 Yi Wu
Yi Wu Xinxin Peng
Xinxin Peng Xiaoya Li
Xiaoya Li Dandan Li1,2
Dandan Li1,2 Zhoujin Tan
Zhoujin Tan- 1College of Chinese Medicine, Hunan University of Chinese Medicine, Changsha, China
- 2Hunan Key Laboratory of Chinese Medicine Prescription and Syndromes Translational Medicine, Changsha, China
- 3Department of Pediatrics, The First Affiliated Hospital of Hunan University of Chinese Medicine, Changsha, China
Sex hormone secretion difference is one of the main reasons for sexually dimorphic traits in animals, which affects the dimorphism of the intestinal microbiota; however, their interaction is still unknown. Intestinal mucosa-associated microbiota (MAM) and intestinal luminal content microbiota (LM) belong to two different habitats according to the difference in interactions between bacteria and host intestinal epithelium/nutrients. To clarify the sexually dimorphic characteristics of MAM and LM and their correlation with sex hormones, 12 specific pathogen-free (SPF) Kunming mice from the same nest were fed separately according to sex. After 8 weeks, samples from the male intestinal mucosa group (MM group), the female intestinal mucosa group (FM group), the male intestinal content group (MC group), and the female intestinal content group (FC group) were collected and then, the next-generation sequencing of 16S ribosomal ribonucleic acid (rRNA) gene was performed. Our results showed that the sexual dimorphism of MAM was more obvious than that of LM and the relative abundance of Muribaculaceae, Turicibacter, and Parasutterella was significantly higher in the FM group than in the MM group (p < 0.001, p < 0.05, p < 0.05). Next, we measured the level of serum sex hormones in mice and calculated the correlation coefficient between major bacteria and sex hormones. The results showed that the correlation between MAM and sex hormones was more prominent, and finally, three bacterial genera (Muribaculaceae, Turicibacter, and Parasutterella) were obtained, which could better represent the relationship between sexual dimorphism and sex hormones. The abundance of Parasutterella is positively and negatively correlated with estradiol and testosterone (T), respectively, which may be related to the differences in the metabolism of bile acid and glucose. A decrease in the abundance of Turicibacter is closely related to autism. Our results show that the abundance of Turicibacter is negatively and positively correlated with T and estradiol, respectively, which can provide a hint for the prevalence of male autism. In conclusion, it is proposed in our study that intestinal microbiota is probably the biological basis of physiological and pathological differences due to sex, and intestinal MAM can better represent the sexual dimorphism of mice.
Introduction
Significant differences in mammalian health and disease exist between males and females, that is, sexual dimorphism (Britannica, 2022). For example, in most mammalian species, males are slightly larger than females (Naqvi et al., 2019), and differences in longevity and aging processes have been observed between males and females (Sampathkumar et al., 2020). Sexually dimorphic traits are also evident in the incidence, epidemic, and mortality of diseases, such as autoimmune disorders and autism (Alshammari, 2021; Manuel and Liang, 2021). In recent years, gut microbiota has become a hotspot for various studies. Coincidentally, these studies found sex differences in gut microbiota composition between humans and rodents (Ding and Schloss, 2014; Falony et al., 2016; Borgo et al., 2018; Sinha et al., 2019). It has also been suggested that sex hormones play an important role in building and maintaining the characteristics of the gut microbiome associated with sex (Zhang X. et al., 2021).
Sexual dimorphism is controlled by sex hormones, which have a bidirectional interaction with the intestinal microbiota. On the one hand, sex hormones affect the intestinal microbiota by regulating the permeability and integrity of an intestinal barrier and adjusting sex hormone receptors, β-glucuronidase, bile acid, intestinal immunity, etc. (Braniste et al., 2009; Looijer-van Langen et al., 2011; Li and Chiang, 2015; Laffont et al., 2017; Pellock and Redinbo, 2017; Miranda-Ribera et al., 2019; Barroso et al., 2020). On the other hand, the intestinal microbiota also influences the secretion of sex hormones, for example, androgen. Testosterone (T) and ovaries are the major production sources of male and female androgen, respectively, and the intestinal microbiota is the major regulator of androgen metabolism in the intestinal tract (Pernigoni et al., 2021). Some bacterial strains have been shown to metabolize androgen in vitro. For example, Aggregatibacter actinomycetecomitans and Porphyromonas gingivalis convert T to dihydrotestosterone (Bélanger et al., 1989). Pathologically, abnormal fluctuations in androgen contribute to the development and progression of diseases by affecting the intestinal microbiota (Yurkovetskiy et al., 2013; Moreno-Indias et al., 2016). Female rats with pathological androgen levels were found to have different intestinal microbiota from normal rats. Studies showed that abnormal androgen levels can lead to intestinal dysbacteria, including enzymes involved in androgen metabolism, which further interfere with androgen metabolism, and are associated with diseases such as polycystic ovary syndrome (Lindheim et al., 2017; Liu et al., 2017; Torres et al., 2018; Zeng et al., 2018), type 1 diabetes (Markle et al., 2013; Yurkovetskiy et al., 2013), and obesity (Kelly and Jones, 2015; Harada et al., 2016). Therefore, intestinal microbiota dimorphism is also a part of sexual dimorphism, in which sex hormones play a crucial role.
In recent years, sufficient animal experiments and clinical trials have confirmed differences in composition and function between luminal content microbiota (LM) and mucosa-associated microbiota (MAM) (Van den Abbeele et al., 2011; Yang et al., 2020). MAM is believed to interact directly or indirectly with the host intestinal epithelium and is therefore critical to the formation of the host immune system. LM is mainly involved in the digestion of nutrients and does not interact directly with the intestinal mucosa. Therefore, the composition and function of MAM is closely related to the host and its immune system, while LM is closely related to nutrients (Van den Abbeele et al., 2011). Due to this difference, LM and MAM have different roles in the initiation and progression of diseases. MAM played the most important role in the pathogenesis of diarrhea-predominant irritable bowel syndrome (IBS-D) (Maharshak et al., 2018), MAM is highly susceptible to disruption in patients with diarrhea because it participates in neurological responses (Zhang C. et al., 2021). Functional MAM and LM differ from each other (He et al., 2019; Wu et al., 2020). According to Francesca Borgo, microbial diversity in these two niches might be influenced by host factors such as body mass index (BMI), diet, and sex. Thus, in our study, we controlled all variables except sex and focused on the effects of sex and sex hormones on intestinal microbiota diversity (Borgo et al., 2018).
In summary, we attempted to clarify the following questions: (i) Are host sex hormones involved in intestinal microbiota diversity? (ii) Is there a difference in the impact of host sex on MAM and LM diversity? (iii) Could the intestinal microbiota be one of the biological bases of sex-associated diseases?
Materials and methods
Materials
Animals and feeding
A total of 12 specific pathogen-free (SPF) Kunming mice (3 weeks of age, half male, half female) were purchased from Hunan Slike Jingda Laboratory Animal Co., Ltd. and fed in a regulated barrier system with light and dark cycles of 12 h, 23–25°C, and 50–70% relative humidity. After 1 week of adaptive feeding, animals were divided into male and female groups and fed for 8 weeks. Mice were fed by the Animal Experiment Center of Hunan University of Chinese Medicine with nutritional standards in line with GB/14924.3 and sanitation standards in line with GB/T149.24.2 to support their growth and reproduction. All animal experiments were licensed by the Animal Experiment of Hunan University of Chinese Medicine (Changsha, China), and the protocol was approved by the Animal Ethics Committee of Hunan University of Chinese Medicine [Facility use permit number: SYXK (Xiang) 2019-0009].
Methods
Serum sex steroid-level testing
Post experiment, all mice were sacrificed by sampling orbital blood after fasting for 12 h; then, small intestinal contents and mucosa were collected according to the method established by our research group (Wu et al., 2021). Four serum sex hormones (estradiol, T, prolactin, and progesterone) were detected using chemiluminescence immunoassay. The operating instrument was the Abbott AXSYM automatic chemiluminescence instrument equipped with matching reagents. The nuclear medicine department of the First Affiliated Hospital of the Hunan University of Chinese Medicine is responsible for the detection. In the female group, intestinal contents were labeled as FC 1, FC 2, FC 3, FC 4, FC 5, and FC 6 and the intestinal mucosa was labeled as FM 1, FM 2, FM 3, FM 4, FM 5, and FM 6. In the male group, intestinal contents were labeled as MC 1, MC 2, MC 3, MC 4, MC 5, and MC 6 and the intestinal mucosa was labeled as MM 1, MM 2, MM 3, MM 4, MM 5, and MM 6. All samples were stored at −80°C for the high-throughput sequencing of the 16S ribosomal ribonucleic acid (rRNA) gene.
Extraction and polymerase chain reaction amplification of total DNA
The total microbial genomic deoxyribonucleic acid (DNA) from each sample was extracted as per the directives of the DNA extraction kit (MN NucleoSpin 96 So), and the steps were included as follows. The sample was precipitated to remove impurities and filtered to remove inhibitors, followed by DNA binding, membrane washing, drying, and elution. Quantity and mass of the extracted DNA were detected using NanoDrop ND-2000 ultramicro spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and Qubit 3.0 Fluorometer (Life Technologies, CA, USA), via agarose gel electrophoresis, respectively. All samples were processed by Beijing Biomac Biotechnology Co., Ltd. (Beijing, China).
Bridge polymerase chain reaction and 16S rRNA gene sequencing
Flow cells are the channels for adsorption of flowing DNA fragments. Adapter-added DNA fragments on a chip containing adapters are bound to flow cells and bridge-amplified. The primers, 338F5'-ACTCCTACGGGAGGCAGCA-3' and 806R5'-GGACTACHVGGGTWTCTAAT-3', were designed according to the conservative region of 16S r DNA V3-V4. The amplification reaction system was as follows: 50 ng of gene DNA, 0.3 μl of Vn F, 0.3 μl of Vn R, 5 μl of KOD FX Neo Buffer, 2 μl of deoxynucleoside triphosphate (dNTP), 0.25 μl of KOD FX Neo, and finally a total reaction volume to 10 μl of double pure water (ddH2O). Polymerase chain reaction (PCR) reaction conditions were given as follows: DNA was rapidly denatured at 95°C for 30 s and rapidly cooled to 50°C for 30 s, and the primers were annealed and bound to the target sequence and rapidly heated to 72°C for 40 s. After the last cycle, the primer strands were extended along the template for 7 min and were maintained at 4°C.
Library preparation and 16S rRNA gene sequencing were performed using the Solexa Genome Analyzer platform. When a complementary chain extends into a lateral column cluster family, each inserted dNTP can release corresponding fluorescence, which is immediately detected by a sequencer and then converted into fragment sequence information. Next-generation sequencing features a bridge polymerase chain reaction along with 16S rRNA gene sequencing. The basic principle is DNA polymerase and fluorescently labeled dNTPs and adapter primers in the amplification reaction system when a complementary chain extends into a lateral column cluster family.
Data processing
The raw sequencing data are processed with quality filtering, double-ended sequence splicing, and chimera elimination. The reads of each sample are spliced using the USEARCH (version 10) (Edgar Robert, 2013) with a minimum overlap length of 10 bp and a maximum mismatch ratio in the overlap area of 0.2 (Default), and the resulting splicing sequence is the Raw Tags. After quality inspection, tags with a length of less than 75% are filtered to get Clean Tags using the Trimmomatic program. To extract the final tag sequence, the chimera was eliminated using UCHIME (version 8.1) (Bokulich et al., 2013).
Bioinformatics analysis and statistical methods
Sequences were clustered using USEARCH (version 10.0) (Edgar Robert, 2013) with a similarity criterion of 97% and the default OTU filtering threshold of 0.005% of all sequences. OTU (operational taxonomic units) were then aligned in the Silva database, and the species were annotated using the blast method. The α and β diversities were demonstrated by Chao 1, Shannon, ACE, Simpson index, non-metric multi-dimensional scaling (NMDS) analysis, and analysis of similarities (ANOSIM). The abundance of microbiota at all levels is calculated based on the OTU and is presented in a histogram. The random forest algorithm is an integrated algorithm that integrates multiple decision trees and can avoid the problem of overfitting a single decision tree. The Gini index calculates the influence of each variable on the observed heterogeneity at each node of the classification tree, and larger values indicate that the variables are more important. Our study uses line discriminant analysis effect size (LEfSe) and random forest algorithm analysis at the same time to find the biomarker for different groups. To further analyze the correlation between the intestinal microbiota and sex hormones, we calculated Spearman's rank correlation coefficient and plotted the heatmap. Compared to PICRUSt 1, PICRUSt 2 (Douglas et al., 2020) has the advantages of richer genomic information, more realistic prediction settings, and more rigorous functional prediction methods. The abovementioned analysis was performed using BMKCloud (www.biocloud.net).
Scatter and linear correlation plots (Pearson correlation method) were drawn using Graph pad Prism 9.0. Statistical data were analyzed using SPSS 24.0 software (IBM, Almonk, NY, USA). The independent sample t-test was used when the two groups of data were in agreement with the normal distribution. Otherwise, the nonparametric test (the Mann–Whitney U test) was used. The test criterion was a p-value < 0.05 or a p-value < 0.01.
Results
DNA sequence and the number of OTUs
As shown in Figure 1A, when the number of species approaches 230, the curve flattens, indicating that the number of species meets the analysis criteria. As shown in Figure 1B, the four groups had 356 shared OTUs; the MC and FC groups found 65 and 28 unique OTUs, respectively. MM and MC groups had 20 and 18 unique OTUs, respectively.
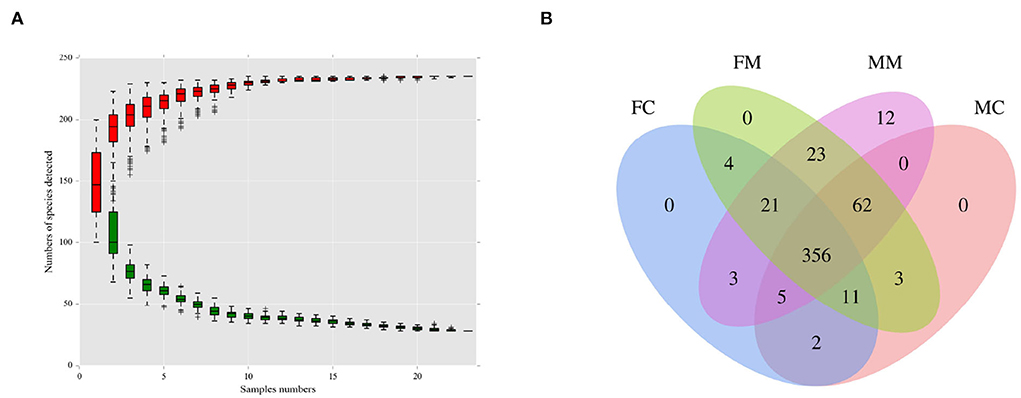
Figure 1. (A) The species accumulation curve was used to evaluate whether the sequencing volume was sufficient to cover all taxa and reflects the species richness of the intestinal microbiota. (B) Venn diagram of each group based on the number of OTUs. FC, luminal content of female mice; FM, mucosa of female mice; MM, mucosa of male mice; MC, luminal content of male mice.
Bacterial diversity analysis
α and β diversities explain the richness and diversity of microbial communities from different dimensions. α diversity refers to the richness and diversity of microbial communities and species within a living territory, expressed in four indicators: Chao 1, Shannon, ACE, and Simpson. As shown in Figure 2, there are no significant sex differences between LM and MM in the Chao 1, Shannon, ACE, and Simpson indices.
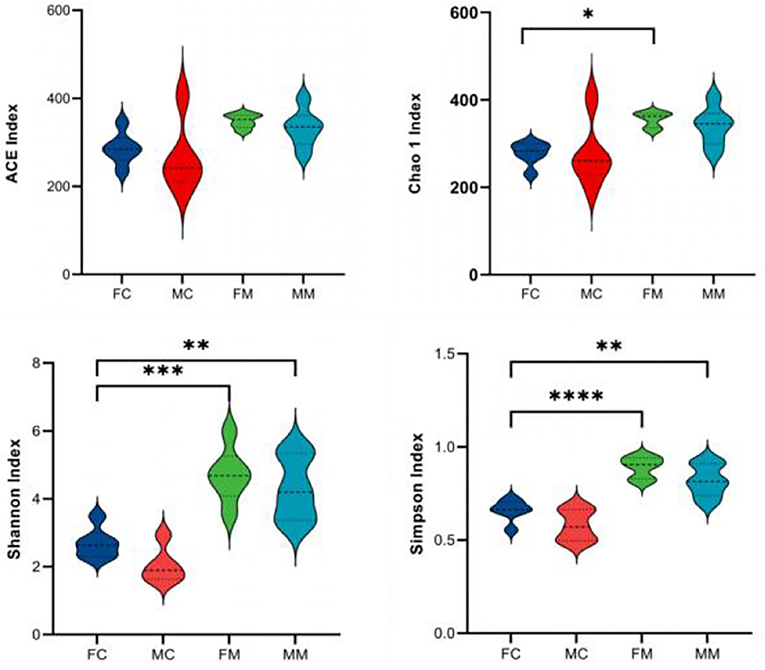
Figure 2. The α diversity index was calculated based on the OTU classification level, consisting of ACE, Chao 1, Shannon, and Simpson. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
β diversity refers to the difference in the number and distribution of species in different environmental communities, reflecting not only the diversity distance between samples but also the degree of differentiation between bacterial communities. As shown in the NMDS analysis (Figure 3A), there was a small distance between the FM and MM groups (stress = 0.1365). The ANOSIM analysis (Figures 3B–D) showed significant differences between the FC and MC groups and the FM and MM groups (p < 0.01). The phylogenetic tree combined with a histogram of species distribution (Figure 3E) intuitively indicates subtle differences among groups, with the MC group being distinguishable from others.
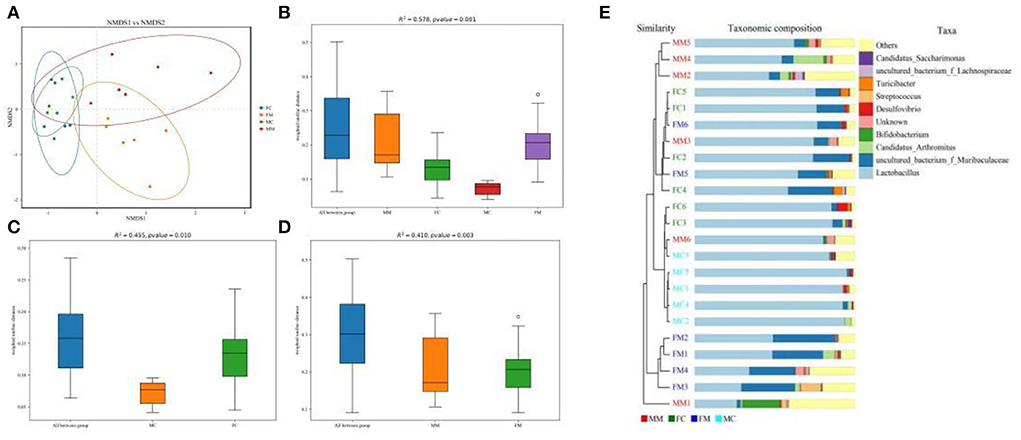
Figure 3. (A) Non-metric multidimensional scaling (NMDS) analysis (based on weighted UniFrac distance). Different groups of samples were colored differently. Distance between the dots presented a difference. NMDS with stress levels of less than 0.2 is acceptable. The closer the sample is to the coordinates, the higher the similarity. (B–D) Analysis of similarities (ANOSIM) (based on weighted UniFrac distance) can test for a significant difference in β-diversity among samples of different groups. The y-axis represents β-distance; the box above “All between *” represents the β-distance data of samples of all groups, while the box above “All within *” represents the β-distance data of samples within all groups. The closer the R-value is to 1, the difference between groups is higher than the difference within groups; the smaller the R-value is, the smaller difference between groups; and the value of p < 0.05 indicates high reliability of the test. (E) Combination of the phylogenetic tree and histogram based on the unweighted group average clustering method.
Bacterial composition analysis of intestinal microbiota
To further investigate the differences in the intestinal microbiota of mice of different sexes, the relative abundance of microbial communities at the phylum and genus level in each group was counted. The combined abundance of Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria exceeded 98% in LM (Figure 4A). The relative abundance of Firmicutes was higher in males than in females (p < 0.05, Figure 4B), while in the case of Bacteroidetes the opposite was true (p < 0.05, Figure 4B). The four phyla were also dominated in MAM, and the relative abundance of Bacteroidetes was also higher in females than in males (p < 0.05, Figure 4B). The Firmicutes/Bacteroidetes (F/B) ratio of MAM and LM was higher in males than in females (p < 0.05, p < 0.05, Figure 4C).
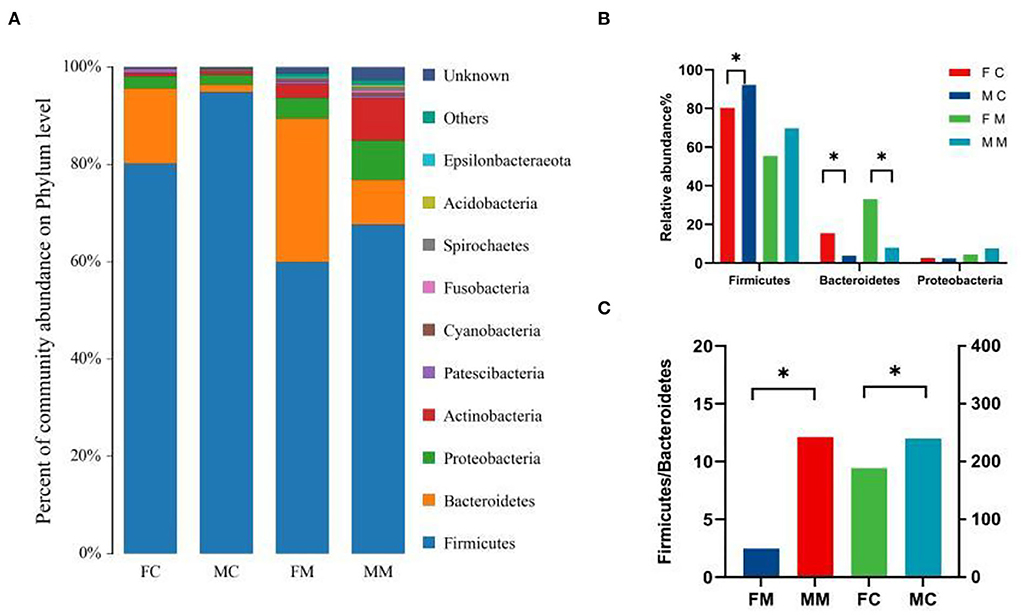
Figure 4. (A) The composition and relative abundance of species at the phylum level of each group, (B) the abundance of Bacteroidetes, Firmicutes, and Proteobacteria in each group (*p < 0.05), and (C) the Firmicutes/Bacteroidetes (F/B) ratio for each group.
Figure 5 shows the relative abundance of each group of bacterial species at the genus level. The results showed that Lactobacillus, Muribaculaceae, Candidatus Arthromitus, Bifidobacterium, and Desulfovibrio were enriched, of which Lactobacillus had the highest relative abundance. The relative abundance of Muribaculaceae, Turicibacter, and Faecalibaculum was significantly higher in the FC group than in the MC group (p < 0.01, p < 0.05, p < 0.05). A total of seven of the top 30 relative abundance had a significant sex difference in MAM. Specifically, the relative abundance of Muribaculaceae, Turicibacter, and Parasutterella was significantly higher in the FM group than in the MM group (p < 0.001, p < 0.05, p < 0.05), while the relative abundance of Bifidobacterium, Gammaproteobacteria, Enterococcus, and Streptococcus was significantly higher in the MM group (p < 0.05) than in the FM group. Overall, sex differences were more prominent in MAM than LM.
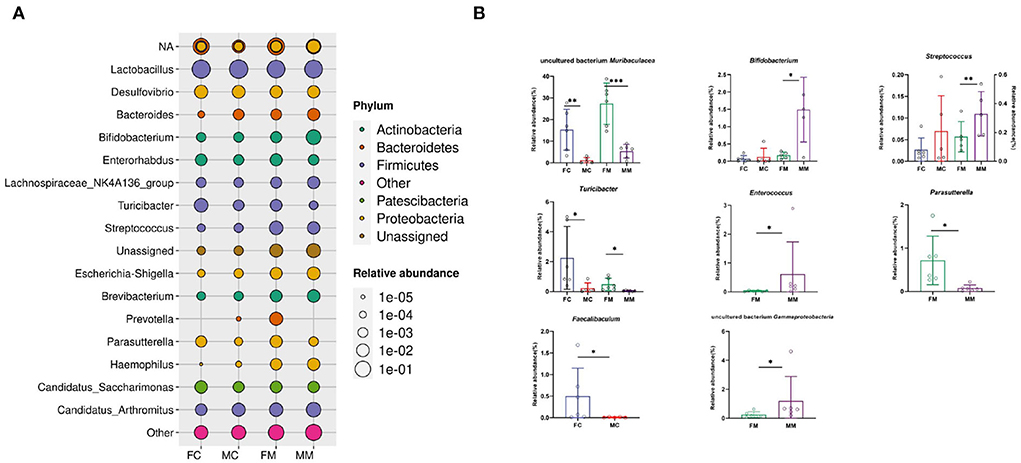
Figure 5. (A) The bacterial species and relative abundance of each group at the genus level, (B) illustration of sex differences in the relative abundance of the top 30 major genera (*p < 0.05, **p < 0.01, ***p < 0.01).
Intestinal differential bacterial species analysis in each group
To further identify the species with the greatest differences in each group, we performed the LESef analysis (SCORE > 4, p < 0.05). The results (Figure 6A) showed that Bifidobacterium longum subsp. was a differential bacterium in the MC group, Prevotella, Muribaculacea, and Bacteroidales were the differential bacteria in the FC group. As shown in Figure 6B, Lactobacillus was a differential bacterium in the MM group and Muribaculaceae, Erysipelotrichaceae, Turicibacter, and Anaerococcus were the differential bacteria in the FM group.
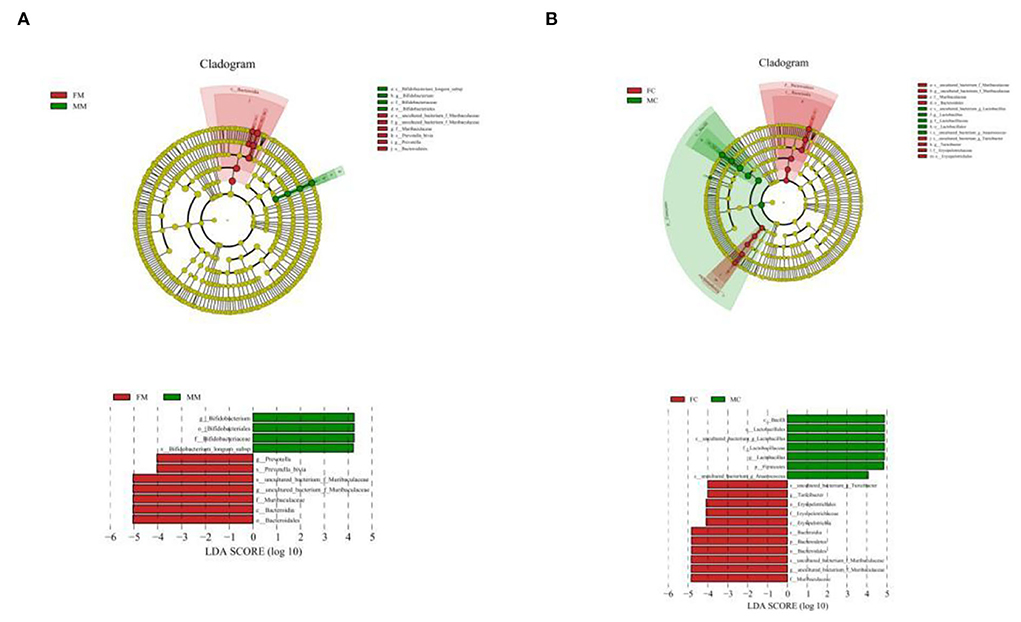
Figure 6. Cladogram based on the line discriminant analysis effect size (LEfSe) analysis and linear discriminant analysis (LDA) value distribution histogram of MAM (A) and LM (B). In this figure, species with no significant differences are colored in yellow, and other colors stand for different groups. The LDA histogram showed the species whose LDA score is higher than the set value (the default is 4.0). The length of the histogram represents the impact of different species (i.e., LDA score), and different colors represent species in different groups. Different colors represent species in different groups, and nodes with different colors represent microbial groups that play an important role in the group represented by the color.
The random forest algorithm analysis showed that Parasutterella, Turicibacter, and Muribaculaceae had the highest Gini index in the MAM group (Figure 7A), and Muribaculaceae and Pleomorphomonadaceae had the highest in the LM group (Figure 7B). In summary, Parasutterella, Muribaculaceae, and Turicibacter were the differential species identified in the FM and MM groups, and Muribaculaceae was the differential species identified in the FC and MC groups, which further confirmed that the sexual dimorphism of MAM was greater than that of LM.
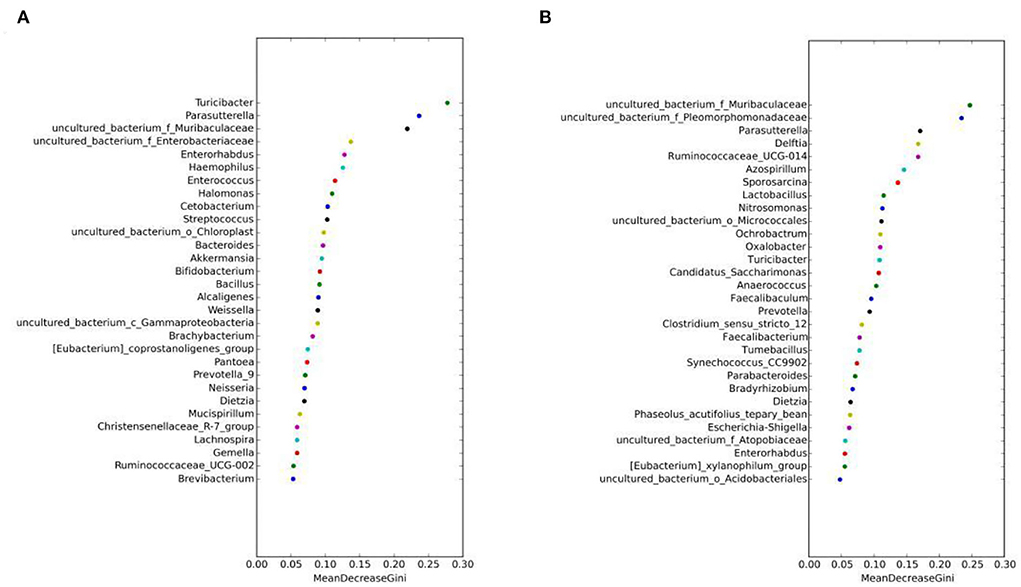
Figure 7. Random forest algorithm analysis of MAM (A) and intestinal LM (B). The horizontal axis is the mean decrease Gini, and the vertical axis is the top 30 genera with the highest relative abundance. The larger the mean decrease Gini, the higher the contribution of bacteria.
Correlation analysis of serum sex steroid hormones and the intestinal microbiota
To confirm whether the serum levels of sex hormones were related to the sexual dimorphism of the intestinal microbiota, we detected four serum sex hormone levels and found (Table 1) significant differences in serum levels of estradiol (E2) and T of male and female mice (p < 0.01). It could be seen in Figures 8A–C that, in MAM, Parasutterella, Muribaculaceae, Enterorhabdus, and E2 were significantly positively correlated (p < 0.05), especially since Muribaculaceae and Parasutterella had extremely significant differences (R > 0.073, p < 0.01; R > 0.089, p < 0.001). Moreover, Brachybacterium, Bacteroides, Gammaproteobacteria, Brevibacterium, and Haemophilus were positively correlated with T (p < 0.05), while Candidatus Saccharimonas, Parasutterella, and Muribaculaceae were negatively correlated with T (p < 0.05). In LM (Figures 8B,D), Faecalibaculum, Turicibacter, Parasutterella, Muribaculaceae, and Lachnospiraceae were positively correlated with E2 (p < 0.05), while Lactobacillus showed the opposite (R < −0.84, p ≤ 0.001) and Turicibacter was negatively correlated with T (R = −0.79, p < 0.05).

Table 1. Differences in serum sex steroid levels in mice ( ± S, n = 6).
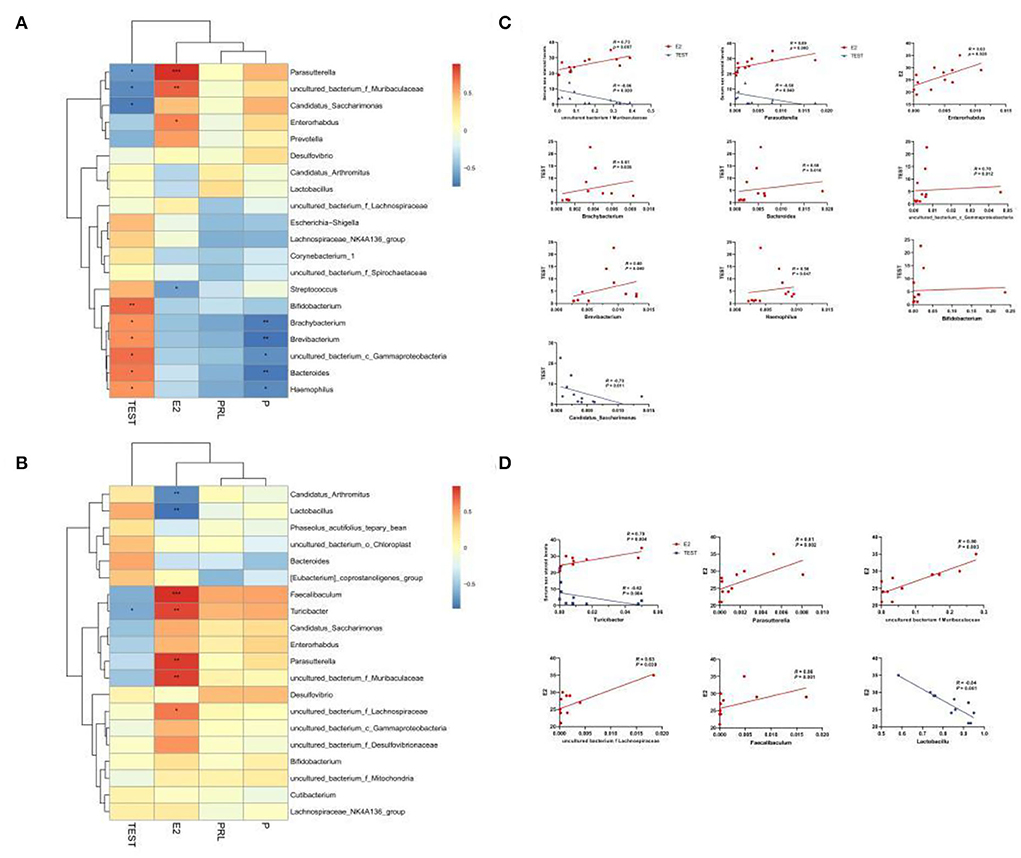
Figure 8. Heatmap of MAM (A) and LM (B) correlations with sex steroid levels. In the heat map, red represents a positive correlation, and blue represents a negative correlation; the closer to red, the closer the R-value is to 1; the closer to blue, the closer the R-value is to −1. In the linear correlation diagram of MAM (C) and LM (D), the closer the R-value is to 1, the more obvious the correlation. *p < 0.05, **p < 0.01, ***p < 0.001.
Functional prediction of MAM in mice of different sexes
To further explore whether the sexual dimorphism of MAM affects the potential function, we performed a functional prediction analysis of each group. As showed in Table 2, the glycosphingolipid biosynthesis—globo and isoglobo series is the only metabolic pathway with significant sex differences in MAM (p < 0.05).

Table 2. Functional prediction table of Kyoto Encyclopedia of Genes and Genomes (KEGG) level 3 of mucosa-associated microbiota (MAM).
Discussion
Differences in the characteristics of the intestinal microbiota in mice of different sexes
One of the important conclusions of this study is that MAM is more sex-sensitive than LM. As reported in the study, sex has a major influence on MAM (Borgo et al., 2018) than on LM, which is further confirmed in our experiment. First of all, β analysis showed sex differences only in the MAM. Secondly, among the top 30 bacteria with relative abundance at the genus level, the number of bacteria with sex differences in LM was 5, while the number of bacteria with sex differences in MAM was 7. Moreover, seven bacterial genera were associated with E2 secretion and only one with T secretion in LM, and four bacterial genera were associated with E2 secretion and nine with T secretion in MAM. In both physiology and pathology, MAM seems to be more susceptible to the host than to LM, which is attributed to the fact that MAM is less affected by food rather than LM. Zhang C. et al. (2021) proved that MAM is more susceptible to repeated stress-related diarrhea in comparison to LM. We conclude that MAM is more influenced by sex hormones, which can better represent sexual dimorphism. In further research, it is best to consider whether MAM is more suitable for the experiment. Unlike other studies, we did not find that the α diversity of female mice was higher than that of male mice, which was probably due to the different locations from which the samples were collected (Roager et al., 2016).
The intestinal microbiota of mice with different sexes provides a new perspective on sexually dimorphic diseases
Our study found that the intestinal microbiota may be one of the important biological bases of sexual dimorphism in physiology and pathology. LEfSe and random forest algorithm analyses showed that Muribaculaceae and Parasutterella were the key genera to distinguish mice with different sexes. The relative abundance analysis of the intestinal microbiota revealed the highest concentration of Muribaculaceae, Parasutterella, and Turicibacter in the intestine of female mice. The correlation analysis of serum sex hormones with the intestinal microbiota illustrated that, in LM, both Muribaculaceae and Parasutterella were positively and negatively correlated with E2 and T, respectively, and Turicibacter was negatively correlated with T. All results demonstrate that Muribaculaceae, Parasutterella, and Turicibacter are the key species of intestinal sexual dimorphism.
Parasutterella may be involved in intestinal bile acid metabolism as the core intestinal microbiota of mice and humans (Ju et al., 2019). Bile acid synthesis rate and bile acid pool have been shown to be higher in females than in males (Turley et al., 1998). Related studies (Org et al., 2016) showed that bile acid metabolism changes significantly with hormonal changes, especially in high-fat and high-sugar diets. In our study, females of Parasutterella were highly abundant than male parasites, further suggesting that sex differences in bile acid metabolism might be related to intestinal Parasutterella. In addition, we found a positive correlation between the abundance of Parasutterella and E2, suggesting that sex hormones influenced bile acid metabolism in females by regulating the abundance of Parasutterella. Type 2 diabetes is a glucose metabolism disorder with significant sexual dimorphism; however, there is no clear mechanism to explain how this sex difference occurs. In animal studies, researchers proved that male mice have lower glucose tolerance compared to females, which is associated with intestinal microbiota and sex hormones (Gao et al., 2021). Parasutterella is a producer of succinate, which improves glucose homeostasis through intestinal gluconeogenesis (De Vadder et al., 2016; Canfora et al., 2019). Therefore, we hypothesize that sex hormones may be involved in the sexual dimorphism of glucose metabolism by regulating the abundance of Parasutterella.
Muribaculaceae is the dominant family in the intestine of mice. Studies showed that Muribaculaceae degrades dietary carbohydrates and rapidly adapts to carbohydrate-enriched diets to resist obesity (Obanda et al., 2018; Lagkouvardos et al., 2019). In our study, the abundance of Bacteroides and Muribaculaceae is significantly higher in females than in males and is positively and negatively correlated with E2 level and T, respectively. In addition, we found that the ratio of F/B (Jasirwan et al., 2021) used to evaluate the energy metabolism capacity was lower in female mice than in male mice (Figure 4C), which indicated that female mice had higher energy metabolism ability than male mice. As mentioned above, how sex hormones successfully and synthetically regulate the body's energy balance with Muribaculaceae, which will help to shed light on the formation mechanism of obesity and its prevalence in specific populations, is shown.
Turicibacter belonging to Firmicutes is involved in the metabolism of bile acids and cholesterol (Kemis et al., 2019). According to studies, Turicibacter may also be related to obesity, its abundance is positively correlated with high-density lipoproteins, and it may be involved in the formation mechanism of obesity by regulating cholesterol metabolism (Zheng et al., 2020). Turicibacter is therefore another important bacterium that reveals the biological basis of sex differences in obesity between males and females. There is a sex bias in mental disorders, which the sex-selective sex hormone theory seeks to uncover (Singh et al., 2021). Similarly, many studies demonstrated that the intestinal microbiota participates in the development of mental diseases through the brain-gut axis (Sharon et al., 2019; Qin et al., 2022). An interesting phenomenon is that males are more likely to exhibit the aggravation of autism symptoms with changes in their intestinal microbiota in times of adversity, but these have a limited impact on females (Rincel et al., 2019). In addition, the intestinal microbiota of male mice with autism showed a decrease in Turicibacter, and the lower the abundance of Turicibacter, the greater the social deficit (Szyszkowicz et al., 2017); however, the abundance of Turicibacter was reversed after intake of Lactobacillus (Kong et al., 2021). In our study, we further found that male mice had a significantly lower abundance of Turicibacter than female mice and were correlated with T. These results suggest that the development of autism in males may be related to congenital hormone levels and the intestinal microbiota, of which Turicibacter is worth exploring.
The abundance of Bifidobacterium, Gammaproteobacteria, and Enterococcus was significantly higher in the MM group than in the FM group and positively correlated with T, indicating that Bifidobacterium, Gammaproteobacteria, and Enterococcus were significantly regulated by sex hormones. Studies showed a marked increase in serum androgens and a lack of intestinal probiotics like Bifidobacterium in polycystic ovarian syndrome, but taking Bifidobacterium can reverse this trend. Unfortunately, the current experiment lacks evidence related to MAM (Zhang et al., 2019). The incidence of Crohn's disease is slightly higher in males than in females, but males had a milder disease severity than females. Studies confirm that this may be related to estrogen (Goodman et al., 2020). Studies showed that proteus plays a key role in the development of Crohn's disease by attacking the intestinal mucosa (Zhang J. et al., 2021). Other research also showed that Gammaproteobacteria are enriched within CD14+ macrophages from the intestinal lamina propria of patients with Crohn's disease vs. mucus (Sekido et al., 2020). However, in our research, Proteus has little correlation with estrogen, and its abundance is higher in the MM group than in the FM group. Enterococcus is a physiologically dominant species in the intestine of mice and humans, with strong tolerance and the ability to colonize the intestinal mucosa and inhibit pathogen damage to intestinal mucosal epithelial cells (O' Shea et al., 2012). So far, however, no link between Enterococcus and sex or sex hormones has been reported.
Conclusion
Our study concluded that MAM is more sexually dimorphic than LM. We identified that various bacteria were prone to a certain sex and were highly correlated with E2 and T levels. Parasutterella, Muribaculacea, and Turicibacter are considered the best representatives of the intestinal microbiota associated with sex hormones. In addition, our study further found that Parasutterella, Muribaculacea, and Turicibacter are the key bacteria that cause intestinal sexual dimorphism. It is speculated that sex hormones may be involved in sexual dimorphism in bile acid metabolism by regulating the abundance of these bacteria. The dimorphism of Turicibacter in the intestinal microbiota also provides insight into how neurological diseases are more common in males than in females. Furthermore, our study also found that the glycosphingolipid metabolism of MAM had significant sex differences, which provided new clues for the mechanisms underlying sex differences in glucose metabolism.
However, our study only compared the differences in the intestinal microbiota in physiological mice and lacked experiments or data analysis for specific sex-associated diseases. Future studies should be based on open databases or more animal experiments to provide some reference for accurate clinical use.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI, PRJNA847180.
Ethics statement
The animal study was reviewed and approved by Animal Ethics Committee of Hunan University of Chinese Medicine. The number of the facility use permit: SYXK (Xiang) 2019-0009.
Author contributions
YW: animal experiments, data analysis, and original draft writing. XP and XL: validation, review, and editing. DL: data analysis. ZT and RY: project administration and funding acquisition. All authors contributed to this article and approved the submitted version.
Funding
This research was financially supported by the National Natural Science Foundation of China (Grant No. 81874460), the National Natural Science Foundation of China Regional Innovation and Development Joint Fund Key Support Project (Grant No. U21A20411), and the Provincial Natural Science Foundation of Hunan (Grant No. 2022JJ30440).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.964847/full#supplementary-material
References
Alshammari, T. K. (2021). Sexual dimorphism in pre-clinical studies of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 105, 110120. doi: 10.1016/j.pnpbp.2020.110120
Barroso, A., Santos-Marcos, J. A., Perdices-Lopez, C., Vega-Rojas, A., Sanchez-Garrido, M. A., Krylova, Y., et al. (2020). Neonatal exposure to androgens dynamically alters gut microbiota architecture. J. Endocrinol. 247, 69–85. doi: 10.1530/JOE-20-0277
Bélanger, B., Bélanger, A., Labrie, F., Dupont, A., Cusan, L., and Monfette, G. (1989). Comparison of residual C-19 steroids in plasma and prostatic tissue of human, rat and guinea pig after castration: unique importance of extratesticular androgens in men. J. Steroid. Biochem. 5, 695–698. doi: 10.1016/0022-4731(89)90514-1
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Method 10, 57–59. doi: 10.1038/nmeth.2276
Borgo, F., Garbossa, S., Riva, A., Severgnini, M., Luigiano, C., Benetti, A., et al. (2018). Body Mass index and sex affect diverse microbial niches within the gut. Front. Microbiol. 9, 213. doi: 10.3389/fmicb.2018.00213
Braniste, V., Leveque, M., Buisson-Brenac, C., Bueno, L., Fioramonti, J., and Houdeau, E. (2009). Oestradiol decreases colonic permeability through oestrogen receptor beta-mediated up-regulation of occludin and junctional adhesion molecule-A in epithelial cells. J. Physiol. 587, 3317–3328. doi: 10.1113/jphysiol.2009.169300
Britannica, The Editors of Encyclopaedia. (2022). “sexual dimorphism”. Encyclopedia Britannica, 14 Feb. 2020, Available online at: https://www.britannica.com/science/sexual-dimorphism (accessed 6 June 2022).
Canfora, E. E., Meex, R. C. R., Venema, K., and Blaak, E. E. (2019). Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 15, 261–273. doi: 10.1038/s41574-019-0156-z
De Vadder, F., Kovatcheva-Datchary, P., Zitoun, C., Duchampt, A., Bäckhed, F., and Mithieux, G. (2016). Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab. 24, 151–157. doi: 10.1016/j.cmet.2016.06.013
Ding, T., and Schloss, P. D. (2014). Dynamics and associations of microbial community types across the human body. Nature 509, 357–360. doi: 10.1038/nature13178
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Edgar Robert, C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Method 10, 996–998. doi: 10.1038/nmeth.2604
Falony, G., Joossens, M., Vieira-Silva, S., Wang, J., Darzi, Y., Faust, K., et al. (2016). Population-level analysis of gut microbiome variation. Science 352, 560–564. doi: 10.1126/science.aad3503
Gao, A., Su, J., Liu, R., Zhao, S., Li, W., Xu, X., et al. (2021). Sexual dimorphism in glucose metabolism is shaped by the androgen-driven gut microbiome. Nat. Commun. 12, 7080. doi: 10.1038/s41467-021-27187-7
Goodman, W. A., Erkkila, I. P., and Pizarro, T. T. (2020). Sex matters: impact on pathogenesis, presentation and treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 17, 740–754. doi: 10.1038/s41575-020-0354-0
Harada, N., Hanaoka, R., Horiuchi, H., Kitakaze, T., Mitani, T., Inui, H., et al. (2016). Castration influences intestinal microflora and induces abdominal obesity in high-fat diet-fed mice. Sci. Rep. 6, 23001. doi: 10.1038/srep23001
He, Y., Tang, Y., Peng, M., Xie, G., Li, W., and Tan, Z. (2019). Influence of Debaryomyces hansenii on bacterial lactase gene diversity in intestinal mucosa of mice with antibiotic-associated diarrhea. PLoS ONE 14, e022580. doi: 10.1371/journal.pone.0225802
Jasirwan, C. O. M., Muradi, A., Hasan, I., Simadibrata, M., and Rinaldi, I. (2021). Correlation of gut Firmicutes/Bacteroidetes ratio with fibrosis and steatosis stratified by body mass index in patients with non-alcoholic fatty liver disease. Biosci. Microbiota. Food Health 40, 50–58. doi: 10.12938/bmfh.2020-046
Ju, T., Kong, J. Y., Stothard, P., and Willing, B. P. (2019). Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 13, 1520–1534. doi: 10.1038/s41396-019-0364-5
Kelly, D. M., and Jones, T. H. (2015). Testosterone and obesity. Obes. Rev. 16, 581–606. doi: 10.1111/obr.12282
Kemis, J. H., Linke, V., Barrett, K. L., Boehm, F. J., Traeger, L. L., Keller, M. P., et al. (2019). Genetic determinants of gut microbiota composition and bile acid profiles in mice. PLoS Genet. 15, e1008073. doi: 10.1371/journal.pgen.1008073
Kong, Q., Wang, B., Tian, P., Li, X., Zhao, J., Zhang, H., et al. (2021). Daily intake of Lactobacillus alleviates autistic-like behaviors by ameliorating the 5-hydroxytryptamine metabolic disorder in VPA-treated rats during weaning and sexual maturation. Food Funct. 12, 2591–2604. doi: 10.1039/D0FO02375B
Laffont, S., Seillet, C., and Guéry, J. C. (2017). Estrogen receptor-dependent regulation of dendritic cell development and function. Front. Immunol. 10, 108. doi: 10.3389/fimmu.2017.00108
Lagkouvardos, I., Lesker, T. R., Hitch, T. C. A., Gálvez, E. J. C., Smit, N., Neuhaus, K., et al. (2019). Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome. 7:28. doi: 10.1186/s40168-019-0637-2
Li, T., and Chiang, J. Y. (2015). Bile acids as metabolic regulators. Curr. Opin. Gastroenterol. 31, 159–165. doi: 10.1097/MOG.0000000000000156
Lindheim, L., Bashir, M., Münzker, J., Trummer, C., Zachhuber, V., Leber, B., et al. (2017). Alterations in gut microbiome composition and barrier function are associated with reproductive and metabolic defects in women with polycystic ovary syndrome (PCOS): a pilot study. PLoS ONE 12, e0168390. doi: 10.1371/journal.pone.0168390
Liu, R., Zhang, C., Shi, Y., Zhang, F., Li, L., Wang, X., et al. (2017). Dysbiosis of gut microbiota associated with clinical parameters in polycystic ovary syndrome. Front. Microbiol. 8, 324. doi: 10.3389/fmicb.2017.00324
Looijer-van Langen, M., Hotte, N., Dieleman, L. A., Albert, E., Mulder, C., and Madsen, K. L. (2011). Estrogen receptor-β signaling modulates epithelial barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 300, 621–626. doi: 10.1152/ajpgi.00274.2010
Maharshak, N., Ringel, Y., Katibian, D., Lundqvist, A., Sartor, R. B., Carroll, I. M., et al. (2018). Fecal and mucosa-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Dig. Dis. Sci. 63, 1890–1899. doi: 10.1007/s10620-018-5086-4
Manuel, R. S. J., and Liang, Y. (2021). Sexual dimorphism in immunometabolism and autoimmunity: Impact on personalized medicine. Autoimmun. Rev. 20, 102775. doi: 10.1016/j.autrev.2021.102775
Markle, J. G., Frank, D. N., Mortin-Toth, S., Robertson, C. E., Feazel, L. M., Rolle-Kampczyk, U., et al. (2013). Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339, 1084–1088. doi: 10.1126/science.1233521
Miranda-Ribera, A., Ennamorati, M., Serena, G., Cetinbas, M., Lan, J., Sadreyev, R. I., et al. (2019). Exploiting the zonulin mouse model to establish the role of primary impaired gut barrier function on microbiota composition and immune profiles. Front. Immunol. 10, 2233. doi: 10.3389/fimmu.2019.02233
Moreno-Indias, I., Sánchez-Alcoholado, L., Sánchez-Garrido, M. Á., Martín-Núñez, G. M., Pérez-Jiménez, F., Tena-Sempere, M., et al. (2016). Neonatal androgen exposure causes persistent gut microbiota dysbiosis related to metabolic disease in adult female rats. Endocrinology 157, 4888–4898. doi: 10.1210/en.2016-1317
Naqvi, S., Godfrey, A. K., Hughes, J. F., Goodheart, M. L., Mitchell, R. N., and Page, D. C. (2019). Conservation, acquisition, and functional impact of sex-biased gene expression in mammals. Science. 365, eaaw7317. doi: 10.1126/science.aaw7317
O' Shea, E. F., Cotter, P. D., Ross, R. P., and Hill, C. (2012). Strategies to improve the bacteriocin protection provided by lactic acid bacteria. Curr. Opin. Biotechnol. 24, 130–134. doi: 10.1016/j.copbio.2012.12.003
Obanda, D., Page, R., Guice, J., Raggio, A. M., Husseneder, C., Marx, B., et al. (2018). CD obesity-prone rats, but not obesity-resistant rats, robustly ferment resistant starch without increased weight or fat accretion. Obesity 26, 570–577. doi: 10.1002/oby.22120
Org, E., Mehrabian, M., Parks, B. W., Shipkova, P., Liu, X., Drake, T. A., et al. (2016). Sex differences and hormonal effects on gut microbiota composition in mice. Gut. Microbes. 7, 313–322. doi: 10.1080/19490976.2016.1203502
Pellock, S. J., and Redinbo, M. R. (2017). Glucuronides in the gut: Sugar-driven symbioses between microbe and host. J. Biol. Chem. 292, 8569–8576. doi: 10.1074/jbc.R116.767434
Pernigoni, N., Zagato, E., Calcinotto, A., Troiani, M., Mestre, R. P., Cal,ì, B., et al. (2021). Commensal bacteria promote endocrine resistance in prostate cancer through androgen biosynthesis. Science 374, 216–224. doi: 10.1126/science.abf8403
Qin, Y., Havulinna, A. S., Liu, Y., Jousilahti, P., Ritchie, S. C., Tokolyi, A., et al. (2022). Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 54, 134–142. doi: 10.1038/s41588-021-00991-z
Rincel, M., Aubert, P., Chevalier, J., Grohard, P. A., Basso, L., Monchaux de Oliveira, C., et al. (2019). Multi-hit early life adversity affects gut microbiota, brain and behavior in a sex-dependent manner. Brain Behav. Immun. 80, 179–192. doi: 10.1016/j.bbi.2019.03.006
Roager, H. M., Hansen, L. B., Bahl, M. I., Frandsen, H. L., Carvalho, V., Gøbel, R. J., et al. (2016). Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat. Microbiol. 1, 16093. doi: 10.1038/nmicrobiol.2016.93
Sampathkumar, N. K., Bravo, J. I., Chen, Y., Danthi, P. S., Donahue, E. K., Lai, R. W., et al. (2020). Widespread sex dimorphism in aging and age-related diseases. Hum. Genet. 139, 333–356. doi: 10.1007/s00439-019-02082-w
Sekido, Y., Nishimura, J., Nakano, K., Osu, T., Chow, C. T., Matsuno, H., et al. (2020). Some Gammaproteobacteria are enriched within CD14+ macrophages from intestinal lamina propria of Crohn's disease patients versus mucus. Sci. Rep. 10, 2988. doi: 10.1038/s41598-020-59937-w
Sharon, G., Cruz, N. J., Kang, D. W., Gandal, M. J., Wang, B., Kim, Y. M., et al. (2019). Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 177, 1600–1618.e17. doi: 10.1016/j.cell.2019.05.004
Singh, R. S., Singh, K. K., and Singh, S. M. (2021). Origin of sex-biased mental disorders: an evolutionary perspective. J. Mol. Evol. 89, 195–213. doi: 10.1007/s00239-021-09999-9
Sinha, T., Vich Vila, A., Garmaeva, S., Jankipersadsing, S. A., Imhann, F., Collij, V., et al. (2019). Analysis of 1135 gut metagenomes identifies sex-specific resistome profiles. Gut. Microbes 10, 358–366. doi: 10.1080/19490976.2018.1528822
Szyszkowicz, J. K., Wong, A., Anisman, H., Merali, Z., and Audet, M. C. (2017). Implications of the gut microbiota in vulnerability to the social avoidance effects of chronic social defeat in male mice. Brain Behav. Immun. 66, 45–55. doi: 10.1016/j.bbi.2017.06.009
Torres, P. J., Siakowska, M., Banaszewska, B., Pawelczyk, L., Duleba, A. J., Kelley, S. T., et al. (2018). Gut microbial diversity in women with polycystic ovary syndrome correlates with hyperandrogenism. J. Clin. Endocrinol. Metab. 103, 1502–1511. doi: 10.1210/jc.2017-02153
Turley, S. D., Schwarz, M., Spady, D. K., and Dietschy, J. M. (1998). Gender-related differences in bile acid and sterol metabolism in outbred CD-1 mice fed low- and high-cholesterol diets. Hepatology 28, 1088–1094. doi: 10.1002/hep.510280425
Van den Abbeele, P., Van de Wiele, T., Verstraete, W., and Possemiers, S. (2011). The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol. Rev. 35, 681–704. doi: 10.1111/j.1574-6976.2011.00270.x
Wu, Y., Tang, Y., Xiao, N., Wang, C., and Tan, Z. (2020). Bacterial lactase gene characteristics in intestinal contents of antibiotic-associated diarrhea mice treated with Debaryomyces hansenii. Med. Sci. Monitor 26, e920879. doi: 10.12659/MSM.920879
Wu, Y., Zhang, C., Shao, H., Luo, H., and Tan, Z. (2021). Characteristics of intestinal microbiota and enzyme activities in mice fed with lily bulb. Biotech. 3, 17. doi: 10.1007/s13205-020-02597-4
Yang, M., Hong, G., Jin, Y., Li, Y., Li, G., and Hou, X. (2020). Mucosal-associated microbiota other than luminal microbiota has a close relationship with diarrhea-predominant irritable bowel syndrome. Front. Cell Infect. Microbiol. 10, 515614. doi: 10.3389/fcimb.2020.515614
Yurkovetskiy, L., Burrows, M., Khan, A. A., Graham, L., Volchkov, P., Becker, L., et al. (2013). Gender bias in autoimmunity is influenced by microbiota. Immunity 39, 400–412. doi: 10.1016/j.immuni.2013.08.013
Zeng, B., Lai, Z., Sun, L., Zhang, Z., Yang, J., Li, Z., et al. (2018). Structural and functional profiles of the gut microbial community in polycystic ovary syndrome with insulin resistance (IR-PCOS): a pilot study. Res. Microbiol. 170, 43–52. doi: 10.1016/j.resmic.2018.09.002
Zhang, C., Peng, X., Shao, H., Li, X., Wu, Y., and Tan, Z. (2021). Gut microbiota comparison between intestinal contents and mucosa in mice with repeated stress-related diarrhea provides novel insight. Front. Microbiol. 12, 626691. doi: 10.3389/fmicb.2021.626691
Zhang, J., Hoedt, E. C., Liu, Q., Berendsen, E., Teh, J. J., Hamilton, A., et al. (2021). Elucidation of proteus mirabilis as a key bacterium in crohn's disease inflammation. Gastroenterology 160, 317–330.e11. doi: 10.1053/j.gastro.2020.09.036
Zhang, J., Sun, Z., Jiang, S., Bai, X., Ma, C., Peng, Q., et al. (2019). Probiotic bifidobacterium lactis v9 regulates the secretion of sex hormones in polycystic ovary syndrome patients through the gut-brain axis. mSystems 4, e00017–19. doi: 10.1128/mSystems.00017-19
Zhang, X., Zhong, H., Li, Y., Shi, Z., Ren, H., Zhang, Z., et al. (2021). Sex-and age-related trajectories of the adult human gut microbiota shared across populations of different ethnicities. Nat. Aging. 1, 87–100. doi: 10.1038/s43587-020-00014-2
Keywords: intestinal mucosa-associated microbiota, intestinal luminal content microbiota, sex hormones, sex dimorphism traits, gender-associated diseases
Citation: Wu Y, Peng X, Li X, Li D, Tan Z and Yu R (2022) Sex hormones influence the intestinal microbiota composition in mice. Front. Microbiol. 13:964847. doi: 10.3389/fmicb.2022.964847
Received: 09 June 2022; Accepted: 26 August 2022;
Published: 31 October 2022.
Edited by:
Tang Zhaoxin, South China Agricultural University, ChinaReviewed by:
José Antonio Moreno Muñoz, Laboratorios Ordesa, SpainMinyi Huang, Hunan University of Humannities, Science and Technology, China
Copyright © 2022 Wu, Peng, Li, Li, Tan and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhoujin Tan, dGFuemhqaW5Ac29odWNvbQ==; Rong Yu, eXVyb25AMjFjbi5jb20=