
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 11 August 2022
Sec. Infectious Agents and Disease
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.966735
This article is part of the Research TopicNew Infectious Agents in Arthropod VectorsView all 9 articles
 Luanying Guo1†
Luanying Guo1† Jun Ma1†
Jun Ma1† Junwei Lin2†
Junwei Lin2† Meiyi Chen1
Meiyi Chen1 Wei Liu1
Wei Liu1 Jin Zha1
Jin Zha1 Qinqin Jin1
Qinqin Jin1 Hongrong Hong1
Hongrong Hong1 Weinan Huang3
Weinan Huang3 Li Zhang4
Li Zhang4 Ketong Zhang5
Ketong Zhang5 Zhengkai Wei1*
Zhengkai Wei1* Quan Liu1,6*
Quan Liu1,6*Tick-borne viruses (TBVs) have increasingly caused a global public health concern. This study collected Rhipicephalus ticks in Guangdong, southern China to identify RNA viruses. Meta-transcriptome analysis revealed the virome in Rhipicephalus ticks, resulting in the discovery of 10 viruses, including Lihan tick virus, Brown dog tick phlebovirus 1 and 2 in the family Phenuiviridae, Mivirus and Wuhan tick virus 2 in the family Chuviridae, Wuhan tick virus 1 in the family Rhabdoviridae, bovine hepacivirus in the family Flaviviridae, Guangdong tick quaranjavirus (GTQV) in the family Orthomyxoviridae, Guangdong tick orbivirus (GTOV) in the family Reoviridae, and Guangdong tick Manly virus (GTMV) of an unclassified family. Phylogenetic analysis showed that most of these TBVs were genetically related to the strains in countries outside China, and GTQV, GTOV, and GTMV may represent novel viral species. These findings provided evidence of the long-distance spread of these TBVs in Guangdong, southern China, suggesting the necessity and importance of TBV surveillance.
Ticks (Acari: Ixodoidea) are considered to be next to mosquitoes as vectors for pathogenic agents. Tick-borne viruses (TBVs) include a large group of viruses of at least 12 genera, nine families, and two orders (Shi et al., 2018 ). In China, tick-borne encephalitis virus (TBEV) in the family Flaviviridae and Crimean-Congo hemorrhagic fever virus (CCHFV) in the family Nairoviridae are responsible for encephalitis and hemorrhagic fever in northeastern and northwestern China, respectively. Emerging TBVs, such as severe fever with thrombocytopenia virus (Yu et al., 2011), Jingmen tick virus (Jia et al., 2019), Alongshan virus (Wang et al., 2019), Songling virus (Ma et al., 2021), Beiji nairovirus (Wang et al., 2021), and Tacheng tick virus 1 and 2 (Liu et al., 2020; Dong et al., 2021), have been reported to be associated with human diseases. In addition, the Nairobi sheep disease virus, another member in the family Nairoviridae that can cause acute hemorrhagic gastroenteritis in sheep and goats, has also been found in ticks in China (Gong et al., 2015).
High-throughput sequencing technology has been widely used to investigate viromes in ticks and diagnose unknown infectious diseases. To date, tick viromes have been reported in Heilongjiang, Liaoning, Hebei, Henan, Hubei, and Yunnan provinces of China (Meng et al., 2019; Zhao et al., 2020; Shi et al., 2021; Xu et al., 2021; Yang et al., 2021), revealing different tick viromes, due to tick species and geographical differences. Guangdong province of China is considered as one of the global hotspots of emerging zoonotic diseases and vector-borne diseases resulting from the specific climate, environment, and biodiversity (Wang et al., 2008; Allen et al., 2017), but the viromes in ticks in Guangdong, southern China remain poorly understood. In this study, we identified RNA viruses in Rhipicephalus sanguineus and Rhipicephalus microplus ticks in Guangdong, southern China using metagenomics and detected 10 viruses, including 3 novel viruses, suggesting a high viral diversity in Rhipicephalus ticks.
From May–June 2020, ticks were collected from cattle and dogs in Zhanjiang and Jieyang, Guangdong, southern China. Tick species were morphologically identified by an experienced technician and confirmed by sequencing the mitochondrial 16S ribosomal RNA (16S rRNA) gene (Liu et al., 2016). The sampled ticks were divided into 6 groups according to the species and collection location. After washing with phosphate buffered saline (PBS), ticks were homogenized in PBS solution, followed by centrifugation at 2,500 × g for 5 min at 4°C. Supernatants were collected for RNA extraction with TRIzol LS reagent (Invitrogen, Carlsbad, CA, United States).
Meta-transcriptome sequencing was conducted as previously described (Shi et al., 2016). In brief, after removing ribosomal RNA (rRNA), RNA was fragmented, reverse-transcribed, and adapted, followed by paired-end (150-bp) sequencing on the Illumina Hiseq 2,500 platform. All library preparation and sequencing were performed by Tianjin Novogene Bioinformatics Technology Co., Ltd., Tianjin, China.
Sequencing reads were adaptor- and quality-trimmed using the FASTP program, followed by de novo assemble by the Megahit program, and the resulting contigs were compared against the nr database using the diamond BLASTX program. The confirmed viral contigs with assembly overlaps or from the same scaffold were merged using the SeqMan program (version 7.1, DNAstar, Madison, WI, United States). Reads were mapped to the target contigs using Bowtie 2, and the integrated genomics viewer (IGV) was used to check for assembly faults in order to validate the assembly results. Gaps between the contigs were filled by RT-PCR and Sanger sequencing. The genomic terminus of targeting viruses was determined using the 5′/3′ RACE kits (TaKaRa, Dalian, China). The complete viral genome was confirmed by Sanger sequencing with overlapping primers that covered the entire viral genome.
The discovered viruses were classified based on the nucleotide (nt) and amino acids (aa) identities. If the species demarcation criteria remain unclear within a genus, a novel viral species is defined if it holds less than 80% nt identity across the complete genome or less than 90% aa identity of the RNA-dependent RNA polymerase (RdRp) domain with known viruses. All the novel viruses were named “Guangdong tick,” followed by common viral names according to their taxonomy. Viruses classified into established taxonomies were marked with “Guangdong (GD)” to distinguish them from other viral strains.
Reference virus sequences were acquired from the GenBank database and examined using Mafft v7.450 to determine the phylogenetic relationships of the identified viruses (Katoh et al., 2019). Phylogenetic trees were constructed using the maximum-likelihood method in PhyML v3.0 with Le and Gascuel substitution model for aa sequence analysis and General Time Reversible substitution model for nt sequence analysis, based on a bootstrap value of 1,000 replicates (Guindon et al., 2005).
A total of 339 ticks were collected from cattle (n = 306) and dogs (n = 33) in Jieyang (n = 242) and Zhanjiang (n = 97) of Guangdong Province, China, including 243 R. sanguineus and 96 R. microplus (Supplementary Table S1; Supplementary Figure S1). Ticks were pooled into 6 groups for RNA library construction and sequencing. After quality control and adapter trimming, a total of 85,275,714 paired-end clean reads were generated in these libraries, resulting in 259,311 viral reads, which accounted for 0.30% of the total RNA reads, and assembled to 190 viral contigs (Supplementary Tables S2, S3). After being aligned by Blast, viral contigs were finally annotated to 10 viruses of six known viral families of Phenuiviridae, Chuviridae, Flaviviridae, Reoviridae, Orthomyxoviridae, Rhabdoviridae, and an unclassified family (Supplementary Table S4).
The complete genomes of representative 23 viral strains were obtained by contig-based PCR and RACE (Supplementary Table S5), including 2 novel viruses, such as Guangdong tick Manly virus (GTMV) and Guangdong tick quaranjavirus (GTQV), 7 known viruses, including Brown dog tick phlebovirus 1 (BDTPV1), Lihan tick virus (LTV), Mivirus sp. (MIV), Wuhan tick virus 1 (WTV1), Hepacivirus N (HNV), Wuhan tick virus 2 (WTV2) and Brown dog tick phlebovirus 2 (BDTPV2) and a virus with partial sequence named Guangdong tick orbivirus (GTOV; Supplementary Table S6). The mean depth of viral sequencing in each pool is shown in Supplementary Table S7, and these viruses were named based on the viral species, the pool name they discovered, and the closest relationships with previously described tick-associated viruses.
Metagenomic analysis demonstrated that bunyavirus reads accounted for 51.4% (133,381/259311) of total viral reads, including 1.04% (2,721/259311), 22.89% (59,367/259311), and 27.49% reads (71,293/259311) mapped to the genomes of LTV, BDTPV1, and BDTPV2, respectively (Supplementary Tables S3, S8). We obtained 10 complete viral genomes and designated LTV GD (JY02-3 and ZJ02), BDTPV1 GD (JY01, JY02-1, JY02-2, JY02-3, and ZJ-0103) and BDTPV2 GD (JY01, JY02-1, and JY02-3; Supplementary Table S4).
The genomes of these viruses contained bi-segments, large (L) and small (S) segments, encoding the putative RdRp and nucleoprotein, respectively (Figure 1). As shown in Supplementary Figure S1; Supplementary Table S8, the S segment of LTV GD contains a 1766 bp putative nucleocapsid ORF, whereas the L segment encodes a 6,614-bp RdRp. The amino acid sequences of LTV GD L proteins were approximately 97.5–99.3%, similar to the L proteins of other LTVs (Figure 1; Supplementary Table S8). The length of L and S segments for BDTPV1 were 6,614 bp and 1,422 bp, sharing 98.9–99.4% aa identity in the RdRp with BDTPV1 strain TTP-Pool-12, which has been reported in Trinidad and Tobago (Sameroff et al., 2019). The length of L and S segments for BDTPV2 was 6,533 bp and 2095 bp. BDTPV2 GD shared 96.7% aa identity with BDTPV2 strain TTP-Pool-5 in the RdRp (Sameroff et al., 2019). Interestingly, both BDTPV1 and BDTPV2 were found to co-exist in five R. sanguineus tick pools.
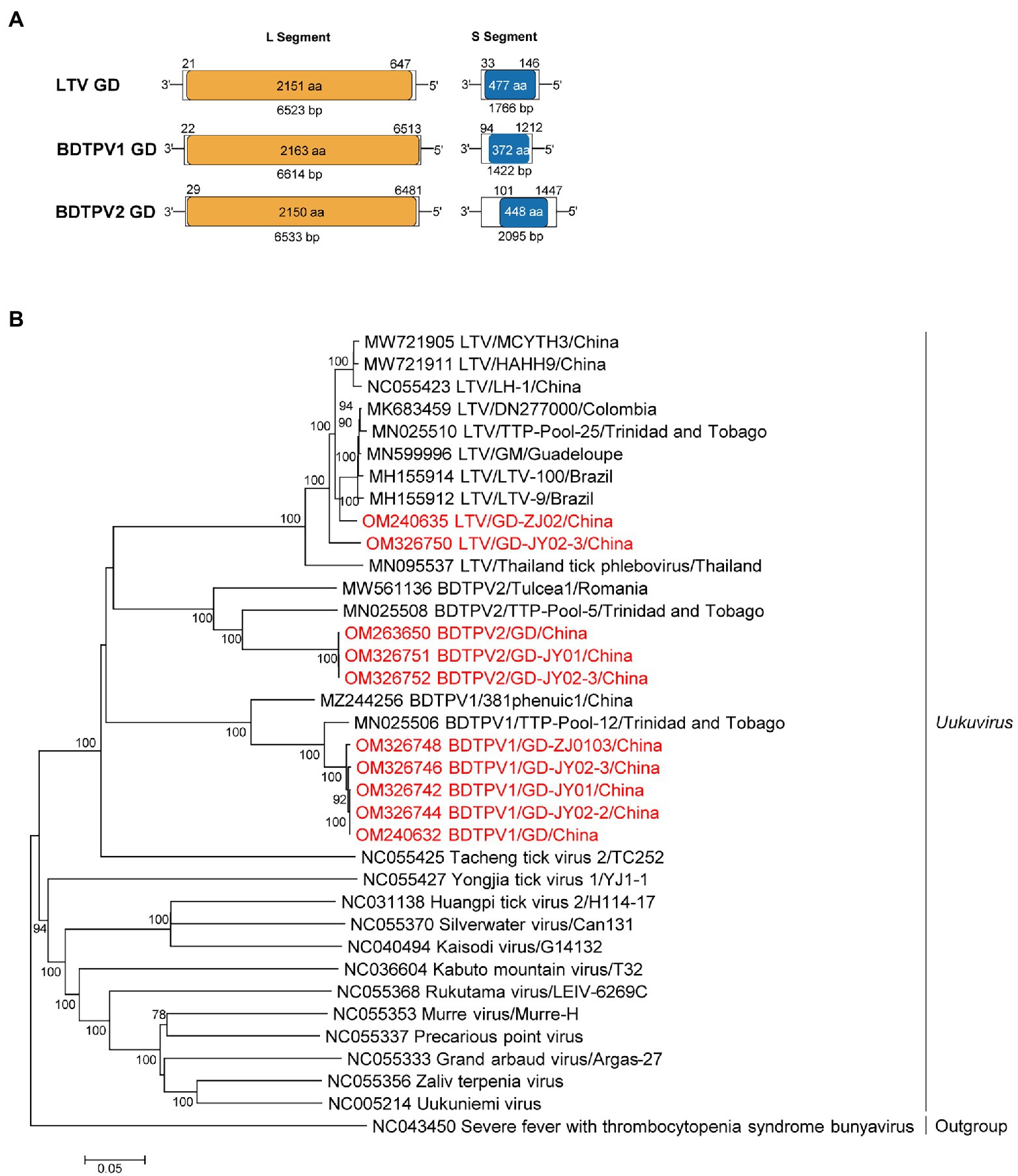
Figure 1. The genome characterization and phylogenetic analysis of uukuviruses. (A) Genome organization and putative coding regions of Lihan tick virus (LTV), Brown dog tick phlebovirus 1 (BDTPV1) and 2 (BDTPV2). The viral genome includes L and S segments that contained the predicted RNA depended RNA polymerase (RdRp) and nucleoprotein (NP) respectively. (B) Phylogenetic analyses of uukuviruses. Phylogenetic trees were constructed based on the L segment nucleotide sequences of representative viruses in the genus Uukuvirus. Viruses obtained in ticks here are highlighted in red. Scale bar indicates nucleotide substitutions per site.
Phylogenetic analyses revealed that all three identified viruses clustered with the other bunyaviruses lacking M segment and shared the common ancestor with the Uukuvirus group. They were genetically related to those identified in Brazil, Thailand, Colombia, or Trinidad and Tobago (Figure 1).
In total, 39,371 reads (15.18%; 39,371/259311) from 3 R. sanguineus tick pools were mapped to the proposed Mivirus, while 20,296 reads (7.83%; 20,296/259311) in 3 pools were matched with WTV2 (Supplementary Table S3).
The complete genomes of miviruses were obtained and named MIV GD and WTV2 GD JY02-3 and ZJ2103. Both the WTV2 and MIV genome of the study were confirmed to belong to the circular structure by performing PCR with the forward primer located in 3′ of the genome and the reverse primer in the 5′ part (Supplementary Table S5). The genome included three ORFs (Figure 2), encoding the putative RdRp, glycoprotein, and nucleoprotein, respectively. The length of MIV GD was 11,271 bp, with the high identity (99.5% of RdRp aa sequence, 97.8% of complete nt sequence) to MIV strain TTP-Pool-7 discovered in Trinidad and Tobago (Sameroff et al., 2019). The length of WTV2 GD (JY02-3 and ZJ2103 strain) were 11,396 bp, sharing 99.5–99.6% aa identity in the RdRp with the strain WTVS-SZWH3 identified in China (Xu et al., 2021; Supplementary Table S9).
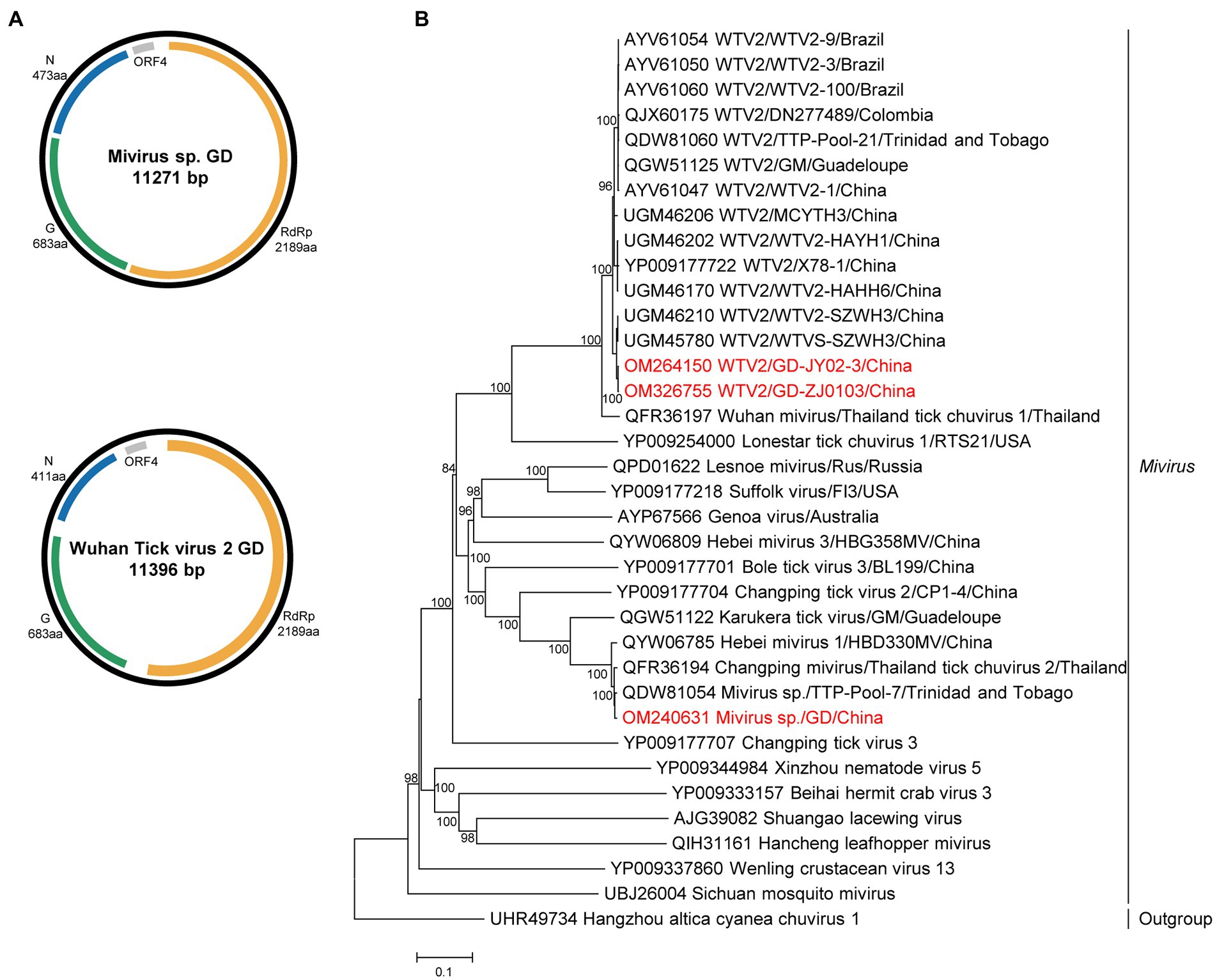
Figure 2. The genome structure and phylogenetic analysis of miviruses. (A) Genome organization and putative coding regions of Mivirus sp. (MIV) and Wuhan tick virus 2 (WTV2). The viral genome contains one segments that encoding the RdRp, glycoprotein (G), nucleoprotein (N) and ORF4. (B) Phylogenetic analyses of miviruses. Phylogenetic trees were constructed based on the RdRp protein sequences of representative viruses in the genus Mivirus. Viruses obtained in ticks here are highlighted in red. Scale bar indicates nucleotide substitutions per site.
Phylogenetically, both chuviruses identified in this study were grouped into the Mivirus clade and closely related to the viral strains discovered in other countries, such as Brazil, Thailand, Colombia, or Trinidad and Tobago (Figure 2).
Metagenomics revealed 3.42% of viral reads (8,861/259311) mapped to WTV1 (Supplementary Table S3), and four complete genomes of WTV1 were obtained and designated WTV1 GD strains ZJ0103, ZJ02, JY02-1, and JY02-3. The full length of WTV1 GD was 10,174 bp, which shared 95.7–99.7% aa identity of RdRp with other strains in China (Supplementary Table S10). WTV1 GD exhibited a non-classic rhabdo-like genome organization, with four open reading frames (ORFs), encoding RdRp, nucleoprotein (N), hypothetical protein 2 (ORF2) and 3 (ORF3). However, the G gene was not found in WTV1 GD (Figure 3). Phylogenetic analysis showed WTV1 had a certain regional difference, though Chinese WTV1 GD strains were clustered with the viral strains identified in Thailand and Turkey (Figure 3).
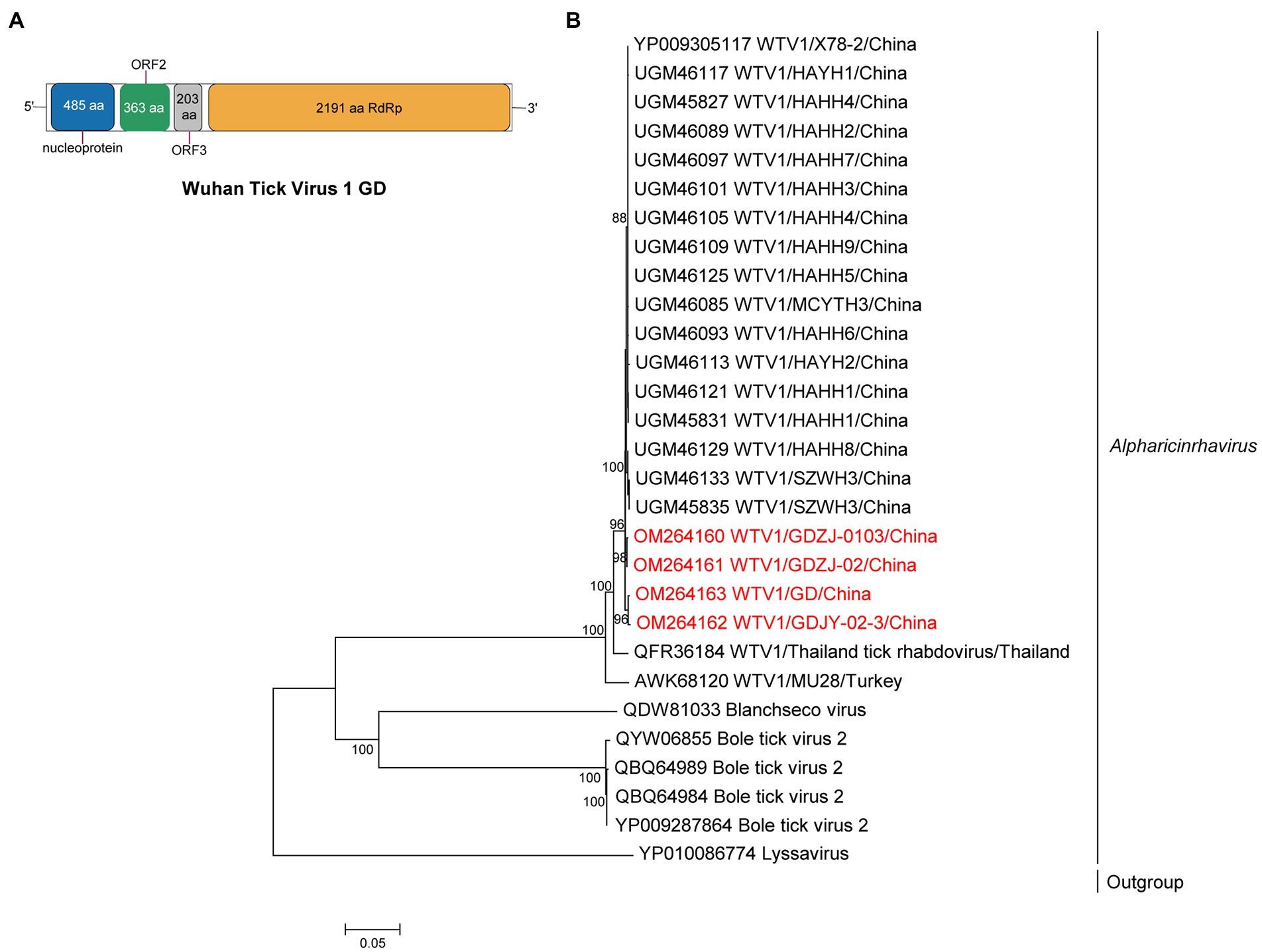
Figure 3. The genome structure and phylogenetic analysis of alpharicinrhaviruses. (A) Genome organization and putative coding regions of Wuhan tick virus 1 (WTV1). The viral genome contains one segments that encoding the nucleoprotein (N), ORF2, ORF3, and RdRp. (B) Phylogenetic analyses of alpharicinrhaviruses. Phylogenetic trees were constructed based on the RdRp protein sequences of representative viruses in the genus Alpharicinrhavirus. Viruses obtained in ticks here are highlighted in red. Scale bar indicates nucleotide substitutions per site.
Approximately 6.1% of reads (233/3642) in GDZJ/02 collected from cattle in Zhanjiang were assigned to BovhepV in the genus Hepacivirus in the family Flaviviridae (Supplementary Table S3). Three strains of BovhepV shared 84.4–84.8% nt identity and 94.8–95.6% aa identity with Brazil strains and formed a novel subtype in genotype 1 (Shao et al., 2021).
Metagenomics showed that 56,128 reads (21.65%; 56,128/2,59,311) in five pools were annotated to GTQV (Supplementary Table S3). We identified a quaranja-like virus in five pools, tentatively named Guangdong tick quaranjavirus (GTQV). The complete genome of GTQV included six segments, PB2, PB1, PA, NP, HA and M, ranging from 930 to 2,422 bp. The VP7 segment of quaranjavirus was likely to be absent in GTQV (Figure 4).
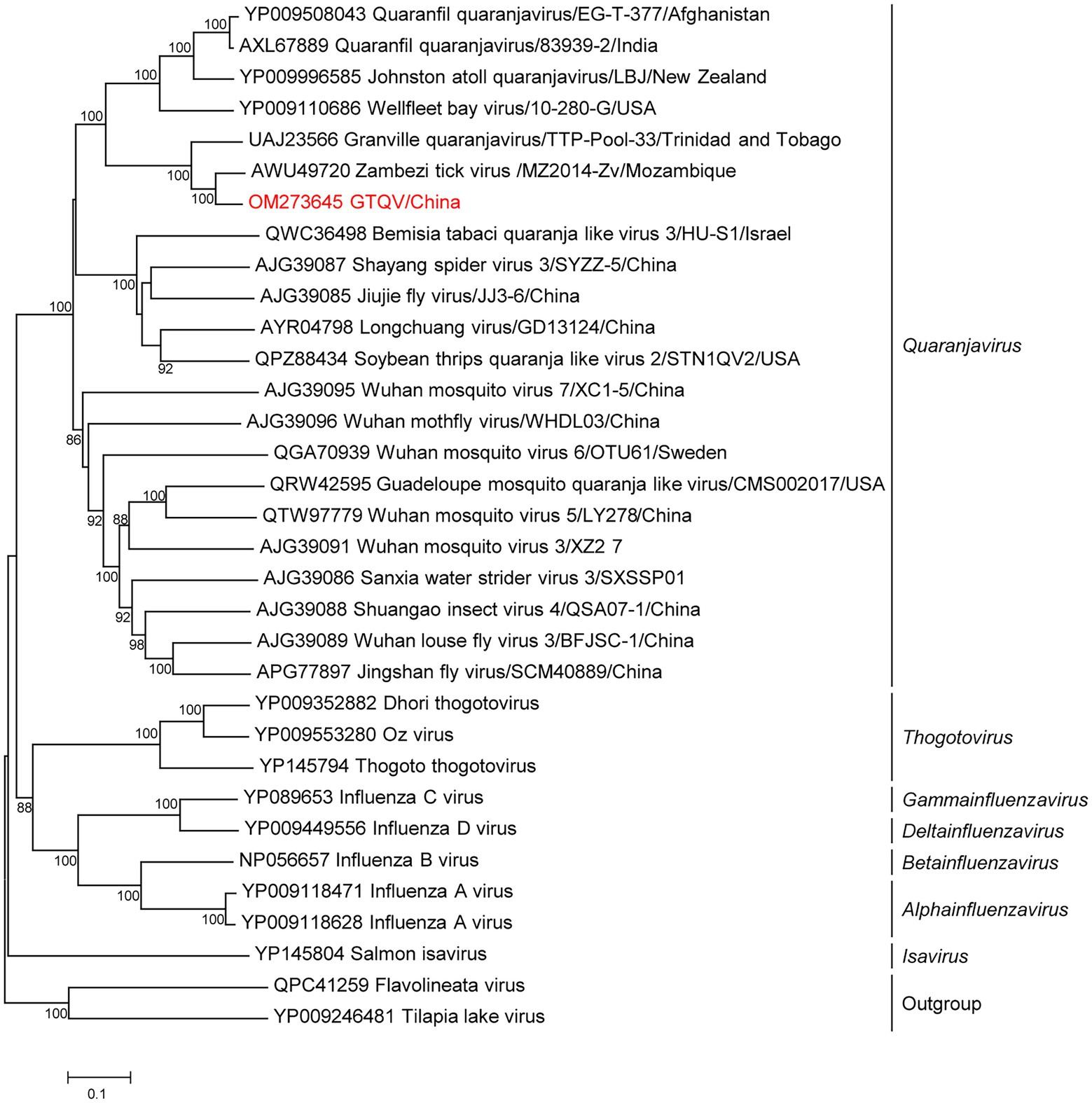
Figure 4. Phylogenetic analysis of orthomyxoviruses. Phylogenetic trees of Guangdong tick quaranjavirus (GTQV) were constructed based on the polymerase basic protein 1 (PB1) protein sequences of representative viruses in the family Orthomyxovirus. Viruses obtained in ticks here are highlighted in red. Scale bar indicates nucleotide substitutions per site.
GTQV shared 57.4–86.2% aa identity to the quaranjaviruses (Supplementary Table S11), whose PB1 had 86.2 and 72.1% aa identity to Zambezi tick virus 1 (Cholleti et al., 2018) and Granville quaranjavirus, respectively (Sameroff et al., 2021). The putative M segment had a similar length to other quaranjaviruses, including two open reading frames (ORF; Supplementary Figure S2). ORF1 had the start codon ATG and encoded a 154-aa protein (Supplementary Figure S3); ORF2 had the start codon GTG and encoded a 79-aa protein. The GTQV genomic segments had a conserve 5′-terminal sequence UGCUGUGGCUGC and 3′-terminal sequence GCTTGTCTCTACT.
Phylogenetic analysis of the complete PB1 protein showed that GTQV was clustered with quaranjaviruses in the family Orthomyxoviridae. Compared with other quaranjaviruses, the GTQV genome had a specific terminal sequence (Supplementary Figure S3).
Metagenomic analysis showed that 391 reads (0.15%, 391/259311) were mapped with the reovirus-like virus in the R. microplus pool of Zhanjiang (Supplementary Table S3). The complete ORF of six segments (VP4, VP5, VP6, NS1, NS2, and NS3) and partial ORF of three segments (VP1, VP2, and VP3) were obtained. Unfortunately, the VP7 gene could not be found in our study. We designated this virus as Guangdong tick orbivirus (GTOV), whose genome shared 73.1–80.5% nt and 70–88.1% aa identity with the known reoviruses (Supplementary Table S4).
Phylogenetic analysis showed that GTOV was genetically related to the known reoviruses (Supplementary Figure S4), in which VP1, VP4 and NS3 were closely related to the Wad Medani virus SUD1952/01 identified in Rhipicephalus in Sudan (Belaganahalli et al., 2015). VP3, VP5, VP6 and NS1 were closely related to the strain Tuva 2012 in Russia (Dedkov et al., 2021), and VP2 was closely related to the strain LEIV-8066Tur in Tajikistan, while NS2 was closely related to the isolate G-673 in India (Yadav et al., 2018).
Mainly virus, first discovered in ticks fed on reptiles in Australia, is an unclassified mononegavirus (Harvey et al., 2019). In this study, a total of 860 (0.33%, 391/259,311) reads were mapped with the mainly virus in pools of Jieyang. We named this virus as Guangdong tick mainly virus (GTMV), which genome had 10,177 nt and shared 44.9% nt and 36.5–65.5% aa similarity with mainly virus.
This study reported the metagenomic analysis of RNA viruses in ticks in Guangdong, southern China, revealing a high viral diversity in these collected ticks and 10 different viruses belonging to six viral families of Phenuiviridae, Chuviridae, Flaviviridae, Reoviridae, Orthomyxoviridae, and Rhabdoviridae and unclassified family, were identified. These viral families were similar to those described in Turkey (Brinkmann et al., 2018), Trinidad and Tobago (Sameroff et al., 2019), Australia (Harvey et al., 2019), Thailand (Temmam et al., 2019), northern Europe (Pettersson et al., 2017), southern Brazil (Souza et al., 2018), and Heilongjiang, Yunnan, and Zhejiang provinces of China (Meng et al., 2019; Shi et al., 2021; He et al., 2022), which may provide the evidence of long-distance spread of tick-borne viruses in Guangdong, southern China through migratory birds or animal movement.
The family Chuviridae includes at least 14 viral genera, genetically located between the segmented and the unsegmented negative-sense RNA viruses. Mivirus, one genus in the family Chuviridae, contained at least 9 viral species discovered in ticks. MIV GD was genetically grouped into the miviruses, closely related to the MIV strain TTP-Pool-7 (Sameroff et al., 2019). Mivirus discovered in this study had a closer relationship with the strains discovered in south America than those identified in China. Wuhan tick virus 2 (WTV2) was firstly discovered in R. microplus ticks from Wuhan, China, and then discovered in South America (Li et al., 2015; Souza et al., 2018; Sameroff et al., 2019; Gomez et al., 2020). WTV2 GD and WTV2-X78-1 were clustered with the strains discovered in South America (Li et al., 2015), but had a higher identity than the strains identified in China. These results confirmed the long-distance viral transmission, but the pathogenicity of MIV GD and WTV2 GD needs further investigation.
The family Orthomyxoviridae is composed of seven genera: types A/B/C and D Influenzavirus, Isavirus, Quaranjavirus, and Thogotovirus. Tick-borne orthomyxoviruses are mainly located in the genus Quaranjavirus and Thogotovirus. The first quaranjavirus was isolated from soft ticks and the blood of children suffering from an unexplained febrile illness (Taylor et al., 1966). Quaranjaviruses are also associated with mass avian die-offs (Allison et al., 2015). GTQV identified here was clustered with other recently identified hard-tick-associated quaranjaviruses, with wide geographic ranges, suggesting that many unidentified novel tick-borne orthomyxoviruses were classified based on the phylogenetic analysis.
LTV was first described in Chinese R. microplus (Li et al., 2015) and reported in Turkish H. marginatum and R. sanguineus ticks (Dincer et al., 2017; Brinkmann et al., 2018), and Colombian Dermacentor nitens ticks (Lopez et al., 2020). LTV GD was closely related to the variants found in China (Xu et al., 2021), France (Gondard et al., 2020), and Brazil (Souza et al., 2018). The discovery of LTVs in various tick species worldwide suggests that it might present a low degree of host restriction. In addition, based on the co-presence of BDTPV1 and BDTPV2 in all five R. Sanguineus tick pools, consistent with a study in Latin America (Sameroff et al., 2019), we hypothesized that BDTPV1 and BDTPV2 may extensively co-infect R. Sanguineus or had a symbiotic relationship with R. Sanguineus. Analysis of additional tick species from different geographic regions will help confirm the evolutionary association of these viruses with their tick hosts.
Rhabdoviruses can infect a wide range of hosts, including vertebrates, arthropods and plants, and the family has 3 subfamilies (Alpharhabdovirinea, Betarhabdovirinae, and Gammarhabdovirinae). Alpharicinrhavirus is a genus of Alpharhabdovirinea, containing three species of viruses discovered in ticks. Wuhan tick virus 1 (WTV1), belonging to the Alpharhabdovirinea subfamily, was first detected in R. microplus from Wuhan, China (Li et al., 2015). WTV1 GD and other reported strains clustered together, showing the conservation of genetic evolution in geographical locations.
Reoviruses have been identified in ticks for decades, in which the Colorado tick fever virus is responsible for an acute systemic febrile illness in humans in the United States and Canada (Klasco, 2002). However, tick-borne reovirus remains to be determined in China. GTOV identified in R. microplus shared 84.6% polymerase and 78.4% inner core protein identity to the Wad Medani virus, suggesting GTOV may represent a novel reovirus species. However, the pathogenicity of GTOV needs to be further investigated. GTOV, first reported in China, showed a close relationship with available WMV strains isolated from Rhipicephalus in Sudan in 1952 (Liu et al., 2016). Previous high-throughput sequencing studies on ticks in China have failed to detect the virus. It is possible that GTOV is not a native virus but instead transmitted to China through livestock trade or migratory birds. Moreover, intersegment reassortment is likely to be as a source of genetic diversity in tick-borne orbiviruses, and multiple reassortment events have also been found within the WMV group.
We also detected bovine hepacivirus in the family Flaviviridae in ticks collected from cattle, where ticks may be the mechanical carrier (Shao et al., 2021). Further investigation is needed to confirm the pathogenicity of bovine hepacivirus and possible transmission routes of bovine hepacivirus.
This study investigated the virome of Rhipicephalus ticks in Guangdong, southern China, and detected 10 viruses, including Lihan tick virus, Brown dog tick phlebovirus 1 and 2 in the family Phenuiviridae, Mivirus and Wuhan tick virus 2 in the family Chuviridae, Wuhan tick virus 1 in the family Rhabdoviridae, Hepacivirus N in the family Flaviviridae, Guangdong tick quaranjavirus (GTQV) in the family Orthomyxoviridae, Guangdong tick orbivirus (GTOV) in the family Reoviridae, and Guangdong tick Manly virus (GTMV) of an unclassified family, in which most of them were genetically related to the virus strains in countries outside China, and GTQV, GTOV, and GTMV may represent novel viral species. These findings provided the evidences of the long-distance spread of these TBVs in Guangdong, southern China, suggesting the necessity and importance of TBV surveillance.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
ZW and QL conceived the project. JM, JL, WH, LZ, and KZ collected the samples. LG, MC, WL, JZ, QJ, HH, and LZ conducted experiment. LG and QL analyzed the data and drafted the manuscript. All authors were involved in critically revised the manuscript and approved the final version.
The study was funded by the Science and Technology Innovation Project in Foshan, Guangdong Province, China (2020001000151), the Pearl River Talent Plan in Guangdong Province of China (2019CX01N111), and Guangzhou Science and Technology Project of China (202103000008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.966735/full#supplementary-material
Allen, T., Murray, K. A., Zambrana-Torrelio, C., Morse, S. S., Rondinini, C., Di Marco, M., et al. (2017). Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 8:1124. doi: 10.1038/s41467-017-00923-8
Allison, A. B., Ballard, J. R., Tesh, R. B., Brown, J. D., Ruder, M. G., Keel, M. K., et al. (2015). Cyclic avian mass mortality in the northeastern United States is associated with a novel orthomyxovirus. J. Virol. 89, 1389–1403. doi: 10.1128/JVI.02019-14
Belaganahalli, M. N., Maan, S., Maan, N. S., Brownlie, J., Tesh, R., Attoui, H., et al. (2015). Genetic characterization of the tick-borne orbiviruses. Viruses 7, 2185–2209. doi: 10.3390/v7052185
Brinkmann, A., Dincer, E., Polat, C., Hekimoglu, O., Hacioglu, S., Foldes, K., et al. (2018). A metagenomic survey identifies Tamdy orthonairovirus as well as divergent phlebo-, rhabdo-, chu- and flavi-like viruses in Anatolia. Ticks Tick Borne Dis. 9, 1173–1183. doi: 10.1016/j.ttbdis.2018.04.017
Cholleti, H., Hayer, J., Mulandane, F. C., Falk, K., Fafetine, J., Berg, M., et al. (2018). Viral metagenomics reveals the presence of highly divergent quaranjavirus in Rhipicephalus ticks from Mozambique. Infect Ecol Epidemiol 8:1478585. doi: 10.1080/20008686.2018.1478585
Dedkov, V. G., Dolgova, A. S., Safonova, M. V., Samoilov, A. E., Belova, O. A., Kholodilov, I. S., et al. (2021). Isolation and characterization of Wad Medani virus obtained in the Tuva Republic of Russia. Ticks Tick Borne Dis. 12:101612. doi: 10.1016/j.ttbdis.2020.101612
Dincer, E., Brinkmann, A., Hekimoglu, O., Hacioglu, S., Foldes, K., Karapinar, Z., et al. (2017). Generic amplification and next generation sequencing reveal Crimean-Congo hemorrhagic fever virus AP92-like strain and distinct tick phleboviruses in Anatolia, Turkey. Parasit. Vectors 10:335. doi: 10.1186/s13071-017-2279-1
Dong, Z., Yang, M., Wang, Z., Zhao, S., Xie, S., Yang, Y., et al. (2021). Human Tacheng tick virus 2 infection, China, 2019. Emerg. Infect. Dis. 27, 594–598. doi: 10.3201/eid2702.191486
Gomez, G. F., Isaza, J. P., Segura, J. A., Alzate, J. F., and Gutierrez, L. A. (2020). Metatranscriptomic virome assessment of Rhipicephalus microplus from Colombia. Ticks Tick Borne Dis. 11:101426. doi: 10.1016/j.ttbdis.2020.101426
Gondard, M., Temmam, S., Devillers, E., Pinarello, V., Bigot, T., Chretien, D., et al. (2020). RNA viruses of Amblyomma variegatum and Rhipicephalus microplus and cattle susceptibility in the French Antilles. Viruses 12, 144. doi: 10.3390/v12020144
Gong, S., He, B., Wang, Z., Shang, L., Wei, F., Liu, Q., et al. (2015). Nairobi sheep disease virus RNA in ixodid ticks, China, 2013. Emerg. Infect. Dis. 21, 718–720. doi: 10.3201/eid2104.141602
Guindon, S., Lethiec, F., Duroux, P., and Gascuel, O. (2005). PHYML online--a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W559. doi: 10.1093/nar/gki352
Harvey, E., Rose, K., Eden, J. S., Lo, N., Abeyasuriya, T., Shi, M., et al. (2019). Extensive diversity of RNA viruses in Australian ticks. J. Virol. 93, e01358–e01418. doi: 10.1128/JVI.01358-18
He, T., Zhu, C., Li, Z., Ai, L., Hu, D., Wang, C., et al. (2022). Virome analysis of ticks in Zhoushan Archipelago, China. J. Vet. Med. Sci. 17, 847–854. doi: 10.1292/jvms.22-0058
Jia, N., Liu, H. B., Ni, X. B., Bell-Sakyi, L., Zheng, Y. C., Song, J. L., et al. (2019). Emergence of human infection with Jingmen tick virus in China: a retrospective study. EBioMedicine 43, 317–324. doi: 10.1016/j.ebiom.2019.04.004
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Klasco, R. (2002). Colorado tick fever. Med. Clin. North Am. 86, 435–440. doi: 10.1016/S0025-7125(03)00096-8
Li, C. X., Shi, M., Tian, J. H., Lin, X. D., Kang, Y. J., Chen, L. J., et al. (2015). Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. elife 4:e05378. doi: 10.7554/eLife.05378
Liu, H., Li, Q., Zhang, X., Li, Z., Wang, Z., Song, M., et al. (2016). Characterization of rickettsiae in ticks in northeastern China. Parasit. Vectors 9:498. doi: 10.1186/s13071-016-1764-2
Liu, X., Zhang, X., Wang, Z., Dong, Z., Xie, S., Jiang, M., et al. (2020). A tentative Tamdy Orthonairovirus related to febrile illness in Northwestern China. Clin. Infect. Dis. 70, 2155–2160. doi: 10.1093/cid/ciz602
Lopez, Y., Miranda, J., Mattar, S., Gonzalez, M., and Rovnak, J. (2020). First report of Lihan tick virus (Phlebovirus, Phenuiviridae) in ticks, Colombia. Virol. J. 17:63. doi: 10.1186/s12985-020-01327-9
Ma, J., Lv, X. L., Zhang, X., Han, S. Z., Wang, Z. D., Li, L., et al. (2021). Identification of a new orthonairovirus associated with human febrile illness in China. Nat. Med. 27, 434–439. doi: 10.1038/s41591-020-01228-y
Meng, F., Ding, M., Tan, Z., Zhao, Z., Xu, L., Wu, J., et al. (2019). Virome analysis of tick-borne viruses in Heilongjiang Province, China. Ticks Tick Borne Dis. 10, 412–420. doi: 10.1016/j.ttbdis.2018.12.002
Pettersson, J. H., Shi, M., Bohlin, J., Eldholm, V., Brynildsrud, O. B., Paulsen, K. M., et al. (2017). Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 7:10870. doi: 10.1038/s41598-017-11439-y
Sameroff, S., Tokarz, R., Charles, R. A., Jain, K., Oleynik, A., Che, X., et al. (2019). Viral diversity of tick species parasitizing cattle and dogs in Trinidad and Tobago. Sci. Rep. 9, 10421. doi: 10.1038/s41598-019-46914-1
Sameroff, S., Tokarz, R., Jain, K., Oleynik, A., Carrington, C. V. F., Lipkin, W. I., et al. (2021). Novel quaranjavirus and other viral sequences identified from ticks parasitizing hunted wildlife in Trinidad and Tobago. Ticks Tick Borne Dis. 12:101730. doi: 10.1016/j.ttbdis.2021.101730
Shao, J. W., Guo, L. Y., Yuan, Y. X., Ma, J., Chen, J. M., and Liu, Q. (2021). A novel subtype of bovine Hepacivirus identified in ticks reveals the genetic diversity and evolution of bovine Hepacivirus. Viruses 13:2206. doi: 10.3390/v13112206
Shi, J., Hu, Z., Deng, F., and Shen, S. (2018). Tick-Borne Viruses. Virol. Sin. 33, 21–43. doi: 10.1007/s12250-018-0019-0
Shi, M., Lin, X. D., Tian, J. H., Chen, L. J., Chen, X., Li, C. X., et al. (2016). Redefining the invertebrate RNA virosphere. Nature 540, 539–543. doi: 10.1038/nature20167
Shi, J., Shen, S., Wu, H., Zhang, Y., and Deng, F. (2021). Metagenomic profiling of viruses associated with Rhipicephalus microplus ticks in Yunnan Province, China. Virol. Sin. 36, 623–635. doi: 10.1007/s12250-020-00319-x
Souza, W. M., Fumagalli, M. J., Torres Carrasco, A. O., Romeiro, M. F., Modha, S., Seki, M. C., et al. (2018). Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 8:16315. doi: 10.1038/s41598-018-34630-1
Taylor, R. M., Hurlbut, H. S., Work, T. H., Kingston, J. R., and Hoogstraal, H. (1966). Arboviruses isolated from Argas ticks in Egypt: Quaranfil, Chenuda, and Nyamanini. Am. J. Trop. Med. Hyg. 15, 76–86. doi: 10.4269/ajtmh.1966.15.76
Temmam, S., Chretien, D., Bigot, T., Dufour, E., Petres, S., Desquesnes, M., et al. (2019). Monitoring silent spillovers before emergence: a pilot study at the tick/human interface in Thailand. Front. Microbiol. 10, 2315. doi: 10.3389/fmicb.2019.02315
Wang, L., Wang, Y., Jin, S., Wu, Z., Chin, D. P., Koplan, J. P., et al. (2008). Emergence and control of infectious diseases in China. Lancet 372, 1598–1605. doi: 10.1016/S0140-6736(08)61365-3
Wang, Z. D., Wang, B., Wei, F., Han, S. Z., Zhang, L., Yang, Z. T., et al. (2019). A new segmented virus associated with human febrile illness in China. N. Engl. J. Med. 380, 2116–2125. doi: 10.1056/NEJMoa1805068
Wang, Y. C., Wei, Z., Lv, X., Han, S., Wang, Z., Fan, C., et al. (2021). A new nairo-like virus associated with human febrile illness in China. Emerg. Microbes Infect. 10, 1200–1208. doi: 10.1080/22221751.2021.1936197
Xu, L., Guo, M., Hu, B., Zhou, H., Yang, W., Hui, L., et al. (2021). Tick virome diversity in Hubei Province, China, and the influence of host ecology. Virus Evol. 7:veab089. doi: 10.1093/ve/veab089
Yadav, P. D., Shete, A. M., Nyayanit, D. A., Albarino, C. G., Jain, S., Guerrero, L. W., et al. (2018). Identification and characterization of novel mosquito-borne (Kammavanpettai virus) and tick-borne (Wad Medani) reoviruses isolated in India. J. Gen. Virol. 99, 991–1000. doi: 10.1099/jgv.0.001102
Yang, Z., Zhang, J., Yang, S., Wang, X., Shen, Q., Sun, G., et al. (2021). Virome analysis of ticks in a forest region of Liaoning, China: characterization of a novel hepe-like virus sequence. Virol. J. 18, 163. doi: 10.1186/s12985-021-01632-x
Yu, X. J., Liang, M. F., Zhang, S. Y., Liu, Y., Li, J. D., Sun, Y. L., et al. (2011). Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 364, 1523–1532. doi: 10.1056/NEJMoa1010095
Keywords: tick-borne viruses, viral diversity, Rhipicephalus ticks, meta-transcriptome, Guangdong
Citation: Guo L, Ma J, Lin J, Chen M, Liu W, Zha J, Jin Q, Hong H, Huang W, Zhang L, Zhang K, Wei Z and Liu Q (2022) Virome of Rhipicephalus ticks by metagenomic analysis in Guangdong, southern China. Front. Microbiol. 13:966735. doi: 10.3389/fmicb.2022.966735
Edited by:
Svetlana Khaiboullina, University of Nevada, Reno, United StatesReviewed by:
Jorge Miranda, Universidad de Córdoba, ColombiaCopyright © 2022 Guo, Ma, Lin, Chen, Liu, Jin, Jin, Hong, Huang, Zhang, Zhang, Wei and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Liu, bGl1cXVhbjE5NzNAaG90bWFpbC5jb20=; Zhengkai Wei, d2VpX3poZW5na2FpQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.