
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 August 2022
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.967949
 Wern Chern Chai1
Wern Chern Chai1 Jonathan J. Whittall1
Jonathan J. Whittall1 Steven W. Polyak1
Steven W. Polyak1 Klyie Foo1Xin Li2
Klyie Foo1Xin Li2 Cameron J. Dutschke1
Cameron J. Dutschke1 Abiodun D. Ogunniyi3
Abiodun D. Ogunniyi3 Shutao Ma2Matthew J. Sykes1
Shutao Ma2Matthew J. Sykes1 Susan J. Semple1,4
Susan J. Semple1,4 Henrietta Venter1*
Henrietta Venter1*Acinetobacter baumannii is a pathogen with high intrinsic antimicrobial resistance while multidrug resistant (MDR) and extensively drug resistant (XDR) strains of this pathogen are emerging. Treatment options for infections by these strains are very limited, hence new therapies are urgently needed. The bacterial cell division protein, FtsZ, is a promising drug target for the development of novel antimicrobial agents. We have previously reported limited activity of cinnamaldehyde analogs against Escherichia coli. In this study, we have determined the antimicrobial activity of six cinnamaldehyde analogs for antimicrobial activity against A. baumannii. Microscopic analysis was performed to determine if the compounds inhibit cell division. The on-target effect of the compounds was assessed by analyzing their effect on polymerization and on the GTPase activity of purified FtsZ from A. baumannii. In silico docking was used to assess the binding of cinnamaldehyde analogs. Finally, in vivo and in vitro safety assays were performed. All six compounds displayed antibacterial activity against the critical priority pathogen A. baumannii, with 4-bromophenyl-substituted 4 displaying the most potent antimicrobial activity (MIC 32 μg/mL). Bioactivity was significantly increased in the presence of an efflux pump inhibitor for A. baumannii ATCC 19606 (up to 32-fold) and significantly, for extensively drug resistant UW 5075 (greater than 4-fold), suggesting that efflux contributes to the intrinsic resistance of A. baumannii against these agents. The compounds inhibited cell division in A. baumannii as observed by the elongated phenotype and targeted the FtsZ protein as seen from the inhibition of polymerization and GTPase activity. In silico docking predicted that the compounds bind in the interdomain cleft adjacent to the H7 core helix. Di-chlorinated 6 was devoid of hemolytic activity and cytotoxicity against mammalian cells in vitro, as well as adverse activity in a Caenorhabditis elegans nematode model in vivo. Together, these findings present halogenated analogs 4 and 6 as promising candidates for further development as antimicrobial agents aimed at combating A. baumannii. This is also the first report of FtsZ-targeting compounds with activity against an XDR A. baumannii strain.
Acinetobacter baumannii has been recognized as one of the most difficult to treat resistant Gram-negative bacterial pathogens (Knight et al., 2018; Abdi et al., 2020; Kyriakidis et al., 2021; Murray et al., 2022). This microorganism is intrinsically resistant to multiple distinct classes of antibiotics (Knight et al., 2018; Abdi et al., 2020). The rise of antimicrobial resistance (AMR) is further compounding the issue (Ambrose et al., 2022), making infections caused by A. baumannii more challenging and expensive to address. Consequently, the World Health Organization (WHO) has prioritized A. baumannii as one of the most critical pathogens in urgent need of new antimicrobial drug discovery and development (World Health Organization, 2017). In line with the WHO Global Action Plan, one of the objectives is to increase the development of new chemotherapies with novel mechanisms of action to combat multidrug resistant pathogens (World Health Organization, 2015), especially A. baumannii.
A promising route toward the development of novel antimicrobial agents necessary to combat AMR is to inhibit bacterial cell division. FtsZ is a key protein that is crucial in cellular division in both Gram-positive and Gram-negative bacteria and is therefore a promising antibacterial drug target (Hurley et al., 2016; Carro, 2019; Kusuma et al., 2019; Casiraghi et al., 2020). During cellular division, FtsZ functions in concert with 11 other essential proteins to form a complex known as the divisome (Bisson-Filho et al., 2017). The divisome is necessary for the formation of two daughter cells by first forming a dynamic ring (Z ring) around the bacterial midcell that ultimately contracts, leading to closure of the septum (Broughton et al., 2015; Bisson-Filho et al., 2017; den Blaauwen and Luirink, 2019). Self-polymerization of FtsZ in a GTP-dependent mechanism is critical to the formation of the Z-ring structure. Given its promise as an antibacterial drug target, multiple classes of small molecules that bind to FtsZ have been identified and reported in the literature (Margalit et al., 2004; Haydon et al., 2008; Kapoor and Panda, 2009; Chan et al., 2013; Kaul et al., 2015; Li and Ma, 2015; Liu et al., 2018; Carro, 2019; Kusuma et al., 2019; Casiraghi et al., 2020; Chai et al., 2020). These include the benzamides (Haydon et al., 2008; Kaul et al., 2015; Bi et al., 2017, 2018; Chai et al., 2020; Straniero et al., 2020); coumarins (de Souza et al., 2005; Yang et al., 2021); berberine derivatives (Domadia et al., 2008); quinazolines and quinolinium compounds (Nair et al., 2017; Sun et al., 2018a,b; Zheng et al., 2018); cinnamaldehydes (Domadia et al., 2007; Li et al., 2015a), zantrins (Margalit et al., 2004), methyl-phenyl-phenanthridium derivatives (Liu et al., 2018) and quinuclidine derivatives (Chan et al., 2013). Whilst several FtsZ inhibitors have efficacy against Gram-positive bacteria, very few display antimicrobial activity against Gram-negative bacteria (Araújo-Bazán et al., 2016; Casiraghi et al., 2020; Gurnani et al., 2022; Rosado-Lugo et al., 2022; Zhong et al., 2022), leaving the development of FtsZ inhibitors with activity against pathogens such as A. baumannii as an area in need of urgent exploration.
The FtsZ protein comprises two globular subdomains, with the GTP-binding site located in the N-terminal domain and a GTPase activating site at the C-terminal end. These subdomains are separated by the central H7 helix (Löwe and Amos, 1998; Matsui et al., 2012). Whilst the N-terminal is generally well conserved between bacterial species, the flexible C-terminal subdomain differs in both length and structure (Huecas et al., 2017). Crystal structures of S. aureus FtsZ in complex with PC190723 and other benzamides have defined the mode of binding for these inhibitors (Matsui et al., 2012; Ferrer-González et al., 2017; Fujita et al., 2017). Since the FtsZ protein plays an essential role in cellular division of prokaryotes (Romberg et al., 2001; Busiek and Margolin, 2015; Aylett and Duggin, 2017), its function is comparable to mammalian tubulin in eukaryotes (Pilhofer et al., 2007; McIntosh et al., 2010; Sundararajan et al., 2015; Battaje and Panda, 2017). However, bacterial FtsZ has distinctive structural differences from mammalian tubulin that permit the development of bacterial-specific inhibitors. Primary structure analysis has revealed the proteins are distinct, with FtsZ from Escherichia coli and human tubulin sharing only 10% homology (Chen and Erickson, 2008). The crystal structures also reveal important differences, for example that the interdomain cleft in FtsZ necessary for inhibitor binding is not observed in tubulin (Kusuma et al., 2019). These key features allow for development of new antibacterial agents that specifically target FtsZ.
The natural product cinnamaldehyde (Figure 1) is one promising lead compound with modest bioactivity against E. coli (MIC 1 mg/mL) (Domadia et al., 2007; Li et al., 2015a). E. coli treated with cinnamaldehyde produces a characteristic enlarged and elongated rod-shaped morphology consistent with the disruption of the divisome and arrest of cell division (Domadia et al., 2007). Furthermore, biochemical analysis focused upon E. coli FtsZ has revealed that cinnamaldehyde inhibited GTPase activity (IC50 5.8 μM) as well as self-polymerization (ED50 6.9 μM) (Domadia et al., 2007). Docking studies have proposed the molecular basis of inhibition is via cinnamaldehyde binding buttressed against the H7 helix, in a binding mechanism analogous to that observed for the benzamides, with the aldehyde group directed into the central cavity of the protein and the benzyl moiety interacting with the T7 loop (Domadia et al., 2007). This structural data suggested that more potent cinnamaldehyde derivatives could be realized through chemical modification of the aldehyde and benzyl moieties. Due to its promising bioactivity toward Gram-negative pathogens, we commenced chemical optimization of cinnamaldehyde with the objective of developing more potent analogs with broad spectrum antimicrobial activity (Li et al., 2015a,b). Indeed, new compounds with superior antibacterial efficacy compared to cinnamaldehyde were produced by appending a methylbenzimidazolyl moiety onto the aldehyde (see compound 1, Figure 1) with modest antimicrobial activity against both E. coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853 (MIC 128 μg/mL). A library of new analogs was subsequently generated by substitution of the aromatic ring with halogen or di-methoxy moieties. The most potent exemplars from the chemical optimization studies are shown in Figure 1 (see compounds 2 to 6) (Li et al., 2015a,b).
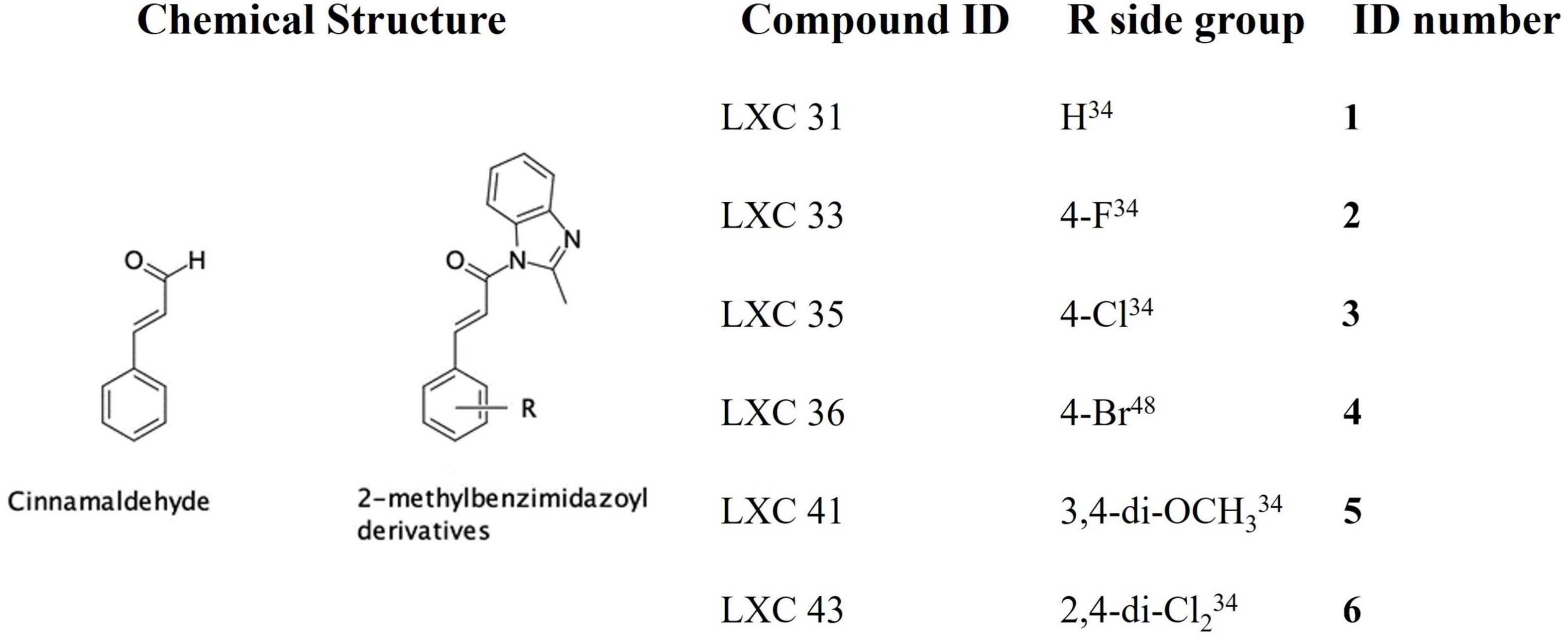
Figure 1. Chemical structures of cinnamaldehyde and analogs.
In this study, we sought to further investigate the biological properties of promising candidates 1 to 6 (the LXC series, Figure 1) against a broad panel of Gram-positive and Gram-negative bacteria, including the ESKAPE pathogens (Rice, 2008). Detailed microbiological and biochemical studies were performed to understand the effect of these compounds on the clinically important and critical priority intrinsically resistant pathogen, A. baumannii (World Health Organization, 2017). These studies demonstrated the efficacy of the compounds in targeting growth inhibition and cell division arrest, and confirmed in vitro activity was indeed through the inhibition of FtsZ from A. baumannii. This is the first study to report the cloning of A. baumannii FtsZ and development of assays required for biochemical characterization of FtsZ inhibitors against this important pathogen. Off-target effects were addressed including activity against mammalian tubulin and potential cytotoxicity using mammalian cell lines and the nematode Caenorhabditis elegans.
All chemicals were obtained from Sigma or Chem-Supply unless otherwise indicated.
Derivatives of cinnamaldehyde 1 to 6 were synthesized, chemically characterized, and assessed for purity, as described previously (Li et al., 2015a,b). Compounds were initially dissolved in DMSO. For the biological assays, a final DMSO concentration of 2.5% (v/v) was used unless otherwise specified.
Reference bacterial strains were obtained from the American Type Culture Collection (ATCC). Vancomycin resistant Enterococcus faecium (VRE 734, wastewater isolate from the Venter Laboratory Collection) was isolated on Bile Esculin agar (Oxoid, Australia) containing 6 μg/mL vancomycin and identified using MALDI-TOF mass spectrometry (Bruker, Preston, Victoria, Australia, Australian Center for Antimicrobial Resistance Ecology). The Pseudomonas aeruginosa WT PAO1 and Escherichia coli with deletions of both acrA and acrB genes encoding drug efflux pumps were obtained from the Venter laboratory stock (Welch et al., 2010). The protocol for microbial growth was adopted from the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (European Committee for Antimicrobial Susceptibility Testing, 2016).
Non-fastidious bacteria and clinical isolates were maintained in Mueller Hinton (MH) broth (Acumedia, United Kingdom) supplemented with 15% (v/v) glycerol (Chem-Supply, Australia), cultured on Mueller Hinton (MH) agar (Acumedia, United Kingdom) plated and incubated at 37°C. Fastidious bacteria were maintained in Brain Heart Infusion (BHI) broth (Acumedia, United Kingdom) supplemented with 15% (v/v) glycerol at −80°C. The fastidious bacteria were cultured on MH agar plates supplemented with 5% (v/v) lysed horse blood and 3% (v/v) fetal bovine serum and incubated at 30°C in the presence of 5% CO2. Drug susceptibility assays were carried out with the non-fastidious cultures grown in cation-adjusted Mueller Hinton (CaMH) broth (Becton Dickinson, France). Streptococcus pyogenes was grown in CaMH broth supplemented with 5% (v/v) lysed horse blood and 3% (v/v) fetal bovine serum whereas Enterococcus faecium was grown in BHI broth.
The minimum inhibitory concentrations (MICs) for all six compounds were determined using the microbroth dilution protocol as per EUCAST with an inoculum of approximately 5 × 106 CFU/mL in an exponential growth phase (European Committee for Antimicrobial Susceptibility Testing, 2016). Bacterial suspension, 100 μL/well, was added to 100 μL/well of drug dilutions, then incubated for 18 h at 37°C. Fastidious bacterial suspensions were instead incubated at 30°C in the presence of 5% CO2. Bacterial growth was assessed by measuring the change in A600 nm between time 0 and 18 h (Cytation5® Cell Imaging Multi-Mode Reader, Bio-Tek®). Levofloxacin was used as the positive control. Assays were completed in duplicate in at least three independent experiments.
To determine if expression of efflux pumps would play a role in the efflux of the cinnamaldehyde derivatives leading to poor antimicrobial activity, interactions between PAβN and the LXC compounds were assessed by checkerboard titration assay. The checkerboard titration assay was performed as previously described (Chai et al., 2020). Briefly, PAβN was added in the first row and serially diluted in CaMH broth along the ordinate of the microwell plate while the LXC compounds were serially diluted along the abscissa. Bacterial suspension for A. baumannii ATCC 19606 or AB UW 5075 100 μL/well were added as described for the MIC assay. Bacterial growth was assessed at A600 nm between time 0 and 18 h (Cytation5® Cell Imaging Multi-Mode Reader, Bio-Tek®).
To determine if the LXC compounds would have bactericidal or bacteriostatic effects, the growth inhibition kinetics were analyzed using the assay as previously described (Chai et al., 2020) with the following modifications. LXC compounds were tested at 1×, 2×, or 4× MIC against A. baumannii ATCC 19606. The bacterial suspension of approximately 1.5 × 108 CFU/mL was prepared in a 1.5 mL Eppendorf® tube. An aliquot was then serially diluted into a microtiter plate before being spot plated onto a fresh MH agar plate. The tube was incubated, and the process was repeated at 1, 3, 6, and 18 h post-exposure to the compounds. All MH agar plates were incubated at 37°C for 18 h. Colonies were counted to calculate the CFU/mL. Dilutions were prepared to ensure the count was kept between 2 and 20 colonies per spot. Levofloxacin was used as a control at sub-MIC (0.25 μg/mL) and 4 × MIC (2 μg/mL) concentrations to produce bacteriostatic and bactericidal effects, respectively. The results from each drug concentration were obtained from at least three independent experiments with different batches of cells.
Morphological changes of bacteria when exposed to the compounds were assessed microscopically using the assay as previously described (Chai et al., 2020). Eppendorf® tubes were set up with A. baumannii ATCC 19606 as described for the time-kill assays. At time 0, 1, 3, 6, and 18 h, an aliquot of the bacterial suspension (1 μL) was transferred onto a glass slide and viewed under a light microscope (Olympus CX33®) at 100 × magnification. Representative images were captured using JENOPTIK GRYPHAX® imaging.
The gene encoding AbFtsZ was obtained via PCR amplification from genomic DNA of A. baumannii ATCC 19606 using forward 5′-AGGGTTTCATATGTTAGAATT TGAACAAGGATTTAATC-3′ and reverse 5′-AGGGTTTCTC GAGACGTCTTGGTTCTTCTTGAACG-3′ oligonucleotides containing NdeI and XhoI sites respectively (underlined). The amplicon was then ligated into NdeI and XhoI sites treated pET-41a(+) (Novagen) vector. The resulting 8-His tagged FtsZ construct was confirmed by Sanger sequencing. This plasmid was transformed into E. coli DH5α™ silver (Bioline) for amplification then subsequently into E. coli BL21(DE3) for recombinant protein production.
Bacterial culture was initially grown to logarithmic phase in prewarmed Luria Bertani (Becton Dickinson, France) broth at 37°C for 5 h before cooling to 25°C. AbFtsZ expression was subsequently induced by the addition of 1 mM isopropyl-β-D-1-thiogalactopyronoside (IPTG; Thermo-Fisher, Australia) for 18 h at 25°C. The cells were harvested by centrifugation at 5,000 g for 20 min and then resuspended in Buffer A (50 mM Tris HCl pH 8.0, 200 mM NaCl and 10% (w/v) glycerol) supplemented with cOmplete™, EDTA-free Protease Inhibitor Cocktail (Roche) and 20 μg/mL DNAse (Sigma, Australia) before lysis using a cell disruptor (Constant Systems E1061, Thermo Scientific, Australia) at 30 kPsi. The lysate was clarified by ultra-centrifugation (Optima XPN-100 Ultracentrifuge, Beckman Colter, Australia) at 200,000 g for 45 min. AbFtsZ was then purified from the supernatant using 2 × 1 mL Histrap HP columns (GE Healthcare) on an Äkta FPLC (GE Healthcare). Lysate was loaded onto the column pre-equilibrated in Buffer A, washed for 10 column volumes with Buffer A containing 20 mM imidazole then eluted with Buffer A at 150 mM imidazole. Fractions containing the desired protein were identified by SDS-PAGE (4–12% NuPAGE Bis-Tris polyacrylamide gels (Invitrogen, Australia). Those fractions containing pure AbFtsZ were pooled and exchanged into Buffer A using a HiTrap desalting column (GE Healthcare) to remove imidazole. Aliquots of the purified FtsZ were snap frozen in liquid nitrogen and stored at −80°C.
The protein concentration was measured using the BioRad™ BCA Protein Assay Standard Kit, according to the manufacturer’s instructions with bovine serum albumin used as a standard. The A750 nm was measured using a Cytation5® Cell Imaging Multi-Mode Reader (Bio-Tek®).
Cinnamaldehyde analogs that inhibited the FtsZ-catalyzed hydrolysis of GTP were assessed using a malachite green-phosphomolybdate colorimetric assay previously described (Chai et al., 2020) with the following modification. The reactions were carried out at 37°C using 0.1 mg/mL of purified AbFtsZ which was mixed with the compounds at a defined concentration in GTP reaction buffer (50 mM Tris HCl pH 7.2, 300 mM KCl and 5 mM MgCl2).
Effects on inhibition of the cinnamaldehyde derivatives on AbFtsZ polymerization were analyzed using the assay as previously described (Chai et al., 2020) with the following modifications. A reaction mix was prepared in a cold Eppendorf® tube where AbFtsZ (0.15 mg/mL) was mixed with the test compound at the desired concentration and a polymerization buffer diluted from a double strength buffer to give a final concentration of 25 mM PIPES pH 6.8, 50 mM KCl and 10 mM MgCl2. The reaction mix was prepared such that the DMSO concentration remained constant at 2% (v/v). The mix was then added into a quartz cuvette (PerkinElmer®, United Kingdom), and placed in a PerkinElmer® LS 55 fluorometer under the fixed condition of excitation/emission at 350/350 nm, slit width < 2 nm, a 1 s read interval. The temperature was calibrated to remain constant at 8°C. The fluorescence was followed for 300 sec to allow equilibration, then polymerization was initiated with the addition of 1 mM GTP. A separate negative control was prepared where 1 mM GDP was added instead of the GTP. The reaction was followed for 600 s.
The chemical structures of compounds 1 to 6 were generated using PICTO (version 4.5.1.0, OpenEye Scientific Software) then saved as SMILES strings before conversion into 3-dimensional conformers (maximum of 200 conformers per compound) via Omega (version 4.1.0.0, OpenEye Scientific Software) using default settings (Hawkins et al., 2010). Four Staphylococcus aureus FtsZ (SaFtsZ) co-complex crystal structures with bound benzamide inhibitors (PDB ID: 3VOB, 5XDT, 5XDU and 6KVP) (Matsui et al., 2012; Ferrer-González et al., 2017; Fujita et al., 2017) served as receptors. The protein receptor structures were prepared using Make Receptor (version 3.0.1, OpenEye Scientific Software), with the bona fide benzamide binding pocket defined as the target binding site. In silico docking of the multi-conformer database into each receptor was performed using the FRED module in OEDocking (version 3.2.0.2, OpenEye Scientific Software) with default settings (McGann, 2011, 2012; Kelley et al., 2015). The docking results were ranked by the Chemgauss 4 scoring function, with the best scoring conformer of each compound retained. An additional 9 poses were subsequently investigated for each molecule by using the FRED flag num_poses. Binding poses were visualized using VIDA (version 4.4.0.4, OpenEye Scientific Software).
The effects of the cinnamaldehyde analogs were analyzed for off-target effects on mammalian tubulin using the protocol as previously described (Chai et al., 2020), using a Tubulin Polymerization Assay Kit (Cytoskeleton Inc., BK011P, Denver, CO, United States), following the protocol described by the manufacturer.
LXC compounds were tested at 1 × MIC with the final concentration of 2.5% (v/v) DMSO maintained in the assay. Controls included 20 μM paclitaxel (promoter of polymerization), 20 μM vinblastine (inhibitor of polymerization) and 2.5% (v/v) DMSO (solvent control). The reaction was initiated by the addition of 1 mM GTP at 37°C. The fluorescence was read for 2,000 s using PerkinElmer Enspire® plate reader.
The effects of the cinnamaldehyde analogs were analyzed for off-target effects on mammalian cells using the protocol as previously described (Chai et al., 2020) with the following modifications. In vitro cytotoxicity was assessed in HepG2 (ATCC HB-8065) cells using the RealTime-Glo™ MT Cell Viability Assay Kit (Promega) as described previously (Wang et al., 2017). The LXC compounds were tested at 1 × and 2 × MIC using a final DMSO concentration of 2% (v/v) in the assay. Two controls, 2% (v/v) DMSO and 50 μg/mL ampicillin were used. The luminescence signal was read at 5-min intervals over 24 h in a Cytation5® Cell Imaging Multi-Mode Reader (Bio-Tek®), at 37°C in the presence of 5% CO2.
The hemolysis assay was performed using fresh human red blood cells (RBCs). PBS solution (137 mM NaCl, 2.7 mM KCl, 1.46 mM KH2PO4, 8.1 mM NaH2PO4 pH 7.4) was used to wash the RBCs three times at 500 g for 5 min, then resuspended in 1% (w/v) PBS solution. Compounds (8 μL) were added to a 96-microwell plate, which were serially diluted from 4 × MIC in 2% (v/v) DMSO. A 1% (v/v) Triton X-100, 2% (v/v) DMSO and 128 μg/mL ampicillin were used as controls. These were performed in quadruplicate. Thereafter, 192 μL RBCs were added into all wells and the plates were incubated at 37°C under constant shaking (100 rpm) for 1 h. RBCs were precipitated by centrifugation of the plates (1,000× g for 3 min). An aliquot (100 μL) of each supernatant was transferred into a new 96-microwell plate and the A450 nm was determined using a PerkinElmer Enspire® plate reader. The fraction of intact RBC for each sample was determined as fraction of the intact RBCs for the sample without the addition of compounds (set at 100%) and plotted as a function of compound concentration.
Protocols for growing, harvesting, and synchronizing C. elegans have been previously described (Porta-de-la-Riva et al., 2012; Chai et al., 2020) and were performed with the following modifications. The nematodes were treated with LXC compounds 1 to 6 at 1 × and 2 × MIC with at least 3 replicates using a final DMSO concentration of 2% (v/v). The plate was incubated at 25°C and the number of live vs dead nematodes was enumerated every 24 h for 72 h. Separate wells containing 50 μg/mL ampicillin and 2% (v/v) DMSO were used as controls. The percentage of living nematodes at each time point across 72 h was calculated to give the percentage of survival.
Compounds 1 to 6 were examined for antimicrobial activity against an extended panel of Gram-positive and Gram-negative bacteria that included isolates of the clinically important ESKAPE pathogens and drug-resistant strains. Critically for this study, A. baumannii ATCC 19606 was the only Gram-negative microorganism broadly susceptible to all of the LXC compounds with the MIC values ranging from 32 to 256 μg/mL (Table 1). Noteworthy were the brominated analog 4 (MIC 32 μg/mL) that had similar potency to gentamicin, and the di-chlorinated analog 6 (MIC 64 μg/mL). All compounds were similarly assayed against the extensively drug resistant (XDR) A. baumannii UW 5075. However, none of the LXC compounds displayed any antibacterial activity against this XDR strain (Table 1). Similarly, no antibacterial activity was observed against other Gram-negative bacteria in the panel, including Klebsiella pneumoniae ATCC 4352, ATCC 13883 and ATCC 33495 and Pseudomonas aeruginosa PAO1 and ATCC 27853. Weak antimicrobial activity was observed against Streptococcus pneumoniae ATCC 6303 (MIC 256 μg/mL for all compounds), but MRSA ATCC 43300 was only susceptible to 6 (MIC 64 μg/mL) (Supplementary Table 1). Due to the urgent and unmet need to develop new antimicrobial agents that target A. baumannii (World Health Organization, 2017), subsequent studies were focused upon this important critical priority Gram-negative bacterium.
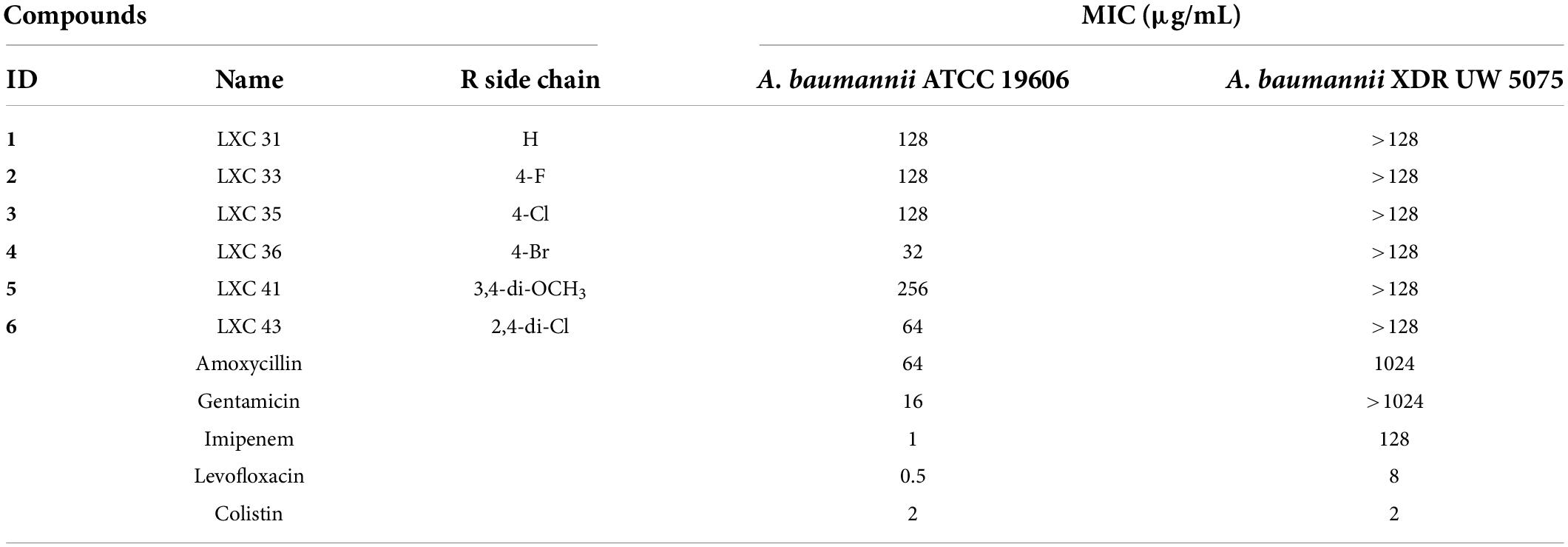
Table 1. Minimum Inhibitory Concentration (MIC) of the LXC compounds and common antimicrobial agents on two strains of Acinetobacter baumannii.
Time-kill studies with A. baumannii ATCC 19606 were conducted to determine if Compounds 1 to 6 were bactericidal or bacteriostatic in their mechanism of action. In all cases, the cell numbers remained constant across 18 h suggesting the compounds are bacteriostatic in their mechanism of action (Supplementary Figure 1). Previous studies have suggested that cinnamaldehyde is bacteriostatic on E. coli (Domadia et al., 2007) however there are no other studies examining the effects of cinnamaldehyde or its derivatives on A. baumannii.
Although A. baumannii is intrinsically more resistant to multiple antibiotics, and much more resistant than other pathogens such as E. coli, we observed higher efficacy for the LXC compounds against A. baumannii compared to E. coli and P. aeruginosa. We have previously observed a similar phenomenon where robenidine only displayed antimicrobial activity against A. baumannii, but not other Gram-negative pathogens (Khazandi et al., 2019). The outer membrane of Gram-negative pathogens acts as a permeability barrier that prevents xenobiotic compounds from penetrating into the bacterial cell (Arzanlou et al., 2017). The outer membrane of A. baumannii is about 100 times more impermeable than that of E. coli as A. baumannii lacks general, non-specific trimeric porins found in E. coli (Nikaido, 2003; Zgurskaya et al., 2015). However, due to discreet differences in the lipid A component, A. baumannii tends to be more permeable to amphiphilic molecules compared to E. coli (Krishnamoorthy et al., 2017).
Poor permeation across cell membranes and active drug efflux of antimicrobial agents are well-known intrinsic resistance mechanisms employed by bacteria (Miki and Hardt, 2013; Venter et al., 2015; Arzanlou et al., 2017; Knight et al., 2018). Hence, we investigated if the absence of whole cell activity noted above for 1 to 6 against the A. baumannii XDR strain could be explained by either of these factors. Antimicrobial susceptibility assays were repeated following permeabilization of the bacterial outer membrane with either EDTA or colistin (Alakomi et al., 2006; Farrag et al., 2019; Khazandi et al., 2019). Neither treatment changed the MIC values (data not shown) indicating that the outer membrane barrier was not responsible for limiting the efficacy of the LXC compounds. Next, the antimicrobial sensitivity assays were performed in the presence of the well-characterized drug efflux pump inhibitor, PAβN (Sharma et al., 2019). The potency of all six compounds against A. baumannii ATCC 19606 was improved when assayed in combination with the efflux pump inhibitor PAβN, with the MIC decreasing to as low as 8 μg/mL for compounds 3 to 6 (Table 2). Importantly, the presence of PAβN bestowed significantly improved antimicrobial activity to the LXC compounds against the XDR strain UW 5075 (Table 2). Noteworthy were compounds 5 and 6 that were inactive alone but displayed an MIC of 32 μg/mL when assayed in combination with PAβN. The improved antimicrobial activities were observed at concentrations of PAβN that did not damage the outer membrane (Supplementary Figure 2) implying that efflux was, indeed, a major factor responsible for impaired activity of the LXC compounds.
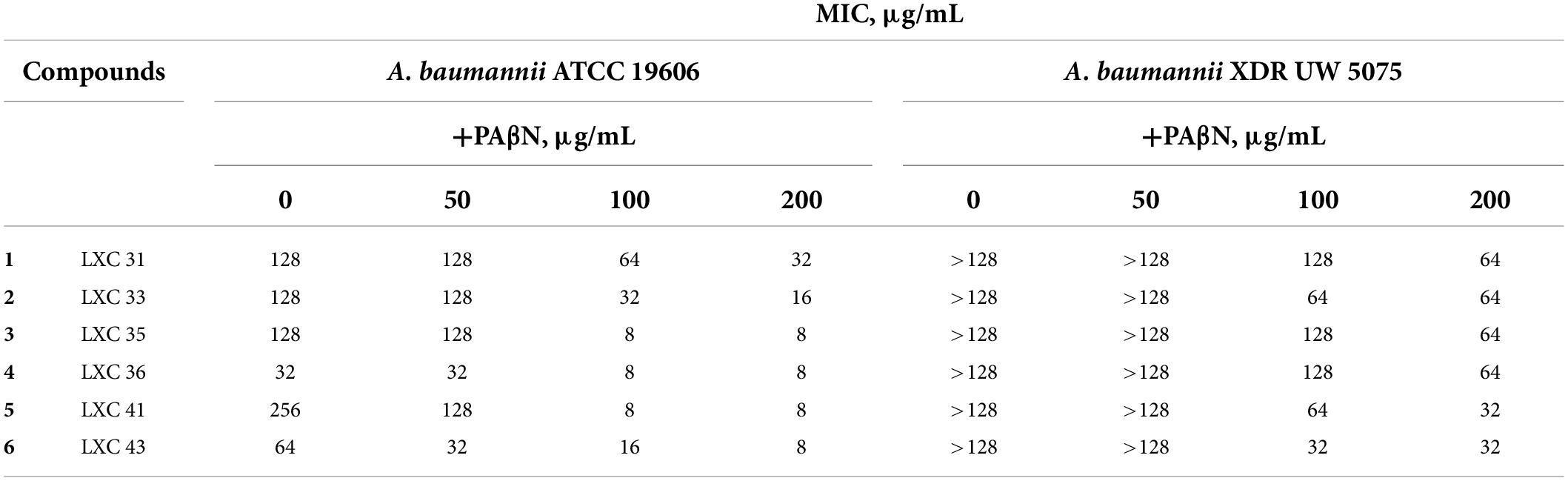
Table 2. MIC of the LXC compounds against A. baumannii ATCC 19606 and XDR UW 5075 in the absence or presence of PAβN.
To confirm that the cinnamaldehyde derivatives disrupted cellular division, microscopy analysis was performed on A. baumannii ATCC 19606 treated with the LXC compounds (Figure 2). The morphology of the bacterium was observed using light microscopy at varying time points. The bacterium was also treated with the non-specific cell division inhibitor divin, which yielded the elongated cell shape characteristic of a rod-shaped bacterium unable to divide (Figures 2A,B). When treated with the LXC compounds at 2 × MIC, all six analogs induced the expected elongated phenotype for A. baumannii within 6 h (Figures 2C–H). Interestingly, this same morphological change was readily visible within 3 h post-treatment for halogenated compounds 2, 3, 4, and 6 but not the unsubstituted compound 1, suggesting that modification of the benzyl ring of cinnamaldehyde was important for increased whole cell activity (Supplementary Figure 3). An expanded version of the microscopy data with all the timepoints and concentrations tested is provided in Supplementary Figure 3. Interestingly, “grooving” (shown with white arrow in Figure 2G) between cells was observed. This is consistent with daughter cells that have undergone division but are unable to separate. Grooving has previously been reported in other rod-shaped bacteria including B. subtilis, E. coli and P. aeruginosa, when cellular division was arrested (Anderson et al., 2004; de Boer, 2010; Annapurna et al., 2015; Pichoff et al., 2018; Araya et al., 2019; Sharavanan et al., 2019). Complementary time-kill studies were also performed upon A. baumannii ATCC 19606 (Supplementary Figure 1). The number of viable cells remained constant throughout the 18 h time-course, indicative of a bacteriostatic mechanism of action. The absence of bactericidal activity was consistent with the lack of cell lysis noted in the microscopy analysis above. Together, these data are consistent with the mechanism of antibacterial action being through the inhibition of cell division.
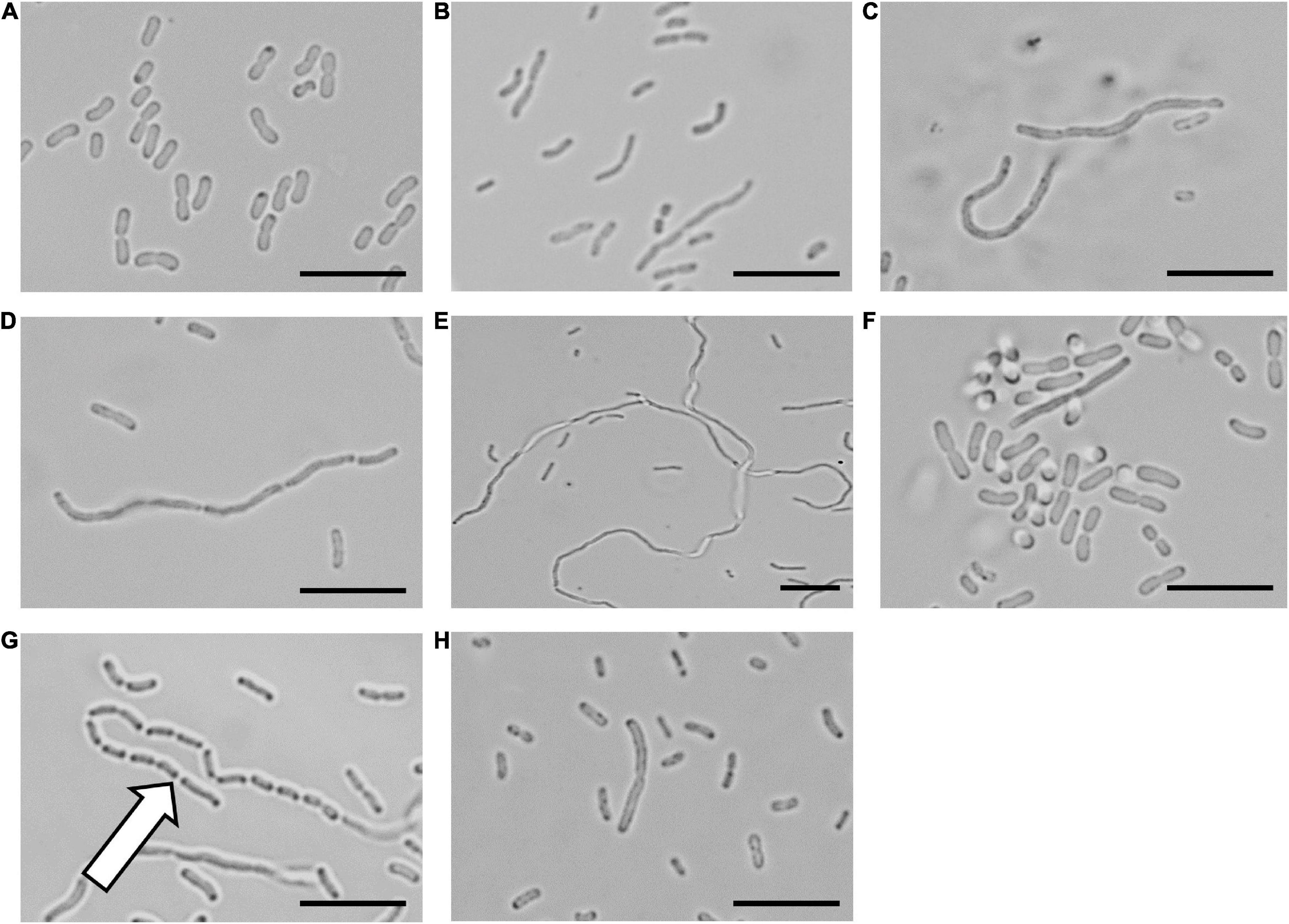
Figure 2. The LXC compounds inhibit cell division in Acinetobacter baumannii ATCC 19606 A. baumannii ATCC 19606 culture was adjusted to a density of approximately 2.7 × 108 CFU/mL in MH broth and incubated at 37°C with the LXC compounds. A 1 μL aliquot was taken out at varying timepoints (0, 1, 3, 6, and 18 h) and imaged by light microscopy. The images for 6 h are depicted here. Images are (A) in the absence of the compounds, (B) with the divisome inhibitor, divin at 64 μg/mL, or with the LXC compounds at 2 × MIC values: (C) compound 1 at 256 μg/mL, (D) compound 2 at 256 μg/mL, (E) compound 3 at 256 μg/mL, (F) compound 4 at 64 μg/mL, (G) compound 5 at 512 μg/mL and (H) compound 6 at 128 μg/mL. The white arrow indicates “grooving” site, an indication of where separation of daughter cells should take place. Scale bar is 50 μm.
Previously, we have shown that cinnamaldehyde analogs inhibit the cellular division of E. coli and P. aeruginosa, creating an elongated phenotype (Li et al., 2015a,b). Here, we observed the same morphological change in A. baumannii. Inhibition of A. baumannii creating this effect is also a feature of the FtsZ targeting compounds. Although this assay is often used as a screening tool to identify FtsZ inhibitors, the on-target effects of these LXC compounds with the FtsZ protein from A. baumannii (AbFtsZ) have not been reported. Therefore, efficacy of these LXC compounds on purified AbFtsZ in vitro had to be verified.
To confirm that the mechanism of action of the LXC compounds was indeed through inhibition of FtsZ, further biochemical studies were performed. The ftsZ gene was obtained by PCR using genomic DNA isolated from A. baumannii ATCC 19606 and ligated in the pET-41a(+) vector for over-expression in E. coli. An 8-His tag was fused onto the C-terminus to facilitate purification by immobilized metal ion chromatography (Figure 3). Conditions for culturing and expression of AbFtsZ in E. coli BL21 (DE3) were optimized for high level production of FtsZ that could be purified to homogeneity in a single chromatography step, with an approximate yield of 20 mg of pure AbFtsZ per liter culture. This protein was subsequently used for in vitro biochemical assays to characterize the cinnamaldehyde analogs.
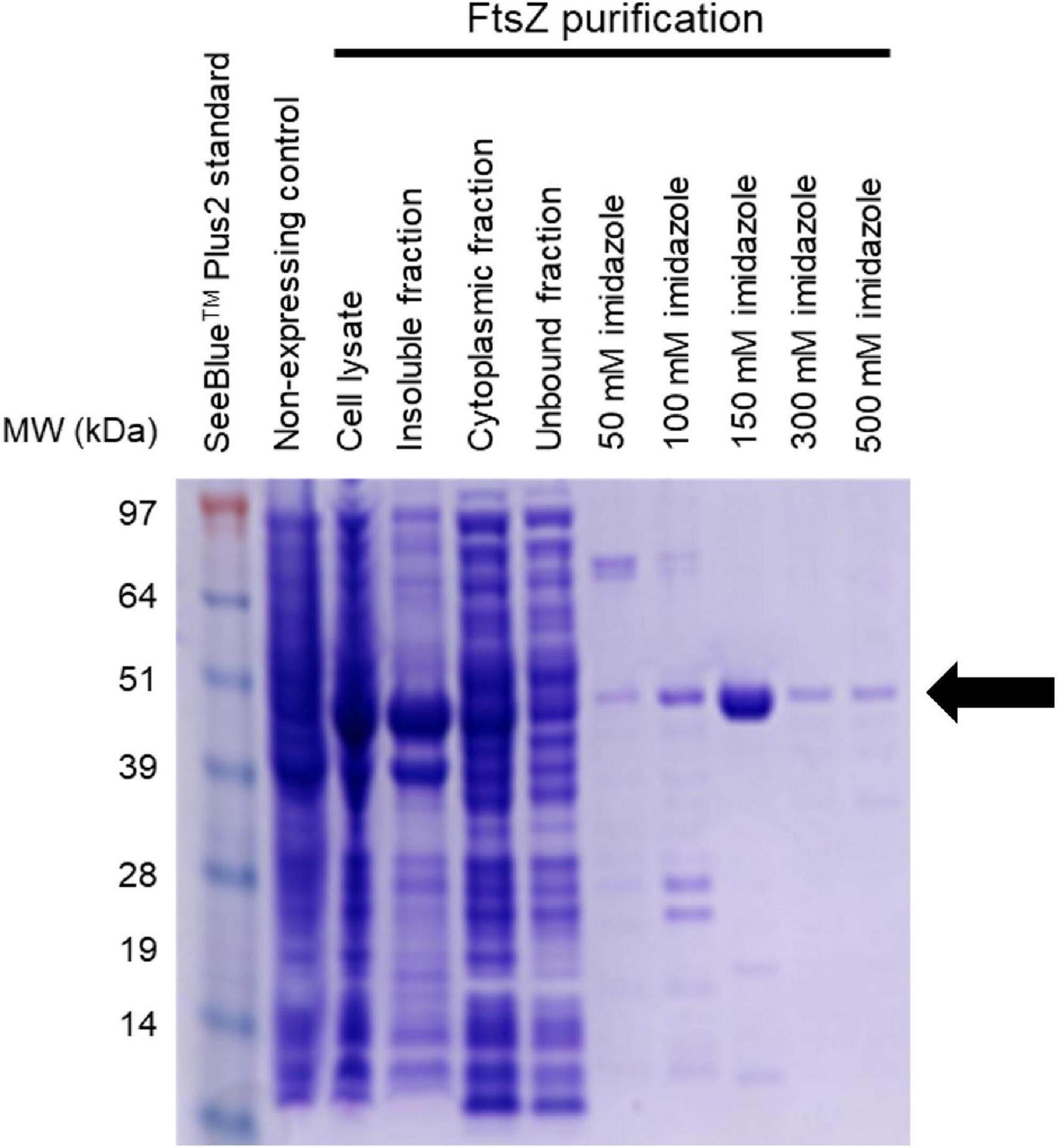
Figure 3. Purification of AbFtsZ AbFtsZ was cloned in the pET-41a(+) vector and expressed in BL21(DE3) E. coli via induction with 1 mM IPTG. The protein was purified from the cytoplasmic fraction using Nickel-affinity column chromatography. The different fractions from the purification process were loaded onto SDS-PAGE (4–12%). Protein was visualized by staining with Coomassie® Brilliant Blue R-250. Purified AbFtsZ (43.4 kDa) is observed as indicated by the arrow. The protein concentration was determined using the standard Bio-Rad™ BCA Protein Assay Kit.
Purified AbFtsZ protein was used to confirm that the LXC compounds exerted their bioactivity directly upon this target. As the hydrolysis of GTP accompanies FtsZ polymerization, the enzymatic activity of AbFtsZ was measured by quantifying the liberation of inorganic phosphate. All six of the LXC compounds inhibit GTPase activity at the highest concentration (128 μg/mL, Figure 4) with IC50 values between 52 and 85 μg/mL. Interestingly, the IC50 of these LXC compounds does not correlate with their respective MIC values (Figure 4). The differences in the biochemical and whole cell activities are consistent with our earlier observation about the role of efflux pumps and the inability of the compounds to obtain suitable cytotoxic concentrations inside the bacteria necessary to inhibit the FtsZ target.

Figure 4. The LXC compounds inhibit GTPase activity in a dose-dependent manner. (A–F) The GTPase activity of purified AbFtsZ was determined from the liberation of Pi from GTP using malachite green. AbFtsZ was incubated with the LXC compounds for 30 min before the reaction was initiated by addition of 1 mM GTP. The reaction was terminated by the addition of citric acid after 10 min. Pi release rate was normalized to the sample without the addition of LXC compounds (86.26 μM Pi/mg AbFtsZ/min) that was taken as 100%. The results are presented in mean ± SEM. Statistical analysis was performed using one-way ANOVA (statistically significance, p < 0.05). A non-linear regression model was used in GraphPad Prism 8.3 to determine the IC50.
Having established that the LXC compounds inhibited GTPase activity, the effect of these compounds on polymerization of AbFtsZ was directly measured using a 90° light scattering assay (Figure 5). This was performed using a fluorescence spectrometer, thermostatically controlled at 8°C, to measure the increased absorbance of scattered light at A350 nm that correlate with changes in protein polymerization. Here, FtsZ was preincubated with either the test compounds at 128 μg/mL (pink curves, Figure 5) or a vehicle control of 2.5% (v/v) DMSO (blue curves, Figure 5). Protein polymerization was then initiated by the addition of GTP. Consistent with earlier reports, polymerization activity was highly specific for GTP as the addition of 1 mM GDP resulted in no change in signal as FtsZ remained within its unpolymerized state (gray curves, Figure 5). All cinnamaldehyde derivatives inhibited protein polymerization to varying extents, with the strongest activity observed with compound 5, followed by compounds 1 and 6 (p < 0.05). We have previously reported inhibition of FtsZ from E. coli (Li et al., 2015a,b) using this light scattering assay. In these previous studies, impaired polymerization was reported for compounds 2, 3 and 6 at 120 μg/mL (Li et al., 2015a). Interestingly, compound 6 displayed a similarly potent inhibition on the polymerization of AbFtsZ.
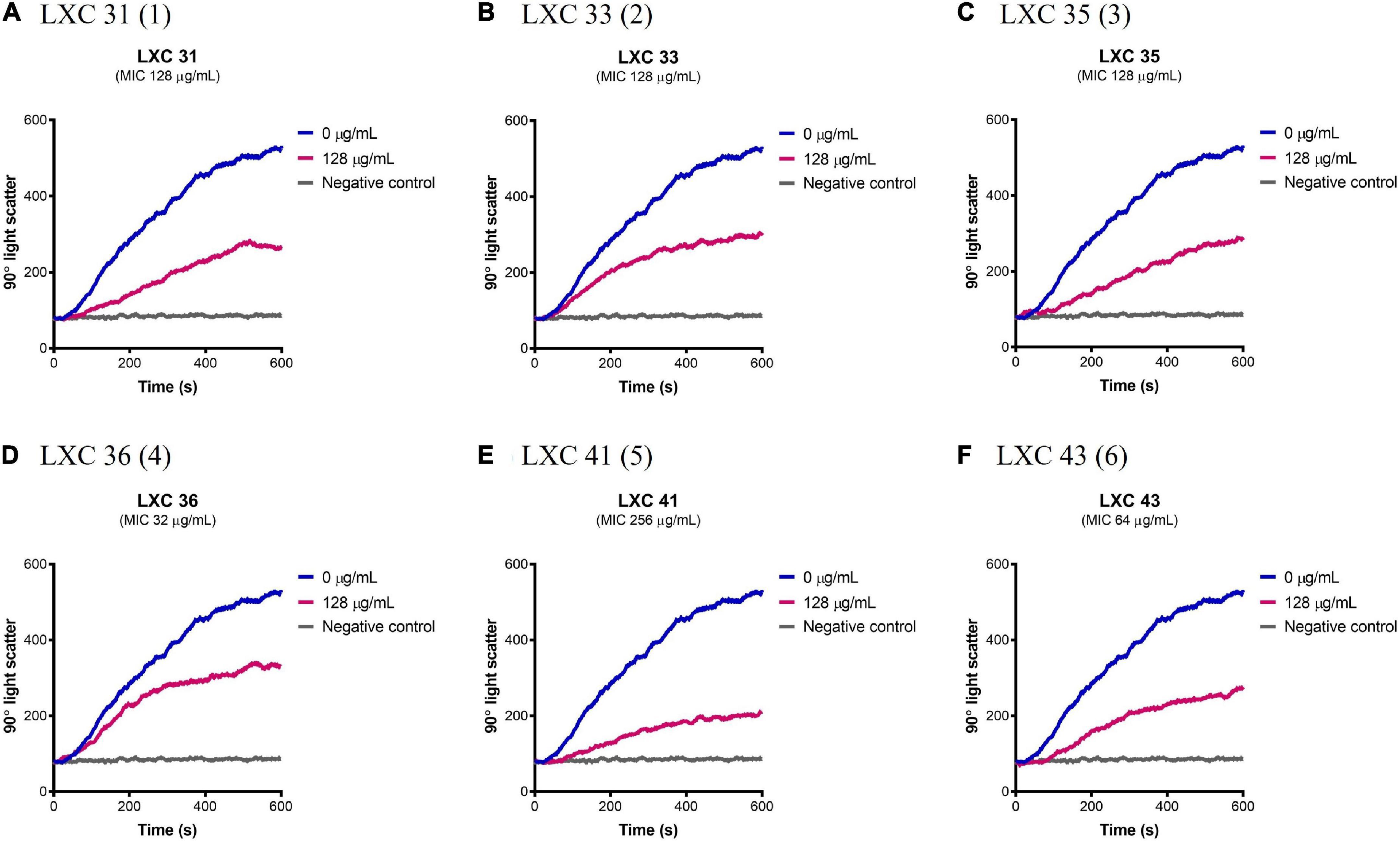
Figure 5. The LXC compounds inhibit AbFtsZ polymerization. (A–F) AbFtsZ (0.15 mg/mL) was pre-incubated in polymerization buffer (25 mM PIPES pH 6.8, 50 mM KCl and 10 mM MgCl2) together with the test compound at 128 μg/mL for 300 s. Polymerization was then initiated with the addition of 1 mM GTP. The fluorescence was measured as a function of time at excitation and emission wavelengths of A350 and A350 nm, respectively, and a slit width of <2 nm. The blue line in each panel indicates the addition of 2.5% (v/v) DMSO, the pink line indicates the test compound tested at 128 μg/mL and the gray line represents the addition of 1 mM GDP (as negative control) instead of GTP.
As bioactive cinnamaldehyde analogs 1 to 6 (Li et al., 2015a,b) targeted A. baumannii specifically through inhibition of AbFtsZ, we were interested to further investigate the potential interactions between the compounds and FtsZ. Docking studies of cinnamaldehyde have previously predicted binding in the interdomain cleft similar to the benzamides (Domadia et al., 2007). Molecular docking studies were performed to see if the cinnamaldehyde analogs were also likely to behave similarly. Crystal structures of FtsZ in complex with di-fluoro-benzamide analogs (Matsui et al., 2012; Ferrer-González et al., 2017; Fujita et al., 2017) were employed for in silico docking studies using the OEDocking module of OpenEye Scientific Software (McGann, 2012). The common inhibitor binding pocket identified in the co-complexes with the benzamides was selected to dock the cinnamaldehydes as the chemical structures of both classes share similar planar, elongated geometries. The docking results with PDB 3VOB (co-crystallized with benzamide PC190723) are shown here as an exemplar of the results obtained. PC190723 was firstly redocked, and the expected X-ray binding orientation was replicated. Subsequent docking analysis of compounds 1 to 6 showed that they were all able to be accommodated in the same inhibitor binding site and adopted binding poses consistent with the binding orientations observed with benzamides (Figure 6). The compounds all buttressed against central helix H7 with the substituted benzyl rings generally orientated toward the center of the protein in a pocket lined by Gln 184, Met 218 and Ile 220. At the other end of the compound, the common imidazolyl moiety was generally oriented toward the solvent exposed T7 loop for the lowest energy pose of each compound. The only exception to this was 6, which was predicted to have an inverted orientation in its lowest energy pose. Analysis of further higher energy poses of 6, showed a mixture of orientations. Compounds 1 to 6 all have an aromatic ring at both ends of their structures, which may give some flexibility in possible orientations within the binding pocket. It was also notable that 6 had the highest degree of shape similarity with PC190723 (data not shown). Analysis of the docking scores revealed that all six test compounds bound with affinities that were comparable with the bona fide benzamide ligands (Supplementary Figure 4 and Supplementary Table 2). Given that the molecular analysis supported the mechanism of action being through the inhibition of FtsZ, the biological properties of 1 to 6 were further investigated.
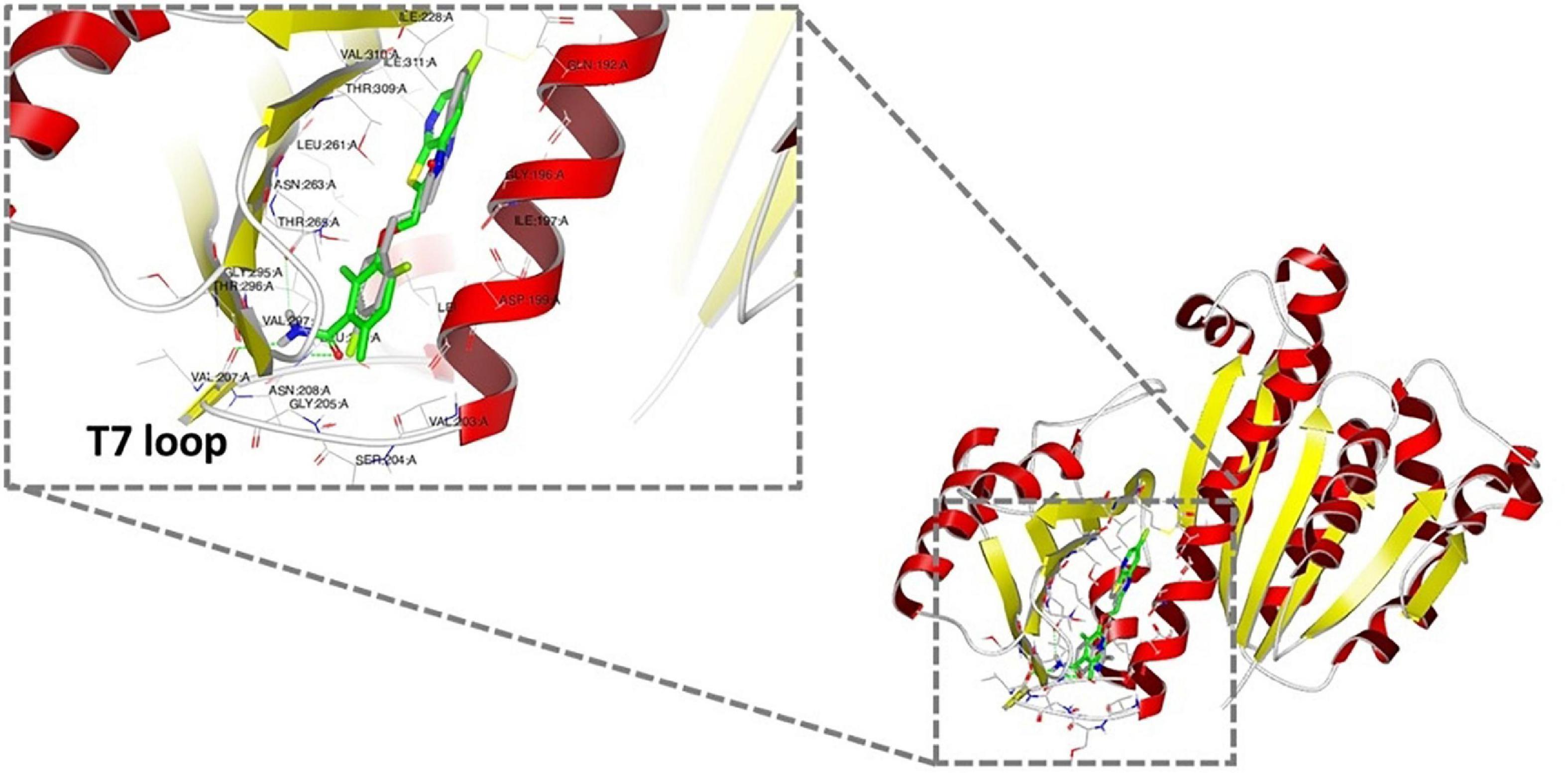
Figure 6. Docking analysis of the predicted binding of cinnamaldehyde analogs to FtsZ In silico docking studies were performed using OpenEye Scientific Software (McGann, 2012). Molecular binding of LXC 31 (6; gray) is shown above alongside co-crystallized FtsZ inhibitor PC190723 (green; PDB 3VOB) (Matsui et al., 2012). Binding poses for compounds 1 to 6 are shown in Supplementary Figure 4. In each case, the lowest energy pose of the lowest energy conformer is depicted.
To investigate the potential of compounds 1 to 6 as preclinical candidates, a series of in vitro and in vivo assays were performed. Here, the aim was to identify those compounds that possess the desirable safety profile in candidates for future drug development. The effect of the compounds on the polymerization of mammalian tubulin was addressed to find those compounds with the necessary selectivity toward the bacterial target. The cytotoxicity of all compounds was then investigated against two mammalian cell lines before assaying for toxicity in vivo in a nematode model. Together, these data provide insight into the preclinical safety profile of our new cinnamaldehyde analogs.
Prokaryotic FtsZ is a structural homolog of eukaryotic tubulin. Consequently, it was essential to assess all preclinical candidates that target bacterial FtsZ for off-target binding to the mammalian tubulin. The effect of all six cinnamaldehyde derivatives on polymerization of human tubulin was analyzed using a commercial tubulin assay kit. The compounds were assayed alongside the known tubulin polymerization stimulator paclitaxel (blue curves, Figure 7) and inhibitor vinblastine (green curves, Figure 7). The compounds were tested at 1 × MIC (pink curves, Figure 7) and compared to a vehicle control of 2.5% (v/v) DMSO (brown curves, Figure 7). The non-substituted analog 1 showed undesirable inhibitory activity, as did mono-halogenated compounds 3 and 4. In contrast, fluorinated 2 and di-substituted analogs 5 and 6 were devoid of tubulin polymerization activity even at concentrations ∼10–20 times higher than that of paclitaxel and vinblastine (Figure 7).
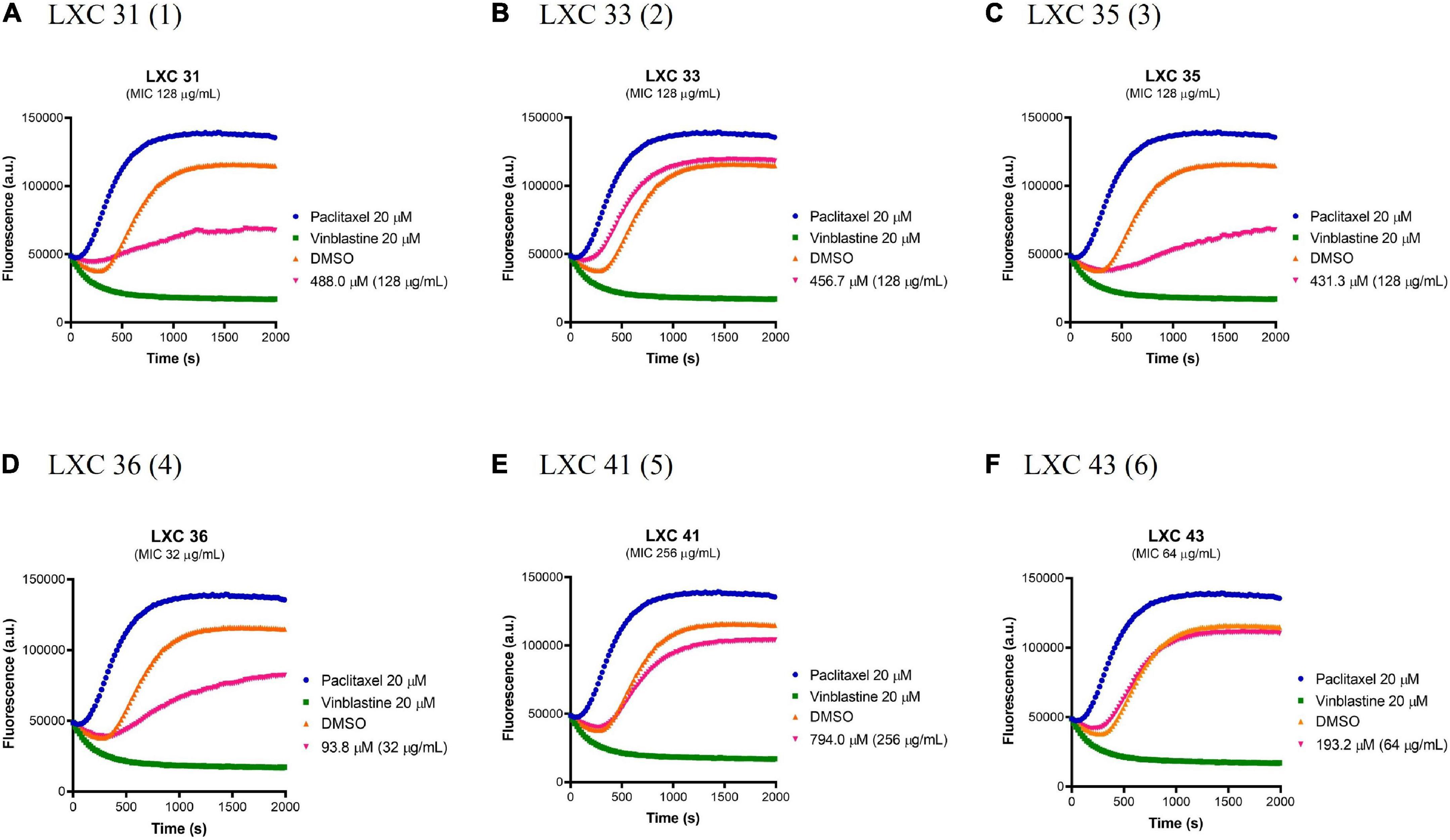
Figure 7. The effect of the LXC compounds on the polymerization of mammalian tubulin. (A–F) The polymerizing activity of the LXC compounds on porcine tubulin was assessed using a commercial Tubulin Polymerization Assay Kit. The LXC compounds at 1 × MIC were pre-incubated with tubulin on ice before the reaction was initiated by the addition of 1 mM GTP. The fluorescence was measured as a function of time at A360 and A420 nm excitation and emission wavelengths, respectively, at the constant temperature of 37°C. DMSO 2.5% (v/v) (brown curves) was used as vehicle control while paclitaxel (polymerization stabilizer; blue curves) and vinblastine (polymerization inhibitor; green curves) at 20 μM each were also included as controls.
The effects of compounds 1 to 6 on the growth and metabolism of HepG2 ATCC HB-8065 mammalian liver cells were investigated using the RealTime-Glo™ MT Cell Viability Assay Kit (Figure 8). This photometric assay measured the ability of metabolically active cells to reduce the NanoLuc substrate to a luminescent product. Cells were treated over 24 h with either 1 × or 2 × MIC concentrations (pink curves and orange curves, respectively, Figure 8) and the resulting growth curves were compared against a vehicle only control of 2.0% (v/v) DMSO (blue curves, Figure 8). At 1 × MIC, the only compound that displayed cytotoxicity was compound 5 (Figure 8E). When the concentration of the compounds was increased to 2 × MIC, cytotoxicity was also observed for 1, 2, 3. Importantly, compounds 4 and 6 were devoid of undesirable toxicity even at 2 × MIC.
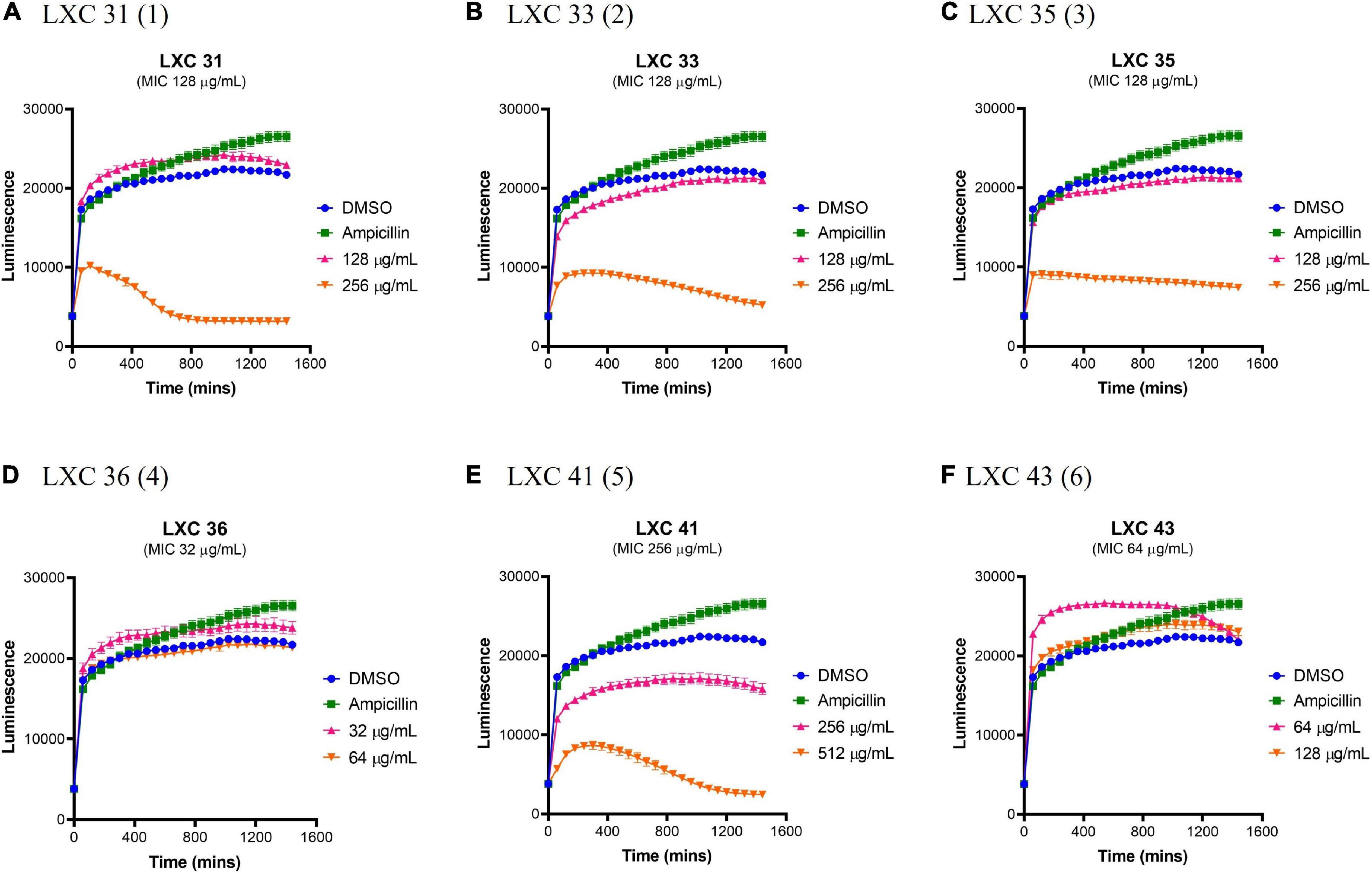
Figure 8. The LXC compounds are generally non-toxic at a concentration of 1 × MIC. (A–F) Real-time liver cell viability measurements for HepG2 after treatment with 1 × (pink line) and 2 × (brown line) MIC values. A 2.0% (v/v) DMSO (vehicle control; blue line) and 50 μg/mL ampicillin (green line) were used as controls. Cell viability was measured every 5 min for 24 h at 37°C and 5% CO2 on a Cytation5® Cell Imaging Multi-Mode Reader using the RealTime-Glo™ MT Cell Viability Assay reagent. The results are presented in mean ± SEM (hourly SEM data was presented).
The lack of cytotoxicity observed in the HepG2 cell viability assay above was also confirmed in a hemolytic assay. Here, fresh human red blood cells were incubated for 1 h with varying concentrations of compounds, after which the release of haem from lysed cells was measured spectroscopically (Figures 9A–F). The non-toxic antibiotic ampicillin served as a non-lytic control (Figure 9G). Treatment of cells with 1% (v/v) Triton X-100 was sufficient to completely disrupt the red blood cells and served as a control for lysis (Figure 9). None of the cinnamaldehyde derivatives showed hemolytic activity at 1 × MIC, except di-methoxy analog 5 which was only non-lytic at a low concentration (0.25 × MIC, Figure 9E). Noteworthy was the observation that cell lysis was observed at 2 × MIC for most compounds except for brominated analog 4 that was devoid of hemolytic activity even at the highest concentration tested (4 × MIC, Figure 9D). Together, the data from the hemolysis and HepG2 cytotoxicity assays help present halogenated analogs 4 and 6 as promising lead compounds.
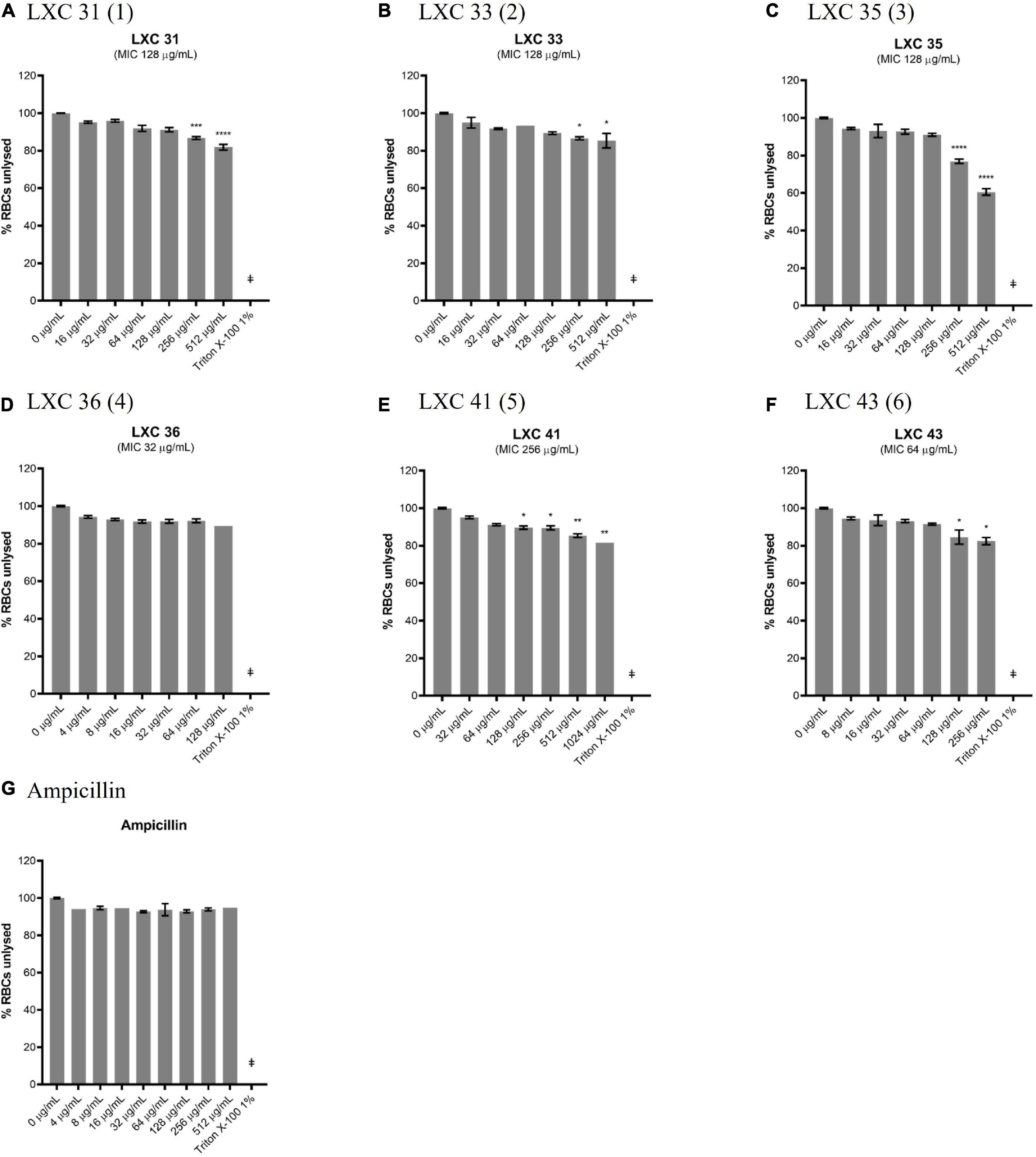
Figure 9. Most of the LXC compounds are not hemolytic up to 128 μg/mL. (A–G) Freshly washed human RBCs in PBS solution (137 mM NaCl, 2.7 mM KCl, 1.46 KH2PO4, 8.1 mM NaH2PO4, pH 7.4) was exposed to 8 μL of LXC compounds with concentrations ranging from 0 μg/mL up to 4× their MIC values in 2.0% (v/v) DMSO. A 1% (v/v) Triton X-100 was used to indicate complete RBC lysis (‡). Ampicillin (0–512 μg/mL) was used as a control or drug that does not cause RBC lysis. The assays were performed in quadruplicates. The plates were incubated at 37°C while constantly shaking at 100 rpm for 1 h. Intact RBCs were removed by centrifugation and the presence of hemolytic products in the supernatant were determined by measuring the absorbance at A450 nm. The results are presented in mean ± SEM. Statistical analysis was performed using one-way ANOVA and the asterisks (*) represent statistical significance (*p < 0.05; **p < 0.005; ***p < 0.0005; ****p < 0.0001).
Potential in vivo toxicity of the LXC compounds was investigated using C. elegans (Figure 10). The nematodes were treated with the LXC compounds at 1 × and 2 × their MIC values (dark and light purple bars, respectively, Figure 10) for up to 3 days. Live nematodes, which are readily distinguished from dead organisms by their physical appearance under the microscope (Chai et al., 2020), were enumerated every 24 h. The survival rates were compared to untreated controls (black bars, Figure 10), a vehicle only control (2% (v/v) DMSO, dark gray bars, Figure 10) and the non-toxic antibiotic ampicillin (light gray bars, Figure 10). Encouragingly, none of the compounds in this series displayed toxicity toward C. elegans, except for di-methoxy analog 5 which showed significant toxicity (p < 0.05) but only after 72 h treatment (Figure 10E). This result agreed with that seen with the cytotoxicity in mammalian liver cells (Figure 8). Compounds 1, 2 and 3 displayed toxicity against mammalian liver cells at 2 × MIC (Figure 8) yet the nematode data suggests these compounds do not have in vivo toxicity in this model at 2 × MICs for up to 72 h (Figure 10).
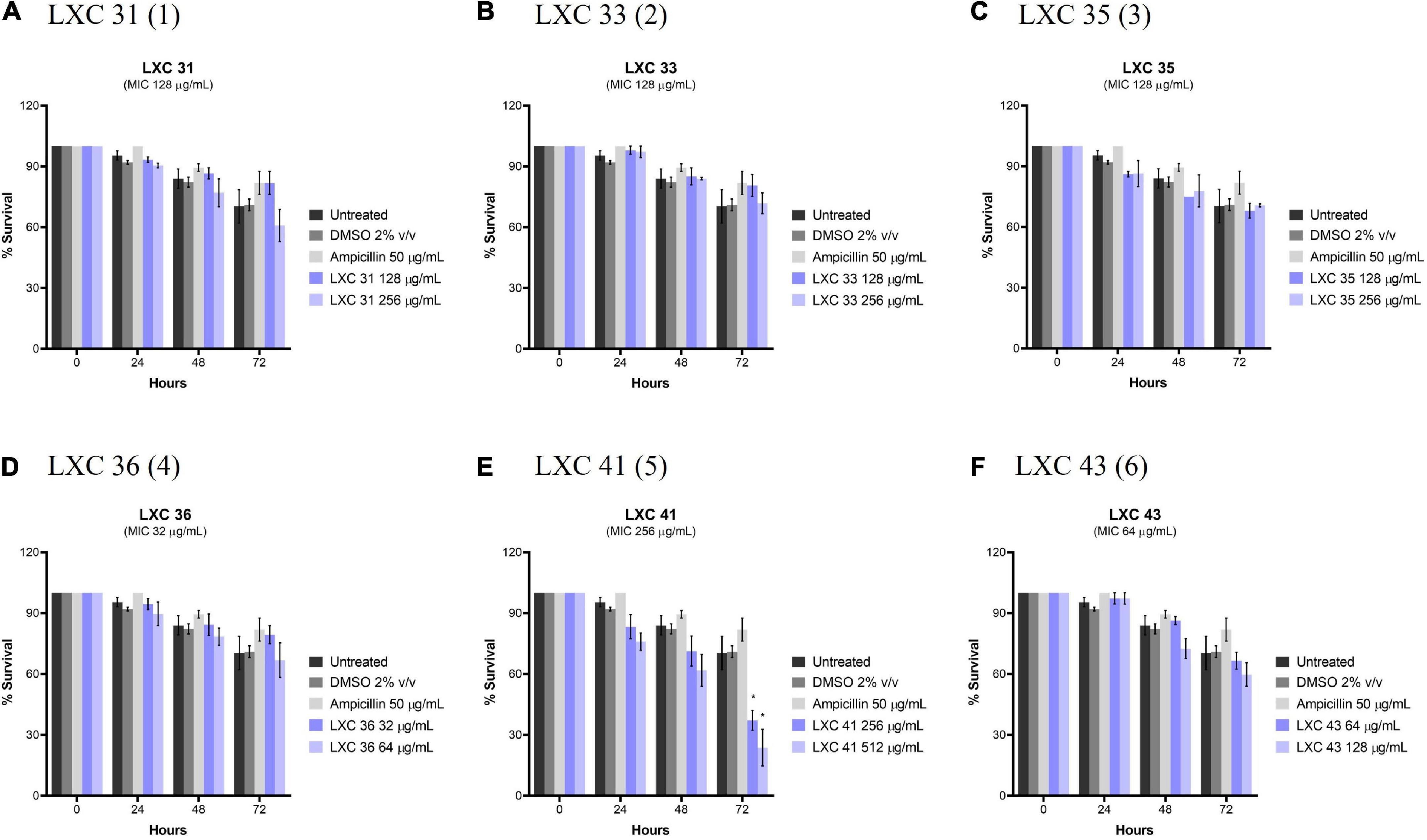
Figure 10. The LXC compounds (apart from compound 5) were not cytotoxic to C. elegans nematodes. (A–F) C. elegans nematodes were cultured on nematode growth media agar plate and harvested in optimized growth media, with E. coli as its primary source of nutrient. Newly harvested adult nematodes were investigated for toxicity in the presence of the LXC compounds at 1× and 2× their MIC values for a timespan of up to 72 h. The nematodes were counted under a light microscope at 400× magnification and the live nematodes at 72 h was indicated as a fraction of the starting number of nematodes (percentage survival). The results are presented in mean percentage of survival ± SEM. Statistical analysis was performed using two-way ANOVA and the asterisks (*) represent statistical significance: LXC 41 (5) at time 72 h (*p < 0.05).
This study explored the antimicrobial potency, on-target activity, and pre-clinical suitability of a panel of 2-methylbenzimidazoyl cinnamaldehyde analogs. In our previous study, cinnamaldehyde derivatives 1 to 6 were identified with antimicrobial activity against methicillin-sensitive S. aureus and modest activity against E. coli (Li et al., 2015a). In the current study, antimicrobial sensitivity screening was expanded using an extended panel of Gram-negative pathogens. Results from this screen revealed the novel observation that 1 to 6 possessed antimicrobial activity against A. baumannii. This finding is most welcome in the fight to combat AMR, especially against this clinically important and difficult to treat pathogen. The antimicrobial activity of the LXC compounds against an XDR strain of A. baumannii is timely and of great significance as A. baumannii is on top of the WHO list of critically important organisms in need of urgent drug discovery (World Health Organization, 2017). The potency of all six compounds was increased when the efflux pump inhibitor, PAβN, was added to the assay. The synergistic activity observed for the combination treatment for the XDR A. baumannii strain that is otherwise only responsive to the last resort antibiotic colistin is of particular significance.
The thorough biological characterization of cinnamaldehyde derivatives 1 to 6 presented here identified the di-chlorinated derivative 6 as a promising pre-clinical candidate for further chemical development. This compound had the requisite activities necessary in a lead compound, including antimicrobial potency, confirmation of on-target activity against FtsZ protein, lack of activity against the tubulin counter-target and good in vitro and in vivo safety profiles. Mono-brominated 4 should also be considered for further study and optimization. Whilst this compound showed undesirable inhibitory activity against tubulin in vitro, it was well-tolerated in both cell-based cytotoxicity assays as well as the nematode model. Interestingly, halogenated derivatives 2, 3, 4, and 6 generally showed superior activity in biological testing relative to the non-substituted parent 1 compound. Together, the findings from this study highlight the potential of halogenated cinnamaldehyde derivatives as a new class of antibacterial that target FtsZ to combat clinically important bacterial pathogens, such as A. baumannii. This work also emphasizes previous literature that suggests that chemical halogenation is a promising route to improve the antibacterial activity of compounds in early stage antibacterial discovery (Domagala, 1994; Brighty and Gootz, 1997; Paparella et al., 2018).
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
HV, SS, and SM: design and concept. WC, JW, SP, AO, CD, KF, and XL: experimental. WC, JW, SP, SS, MS, and HV: writing. All authors have approved the final article.
This research was supported financially by the National Health and Medical Research Council of Australia (GN1147538) to HV and SM, the National Natural Science Foundation of China (81973179 and 81673284) to SM and HV, and a China-Australia Center for Health Sciences Research grant to HV, SM, and SS. WC is the recipient of a Research Training Program (RTP) Domestic Offset Scholarship.
We thank Bart Eijkelkemp, Flinders University, for the provision of XDR A. baumannii UW 5075 and summer vacation scholarship students, UniSA, Dona Babu, and Milena Rizovski for their assistance with microscopy. MS acknowledges the receipt of a no-cost academic license from Openeye Scientific Software.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.967949/full#supplementary-material
Abdi, S. N., Ghotaslou, R., Ganbarov, K., Mobed, A., Tanomand, A., Yousefi, M., et al. (2020). Acinetobacter baumannii efflux pumps and antibiotic resistance. Infect. Drug Resist. 13, 423–434. doi: 10.2147/IDR.S228089
Alakomi, H. L., Paananen, A., Suihko, M. L., Helander, I. M., and Saarela, M. (2006). Weakening effect of cell permeabilizers on gram-negative bacteria causing biodeterioration. Appl. Environ. Microbiol. 72, 4695–4703. doi: 10.1128/AEM.00142-06
Ambrose, S. J., Hamidian, M., and Hall, R. M. (2022). Extensively resistant Acinetobacter baumannii isolate RCH52 carries several resistance genes derived from an IncC plasmid. J. Antimicrob. Chemother. 77, 930–933. doi: 10.1093/jac/dkab473
Anderson, D. E., Gueiros-Filho, F. J., and Erickson, H. P. (2004). Assembly dynamics of FtsZ rings in Bacillus subtilis and Escherichia coli and effects of FtsZ-regulating proteins. J. Bacteriol. 186, 5775–5781. doi: 10.1128/JB.186.17.5775-5781.2004
Annapurna, S. M., Harikrishna, S. S., Aravind, K. B., and Pawar, P. D. (2015). Targeting FtsZ as novel therapeutic strategy against multi-drug resistant Staphylococcus aureus. Asian J. Microbiol. Biotechnol. Environ. Sci. 17, 131–138.
Araújo-Bazán, L., Ruiz-Avila, L. B., Andreu, D., Huecas, S., and Andreu, J. M. (2016). Cytological profile of antibacterial FtsZ inhibitors and synthetic peptide MciZ. Front. Microbiol. 7:1558. doi: 10.3389/fmicb.2016.01558
Araya, G., Benites, J., Reyes, J. S., Marcoleta, A. E., Valderrama, J. A., Lagos, R., et al. (2019). Inhibition of Escherichia coli and Bacillus subtilis FtsZ polymerization and Bacillus subtilis growth by dihydroxynaphtyl aryl ketones. Front. Microbiol. 10:1225. doi: 10.3389/fmicb.2019.01225
Arzanlou, M., Chai, W. C., and Venter, H. (2017). Intrinsic, adaptive and acquired antimicrobial resistance in Gram-negative bacteria. Essays Biochem. 61, 49–59. doi: 10.1042/EBC20160063
Aylett, C. H. S., and Duggin, I. G. (2017). The tubulin superfamily in archaea. Subcell. Biochem. 84, 393–417. doi: 10.1007/978-3-319-53047-5_14
Battaje, R. R., and Panda, D. (2017). Lessons from bacterial homolog of tubulin FtsZ for microtubule dynamics. Endocr. Relat. Cancer 24, T1–T21. doi: 10.1530/ERC-17-0118
Bi, F., Guo, L., Wang, Y., Venter, H., Semple, S. J., Liu, F., et al. (2017). Design, synthesis and biological activity evaluation of novel 2,6-difluorobenzamide derivatives through FtsZ inhibition. Bioorg. Med. Chem. Lett. 27, 958–962. doi: 10.1016/j.bmcl.2016.12.081
Bi, F., Song, D., Zhang, N., Liu, Z., Gu, X., Hu, C., et al. (2018). Design, synthesis and structure-based optimization of novel isoxazole-containing benzamide derivatives as FtsZ modulators. Eur. J. Med. Chem. 159, 90–103. doi: 10.1016/j.ejmech.2018.09.053
Bisson-Filho, A. W., Hsu, Y. P., Squyres, G. R., Kuru, E., Wu, F., Jukes, C., et al. (2017). Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355, 739–743. doi: 10.1126/science.aak9973
Brighty, K. E., and Gootz, T. D. (1997). The chemistry and biological profile of trovafloxacin. J. Antimicrob. Chemother. 39, 1–14. doi: 10.1093/jac/39.suppl_2.1
Broughton, C. E., Roper, D. I., van den Berg, H. A., and Rodger, A. (2015). Bacterial cell division: Experimental and theoretical approaches to the divisome. Sci. Prog. 98, 313–345. doi: 10.3184/003685015X14461391862881
Busiek, K. K., and Margolin, W. (2015). Bacterial actin and tubulin homologs in cell growth and division. Curr. Biol. 25, R243–R254. doi: 10.1016/j.cub.2015.01.030
Carro, L. (2019). Recent progress in the development of small-molecule FtsZ inhibitors as chemical tools for the development of novel antibiotics. Antibiotics 8:217. doi: 10.3390/antibiotics8040217
Casiraghi, A., Suigo, L., Valoti, E., and Straniero, V. (2020). Targeting bacterial cell division: A binding site-centered approach to the most promising inhibitors of the essential protein FtsZ. Antibiotics 9:69. doi: 10.3390/antibiotics9020069
Chai, W. C., Whittall, J. J., Song, D., Polyak, S. W., Ogunniyi, A. D., Wang, Y., et al. (2020). Antimicrobial action and reversal of resistance in MRSA by difluorobenzamide derivatives targeted at FtsZ. Antibiotics 9:873. doi: 10.3390/antibiotics9120873
Chan, F. Y., Sun, N., Neves, M. A. C., Lam, P. C. H., Chung, W. H., Wong, L. K., et al. (2013). Identification of a new class of FtsZ inhibitors by structure-based design and in vitro screening. J. Chem. Inf. Model. 53, 2131–2140. doi: 10.1021/ci400203f
Chen, Y., and Erickson, H. P. (2008). In vitro assembly studies of FtsZ/tubulin-like proteins (TubZ) from Bacillus plasmids: Evidence for a capping mechanism. J. Biol. Chem. 283, 8102–8109. doi: 10.1074/jbc.M709163200
de Boer, P. A. J. (2010). Advances in understanding E. coli cell fission. Curr. Opin. Microbiol. 13, 730–737. doi: 10.1016/j.mib.2010.09.015
de Souza, S. M., Delle Monache, F., and Smânia, A. Jr. (2005). Antibacterial activity of coumarins. Z. Naturforsch. C J. Biosci. 60, 693–700. doi: 10.1515/znc-2005-9-1006
den Blaauwen, T., and Luirink, J. (2019). Checks and balances in bacterial cell division. mBio 10, e149–e119.
Domadia, P., Bhunia, A., Sivaraman, J., Swarup, S., and Dasgupta, D. (2008). Berberine targets assembly of Escherichia coli cell division protein FtsZ. Biochemistry 47, 3225–3234. doi: 10.1021/bi7018546
Domadia, P., Swarup, S., Bhunia, A., Sivaraman, J., and Dasgupta, D. (2007). Inhibition of bacterial cell division protein FtsZ by cinnamaldehyde. Biochem. Pharmacol. 74, 831–840.
Domagala, J. M. (1994). Structure-activity and structure-side-effect relationships for the quinolone antibacterials. J. Antimicrob. Chemother. 33, 685–706.
European Committee for Antimicrobial Susceptibility Testing (2016). Media for MIC Determination by the Broth Microdilution Method. Vaxjo: European Committee for Antimicrobial Susceptibility Testing.
Farrag, H. A., Abdallah, N., Shehata, M. M. K., and Awad, E. M. (2019). Natural outer membrane permeabilizers boost antibiotic action against irradiated resistant bacteria. J. Biomed. Sci. 26:69. doi: 10.1186/s12929-019-0561-6
Ferrer-González, E., Kaul, M., Parhi, A. K., LaVoie, E. J., and Pilch, D. S. (2017). β-lactam antibiotics with a high affinity for PBP2 act synergistically with the FtsZ-targeting agent TXA707 against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 61, e863–e817. doi: 10.1128/AAC.00863-17
Fujita, J., Maeda, Y., Mizohata, E., Inoue, T., Kaul, M., Parhi, A. K., et al. (2017). Structural flexibility of an inhibitor overcomes drug resistance mutations in Staphylococcus aureus FtsZ. ACS Chem. Biol. 12, 1947–1955. doi: 10.1021/acschembio.7b00323
Gurnani, M., Chauhan, A., Ranjan, A., Tuli, H. S., Alkhanani, M. F., Haque, S., et al. (2022). Filamentous Thermosensitive Mutant Z: An Appealing Target for Emerging Pathogens and a Trek on Its Natural Inhibitors. Biology 11:624. doi: 10.3390/biology11050624
Hawkins, P. C. D., Skillman, A. G., Warren, G. L., Ellingson, B. A., and Stahl, M. T. (2010). Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 50, 572–584. doi: 10.1021/ci100031x
Haydon, D. J., Stokes, N. R., Ure, R., Galbraith, G., Bennett, J. M., Brown, D. R., et al. (2008). An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 321, 1673–1675.
Huecas, S., Ramírez-Aportela, E., Vergoñós, A., Núñez-Ramírez, R., Llorca, O., Díaz, J. F., et al. (2017). Self-organization of FtsZ polymers in solution reveals spacer role of the disordered C-terminal tail. Boiophys. J. 113, 1831–1844. doi: 10.1016/j.bpj.2017.08.046
Hurley, K. A., Santos, T. M. A., Nepomuceno, G. M., Huynh, V., Shaw, J. T., and Weibel, D. B. (2016). Targeting the bacterial division protein FtsZ. J. Med. Chem. 59, 6975–6998.
Kapoor, S., and Panda, D. (2009). Targeting FtsZ for antibacterial therapy: A promising avenue. Expert Opin. Ther. Targets 13, 1037–1051.
Kaul, M., Mark, L., Zhang, Y., Parhi, A. K., Lyu, Y. L., Pawlak, J., et al. (2015). TXA709, an FtsZ-targeting benzamide prodrug with improved pharmacokinetics and enhanced in vivo efficacy against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 59, 4845–4855. doi: 10.1128/AAC.00708-15
Kelley, B. P., Brown, S. P., Warren, G. L., and Muchmore, S. W. (2015). POSIT: Flexible Shape-Guided Docking For Pose Prediction. J. Chem. Inf. Model. 55, 1771–1780. doi: 10.1021/acs.jcim.5b00142
Khazandi, M., Pi, H., Chan, W. Y., Ogunniyi, A. D., Sim, J. X. F., Venter, H., et al. (2019). In vitro antimicrobial activity of robenidine, ethylenediaminetetraacetic acid and polymyxin B nonapeptide against important human and veterinary pathogens. Front. Microbiol. 10:837. doi: 10.3389/fmicb.2019.00837
Knight, D., Rudin, S., Bonomo, R. A., and Rather, P. (2018). Acinetobacter nosocomialis: Defining the role of efflux pumps in resistance to antimicrobial therapy, surface motility, and biofilm formation. Front. Microbiol. 9:1902. doi: 10.3389/fmicb.2018.01902
Krishnamoorthy, G., Leus, I. V., Weeks, J. W., Wolloscheck, D., Rybenkov, V. V., and Zgurskaya, H. I. (2017). Synergy between active efflux and outer membrane diffusion defines rules of antibiotic permeation into Gram-negative bacteria. mBio 8, e1172–e1117. doi: 10.1128/mBio.01172-17
Kusuma, K. D., Payne, M., Ung, A. T., Bottomley, A. L., and Harry, E. J. (2019). FtsZ as an antibacterial target: Status and guidelines for progressing this avenue. ACS Infect. Dis. 5, 1279–1294. doi: 10.1021/acsinfecdis.9b00055
Kyriakidis, I., Vasileiou, E., Pana, Z. D., and Tragiannidis, A. (2021). Acinetobacter baumannii Antibiotic Resistance Mechanisms. Pathogens 10:373. doi: 10.3390/pathogens10030373
Li, X., and Ma, S. (2015). Advances in the discovery of novel antimicrobials targeting the assembly of bacterial cell division protein FtsZ. Eur. J. Med. Chem. 95, 1–15. doi: 10.1016/j.ejmech.2015.03.026
Li, X., Sheng, J., Huang, G., Ma, R., Yin, F., Song, D., et al. (2015a). Design, synthesis and antibacterial activity of cinnamaldehyde derivatives as inhibitors of the bacterial cell division protein FtsZ. Eur. J. Med. Chem. 97, 32–41. doi: 10.1016/j.ejmech.2015.04.048
Li, X., Sheng, J., Song, D., Guo, L., and Ma, S. (2015b). Phenylacrylamides as novel FtsZ-targeted potential antimicrobials. Lett. Drug. Des. Discov. 12, 234–240.
Liu, J., Ma, R., Bi, F., Zhang, F., Hu, C., Venter, H., et al. (2018). Novel 5-methyl-2-phenylphenanthridium derivatives as FtsZ-targeting antibacterial agents from structural simplification of natural product sanguinarine. Bioorg. Med. Chem. Lett. 28, 1825–1831. doi: 10.1016/j.bmcl.2018.04.015
Löwe, J., and Amos, L. A. (1998). Crystal structure of the bacterial cell-division protein FtsZ. Nature 391, 203–206.
Margalit, D. N., Romberg, L., Mets, R. B., Hebert, A. M., Mitchison, T. J., Kirschner, M. W., et al. (2004). Targeting cell division: Small-molecule inhibitors of FtsZ GTPase perturb cytokinetic ring assembly and induce bacterial lethality. Proc. Natl. Acad. Sci. 101, 11821–11826. doi: 10.1073/pnas.0404439101
Matsui, T., Yamane, J., Mogi, N., Yamaguchi, H., Takemoto, H., Yao, M., et al. (2012). Structural reorganization of the bacterial cell-division protein FtsZ from Staphylococcus aureus. Acta Crystallogr. Sect. D Biol. Crystallogr. 68, 1175–1188. doi: 10.1107/S0907444912022640
McGann, M. (2011). FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 51, 578–596. doi: 10.1021/ci100436p
McGann, M. (2012). FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 26, 897–906. doi: 10.1007/s10822-012-9584-8
McIntosh, J. R., Volkov, V., Ataullakhanov, F. I., and Grishchuk, E. L. (2010). Tubulin depolymerization may be an ancient biological motor. J. Cell Sci. 123, 3425–3434.
Miki, T., and Hardt, W. D. (2013). Outer membrane permeabilization is an essential step in the killing of Gram-negative bacteria by the Lectin RegIIIβ. PLoS One 8:e69901. doi: 10.1371/journal.pone.0069901
Murray, C. J. L., Ikuta, K. S., Sharara, F., Swetschinski, L., Robles Aguilar, G., and Gray, A. (2022). Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 399, 629–655.
Nair, D. R., Chen, J., Monteiro, J. M., Josten, M., Pinho, M. G., Sahl, H. G., et al. (2017). A quinolinol-based small molecule with anti-MRSA activity that targets bacterial membrane and promotes fermentative metabolism. J. Antibiot. 70, 1009–1019. doi: 10.1038/ja.2017.79
Nikaido, H. (2003). Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 67, 593–656. doi: 10.1128/MMBR.67.4.593-656.2003
Paparella, A. S., Lee, K. J., Hayes, A. J., Feng, J., Feng, Z., Cini, D., et al. (2018). Halogenation of Biotin Protein Ligase Inhibitors Improves Whole Cell Activity against Staphylococcus aureus. ACS Infect. Dis. 4, 175–184. doi: 10.1021/acsinfecdis.7b00134
Pichoff, S., Du, S., and Lutkenhaus, J. (2018). Disruption of divisome assembly rescued by FtsN–FtsA interaction in Escherichia coli. Proc. Natl. Acad. Sci. 115, E6855–E6862. doi: 10.1073/pnas.1806450115
Pilhofer, M., Rosati, G., Ludwig, W., Schleifer, K. H., and Petroni, G. (2007). Coexistence of tubulins and FtsZ in different Prosthecobacter species. Mol. Biol. Evol. 24, 1439–1442. doi: 10.1093/molbev/msm069
Porta-de-la-Riva, M., Fontrodona, L., Villanueva, A., and Cerón, J. (2012). Basic Caenorhabditis elegans methods: Synchronization and observation. J. Vis. Exp. 2012, e4019–e4019. doi: 10.3791/4019
Rice, L. B. (2008). Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 197, 1079–1081. doi: 10.1086/533452
Romberg, L., Simon, M., and Erickson, H. P. (2001). Polymerization of FtsZ, a bacterial homolog of tubulin. Is assembly cooperative? J. Biol. Chem. 276, 11743–11753.
Rosado-Lugo, J. D., Sun, Y., Banerjee, A., Cao, Y., Datta, P., Zhang, Y., et al. (2022). Evaluation of 2,6-difluoro-3-(oxazol-2-ylmethoxy)benzamide chemotypes as Gram-negative FtsZ inhibitors. J. Antibiot. 75, 385–395. doi: 10.1038/s41429-022-00531-9
Sharavanan, V. J., Sivaramakrishnan, M., Kothandan, R., Muthusamy, S., and Kandaswamy, K. (2019). Molecular docking studies of phytochemicals from leucas aspera targeting Escherichia coli and Bacillus subtilis subcellular proteins. Pharmacogn. J. 11, 278–285.
Sharma, A., Gupta, V. K., and Pathania, R. (2019). Efflux pump inhibitors for bacterial pathogens: From bench to bedside. Indian J. Med. Res. 149, 129–145. doi: 10.4103/ijmr.IJMR_2079_17
Straniero, V., Suigo, L., Casiraghi, A., Sebastián-Pérez, V., Hrast, M., Zanotto, C., et al. (2020). Benzamide derivatives targeting the cell division protein FtsZ: Modifications of the linker and the benzodioxane scaffold and their effects on antimicrobial activity. Antibiotics 9:160. doi: 10.3390/antibiotics9040160
Sun, N., Ban, L., Li, M., Fang, Z., Li, X., Yao, W., et al. (2018a). Probing the benzofuroquinolinium derivative as a potent antibacterial agent through the inhibition of FtsZ activity. J. Pharmacol. Sci. 138, 83–85. doi: 10.1016/j.jphs.2018.09.001
Sun, N., Du, R. L., Zheng, Y. Y., Guo, Q., Cai, S. Y., Liu, Z. H., et al. (2018b). Antibacterial activity of 3-methylbenzo[d]thiazol-methylquinolinium derivatives and study of their action mechanism. J. Enzyme Inhib. Med. 33, 879–889. doi: 10.1080/14756366.2018.1465055
Sundararajan, K., Miguel, A., Desmarais, S. M., Meier, E. L., Casey Huang, K., and Goley, E. D. (2015). The bacterial tubulin FtsZ requires its intrinsically disordered linker to direct robust cell wall construction. Nat. Commun. 6:7281. doi: 10.1038/ncomms8281
Venter, H., Mowla, R., Ohene-Agyei, T., and Ma, S. (2015). RND-type drug efflux pumps from Gram-negative bacteria: Molecular mechanism and inhibition. Front. Microbiol. 6:377. doi: 10.3389/fmicb.2015.00377
Wang, Y., Mowla, R., Guo, L., Ogunniyi, A. D., Rahman, T., De Barros Lopes, M. A., et al. (2017). Evaluation of a series of 2-napthamide derivatives as inhibitors of the drug efflux pump AcrB for the reversal of antimicrobial resistance. Bioorg. Med. Chem. Lett. 27, 733–739. doi: 10.1016/j.bmcl.2017.01.042
Welch, A., Awah, C. U., Jing, S. H., Van Veen, H. W., and Venter, H. (2010). Promiscuous partnering and independent activity of MexB, the multidrug transporter protein from Pseudomonas aeruginosa. Biochem. J. 430, 355–364. doi: 10.1042/BJ20091860
World Health Organization (2015). Global Action Plan on Antimicrobial Resistance. Geneva: World Health Organization.
World Health Organization (2017). WHO Priority Pathogens List for Research and Development of New Antibiotics. Geneva: World Health Organization.
Yang, L., Wang, Y., He, X., Xiao, Q., Han, S., Jia, Z., et al. (2021). Discovery of a novel plant-derived agent against Ralstonia solanacearum by targeting the bacterial division protein FtsZ. Pestic. Biochem. Physiol. 177:104892. doi: 10.1016/j.pestbp.2021.104892
Zgurskaya, H. I., López, C. A., and Gnanakaran, S. (2015). Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infect. Dis. 1, 512–522. doi: 10.1021/acsinfecdis.5b00097
Zheng, Y. Y., Du, R. L., Cai, S. Y., Liu, Z. H., Fang, Z. Y., Liu, T., et al. (2018). Study of benzofuroquinolinium derivatives as a new class of potent antibacterial agent and the mode of inhibition targeting FtsZ. Front. Microbiol. 9:1937. doi: 10.3389/fmicb.2018.01937
Keywords: antimicrobial resistance, antimicrobial drug development, FtsZ, FtsZ inhibitor, cinnamaldehyde, gram-negative, XDR Acinetobacter baumannii
Citation: Chai WC, Whittall JJ, Polyak SW, Foo K, Li X, Dutschke CJ, Ogunniyi AD, Ma S, Sykes MJ, Semple SJ and Venter H (2022) Cinnamaldehyde derivatives act as antimicrobial agents against Acinetobacter baumannii through the inhibition of cell division. Front. Microbiol. 13:967949. doi: 10.3389/fmicb.2022.967949
Received: 13 June 2022; Accepted: 05 August 2022;
Published: 29 August 2022.
Edited by:
Silvia Buroni, University of Pavia, ItalyCopyright © 2022 Chai, Whittall, Polyak, Foo, Li, Dutschke, Ogunniyi, Ma, Sykes, Semple and Venter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henrietta Venter, UmlldGllLlZlbnRlckB1bmlzYS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.