 Yuanyuan Li1,2
Yuanyuan Li1,2 Ximing Xu3,4
Ximing Xu3,4 Ziyao Guo1,2Qinyuan Li1,2Yiying Wang1,2Ding Jian1,2
Ziyao Guo1,2Qinyuan Li1,2Yiying Wang1,2Ding Jian1,2 Guangli Zhang1Xiaoyin Tian1Shiyi Chen1,2
Guangli Zhang1Xiaoyin Tian1Shiyi Chen1,2 Zhengxiu Luo1*
Zhengxiu Luo1*- 1Department of Respiratory Medicine of Children’s Hospital of Chongqing Medical University, National Clinical Research Center for Child Health and Disorders, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing, China
- 2Chongqing Key Laboratory of Pediatrics, Chongqing Key Laboratory of Child Infection and Immunity, Chongqing, China
- 3Department of Medical Record Statistics of Children’s Hospital of Chongqing Medical University, National Clinical Research Center for Child Health and Disorders, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing, China
- 4Chongqing Higher Institution Engineering Research Center of Children’s Medical Big Data Intelligent Application, Chongqing, China
Early life is a “critical window” for gut microbiota development, antibiotic use during this period exerts a profound effect on gut microbial dysbiosis and asthma. In clinical practice, antibiotics are usually used in patients with bacterial infections, we previously showed that neonatal S. pneumoniae pneumonia promoted adult-onset asthma in mice model, while it remains unclear whether neonatal S. pneumoniae infection have long-term effects on gut microbiota. Neonatal BALB/c mice were inoculated with 5*106 CFU D39 to establish non-lethal S. pneumoniae pneumonia model. At 2, 3, 8 weeks of age, feces in the cecum were prepared for 16S rRNA sequencing, lungs were collected for histopathologic and lung function analysis. S. pneumoniae-infected neonatal mice exhibited histopathologic lesions in their lungs and increased airway hyperresponsiveness, obvious alterations in alpha and beta diversities in the entire gut microbiota, and changes of the community structure during the breastfeeding period, infancy, and adulthood. Furthermore, gut microbial composition was modified after neonatal S. pneumoniae infection, with a decreased relative abundance of Lactobacillus in the breastfeeding period and infancy; in adulthood, the relative abundance of Allobaculum diminished while that of Proteobacteria was augmented. Neonatal S. pneumoniae infection induced a long-term alteration in microbial community composition.
Introduction
The microbiome consists of organizations of bacteria, viruses, archaea, and fungal species, majority of which reside in the gut. The acquisition of the gut microbiota begins at birth and reaches a relatively stable level at nearly 3 years of age in humans and at 8 weeks of age in mice (Laukens et al., 2016); the gut microbiota play key roles in homeostasis, metabolism, immunity, and physiologic or pathologic situations. The formation of the gut microbiota reflects several stages, and early life is hypothesized to be the “critical window” for gut microbiome development, that is, when gut microbial dysbiosis exerts the greatest influence on immune and metabolic systems (Cunningham et al., 2014; Houghteling and Walker, 2015). Microbial dysbiosis during this period is linked to various types of immune-related diseases such as asthma (van Nimwegen et al., 2011; Arrieta et al., 2015; Depner et al., 2020; Gürdeniz et al., 2022), eczema, and food allergies (Azad et al., 2015). Microbial dysbiosis in the early years of human life is associated with a variety of factors—including delivery mode, feeding pattern, and antibiotic use. Accumulating evidence reveals that early-life use of antibiotics is a risk factor for microbial dysbiosis in both human trials and in mouse models (Penders et al., 2011; Nobel et al., 2015; Stokholm et al., 2018). In neonates, the development of the gut microbiota differs between antibiotic-treated and non-treated infants (Tanaka et al., 2009; Fouhy et al., 2012), indicating a long-lasting alteration in the microbiome due to antibiotic use, and antibiotics are typically administered clinically to patients with bacterial infections. However, whether or not bacterial infection itself influences the microbiota composition needs to be further explored. Studies have indicated that influenza (Ding et al., 2019), HIV (Gosmann et al., 2017), RSV (Groves et al., 2020), and Candida albicans (d’Enfert et al., 2021) infection influence disease severity through modifications in the composition of the gut microbiota. Yet, whether early-life bacterial infection affects gut microbiota development is less well understood.
Streptococcus pneumoniae (S. pneumoniae) is the most common bacterial pathogen of community-acquired respiratory infection in childhood (Fritz et al., 2019). Our previous studies showed that even when S. pneumoniae was eliminated from the lung tissue 7 days post-infection, neonatal S. pneumoniae infection still promoted asthma development in adults (Yang et al., 2015) by alerting CD4+T cell subsets (Tian et al., 2020), indicating that neonatal infection with S. pneumoniae induced long-term effects on pulmonary immune function. However, whether neonatal S. pneumoniae infection promoted long-term immune alterations in the lungs by perturbing the gut microbial community remains unclear.
Against the backdrop of early-life events in the development of gut microbiota and asthma, we herein investigated the dynamic changes in gut microbiota after neonatal S. pneumoniae infection in a mouse model. We conducted 16S rRNA gene high-throughput sequencing in feces collected from mice 1 week post-infection (1 wpi, breastfeeding period), 2 weeks post-infection (2 wpi, infancy), and 7 weeks post-infection (7 wpi, adulthood) after nasal inoculation. Our results indicated that neonatal S. pneumoniae infection induced a marked perturbation in the microbiota at different developmental stages, especially in infancy. With this study, we first advanced the concept that early-life bacterial infection promoted a gradual but long-lasting influence on gut microbiota. We expect that these results will be helpful in better understanding early-life bacterial infection as related to microbiome disturbances and in providing guidance for developing potential interventions in infection-induced asthma.
Materials and methods
Animals
Parturient BALB/C mice and their newborn pups were purchased from Ensiweier Biotechnology Co., Ltd. (Chongqing, China). All experimental protocols were approved by the Institutional Animal Care and Research Advisory Committee of the Chongqing Medical University, and all animals were treated in accordance with the guidelines issued by the Chinese Council on Animal Care. Newborn mice were housed with their mother at 25°C under a 12-h light/12-h dark cycle with a normal diet and water.
Study design
Totally 32 neonatal mice at 1 week of age were randomly allocated to two groups: the mock-infected control group (mean weight, 5.22 ± 0.54 g; F/M, 8/7; 10 ul of PBS) and the S. pneumoniae infection (S. pp) group (mean weight, 5.30 ± 0.36 g; F/M, 8/7; S. pneumonia = 5*106 CFU in 10 ul of PBS). Forty-eight hours post-infection, two infected mice were randomly selected for a model verification test. Then, at 1 wpi (2 weeks old, breastfeeding period), 2 wpi (3 weeks old, infant), and 7 wpi (8 weeks old, adulthood), five mice were randomly selected from each group and euthanized, and the feces in their cecum and lungs were collected. The flow diagram of the experimental design is depicted in Figure 1. Our infectious model was verified with tissue homogenates cultured on blood agar (Supplementary Figure 1) (Dong et al., 2020). The fecal samples were collected for 16S rRNA sequencing, and lungs were collected for histopathologic staining.
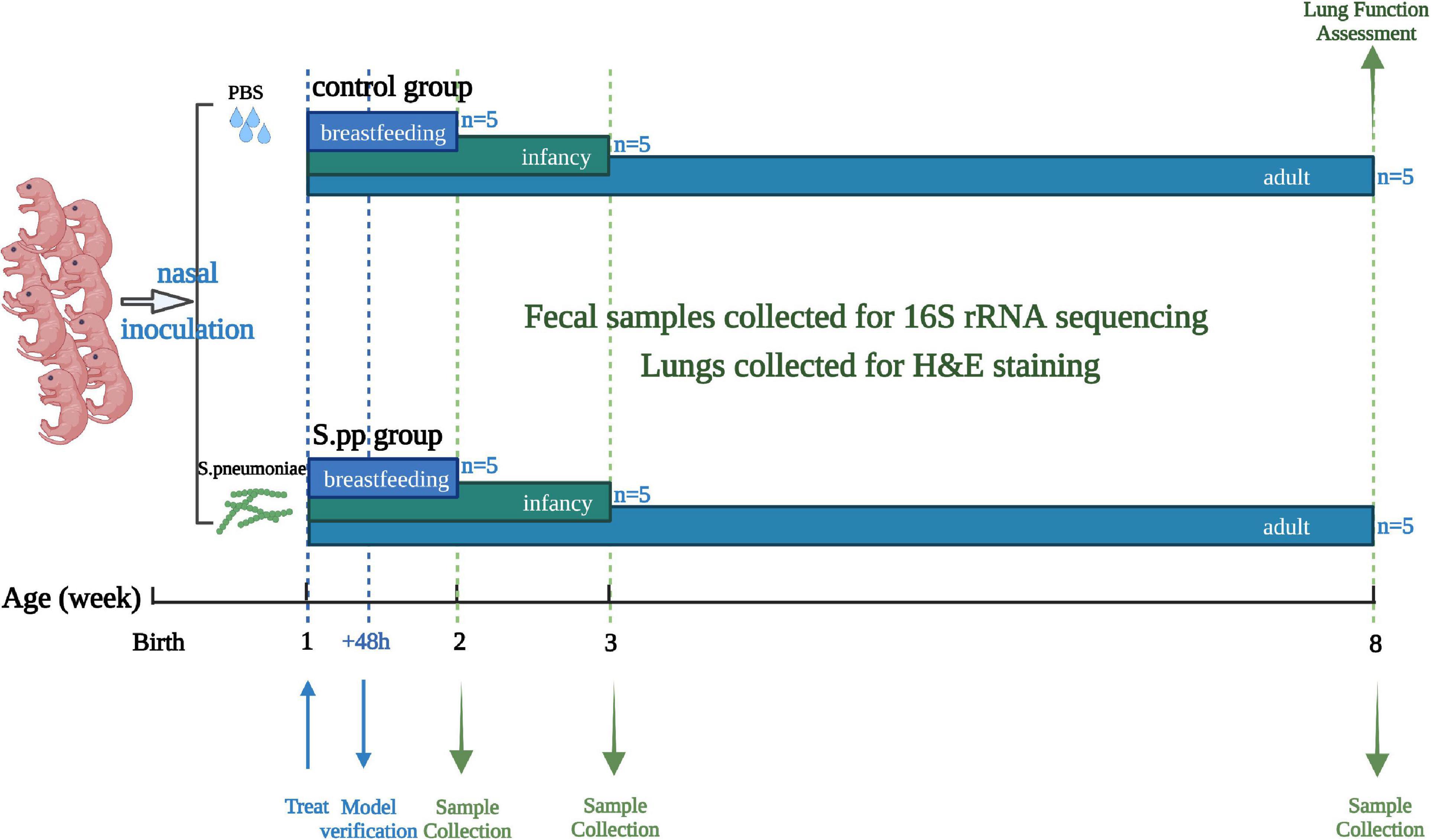
Figure 1. Experimental design and sample-collection time. In brief, mice were intranasally instilled with PBS (Control) or Streptococcus pneumoniae (S.pp) at 1 week of age, and a lung homogenate was used for model verification 48 h post-infection. Fecal and lung samples were collected at 1 wpi (breastfeeding period), 2 wpi, (infancy), and 7 wpi, (adulthood) for 16S rRNA sequencing and H&E staining, lung function were assessed in adulthood.
Establishment of a non-lethal neonatal Streptococcus pneumoniae infection model
The non-lethal neonatal S. pneumonia infection model was established according to our previous study (Yang et al., 2015; Tian et al., 2020). Briefly, the S. pneumoniae D39 strain was plated onto blood agar (Bio-Caring, P0901, China) and incubated for 12–14 h at 37°C in 5% CO2 in air. Bacteria were then transferred to Todd-Hewitt (BD, BX04921, United States) broth supplemented with 0.5% yeast extract (THY) and incubated at 37°C until the mid-logarithmic phase (OD600 = 0.50–0.55). Neonatal (1-week-old) BALB/C mice were inoculated intranasally with 10 μL bacteria suspension (5*106 colony-forming units (CFU) of S. pneumoniae in PBS) without anesthetic, while the mock-infected controls received only the same volume of sterile PBS. Forty-eight hours post-infection, two infected mice were randomly selected, and their lungs were removed and homogenized; the tissue homogenates were then cultured on blood agar for 12–16 h at 37°C under 5% CO2 in air to determine the bacterial load.
Sample collection, DNA extraction, and amplification
Mock-infected controls and neonatal S. pneumoniae-infected mice were euthanized by cervical dislocation at 2 weeks (1 wpi, breastfeeding period), 3 weeks (2 wpi, infancy), 8 weeks (7 wpi, adulthood) of age; and the mouse age brackets were classified according to a related review (Sengupta, 2013). The contents of the cecum were collected and stored at –80°C for analysis of microbial composition. Total genomic DNA was extracted from the collected fecal samples (∼0.1 g) using the OMEGA Soil DNA Kit (M5635-02) (Omega Bio-Tek, Norcross, GA, United States) following the manufacturer’s instructions and immediately stored at –20°C for further analysis. The hypervariable V3–V4 region of the bacterial 16S rRNA genes was amplified by PCR using the forward primer 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and the reverse primer 806R (5′-GGACTACHVGGGTWTCTAAT-3′). Each 25-μL PCR reaction contained 5 μl of buffer (5 ×), 0.25 μl of Fast pfu DNA Polymerase (5 U/μl), 2 μl (2.5 mM) of dNTPs, 1 μl (10 uM) of each forward and reverse primer, 1 μl of DNA template, and 14.75 μl of ddH2O. The thermal cycling conditions consisted of an initial denaturation at 98°C for 5 min, followed by 25 cycles of denaturation at 98°C for 30 s, annealing at 53°C for 30 s, and primer template extension at 72°C for 45 s, with a final extension of 5 min at 72°C.
16S rRNA sequencing and taxonomic classification
PCR amplicons were purified and quantified using the Illlumina NovaSeq platform with the NovaSeq 6000 SP Reagent Kit (500 cycles) at Shanghai Personal Biotechnology Co., Ltd., Shanghai, China. QIIME2 was slightly modified according to the official tutorials and used for quality filtering of DNA sequences, demultiplexing, taxonomic assignment, and alpha and beta diversity calculations. Non-singleton amplicon sequence variants (ASVs) were aligned and used to construct a phylogeny with fasttree2. Taxonomy was assigned to ASVs using the classify-SciKit Learn’s sklearn naiïve Bayes taxonomy classifier in the feature-classifier plugin against the Greengenes Release 13.8 Database.
Bioinformatics and statistical analyses
Data analyses were executed using QIIME2 and R packages (v3.2.0), and the microbiota alpha-diversity indices were calculated using the ASV table in QIIME2 and visualized as box-plots. To investigate the structural variations in microbial communities across samples, beta-diversity analysis was employed and visualized via principal coordinate analysis (PCoA) and non-metric multidimensional scaling (NMDS). Differentiation of microbiota structure among groups was assessed by permutational multivariate analysis of variance (PERMANOVA). The taxonomic compositions and abundances were visualized using MEGAN and GraPhlAn. Linear discriminant analysis effect size (LEfSe) was exploited to detect differentially abundant taxa across groups using the default parameters. We implemented co-occurrence network analysis with SparCC, and the pseudocount value in SparCC was set to 10-6. The cut-off value for correlation coefficients was determined to be 70 through random matrix theory-based methods as implemented in the R package RMThreshold. Based on the correlation coefficients, we constructed a co-occurrence network with nodes that represented ASVs and edges that represented correlations between the ASVs. We visualized the network using the R packages igraph and ggraph. Microbial functions were predicted by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2; Douglas et al., preprint) with MetaCyc. One-way analysis of variance (ANOVA) and Kruskal–Wallis tests were used to compare the relative abundances among groups with Graphpad Prism 9.0.0.
Histology of the lungs
After collection, the lungs were fixed in formaldehyde for 24 h and then dehydrated and paraffin embedded. The lung tissues were sectioned at 4 microns and then stained with hematoxylin and eosin stain (H&E; Sigma-Aldrich). At least three views were then selected from each mouse for analysis. Images were captured under a Nikon Eclipse E200 microscope connected to a Nikon Coolpix 995 camera (Nikon, Tokyo, Japan).
Airway hyperresponsiveness measurement
As previously described, airway hyperresponsiveness (AHR) was measured in vivo by Penh using whole-body plethysmograph method. Penh is a dimensionless value represents pulmonary airflow resistance, which also reflects a function of the timing of expiration, and the ratio of peak expiratory flow to peak inspiratory flow. Briefly, each conscious adult mouse was exposed to aerosolized normal saline followed by increasing concentrations of aerosolized methacholine (Sigma-Aldrich, St. Louis, MO, United States) solution (3.125, 6.25, 12.5, and 25 mg/ml; Sigma-Aldrich, St. Louis, MO, United States) in saline for 3 min and then rested for 2 min. The average Penh for each concentration was calculated from the continuously recorded pressure and flow data for 5 min.
Results
Neonatal Streptococcus pneumoniae infection induces pulmonary histopathologic changes
As we previously found, S. pneumoniae was eliminated from the lung 7 days post-infection. Simultaneously, infected mice gained transient body and lung weight losses along with S. pneumoniae infection within the first-week post-infection, while restored at 14-days post-infection (Peng et al., 2019). The lungs were further collected during the breastfeeding period (1 wpi), infancy (2 wpi), and adulthood (7 wpi). As shown in Figure 2, compared with the mock-infected control group, lung sections of the neonatal S. pneumoniae infection group during infancy and adulthood showed histopathologic lesions that included increased inflammatory cell infiltration and thickened alveolar septa. These results indicated that neonatal S. pneumoniae infection induced long-lasting structural changes in the pulmonary tissue.
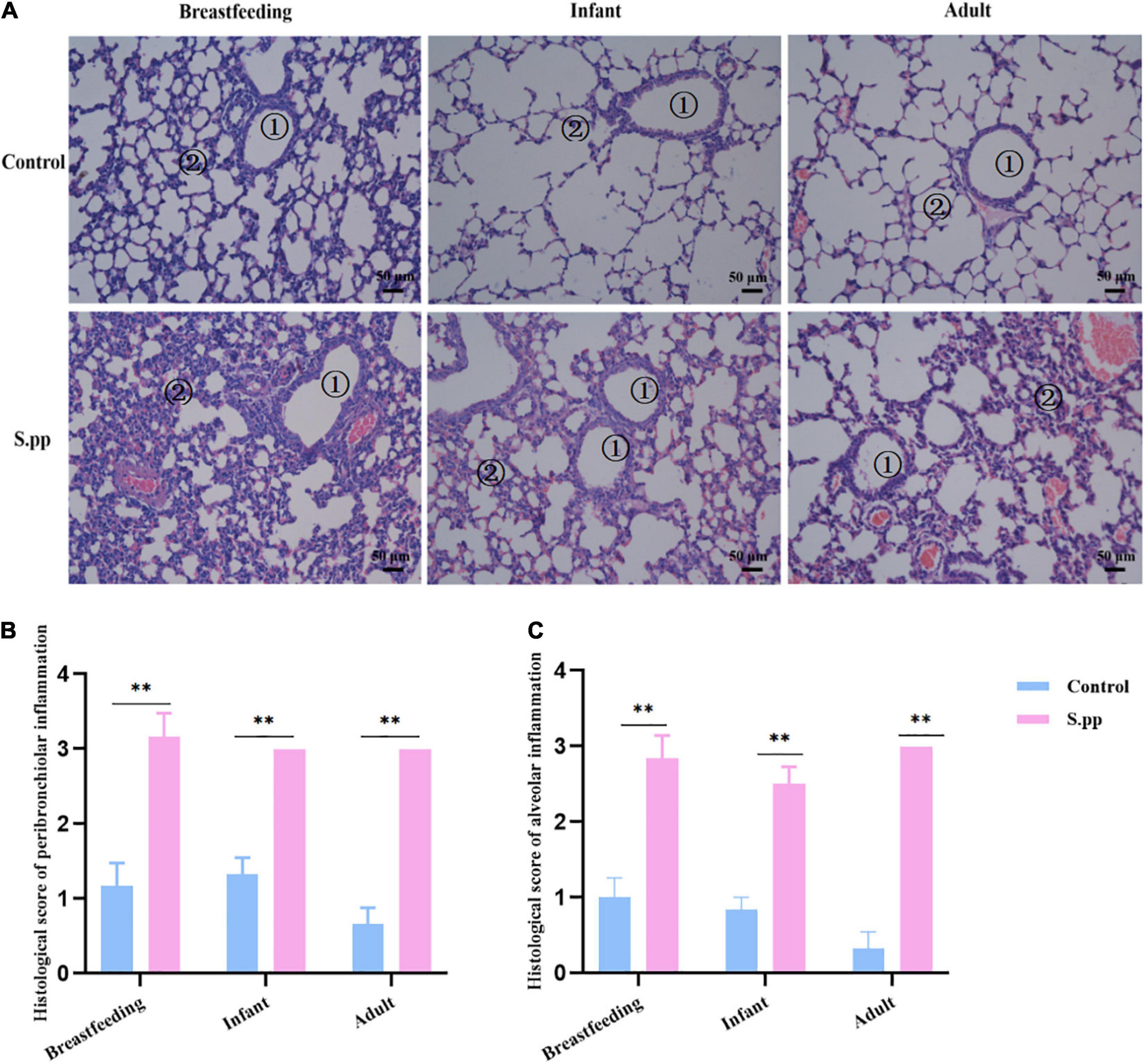
Figure 2. Neonatal S. pneumoniae infection induces inflammatory cell infiltration in lung tissues at different stages. (A) Representative H&E staining in lung slices from mice in the control and S. pp groups during the breastfeeding period, infancy, and adulthood (200 × magnification). The numbers in the figures indicate bronchioles ➀, alveoli ➁. (B) Histological scores of pulmonary peribronchiolar inflammation. (C) Histological scores of pulmonary alveolar inflammation. n = 3–6 mice/group. **P < 0.01, compared with control group.
Neonatal Streptococcus pneumoniae infection promotes long-lasting airway hyperresponsiveness
Seven weeks after S.pneumoniae infection, AHR was measured by the calculation of Penh. The airway responsiveness appeared no significant differences following normal saline challenge. While with the methacholine concentration increase, neonatal S. pneumoniae infection significantly increased AHR when compared to the controls at methacholine concentrations of 6.25,12.5, 25.00, and 50.00 mg/ml (Figure 3). These indicated that neonatal S. pneumoniae infection promoted long-lasting airway hyperresponsiveness.
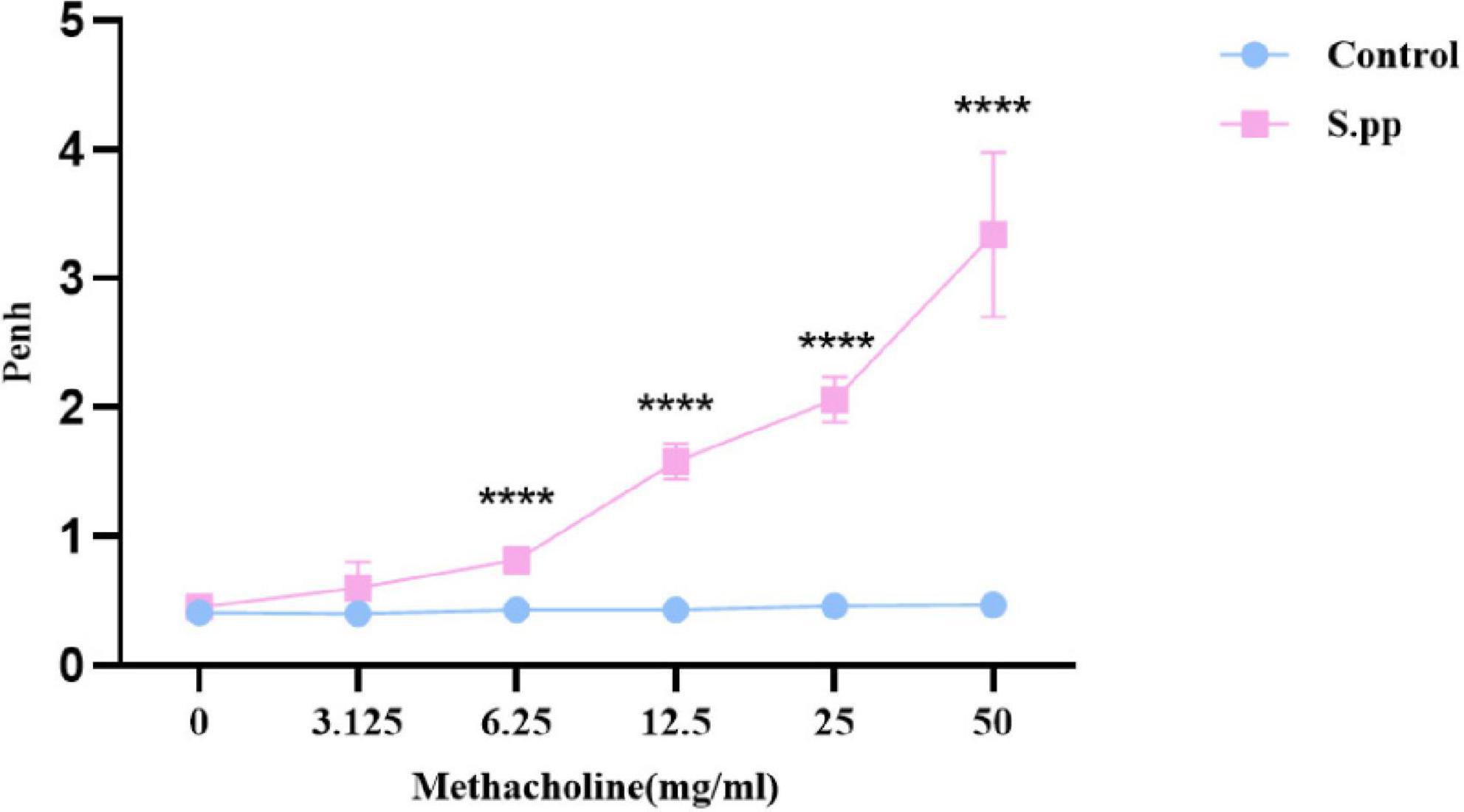
Figure 3. Neonatal S. pneumoniae infection promotes long-lasting AHR. Wholebody plethysmography was conducted in mock-infected (control) and neonatal S. pneumoniae infection mice (S.pp) with methacholine challenge. All data are presented as mean ± SD (n = 5 mice/group). ****P < 0.0001, compared with control group.
Neonatal Streptococcus pneumoniae infection alters gut microbial diversity and community structure
Based on our high-throughput sequencing, a total of 2,194,529 raw reads were generated for 30 fecal samples. After removing the low-quality sequences, 1,696,576 high-quality reads were obtained for abundance analysis, diversity analysis, and taxonomic comparisons.
Alpha diversity
As evident from both Shannon and Simpson indices, the alpha diversities across different age groups exhibited an “inverse U” trend that significantly increased during infancy (2 wpi) and then slightly diminished during adulthood (7 wpi), with a significantly altered overall Shannon index (p = 0.0022, Figure 4A) and Simpson index (p = 0.09, Figure 4B). When compared with the mock-infected control mice at the same age, the reducing trends were observed in the neonatal S. pneumoniae infected group, further PERMANOVA test revealed that not only age but also S. pneumoniae infection induced significant differences in alpha diversity indices (p = 0.001, Supplementary Figure 2 and Supplementary Table 1).
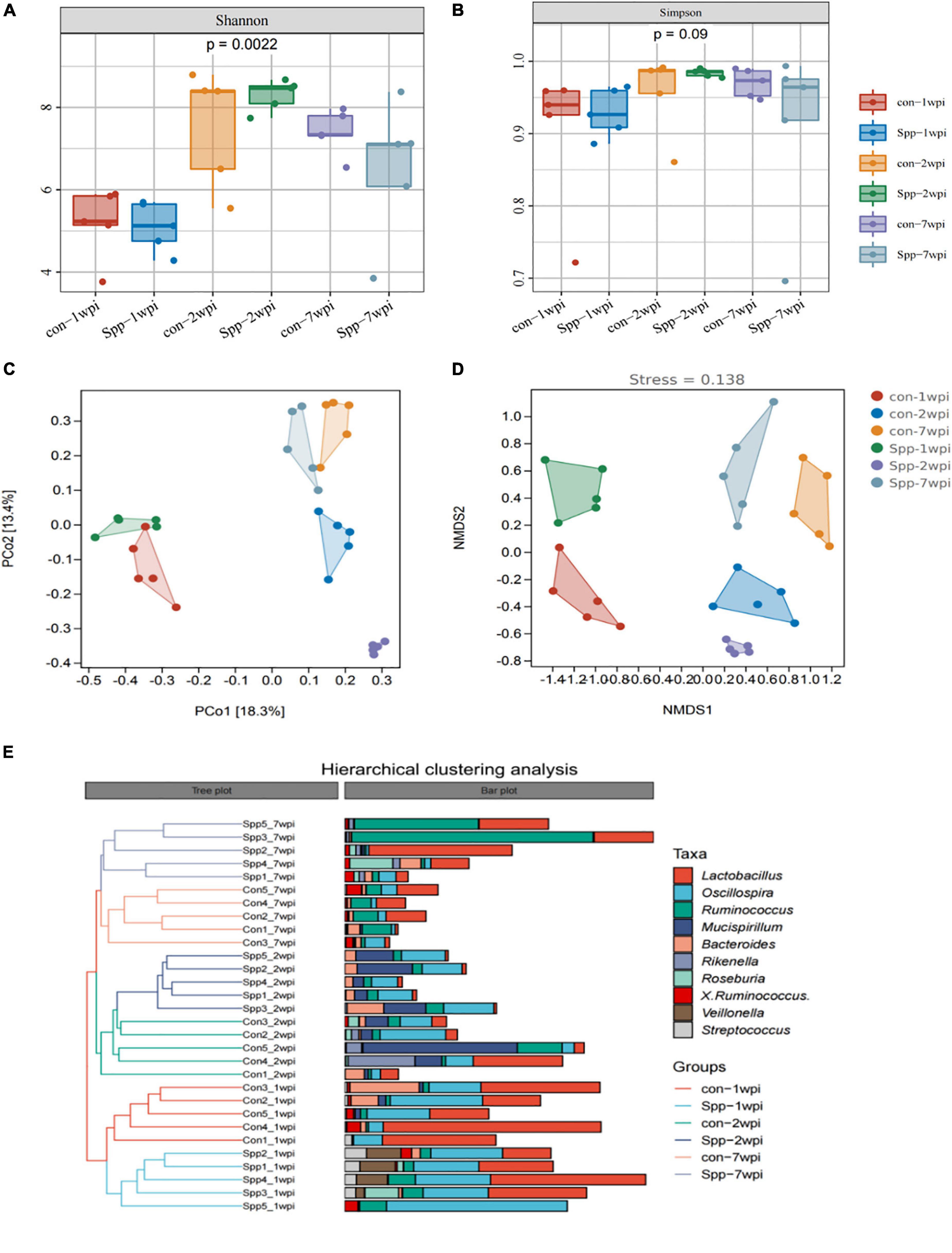
Figure 4. Neonatal S. pneumoniae infection alters gut microbial community structure. (A) The alpha diversity of gut microbiota from the mock-infected control- and neonatal S. pneumoniae infection (S. pp)-treated mice in the breastfeeding period (1 wpi), infancy (2 wpi), and adulthood (7 wpi) was represented by Shannon indices. (B) The alpha diversity of gut microbiota from the mock-infected control- and neonatal S. pneumoniae infection (S. pp)-treated mice was represented by Simpson indices at different stages. (C) Beta-diversity analysis of relative abundances of the entire microbiota using principal coordinate analysis (PCoA) with Bray-Curtis dissimilarity index, followed by the PERMANOVA significance test. (D) Beta-diversity analysis of whole-microbiota relative abundance with two-dimensional non-metric multidimensional scaling of Bray-Curtis dissimilarity distance metric (2D stress value = 0.138). (E) Hierarchical clustering of mock-infected control- and S. pp-treated mice in the breastfeeding period, infancy, and adulthood.
Beta diversity
Principal coordinate analysis and NMDS analyses were applied based on ASV/OUT for beta diversity analysis. As indicated, the plots reflecting the samples taken during the breastfeeding period (1 wpi), infancy (2 wpi), and adulthood (7 wpi) were discretely separate, as were the plots for the neonatal S. penumoniae infection group relative to the mock-infected controls at the same age (Figures 4C,D). PCoA was also implemented based on the phylum, order, class, family, genus, and species, and we demonstrated that the more granular the hierarchy, the more pronounced the distinctions (Supplementary Figure 3). The plots of the samples in infancy were most markedly disparate for each hierarchy.
Hierarchical cluster analysis
We subsequently constructed a dendrogram using hierarchical cluster analysis with Bray-Curtis dissimilarity indices, and as shown in Figure 4E, the samples from breastfeeding mice were grouped into one major branch of the sample tree, while the subgroups of infant and adult mice co-clustered as another major branch. Moreover, for every sub-branch for age, the neonatal S. pneumoniae infected group and the mock-infected controls were clustered discretely. These results portrayed neonatal S. pneumoniae infection as exerting a significant effect on the structure of the gut microbial community.
Neonatal Streptococcus pneumoniae infection affects gut microbial composition
As a consequence of the altered microbial-community structure, we further explored microbial composition, and our analysis of taxonomic composition revealed that the most prevalent phyla were Firmicutes (74.15%) and Bacteroidetes (18.59%) (Supplementary Figure 4), while Lactobacillus (12.40%) was the most prevalent genus in the microbial community. Lactobacillus, which is considered to be an probiotic genus, showed the highest relative abundance in the breastfeeding period, was attenuated in infancy, and then remained relatively stable until adulthood. When compared with their respective mock-infected control group, samples of neonatal S. pneumoniae infected groups exhibited a reduced trend for Lactobacillus in the breastfeeding period and a significant reduction in relative abundance in infancy, even when the brief changes in composition recovered in adulthood (Figure 5A). These modulation patterns indicated the long-lasting disruption of gut microbial community composition by neonatal S. pneumoniae infection and one that occurred gradually.
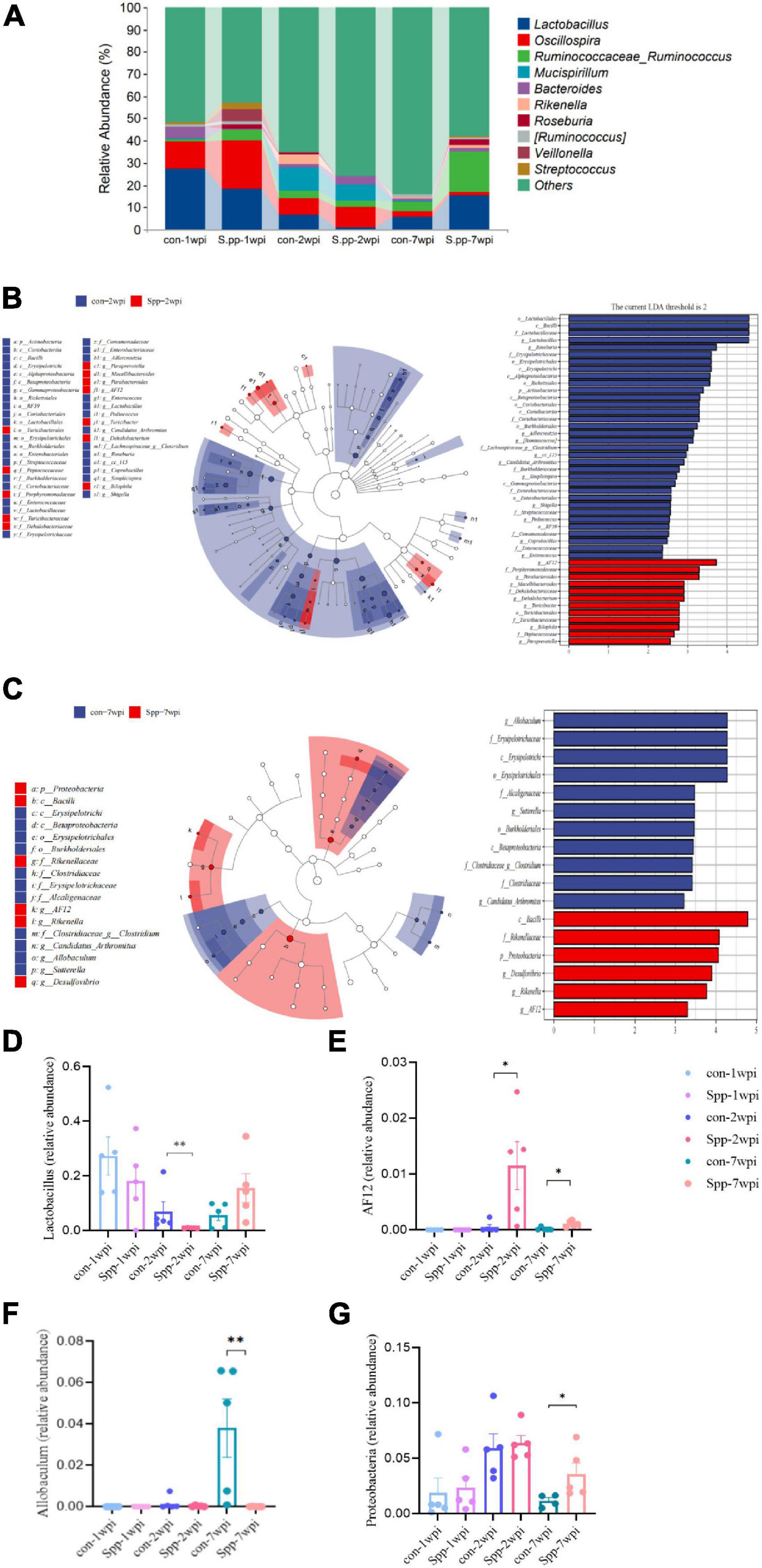
Figure 5. Effects of neonatal S. pneumoniae infection on gut microbial composition. (A) Relative abundances at the genus level of the mock-infected control and neonatal S. pneumoniae infected (S. pp) groups in the breastfeeding period (1 wpi), infancy (2 wpi), and adulthood (7 wpi). (B) LEfSe analysis based on the samples of the control and S. pp groups in infancy in various abundant taxa. The left panel shows the cladogram and the right panel shows the linear discriminant analysis (LDA) score (LDA > 2 shown in the Figure) for indicator taxa of the control and S. pp groups (P < 0.05). (C) LEfSe analysis based on the samples of the control and S. pp groups in adulthood in various abundant taxa. (D) Relative abundance of the genus Lactobacillus was significantly decreased in infancy (**P < 0.01, con-2wpi vs. Spp-2wpi); (E) Relative abundances of the genus AF12 were significantly increased in infancy and adulthood (*P < 0.05, con-2wpi vs. Spp-2wpi, con-7wpi vs. Spp-7wpi; (F) Relative abundance of the genus Allobaculum was significantly reduced in adulthood (P < 0.01); (G) Relative abundance of the phylum Proteobacteria was significantly reduced in adulthood (P < 0.05).
To explore the contributions of the differential flora on gut microbial alterations, the differences in the relative microbial abundances within the gut microbiota in samples were determined using LEfSe analysis (Figure 5B). The results revealed that the most notably enriched taxa were Lactobacillales bacilli, Lactobacillaceae, and Lactobacillus (all belong to Firmicutes) in the mock-infected control group, while AF12 (belonging to Bacteroidetes) was the most enriched taxa in the neonatal S. pneumoniae-infected group in infancy. In adulthood, the most obviously predominant taxa in the control group were Allobaculum, Erysipelotrichales, Erysipelotrichi, and Erysipelotrichales (all belong to Firmicutes) (Figure 5C). We then compared the relative abundances of Lactobacillus, AF12, Allobaculum and Proteobacteria among the six groups and found a significant reduction in Lactobacillus and enrichment in AF12 in the neonatal S. pneumoniae-infected group during infancy (Figure 5D,E), and a marked abridgment of Allobaculum and enrichment of AF12 and Proteobacteria in adulthood (Figures 5D–G). Those results demonstrated that neonatal S. pneumoniae infection induced a significant transformation in gut microbial composition—primarily in infancy—and that the gut microbiome partially recovered by adulthood.
Gut microbial dysbiosis-related pathway prediction
To determine the importance of neonatal S. pneumoniae infection-induced microbial dysbiosis in infancy, we exploited Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) to conduct functional predictions for gut microbiota. When we compared the genetic composition of the gut microbiota of the mock-infected control and neonatal S. pneumoniae-infected mice in infancy, our results indicated that the metabolism- and degradation-related pathways were visibly enriched (Supplementary Figure 5). We also ascertained that the beta-alanine metabolism- and toluene-degradation pathways were significantly affected by neonatal S. pneumoniae infection in infancy relative to the control group.
Discussion
Early life is a critical developmental window for gut microbial development, the disruption of which during maturation substantially perturbs immune- and metabolic- system evolution (Cunningham et al., 2014; Houghteling and Walker, 2015). Although exposure to antibiotics has been widely accepted to be a risk factor in microbial dysbiosis and asthma genesis (Tanaka et al., 2009; Ding et al., 2019), the effect of bacterial infection as an indication of antibiotic use has been neglected. Recently, the cross-talk between respiratory system and commensal organism, and it’s role in pulmonary disease are hot off the press. It is thought that gut microbial dysbiosis could produce adverse effect to innate lung immunity and increasing lung inflammation (Samuelson et al., 2015), contributing to asthma development (Depner et al., 2020). On the other side, infant respiratory infection (Harding et al., 2020) and neonatal hyperoxia exposure (Althouse et al., 2019) were reported associated with gut microbial community alteration. Those indicated that gut-lung axis is essential for homeostasis and host immune system training, may associated with chronic disease development and long-term health. We therefore adopted a conventional mouse model of neonatal S. pneumoniae infection that was reported to induce airway hyperresponsiveness (AHR) (Peng et al., 2019; Li et al., 2021) and experimental asthma (Yang et al., 2015; Tian et al., 2020), so as to explore the effect of neonatal bacterial infection on the gut microbiota. Although little influence was found on the total richness or uniformity of gut microbiota after neonatal S. pneumoniae infection, we observed a clear difference in gut microbial community structure and composition. This study presents a new perspective on the impact of early-life bacterial respiratory infections on gut microbial development and may lead to a better elucidation of the gut microbiome’s effect on lifelong health.
Gut microbial colonization begins at the moment of birth and fluctuates with age (Del Chierico et al., 2015). In this study we thus evaluated the dynamic changes of the gut microbiota in control mice and mice neonatally infected with S. pneumoniae at different developmental stages. Without detectable S. pneumoniae in gut (data not shown), we ascertained that alpha diversity increased with age during development and reached stability in adulthood, as revealed by the increased Shannon indices. The relative abundance of Bacteroidetes increased with age, and Firmicutes and Bacteroidetes ranked as the top two phyla. These results thus confirmed the rules governing gut microbial development, and were consistent with other studies in mice and humans (Tasnim et al., 2021).
After adjusting for age, we determined that neonatal S. pneumoniae infection disrupted gut microbial structure and composition using PCoA and LEFSe analyses, and found that this disruption occurred gradually but endured. One week after infection, Lactobacillus only showed a decreased trend in the breastfeeding period, while this reduction was amplified in infancy and then recovered until adulthood. Indeed, the reduction in Lactobacillus was an indication of gut microbial dysbiosis. Lactobacillus belongs to the larger lactic-acid bacterial group, which potentially influences the behavior of immune cells, and acts as an immune adjuvant and cellular forge that synthesizes and secretes bioactive molecules (Cho et al., 2020). In the presence of L. plantarum (a species of Lactobacillus), macrophages produced lesser amounts of TNF-α, IL-1β, and IL-17; and peripheral blood mononuclear cells (PBMCs) produced more IL-10 (Ferreira Dos Santos et al., 2016). Other than their anti-inflammatory activities, Lactobacillus bacilli also influence immune-cell maturation and differentiation. L. rhamnosus and L. plantarum (two species of Lactobacillus) increased the population of Th1 cells and Tregs in systemic lupus erythematosus-mouse models to reverse the disease profiles (Ivanovic et al., 2015). However, in chronic inflammatory disease, Lactobacillus bacilli tend to reduce Th activity, including Th17 cells (Chen et al., 2016). In addition, a positive role for early-life Lactobacillus abundance in asthma development has been confirmed in a large number of cohort studies of children (Björkander et al., 2020). These investigators determined that the absence of Lactobacillus species at six months of age induced elevated levels of chemokines that preceded allergic disease, and was associated with lower IFN-γ concentrations, higher fractional exhaled nitric oxide, and a higher incidence of allergic diseases. In our previous study we demonstrated that neonatal S. pneumoniae infection attenuated IL-10 and elevated IL-17 cytokines in bronchoalveolar lavage fluid (BALF), and reduced Th1 cells but enhanced pulmonary Th17 populations in adulthood, and that these alterations were confirmed to be contributors to the development of allergic airway disease (AAD) in adulthood (Yang et al., 2015; Tian et al., 2020). This may indicate that neonatal S. pneumoniae infection-induced Lactobacillus reduction in infancy comprises an essential component of gut microbial dysbiosis, which may in turn contribute to AAD development in adulthood.
Dysbiosis in infancy, however, does not impede the influence of S. pneumoniae infection on the gut microbiota, as we also found significant alterations in the gut microbial community during adulthood; this included increased Proteobacteria and decreased Allobaculum in neonatal S. pneumoniae-infected mice. Proteobacteria comprises a type of microbial dysbiosis signature that is reported to explain multiple pathogenic mechanisms (Lin et al., 2020). The phylum Proteobacteria is represented by potentially pathogenic bacteria—including Haemophilus, Moraxella, and Neisseria genera—that delineate toxin- and biofilm-producing opportunistic pathogens (Huang et al., 2015; Caverly et al., 2019). A higher abundance of Proteobacteria was uncovered in severely asthmatic patients, and this expansion was correlated with a Th17-cell epithelial-gene signature and was further correlated with asthma severity (Huang et al., 2015). Similar characteristics were unveiled in a mouse model, as Moraxella-infected mice showed augmented levels of IL-6, IL-1β, IL-17, and tumor necrosis factor α (TNF-α) in lungs, and neutralization of IL-17 and TNF-α was sufficient to accelerate airway inflammation and reduce the risk of AAD (Alnahas et al., 2017). Our previous work suggested that IL-17 acted as an essential cytokine in AAD development, indicating that proteobacterial expansion in gut microbiota could facilitate airway inflammation and AAD development. In contrast, Allobaculum is a genus of mouse commensal bacteria similar to Faecalibacterium, manifests the ability to assimilate glucose and produce short-chain fatty acids (SCFAs) (Greetham et al., 2004; Balakrishnan et al., 2021), such as butyrate, to exhibit immunoregulatory ability. Allobaculum was recently reported benefit for arthritis, which maintains immune and gut homeostasis through butyrate production augment (Balakrishnan et al., 2021). Functional prediction also consistently showed that metabolic and degradation-related pathways were markedly enriched in our model. Additionally, beta-alanine and toluene were both reported related with airway obstruction or asthma development (Wang et al., 2021), which might be an indication for our further mechanism exploration. Thus, the changes we noted in mice neonatally infected with S. pneumoniae suggest long-lasting disruption of gut microbiota by bacterial respiratory infection, and may correlate with health conditions in later life.
The findings from this study suggested that bacterial respiratory infection in mouse neonates produced a profound effect on the gut microbiota, and that although this effect occurred gradually, it continued into adulthood. In the context of respiratory disease, most of the studies focused on the effect of gut microbial dysbiosis to lung immunity, Alterations of the gut microbial community may be essential for the immune and metabolic systems, and this might explain diseases that occur in later life. On the other side, with the hypothesis of gut-lung axis, investigators come to realize the influence of virus respiratory infection on gut microbiota (Deriu et al., 2016; Bartley et al., 2017). Groves et al. (2020) reported airway RSV infection induced CD8 + cytokine production, which may inhibit appetite of mice and contribute to gut microbial alteration. Consistent with that, we previously found loss of appetite and weight in our neonatal S.pneumoniae infection model, indicating the possible mechanism in our investigation.
There were several limitations to this study. First, although 16S rRNA high-throughput gene sequencing identified the majority of bacteria, genome sequencing and metabonomics are still needed to allow further progress in understanding the underlying mechanisms of action. We also found a significant gut microbiota dysbiosis after neonatal S. pneumoniae infection, and further microbial community restoration is therefore required to fully investigate the relationships between microbial disruption and AAD development. Finally, due to medical ethics, this hypothesis could only be applied to a mouse model, even though mice have been thought to constitute a well-recognized model for human trials. As there are, of course, species differences in the gut microbiota of humans and mice, additional studies are required to better clarify the effects of neonatal bacterial respiratory infection on microbiome development in humans.
Conclusion
In summary, our results first highlighted the substantial influence of neonatal bacterial respiratory infection in neonates on the gut microbiota. Neonatal S. pneumoniae infection not only influenced gut microbial diversity and community, it also promoted a long-lasting modification of microbial community composition, and this was particularly true for the reduction in immune/metabolic regulatory flora such as Lactobacillus and Allobaculum. These findings will better elucidate bacterial respiratory infection in early life on gut microbiota development, and clarify its effects on lifelong health.
Data availability statement
The data presented in this study are deposited in the NCBI repository, accession number: PRJNA858952 (https://submit.ncbi.nlm.nih.gov/subs/sra/SUB11796996/overview).
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Research Advisory Committee of the Chongqing Medical University.
Author contributions
ZL conceived and designed the experiments. YL drafted the manuscript. YL, XX, and ZL revised the manuscript. YL, YW, DJ, and ZG performed the experiments. YL, GZ, XT, and XX analyzed the data. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the Department of Laboratory Medicine, Key Laboratory of Diagnostic Medicine, Chongqing Medical University, for providing the S. pneumoniae D39 strain and LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. We also thank BioRender.com for creating the diagram of our experimental design.
Funding
This work was supported by the Science and Technology Department of Chongqing (cstc2020jcyj-msxmX0677) and the Health Committee of Chongqing (CQYC2020030393).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.961684/full#supplementary-material
References
Alnahas, S., Hagner, S., Raifer, H., Kilic, A., Gasteiger, G., Mutters, R., et al. (2017). IL-17 and TNF-α are key mediators of moraxella catarrhalis triggered exacerbation of allergic airway inflammation. Front. Immunol. 8:1562. doi: 10.3389/fimmu.2017.01562
Althouse, M. H., Stewart, C., Jiang, W., Moorthy, B., and Lingappan, K. (2019). Impact of early life antibiotic exposure and neonatal hyperoxia on the murine microbiome and lung injury. Sci. Rep. 9:14992. doi: 10.1038/s41598-019-51506-0
Arrieta, M. C., Stiemsma, L. T., Dimitriu, P. A., Thorson, L., Russell, S., Yurist-Doutsch, S., et al. (2015). Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 7:307ra152. doi: 10.1126/scitranslmed.aab2271
Azad, M. B., Konya, T., Guttman, D. S., Field, C. J., Sears, M. R., HayGlass, K. T., et al. (2015). Infant gut microbiota and food sensitization: associations in the first year of life. Clin. Exp. Allergy 45, 632–643. doi: 10.1111/cea.12487
Balakrishnan, B., Luckey, D., Bodhke, R., Chen, J., Marietta, E., Jeraldo, P., et al. (2021). Prevotella histicola protects from arthritis by expansion of allobaculum and augmenting butyrate production in humanized mice. Front. Immunol. 12:609644. doi: 10.3389/fimmu.2021.609644
Bartley, J. M., Zhou, X., Kuchel, G. A., Weinstock, G. M., and Haynes, L. (2017). Impact of age, caloric restriction, and influenza infection on mouse gut microbiome: an exploratory study of the role of age-related microbiome changes on influenza responses. Front. Immunol. 8:1164. doi: 10.3389/fimmu.2017.01164
Björkander, S., Carvalho-Queiroz, C., Hallberg, J., Persson, J. O., Johansson, M. A., Nussbaum, B., et al. (2020). Childhood allergy is preceded by an absence of gut lactobacilli species and higher levels of atopy-related plasma chemokines. Clin. Exp. Immunol. 202, 288–299. doi: 10.1111/cei.13494
Caverly, L. J., Huang, Y. J., and Sze, M. A. (2019). Past, present, and future research on the lung microbiome in inflammatory airway disease. Chest 156, 376–382. doi: 10.1016/j.chest.2019.05.011
Chen, R. C., Xu, L. M., Du, S. J., Huang, S. S., Wu, H., Dong, J. J., et al. (2016). Lactobacillus rhamnosus GG supernatant promotes intestinal barrier function, balances Treg and TH17 cells and ameliorates hepatic injury in a mouse model of chronic-binge alcohol feeding. Toxicol. Lett. 241, 103–110. doi: 10.1016/j.toxlet.2015.11.019
Cho, S. W., Yim, J., and Seo, S. W. (2020). Engineering tools for the development of recombinant lactic acid bacteria. Biotechnol. J. 15:e1900344. doi: 10.1002/biot.201900344
Cunningham, S. A., Kramer, M. R., and Narayan, K. M. (2014). Incidence of childhood obesity in the united states. N. Engl. J. Med. 370, 403–411. doi: 10.1056/NEJMoa1309753
d’Enfert, C., Kaune, A. K., Alaban, L. R., Chakraborty, S., Cole, N., Delavy, M., et al. (2021). The impact of the fungus-host-microbiota interplay upon candida albicans infections: current knowledge and new perspectives. FEMS Microbiol. Rev. 45:60. doi: 10.1093/femsre/fuaa060
Del Chierico, F., Vernocchi, P., Petrucca, A., Paci, P., Fuentes, S., Praticò, G., et al. (2015). Phylogenetic and metabolic tracking of gut microbiota during perinatal development. PLoS One 10:e0137347. doi: 10.1371/journal.pone.0137347
Depner, M., Taft, D. H., Kirjavainen, P. V., Kalanetra, K. M., Karvonen, A. M., Peschel, S., et al. (2020). Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat. Med. 26, 1766–1775. doi: 10.1038/s41591-020-1095-x
Deriu, E., Boxx, G. M., He, X., Pan, C., Benavidez, S. D., Cen, L., et al. (2016). Influenza virus affects intestinal microbiota and secondary salmonella infection in the gut through type i interferons. PLoS Pathog. 12:e1005572. doi: 10.1371/journal.ppat.1005572
Ding, T., Song, T., Zhou, B., Geber, A., Ma, Y., Zhang, L., et al. (2019). Microbial composition of the human nasopharynx varies according to influenza virus type and vaccination status. mBio 10:19. doi: 10.1128/mBio.01296-19
Dong, W., Rasid, O., Chevalier, C., Connor, M., Eldridge, M., and Hamon, M. A. (2020). Streptococcus pneumoniae infection promotes histone h3 dephosphorylation by modulating host pp1 phosphatase. Cell Rep. 30, 4016.e–4026.e. doi: 10.1016/j.celrep.2020.02.116
Ferreira Dos Santos, T., Alves Melo, T., Almeida, M. E., Passos Rezende, R., and Romano, C. C. (2016). Immunomodulatory effects of lactobacillus plantarum lp62 on intestinal epithelial and mononuclear cells. Biomed. Res. Int. 2016:8404156. doi: 10.1155/2016/8404156
Fouhy, F., Guinane, C. M., Hussey, S., Wall, R., Ryan, C. A., Dempsey, E. M., et al. (2012). High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother. 56, 5811–5820. doi: 10.1128/AAC.00789-12
Fritz, C. Q., Edwards, K. M., Self, W. H., Grijalva, C. G., Zhu, Y., Arnold, S. R., et al. (2019). Prevalence, risk factors, and outcomes of bacteremic pneumonia in children. Pediatrics 144:3090. doi: 10.1542/peds.2018-3090
Gosmann, C., Anahtar, M. N., Handley, S. A., Farcasanu, M., Abu-Ali, G., Bowman, B. A., et al. (2017). Lactobacillus-deficient cervicovaginal bacterial communities are associated with increased hiv acquisition in young south african women. Immunity 46, 29–37. doi: 10.1016/j.immuni.2016.12.013
Greetham, H. L., Gibson, G. R., Giffard, C., Hippe, H., Merkhoffer, B., Steiner, U., et al. (2004). Allobaculum stercoricanis gen. nov., sp. nov., isolated from canine feces. Anaerobe 10, 301–307. doi: 10.1016/j.anaerobe.2004.06.004
Groves, H. T., Higham, S. L., Moffatt, M. F., Cox, M. J., and Tregoning, J. S. (2020). Respiratory viral infection alters the gut microbiota by inducing inappetence. mBio 11:19. doi: 10.1128/mBio.03236-19
Gürdeniz, G., Ernst, M., Rago, D., Kim, M., Courraud, J., Stokholm, J., et al. (2022). Neonatal metabolome of caesarean section and risk of childhood asthma. Eur. Respir. J. 59:2021. doi: 10.1183/13993003.02406-2021
Harding, J. N., Siefker, D., Vu, L., You, D., DeVincenzo, J., Pierre, J. F., et al. (2020). Altered gut microbiota in infants is associated with respiratory syncytial virus disease severity. BMC Microbiol. 20:140. doi: 10.1186/s12866-020-01816-5
Houghteling, P. D., and Walker, W. A. (2015). Why is initial bacterial colonization of the intestine important to infants’ and children’s health. J. Pediatr. Gastroenterol. Nutr. 60, 294–307. doi: 10.1097/MPG.0000000000000597
Huang, Y. J., Nariya, S., Harris, J. M., Lynch, S. V., Choy, D. F., Arron, J. R., et al. (2015). The airway microbiome in patients with severe asthma: associations with disease features and severity. J. Allergy Clin. Immunol. 136, 874–884. doi: 10.1016/j.jaci.2015.05.044
Ivanovic, N., Minic, R., Dimitrijevic, L., Radojevic Skodric, S., Zivkovic, I., and Djordjevic, B. (2015). Lactobacillus rhamnosus LA68 and Lactobacillus plantarum WCFS1 differently influence metabolic and immunological parameters in high fat diet-induced hypercholesterolemia and hepatic steatosis. Food Funct 6, 558–565. doi: 10.1039/c4fo00843j
Laukens, D., Brinkman, B. M., Raes, J., De Vos, M., and Vandenabeele, P. (2016). Heterogeneity of the gut microbiome in mice: guidelines for optimizing experimental design. FEMS Microbiol. Rev. 40, 117–132. doi: 10.1093/femsre/fuv036
Li, Y., Guo, Z., Zhang, G., Tian, X., Li, Q., and Luo, Z. (2021). Neonatal Streptococcus Pneumoniae pneumonia induces airway SMMHC expression through HMGB1/TLR4/ERK. Immunol. Lett. 240, 149–158. doi: 10.1016/j.imlet.2021.10.005
Lin, H., Wang, Q., Yuan, M., Liu, L., Chen, Z., Zhao, Y., et al. (2020). The prolonged disruption of a single-course amoxicillin on mice gut microbiota and resistome, and recovery by inulin, Bifidobacterium longum and fecal microbiota transplantation. Environ. Pollut. 265:114651. doi: 10.1016/j.envpol.2020.114651
Nobel, Y. R., Cox, L. M., Kirigin, F. F., Bokulich, N. A., Yamanishi, S., Teitler, I., et al. (2015). Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 6:7486. doi: 10.1038/ncomms8486
Penders, J., Kummeling, I., and Thijs, C. (2011). Infant antibiotic use and wheeze and asthma risk: a systematic review and meta-analysis. Eur. Respir. J. 38, 295–302. doi: 10.1183/09031936.00105010
Peng, X., Wu, Y., Kong, X., Chen, Y., Tian, Y., Li, Q., et al. (2019). Neonatal streptococcus pneumoniae pneumonia induces an aberrant airway smooth muscle phenotype and ahr in mice model. Biomed. Res. Int. 2019:1948519. doi: 10.1155/2019/1948519
Samuelson, D. R., Welsh, D. A., and Shellito, J. E. (2015). Regulation of lung immunity and host defense by the intestinal microbiota. Front. Microbiol. 6:1085. doi: 10.3389/fmicb.2015.01085
Sengupta, P. (2013). The laboratory rat: relating its age with human’s. Int. J. Prev. Med. 4, 624–630.
Stokholm, J., Blaser, M. J., Thorsen, J., Rasmussen, M. A., Waage, J., Vinding, R. K., et al. (2018). Maturation of the gut microbiome and risk of asthma in childhood. Nat. Commun. 9:141. doi: 10.1038/s41467-017-02573-2
Tanaka, S., Kobayashi, T., Songjinda, P., Tateyama, A., Tsubouchi, M., Kiyohara, C., et al. (2009). Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 56, 80–87. doi: 10.1111/j.1574-695X.2009.00553.x
Tasnim, N., Quin, C., Gill, S., Dai, C., Hart, M., and Gibson, D. L. (2021). Early life environmental exposures have a minor impact on the gut ecosystem following a natural birth. Gut Microbes 13, 1–15. doi: 10.1080/19490976.2021.1875797
Tian, Y., Tian, Q., Wu, Y., Peng, X., Chen, Y., Li, Q., et al. (2020). Vitamin A supplement after neonatal Streptococcus pneumoniae pneumonia inhibits the progression of experimental asthma by altering CD4(+)T cell subsets. Sci. Rep. 10:4214. doi: 10.1038/s41598-020-60665-4
van Nimwegen, F. A., Penders, J., Stobberingh, E. E., Postma, D. S., Koppelman, G. H., Kerkhof, M., et al. (2011). Mode and place of delivery, gastrointestinal microbiota, and their influence on asthma and atopy. J. Allergy Clin. Immunol. 128, e1–.e3. doi: 10.1016/j.jaci.2011.07.027
Wang, C., Jiang, S., Zhang, S., Ouyang, Z., Wang, G., and Wang, F. (2021). Research progress of metabolomics in asthma. Metabolites 11:11090567. doi: 10.3390/metabo11090567
Keywords: long-lasting dysbiosis, mice, gut microbiota, neonatal infection, Streptococcus pneumoniae
Citation: Li Y, Xu X, Guo Z, Li Q, Wang Y, Jian D, Zhang G, Tian X, Chen S and Luo Z (2022) Neonatal Streptococcus pneumoniae infection induces long-lasting dysbiosis of the gut microbiota in a mouse model. Front. Microbiol. 13:961684. doi: 10.3389/fmicb.2022.961684
Received: 10 June 2022; Accepted: 19 July 2022;
Published: 18 August 2022.
Edited by:
George Grant, University of Aberdeen, United KingdomReviewed by:
Nabil Bosco, Lesaffre Group, FranceRachel McMahan, University of Colorado Anschutz Medical Campus, United States
Copyright © 2022 Li, Xu, Guo, Li, Wang, Jian, Zhang, Tian, Chen and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengxiu Luo, bHVvemhlbmd4aXU4MTZAaG9zcGl0YWwuY3FtdS5lZHUuY24=