 Maria Rosilene Alves Damasceno1
Maria Rosilene Alves Damasceno1 Camila Gracyelle de Carvalho Lemes2
Camila Gracyelle de Carvalho Lemes2 Lucélia Sandra Silva Barbosa Braga3
Lucélia Sandra Silva Barbosa Braga3 Polyana Cristine Tizioto4
Polyana Cristine Tizioto4 Horácio Montenegro4
Horácio Montenegro4 Marcela Paduan4Josielda Gomes Pereira5Isabella Ferreira Cordeiro2
Marcela Paduan4Josielda Gomes Pereira5Isabella Ferreira Cordeiro2 Lorrana Cachuite Mendes Rocha2Sibele Aryadne da Silva2
Lorrana Cachuite Mendes Rocha2Sibele Aryadne da Silva2 Angélica Bianchini Sanchez2
Angélica Bianchini Sanchez2 Wanderson Geraldo Lima2,6
Wanderson Geraldo Lima2,6 Gabriel Menezes Yazbeck5
Gabriel Menezes Yazbeck5 Leandro Marcio Moreira2,6
Leandro Marcio Moreira2,6 Camila Carrião Machado Garcia1,2,6*
Camila Carrião Machado Garcia1,2,6*- 1Programa de Pós-Graduação em Ciências Biológicas, Núcleo de Pesquisas em Ciências Biológicas, Universidade Federal de Ouro Preto, Ouro Preto, Brazil
- 2Programa de Pós-Graduação em Biotecnologia, Núcleo de Pesquisas em Ciências Biológicas, Universidade Federal de Ouro Preto, Ouro Preto, Brazil
- 3Instituto Federal de Educação, Ciência e Tecnologia do Norte de Minas Gerais, Januária, Brazil
- 4NGS Soluções Genômicas, Piracicaba, Brazil
- 5DEZOO, Universidade Federal de São João del-Rei, São João del-Rei, Brazil
- 6Departamento de Ciências Biológicas, Instituto de Ciências Exatas e Biológicas, Universidade Federal de Ouro Preto, Ouro Preto, Brazil
The São Francisco River (SFR), one of the main Brazilian rivers, has suffered cumulative anthropogenic impacts, leading to ever-decreasing fish stocks and environmental, economic, and social consequences. Rhinelepis aspera and Prochilodus argenteus are medium-sized, bottom-feeding, and rheophilic fishes from the SFR that suffer from these actions. Both species are targeted for spawning and restocking operations due to their relevance in artisanal fisheries, commercial activities, and conservation concerns. Using high-throughput sequencing of the 16S rRNA gene, we characterized the microbiome present in the gills and guts of these species recruited from an impacted SFR region and hatchery tanks (HT). Our results showed that bacterial diversity from the gill and gut at the genera level in both fish species from HT is 87% smaller than in species from the SFR. Furthermore, only 15 and 29% of bacterial genera are shared between gills and guts in R. aspera and P. argenteus from SFR, respectively, showing an intimate relationship between functional differences in organs. In both species from SFR, pathogenic, xenobiont-degrading, and cyanotoxin-producer bacterial genera were found, indicating the critical pollution scenario in which the river finds itself. This study allowed us to conclude that the conditions imposed on fish in the HT act as important modulators of microbial diversity in the analyzed tissues. It also raises questions regarding the effects of these conditions on hatchery spawn fish and their suitability for restocking activities, aggravated by the narrow genetic diversity associated with such freshwater systems.
Introduction
Human impacts on fish biodiversity affect more than 50% of the world’s freshwater systems (Su et al., 2021). The Neotropical region harbors over 6.200 named freshwater fish species, making it the most diverse continental vertebrate fauna on the planet (Albert et al., 2020). The São Francisco River (SFR) is the fifth-largest river in South America, more than 2,700 km in length, enclosed in a 639.219 km2 eponymous basin, and it constitutes the most extensive river residing entirely inside Brazilian territory (Sun et al., 2016) (Figure 1). It cuts through two major biomes, the Brazilian savannah, Cerrado, and the Caatinga, allowing for agricultural operations around it in otherwise dry regions, as well as inland artisanal and commercial fisheries (Holanda et al., 2009; Maneta et al., 2009; Torres et al., 2012).
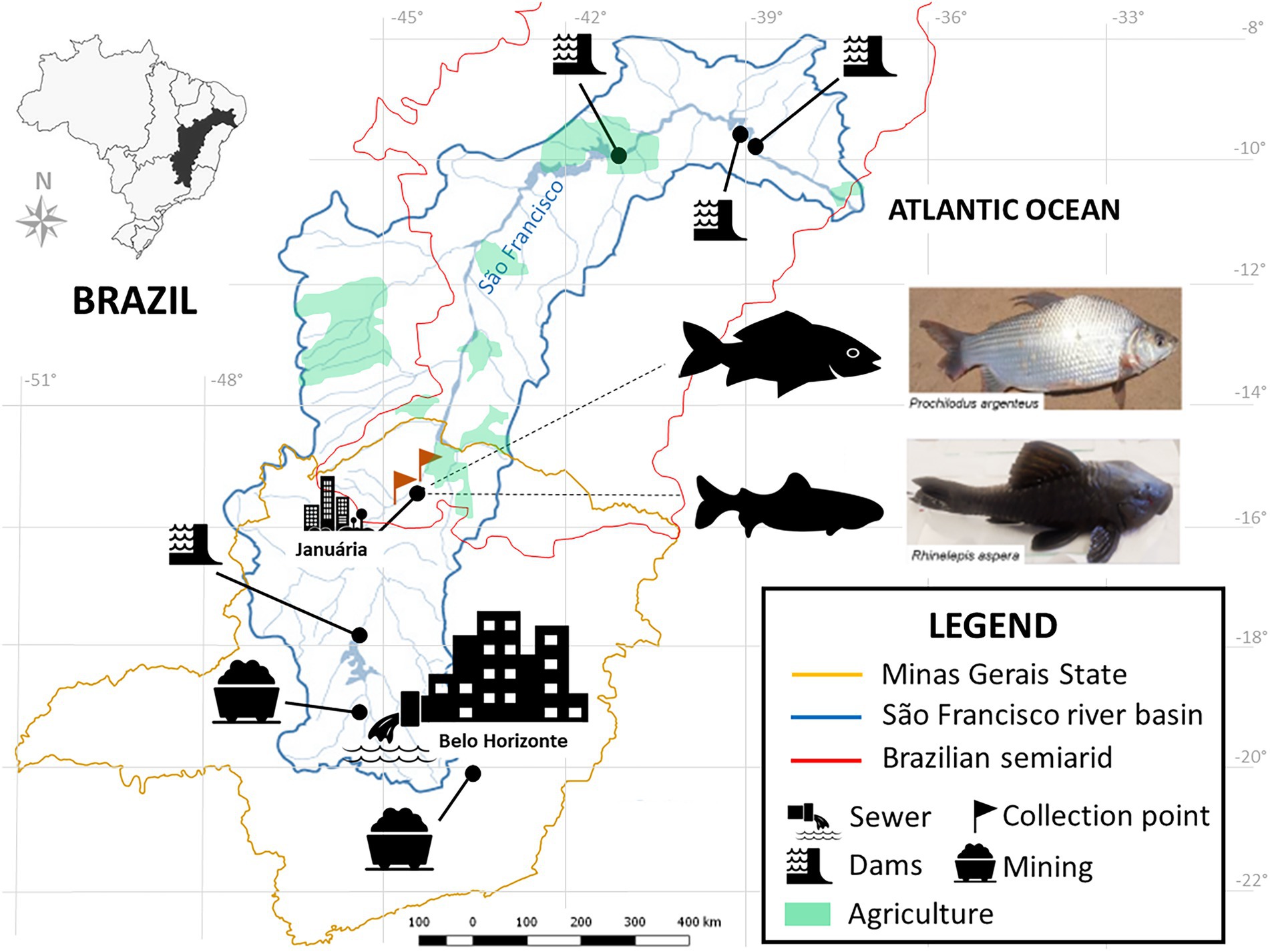
Figure 1. Location of the SFR Basin, highlighting the region of collection and the species of ichthyofauna studied. Fish collection site in the middle SFR region, upstream and downstream of the city of Januária, North of Minas Gerais. Brazil. Google. The map was obtained from the Pristine Institute: digital geoenvironmental atlas. WebGis (Web Geographical Information System) with “free access to the environmental database.” Available at <https://institutopristino.org.br/atlas/>. Access on 25/Jun/2020.
The SFR is widely afflicted by the cumulative effects of several anthropogenic impact sources, such as the introduction of invasive species (Linares et al., 2019); suppression of riverine vegetation (Holanda et al., 2009); mining and damming for hydropower generation activities (Nascimento and Becker, 2010), and urban and rural pollution (Ribeiro et al., 2012) (Figure 1). In addition, during the last decade, unstable hydrological regimes in dry regions (Otto et al., 2015; Getirana, 2016; Gudmundsson et al., 2021) and overfishing have contributed to ever-decreasing fish stocks (Barbosa et al., 2017; Figueiredo, 2018). Thus, sustainability in the SFR is threatened, jeopardizing the livelihood of its communities of part-time and full-time fishing folk (Gutberlet et al., 2007; Alves and Leal, 2010; Barbosa et al., 2017).
Fish spawning and stocking operations have been set as hatchery tanks (HT) for fish farming, employed to produce angling opportunities, commercial sales, and to counteract environmental pressures (Arantes et al., 2011). However, its efficiency remains to be shown, especially if disconnected from broader policy actions to address the root causes of fish stock decline in the SFR (Araki and Schmid, 2010; González-Wangüemert et al., 2012; Klinger and Naylor, 2012; Savary et al., 2017). In addition, there are concerns related to the potentially deleterious effects of genetic diversity impoverishment in spawned fish and sanitary hazards observed in hatcheries (González-Wangüemert et al., 2012; Carmo et al., 2015; Benson et al., 2016; Pavlova et al., 2017; Savary et al., 2017; FAO, 2020).
Environmental factors, both biotic and abiotic, modulate the assemblage of microorganisms (microbiome) associated with organisms and their different morphophysiological features (Sullam et al., 2012; Duarte et al., 2014; Ringo et al., 2016; Sylvain et al., 2016; Guivier et al., 2017).
Few studies are addressing the morphophysiological changes caused to Neotropical fish by chronic exposure to environmental toxins in their natural habitat (Paschoalini and Bazzoli, 2021). Also, the presented data are not robust enough to link aquatic contamination to mortality risks of the ichthyofauna species (Molbert et al., 2021). However, there are reports of histological and molecular changes in the liver, spleen, and gills of P. argenteus recruited from regions impacted by heavy metals in the SFR basin. Among them, a significant increase in melanomacrophage centers in the liver and spleen, a high incidence of fibrosis in the spleen, alterations in the number and morphology of germ cells, and also endocrine and reproductive dysfunctions, with a negative effect on the gonadal development of females (Paschoalini et al., 2019). In the gills, increased inflammatory foci, hyperplasia, edema, and lamellar fusion were reported (Arantes et al., 2011; Procópio et al., 2014). Additionally, toxicogenetic biomarkers positively correlated the increased frequency of micronuclei in erythrocytes (Shirmohammadi et al., 2018; Delcorso et al., 2020) with the speed of telomere shortening (Molbert et al., 2021).
Changes in microbiome composition have been implicated in increased disease occurrence, population size decline, and other adverse effects in animals (Sylvain et al., 2016; Pękala-Safińska, 2018). Also, the microbial composition is related to vital metabolic processes in the host (Izvekova et al., 2007; Lowrey et al., 2015; Liu et al., 2016; Sylvain et al., 2016; van Kessel et al., 2016).
Significant advances toward a complete understanding of microbial-environmental interactions have been strengthened by the development of culture-free methods in microbiology, such as high-throughput sequencing of specific genes or the assembly of complete microbial genomes (Faust and Raes, 2012; Blainey, 2013; van Dijk et al., 2014), which allows the identification of a myriad of prokaryotic species, more significant in scope than with the few species eventually detected by growth on culture media.
The importance of fish-microbiome interaction, its relationship to aquaculture, and its role in survival and performance has been acknowledged in this century (van Kessel et al., 2011; Ringo et al., 2016; Tarnecki et al., 2016; Perry et al., 2020). Characterizing microbiome composition could help craft ways to facilitate the functional enhancement of nutritional, immunologic, and other beneficial traits in reared fish (Perry et al., 2020). Studies involving fish have provided insights into the importance of microbiome associated with the skin, gills, and gut, allowing for the characterization of factors influencing microbial diversity in both wild and reared species; at least 145 fish gut microbiomes have been analyzed to date (Roeselers et al., 2011; Sylvain et al., 2016, 2019; Tarnecki et al., 2016; Uren Webster et al., 2018; Krotman et al., 2020; Nolorbe-Payahua et al., 2020; Perry et al., 2020; Riiser et al., 2020). Nevertheless, most of these studies have been performed on economically relevant fish such as Cyprinus carpio (van Kessel et al., 2016), Ctenopharyngodon idellus (Wu et al., 2012), Salmo salar (Gajardo et al., 2016; Schmidt et al., 2016), Oreochromis niloticus (Yu et al., 2019; Xia et al., 2020), Oncorhynchus mykiss (Lowrey et al., 2015), Colossoma macropomum (Sylvain et al., 2016), Arapaimas gigas (Ramírez et al., 2018), Seriola lalandi (Legrand et al., 2018) and Ictalurus punctatus (Larsen et al., 2014). Thus, there is still a relatively low number of studies focusing on microbiomes in wild living fish. Studies involving fish microbiomes from the SFR are still scarce (Silva et al., 2005; Makino et al., 2012; Santos et al., 2014; Da Silva et al., 2020). Even though important, they are traditionally limited by culture-dependent methods.
So far, most metagenome research has focused on the fish gut microbiota. These studies have shown that diet and physicochemical variations of water are strong modulators of microbial diversity, leading to metabolic dysbiosis, increased susceptibility to infections, and reduced environmental adaptability (Ning et al., 2020). Such alterations were observed in Gambusia affinis and Danio rerio exposed to Benzo(a)pyrene, which induced intestinal dysbiosis and the increased expression of genes related to the inflammatory response (Xie et al., 2020). Exposure of D. rerio to titanium dioxide nanoparticles and Bisphenol A induced an increase in the Lawsonia genera, a pathogen of the gastrointestinal tract of animals, and a decrease in Hyphomicrobium associated with denitrification metabolism (Chen et al., 2018). Reported as polycyclic aromatic hydrocarbons (PAHs) degraders, the bacterial genera Novosphingobium, Sphingobium, and Sphingomonas were found in the intestinal microbiota of Gadus morhua collected in impacted environments by oil contamination while absent in unimpacted environments (Walter et al., 2019).
This work aimed to characterize the microbiomes present in the gills and guts of two relevant bottom-feeding species from the SFR (R. aspera and P. argenteus) and to establish a comparison with the same species from an HT for fish farming intended for restocking activities through a culture-free approach through high-throughput sequencing of the 16S rRNA gene.
In this study, we hypothesized that benthic fish, iliophagous detritivores, which differ in behavior in their natural habitat, being one a long-distance migrator (P. argenteus) and the other a sedentary one (R. aspera), are exposed to different levels of environmental stress and physicochemical gradients of water, differentially modulating the structure and diversity of the intestinal and gill microbiome of these species. Additionally, this study was the first to characterize the microbiomes of these two ichthyic species recruited in the SFR, providing important insights into the effects of confinement on the structure and diversity of the investigated microbiomes, pointing out vulnerabilities associated with commercial aquaculture and restocking programs, and aiming at the restoration of natural ichthyofauna stocks.
Methodology
The complete methodological workflow of this study is presented in Figure 2A. Two species (R. aspera and P. argenteus) were selected based on relevance in fisheries, drastic stock reduction, and vulnerability. In addition, as bottom feeders, both species constitute adequate water column indicators and sediment contamination (Camargo et al., 2009; Islam et al., 2015; Paschoalini et al., 2019).

Figure 2. (A) Experimental design. The planning steps are on a yellow background, the collection and processing steps on a salmon background, and the steps to obtain results and data analysis on a green background. Photos that, respectively, denote degradation of riparian forests (B), which causes landslides (C) that in turn contribute to silting up of the SFR bed (D). (E) Evident eutrophication of the SFR waters at the time of the first collection is best proven when trawls are used (F). (G,H) HT structure where they were collected.
Morphophysiological and behavioral characterization of species
Rhinelepis aspera (Spix and Agassiz, 1829) is a medium-sized, stream-dwelling, melanic, armored catfish locally known as acari or cascudo preto (Siluriformes order, the Loricariidae family (Britski et al., 1988; Buckup et al., 2007). It is among the single largest species (> 4 kg) within this family, which, in turn, is characterized by the presence of thick bony plates covering the skin, ventral suckermouth, and round eye’s iris (Covain and Fisch-Muller, 2007). The specie R. aspera exhibits obligatory male parental care of young adults and lacks long migratory movements associated with reproduction (wet season), which is common in large Neotropical catfish (Suzuki et al., 2000; Sato et al., 2003). The International Union for Conservation of Nature does not assess its conservation status (International Union for Conservation of Nature and Natural Resources –IUCN, 2022). Still, it was deemed a species endangered at the end of the 20th century, and it has been reported as rare in the main body of the SFR basin, although abundant in its main tributary, the Paracatu River (Sato et al., 1998). It shows a specialized mouth, adapted to its iliophagous lifestyle, being an important organic matter and nutrient recycling player (Roxo et al., 2019). This fish is highly adaptable to lentic environments, and its high fecundity makes it a natural target for fish farming initiatives in the SFR region.
The specie P. argenteus, Spix and Agassiz, 1829, is one of the two species of the Prochilodus genera (Characiformes order, Prochilodontidae family (Britski et al., 1988; Castro and Vari, 2004) occurring in the SFR basin. This region could have served as a dispersal departure point for the evolutionary radiation of the five species in southeastern Brazil (Santos et al., 2021). P. argenteus is the largest species from its family (Sato et al., 2003) and is popularly known as zulega, curimatã-pacu, curimba, or papa-terra (Perini et al., 2021). It can reach approximately 45 cm in length, up to 15 kg, and it is one of the largest and most prominent species for artisanal and commercial fisheries in the SFR (Godinho and Godinho, 2003; Sato et al., 2005; Godinho et al., 2017). It is a typical potamodromous migratory Neotropical fish that performs long upstream movements in the wet season (Sato et al., 2003, 2005), with high tolerance and suitability for aquaculture (Almeida et al., 2015).
Collection sites and experimental design
This study was conducted in the middle portion of the SFR, immediately upstream and downstream of the city of Januária (S 15°29.576’ W 044°21°0.069′), in the north of the Minas Gerais estate, Brazil (Figure 1). It is a major regional center, with a population of approximately 65.463 and a human development index of 0.658, with only 37.2% of households served by sewage (IBGE, 2010).
Rhinelepis aspera and P. argenteus specimens were sampled from the SFR (Figures 2B–G) and an HT from Januária (S 15°28′ 48.94752” W 044°21′42.50952″). This tank was built in tiled masonry above the ground, supplied with water from the river after being treated by the Companhia de Saneamento de Minas Gerais (COPASA).
The Instituto Mineiro de Gestão das Águas em Minas Gerais (IGAM, 2018) has classified the water from the sampling point as Class II, with several diagnostic features above the limits established by Brazilian legislation (BRASIL, 2005) like dissolved aluminum, total manganese, lead, total solids, turbidity, biochemical oxygen demand (BOD), and cyanobacteria density (visually confirmed during sampling; Supplementary Table 1 and Figures 2E,F).
Three fish were collected from each species in the SFR and HT, totaling 12 individuals. Fish were trapped with a dragnet (Figure 2F) by professional fishers from the Associação de Pescadores de Januária in July 2018. Two male and one female specimen of the fish were taken alive to the Laboratório de Zoologia do Instituto Federal de Educação, Ciência e Tecnologia do Norte de Minas Gerais, campus Januária. The fish identification was confirmed according to Britski et al. (1988). One voucher specimen for each species was stored in the laboratory collection mentioned before. The fishes were euthanized to remove the medial portions of the gills and guts using sterile instrumentation under a laminar flow hood.
Total DNA extraction
For associated microbiome DNA extraction, two samples from each fish were collected: gills (G) and medium gut (I) from each species P. argenteus (Pa) and R. aspera (Ra), from SFR or HT (TQ), totaling 24 samples (Table 1). According to the manufacturer’s guidelines, small sections of approximately 3 g of gill and gut of each animal were used for DNA extraction using the NucleoSpin™ Tissue (Macherey-Nagel, Germany) commercial kit followed by NanoDrop™ (Thermo Fisher Scientific, USA) quantification at 260 nm. In addition, DNA integrity was verified using 0.8% agarose gel electrophoresis.
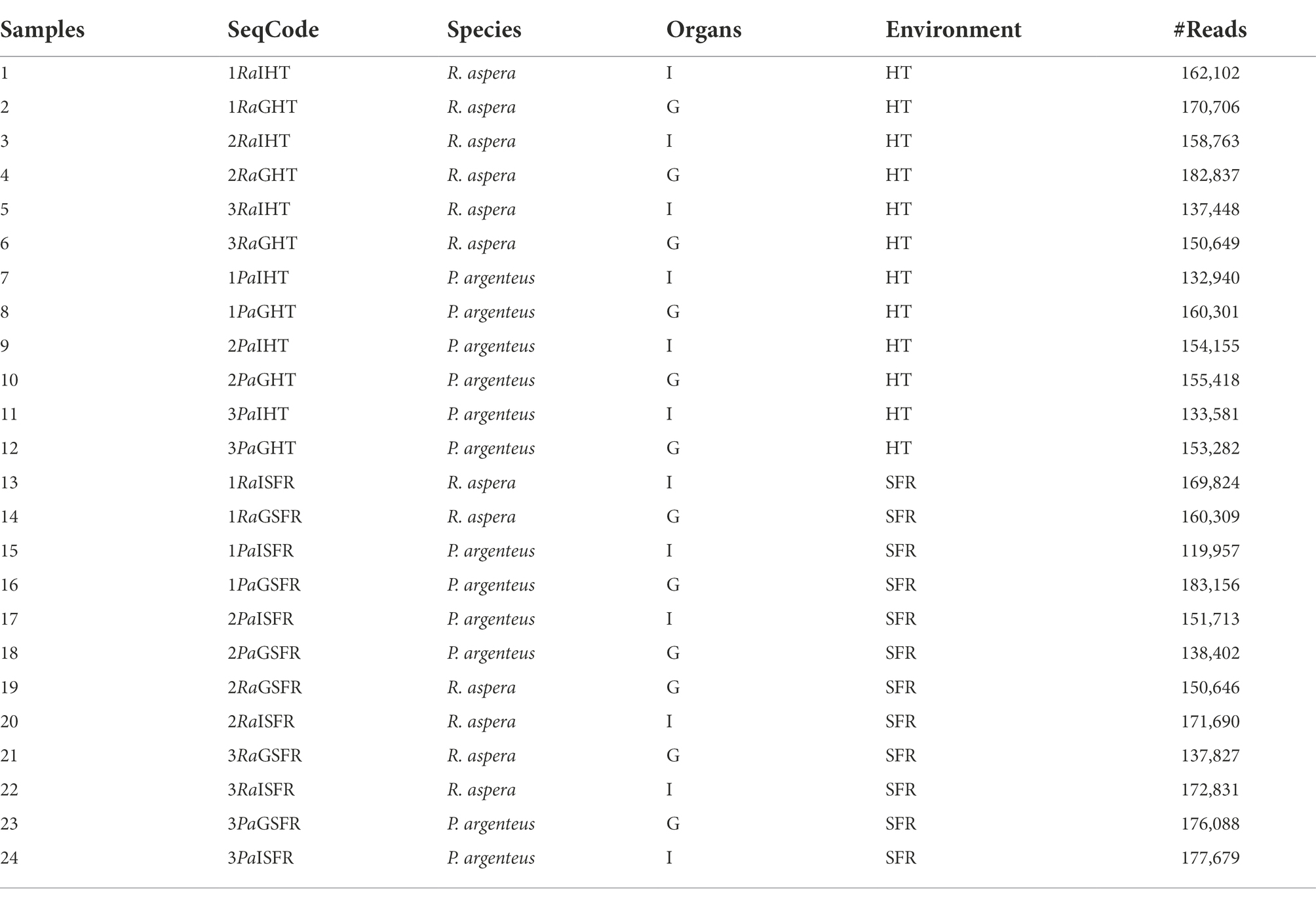
Table 1. Number and characteristics of samples obtained from R. aspera and P. argenteus collected in the SFR and HT.
Partial amplification of the 16S ribosomal gene
The preparation of the libraries was performed following the recommendations for sequencing on the Illumina platform. Oligonucleotides 341F and 785R (Klindworth et al., 2013) were used, which amplify the V3/V4 region of the 16S ribosomal gene with the included overhang adapter sequences. The sequences of the primers (in uppercase) plus adapters (in lowercase) are 341F: (5’tcgtcggcagcgtcagatgtgtataagagacagCCTACGGGNGGCWGCAG) and 785R: (5’gtctcgtgggctcggagatgtgtataagagacagGACTACHVGGGTATCTAATCC). One μL of DNA was used for the amplification, in addition to 0.5 μl of each primer at 10 μM (0.2 μM final concentration in the reaction, in a final volume of 25 μl), 10 μl of the 2X Ultra Mix PCRBio master mix™ (PCR BioSystems, United Kingdom), topping up with ultrapure water for the final volume of 25 μl. The PCR conditions were: 1 cycle of 95°C for 3 min; 25 cycles: 95°C for 30 s, 55°C for 30 s, 72°C for 30 s; 1 cycle of 72°C for 5 min; keep at 4°C indefinitely. The PCR product was applied on a 1.5% agarose gel and then purified with AMPure XP Beads (Beckmann-Coulter, USA). To connect the adapters, 2.5 μl of the amplification product, 2.5 μl of each index (i5 and i7), 12.5 μl of the 2X Ultra Mix PCRBio master mix™ (PCR BioSystems, USA), and 5 μl of ultra-water were used to adjust the final volume at 25 μl. The PCR conditions for connecting the adapters were: 95°C for 3 min; 8 cycles: 95°C for 30 s, 55°C for 30 s, 72°C for 30 s; 1 cycle of 72°C for 5 min; keep at 4°C indefinitely. The ligation product was purified with beads and then applied on a 1.5% agarose gel. The libraries were quantified with a NanoDrop@ instrument, and their concentration was normalized. An equimolar pool was made with all of them. The pool was quantified by qPCR using the KAPA Biosystems kit™ (KAPA Biosystems, USA) to estimate its concentration and load the MiSeq with 20% Phix. The sequencing was performed with paired readings of 2× 250 bp.
Sequencing analysis
The data were analyzed according to Callahan et al. (2016b) using a set of R packages available through the BioConductor project (Gentleman et al., 2004; Huber et al., 2015). The DADA2 package (Callahan et al., 2016a) was used for trimming and filtering low-quality reads, modeling and correcting amplicon errors, and deduplicating and overlapping the forward and reverse reads. After the removal of chimeric sequences, taxonomies were assigned to each ASV (Amplicon Sequencing Variant) using the DADA2 implementation of the naive Bayesian classifier (Wang et al., 2007), using the Genome Taxonomy Database (GTDB) release 95 as reference (Parks et al., 2017).
Taxonomic classifications generated by DADA2 and their quantifications were imported into the Phyloseq R package (McMurdie and Holmes, 2013). ASVs were grouped at the genera level, and ASVs that were not classified at the family level were filtered out. After these filters, the tables of counts, relative abundance, and Alpha and Beta diversity were obtained. The limma R package (Ritchie et al., 2015) was used to perform the differential abundance analysis. In short, counts were transformed to log2 (counts per million) (Law et al., 2014), which allows the linear models implemented in the limma package to be used to count data. Factors were created for each combination of species + tissue + environment, and the differential taxonomic abundance was tested for each contrast of interest with moderate t-tests (Smyth, 2004). The ordinate function from phyloseq was then used to calculate the Unifrac distance (Hamady et al., 2010), and the graphs of the Principal Coordinate Analysis (PCoA) were generated from these distances (Lozupone and Knight, 2005). Ellipses were computed for the ordination plot with stat_ellipse function from ggplot2 v.3.3.3 (Wickham, 2016), considering a multivariate t-distribution with a 0.95 level. The heatmap analysis was generated using the HTML server http://www.heatmapper.ca/ (Babicki et al., 2016).
Raw sequencing reads were deposited at NCBI Sequence Read Archive under BioProject PRJNA768447. The datasets generated for this study can be found in the NCBI under BioSample accessions SAMN22044346 - SAMN22044369.
Results
Characterization of α-diversity
The microbiomes associated with the gills and guts of R. aspera and P. argenteus from the SFR and HT were compared (Figure 3A). Different microbial diversity levels were observed between the gills of fish from the same species originating from the SFR and HT (Shannon’s index [F = 0.0006] and Simpson’s index [F = 0.01974]). Also, microbiomes from the gills and guts showed higher α-diversity in fish from the river than in the hatchery counterparts (Figure 3B and Supplementary Table 2).
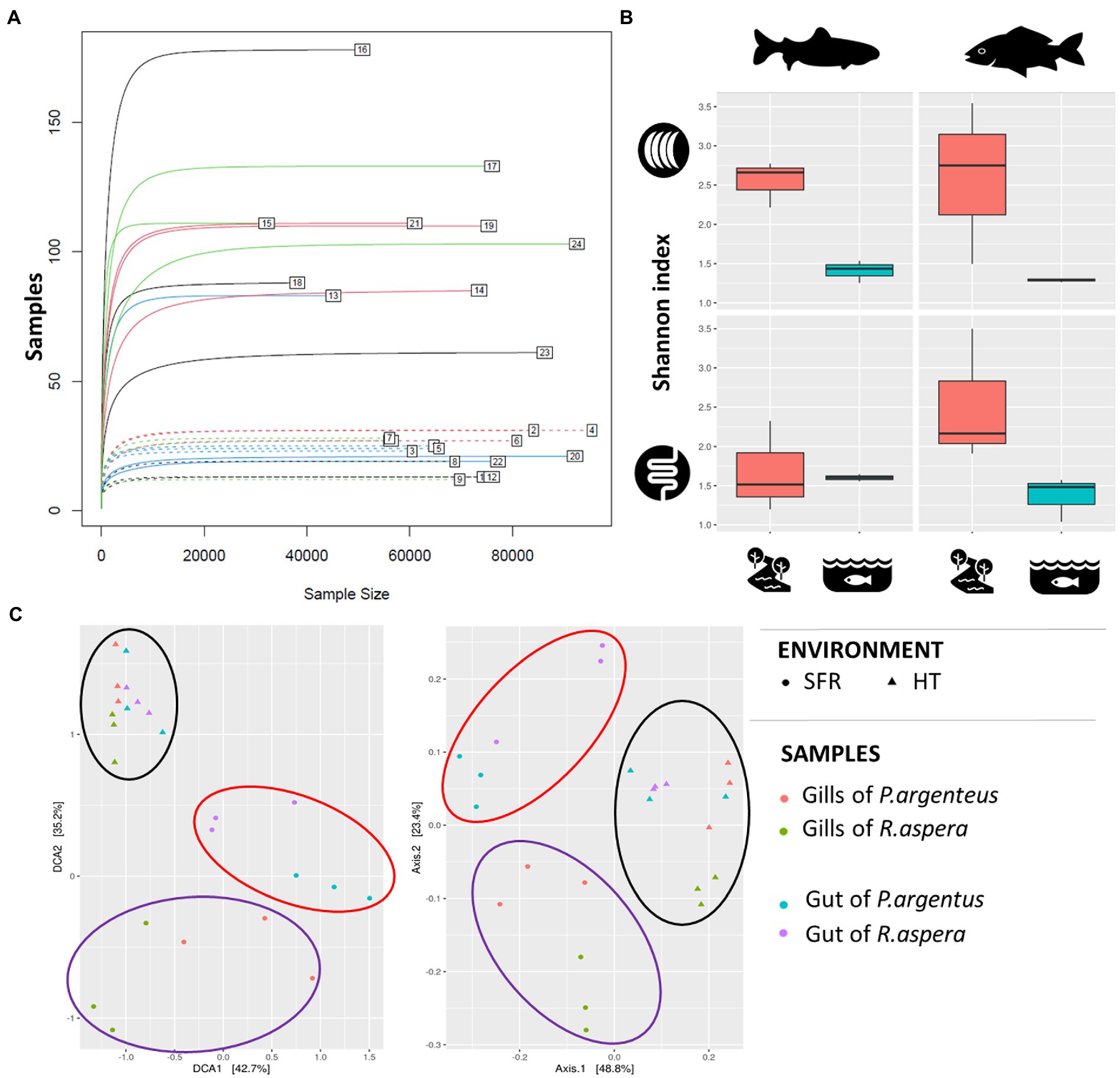
Figure 3. (A) Rarefaction curve. The numbers indicate each of the samples described in Table 1. (B) Alpha-diversity indices of the microbiota associated with the gills and guts of R. aspera and P. argenteus from SFR and HT. (C) Diversity β is expressed as dispersion, given by the metric of the weighted UniFrac distance to evaluate the similarity between the different species tissues and habitats. Each sample is represented by a triangle (from HT) or a circle (from SFR). The red and blue triangles and circles inherit from the gills and guts of P. argenteus, while those from green and purple nuclei inherit from the gills and guts of R. aspera.
Characterization of β-diversity
Scatter analysis revealed that the clustering pattern of the microbiome was markedly affected by fish origin – SFR or HT (Figure 3C). In addition, there was a sharp distinction between microbial communities associated with each environmental source of fish for each analyzed organ in both species.
Taxonomic classification
A total of 33 phyla, 61 classes, 133 orders, 220 families, and 295 genera distributed within the gills and guts of R. aspera and P. argenteus from the SFR and hatchery systems were resolved. The most abundant phyla were Proteobacteria, Fusobacteriota, Bacteroidota, Planctomycetes, Actinobacteria, Cyanobacteria, and Verrumicrobia. Among these, Proteobacteria and Fusobacteriota were predominant in both fish species (Supplementary Table 3 and Supplementary Figure 1), regardless of the environmental source, with the former being more abundant in the SFR (45.11 and 51.3% from R. aspera and 74 and 74.75% from P. argenteus), respectively in gills and gut and the latter being more abundant in the HT environment (46 and 49% from R. aspera and 42 and 68% from P. argenteus, respectively in gills and gut. The Bacteroidota phylum was abundant in the gills of R. aspera from SFR (16%) and the guts of P. argenteus from HT (8.8%). Besides these shared phyla, an exclusive phylum was detected for the SFR: Cyanobacteria, whose abundance varied from 0.87 to 3% in the gills and guts of both species. Other rare phyla were associated with the gills and gut of both species (Armatimonadota, Firmicutes_C, Gemmatimonadota and Spirochaetota), only with the gut of both species (Acidobacteriota, Chloroflexota-A, Dependentiae, and Desulfobacteriota, Myxococcota), only with the gills of P. argenteus (Chloroflexota, Elusimicrobiota, and Desulfuromonadota); only the gut of P. argenteus (Binatota and Dormibacterota); only the gut of R. aspera (Firmicutes_B); associated with the gills of both species and the gut of P. argenteus (Deinoccota) (Supplementary Table 3 and Supplementary Figure 1).
At the genera level, the highest diversity was recorded in fish from the SFR (289) when compared with those from the HT (39) (Supplementary Table 3 and Supplementary Figure 2). Furthermore, among the fish from the SFR, genera diversity was higher in both the gut (138) and gills (155) of P. argenteus than in the counterparts (54 and 119, respectively) of R. aspera (Supplementary Table 3 and Supplementary Figure 2).
Microbiome characterization of Rhinelepis aspera and Prochilodus argenteus × environment
The number of unique and shared genera of microorganisms among species, environments, and organs can be viewed in Figures 4A–D. Taxonomic details are presented in Table 2. The gills and gut microbiomes of R. aspera from both environments showed 155 bacterial taxa for the genera, whereas those of P. argenteus had 234 identified taxa (Figures 4A,B). Only 6.45% (10 genera) were shared among organs and environments for the sampled R. aspera. In contrast, only 3.41% were common among the 234 identified taxa in the sampled P. argenteus (Figures 4A,B).
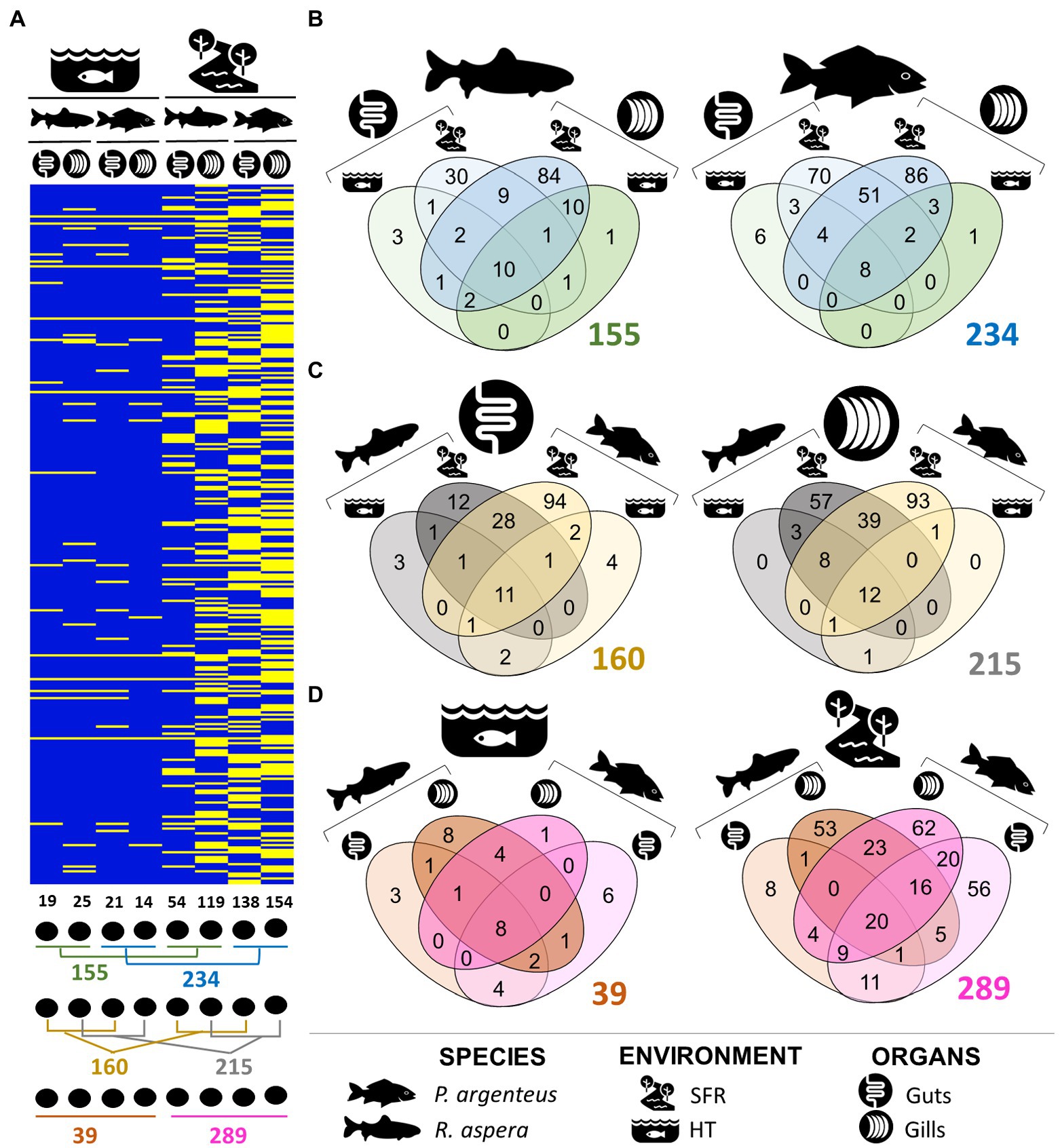
Figure 4. (A) Presence and absence of ASVs that attributed taxonomies at the genera level, illustrating the differences in the structure of the gill and intestinal microbiomes of R. aspera and P. argenteus collected in SFR and HT. Below the heatmap are shown the ways of comparison whose numbers are expressed in Venn diagrams (B–D) indicating the number of total, shared, and unique phylotypes in terms of genera level of the microorganisms associated with samples obtained from fish, according to the host species resident in different ecological systems (B), different colonized body niches (C) and the host habitats (D).

Table 2. Unique communities at the genera level of the gill and gut microbiomes of species recruited from the SFR and HT.
Microbiome characterization of Rhinelepis aspera and Prochilodus argenteus × organs
The abundance of exclusive genera belonging to the microbiomes associated with different organs in each host species (Figure 4C) indicates the occurrence of interspecific phylogenetic variability that distinguishes the microbiome present in these structures. Furthermore, the distinction between bacterial communities in different organs of P. argenteus and R. aspera in the SFR was supported by the shared genera proportions of 28.63 and 14.56%, respectively. The same trend of microbial diversity reduction was observed in P. argenteus from HT (Figure 4D).
Microbiota associated only with SFR fishes
The R. aspera gill microbiome comprised 21 phyla, 30 classes, 98 families, and 119 genera. Among them, 84 exclusive (54.2%) to SFR (Figure 4B). Proteobacteria (45.41%), Fusobacteriota (11.66%), and Bacteroidota (16%) were the dominant phyla (Supplementary Figure 1). The phylum Fibrobacteriota (<1%) was exclusively associated with the gill microbiota of this species. The phylum Proteobacteria was identified by 68 ASVs that attributed taxonomical classification to 58 genera, with 19 affiliated with Betaproteobacteria, 16 with Gammaproteobacteria, and 13 with Alphaproteobacteria.
The most diverse orders were Burkholderiales, Enterobacterales, and Pseudomonadales. The phylum Bacteroidota was identified by 44 ASVs that classified 33 genera, distributed mainly by the orders Cytophagales, Flavobacteriales, Bacteroidales, and Chitinophagales (Supplementary Figure 1). Interestingly, the phylum Fusobacteriota was identified only by two ASVs, and by an integrated gill microbiome with an average relative abundance of approximately 20%. The phyla Bacteroidota and Proteobacteria comprised 38 exclusive genera to the gill microbiome of R. aspera, including Taibaiella_B, Fluviicola, Runella, Flexibacter, Pseudopedobacter, Solirubrum, Pseudoxanthomonas_A, Porphyrobacter, Cellvibrio, Dechloromonas, and Methylophilus. The most abundant genera were Aeromonas (18%), Cetobacterium_A (19.6%), Flavobacterium (16.1%), Shewanella (5.7%), Pseudoduganella (4.34%), Vogesella (4.23%), Acinetobacter (3%), and Plesiomonas (3%) (Supplementary Figure 2).
The gut microbiome of R. aspera from the SFR exhibited the lowest observed diversity among other microbiomes in this environment (Figures 4A–C), with 18 phyla, 22 classes, 49 orders, 66 families, and 54 genera, with 30 unique phylotypes (55.55%). The dominant phyla (Supplementary Figure 1) were Proteobacteria (55.44%), identified by 29 ASVs, followed by Fusobacteriota, identified by only three ASVs, representing approximately 28%. Other less abundant phyla included Actinobacteriota (0.72%), Bacteroidota (0.7%), Firmicutes (0.04%), Cyanobacteria (3%), and Planctomycetota (3%). In contrast to the gill microbiome of this species, the phylum Bacteroidota was represented by the families Bacteroidaceae, Weeksellaceae, and Tannerellaceae. The phylum Proteobacteria was diverse, with microorganisms from the class Gammaproteobacteria, from the order Enterobacteriales, and the Aeromonadaceae, Shewanellaceae, and Enterobacteriaceae families, with Shewanella (46.17%), Plesiomonas (19.48%), and Aeromonas (5.74%) standing out in abundance (Supplementary Figure 1). The phyla Firmicutes_A and Firmicutes, represented by the classes Clostridia and Bacilli, respectively, constituted 3.1% of the gut microbiome for this species.
Tenant microbial communities of Prochilodus argenteus from the SFR
The tenant gill microbiome of P. argenteus was composed of 23 phyla, 46 classes, 99 orders, 129 families, and 154 genera (Figure 4A), of which 86 (55.84%) were exclusive to this organ (Figure 4B). The dominant phyla were Proteobacteria (74.3%) and Fusobacteriota (14.63%) (Supplementary Figure 1). Some poor abundant phyla were identified by numerically conspicuous sequences, such as Bacteroidota (30 ASVs), Actinobacteria (31 ASVs), and Firmicutes (26 ASVs), which together comprised 58 genera, undoubtedly contributing to increasing microbial diversity but adding less abundant microorganisms to the gill microbiome. Three phyla exclusive to the gill microbiome were observed: Chloroflexota, Desulfuromonadota, and Elusimicrobiota. The most diverse taxa included the genera Aeromonas (28.84%), Cetobacterium_A (14.63%), Plesiomonas (13.33%), Acinetobacter (2.94%), Vogesella (1.55%), Polynucleobacter (1.13%), and other unidentified genera belonging to the Enterobacteriaceae (4%) and Burkhoderiaceae (1.87%) families, in addition to microorganisms exclusive to this organ, Caedimonas (6.22%), and Caedibacter (2.44%).
The gut microbiome was composed of 26 phyla, 36 classes, 93 orders, 136 families, and 138 genera, with 70 of these genera (50.7%) occurring only for this niche in the host (Figure 4B). The phyla Dormibacteriota and Binatota were present only in the gut microbiome of P. argenteus. The dominant phyla were Proteobacteria (47%), Fusobacteriota (25.97%), Planctomycetota (6.2%), Verrumicrobiota (1.7%), and Cyanobacteria (1.46%). Although 31 sequences (ASVs) have been attributed to the phylum Actinobacteria, with 22 genera, this taxon integrated the microbiome with low abundance (<1%). The order Enterobacterales and Fusobacteriales contained the most abundant genera, Aeromonas (7.55%), Plesiomonas (23.24%), and Cetobacterium_A (25.9%), respectively. From the phylum Spirochaetes, the family Brevinemataceae was recorded only in the gut microbiome of P. argenteus. The phylum Dependentiae was observed as part of the intestinal microbiome of both R. aspera and P. argenteus, collected from the SFR.
Microbiota associated only with HT fishes
The gill microbiota of R. aspera reared in HT consisted of 8 phyla, 24 families, 15 orders, 9 classes, and 25 genera. The dominant phyla (Supplementary Figure 1) were Fusobacteriota (49%), Proteobacteria (24.6%), and Bacteroidota (0.76%). Less abundant phyla (<1%) included Actinobacteria, Bdellovibrionata, Firmicutes, Patescibacteria, and Verrumicrobiota. The phylum Firmicutes was represented by a single family, Erysipelotrichaceae, and by a single genus, Anaerorhabdus. The phylum Campylobacterota showed only the genera Aliarcobacter, constituting an exclusive taxon to the gill microbiome of R. aspera and P. argenteus. We found 9 phyla, 10 classes, 16 orders, 21 families, and 19 genera in the gut microbiome. In addition, there were two unique and less abundant genera (<1%), Bosea and Desulfovibrio (Table 2). The samples isolated from both niches were dominated by Fusobacteriaceae and Enterobacteriaceae families. At the genera level, similarly to the fish obtained from the SFR, the gills and gut microbiome were dominated by Cetobacterium_ A (49 and 46%), Plesiomonas (15.7 and 27.9%), and Aeromonas (24 and 5%), Edwardsiella (1.12 and 1.59%) and Bacteroides (0.82 and 1.9%), respectively (Supplementary Figure 2). The Anderseniellaceae family was observed at a higher relative abundance (7.5%), integrating the gut microbiome of R. aspera.
The gill microbiota consisted of 7 phyla, 7 classes, 11 orders, 15 families, and 14 genera (Supplementary Figure 1). The structure of the gill and gut microbiomes was strongly modulated by the environmental conditions imposed by HT (Figure 4A). However, the gill and gut microbial communities did not differ significantly in their taxonomic ASV identities.
The intestinal microbiota was composed of 7 phyla, 10 classes, 17 orders, 24 families, and 21 genera, and among these, 6 exclusive genera were identified (Figure 4B). The dominant and most diverse phyla on the gill microbiota and the gut microbiota of P. argenteus were Proteobacteria (51 and 12.79%) and Fusobacteriota (44 and 60.82%, respectively). In the gills, the genera Aeromonas (16.9%), Cetobacterium_A (42%), and Plesiomonas (33.40%) dominated the microbial assemblage (Supplementary Figure 2). The gut presented a reduced relative abundance of the genera Aeromonas (3.66%) and Plesiomonas (12.79%), as well as an increase in Bacteroides (8.37%) and Edwardsiella (3.96%) (Figure 5).
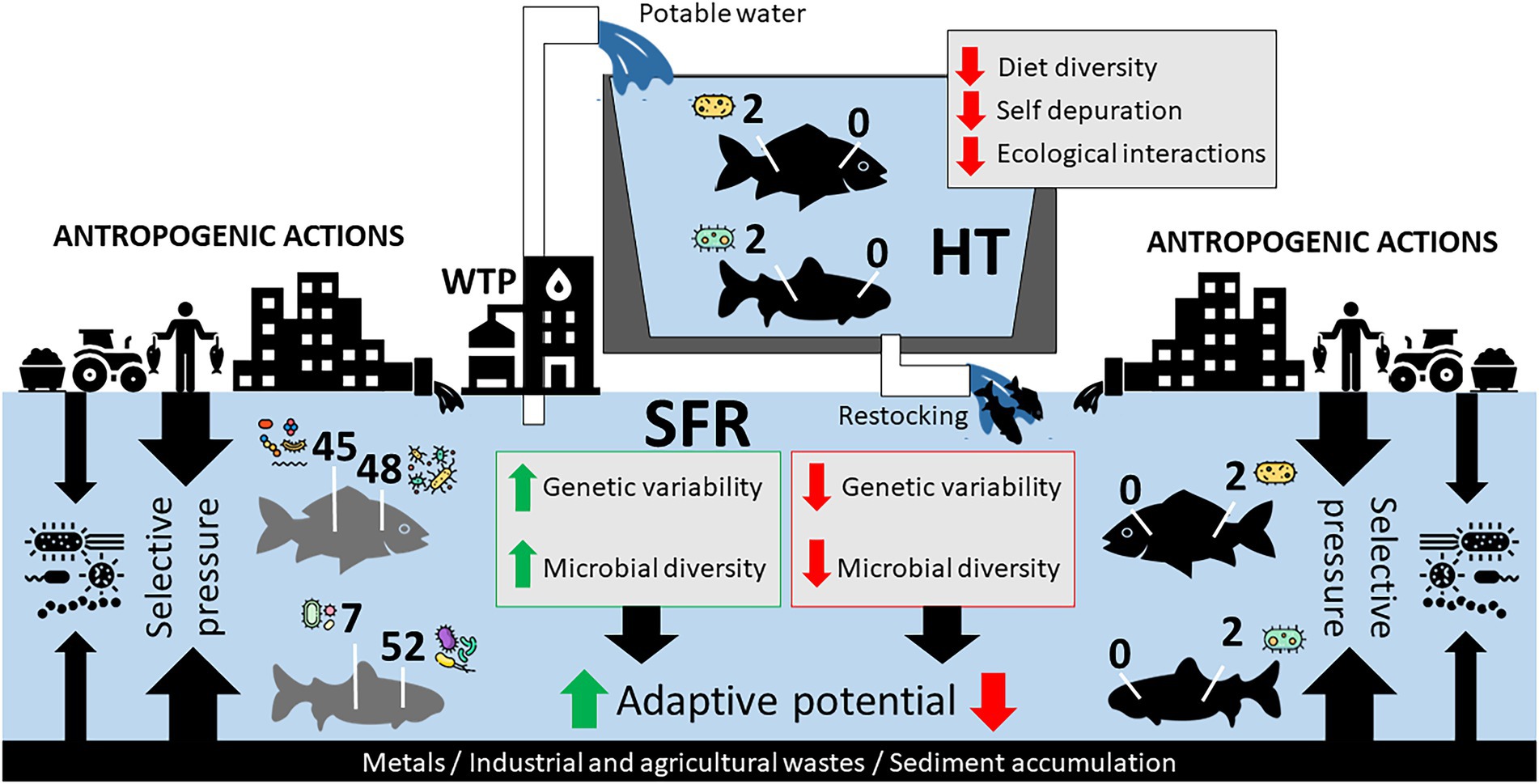
Figure 5. Hypothetical model of the adaptive relationship of species in SFR, in HT, and after being returned to SFR after restocking. Impacts of anthropogenic actions in SFR and zootechnical management in HT on the constitution of R. aspera and P. argenteus microbiomes should contribute to microbiota structure. The reduction of genetic diversity and the gill and gut microbiota can massively interfere with the adaptation of fish after restocking.
Unique microbiota associated with organs
Of the 295 bacterial genera identified among all the different samples, 215 were reported to be associated with gills (Figure 4C), with 12 genera (5.58%) shared among all samples originating from the gills (SFR and HT). Of these, 214 were associated with the gill microbiomes of the fish recruited from the SFR (119 in R. aspera and 154 in P. argenteus), both of which shared 27.5% in common. The interspecific difference in the gill microbial community composition was evidenced by the number of exclusive genera in R. aspera (54) and P. argenteus (93). The dominant genera in both species included Cetobacterium_A, Aeromonas, and Plesiomonas (Supplementary Figure 2).
Although present in both species, the genera Flavobacterium, Shewanella, Vogesella, and Pseudoduganella were more abundant in the gills of R. aspera from the SFR (Supplementary Figure 2). Some of the exclusive taxa from the gill microbiome (Table 2) were mainly classified within the Bacteroidetes phylum (Fluviicola, Flavobacterium, Soonwooa, Solirubrum, Haliscomenobacter, Taibaiella, Pedobacter, Sphingobacterium, and Flexibacter), Betaproteobacteria (Rhodoferax, Hydrogenophaga, Paucibacter, Sphaerotilus, Limnobacter) and Gammaproteobacteria (Alkanindiges, Cellvibrio, Pseudoxanthomonas, Thiolinea).
Some of the unique microbial taxa associated with the gills of P. argenteus included Caedimonas, Caedibacter, Vibrionimonas, Porphyromonas, Nanopelagicus, Planktophila, Scytonema, Dermacoccus, Dietzia, Granulicatella, Rufibacter, Leucobacter, Curtobacterium, Moraxella_C, Acetobacterium, Haemophilus, Chromobacterium).
Reduced microbial diversity was associated with gills from both species recruited from the HT, with more remarkable interspecific similarity (53.84% of shared genera) of the identified microorganisms. The proportion of commonly shared genera in comparing the same species (SFR vs. HT) was 9% for P. argenteus and 19% for R. aspera (Figure 4C). The gill microbiome was dominated by the Fusobacteriaceae, Enterobacteriaceae, Aeromonadaceae, and Bacteroidaceae families.
Microbiota communities associated with medium gut
The patterns of interspecific variations became conspicuous when comparing bacterial communities colonizing the medium gut of P. argenteus and R. aspera from SFR, which amounted to the lowest microbial diversity of the characterized microbiomes from this environment. We registered 151 genera, with 7.28% of this total shared among the two host fish species (Figure 4C).
The microbial community in the guts of P. argenteus comprised 138 genera, of which 94 (67.4%) were exclusive to that host (Figure 4C). These results clearly distinguish the intestinal microbiota of the evaluated fish species. The intestinal microbiota in both fish, recruited from SFR and HT, was predominantly composed of the microorganisms Cetobacterium_A, Aeromonas, and Plesiomonas. Differences were noted in the higher abundance of the Shewanella genera (46.17%), integrating the intestinal microbiota of R. aspera from the SFR. The Edwardsiella genera was observed over the gills and guts of both species, varying in abundance from 0.45 to 3.96%, with more significant proportions found in the samples of hosts reared in HT. Among the unique genera associated with the enteric microbiota of P. argenteus (Table 2), we observed microorganisms affiliated with the classes Alphaproteobacteria (Rubellimicrobium, Hyphomicrobium, Phenylobacterium, Sphingomonas_A, Microvirga) and Betaproteobacteria (Nitrosomonas, Cupriavidus, Sulfuritalea, Lautropia).
The HT environment also negatively affected the gut microbiome composition in both species, with only 19 and 21 genera colonizing the guts of R. aspera and P. argenteus, respectively. However, the relatively small number of exclusive taxa (Figure 4C) in R. aspera (3) and P. argenteus (4) indicates that there is little interspecific variability in the microbial diversity of the guts of both host species.
Phylogenetically distinct host species share a central microbiome in different ecological systems
The central microbiota comprised the proportion of the microbial community shared among all samples. The gill and gut microbiomes of both species share many broader taxonomic groups. At the genera level, the gill and gut microbiomes shared between R. aspera and P. argenteus were remarkably low, accounting for only 3.4 to 6.92% of the total when compared between fish species and environments. Interestingly, most diverse genera, like Aeromonas (3.6 to 28.84%), Cetobacterium (19 to 60.82%), and Plesiomonas (3 to 33.40%), were shared between organs and species of the SFR and the HT (Supplementary Figure 2). Other taxa shared at the genera level at least 75% of the total biological samples included Bacteroides, Akkermansia, Edwardsiella, Pseudomonas, and Shewanella, among other non-identified associated with Burkholderiaceae, Enterobacteriaceae e Fusobacteriaceae families.
Discussion
SFR is one of the most important Brazilian rivers. It contributes to the national energy matrix, favoring tourism and agriculture in its surroundings and providing fish to those who depend on it (Sun et al., 2016; Castro and Pereira, 2019). However, throughout their extension, these activities have contributed to the decline of the ichthyofauna, especially related to endemic and native species to this watershed (Holanda et al., 2009; Barbosa et al., 2017; Figueiredo, 2018).
This worrying scenario, which repeats in other basins around the world, puts aquaculture as a positive contributor to increasing the supply of fish as a source of animal protein (FAO, 2016). That minimizes the pressure on population stocks at the same time that it can support stochastic re-population strategies to preserve biodiversity (Klinger and Naylor, 2012).
However, studies indicate that the release of fish cultivated in re-population programs can pose risks of disease transmission to native populations due to the decrease in genetic variability and the possible introduction of exotic alleles that can compromise wild populations (Araki and Schmid, 2010; Cheng et al., 2015; Pavlova et al., 2017; Savary et al., 2017). Additionally, very little is known about the microbiomes associated with wildlife. Therefore, the first characterization of the structure of not yet known microbiomes associated with fish species may reveal an as-yet undescribed and particular microbiota (Levin et al., 2021). Furthermore, there is limited research on the relationship between species confinement and the modulation of microbiota associated with important tissues such as skin, gills, and guts.
In an attempt to reduce this knowledge gap and effectively contribute to studies involving fish species of ecological and commercial interest, this work characterized and compared for the first time the intestinal and gill microbiota of the species P. argenteus and R. aspera from the SFR and kept in an HT, both endemic to SFR natives, by high-throughput DNA sequencing. This culture-free method allows a broader view of microbial diversity associated with organisms, organs, and environments.
The present research findings indicate that fish-rearing conditions significantly altered the structure of the gut and gill microbiome of R. aspera and P. argenteus from the HT, suggesting that it was strongly modulated by the environmental conditions imposed by the host habitat. The microbiomes of the confined fish showed an 87% reduction in the diversity of taxa compared with the same species collected from the river. Studies show that the composition of microbiomes in teleost fish can differ under the influence of several factors such as phylogenetic position, trophic level and diet composition, the microhabitat used by hosts, its environment, and the stocking and rearing densities in hatchery environments (Sullam et al., 2012; Ursell et al., 2012; Boutin et al., 2013; Sylvain et al., 2016; Uren Webster et al., 2018, 2020; Tarnecki et al., 2019). In this context, dysbiosis caused by the drastic decrease in microbial diversity due to environmental variables and changes in diet can affect the nutritional and immunological status of fish, making them more vulnerable and susceptible to opportunistic pathogens (Boutin et al., 2013; Pękala-Safińska, 2018) and compromising aquaculture and food security.
The structure of microbiomes from fish confined in HT showed greater uniformity, with the predominance of the phyla Fusobacteriota and Proteobacteria, which affiliated with the majority (> 95%) of the identified taxonomic signatures. These results are consistent with other findings in other fish species evaluated in a confinement system, such as the cultivation of Panaque sp., Lates calcarifer, Oreochromis niloticus, and Arapaimas gigas (Di Maiuta et al., 2013; Zhai et al., 2017; Ramírez et al., 2018; Xia et al., 2020). In contrast, the wild fish microbiomes (SFR) showed greater richness and diversity, mainly composed of Proteobacteria of the classes Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria, followed by Fusobacteriota, Bacteroidota, Planctomycetota, and Cyanobacteria. The environmental heterogeneity expected from the river (more so given its diffuse anthropic impacts) should impact fish-associated microbiomes (Krotman et al., 2020). Being illiophagous, both fish grate the substrate and feed over fresh and decomposing algae and also have an intimate relationship with a more anoxic layer of the benthos, where a whole community of specialized microorganisms would be expected.
It is increasingly evident that microbiomes associated with wild animals provide these hosts with metabolic adaptations for survival in environments contaminated by recalcitrant pollutants and the consumption of toxic foods or those deteriorated by pathogens (Tarnecki et al., 2019; Levin et al., 2021). At the same time, some taxa can be characterized as possible biomarkers of environmental pollution (Caruso, 2016; Sylvain et al., 2016; Ventorino et al., 2018; Tarnecki et al., 2019; Nolorbe-Payahua et al., 2020). Cetobacterium, Aeromonas, and Plesiomonas also enriched SFR and HT fish microbiomes. The predominance of Cetobacterium was notably observed as associated with the gills and gut of P. argenteus and R. aspera collected from HT and has also been reported for other commonly cultivated fish species such as Cyprinus carpio (van Kessel et al., 2011), Panaque nigrolineatus (Di Maiuta et al., 2013), Ictalurus punctatus, Micropterus salmoides, Lepomis macrochirus (Larsen et al., 2014), Arapaimas gigas (Ramírez et al., 2018) and Oreochromis niloticus (Yu et al., 2019). The genera Aeromonas and Plesiomonas were prevalent in all samples, with a higher relative abundance of Aeromonas in the gills of R. aspera (24%) and P. argenteus (28.43%) from the HR and SFR, respectively, contrasting with the reduced abundance associated with intestinal samples (between 3.66 and 7.55%) in both hosts recruited from the SFR and HT. The genera Aeromonas is highly pathogenic, comprising 36 genera that establish symbiotic and pathogenic interactions with their hosts (Fernández-Bravo and Figueras, 2020). They carry multiple virulence factors (Nam and Joh, 2007) and are resistant to antimicrobials (Cízek et al., 2010; Nguyen et al., 2014) and heavy metals (Akinbowale et al., 2007). Plesiomonas is commonly found in the aquatic environment and associated with fish, being recognized as potential enteropathogens (Pękala-Safińska, 2018). Plesiomonas integrated the SFR and HT fish microbiota in relative abundance from 3% gill of R. aspera from SFR to 33.4%. These results corroborate those reported for Plesiomonas commonly found in fish microbiota (Duarte et al., 2014; Mohammed and Arias, 2014), with a higher prevalence rate in fish spawn in aquaculture.
The most diverse taxa of the microbiomes characterized in this study for R. aspera and P. argenteus were Aeromonas, Flavobacterium, Acinetobacter, Plesiomonas, and Shewanella, which frequently colonize the mucous surfaces of fish and are associated with severe pathologies in these hosts (Wu et al., 2012; Latha and Mohan, 2013; Liu et al., 2016; Tarnecki et al., 2016; Pękala-Safińska, 2018; Pratte et al., 2018). In this context, Sylvain et al. (2016) considered Flavobacterium a natural biomarker of environmental stress, susceptible to acidic pH, in the context of its abundance decrease, in the skin mucus and feces microbiome of Colossoma macropomum while exposed to ever-increasing acid concentrations. In contrast, Yu et al. (2019) demonstrated that exposure of Nile tilapia to aluminum significantly increased the abundance of Flavobacterium and decreased Plesiomonas. Acinetobacter was detected in greater quantities, integrating both wild species’ gill and gut microbiomes. They are reported as emerging pathogens, often associated with polluted environments, carrying antimicrobial resistance genes, which have also been reported as alkane degraders (Manchanda et al., 2010; Costa et al., 2015) and have been isolated from Oncorhynchus mykiss and Cyprinus carpio, causing severe damage to organisms (Pękala-Safińska, 2018). The genera Shewanella was observed in 46% of the guts of wild R. aspera, which was different from that reported by Pratte et al. (2018), where it was among the most diverse OTUs in gill microbiomes of 15 species of teleost fish recruited from coral reefs. Bacterial strains of the genera Shewanella have been intensively researched as candidates for biotechnological applications, especially in the bioremediation of hydrocarbons and metallic pollutants (Lemaire et al., 2020), and also have been reported to cause infections in popular freshwater-reared species Cyprinus carpio and Oncorhynchus mykiss (Pękala et al., 2015). The genera Edwardsiella is frequently reported to be associated with fish microbiota and includes pathogens such as E. tarda and E. ictaluri (Pękala-Safińska, 2018); and is also related to the massive mortality of Myleus micans (pacu), an important fisheries resource in the SFR, that occurred in the turn of 2005/2006 (Lima et al., 2008). The present study shows that the microbiota of these fish confined in this hatchery system had an increased abundance of Edwardsiella spp. (0.84% a 3.96%); therefore, it is a potential genus to risk the aquaculture production of the investigated species.
The phylum Cyanobacteria was observed only in samples originating from wild fish, aggregating 11 genera, among which Planktothrix and Pseudanabaena are frequently related to flowering and production of cyanotoxins (Su et al., 2015) in eutrophic environments with high concentrations of nitrogen and phosphorus (Paerl, 2018). These results corroborate those presented by the Instituto de Gestão da Águas de Minas Gerais (IGAM, 2018), indicating frequent events of bacterial blooms in the middle SFR. Bacteroidetes of the order Chitinophagales and Cytophagales were prevalent and more abundant in the gill microbiota of R. aspera in the SFR, and together, they affiliated with nine unique genera. The Chitinophagales family was associated with environments contaminated by heavy metals and impacted by agricultural activities (Costa et al., 2015; Júnior et al., 2021), which are also configured as common scenarios for the SFR.
Some taxonomic signatures were prevalent in the different body niches of both species, recruited from their natural habitat and captivity under the microbiomes characterized in this study. Therefore, we corroborate the hypothesis that phylogenetically diverse species may share basal or nuclear microbiomes (Roeselers et al., 2011; Lowrey et al., 2015; Hennersdorf et al., 2016; Liu et al., 2016). Interestingly, these shared taxonomic signatures correspond to 2.65% of the total chains from the R. aspera and P. argenteus microbiome, the most abundant taxa found in the samples: Cetobacterium, Plesiomonas, and Aeromonas. Finally, in this study, no significant differences were found in the diversity index α (Shannon and Simpson) between the compared microbiomes, considering species and organs of fish collected in the SFR. This result suggests that the sympatric occurrence of R. aspera and P. argenteus is associated with the iliophagous ecological convergence and has contributed to the microbial similarity observed in phylogenetically distinct fish species. Previous studies on the characterization of bird, mammal, and fish microbiomes have described the overlap of the ecological niche with increased interspecific microbial convergence (van Veelen et al., 2017; Baldo et al., 2019; Song et al., 2020).
However, a particular distinction between the microbiomes of different tissues of wild fish can be supported by the expressive number of unique taxa found only in the hosts’ gills and guts and by the percentage of shared taxa. In P. argenteus, only 28.6% of the taxa at the genera level were shared between gills and guts. This result corroborates the findings of Pratte et al. (2018), which characterized microbiomes of different populations of fish recruited from coral reefs, reporting between 20 to 25% of taxa shared between the gill and intestinal microbiota of individuals of the same species. The intestinal microbiota of R. aspera obtained from the SFR harbored a lower richness of bacterial consortia considering this environment compared to P. argenteus, also showing a high intraspecific variation between the gill and gut microbiota by sharing only 14.56% of genera. A similar result was reported for other Siluriformes related to the enteric microbiota, mucus, and gut content of P. nigrolineatus, Hypostomus auroguttatus, and Pelteobagrus fulvidraco (Wu et al., 2010, 2012; Duarte et al., 2014). Therefore, the observed variations may be directly related to the phylogeny and diet of the hosts, as well as the role that these tissues play in fish (Ngugi et al., 2017; Pratte et al., 2018; Levin et al., 2021).
From the perspective of fish farming for the repopulation of endangered species, our study brings a new discussion to the debate. The confinement of fish of the species R. aspera and P. argenteus resulted in a considerable discrepancy between the microbiota from wild and domesticated animals, with a massive reduction of bacterial diversity both in the gills and in the gut of HT fish. This effect probably reflects the contrasting selective pressures of the farming system and the natural habitat and points to vulnerabilities to the aquaculture of these species. Microbiomes as small as those observed in this study in confined fish can negatively impact the hosts’ health, resulting in economic losses and a possible reduction in the ability of cultured fish to be reintroduced into nature to rebuild wild populations (Figure 5). This effect, associated with a decrease in the genetic variability of individuals in confinement, can lead to high production costs with low adaptive capacity in the wild, thus opening new investigative windows in proposals for reintroducing native and endemic species.
One of the biggest challenges to the growth of industrial aquaculture is disease control (Zheng et al., 2020; Karvonen et al., 2021). In this context, the commensal microbiota is important in the innate and adaptive immunity of animals (Vadstein et al., 2018; Sehnal et al., 2021; Huang et al., 2022), whose change in composition can promote or mitigate disease states in hosts (Sehnal et al., 2021). Thus, the reduction in gut and gill microbiome diversity reported in fish farmed in closed systems may have epidemiological relevance and should not be overlooked (Pękala-Safińska, 2018; Tarnecki et al., 2019; Amillano-Cisneros et al., 2022). This perspective is supported by similar studies in Cyprinus carpio (Ruzauskas et al., 2021), Chirostoma estor (Amillano-Cisneros et al., 2022), Tor tambroides (Tan et al., 2019), Salmo salar (Uren Webster et al., 2018; Lavoie et al., 2021), Centropomus undecimalis (Tarnecki et al., 2019), and Paralichthys adspersus (Salas-Leiva et al., 2017).
Recent research, conducted mainly in mammals, has demonstrated the importance of microbiota-host interactions, including the development and regulation of the host’s immune response in the fight against infections and diseases. Reports in the literature relate the loss of microbial diversity in fish exposed to stress or disease (Legrand et al., 2020). In this context, exposure of D. rerio to the pathogen Aeromonas hydrophila decreased the abundance of beneficial microbial species and increased the abundance of harmful microorganisms and reactive oxygen species (Yang et al., 2017). Inflammatory disorders such as intestinal enteritis have been affecting the rearing of several fish species and were positively correlated with dietary supplements enriched with soybean meal and changes in the structure and diversity of the host microbiota (Urán et al., 2008; Legrand et al., 2020). Intestinal enteritis-induced Mycoplasmataceae enrichment and drastic loss of skin diversity were found in sick yellow-tailed kingfish (Legrand et al., 2020). Such changes can lead to functional dysbiosis in the host and loss of resistance to the invasion of opportunistic pathogens (De Schryver and Vadstein, 2014), demonstrating that the disruption of the host’s microbial flora homeostasis makes it more susceptible to diseases. Bacterial phyla associated with the gut microbiota that metabolize steroid hormones, including progesterone, estradiol, and testosterone, have also been reported in several animal models (Hussain et al., 2021), thus evidencing the involvement of the intestinal microbiota in reproduction. Lavoie et al., 2021 pointed out the relevance of monitoring bacterial communities of fish kept in captivity for repopulation programs, as they observed that the microbiota of confined fish remained different from that of wild fish, even after 2 years of translocation, with resistance to colonization by bacterioplankton bacteria in the natural habitat. Even though this study resolves fish-associated microbial diversity at the genera level, it is reasonable to assume many of the ASVs listed here represent new species. Thus, this line of inquiry would benefit from the whole assembly metagenome in the future to reveal this hidden biodiversity (Levin et al., 2021).
Our study revealed that wild species harbor highly diverse microbial communities. This repertoire of information will contribute to the understanding of microbiota-host bioecological interactions. Additional studies are needed to investigate the functional profile of wild microbiota on fish health and assess the impacts of reduced microbial diversity on farmed fish. Such information will constitute a reference for future studies, either as an attribute of comparing information to other systems and biological models or for implementing improvements in fish production for consumption or repopulation of natural environments.
In conclusion, the stress conditions the fish from HT are subjected to negatively modify the composition of the microbiomes of the host fish from both species, with a significant loss of diversity compared to the microbiomes of their wild counterparts. The set of bacterial communities associated with the body niches of both fish species reared in tanks points to a picture of dysbiosis with a possible decrease in the immune response of host fish and low resistance to colonization by pathogens, which can cause significant losses to aquaculture. We also identified taxa where the investigated species can establish important symbiotic interactions such as nutrition, detoxification, and protection. However, genera frequently reported as opportunistic pathogens were also associated with the investigated tissues, causing public health risks because they are conveyed in water and various foods. This study is the first to characterize the structure and composition of microbial diversity in R. aspera and P. argenteus via high-throughput sequencing of the 16S rRNA gene. These results can be used as a departure point to evaluate the putative effects of stocking impacted environments with fish spawn in hatcheries in the context of conservation, fisheries, and fish production in the SFR.
Data availability statement
The datasets generated for this study can be found in the NCBI under BioSample accessions SAMN22044346 - SAMN22044369 - https://www.ncbi.nlm.nih.gov/biosample/22044369 to https://www.ncbi.nlm.nih.gov/biosample/22044369.
Ethics statement
The animal study was reviewed and approved by this work has an environmental licensing nos. 45371-2 and 59845-1 issued by the Ministry of the Environment–MMA, Chico Mendes Institute for Biodiversity Conservation–ICMBio, through the Biodiversity Authorization and Information System-SISBIO and was approved by the Ethics Committee at Use of Animals CEUA / UFOP by protocol no. 2017/54 and enrolled in SISGEN A68C86F.
Author contributions
MD, CC, LB, and CL collected fish from the São Francisco River and the polyculture system in a suspended tank. MD, CL, LB, CG, and LM conceived and designed all the experiments. PT, HM, LM, CG, MD, and CL analyzed the biological data. NGS genomic solutions, Microbiology Laboratory of the Federal Institute of Education, Science, and Technology of Northern Minas Gerais contributed reagents, materials, and analysis tools. PT, HM, LM, and MD prepared the figures and tables. MD, CL, CG, LM, PT, HM, AS, and LR wrote, revised, and approved the final version of the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (CAPES) – Finance Code 001 - (the BIGA Project, CFP 51/2013, process 3385/2013 and DINTER Project Biological Sciences 223616/2014), National Council of Technological and Scientific Development (CNPq Process 481226/2013-3), and by the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG process APQ-02357-17). LM has a research fellowship from CNPq. CG and LM have UFOP grants.
Acknowledgments
We want to thank all the members of the Zoology Laboratory of the Federal Institute of Education, Science and Technology of Northern Minas Gerais (IFNMG), campus Januária. Special thanks to all riverine fishermen who have contributed to this work’s success. Special thanks to Diego Marçal Guedes, owner of the HTs, who provided the samples for analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.966436/full#supplementary-material
References
Akinbowale, O. L., Peng, H., Grant, P., and Barton, M. D. (2007). Antibiotic and heavy metal resistance in motile aeromonads and pseudomonads from rainbow trout (Oncorhynchus mykiss) farms in Australia. Int. J. Antimicrob. Agents 30, 177–182. doi: 10.1016/j.ijantimicag.2007.03.012
Albert, J. S., Tagliacollo, V. A., and Dagosta, F. (2020). Diversification of Neotropical freshwater fishes. Annu. Rev. Ecol. Evol. Syst. 51, 27–53. doi: 10.1146/annurev-ecolsys-011620-031032
Almeida, E., Santos, R., Filho, P. C., Souza, A., and Soares, E. (2015). Polyculture of curimatã pacu and freshwater prawn. Bol. Inst. Pesca 41, 271–278. doi: 10.13140/RG.2.1.2020.7204
Alves, C. B., and Leal, C. (2010). Aspectos da conservação da fauna de peixes da bacia do rio São Francisco em Minas Gerais. MG-Biota 2, 26–50.
Amillano-Cisneros, J. M., Hernández-Rosas, P. T., Gomez-Gil, B., Navarrete-Ramírez, P., Ríos-Durán, M. G., Martínez-Chávez, C. C., et al. (2022). Loss of gut microbial diversity in the cultured, agastric fish, Mexican pike silverside (Chirostoma estor: Atherinopsidae). PeerJ 10:e13052. doi: 10.7717/peerj.13052
Araki, H., and Schmid, C. (2010). Is hatchery stocking a help or harm?: evidence, limitations and future directions in ecological and genetic surveys. Aquaculture 308, S2–S11. doi: 10.1016/j.aquaculture.2010.05.036
Arantes, F., Santos, H., Rizzo, E., Sato, Y., and Bazzoli, N. (2011). Collapse of the reproductive process of two migratory fish (Prochilodus argenteus and Prochilodus costatus) in the Três Marias reservoir, São Francisco River, Brazil. J. Appl. Ichthyol. 27, 847–853. doi: 10.1111/j.1439-0426.2010.01583.x
Babicki, S., Arndt, D., Ana Marcu, A., Liang, Y., Grant, J. R., Maciejewski, A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res., W147–W153. doi: 10.1093/nar/gkw419
Baldo, L., Riera, J. L., Salzburger, W., and Barluenga, M. (2019). Phylogeography and ecological niche shape the cichlid fish gut microbiota in central American and African Lakes. Front. Microbiol. 10, 1–19. doi: 10.3389/fmicb.2019.02372
Barbosa, J. M., Soares, E. C., Cintra, I. H. A., Hermann, M., and Araújo, A. R. R. (2017). Perfil da ictiofauna da bacia do rio São Francisco / profile of the fish fauna of the São Francisco river basin. Acta Fisher. Aquat. Resource. 5, 70–90. doi: 10.2312/Actafish.2017.5.1.70-90
Benson, J. F., Mahoney, P. J., Sikich, J. A., Serieys, L. E. K., Pollinger, J. P., Ernest, H. B., et al. (2016). Interactions between demography, genetics, and landscape connectivity increase extinction probability for a small population of large carnivores in a major metropolitan area. Proc. Biol. Sci. 283:20160957. doi: 10.1098/rspb.2016.0957
Blainey, P. C. (2013). The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 37, 407–427. doi: 10.1111/1574-6976.12015
Boncompagni-Júnior, O., Normando, F. T., Brito, M. F. G., and Bazzoli, N. (2013). Reproductive biology of Prochilodus argenteus Agassiz, 1829 (Pisces: Prochilodontidae) in São Francisco River, Brazil. J. Appl. Ichthyol. 29, 132–138. doi: 10.1111/jai.12018
Boutin, S., Bernatchez, L., Audet, C., and Derôme, N. (2013). Network analysis highlights complex interactions between pathogen, host and commensal microbiota. PLoS One 8:e84772. doi: 10.1371/journal.pone.0084772
BRASIL (2005). Conselho nacional do meio ambiente (CONAMA). Available at: https://www.gov.br/mma/pt-br (Accessed March 12, 2019).
Britski, H. A., Sato, Y., and ROSA, A. B. S. (1988). Manual de identificação de peixes da região de Três Marias (Com chaves de identificação para os peixes da bacia do São Francisco). 3rd. Brasília: CODEVASF.
Buckup, P. A., Menezes, N. A., Ghazzi, M. S., and Nacional, Museu (2007). Catálogo das espécies de peixes de água doce do Brasil. Rio de Janeiro: Museu Nacional.
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016a). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Callahan, B. J., Sankaran, K., Fukuyama, J. A., McMurdie, P. J., and Holmes, S. P. (2016b). Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000Res 5:1492. doi: 10.12688/f1000research.8986.2
Camargo, M. M. P., Fernandes, M. N., and Martinez, C. B. R. (2009). How aluminum exposure promotes osmoregulatory disturbances in the neotropical freshwater fish Prochilodus lineatus. Aquat. Toxicol. 94, 40–46. doi: 10.1016/j.aquatox.2009.05.017
Carmo, F. M., Da, S., Polo, É. M., Silva, M. A.Da, and Yazbeck, G. De M. (2015). Optimization of heterologous microsatellites in Piracanjuba. Pesq. Agropec. Bras. 50, 1236–1239. doi:doi: 10.1590/S0100-204X2015001200015.
Caruso, G. (2016). Antibiotic resistance in fish farming environments: a global concern. J. Fisher. Sci. 10, 9–13.
Castro, C. N., and Pereira, C. N. (2019). Revitalização da Bacia Hidrográfica do rio São Francisco: Histórico, Diagnóstico e Desafios. Brasília: IPEA.
Castro, R. M. C., and Vari, R. P. (2004). Detritivores of the south American fish family Prochilodontidae (Teleostei:Ostariophysi:Characiformes): a phylogenetic and revisionary study. 189.
Chen, L., Guo, Y., Hu, C., Lam, P. K. S., Lam, J. C. W., and Zhou, B. (2018). Dysbiosis of gut microbiota by chronic co exposure to titanium dioxide nanoparticles and bisphenol a: implications for host health in zebrafish. Environ. Pollut. 234, 307–317. doi: 10.1016/j.envpol.2017.11.074
Cheng, Y., Fox, S., Pemberton, D., Hogg, C., Papenfuss, A. T., and Belov, K. (2015). The Tasmanian devil microbiome-implications for conservation and management. Microbiome 3:76. doi: 10.1186/s40168-015-0143-0
Cízek, A., Dolejská, M., Sochorová, R., Strachotová, K., Piacková, V., and Veselý, T. (2010). Antimicrobial resistance and its genetic determinants in aeromonads isolated in ornamental (koi) carp (Cyprinus carpio koi) and common carp (Cyprinus carpio). Vet. Microbiol. 142, 435–439. doi: 10.1016/j.vetmic.2009.10.001
Costa, P. S., Reis, M. P., Ávila, M. P., Leite, L. R., de Araújo, F. M. G., Salim, A. C. M., et al. (2015). Metagenome of a microbial community inhabiting a metal-rich tropical stream sediment. PLoS One 10:e0119465. doi: 10.1371/journal.pone.0119465
Covain, R., and Fisch-Muller, S. (2007). The genera of the Neotropical armored catfish subfamily Loricariinae (Siluriformes: Loricariidae): a practical key and synopsis. Zootaxa 1462, 1–40.doi: 10.11646/ZOOTAXA.1462.1.1,
Da Silva, F. F., Silva, J. M.Da, Silva, T. De. J. Da, Tenorio, B. M., Tenorio, F. Das C. A. M., and Santos, E. L., et al. (2020). Evaluation of Nile tilapia (Oreochromis niloticus) fingerlings exposed to the pesticide pyriproxyfen. Lat. Am. J. Aquat. Res. 48, 826–835. doi:doi: 10.3856/vol48-issue5-fulltext-2556.
De Schryver, P., and Vadstein, O. (2014). Ecological theory as a foundation to control pathogenic invasion in aquaculture. ISME J. 8, 2360–2368. doi: 10.1038/ismej.2014.84
Delcorso, M. C., de Paiva, P. P., Grigoleto, M. R. P., Queiroz, S. C. N., Collares-Buzato, C. B., and Arana, S. (2020). Effects of sublethal and realistic concentrations of the commercial herbicide atrazine in Pacu (Piaractus mesopotamicus): long-term exposure and recovery assays. Vet World 13, 147–159. doi: 10.14202/vetworld.2020.147-159
Di Maiuta, N., Schwarzentruber, P., Schenker, M., and Schoelkopf, J. (2013). Microbial population dynamics in the feces of wood-eating loricariid catfishes. Lett. Appl. Microbiol. 56, 401–407. doi: 10.1111/lam.12061
Duarte, S., Silva, F. C. D. P. E., Zauli, D. A. G., Nicoli, J. R., and Araújo, F. G. (2014). Gram-negative intestinal indigenous microbiota from two Siluriform fishes in a tropical reservoir. Braz. J. Microbiol. 45, 1283–1292. doi: 10.1590/S1517-83822014000400019
FAO (2016). Contributing to food Security and Nutrition for all. The state of world fisheries and aquaculture Rome: FAO.
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Fernández-Bravo, A., and Figueras, M. J. (2020). An update on the genera Aeromonas: taxonomy, epidemiology, and pathogenicity. Microorganisms 8:129. doi: 10.3390/microorganisms8010129
Figueiredo, A. V. A. (2018). Efeitos da Regulação da vazão sobre os atributos ecológicos da ictiofauna no baixo curso do rio São Francisco. Revista de Estudos Ambientais. 22, 6–21. doi: 10.7867/1983-1501.2020v22n2p6-21
Gajardo, K., Rodiles, A., Kortner, T. M., Krogdahl, Å., Bakke, A. M., Merrifield, D. L., et al. (2016). A high-resolution map of the gut microbiota in Atlantic salmon (Salmo salar): a basis for comparative gut microbial research. Sci. Rep. 6, 1–10. doi: 10.1038/srep30893
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. doi: 10.1186/gb-2004-5-10-r80
Getirana, A. (2016). Extreme water deficit in Brazil detected from space. J. Hydrometeorol. 17, 591–599. doi: 10.1175/JHM-D-15-0096.1
Godinho, H. P., and Godinho, A. L. (2003). Águas, peixes e pescadores do São Francisco das Minas Gerais. Belo Horizonte, Brazil: Editora PUC Minas.
Godinho, A. L., Silva, C. C. F., and Kynard, B. (2017). Spawning calls by zulega, Prochilodus argenteus, a Brazilian riverine fish. Environ. Biol. Fish 100, 519–533. doi: 10.1007/s10641-017-0582-5
González-Wangüemert, M., Fernández, T. V., Pérez-Ruzafa, A., Giacalone, M., D’Anna, G., and Badalamenti, F. (2012). Genetic considerations on the introduction of farmed fish in marine protected areas: the case of study of white seabream restocking in the Gulf of Castellammare (southern Tyrrhenian Sea). J. Sea Res. 68, 41–48. doi: 10.1016/j.seares.2011.12.005
Gudmundsson, L., Boulange, J., Faça, H. X., Gosling, S. N., Grillakis, M. G., Koutroulis, A. G., et al. (2021). Globally observed trends in mean and extreme river flow attributed to climate change. Science 371, 1159–1162. doi: 10.1126/science.aba3996
Guivier, E., Martin, J.-F., Pech, N., Ungaro, A., Chappaz, R., and Gilles, A. (2017). Microbiota diversity within and between the tissues of two wild interbreeding species. Microb. Ecol. 75, 799–810. doi: 10.1007/s00248-017-1077-9
Gutberlet, J., Seixas, C. S., Thé, A. P. G., and Carolsfeld, J. (2007). Resource conflicts: challenges to fisheries Management at the São Francisco River, Brazil. Hum. Ecol. 35, 623–638. doi: 10.1007/s10745-007-9132-7
Hamady, M., Lozupone, C., and Knight, R. (2010). Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4, 17–27. doi: 10.1038/ismej.2009.97
Hennersdorf, P., Kleinertz, S., Theisen, S., Abdul-Aziz, M. A., Mrotzek, G., Palm, H. W., et al. (2016). Microbial diversity and parasitic load in tropical fish of different environmental conditions. PLoS One 11:e0151594. doi: 10.1371/journal.pone.0151594
Holanda, F. S. R., Ismerim, S. S., Da Rocha, I. P., De Jesus, A. S., De Filho, R. N., and De Júnior, A. V. (2009). Environmental perception of the São Francisco riverine population in regards to flood impact. J. Hum. Ecol. 28, 37–46. doi: 10.1080/09709274.2009.11906216
Huang, Z., Zhan, M., Cheng, G., Lin, R., Zhai, X., Zheng, H., et al. (2022). IHNV infection induces strong mucosal immunity and changes of microbiota in trout gut. Viruses 14, 1–17. doi: 10.3390/v14081838
Huber, W., Carey, V. J., Gentleman, R., Anders, S., Carlson, M., Carvalho, B. S., et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. doi: 10.1038/nmeth.3252
Hussain, T., Murtaza, G., Kalhoro, D. H., Kalhoro, M. S., Metwally, E., Chughtai, M. I., et al. (2021). Relationship between gut microbiota and host-metabolism: emphasis on hormones related to reproductive function. Anim Nutr 7, 1–10. doi: 10.1016/j.aninu.2020.11.005
IBGE (2010). INSTITUTO BRASILEIRO DE GEOGRAFIA E ESTATÍSTICA | IBGE Cidades@ | Minas Gerais | Januária | Panorama. Available at: https://cidades.ibge.gov.br/brasil/mg/januaria/panorama (Accessed August 8, 2021).
IGAM (2018). “INSTITUTO MINEIRO DE GESTÃO DAS ÁGUAS” in Monitoramento da Qualidade das Águas Superficiais no Estado de Minas Gerais - Relatório Trimestral - 2° Trimestre de 2018 (Bairro Serra Verde: SEMAD)
International Union for Conservation of Nature and Natural Resources –IUCN (2022). The IUCN red list of threatened species. Available at: https://www.iucnredlist.org/en (Accessed October 26, 2022).
Islam, M. S., Ahmed, M. K., Raknuzzaman, M., Habibullah-Al-Mamun, M., and Islam, M. K. (2015). Heavy metal pollution in surface water and sediment: a preliminary assessment of an urban river in a developing country. Ecol. Indic. 48, 282–291. doi: 10.1016/j.ecolind.2014.08.016
Izvekova, G. I., Izvekov, E. I., and Plotnikov, A. O. (2007). Symbiotic microflora in fishes of different ecological groups. Biol Bull Russ Acad Sci 34, 610–618. doi: 10.1134/S106235900706012X
Júnior, M. D. P., Silva, T. A. D. C., Lacorte, G. A., and Barezani, A. S. A. (2021). Biodiversidade das comunidades microbianas do sedimento do leito do rio São Francisco na serra da Canastra em Minas Gerais. Revista Multidisciplinar de Educação e Meio Ambiente 2:46. doi: 10.51189/rema/1061
Karvonen, A., Räihä, V., Klemme, I., Ashrafi, R., Hyvärinen, P., and Sundberg, L.-R. (2021). Quantity and quality of aquaculture enrichments influence disease epidemics and provide ecological alternatives to antibiotics. Antibiotics (Basel) 10, 1–10. doi: 10.3390/antibiotics10030335
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Klinger, D., and Naylor, R. (2012). Searching for solutions in aquaculture: charting a sustainable course. Annu. Rev. Environ. Resour. 37, 247–276. doi: 10.1146/annurev-environ-021111-161531
Krotman, Y., Yergaliyev, T. M., Alexander Shani, R., Avrahami, Y., and Szitenberg, A. (2020). Dissecting the factors shaping fish skin microbiomes in a heterogeneous inland water system. Microbiome 8:9. doi: 10.1186/s40168-020-0784-5
Larsen, A. M., Mohammed, H. H., and Arias, C. R. (2014). Characterization of the gut microbiota of three commercially valuable warmwater fish species. J. Appl. Microbiol. 116, 1396–1404. doi: 10.1111/jam.12475
Latha, N., and Mohan, M. R. (2013). The bacterial microflora in the fish organs-a public health aspect. Indian J. Adv. Chem. Sci. 1, 139–143.
Lavoie, C., Wellband, K., Perreault, A., Bernatchez, L., and Derome, N. (2021). Artificial rearing of Atlantic Salmon juveniles for supportive breeding programs induces long-term effects on gut microbiota after stocking. Microorganisms 9:1932. doi: 10.3390/microorganisms9091932
Law, C. W., Chen, Y., Shi, W., and Smyth, G. K. (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15:R29. doi: 10.1186/gb-2014-15-2-r29
Legrand, T. P. R. A., Catalano, S. R., Wos-Oxley, M. L., Stephens, F., Landos, M., Bansemer, M. S., et al. (2018). The inner workings of the outer surface: skin and gill microbiota as indicators of changing gut health in yellowtail kingfish. Front. Microbiol. 8:2664. doi: 10.3389/fmicb.2017.02664
Legrand, T. P. R. A., Wynne, J. W., Weyrich, L. S., and Oxley, A. P. A. (2020). A microbial sea of possibilities: current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 12, 1101–1134. doi: 10.1111/raq.12375
Lemaire, O. N., Méjean, V., and Iobbi-Nivol, C. (2020). The Shewanella genera: ubiquitous organisms sustaining and preserving aquatic ecosystems. FEMS Microbiol. Rev. 44, 155–170. doi: 10.1093/femsre/fuz031
Levin, D., Raab, N., Pinto, Y., Rothschild, D., Zanir, G., Godneva, A., et al. (2021). Diversity and functional landscapes in the microbiota of animals in the wild. Science 372:254. doi: 10.1126/science.abb5352
Lima, L. C., Fernandes, A. A., Costa, A. A. P., Velasco, F. O., Leite, R. C., and Hackett, J. L. (2008). Isolation and characterization of Edwardsiella tarda from pacu Myleus micans. Arq. Bras. Med. Vet. Zootec. 60, 275–277. doi: 10.1590/S0102-09352008000100040
Linares, M. S., Assis, W., de Castro Solar, R. R., Leitão, R. P., Hughes, R. M., and Callisto, M. (2019). Small hydropower dam alters the taxonomic composition of benthic macroinvertebrate assemblages in a neotropical river. River Res. Appl. 35, 725–735. doi: 10.1002/rra.3442
Liu, H., Guo, X., Gooneratne, R., Lai, R., Zeng, C., Zhan, F., et al. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 6:24340. doi: 10.1038/srep24340
Lowrey, L., Woodhams, D. C., Tacchi, L., and Salinas, I. (2015). Topographical mapping of the rainbow trout (Oncorhynchus mykiss) microbiome reveals a diverse bacterial community with antifungal properties in the skin. Appl. Environ. Microbiol. 81, 6915–6925. doi: 10.1128/AEM.01826-15
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Makino, L. C., Faustino, F., Paes, M. C. F., Beraldo-Massoli, M. C., Cardozo, M. V., Schocken-Iturrino, R. P., et al. (2012). Morfologia e quantificação da microbiota intestinal do curimbatá (Prochilodus lineatus) e do cascudo cinza (Pterygoplichthys anisitsi) cultivados em cativeiro. Arquivo Brasileiro de Medicina Veterinária e Zootecnia 64, 916–926. doi: 10.1590/S0102-09352012000400019
Manchanda, V., Sanchaita, S., and Singh, N. (2010). Multidrug Resistant Acinetobacter. J Glob Infect Dis 2, 291–304. doi: 10.4103/0974-777X.68538
Maneta, M. P., Torres, M., Wallender, W. W., Vosti, S., Kirby, M., Bassoi, L. H., et al. (2009). Water demand and flows in the São Francisco River basin (Brazil) with increased irrigation. Agric. Water Manag. 96, 1191–1200. doi: 10.1016/j.agwat.2009.03.008
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Mohammed, H. H., and Arias, C. R. (2014). Epidemiology of columnaris disease affecting fishes within the same watershed. Dis. Aquat. Org. 109, 201–211. doi: 10.3354/dao02739
Molbert, N., Angelier, F., Alliot, F., Ribout, C., and Goutte, A. (2021). Fish from urban rivers and with high pollutant levels have shorter telomeres. Biol. Lett. 17:20200819. doi: 10.1098/rsbl.2020.0819
Nam, I.-Y., and Joh, K.-S. (2007). Rapid detection of virulence factors of Aeromonas isolated from a trout farm by Hexaplex-PCR. J. Microbiol. 45, 297–304.
Nascimento, L. N., and Becker, M. L. (2010). Hydro-businesses: national and global demands on the São Francisco River basin environment of Brazil. Int Rev of Soc His 55, 203–233. doi: 10.1017/S0020859010000556
Ngugi, D. K., Miyake, S., Cahill, M., Vinu, M., Hackmann, T. J., Blom, J., et al. (2017). Genomic diversification of giant enteric symbionts reflects host dietary lifestyles. PNAS 114, E7592–E7601. doi: 10.1073/pnas.1703070114
Nguyen, F., Starosta, A. L., Arenz, S., Sohmen, D., Dönhöfer, A., and Wilson, D. N. (2014). Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 395, 559–575. doi: 10.1515/hsz-2013-0292
Ning, Y., Qi, J., Dobbins, M. T., Liang, X., Wang, J., Chen, S., et al. (2020). Comparative analysis of microbial community structure and function in the gut of wild and captive Amur Tiger. Front. Microbiol. 11:1665. doi: 10.3389/fmicb.2020.01665
Nolorbe-Payahua, C. D., de Freitas, A. S., Roesch, L. F. W., and Zanette, J. (2020). Environmental contamination alters the intestinal microbial community of the livebearer killifish Phalloceros caudimaculatus. Heliyon 6:e04190. doi: 10.1016/j.heliyon.2020.e04190
Otto, F. E. L., Haustein, K., Uhe, P., Coelho, C. A. S., Aravequia, J. A., Almeida, W., et al. (2015). Factors other than climate change, Main drivers of 2014/15 water shortage in Southeast Brazil. Bull. Amer. Meteor. Soc. 96, S35–S40. doi: 10.1175/BAMS-D-15-00120.1
Paerl, H. W. (2018). Mitigating toxic planktonic cyanobacterial blooms in aquatic ecosystems facing increasing anthropogenic and climatic pressures. Toxins (Basel) 10:E76. doi: 10.3390/toxins10020076
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P.-A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-7
Paschoalini, A. L., and Bazzoli, N. (2021). Heavy metals affecting Neotropical freshwater fish: a review of the last 10 years of research. Aquat. Toxicol. 237:105906. doi: 10.1016/j.aquatox.2021.105906
Paschoalini, A. L., Savassi, L. A., Arantes, F. P., Rizzo, E., and Bazzoli, N. (2019). Heavy metals accumulation and endocrine disruption in Prochilodus argenteus from a polluted neotropical river. Ecotoxicol. Environ. Saf. 169, 539–550. doi: 10.1016/j.ecoenv.2018.11.047
Pavlova, A., Beheregaray, L. B., Coleman, R., Gilligan, D., Harrisson, K. A., Ingram, B. A., et al. (2017). Severe consequences of habitat fragmentation on genetic diversity of an endangered Australian freshwater fish: a call for assisted gene flow. Evol. Appl. 10, 531–550. doi: 10.1111/eva.12484
Pękala, A., Kozińska, A., Paździor, E., and Głowacka, H. (2015). Phenotypical and genotypical characterization of Shewanella putrefaciens strains isolated from diseased freshwater fish. J. Fish Dis. 38, 283–293. doi: 10.1111/jfd.12231
Pękala-Safińska, A. (2018). Contemporary threats of bacterial infections in freshwater fish. J. Vet. Res. 62, 261–267. doi: 10.2478/jvetres-2018-0037
Perini, V. R., Paschoalini, A. L., Bazzoli, N., Rizzo, E., and Carvalho, D. C. (2021). Metapopulation dynamics of the migratory fish Prochilodus lineatus (Characiformes: Prochilodontidae) in a lotic remnant of the Grande River. Southeastern Brazil. Neotrop. ichthyol. 19, 1–15. doi: 10.1590/1982-0224-2020-0046
Perry, W. B., Lindsay, E., Payne, C. J., Brodie, C., and Kazlauskaite, R. (2020). The role of the gut microbiome in sustainable teleost aquaculture. Proc. R. Soc. B Biol. Sci. 287:20200184. doi: 10.1098/rspb.2020.0184
Pratte, Z. A., Besson, M., Hollman, R. D., and Stewart, F. J. (2018). The gills of reef fish support a distinct microbiome influenced by host-specific factors. Appl. Environ. Microbiol. 84, e00063–e00018. doi: 10.1128/AEM.00063-18
Procópio, M. S., Ribeiro, H. J., Pereira, L. A., Oliveira Lopes, G. A., Santana Castro, A. C., Rizzo, E., et al. (2014). Sex-response differences of immunological and histopathological biomarkers in gill of Prochilodus argenteus from a polluted river in Southeast Brazil. Fish Shellfish Immunol. 39, 108–117. doi: 10.1016/j.fsi.2014.04.010
Ramírez, C., Coronado, J., Silva, A., and Romero, J. (2018). Cetobacterium is a major component of the microbiome of Giant Amazonian fish (Arapaima gigas) in Ecuador. Animals (Basel) 8, 1–6. doi: 10.3390/ani8110189
Ribeiro, E. V., Junior, A. P. M., Horn, A. H., and Trindade, W. M. (2012). Metais pesados e qualidade da água do rio São Francisco no segmento entre Três Marias e Pirapora/MG: Índice de Contaminação. Geonomos 20, 49–63. doi: 10.18285/geonomos.v20i1.27
Riiser, E. S., Haverkamp, T. H. A., Varadharajan, S., Borgan, Ø., Jakobsen, K. S., Jentoft, S., et al. (2020). Metagenomic shotgun analyses reveal complex patterns of intra-and interspecific variation in the intestinal microbiomes of codfishes. Appl. Environ. Microbiol. 86, 1–16. doi: 10.1128/AEM.02788-19
Ringo, E., Zhou, Z., Vecino, J. L. G., Wadsworth, S., Romero, J., Krogdahl, Å., et al. (2016). Effect of dietary components on the gut microbiota of aquatic animals. A never-ending story? Aquac. Nutr. 22, 219–282. doi: 10.1111/anu.12346
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010a). EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Robinson, M. D., and Oshlack, A. (2010b). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11:R25. doi: 10.1186/gb-2010-11-3-r25
Roeselers, G., Mittge, E. K., Stephens, W. Z., Parichy, D. M., Cavanaugh, C. M., Guillemin, K., et al. (2011). Evidence for a core gut microbiota in the zebrafish. ISME J. 5, 1595–1608. doi: 10.1038/ismej.2011.38
Roxo, F. F., Ochoa, L. E., Sabaj, M. H., Lujan, N. K., Covain, R., Silva, G. S. C., et al. (2019). Phylogenomic reappraisal of the Neotropical catfish family Loricariidae (Teleostei: Siluriformes) using ultraconserved elements. Mol. Phylogenet. Evol. 135, 148–165. doi: 10.1016/j.ympev.2019.02.017
Ruzauskas, M., Armalytė, J., Lastauskienė, E., Šiugždinienė, R., Klimienė, I., Mockeliūnas, R., et al. (2021). Microbial and antimicrobial resistance profiles of microbiota in common carps (Cyprinus carpio) from Aquacultured and wild fish populations. Animals (Basel) 11, 1–14. doi: 10.3390/ani11040929
Salas-Leiva, J., Opazo, R., Remond, C., Uribe, E., Velez, A., and Romero, J. (2017). Characterization of the intestinal microbiota of wild-caught and farmed fine flounder (Paralichthys adspersus). Lat. Am. J. Aquat. Res. 45, 370–378. doi: 10.3856/vol45-issue2-fulltext-12
Santos, F. G. B., Gouveia, G. V., França, C. A., Souza, M. G., and Costa, M. M. (2014). Microbiota bacteriana com potencial patogênico em Pacamã e perfil de sensibilidade a antimicrobianos. Revista Caatinga 27, 176–183.
Santos, R. P., Melo, B. F., Yazbeck, G. M., Oliveira, S., Hilário, H. O., Prosdocimi, F., et al. (2021). Diversification of Prochilodus in the eastern Brazilian shield: evidence from complete mitochondrial genomes (Teleostei, Prochilodontidae). J. Zool. Syst. Evol. Res. 59, 1053–1063. doi: 10.1111/jzs.12475
Sato, Y., Bazzoli, N., Rizzo, E., Boschi, M. B., and Miranda, M. O. T. (2005). Influence of the Abaeté River on the reproductive success of the neotropical migratory teleost Prochilodus argenteus in the São Francisco River, downstream from the Três Marias dam, southeastern Brazil. River Res. Appl. 21, 939–950. doi: 10.1002/rra.859
Sato, Y., Fenerich-Verani, N., and Godinho, H. (2003). “Padrões reprodutivos de peixes da bacia do São Francisco”, in Águas, peixes e pescadores do São Francisco das Minas Gerais. eds. H. P. Godinho and A. L. Godinho (Brazil: Belo Horizonte), 229–274.
Sato, Y., Fenerich-Verani, N., Verani, J. R., Godinho, H. P., and Sampaio, E. V. (1998). Induced reproduction and reproductive characteristics of Rhinelepis aspera Agassiz, 1829 (osteichthyes: Siluriformes, Loricariidae). Braz. Arch. Biol. Technol. 41, 309–314. doi: 10.1590/S1516-89131998000300006
Savary, R., Dufresnes, C., Champigneulle, A., Caudron, A., Dubey, S., Perrin, N., et al. (2017). Stocking activities for the Arctic charr in Lake Geneva: genetic effects in space and time. Ecol. Evol. 7, 5201–5211. doi: 10.1002/ece3.3073
Schmidt, V., Amaral-Zettler, L., Davidson, J., Summerfelt, S., and Good, C. (2016). Influence of fishmeal-free diets on microbial communities in Atlantic Salmon (Salmo salar) recirculation aquaculture systems. Appl. Environ. Microbiol. 82, 4470–4481. doi: 10.1128/AEM.00902-16
Sehnal, L., Brammer-Robbins, E., Wormington, A. M., Blaha, L., Bisesi, J., Larkin, I., et al. (2021). Microbiome composition and function in aquatic vertebrates: small organisms making big impacts on aquatic animal health. Front. Microbiol. 12:567408. doi: 10.3389/fmicb.2021.567408
Shirmohammadi, M., Salamat, N., Ronagh, M. T., Movahedinia, A., and Hamidian, G. (2018). Using cell apoptosis, micronuclei and immune alternations as biomarkers of phenanthrene exposure in yellowfin seabream (Acanthopagrus latus). Fish Shellfish Immunol. 72, 37–47. doi: 10.1016/j.fsi.2017.10.039
Silva, F. C. P., Brito, M. F. G., Farias, L. M., and Nicoli, J. R. (2005). Composition and antagonistic activity of the indigenous intestinal microbiota of Prochilodus argenteus Agassiz. J. Fish Biol. 67, 1686–1698. doi: 10.1111/j.1095-8649.2005.00877.x
Silva da Costa, R., Okada, E., Agostinho, A., and Gomes, L. (2012). Temporal variation in yield and composition of the artisanal fishery of the upper Paraná River, PR - Brazil: chronic effects of impoundments. Bol. Inst. Pesca 38, 199–213.
Smyth, G. K. (2004). Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article3. doi: 10.2202/1544-6115.1027
Song, S. J., Sanders, J. G., Delsuc, F., Metcalf, J., Amato, K., Taylor, M. W., et al. (2020). Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. MBio 11, 1–14. doi: 10.1128/mBio.02901-19
Su, G., Logez, M., Xu, J., Tao, S., Villéger, S., and Brosse, S. (2021). Human impacts on global freshwater fish biodiversity. Science 371, 835–838. doi: 10.1126/science.abd3369
Su, M., Yu, J., Zhang, J., Chen, H., An, W., Vogt, R. D., et al. (2015). MIB-producing cyanobacteria (Planktothrix sp.) in a drinking water reservoir: distribution and odor producing potential. Water Res. 68, 444–453. doi: 10.1016/j.watres.2014.09.038
Sullam, K. E., Essinger, S. D., Lozupone, C. A., O’Connor, M. P., Rosen, G. L., Knight, R., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21, 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Sun, T., Ferreira, V., He, X., and Andam-Akorful, S. (2016). Water availability of São Francisco River basin based on a space-borne geodetic sensor. Water 8:213. doi: 10.3390/w8050213
Suzuki, H. I., Agostinho, A. A., and Winemiller, K. O. (2000). Relationship between oocyte morphology and reproductive strategy in loricariid catfishes of the Paraná River, Brazil. J. Fish Biol. 57, 791–807. doi: 10.1111/j.1095-8649.2000.tb00275.x
von Spix, J. B., and Agassiz, J. L. R. (1829). Prochilodus argenteus Spix & Agassiz, 1829. Available at: https://www.gbif.org/pt/species/2352177 (Accessed November 8, 2022).
Sylvain, F.-É., Cheaib, B., Llewellyn, M., Gabriel Correia, T., Barros Fagundes, D., Luis Val, A., et al. (2016). pH drop impacts differentially skin and gut microbiota of the Amazonian fish tambaqui (Colossoma macropomum). Sci. Rep. 6, 1–10. doi: 10.1038/srep32032
Sylvain, F.-É., Holland, A., Audet-Gilbert, É., Luis Val, A., and Derome, N. (2019). Amazon fish bacterial communities show structural convergence along widespread hydrochemical gradients. Mol. Ecol. 28, 3612–3626. doi: 10.1111/mec.15184
Tan, C. K., Natrah, I., Suyub, I. B., Edward, M. J., Kaman, N., and Samsudin, A. A. (2019). Comparative study of gut microbiota in wild and captive Malaysian Mahseer (Tor tambroides). Microbiology. doi:doi: 10.1002/mbo3.734, 8:e00734.
Tarnecki, A. M., Brennan, N. P., Schloesser, R. W., and Rhody, N. R. (2019). Shifts in the skin-associated microbiota of hatchery-reared common Snook Centropomus undecimalis during acclimation to the wild. Microb. Ecol. 77, 770–781. doi: 10.1007/s00248-018-1252-7
Tarnecki, A. M., Patterson, W. F., and Arias, C. R. (2016). Microbiota of wild-caught red snapper Lutjanus campechanus. BMC Microbiol. 16:245. doi: 10.1186/s12866-016-0864-7
Torres, M. O., Maneta, M., Howitt, R., Vosti, S. A., Wallender, W. W., Bassoi, L. H., et al. (2012). Economic impacts of regional water scarcity in the São Francisco River basin, Brazil: an application of a linked hydro-economic model. Envir. Dev. Econ. 17, 227–248. doi: 10.1017/S1355770X11000362
Urán, P. A., Gonçalves, A. A., Taverne-Thiele, J. J., Schrama, J. W., Verreth, J. A. J., and Rombout, J. H. W. M. (2008). Soybean meal induces intestinal inflammation in common carp (Cyprinus carpio L.). Fish Shellfish Immunol. 25, 751–760. doi: 10.1016/j.fsi.2008.02.013
Uren Webster, T. M., Consuegra, S., Hitchings, M., and Garcia de Leaniz, C. (2018). Interpopulation variation in the Atlantic Salmon microbiome reflects environmental and genetic diversity. Appl. Environ. Microbiol. 84, 1–14. doi: 10.1128/AEM.00691-18
Uren Webster, T. M., Rodriguez-Barreto, D., Castaldo, G., Gough, P., Consuegra, S., and Garcia de Leaniz, C. (2020). Environmental plasticity and colonization history in the Atlantic salmon microbiome: a translocation experiment. Mol. Ecol. 29, 886–898. doi: 10.1111/mec.15369
Ursell, L. K., Clemente, J. C., Rideout, J. R., Gevers, D., Caporaso, J. G., and Knight, R. (2012). The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J. Allergy Clin. Immunol. 129, 1204–1208. doi: 10.1016/j.jaci.2012.03.010
Vadstein, O., Attramadal, K. J. K., Bakke, I., Forberg, T., Olsen, Y., Verdegem, M., et al. (2018). Managing the microbial Community of Marine Fish Larvae: a holistic perspective for Larviculture. Front. Microbiol. 9:1820. doi: 10.3389/fmicb.2018.01820
van Dijk, E. L., Auger, H., Jaszczyszyn, Y., and Thermes, C. (2014). Ten years of next-generation sequencing technology. Trends Genet. 30, 418–426. doi: 10.1016/j.tig.2014.07.001
van Kessel, M. A., Dutilh, B. E., Neveling, K., Kwint, M. P., Veltman, J. A., Flik, G., et al. (2011). Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.). AMB Express 1:41. doi: 10.1186/2191-0855-1-41
van Kessel, M., Mesman, R. J., Arshad, A., Metz, J. R., Spanings, F. A. T., van Dalen, S. C. M., et al. (2016). Gill nitrogen cycle symbionts can remove ammonia in fish gills. Environ. Microbiol. Rep. 8, 590–594. doi: 10.1111/1758-2229.12407
van Veelen, H. P. J., Falcao Salles, J., and Tieleman, B. I. (2017). Multi-level comparisons of cloacal, skin, feather and nest-associated microbiota suggest considerable influence of horizontal acquisition on the microbiota assembly of sympatric woodlarks and skylarks. Microbiome 5:156. doi: 10.1186/s40168-017-0371-6
Ventorino, V., Pascale, A., Adamo, P., Rocco, C., Fiorentino, N., Mori, M., et al. (2018). Comparative assessment of autochthonous bacterial and fungal communities and microbial biomarkers of polluted agricultural soils of the terra dei Fuochi. Sci. Rep. 8:14281. doi: 10.1038/s41598-018-32688-5
Walter, J. M., Bagi, A., and Pampanin, D. M. (2019). Insights into the potential of the Atlantic cod gut microbiome as biomarker of oil contamination in the marine environment. Microorganisms 7:E209. doi: 10.3390/microorganisms7070209
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wickham, H. (2016). Ggplot2: Elegant Graphics for Data Analysis, 2nd, New York: Springer International Publishing.
Wu, S., Gão, T., Wang, W., Cheng, Y., and Wang, G. (2010). Microbial diversity of intestinal contents and mucus in yellow catfish (Pelteobagrus fulvidraco). Aquaculture 303, 1–7. doi: 10.1016/j.aquaculture.2009.12.025
Wu, S., Wang, G., Angert, E. R., Wang, W., Li, W., and Zou, H. (2012). Composition, diversity, and origin of the bacterial Community in Grass Carp gut. PLoS One 7, 1–11. doi: 10.1371/journal.pone.0030440
Xia, Y., Wang, M., Gao, F., Lu, M., and Chen, G. (2020). Effects of dietary probiotic supplementation on the growth, gut health and disease resistance of juvenile Nile tilapia (Oreochromis niloticus). Anim. Nutr. 6, 69–79. doi: 10.1016/j.aninu.2019.07.002
Xie, S., Zhou, A., Xu, N., Feng, Y., Pan, Z., Junaid, M., et al. (2020). Benzo[a]pyrene induces microbiome dysbiosis and inflammation in the intestinal tracts of western mosquitofish (Gambusia affinis) and zebrafish (Danio rerio). Fish Shellfish Immunol. 105, 24–34. doi: 10.1016/j.fsi.2020.06.041
Yang, H.-T., Zou, S.-S., Zhai, L.-J., Wang, Y., Zhang, F.-M., An, L.-G., et al. (2017). Pathogen invasion changes the intestinal microbiota composition and induces innate immune responses in the zebrafish gut. Fish Shellfish Immunol. 71, 35–42. doi: 10.1016/j.fsi.2017.09.075
Yu, L., Qiao, N., Li, T., Yu, R., Zhai, Q., Tian, F., et al. (2019). Dietary supplementation with probiotics regulates gut microbiota structure and function in Nile tilapia exposed to aluminum. PeerJ 7:e6963. doi: 10.7717/peerj.6963
Zhai, Q., Yu, L., Li, T., Zhu, J., Zhang, C., Zhao, J., et al. (2017). Effect of dietary probiotic supplementation on intestinal microbiota and physiological conditions of Nile tilapia (Oreochromis niloticus) under waterborne cadmium exposure. Antonie Van Leeuwenhoek 110, 501–513. doi: 10.1007/s10482-016-0819-x
Keywords: São Francisco River, Neotropical fish, microbiome, next-generation sequencing, aquaculture, conservation
Citation: Damasceno MRA, Lemes CGC, Braga LSSB, Tizioto PC, Montenegro H, Paduan M, Pereira JG, Cordeiro IF, Rocha LCM, da Silva SA, Sanchez AB, Lima WG, Yazbeck GM, Moreira LM and Garcia CCM (2022) Hatchery tanks induce intense reduction in microbiota diversity associated with gills and guts of two endemic species of the São Francisco River. Front. Microbiol. 13:966436. doi: 10.3389/fmicb.2022.966436
Edited by:
Carlos Puch Hau, Centro de Investigación y de Estudios Avanzados del IPN, MexicoReviewed by:
Luis Alberto Lara-Perez, Instituto Tecnológico de la Zona Maya, (ITZM), Chetumal, MexicoT. T. Ajith Kumar, National Bureau of Fish Genetic Resources, India
Copyright © 2022 Damasceno, Lemes, Braga, Tizioto, Montenegro, Paduan, Pereira, Cordeiro, Rocha, da Silva, Sanchez, Lima, Yazbeck, Moreira and Garcia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Camila Carrião Machado Garcia, Y2Fycmlhb0B1Zm9wLmVkdS5icg==