous study (unpublished), the positivity rate of Rickettsia in M. ovinus in Xinjiang province, China is also high. Strangely, Rickettsia was not detected in M. ovinus from Tibet in this study, implying that the Rickettsia carriage in M. ovinus could be based on the geographical region.
Bartonella mainly infects mammals and causes intraerythrocytic bacteremia. At least 30 Bartonella species have been reported to cause salonica fever, cat-scratch fever, and Carrion’s disease in humans (Duan et al., 2017, 2020). The main vectors are blood-sucking parasitic arthropods, such as ticks, chiggers and lice (Duan et al., 2017). In 2004, the presence of Bartonella in ruminant blood-sucking flies (Lipoptena cervi, Hippobosca equina, M. ovinus) was first reported. Bartonella can spread through hematophagous arthropods and is considered a symbiont in some insects, of which M. ovinus is a potential vector (Halos et al., 2004; Duan et al., 2020). Bartonella is common in M. ovinus, and several Bartonella species were previously identified in M. ovinus. However, Ba. melophagi is the most common (Halos et al., 2004; Kosoy et al., 2004, 2016; Reeves et al., 2006; Bemis and Kania, 2007; Kumsa et al., 2014; Rudolf et al., 2016; Liu et al., 2018; Boucheikhchoukh et al., 2019; Duan et al., 2020; Flores-Mendoza et al., 2021; Werszko et al., 2021). Ba. chomelii was first discovered in French domestic cows and is the 4th Bartonella species to be isolated from the ruminants. The species was named in honor of Bruno B. Chomel (Maillard et al., 2004). Ba. chomelii is the most common species of beef cattle infection in the Basque Country in Spain (Antequera-Gómez et al., 2015). Ba. chomelii was first identified in M. ovinus from the Gansu province, China, and is a dominant bacterial species. It has also been reported that the abundance of Ba. chomelii in fully engorged adult female samples (46.5 and 16.4%) was significantly higher than that in newly hatched and unfed adult female samples (1.19 and 1.17%, respectively), where the proliferation of Ba. chomelii was enhanced eventually with blood-feeding (Duan et al., 2020). This was consistent with the test results of engorged M. ovinus in Tibet, China, in this study, and the relative abundances of the three samples were 97.31, 95.64, and 69.33%, respectively. Considering the aforementioned reasons, it is necessary to further examine the specific role of Ba. chomelii, with high abundance, in M. ovinus, and the risk of transmission of M. ovinus-derived and ruminant-derived Bartonella to humans should not be underestimated.
Wolbachia is an obligate intracellular bacterium that can infect many hosts, such as ticks, sand flies, tsetse flies, mosquitos, fleas and mites, and cause reproductive manipulation and parthenogenesis through cytoplasmic incompatibility, thereby regulating reproductive behavior (Duan et al., 2017, 2020). Previously, it was reported that M. ovinus carries Wolbachia (Duan et al., 2017, 2020; Liu et al., 2018; Boucheikhchoukh et al., 2019), and Wolbachia is also a common dominant bacterial genus among the three samples. However, the role of Wolbachia remains to be determined. Arsenophonus is an intracellular symbiotic bacterium with a broad host range and infects several arthropods (Duan et al., 2017). Arsenophonus is also present in M. ovinus (Nováková et al., 2015; Duan et al., 2017, 2020; Husnik et al., 2020), which plays an important role in killing male hosts and provides vitamins and other nutrients to host insects (Duan et al., 2017). Further studies are required to determine the role of Arsenophonus in M. ovinus and to understand whether it is related to nutrient supply. A more challenging question is whether Wolbachia and Arsenophonus should be considered endosymbionts or sheep parasites of M. ovinus. The answer to this question is not straightforward and requires confirmation through a large number of experiments by researchers.
Kluyvera intermedia was previously reported to be isolated from patients (Thele et al., 2017; Paskova et al., 2018), and K. intermedia originating from spiders has been reported to be pathogenic to humans (Dunbar et al., 2020). The genus Kluyvera has the potential to act as a rare infectious agent and causes soft tissue infections, urinary tract infections, intra-abdominal abscesses, catheter-associated bloodstream infections and septic shock (Thele et al., 2017). This study was the first to identify K. intermedia in M. ovinus, with a relatively high abundance. The risk of K. intermedia transmission by M. ovinus to sheep or humans is worthy of close attention by researchers.
The results of viral metagenomic sequencing revealed that all three samples were annotated to four identical virus orders (Caudovirales, o__norank_d__Viruses, Herpesvirales, and Ortervirales). At the family level, after excluding phage-related Caudovirales, which was not the focus of this study, viruses associated with vertebrates and insects that were annotated in o__norank_d__Viruses included Mimiviridae, Marseilleviridae, Poxviridae, Ascoviridae, Iridoviridae, Baculoviridae, Hytrosaviridae, Nudiviridae, Polydnaviridae, Adomaviridae, Asfarviridae, Hepeviridae, and some plant viruses and algal viruses were annotated. The three samples were mainly annotated to Herpesviridae and Retroviridae in Herpesvirales and Ortervirales, respectively. The abovementioned viruses were not reported in M. ovinus in Diptera. Mimiviridae is one of the largest and most diverse families of eukaryotic viruses isolated from aquatic environments. This family of viruses can infect a broad range of eukaryotic unicellular organisms, ranging across six major phyla: Amoebozoa, Chlorophyta, Haptophyta, Heterokonta, Excavata, and perhaps Opisthokonta (Claverie and Abergel, 2018). In this family, we identified three genera with high abundance, namely Tupanvirus, Klosneuvirus, and Indivirus. The family Marseilleviridae is a taxon for giant viruses that infect amoeba, where this family along with the family Mimiviridae, as well as the families Poxviridae, Iridoviridae, Ascoviridae, Asfarviridae all belong to a viral family among nucleo-cytoplasmic large DNA viruses (Colson et al., 2013). This list includes the genus Pandoraviruses with a high abundance, which was annotated to the f__norank_d__Viruses family—giant viruses that infect amoeba (Takemura, 2020). It is still unknown whether these annotated giant viruses are related to protozoal infection in M. ovinus. Currently, large dsDNA viruses that are known to circulate in insects include Poxviridae, Ascoviridae, Iridoviridae, Baculoviridae, Hytrosaviridae, Nudiviridae, where Iridoviruses, Poxviruses, and Nudiviruses are ubiquitous in several insect orders; Baculoviruses is reported to be present in Lepidoptera, Hemiptera, and Diptera; and Ascoviruses infects lepidopteran larvae (Gilbert and Belliardo, 2022). The Hytrosaviruses (family: Hytrosaviridae) are viruses identified in different dipteran species. The family includes two genera (Muscavirus and Glossinavirus). Hytrosaviruses can cause salivary gland hypertrophy and testicular and ovarian malformation leading to sterility in adult dipterans. Currently identified Hytrosaviruses only include three virus species from the three genera of Diptera: the tsetse fly Glossina pallidipes, the housefly Musca domestica, and the narcissus bulb fly Merodon equestris (Abd-Alla et al., 2009; Lacey, 2012; Omkar, 2016; Kariithi et al., 2017). Two Hytrosaviridae genera, Muscavirus and Glossinavirus were detected in our three samples as well as other insect-related virus families Poxviridae, Iridoviridae, Baculoviridae, and Nudiviridae. Insects-associated viruses Ascoviridae were also detected in the SK3 samples. The virus families Polydnaviridae, Asfarviridae, Herpesviridae, and Retroviridae were annotated in three samples, Hepeviridae was annotated in two samples, and several viruses were annotated in Poxviridae. Although the abundances of some annotated viruses are low, their presence is not a complete coincidence. This is the first discovery of these viruses in M. ovinus from the order Diptera. Particularly, Hytrosaviridae could be a potential biocontrol agent, warranting further in-depth research. In addition, adomaviruses, adenoviruses, papillomaviruses, parvoviruses, and polyomaviruses are collectively known as small DNA tumor viruses. Small DNA tumor virus oncoproteins and capsid proteins demonstrated structural and functional similarity (Welch et al., 2019). Further evaluation is warranted to examine whether these viruses can proliferate in M. ovinus, play a role in pathogen dissemination in M. ovinus and are involved in the risk of causing human diseases. Finally, we would like to mention one of the many viruses that were annotated in three samples—the African swine fever virus (ASFV). In 2018, an outbreak of African swine fever (ASF) occurred in China, which was the site of the first outbreak of Asian ASF (Abd-Alla et al., 2009). Countries that experienced ASF outbreaks had to undergo catastrophic pig culling. More importantly, soft ticks are biological transmission vectors of ASFV (Cheng and Ward, 2022). It has been previously reported that M. ovinus is a mechanical vector for the blue-tongue virus (Luedke et al., 1965) and that M. ovinus may or may not be a biological or mechanical vector for ASFV.
Several microorganisms colonize in insect vectors, including endosymbionts, potentially pathogenic pathogens, and non-pathogenic pathogens. Microorganisms in insect vectors play important roles in pathogen survival, transmission, modulating vector competence, reproductive fitness, and nutrient supply (Duan et al., 2020). Evaluating microorganisms in insect vectors could help in the further study on how microorganisms interact with vectors of animal parasite and on ways to develop biological control tools. M. ovinus is a blood-sucking insect that was directly collected from sheep in our study. This finding implies that microorganisms found in this study may infect M. ovinus or sheep or circulate between M. ovinus and sheep (as an insect-borne pathogen). This study found that several pathogens are present in other insects, other species in Diptera, or M. ovinus, implying that some of the pathogens found in this study may be specific to arthropods. However, the possibility of these pathogens being sheep pathogens or insect-borne pathogens cannot be ignored. Although we noted the diversity of microorganisms carried by M. ovinus, the report may be the tip of the iceberg with regards to the actual number of pathogens carried and transmitted by M. ovinus. In addition, similar to the long period needed to confirm human infection after discovering potential pathogenic factors in ticks, tick-borne pathogens that infect livestock may ultimately cause human disease (Fang et al., 2015). Therefore, we require a large volume of studies to demonstrate the pathogens carried by M. ovinus and the pathogenicity of these pathogens. In summary, we mined microorganisms in M. ovinus and expanded the spectrum of pathogens that could be present in M. ovinus. Our study has veterinary and medical significance and can facilitate prediction of the emerging pathogens, evaluation of potential risks to animal and public health, development of biological control tools, and control management of insect-borne diseases.
Conclusion
Proteobacteria is the dominant bacterial phylum in M. ovinus from Tibet, China, and 29 bacteria genera were annotated. The dominant bacterial genera included Bartonella, Wolbachia, and Arsenophonus and this is the first report of 23 bacteria genera in M. ovinus. In addition, this is also the first report of Kluyvera intermedia, Corynebacterium maris DSM 45190, Planomicrobium okeanokoites, and Rhodococcus erythropolis. All DNA viruses reported in this study are the first to be reported in M. ovinus.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: The raw tags have been deposited in Sequence Read Archive (SRA) from the NCBI under BioProject accession numbers: PRJNA849748 and PRJNA852367. The individual run files received the accession numbers SRR19668428, SRR19668429, SRR19668430, SRR19834166, SRR19834167, and SRR19834168.
Ethics statement
The animal study was reviewed and approved by the biomedical research ethics committee of Inner Mongolia Agricultural University specifically approved this study [No. 2020 (080)]. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
Y-HL and LZ conceived and designed the study and critically revised the manuscript. Y-LD, LZ, BY, and W-XH performed the sheep ked collection. Y-HL, Y-MM, H-OT, W-HZ, H-LC, Z-SZ, L-FW, and Y-LD conducted the laboratory experiments. Y-HL, LZ, Y-MM, YX, and LC conducted sequencing and participated in sequence analysis. All authors read and approved the final manuscript.
Funding
This study was funded by the National Natural Science Foundation of China (31860698), Inner Mongolia Agricultural University High-level Talents Research Initiation Fund Project (NDYB2019-3 and NDYB2018-5), National Natural Science Foundation of Inner Mongolia (2022MS03023), and Supported by State Key Laboratory of Veterinary Biotechnology Foundation (SKLVBF202204).
Conflict of interest
W-XH was employed by Inner Mongolia Saikexing Reproductive Biotechnology (Group) Co., Ltd., Hohhot, China. YX and LC were employed by Shanghai Origingene Bio-pharm Technology Co., Ltd., Shanghai, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ http://drive5.com/uparse/
- ^ http://sourceforge.net/projects/rdp-classifier/
- ^ http://www.mothur.org/wiki/Chao
- ^ http://www.mothur.org/wiki/Shannon
- ^ http://www.mothur.org/wiki/Simpson
- ^ http://www.mothur.org/wiki/Coverage
- ^ http://qiime.org/scripts/assign_taxonomy.html
- ^ https://cutadapt.readthedocs.io/en/stable/
- ^ http://bio-bwa.sourceforge.net/
- ^ http://i.cs.hku.hk/~alse/hkubrg/projects/idba_ud/
- ^ https://github.com/gigascience/bgi-soap1
- ^ http://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ http://ab.inf.uni-tuebingen.de/software/megan/
References
Abd-Alla, A. M., Vlak, J. M., Bergoin, M., Maruniak, J. E., Parker, A., Burand, J. P., et al. (2009). Hytrosaviridae: A proposal for classifification and nomenclature of a new insect virus family. Arch. Virol. 154, 909–918. doi: 10.1007/s00705-009-0398-5
Antequera-Gómez, M. L., Lozano-Almendral, L., Barandika, J. F., González-Martín-Niño, R. M., Rodríguez-Moreno, I., García-Pérez, A. L., et al. (2015). Bartonella chomelii is the most frequent species infecting cattle grazing in communal mountain pastures in Spain. Appl. Environ. Microb. 81, 623–629. doi: 10.1128/AEM.03159-14
Arkwright, J. A., Bacot, A., and Duncan, F. M. (1919). The association of Rickettsia with trench fever. J. Hygiene 18, 76–94.3. doi: 10.1017/S0022172400007397
Bemis, D. A., and Kania, S. A. (2007). Isolation of Bartonella sp. from sheep blood. Emerg. Infect. Dis. 13, 1565–1567. doi: 10.3201/eid1310.070570
Bequaert, J. (1942). A monograph of the Melophaginae, or ked-flies, of sheep, goats, deer and antelopes (Diptera, Hippoboscidae). Entomol. Am. 22, 1–220.
Bezerra-Santos, M. A., and Otranto, D. (2020). Keds, the enigmatic flies and their role as vectors of pathogens. Acta Trop. 209:105521. doi: 10.1016/j.actatropica.2020.105521
Boucheikhchoukh, M., Mechouk, N., Benakhla, A., Raoult, D., and Parola, P. (2019). Molecular evidence of bacteria in Melophagus ovinus sheep keds and Hippobosca equina forest flies collected from sheep and horses in northeastern Algeria. Comp. Immunol. Microb. 65, 103–109. doi: 10.1016/j.cimid.2019.05.010
Boyd, B. M., Allen, J. M., Koga, R., Fukatsu, T., Sweet, A. D., Johnson, K. P., et al. (2016). Two bacterial genera, Sodalis and Rickettsia, associated with the seal louse Proechinophthirus fluctus (Phthiraptera: Anoplura). Appl. Environ. Microb. 82, 3185–3197. doi: 10.1128/AEM.00282-16
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cheng, J. M., and Ward, M. P. (2022). Risk factors for the spread of African swine fever in China: A systematic review of Chinese-language literature. Transbound. Emerg. Dis. ahead of print]. doi: 10.1111/tbed.14573[Epub
Chrudimský, T., Husnik, F., Novakova, E., and Hypša, V. (2012). Candidatus Sodalis melophagi sp. nov.: Phylogenetically independent comparative model to the tsetse fly symbiont Sodalis glossinidius. PLoS One. 7:e40354. doi: 10.1371/journal.pone.0040354
Chu, C. Y., Jiang, B. G., Qiu, E. C., Zhang, F., Zuo, S. Q., Yang, H., et al. (2011). Borrelia burgdorferi sensu lato in sheep keds (Melophagus ovinus), Tibet, China. Vet. Microbiol. 149, 526–529. doi: 10.1016/j.vetmic.2010.11.031
Claverie, J. M., and Abergel, C. (2018). Abergel C. Mimiviridae: A rising family of highly diverse large aquatic dsDNA viruses infecting a wide variety of eukaryotes. Viruses 10:506. doi: 10.3390/v10090506
Colson, P., Pagnier, I., Yoosuf, N., Fournous, G., La Scola, B. L., and Raoult, D. (2013). ‘Marseilleviridae’, a new family of giant viruses infecting amoebas. Arch. Virol. 158, 915–920. doi: 10.1007/s00705-012-1537-y
Cowdry, E. V. (1923). The distribution of Rickettsia in the tissues of insects and arachnids. J. Exp. Med. 37, 431–456. doi: 10.1084/jem.37.4.431
Duan, D. Y., Liu, G. H., Cheng, T. Y., and Wang, Y. Q. (2017). Microbial population analysis of the midgut of Melophagus ovinus via high-throughput sequencing. Parasit. Vector. 10:382. doi: 10.1186/s13071-017-2323-1
Duan, D. Y., Zhou, H. M., and Cheng, T. Y. (2020). Comparative analysis of microbial community in the whole body and midgut from fully engorged and unfed female adult Melophagus ovinus. Med. Vet. Entomol. 34, 215–224. doi: 10.1111/mve.12424
Dunbar, J. P., Khan, N. A., Abberton, C. L., Brosnan, P., Murphy, J., Afoullouss, S., et al. (2020). Synanthropic spiders, including the global invasive noble false widow Steatoda nobilis, are reservoirs for medically important and antibiotic resistant bacteria. Sci. Rep. 10:20916. doi: 10.1038/s41598-020-77839-9
Edgar, R. C. (2013). Uparse: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Fang, L. Q., Liu, K., Li, X. L., Liang, S., Yang, Y., Yao, H. W., et al. (2015). Emerging tick-borne infections in mainland China: An increasing public health threat. Lancet Infect. Dis. 15, 1467–1479. doi: 10.1016/S1473-3099(15)00177-2
Flores-Mendoza, C., Loyola, S., Jiang, J., Farris, C. M., Mullins, K., Estep, A. S., et al. (2021). Molecular characterization of Bartonella species discovered in ectoparasites collected from domestic animals, Cuzco, Peru. Vector Borne Zoonot. 21, 330–341. doi: 10.1089/vbz.2020.2697
Fouts, D. E., Szpakowski, S., Purushe, J., Torralba, M., Waterman, R. C., MacNeil, M. D., et al. (2012). Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS One 7:e48289. doi: 10.1371/journal.pone.0048289
Gibson, W., Pilkington, J. G., and Pemberton, J. M. (2010). Trypanosoma melophagium from the sheep ked Melophagus ovinus on the island of St Kilda. Parasitology 137, 1799–1804. doi: 10.1017/S0031182010000752
Gilbert, C., and Belliardo, C. (2022). The diversity of endogenous viral elements in insects. Curr. Opin. Insect Sci. 49, 48–55. doi: 10.1016/j.cois.2021.11.007
Hadgu, A., Lemma, A., Yilma, T., and Fesseha, H. (2021). Major causes of calf and lamb mortality and morbidity and associated risk factors in the mixed crop-livestock production system in Jamma District, South Wollo, Ethiopia. Vet. Med. Int. 2021:6689154. doi: 10.1155/2021/6689154
Halos, L., Jamal, T., Maillard, R., Girard, B., Guillot, J., Chomel, B., et al. (2004). Role of Hippoboscidae flies as potential vectors of Bartonella spp. infecting wild and domestic ruminants. Appl. Environ. Microbiol. 70, 6302–6305. doi: 10.1128/AEM.70.10.6302-6305.2004
Hao, L. L., Yuan, D. B., Li, S. H., Jia, T., Guo, L., Hou, W., et al. (2020). Detection of Theileria spp. in ticks, sheep keds (Melophagus ovinus), and livestock in the eastern Tibetan Plateau. China. Parasitol. Res. 119, 2641–2648. doi: 10.1007/s00436-020-06757-6
Hertig, M., and Wolbach, S. B. (1924). Studies on rickettsia-like micro-organisms in insects. J. Med. Res. 44, 329–374.7. doi: 10.1002/path.1700280417
Hoare, C. A. (1923). An experimental study of the sheep-trypanosome (T. melophagium Flu, 1908), and its transmission by the sheep-ked (Melophagus ovinus L.). Parasitology 15, 365–424. doi: 10.1017/S0031182000014888
Hoare, C. A. (1972). The Trypanosomes of Mammals. A Zoological Monograph. Hoboken, NJ: Blackwell Scientific Publications.
Hornok, S., de la Fuente, J., Biró, N., Fernández de Mera, I. G., Meli, M. L., Elek, V., et al. (2011). First molecular evidence of Anaplasma ovis and Rickettsia spp. in keds (Diptera: Hippoboscidae) of sheep and wild ruminants. Vector Borne Zoonot. 11, 1319–1321. doi: 10.1089/vbz.2011.0649
Hubálek, Z., Černý, V., Mittermayer, T., Kilík, J., Halouzka, J., Juřicová, Z., et al. (1986). Arbovirological survey in Silica plateau area, Roznava district, Czechoslovakia. J. Hyg. Epidem. Microbiol. Immunol. 30, 87–98.
Husnik, F., Hypsa, V., and Darby, A. (2020). Insect-symbiont gene expression in the midgut bacteriocytes of a blood-sucking parasite. Genome Biol. Evol. 12, 429–442. doi: 10.1093/gbe/evaa032
Huson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Izdebska, J. N. (2001). European bison arthropod parasites from closed Polish breeding facilities. Acta Parasitol. 46, 135–137. doi: 10.1016/S0304-4017(00)00435-0
Kariithi, H. M., Meki, I. K., Boucias, D. G., and Abd-Alla, A. M. (2017). Hytrosaviruses: Current status and perspective. Curr. Opin. Insect Sci. 22, 71–78. doi: 10.1016/j.cois.2017.05.009
Kligler, I. J., and Aschner, M. (1931). Cultivation of rickettsia-like microorganisms from certain blood-sucking Pupipara. J. Bacteriol. 22, 103–117. doi: 10.1128/jb.22.2.103-117.1931
Kosoy, M., Bai, Y., Enscore, R., Rizzo, M. R., Bender, S., Popov, V., et al. (2016). Bartonella melophagi in blood of domestic sheep (Ovis aries) and sheep keds (Melophagus ovinus) from the southwestern US: Cultures, genetic characterization, and ecological connections. Vet. Microbiol. 190, 43–49. doi: 10.1016/j.vetmic.2016.05.009
Kosoy, M., Sheff, K., Bai, Y., Enscore, R., Dodge, J., Irkhin, A., et al. (2004). “Isolation and identification of a new Bartonella species from a sheep ked,” in The 4th International Conference on Bartonella as Emerging Pathogens, (Uppsala: Uppsala University), 28.
Kumsa, B., Parola, P., Raoult, D., and Socolovschi, C. (2014). Bartonella melophagi in Melophagus ovinus (sheep ked) collected from sheep in northern Oromia, Ethiopia. Comp. Immunol. Microb. 37, 69–76. doi: 10.1016/j.cimid.2013.11.001
Kumsa, B., Socolovschi, C., Parola, P., Rolain, J. M., and Raoult, D. (2012). Molecular detection of Acinetobacter species in lice and keds of domestic animals in Oromia regional state, Ethiopia. PLoS One. 7:e52377. doi: 10.1371/journal.pone.0052377
Lacey, L. A. (2012). Manual Of Techniques In Invertebrate Pathology, 2nd Edn. Cambridge, MA: Academic Press, doi: 10.1016/C2010-0-66784-8
Lassnig, H., Prosl, H., and Hinterdorfer, F. (1998). Parasites of the red fox (Vulpes vulpes) in Styria. Wien Tierarztl Monat. 85, 116–122. doi: 10.2307/3440796
Litov, A. G., Belova, O. A., Kholodilov, I. S., Gadzhikurbanov, M. N., Gmyl, L. V., Oorzhak, N. D., et al. (2021). Possible arbovirus found in virome of melophagus ovinus. Viruses 13:2375. doi: 10.3390/v13122375
Liu, D., Wang, Y. Z., Zhang, H., Liu, Z. Q., Wureli, H. Z., Wang, S. W., et al. (2016). First report of Rickettsia raoultii and R. slovaca in Melophagus ovinus, the sheep ked. Parasite Vector 9:600. doi: 10.1186/s13071-016-1885-7
Liu, Y. H., He, B., Li, F., Li, K. R., Zhang, L. Y., Li, X. Q., et al. (2018). Molecular identification of Bartonella melophagi and Wolbachia supergroup F from sheep keds in Xinjiang, China. Korean J. Parasitol. 56, 365–370. doi: 10.3347/kjp.2018.56.4.365
Liu, Y. H., He, B., Li, K. R., Li, F., Zhang, L. Y., Li, X. Q., et al. (2019). First report of border disease virus in Melophagus ovinus (sheep ked) collected in Xinjiang, China. PLoS One 14:e0221435. doi: 10.1371/journal.pone.0221435
Liu, Z. Q., Kuermanali, N., Li, Z., Chen, S. J., Wang, Y. Z., Tao, H., et al. (2017). The complete mitochondrial genome of the parasitic sheep ked Melophagus ovinus (Diptera: Hippoboscidae). Mitochondrial DNA. B Resour. 2, 432–434. doi: 10.1080/23802359.2017.1347832
Luedke, A. J., Jochim, M. M., and Bowne, J. G. (1965). Preliminary bluetongue transmission with the sheep ked Melophagus ovinus (L.). Can. J..Comp. Med. and Vet. Sci. 29, 229–231.
Maillard, R., Riegel, P., Barrat, F., Bouillin, C., Thibault, D., Gandoin, C., et al. (2004). Bartonella chomelii sp. nov., isolated from French domestic cattle (Bos taurus). Int. J. Syst. Evol. Micr. 54, 215–220. doi: 10.1099/ijs.0.02770-0
Martinković, F., Matanović, K., Rodrigues, A. C., Garcia, H. A., and Teixeira, M. M. G. (2012). Trypanosoma (Megatrypanum) melophagium in the sheep ked Melophagus ovinus from organic farms in Croatia: Phylogenetic inferences support restriction to sheep and sheep keds and close relationship with trypanosomes from other ruminant species. J. Eukaryot. Microbiol. 59, 134–144. doi: 10.1111/j.1550-7408.2011.00599.x
Nelder, M. P., Lloyd, J. E., Loftis, A. D., and Reeves, W. K. (2008). Coxiella burnetii in wild-caught filth flies. Emerg. Infect. Dis. 14, 1002–1004. doi: 10.3201/eid1406.071691
Nováková, E., Husník, F., Šochová, E., and Hypša, V. (2015). Arsenophonus and Sodalis symbionts in louse flies: An analogy to the Wigglesworthia and Sodalis system in tsetse flies. Appl. Environ. Microbiol. 81, 6189–6199. doi: 10.1128/AEM.01487-15
Omkar, O. (2016). Ecofriendly Pest Management For Food Security. Cambridge, MA: Academic Press, doi: 10.1016/C2014-0-04228-1
Paskova, V., Medvecky, M., Skalova, A., Chudejova, K., Bitar, I., Jakubu, V., et al. (2018). Characterization of NDM-encoding plasmids from Enterobacteriaceae recovered from Czech hospitals. Front. Microbiol. 9:1549. doi: 10.3389/fmicb.2018.01549
Pavilanis, V., Duval, L., Foley, A. R., and L’heureux, M. (1958). An epidemic of Q fever at Princeville, Quebec. Can. J. Public Health 49, 520–529. doi: 10.2307/41981334
Payzin, S. (1953). Epidemiological investigations on Q fever in Turkey. Bull.World Health Organ. 9, 553–558.
Reeves, W. K., Nelder, M. P., Cobb, K. D., and Dasch, G. A. (2006). Bartonella spp. in deer keds, Lipoptena mazamae (Diptera: Hippoboscidae), from Georgia and South Carolina, USA. J. Wildl. Dis. 42, 391–396. doi: 10.7589/0090-3558-42.2.391
Rudolf, I., Betášová, L., Bischof, V., Venclíková, K., Blažejová, H., Mendel, J., et al. (2016). Molecular survey of arthropod-borne pathogens in sheep keds (Melophagus ovinus), central Europe. Parasitol. Res. 115, 3679–3682. doi: 10.1007/s00436-016-5175-2
Setién, ÁA., Baltazar, A. G., Leyva, I. O., Rojas, M. S., Koldenkova, V. P., García, M. P. P., et al. (2017). Ectoparasitic hematophagous dipters: Potential reservoirs of dengue virus? Gac. Med. Mex 153:S82–S90. doi: 10.24875/GMM.M17000009
Seyoum, Z., Tadesse, T., and Addisu, A. (2015). Ectoparasites prevalence in small ruminants in and around Sekela, Amhara regional state, Northwest Ethiopia. J. Vet. Med. 2015:216085. doi: 10.1155/2015/216085
Small, R. W. (2005). A review of Melophagus ovinus (L.), the sheep ked. Vet. Parasitol. 130, 141–155. doi: 10.1016/j.vetpar.2005.03.005
Takemura, M. (2020). Medusavirus ancestor in a proto-eukaryotic cell: Updating the hypothesis for the viral origin of the nucleus. Front. Microbiol. 11:571831. doi: 10.3389/fmicb.2020.571831
Tang, J. M., Li, F., Cheng, T. Y., Duan, D. Y., and Liu, G. H. (2018). Comparative analyses of the mitochondrial genome of the sheep ked Melophagus ovinus (Diptera: Hippoboscidae) from different geographical origins in China. Parasitol. Res. 117, 2677–2683. doi: 10.1007/s00436-018-5925-4
Tetley, J. H. (1958). The sheep ked, Melophagus ovinus L. I Dissemination potential. Parasitology 48, 353–363. doi: 10.1017/s0031182000021302
Thele, R., Gumpert, H., Christensen, L. B., Worning, P., Schønning, K., Westh, H., et al. (2017). Draft genome sequence of a Kluyvera intermedia isolate from a patient with a pancreatic abscess. J. Glob. Antimicrob. Resist. 10, 1–2. doi: 10.1016/j.jgar.2017.05.007
Ulicsni, V., Svanberg, I., and Molnár, Z. (2016). Folk knowledge of invertebrates in Central Europe – Folk taxonomy, nomenclature, medicinal and other uses, folklore, and nature conservation. J. Ethnobiol. Ethnomed. 12:47. doi: 10.1186/s13002-016-0118-7
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Welch, N. L., Tisza, M. J., Belford, A., Pastrana, D. V., Pang, Y. S., Schiller, J. T., et al. (2019). Identification of “missing link” families of Small DNA tumor viruses. bioRxiv 11:697771. doi: 10.1101/697771
Werszko, J., Asman, M., Witecka, J., Steiner-Bogdaszewska, Ż, Szewczyk, T., Kuryło, G., et al. (2021). The role of sheep ked (Melophagus ovinus) as potential vector of protozoa and bacterial pathogens. Sci. Rep. 11:15468. doi: 10.1038/s41598-021-94895-x
Zaugg, J. L., and Coan, M. E. (1986). Test of the sheep ked Melophagus ovinus (L.) as a vector of Anaplasma ovis Lestoquard. Am. J. Vet. Res. 47, 1060–1062. doi: 10.2307/20094798
Zhang, Q. X., Wang, Y., Li, Y., Han, S. Y., Wang, B., Yuan, G. H., et al. (2021). Vector-borne pathogens with veterinary and public health significance in melophagus ovinus (sheep ked) from the Qinghai-Tibet Plateau. Pathogens 10:249. doi: 10.3390/pathogens10020249
Zhao, G. P., Wang, Y. X., Fan, Z. W., Ji, Y., Liu, M. J., Zhang, W. H., et al. (2021). Mapping ticks and tick-borne pathogens in china. Nat. Commun. 12:1075. doi: 10.1038/s41467-021-21375-1
Zhao, L., He, B., Li, K. R., Li, F., Zhang, L. Y., Li, X. Q., et al. (2018). First report of Anaplasma ovis in pupal and adult Melophagus ovinus (sheep ked) collected in South Xinjiang, China. Parasite. Vector. 11:258. doi: 10.1186/s13071-018-2788-6
Zhao, L., Wang, J. L., Ding, Y. L., Li, K. R., He, B., Li, F., et al. (2020). Theileria ovis (Piroplasmida: Theileriidae) detected in Melophagus ovinus (Diptera: Hippoboscoidea) and Ornithodoros lahorensis (Ixodida: Argasidae) removed from sheep in Xinjiang, China. J. Med. Entomol. 57, 631–635. doi: 10.1093/jme/tjz193
Keywords: Melophagus ovinus, sheep ked, Tibet, microbial population, viral metagenomics
Citation: Liu Y-H, Ma Y-M, Tian H-O, Yang B, Han W-X, Zhao W-H, Chai H-L, Zhang Z-S, Wang L-F, Chen L, Xing Y, Ding Y-L and Zhao L (2022) First determination of DNA virus and some additional bacteria from Melophagus ovinus (sheep ked) in Tibet, China. Front. Microbiol. 13:988136. doi: 10.3389/fmicb.2022.988136
Received: 07 July 2022; Accepted: 16 August 2022;
Published: 06 September 2022.
Edited by:
Peirong Jiao, South China Agricultural University, ChinaReviewed by:
Bhanuprakash V, Indian Veterinary Research Institute, IndiaBeyhan Sareyyüpoğlu, Ministry of Agriculture and Forestry (Turkey), Turkey
Copyright © 2022 Liu, Ma, Tian, Yang, Han, Zhao, Chai, Zhang, Wang, Chen, Xing, Ding and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhao, emhhb2xpZGt5QDEyNi5jb20=
†ORCID: Yong-Hong Liu, orcid.org/0000-0003-4432-9433; Li Zhao, orcid.org/0000-0001-8538-5605