 Xu Zhang1†
Xu Zhang1† Hang-Yuan Li1†
Hang-Yuan Li1† Jian-Wei Shao1
Jian-Wei Shao1 Ming-Chao Pei1Chong Cao2,3
Ming-Chao Pei1Chong Cao2,3 Fu-Qiang Huang1*
Fu-Qiang Huang1* Ming-Fei Sun4,5,6,7*
Ming-Fei Sun4,5,6,7*- 1School of Life Science and Engineering, Foshan University, Foshan, China
- 2Fujian Provincial Key Laboratory for the Prevention and Control of Animal Infectious Diseases and Biotechnology, Longyan, China
- 3Key Laboratory of Preventive Veterinary Medicine and Biotechnology, Longyan University, Longyan, China
- 4Zhaoqing/Maoming Branch Center of Guangdong Laboratory for Lingnan Modern Agricultural Science and Technology, Zhaoqing, China
- 5Key Laboratory of Livestock Disease Prevention of Guangdong Province, Guangzhou, China
- 6Key Laboratory of Avian Influenza and Other Major Poultry Diseases Prevention and Control, Ministry of Agriculture and Rural Affairs, Guangzhou, China
- 7Institute of Animal Health, Guangdong Academy of Agricultural Sciences, Guangzhou, China
The increasing prevalence and transmission of tick-borne diseases, especially those emerging ones, have posed a significant threat to public health. Thus, the discovery of neglected pathogenic agents carried and transmitted by ticks is urgently needed. Using unbiased high-throughput sequencing, a novel Orthonairovirus designated as Meihua Mountain virus (MHMV), was identified in bloodsucking ticks collected from cattle and wild boars in Fujian province, Southeastern China. The full-length genome was determined by RT-PCR and RACE. Genomic architecture of MHMV shares typical features with orthonairoviruses. Phylogenetic analyses suggested that MHMV is clustered into the Nairobi sheep disease (NSD) genogroup of the genus Orthonairovirus and is closely related to the Hazara virus. The RdRp, GPC, and N protein of MHMV shares 62.3%–83.5%, 37.1%–66.1%, and 53.4%–77.3% amino acid identity with other NSD genogroup viruses, respectively, representing a novel species. The overall pooled prevalence of MHMV in ticks was 2.53% (95% CI: 1.62%–3.73%, 22 positives of 134 tick pools), with 7.38% (95% CI: 3.84%–12.59%, 11 positives of 18 pools) in Haemaphysalis hystricis, 6.02% (95% CI: 1.85%–14.22%, four positives of eight pools) in H. formosensis, 25.03% (95% CI: 9.23%–54.59%, six positive of eight pools) in Dermacentor taiwanensis, and 0.16% (95% CI: 0.01%–0.72%, one positive of 100 pools) in Rhipicephalus microplus. This study presented the first report of tick-carried Orthonairovirus in Fujian province and highlighted the broad geographic distribution and high genetic diversity of orthonairoviruses in China.
Introduction
Ticks are one of the main vectors transmitting viral, bacterial, and parasitic pathogens that cause severe illnesses to animals and humans. The increasing prevalence and transmission of tick-borne diseases, especially those emerging ones such as severe fever with thrombocytopenia syndrome and Heartland virus diseases, are keeping challenging our preparedness and have posed a significant threat to public health (Madison-Antenucci et al., 2020). Thus, unbiased discovery and detection of emerging human-pathogenic agents carried and transmitted by ticks are urgently needed.
Members of the genus Orthonairovirus are all transmitted by ticks, distinguishing themselves from most of the other members of the family Nairoviridae (Lasecka and Baron, 2014). Based on the phylogenetic relationships, tick-associated orthonairoviruses were classified into nine genogroups, of which the most important is the Nairobi sheep disease (NSD) genogroup (Walker et al., 2016). The NSD genogroup includes Crimean-Congo hemorrhagic fever virus (CCHFV), Nairobi sheep disease virus (NSDV), Hazara virus (HAZV), Dugbe virus (DUGV), and Kupe virus (KUPV; Walker et al., 2016). The recently identified Tofla virus (TFLV) also belongs to this genogroup (Shimada et al., 2016). Among these, the fatal human pathogen CCHFV can cause hemorrhagic fever with mortality up to 30% (Shahhosseini et al., 2021), and the NSDV causes severe hemorrhagic gastroenteritis in sheep and goats with mortality up to 90% (Krasteva et al., 2020). Both CCHFV and NSDV endemic foci can be found in Africa, Asia, and the Middle East, suggesting the wide geographic distribution of orthonairoviruses. Except for CCHFV and NSDV, the vertebrate host has not been established for other members of the NSD genogroup. Meanwhile, human infection has not been reported, thus their relevance to public health remains unclear. However, infection with these viruses can be lethal to immunocompromised mice, emphasizing their pathogenic potential (Dowall et al., 2012; Shimada et al., 2016). Importantly, several recently discovered tick-borne orthonairoviruses in Eastern Asia, such as the Yezo virus (Kodama et al., 2021), Songling virus (Ma et al., 2021), and Tacheng tick virus 1 (Liu et al., 2019), were associated with human febrile illness with different severity, suggesting the high diversity of orthonairoviruses in nature and their potential risk to public health.
With the expansion of tick habitats, it is necessary to monitor the agents they carried (Dong and Soong, 2021). Fujian, a coastal mountainous province, locates in Southeastern China and possesses a middle subtropical climate. The suitable climatic conditions and abundant wildlife in mountains shaped a perfect environment for several tick species, such as Ixodes granulatus, Rhipicephalus microplus, and Dermacentor auratus (Zhang et al., 2019). The Meihua Mountain Nature Reserve in Fujian province is an ecological forest area, in which a variety of wild animals provide abundant host resources for ectoparasitic ticks. However, little is known about the background information of tick-carried pathogenic agents. In this study, we performed unbiased high-throughput meta-transcriptomic sequencing to profile RNA virus in ticks. A novel member of NSD genogroup Orthonairovirus, designated as Meihua Mountain virus (MHMV), was identified in bloodsucking ticks collected from cattle and wild boar. The complete genome sequence, genomic characteristics, phylogenetic relationship, and molecular prevalence of MHMV were analyzed. This study presented the first report of tick-borne Orthonairovirus in Fujian province and highlighted the broad geographic distribution and genetic diversity of orthonairoviruses in China.
Materials and methods
Tick collection and RNA extraction
From 2019 to 2020, 988 bloodsucking ticks were collected from cattle and wild boar in seven villages of Meihua Mountain Nature Reserve, Fujian province, Southeastern China. The tick species were morphologically identified by microscopy, and were further confirmed by 16S ribosomal RNA gene sequencing as described (The primers used are listed in Supplementary Table S1; Sameroff et al., 2019). Subsequently, the same species of ticks were pooled and stored at −80°C until further processing.
Before extracting nucleic acid, pooled samples were washed in 1 ml of 70% ethanol followed by three washes with nuclease-free water and then dried. Each pool was homogenized in 400 μl Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, United States) using TissueLyser II (Qiagen, Hilden, Germany). Homogenates were then centrifuged at 12,000 rpm for 1 min to precipitate debris. To extract RNA, 100 μl supernatant was mixed with 300 μl TRIzol LS reagent (Invitrogen, Carlsbad, CA, United States) and purified according to the manufacturer’s instructions. For meta-transcriptomic sequencing, a total of 34 pools were selected based on tick species and host animals (Supplementary Table S2). The total RNA was extracted using the homogenate mixture from 34 pools according to tick numbers in the pools to make sure that the RNA from each tick is equal. All purified RNA was quantified using Qubit 3.0 fluorometer (Thermo Fisher Scientific) and stored at −80°C until further use.
Meta-transcriptome sequencing
RNA library preparation was conducted following the library preparation protocol provided by Illumina, with slight modification. Briefly, ribosomal RNA in total RNA was removed by a Ribo-Zero Plus rRNA Depletion Kit (Illumina, San Diego, CA, United States). The remaining RNA was then fragmented, reverse-transcribed, adapted, purified, and analyzed with the Agilent 2100 Bioanalyzer. Paired-end (150 bp) sequencing was conducted on the Illumina HiSeq 2500 platform (Illumina). The library preparation and sequencing were performed at Novogene (Tianjin, China).
Bioinformatics analysis and full-length genome determination
The sequencing reads (raw data) were quality-controlled with SOAPnuke (v2.0.5) software to generate clean reads (Chen et al., 2018). The rRNA and host reads were then filtered by BWA (v0.7.17) with default settings (Li and Durbin, 2009). De novo assembly was performed with Megahit (v1.1.2; Li et al., 2015b) and the resulting contigs were blasted against reference virus nucleotides and proteins by BLASTN as well as Diamond BLASTX, respectively (Buchfink et al., 2015). The viral contigs mapped to the same segment were further assembled using ContigExpress, and subsequently mapped to a Hazara virus reference genome (GenBank accession numbers are listed in Supplementary Table S3) using Geneious (Biomatters, Ltd., New Zealand).
To fill the gaps between contigs, primers were designed according to corresponding contigs. Then nested reverse transcription polymerase chain reaction (RT-PCR) was done and followed by Sanger sequencing. For the genome termini, rapid amplification of cDNA ends (RACE) was performed using a SMARTer RACE 5′/3′ Kit (Takara, Beijing, China), as described previously (Li et al., 2015a).
Molecular screening of MHMV in individual tick pools
Primer pairs targeting the coding region of the L segment were designed. Primers are listed in Supplementary Table S1. Total RNA extracted from individual tick pools (134) was subjected to the screening of MHMV using nested RT-PCR and confirmed by sequencing. The nested PCR was performed according to the manufacturer’s instructions. Briefly, RT-PCR was performed with the PrimeScript One Step RT-PCR kit (TaKaRa). The 50 μl reaction contained 18 μl RNase-Free ddH2O, 25 μl 2× buffer, 2 μl PrimeScript Enzyme, 2 μl of forward and reverse primers, and 2 μg total RNA. The cycling program was set at 50°C for 30 min and 94°C for 2 min, followed by 35 cycles of standard PCR and a final extension step at 72°C for 5 min. The second round PCR was performed with ExTaq Premix (TaKaRa). The reaction was assembled by combining 19 μl RNase-Free ddH2O, 25 μl Taq Premix, 2 μl of forward and reverse primers each, and 2 μl PCR product from RT-PCR reaction. The following cycles were set as 98°C for 1 min, 35 cycles of standard PCR, and 72°C for 5 min. The annealing temperature of both RT-PCR and the second round PCR was set as 55°C.
Prevalence of MHMV was estimated assuming perfect sensitivity and specificity of molecular detection using the EPITOOLS online statistical program “Pooled prevalence for variable pool size and perfect tests”1 that based on the model established by Williams and Moffitt (2001). The input data were prepared as instructed by the EPITOOLS user guide.
Virus isolation
The homogenate from positive tick samples was centrifuged at 6,000 g for 10 min and the supernatant was collected. The 100 μl of undiluted and diluted (1:5 and 1:10) supernatant was inoculated onto monolayers of Vero E6, SW-13, and BHK-21 cell in 6-well plate. After 2 h of inoculation, the supernatant was aspirated out and replaced with medium containing 2% fetal bovine serum. The cytopathic effect (CPE) was examined daily and each passage was tested for the presence of MHMV RNA using RT-PCR as described in section 2.4. Unfortunately, neither apparent CPE nor the existence of viral RNA was observed throughout eight blind passages, indicating that MHMV may not be able to propagate in the above cell lines.
Genomic characterization and phylogenetic analysis
The potential open reading frames (ORFs) were predicted in ORFfinder and compared with other Orthonairovirus sequences (GenBank accession numbers are listed in Supplementary Table S2). Analysis of the glycoprotein precursor (GPC) domains and post-translational modification features was conducted with TMHMM (for transmembrane protein prediction; Krogh et al., 2001), ProP−1.0 (for signal peptidase cleavage site prediction; Petersen et al., 2011), NetNGlyc−1.0 (for N-linked glycosylation site prediction), and NetOGlyc (for mucin-type O-glycosylation site prediction; Steentoft et al., 2013).
To determine the phylogenetic relationship among orthonairoviruses, amino acid alignments were conducted using the E-INS-i algorithm in MAFFT 7.0 (Katoh et al., 2019). TrimAI 1.2 was used to trim off ambiguous positions and sequences were re-aligned in MEGA X (MUSCLE; Capella-Gutiérrez et al., 2009; Kumar et al., 2018). Phylogenetic trees were constructed using the maximum likelihood (ML) method (LG substitution model, Gamma-distributed rate variation) with 100 bootstrap replications (Le and Gascuel, 2008).
The amino acid and nucleotide sequence identity was determined with Clustal Omega (Sievers and Higgins, 2018). Both the putative Gn and Gc protein amino acid sequence alignment of NSD genogroup viruses were performed with MAFFT version 7.0 online server2 (Katoh et al., 2019). G-INS-i algorithm was used and the Unaligned level value was set as 0.4 with other parameters that were set as default. Visualization of the resulting alignments was done with the ESpriPt 3.03 (Robert and Gouet, 2014) and edited in Adobe Illustrator CC.
Ethical approval
This study did not involve the purposeful killing of animals. All samples were collected by passive surveillance under Chinese legislation. No ethical approval was necessary.
Results
Identification of MHMV in ticks
From 2019 to 2020, a total of 988 bloodsucking ticks were collected from cattle and wild boar in seven villages of the Meihua Mountain Nature Reserve, Fujian province, Southeastern China. The tick species were determined as Dermacentor taiwanensis (59, 8 pools), Haemaphysalis hystricis (214, 18 pools), Haemaphysalis formosensis (103, 8 pools), and Rhipicephalus microplus (612, 100 pools). The 988 ticks were divided into 134 individual pools based on tick species and host animals (Figure 1).
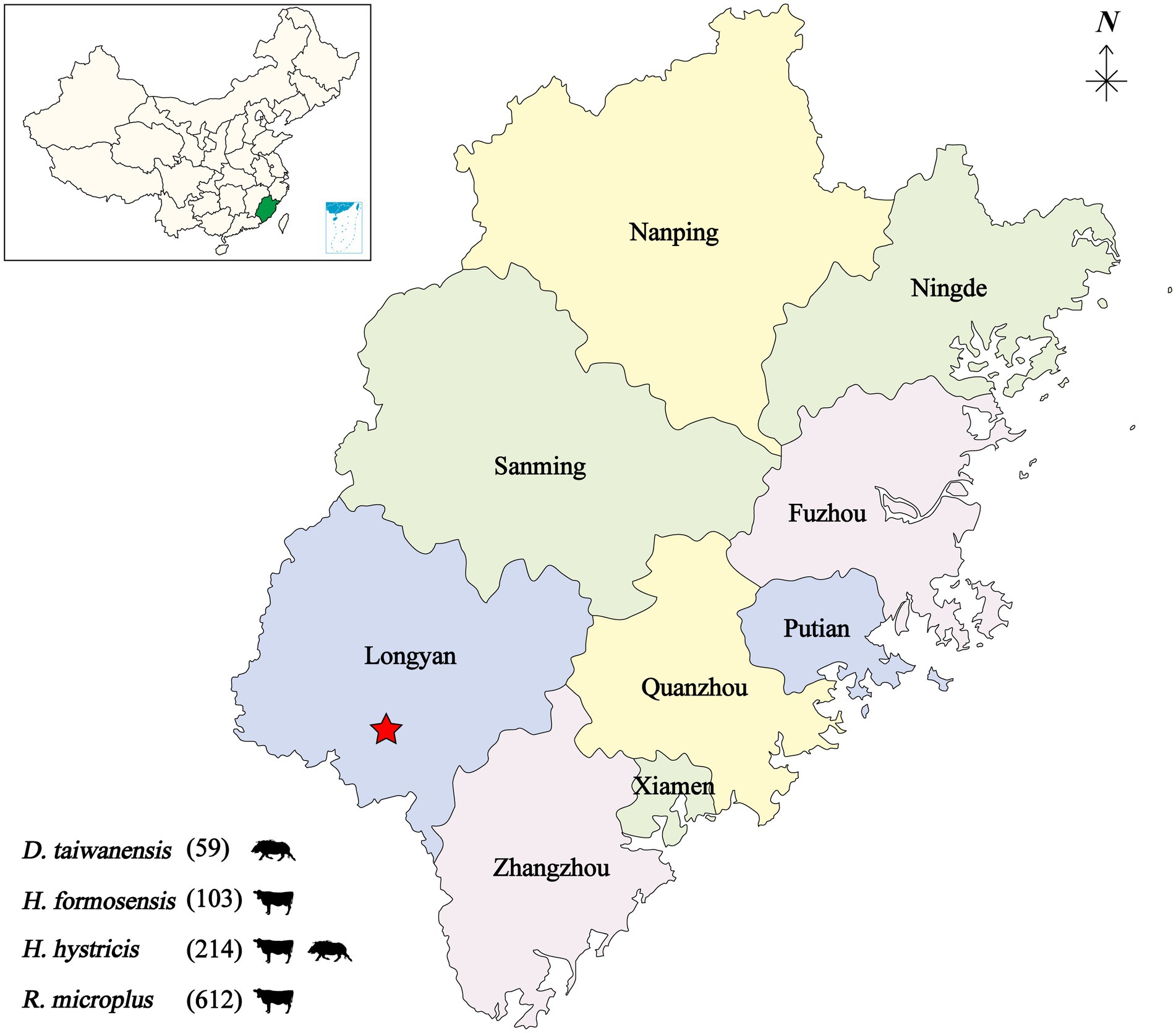
Figure 1. Geographic map showing the tick sampling information. The tick collection site is indicated by a red star, annotated with tick species and infesting animals. D, Dermacentor; H, Haemaphysalis; R, Rhipicephalus.
RNA from 34 pools was selected based on tick species and host animals, mixed, and subjected to meta-transcriptomic sequencing. A total of 41,271,206 paired-end clean reads were generated. After removing the ribosomal and tick host reads, 14,569,764 reads were obtained and assembled. The resulting contigs were blasted against virus nucleotides and proteins, which identified 706,102 viral reads, accounting for 1.71% of total clean reads. Among the assembled contigs, 59 viral contigs were annotated as the Large (L) (32), Middle (M) (21), and Small (S) (6) segments of orthonairoviruses within the family Nairoviridae. The novel virus was tentatively designated as MHMV. The full-length genome was determined by RT-PCR to fill the gaps between assembled contigs and RACE to obtain the genome termini. Thereafter, a total of 94,802 reads were re-mapped to the determined MHMV genome, revealing an overall >99% genome coverage with a mean depth of >350 × (Figure 2). MHMV has a tripartite negative-sense RNA genome that comprises three segments, including an L segment encoding an RNA-dependent RNA polymerase (RdRp), an M segment encoding a GPC, and an S segment encoding a nucleocapsid protein. The nucleotide sequences of L, M, and S segments of MHMV have been deposited in GenBank under accession numbers ON184084−ON184086, respectively.
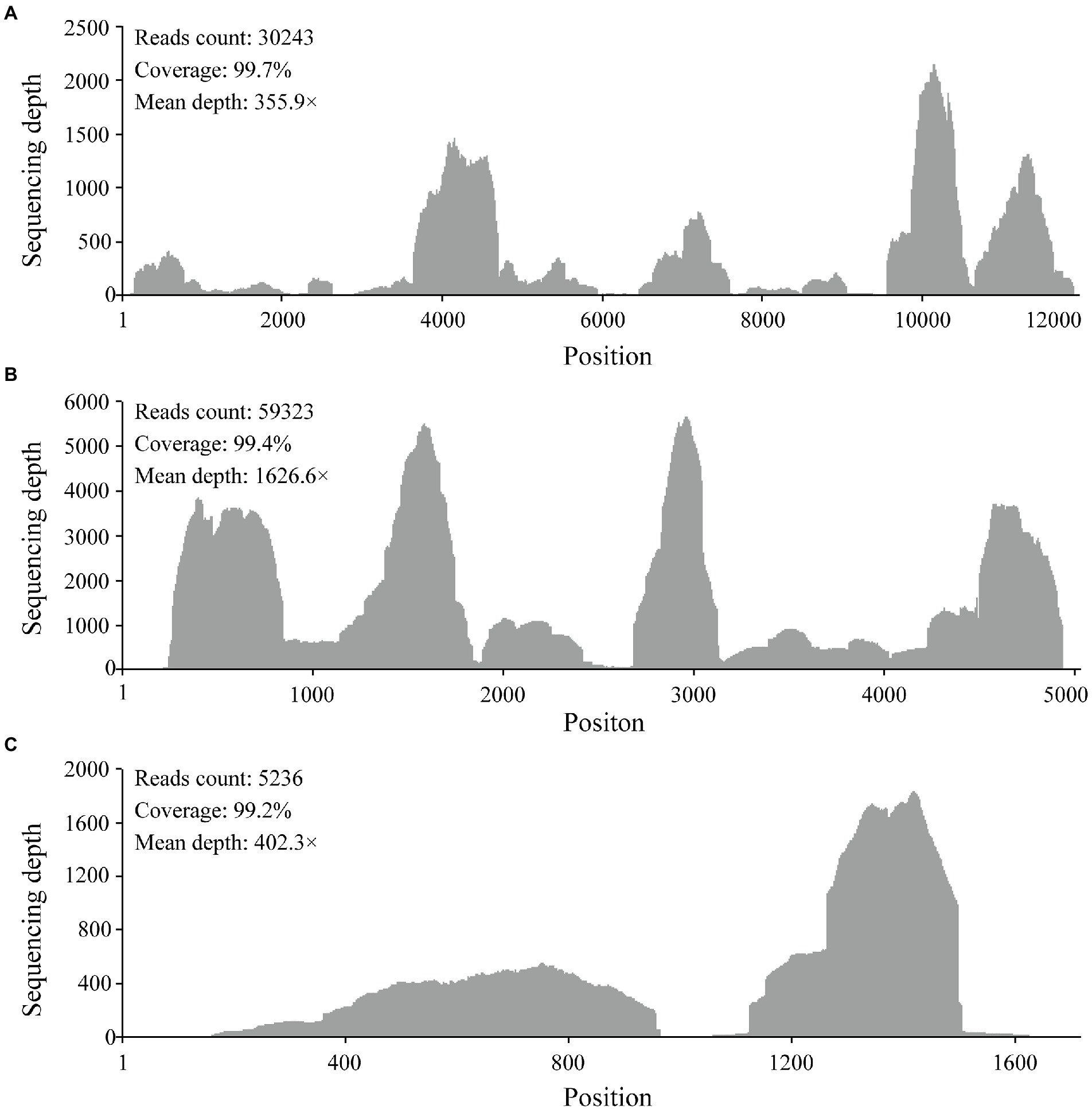
Figure 2. Mapped reads count plot of the MHMV genome. (A) L segment; (B) M segment; (C) S segment. The histograms show the depth at a given base position.
Phylogenetic analysis of MHMV
The virus sequence was preliminarily compared against Viruses reference sequences with blastn suite. The nucleotide sequences of L segment, M segment, and S segment of MHMV have the highest sequence similarity with HAZV with identities of 71.9%, 64.4%, and 69.4%, respectively (Table 1). These results support that MHMV is a novel species in genus Orthonairovirus according to the criteria suggested (He et al., 2022). ML phylogenetic trees reconstructed based on the amino acid sequences from L, M, and S segments of orthonairoviruses confirmed that members of the genus Orthonairovirus have been assigned into nine genogroups (Figure 3; Walker et al., 2016). The RdRp, GPC, and N protein trees all showed that MHMV was closely clustered with HAZV and a recently identified Orthonairovirus, TFLV (Figure 3). These three viruses formed a well-supported (100% bootstrap value at the node) small clade in the NSD genogroup, indicating MHMV is a novel member of the NSD genogroup.
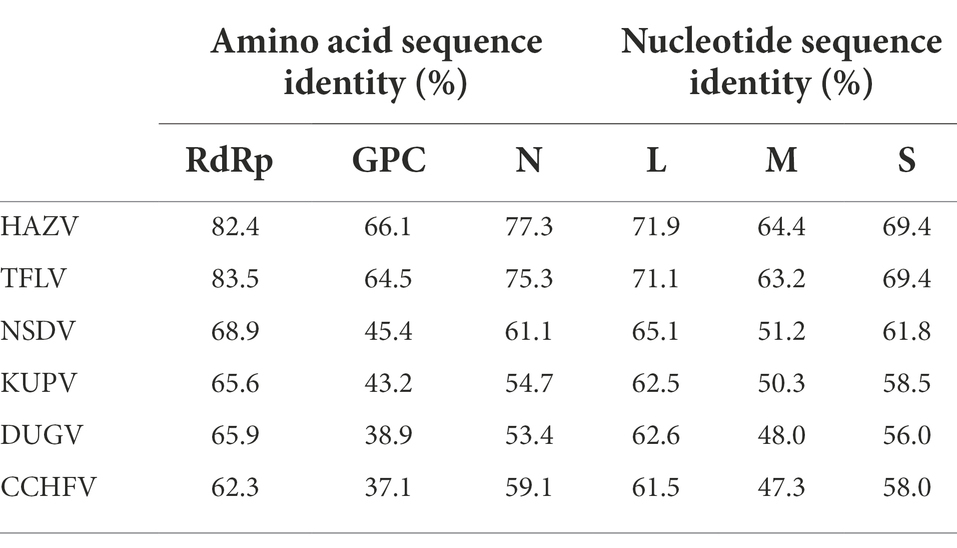
Table 1. Amino acid sequence and nucleotide identity of MHMV RdRp (L segment), GPC (M segment), and N protein (S segment) with the NSD genogroup orthonairoviruses.
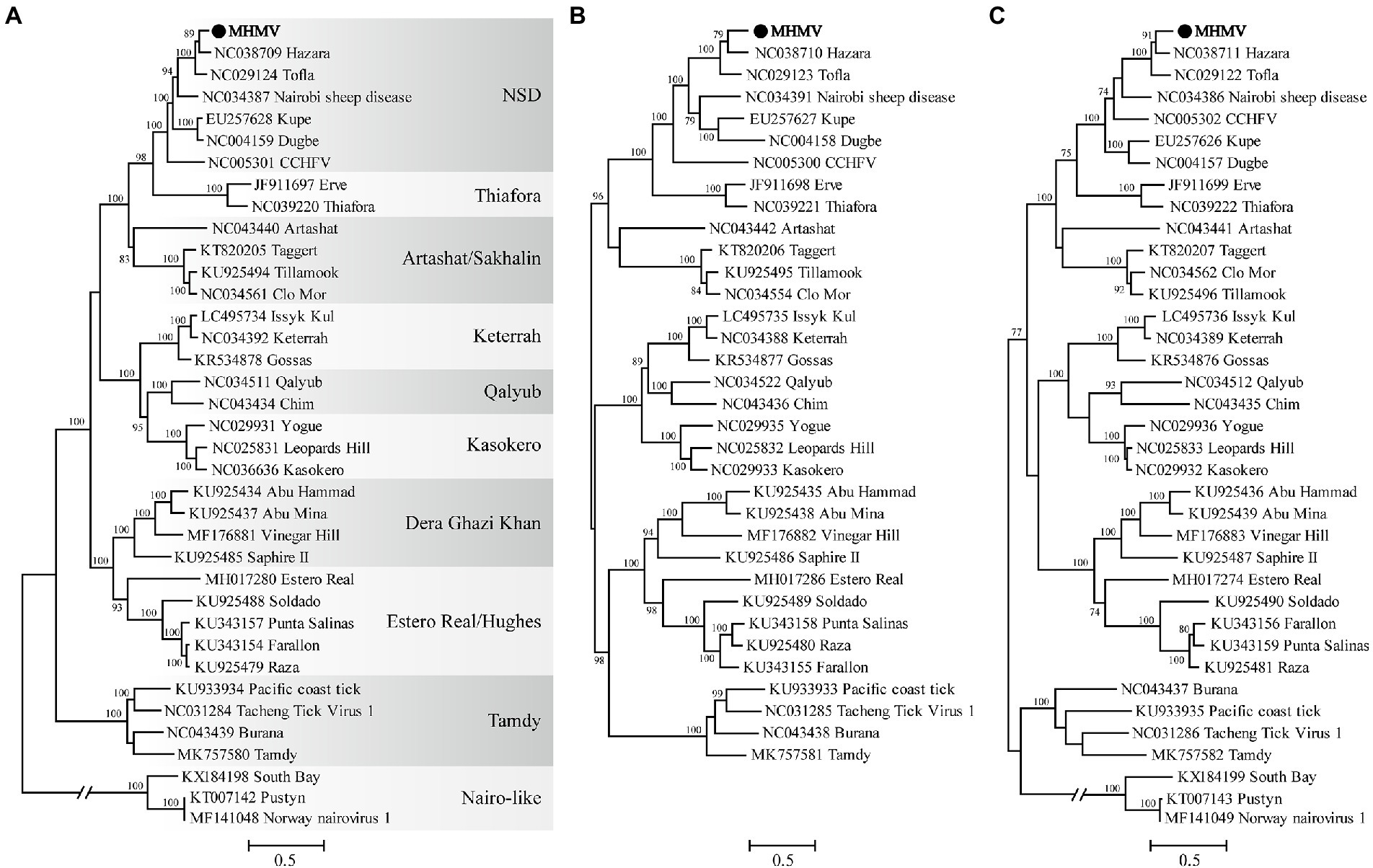
Figure 3. ML phylogenetic trees based on the amino acid sequences deduced from L, M, and S segments of Meihua Mountain Virus (Black dots) and other representative orthonairoviruses. (A) Tree inferred from the RdRp amino acid sequences. (B) Tree inferred from the GPC amino acid sequences. (C) Tree inferred from the N protein amino acid sequences. Genogroup assignments are indicated with differential shading patterns. Bootstrap support values ≥70% are indicated. Sequences are identified by their GenBank accession numbers, followed by the virus name. GenBank accession numbers of sequences used in the alignments are listed in Supplementary Table S2.
Genome characteristics of the novel MHMV
The MHMV has a tripartite negative-sense RNA genome that comprises three segments as other orthonairoviruses. The L segment is 11,897 nt in length encoding an RdRp (3,923 aa). The M segment (4,853 nt) encodes a GPC (1,482 aa), and an S segment (1,682 nt) encodes an N protein (485 aa). The RdRp ORF is flanked by an 85 bp 5′-non-coding region (NCR) and a 40 bp 3′-NCR in the L segment. The M segment is flanked by a 337 bp 5′-NCR and a 67 bp 3′-NCR, while the S segment has a 143 bp 5′-NCR and an 81 bp 3′-NCR. All three segments shared the conserved terminal sequences with 3′-AGAGUUUCU and a reverse complementary 5′-AGAAACUCU, the typical feature of the genus Orthonairovirus (Kuhn et al., 2016).
The RdRp of nairoviruses contains several presumed regions and domains, including ovarian tumor-like (OTU-like) protease domain, polymerase module (pre-Motif A, motif A-E), and the Region I, Region II (a recently identified N-terminal localized Influenza-Like endonuclease domain which had a cap-snatching activity) and Region IV (Honig et al., 2004; Kinsella et al., 2004; Reguera et al., 2010). Amino acid sequence alignment of these regions confirmed that the RdRp of MHMV contains all the aforementioned highly conserved regions as other NSD genogroup members (Supplementary Figure S1).
Moreover, protein domain and post-translational modification analysis of MHMV GPC showed typical organizational characteristics in terms of three predicted signal peptidase cleavage sites, three subtilisin/kexin-isozyme-1 (SKI-1) protease cleavage sites (RRKL51, RRLM310, and RKLL837), 5 transmembrane domains, presumable mucin-like domain located at the N-terminus, glycoprotein (Gn), a non-structural protein (NSm), and glycoprotein (Gc; Figure 4). Notably, MHMV has similar Gn and Gc structure and protein size, although a relatively lower amino acid identity with other NSD genogroup viruses (Figure 4; Table 1). Interestingly, MHMV, HAZV, and TFLV contain much shorter mucin-like domains and fewer O-glycosylation sites in comparison with NSDV, KUPV, DUGV, and CCHFV (Figure 4). The Gn protein of MHMV contains two N-glycosylation sites, two transmembrane domains, and a pair of conserved zinc-finger domains that have also been found in CCHFV (Supplementary Figure S2; Estrada and De Guzman, 2011). Similarly, three conserved N-glycosylation sites have been found in Gc of MHMV and other NSD group viruses (Supplementary Figure S3). A C-terminus localized transmembrane domain and two conserved N-glycosylation sites were also predicted in other NSD genogroup members (Supplementary Figure S3). Moreover, MHMV possesses conserved fusion loop regions as other Orthonairovirus (Supplementary Figure S3; Li et al., 2022).
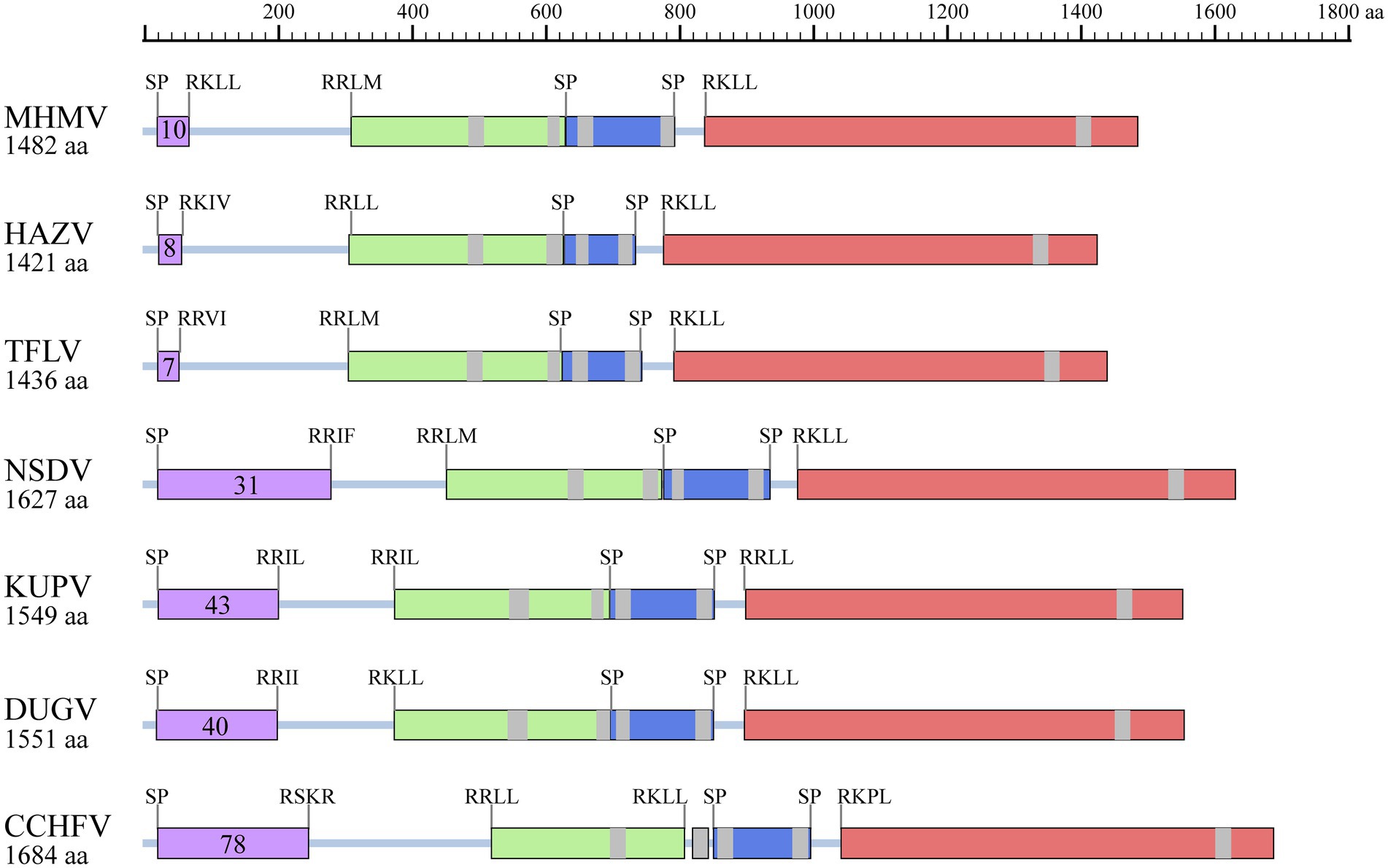
Figure 4. Illustration of GPC structures of the NSD genogroup viruses. Mucin-like domain (purple), Gn (light green), NSm (dark blue), and Gc (pink) are shaded correspondingly. Predicted signal peptidase (SP) cleavage sites and putative furin-like and subtilisin/kexin-isozyme-1 (SKI-I) cleavage sites are labeled. The number of predicted O-linked glycosylation sites in the mucin-like domain is also shown in the purple shaded box. The transmembrane domains are shaded with grey. The protein length is scaled. HAZV, Hazara virus; TFLV, Tofla virus; NSDV, Nairobi sheep disease virus; KUPV, Kupe virus; DUGV, Dugbe virus; CCHFV, Crimean-Congo hemorrhagic fever virus.
Structural biology studies of N protein of nairoviruses identified two major domains, a globular head and an extended stalk, with RNA/DNA binding-associated sites. The head domains of the MHMV and other NSD genogroup viruses showed high conservation while apparent high flexibility can be seen in the stalk domain as previously reported (Supplementary Figure S4; Wang et al., 2015). In addition, the DEVD caspase-3 cleavage site motif that has been identified in CCHFV is not present in MHMV N protein (Wang et al., 2012). These results showed that MHMV shares conserved genome organization and protein structures with other NSD genogroup orthonairoviruses.
Sequence comparison of MHMV with other NSD genogroup members
The NSD genogroup now consists of six representative species, including HAZV, TFLV, NSDV, KUPV, DUGV, and CCHFV. The pairwise alignment showed that the RdRp, GPC, and N protein of MHMV share 62.3%–83.5%, 37.1%–66.1%, and 53.4%–77.3% amino acid identity with other NSD genogroup viruses, respectively (Table 1). The RdRp amino acid sequence of MHMV has the highest identity (83.5%) with TFLV, while GPC and N both showed the highest identity to HAZV with the identity of 66.1 and 77.3%, respectively (Table 1).
Notably, the Gn protein of the NSD genogroup viruses has a similar percent identity (38.4%–66.5%) with GPC polyprotein (35.8%–66.1%; Table 2). However, the Gc part showed a significantly higher percent identity range from 44.8% to 78.0% within the genogroup (Table 2). To date, an objective species demarcation criterion of Orthonairovirus stays unestablished. The species demarcation criteria for Nairoviridae are recently proposed to be <93% identity in L protein, considering that available CCHFV strains possess a maximum amino acid distance of 6.8% (Walker et al., 2016). Thus, we tentatively designated the newly identified virus, MHMV, as a novel species of the NSD genogroup in the genus Orthonairovirus.
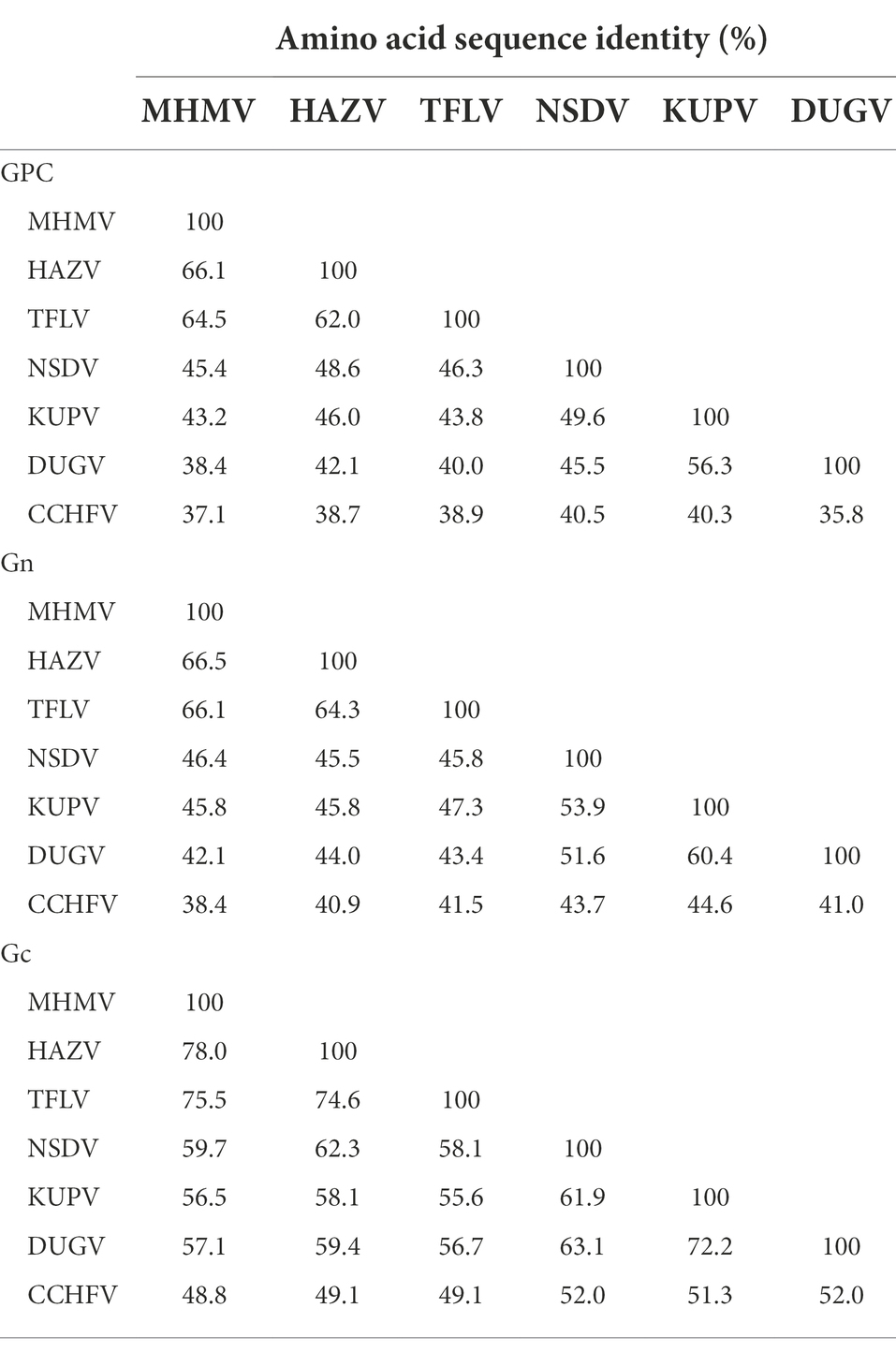
Table 2. Pairwise comparisons of GPC, Gn, and Gc amino acid sequences among the NSD genogroup orthonairoviruses.
Prevalence of MHMV in ticks
To estimate the prevalence of MHMV in ticks, the collected 134 individual tick pools were subjected to molecular screening of MHMV. The amplicon of RT-PCR was sequenced and two variant sequences have been deposited to Genbank under accession numbers of OP056094 and OP056095, respectively. The positive rate was 7.38% (95% CI: 3.84%–12.59%, 18 pools) in H. hystricis, 6.02% (95% CI: 1.85%–14.22%, eight pools) in H. formosensis, and 25.03% (95% CI: 9.23%–54.59%, eight pools) in D. taiwanensis, while only one pool of R. microplus was tested positive out of 100 (Table 3), suggesting that the hard ticks, Haemaphysalis spp. and Dermacentor spp., might be the predominant vectors of MHMV. However, isolation of MHMV with commonly used cell lines for NSD genogroup virus isolation was not successful.
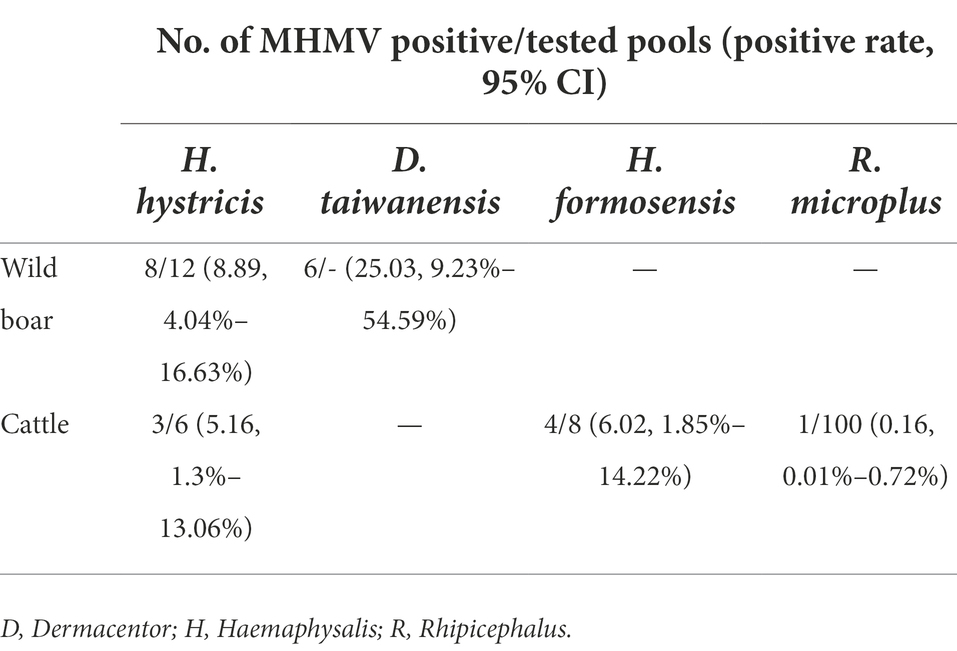
Table 3. Prevalence of MHMV in tick pools screened by RT-PCR.
Discussion
Tick-transmitted viral pathogens can cause deadly illnesses in animals and humans, which has posed a significant threat to public health (Madison-Antenucci et al., 2020). With the expansion of tick habitats, it is necessary to survey the pathogenic agents they carried. In this study, we took unbiased high throughput meta-transcriptomic sequencing to profile viruses carried by ticks collected from Fujian, a coastal mountainous province, Southeastern China. A novel Orthonairovirus, MHMV, was identified in bloodsucking ticks collected from cattle and wild boar. Since the viral structural proteins possess relatively low identity (<83.5% in L, <66.1% in GPC, and <77.3% in N) to its closest Orthonairovirus relatives, we herein suggest MHMV as a novel species of genus Orthonairovirus. Phylogenetic analysis suggested that MHMV is closely related to HAZV and belongs to the NSD genogroup in the genus Orthonairovirus. It shares typical genomic characteristics with other orthonairoviruses. Molecular surveillance showed that MHMV is mainly carried by the ticks of Haemaphysalis spp. and Dermacentor spp.
The expanding genus Orthonairovirus is now a large group with 15 highly divergent species and several unclassified nairoviruses (Garrison et al., 2020). Among them, CCHFV, the most pathogenic one, has been found in China by molecular and serological detection in ticks and humans, respectively (Sun et al., 2009; Moming et al., 2018). Recently, some novel Orthonairovirus, such as Songling virus (Ma et al., 2021), Tacheng tick virus 1(Liu et al., 2019), and Tamdy virus (strain XJ01/TAMV/China/2018; Zhou et al., 2019) have been identified in ticks collected in Northeastern and Northwestern China, respectively. The identification of MHMV in Fujian province, Southeastern China, suggested the genetic diversity and wide geographic distribution of orthonairoviruses in China. More importantly, songling virus and Tacheng tick virus 1 have been confirmed associated with human febrile illness (Liu et al., 2019; Ma et al., 2021), indicating the public health significance of Orthonairovirus. Although the pathogenicity of MHMV needs further investigation, the close phylogenetic relationship with other pathogenic Orthonairovirus still indicates its pathogenic potential to human or animal.
Previously, it has been established that the classification of the genus Orthonairovirus was well supported by phylogenetic relationships and structural features of GPC (Walker et al., 2016). In this study, we compared the sequence conservativity and protein structure of the NSD genogroup members. Both sequence alignment and protein structure analysis supported the genogroup classification proposed by Walker et al. (2016). Notably, we showed that the Gn amino acid sequence has a similar level of identity with the whole GPC polyprotein, while the Gc part shared an apparently higher identity. If these observations infer functional consequences in terms of pathogenesis and viral biology of the NSD genogroup members remains unclear.
We have attempted to isolate MHMV with Vero E6, SW-13, and BHK-21 cell lines that have been used for isolation of NSD genogroup viruses (CCHFV, HAZV, and TFLV; Shimada et al., 2016; Fuller et al., 2020; Dai et al., 2021). However, neither CPE nor viral RNA has been detected across several passages in cell cultures. This could be explained by that MHMV is a novel virus with low identity with known NSD genogroup viruses. Tick cell line may be a better alternative for MHMV isolation in the future since it has been successfully applied in the isolation of several novel viruses from ticks (Jia et al., 2019; Kholodilov et al., 2020).
Taken together, a novel Orthonairovirus was identified in ticks collected in the Fujian province of China through unbiased RNA sequencing, which expands the knowledge about the genetic diversity of orthonairoviruses in China.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, ON184084, ON184085, and ON184086.
Author contributions
XZ and CC sampled the ticks. XZ, H-YL, and M-CP prepared the nucleic acid samples and gathered the molecular data. XZ, J-WS, and F-QH performed the bioinformatics analyses and drafted the manuscript. F-QH and M-FS conceived the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Guangdong Basic and Applied Basic Research Foundation (2020A1515011575, 2021A1515110450) and the Natural Science Foundation of Fujian Province (2021J05235).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.977405/full#supplementary-material
Footnotes
References
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7, 1–6. doi: 10.1093/gigascience/gix120
Dai, S., Wu, Q., Wu, X., Peng, C., Liu, J., Tang, S., et al. (2021). Differential cell line susceptibility to Crimean-Congo hemorrhagic fever virus. Front. Cell. Infect. Microbiol. 11:648077. doi: 10.3389/fcimb.2021.648077
Dong, X. P., and Soong, L. (2021). Emerging and re-emerging zoonoses are major and global challenges for public health. Zoonoses 1, 1–2. doi: 10.15212/ZOONOSES-2021-0001
Dowall, S. D., Findlay-Wilson, S., Rayner, E., Pearson, G., Pickersgill, J., Rule, A., et al. (2012). Hazara virus infection is lethal for adult type I interferon receptor-knockout mice and may act as a surrogate for infection with the human-pathogenic Crimean–Congo hemorrhagic fever virus. J. Gen. Virol. 93, 560–564. doi: 10.1099/vir.0.038455-0
Estrada, D. F., and De Guzman, R. N. (2011). Structural characterization of the Crimean-Congo hemorrhagic fever virus Gn tail provides insight into virus assembly. J. Biol. Chem. 286, 21678–21686. doi: 10.1074/jbc.M110.216515
Fuller, J., Álvarez-Rodríguez, B., Todd, E., Mankouri, J., Hewson, R., and Barr, J. N. (2020). Hazara nairovirus requires COPI components in both Arf1-dependent and Arf1-independent stages of its replication cycle. J. Virol. 94:e00766-20. doi: 10.1128/jvi.00766-20
Garrison, A. R., Alkhovsky, S. V., Avsic-Zupanc, T., Bente, D. A., Bergeron, E., Burt, F., et al. (2020). ICTV virus taxonomy profile: nairoviridae. J. Gen. Virol. 101, 798–799. doi: 10.1099/jgv.0.001485
He, W. T., Hou, X., Zhao, J., Sun, J., He, H., Si, W., et al. (2022). Virome characterization of game animals in China reveals a spectrum of emerging pathogens. Cell 185, 1117.e8–1129.e8. doi: 10.1016/j.cell.2022.02.014
Honig, J. E., Osborne, J. C., and Nichol, S. T. (2004). Crimean-Congo hemorrhagic fever virus genome L RNA segment and encoded protein. Virology 321, 29–35. doi: 10.1016/j.virol.2003.09.042
Jia, N., Liu, H. B., Ni, X. B., Bell-Sakyi, L., Zheng, Y. C., Song, J. L., et al. (2019). Emergence of human infection with Jingmen tick virus in China: a retrospective study. EBioMedicine 43, 317–324. doi: 10.1016/j.ebiom.2019.04.004
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kholodilov, I. S., Litov, A. G., Klimentov, A. S., Belova, O. A., Polienko, A. E., Nikitin, N. A., et al. (2020). Isolation and characterisation of Alongshan virus in Russia. Viruses 12:362. doi: 10.3390/v12040362
Kinsella, E., Martin, S. G., Grolla, A., Czub, M., Feldmann, H., and Flick, R. (2004). Sequence determination of the Crimean-Congo hemorrhagic fever virus L segment. Virology 321, 23–28. doi: 10.1016/j.virol.2003.09.046
Kodama, F., Yamaguchi, H., Park, E., Tatemoto, K., Sashika, M., Nakao, R., et al. (2021). A novel nairovirus associated with acute febrile illness in Hokkaido. Japan. Nat. Commun. 12:5539. doi: 10.1038/s41467-021-25857-0
Krasteva, S., Jara, M., Frias-De-Diego, A., and Machado, G. (2020). Nairobi sheep disease virus: a historical and epidemiological perspective. Front. Vet. Sci. 7:419. doi: 10.3389/fvets.2020.00419
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kuhn, J. H., Wiley, M. R., Rodriguez, S. E., Bào, Y., Prieto, K., Travassos da Rosa, A. P. A., et al. (2016). Genomic characterization of the genus Nairovirus (family Bunyaviridae). Viruses 8:164. doi: 10.3390/v8060164
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lasecka, L., and Baron, M. D. (2014). The molecular biology of nairoviruses, an emerging group of tick-borne arboviruses. Arch. Virol. 159, 1249–1265. doi: 10.1007/s00705-013-1940-z
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015b). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, N., Rao, G., Li, Z., Yin, J., Chong, T., Tian, K., et al. (2022). Cryo-EM structure of glycoprotein C from Crimean-Congo hemorrhagic fever virus. Virol. Sin. 37, 127–137. doi: 10.1016/j.virs.2022.01.015
Li, C. X., Shi, M., Tian, J. H., Lin, X. D., Kang, Y. J., Chen, L. J., et al. (2015a). Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. elife 4:e05378. doi: 10.7554/eLife.05378
Liu, X., Zhang, X., Wang, Z., Dong, Z., Xie, S., Jiang, M., et al. (2019). A tentative Tamdy Orthonairovirus related to febrile illness in northwestern China. Clin. Infect. Dis. 70, 2155–2160. doi: 10.1093/cid/ciz602
Ma, J., Lv, X. L., Zhang, X., Han, S. Z., Wang, Z. D., Li, L., et al. (2021). Identification of a new orthonairovirus associated with human febrile illness in China. Nat. Med. 27, 434–439. doi: 10.1038/s41591-020-01228-y
Madison-Antenucci, S., Kramer, L. D., Gebhardt, L. L., and Kauffman, E. (2020). Emerging tick-borne diseases. Clin. Microbiol. Rev. 33, e00083–e00018. doi: 10.1128/cmr.00083-18
Moming, A., Yue, X., Shen, S., Chang, C., Wang, C., Luo, T., et al. (2018). Prevalence and phylogenetic analysis of Crimean-Congo hemorrhagic fever virus in ticks from different ecosystems in Xinjiang. China. Virol. Sin. 33, 67–73. doi: 10.1007/s12250-018-0016-3
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Reguera, J., Weber, F., and Cusack, S. (2010). Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 6:e1001101. doi: 10.1371/journal.ppat.1001101
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. doi: 10.1093/nar/gku316
Sameroff, S., Tokarz, R., Charles, R. A., Jain, K., Oleynik, A., Che, X., et al. (2019). Viral diversity of tick species parasitizing cattle and dogs in Trinidad and Tobago. Sci. Rep. 9:10421. doi: 10.1038/s41598-019-46914-1
Shahhosseini, N., Wong, G., Babuadze, G., Camp, J. V., Ergonul, O., Kobinger, G. P., et al. (2021). Crimean-Congo hemorrhagic fever virus in Asia, Africa and Europe. Microorganisms 9:1907. doi: 10.3390/microorganisms9091907
Shimada, S., Aoki, K., Nabeshima, T., Fuxun, Y., Kurosaki, Y., Shiogama, K., et al. (2016). Tofla virus: a newly identified Nairovirus of the Crimean-Congo hemorrhagic fever group isolated from ticks in Japan. Sci. Rep. 6:20213. doi: 10.1038/srep20213
Sievers, F., and Higgins, D. G. (2018). Clustal omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145. doi: 10.1002/pro.3290
Steentoft, C., Vakhrushev, S. Y., Joshi, H. J., Kong, Y., Vester-Christensen, M. B., Schjoldager, K. T., et al. (2013). Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32, 1478–1488. doi: 10.1038/emboj.2013.79
Sun, S., Dai, X., Aishan, M., Wang, X., Meng, W., Feng, C., et al. (2009). Epidemiology and phylogenetic analysis of crimean-Congo hemorrhagic fever viruses in Xinjiang, China. J. Clin. Microbiol. 47, 2536–2543. doi: 10.1128/JCM.00265-09
Walker, P. J., Widen, S. G., Wood, T. G., Guzman, H., Tesh, R. B., and Vasilakis, N. (2016). A global genomic characterization of Nairoviruses identifies nine discrete Genogroups with distinctive structural characteristics and host-vector associations. Am. J. Trop. Med. Hyg. 94, 1107–1122. doi: 10.4269/ajtmh.15-0917
Wang, Y., Dutta, S., Karlberg, H., Devignot, S., Weber, F., Hao, Q., et al. (2012). Structure of Crimean-Congo hemorrhagic fever virus nucleoprotein: superhelical homo-oligomers and the role of caspase-3 cleavage. J. Virol. 86, 12294–12303. doi: 10.1128/jvi.01627-12
Wang, W., Liu, X., Wang, X., Dong, H., Ma, C., Wang, J., et al. (2015). Structural and functional diversity of Nairovirus-encoded nucleoproteins. J. Virol. 89, 11740–11749. doi: 10.1128/jvi.01680-15
Williams, C. J., and Moffitt, C. M. (2001). A critique of methods of sampling and reporting pathogens in populations of fish. J. Aquat. Anim. Health 13, 300–309. doi: 10.1577/1548-8667(2001)013<0300:ACOMOS>2.0.CO;2
Zhang, G., Zheng, D., Tian, Y., and Li, S. (2019). A dataset of distribution and diversity of ticks in China. Sci. Data 6:105. doi: 10.1038/s41597-019-0115-5
Keywords: Meihua Mountain virus, novel Orthonairovirus, NSD genogroup, metatranscriptomics, China
Citation: Zhang X, Li H-Y, Shao J-W, Pei M-C, Cao C, Huang F-Q and Sun M-F (2022) Genomic characterization and phylogenetic analysis of a novel Nairobi sheep disease genogroup Orthonairovirus from ticks, Southeastern China. Front. Microbiol. 13:977405. doi: 10.3389/fmicb.2022.977405
Edited by:
Wei Wang, Jiangsu Institute of Parasitic Diseases (JIPD), ChinaReviewed by:
Sabri Hacioglu, Veterinary Control Central Research Institute, TurkeyWen-Ping Guo, Northwest A&F University, China
Jia-Fu Jiang, Beijing Institute of Microbiology and Epidemiology, China
Katsunori Okazaki, Health Sciences University of Hokkaido, Japan
Copyright © 2022 Zhang, Li, Shao, Pei, Cao, Huang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fu-Qiang Huang, cXhodWFuZ2Z1cWlhbmdAZm9zdS5lZHUuY24=; Ming-Fei Sun, c21mNzgxMEAxMjYuY29t
†These authors have contributed equally to this work