 Xi Cheng
Xi Cheng Tingting Zhou
Tingting Zhou Yanqiu He
Yanqiu He Yumei Xie
Yumei Xie Yong Xu
Yong Xu Wei Huang
Wei Huang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 09 August 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.961536
This article is part of the Research TopicDietary Polyphenols, Gut Microbiota, and Human HealthView all 7 articles
Diabetic kidney disease (DKD) remains the leading cause of the end-stage renal disease and is a major burden on the healthcare system. The current understanding of the mechanisms responsible for the progression of DKD recognizes the involvement of oxidative stress, low-grade inflammation, and fibrosis. Several circulating metabolites that are the end products of the fermentation process, released by the gut microbiota, are known to be associated with systemic immune-inflammatory responses and kidney injury. This phenomenon has been recognized as the “gut–kidney axis.” Butyrate is produced predominantly by gut microbiota fermentation of dietary fiber and undigested carbohydrates. In addition to its important role as a fuel for colonic epithelial cells, butyrate has been demonstrated to ameliorate obesity, diabetes, and kidney diseases via G-protein coupled receptors (GPCRs). It also acts as an epigenetic regulator by inhibiting histone deacetylase (HDAC), up-regulation of miRNAs, or induction of the histone butyrylation and autophagy processes. This review aims to outline the existing literature on the treatment of DKD by butyrate in animal models and cell culture experiments, and to explore the protective effects of butyrate on DKD and the underlying molecular mechanism.
Diabetic kidney disease (DKD) is a serious microvascular complication of diabetes mellitus and is also the leading cause of the end-stage renal disease (Marshall, 2016). Based on data from International Diabetes Federation, over 40% of patients with diabetes will develop DKD. This causes an increase in health care costs, cardiovascular events risks, and mortality. The etiology and pathogenesis of DKD are complex and have not been completely understood. It mainly includes hemodynamic changes, oxidative stress, insulin resistance, and the release of pro-inflammatory cytokines. All these events lead to glomerulosclerosis, tubular atrophy, fibrosis, and irreversible renal injury (Kawanami et al., 2016). Thus, the prevention and effective treatment of DKD is crucial and the most challenging aspect of clinical studies.
Short-chain fatty acids (SCFAs) are the main metabolic products by the bacterial fermentation of macro-fibrous material that escapes digestion and enters the colon. 90 to 95% of SCFAs in the colon are composed of acetate, propionate, and butyrate (Cummings et al., 1987). Among these SCFAs, butyrate is of particular concern due to its positive influence on cell energy metabolism and intestinal environmental stability (Guilloteau et al., 2010). It also relieves oxidative stress, inflammation, and fibrosis in diabetes and kidney diseases via G-protein coupled receptors (GPCRs) or serves as an epigenetic regulator by inhibiting histone deacetylase (HDAC), up-regulation of miRNAs, or induction of the histone butyrylation, a novel histone post-translational modification (Lin et al., 2015; Lu et al., 2016; Xu et al., 2018; Gonzalez et al., 2019; Sanna et al., 2019; Elgamal et al., 2020; Noureldein et al., 2020; Rodríguez-Carlos et al., 2020). Studies have already reported the therapeutic effects of butyrate or sodium butyrate, demonstrating that butyrate metabolic pathway could be a new therapeutic target for DKD (Du et al., 2020a,b; Huang et al., 2020; Li et al., 2020). This article aims to provide an overview of the role and the latent mechanism of butyrate action on DKD.
As shown in Figure 1, butyrate is generally thought to be generated by bacterial fermentation of dietary fiber that enters the colon after digestion in the upper gastrointestinal tract (Bergman, 1990; Simpson and Campbell, 2015). The bacteria involved in butyrate production are widespread, the two most important clusters being Faecalibacterium prausnitzii in the Clostridium leptum cluster and Eubacterium rectale/Roseburia spp. in the Clostridium coccoides cluster of Firmicutes (Mokhtari et al., 2017). In a normal human feces sample, each typically accounts for 5–10% of the total microbial load that may be detected (Haenen et al., 2013). Some of these butyrate-producing bacteria are also scattered in clusters IX, XV, XVI, and XVII (Haenen et al., 2013).
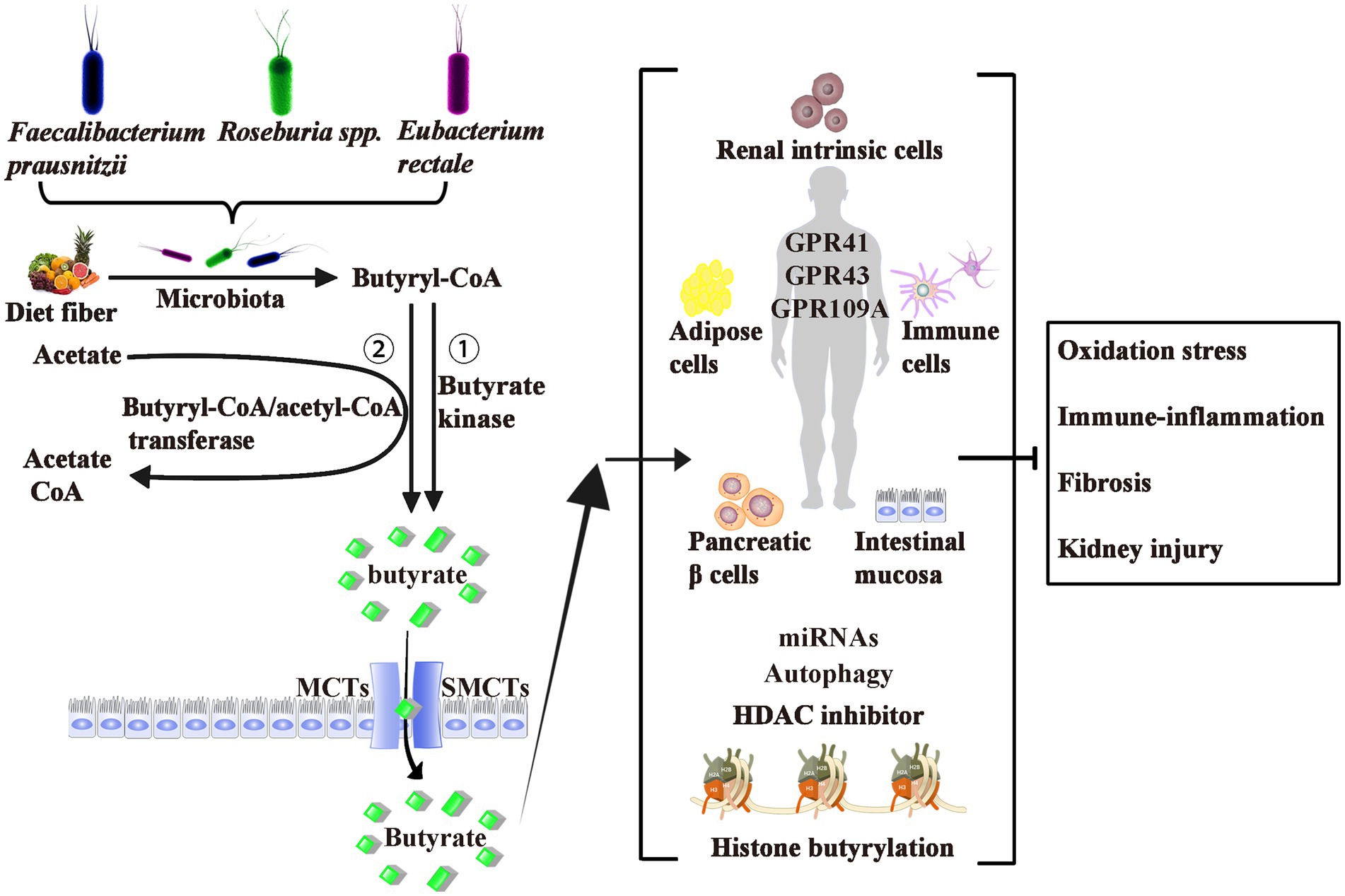
Figure 1. Origin, Production, Transport, Effects, and Mechanism of Butyrate. Butyrate is produced from dietary fiber by bacterial fermentation through two metabolic pathways: (1) butyryl-CoA is transformed to butyrate via butyrate kinase and (2) the CoA moiety of butyryl-CoA is transferred to butyrate and acetyl-CoA via butyryl-CoA: acetate CoA-transferase. The two most important butyrate-producing bacteria are Faecalibacterium prausnitzii and Eubacterium rectale/Roseburia spp. Butyrate is absorbed by colonic epithelial cells as energy sources mainly through MCTs and SMCTs. About three of the de-orphanized GPCRs (GPR41, GPR43, and GPR109A) have been identified as butyrate receptors in the human intestinal mucosa, renal intrinsic cells, immune cells, pancreatic β cells, and adipose tissues. Butyrate act as epigenetic regulators by the inhibition of HDAC, the upregulation of miRNAs, or induction of the histone butyrylation and autophagy. Although controversial, most studies believe that exogenous or endogenous butyrate improves inhibits oxidative stress, and ameliorates diabetic inflammation. GPCRs, G-protein coupled receptors; MCTs, monocarboxylate transporters; SMCTs, sodium-coupled monocarboxylate transporters; CoA, coenzyme A; HDAC, histone deacetylase; miRNAs, microRNAs.
Butyrate is produced via two metabolic pathways: firstly, butyryl-coenzyme A (CoA) is phosphorylated to form butyryl-phosphate, which is then converted to butyrate via butyrate kinase (Louis and Flint, 2017). Secondly, the CoA moiety of butyryl-CoA is transferred to butyrate and acetyl-CoA via butyryl-CoA: acetate CoA-transferase (Trachsel et al., 2016). The butyrate’s absorption mechanisms through the apical membrane of colon cells include monocarboxylate transporters and sodium-coupled monocarboxylate transporters (Kumar et al., 2015; Counillon et al., 2016). These transporters are diffusely expressed from the central nervous system to peripheral tissues, including liver, fat, heart, and kidney tissues.
Studies have demonstrated a protective effect of butyrate by improving body weight, blood glucose, lipid distribution, and insulin sensitivity in animal models of obesity and diabetes, which can be postulated as new therapeutic strategy to counteract obesity and insulin resistance (Gao et al., 2009; Khan and Jena, 2016; Mollica et al., 2017; Hu et al., 2018). The butyrate-mediated secretion of glucagon-like peptide-1 may mediate the reduction of insulin sensitivity and diabetes (Bridgeman et al., 2021). Glucagon-like peptide-1 has been shown to reduce hepatic gluconeogenesis and stimulate insulin secretion (Jin and Weng, 2016). Increased levels of butyrate-induced glucose transporter 4 in adipose tissues is also a crucial factor hypothesized to be responsible for the improvement in levels of glucose metabolites (Si et al., 2018). The current studies suggest that butyrate improves acute-chronic kidney injury that has been induced by ischemia–reperfusion (Zheng et al., 2019; Sun et al., 2022), contrast agent (Machado et al., 2012), gentamicin (Sun et al., 2013), doxorubicin (Felizardo et al., 2019), hypertension (Wu et al., 2021), and lipopolysaccharide (LPS; Huang et al., 2017; Dou et al., 2022). It acts on kidney inflammation, immunity, fibrosis, and energy metabolism and has a protective effect on the kidneys. Thus, the role of butyrate in kidney disease is an intriguing area of research.
A growing number of studies have suggested that kidney inflammation is related to bacterial load and endotoxins produced by gastrointestinal dysbiosis, which increases gut permeability and contributes to infection-related acute renal failure and chronic accelerated development of kidney disease (Li et al., 2019; Yang et al., 2019; Cai et al., 2020; Snelson et al., 2021). Several studies have observed significant differences in gut microbiota richness and populations between DKD patients and healthy controls (Jiang et al., 2016; Castillo-Rodriguez et al., 2018; Hu et al., 2020). This connection between the gut and kidney has been termed the “gut-kidney axis” (Leonel and Alvarez-Leite, 2012; Evenepoel et al., 2017).
A study by Terpstra ML (Terpstra et al., 2019) did not find significant differences in the number or capacity of the three most abundant butyrate-producing bacteria (F. prausnitzii, E. rectale, and Roseburia spp) between end-stage renal disease patients and healthy kidney donors. However, butyrate has been shown to make a positive contribution to kidney disease in humans. A recent clinical study by Wang et al. (2019) indicated reduced SCFA levels in patients with chronic kidney disease (CKD), suggesting that butyrate supplementation may delay the progression of CKD. Of further interest is a recent study that showed that quantitative reduction in SCFAs, especially butyrate, contributed to the progression of CKD (Wang et al., 2019). In a study by Cai et al. (2022), decreased serum butyrate levels were noted in the DKD group. Also, dysbiosis was evident in the gut microbiota of DKD patients, particularly associated with the lower abundance of SCFAs-producing bacteria belonging to the Ruminococcaceae, Lachnospiraceae, and Butyricicoccus groups. Li et al. (2022) also observed that the butyrate in serum was inversely connected with DKD. Given the substantial association between gut microbiota and butyrate, randomized controlled studies are necessary to examine whether alteration in the gut microbiota prevents DKD. Despite the controversy, the relationship between butyrate levels and renal injury has been proposed, indicating that butyrate may be a prospective target for the treatment of DKD. New mechanisms are being uncovered demanding the investigation of the role of endogenous butyrate on the “gut-kidney axis.”
Butyrate modulates the host’s biological responses mainly through the following mechanisms: (1) Butyrate directly inhibits HDAC (enzymes that remove acetyl groups from histone tails and regulate gene expression; Bose et al., 2014; Bridgeman et al., 2021). (2) Butyrate is involved in signalling through metabolite-sensing GPCRs. Three butyrate receptors have been identified, namely GPR41, GPR43, and GPR109A, out of the ten trophic receptor GPCRs (Miyamoto et al., 2016). (3) Butyrate may increase autophagy by switching on the AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) pathway (Cai et al., 2022). (4) The epigenetic mechanisms such as butyrate-mediated histone butyrylation (Goudarzi et al., 2016) and microRNAs (miRNAs; Yang et al., 2021) also modulate biological processes and are being extensively scrutinized. The effects and mechanisms of butyrate metabolism relevant to the “gut-kidney axis” and DKD will be discussed further in this part of the review.
GPR41 and GPR43 have been identified as receptors for SCFAs, hence named free fatty acid receptors 3 and free fatty acid receptors 2, respectively (Brown et al., 2003). A previous study (Huang et al., 2020) by our research group evaluated the effects of three major SCFAs (acetate, propionate, and butyrate) on a high-fat diet and streptozotocin (STZ)-induced type 2 diabetes and DKD mouse models. The role and mechanism of butyrate in high glucose (HG)-induced mouse glomerular mesangial cells were explored to decipher new therapeutic strategies and molecular targets for DKD. We demonstrated that exogenous SCFAs, especially butyrate, partially ameliorated type 2 diabetes-induced kidney injury through GPR43-mediated inhibition of oxidative stress and nuclear factor kappa B (NF-κB) signalling. Our study suggested that butyrate may be a potential therapeutic agent for the prevention and treatment of DKD (Huang et al., 2020).
Both GPR41 and GPR43 activate the heterotrimeric G protein and subsequently unite with β-arrestins (β-arrs), which regulate the desensitization, internalization, intracellular signal transduction, and recirculation of GPCRs, and are responsible for the inflammation signalling pathway (Lee et al., 2013; Eichel et al., 2018). Huang et al. (2020) showed that HG induced the expression of βarr-2, but not βarr-1, and HG reduced the interaction between βarr-2 and I-κBα. However, this effect was reversed by butyrate via GPR43, suggesting that GPR43-β-arrestin-2 signalling could be a prospective target for DKD treatment. Nevertheless, numerous questions regarding the functionality of butyrate remain unanswered and disputed. Future research on GPR41 and GPR43 will require more effective and selective tools.
GPR109A was first identified as a niacin receptor activated by β-hydroxybutyrate and butyrate (Walters et al., 2009). As a ligand for GPR109A, butyrate reduces intestinal inflammation and promotes the integrity of the intestinal epithelial barrier, so activation of GPR109A is thought to have a protective effect (Macia et al., 2015; Feng et al., 2018). One study (Li et al., 2020) suggested that dietary fiber prevents DKD by regulating the intestinal flora, enriching SCFAs producing bacteria, and increasing SCFAs production, GPR43−/− and GPR109A−/− mice were sensitive to STZ-induced diabetes, indicating that GPR43 and GPR109A are indispensable for fiber and butyrate mediated protection against DKD. However, the mechanism of GPR109A in the pathogenesis and treatment of DKD needs further characterization.
Acetate, propionate, and butyrate have all been reported as HDAC inhibitors, with butyrate being the most widely studied. Also, amongst all SCFAs, butyrate was mentioned to have the most potent inhibitory effect on HDAC activity in vitro and in vivo. The maximum inhibition effectiveness of about 80% was observed for HDAC1/2, while that of propionate was about 60% (Corfe, 2012). Several reported effects of butyrate have been attributed to epigenetic effects that regulate the expression of genes in activated T cells, including NF-κB, myoblast antigens, p53, and nuclear factors, by inhibiting HDAC, increasing histone acetylation, and reducing histone densification (Wang and Friedman, 2000; Cruz-Bravo et al., 2014).
The mechanism of inhibition of HDAC activity by butyrate remains unclear. However, its benefits may be related to its anti-fibrosis, anti-inflammatory, and immunosuppressive effects in polycystic kidney disease (Vinolo et al., 2011; Manson et al., 2014; Khan et al., 2015). Oxidative stress contributes to the pathogenesis of DKD (Zhang et al., 2012; Keshari et al., 2015). Du et al. (2020a) found that sodium butyrate acts as an antioxidant and suppresses the HG-induced apoptosis in normal rat kidney tubular epithelial cells by inhibition of HDAC2. Sodium butyrate is also an activator of nuclear factor erythroid 2-related factor 2 (Nrf2, Yaku et al., 2012; Wu et al., 2018). Dong et al. (2017) first noted that Nrf2 was an important aspect of sodium butyrate’s mediated protection against DKD. Sodium butyrate may inhibit HDAC activity, facilitate the expression of the Nrf2 gene, which might enter the nucleus, and upregulate downstream targets-HO1 (heme oxygenase 1) and NQO1 (NAD (P) H dehydrogenase quinone 1). It also inhibits oxidative stress and inflammation in DKD. In contrast, in the absence of the Nrf2 gene, the role of sodium butyrate is eliminated. Although sodium butyrate seems to play a significant role via the activation of Nrf2, further studies are necessary before sodium butyrate can be suggested for use in humans.
The coagulation protease-activated protein C studies showed cytoprotective effects in vitro and in vivo disease models, including DKD (Madhusudhan et al., 2020). Activated protein C reduced glucose-induced hypomethylation and hyperacetylation of the p66Shc promoter in podocytes, sodium butyrate eliminated the protective effect of protease-activated protein C (Bock et al., 2013). Therefore, the role of butyrate as an HDAC modulator in the DKD is still controversial, and it is necessary to understand the mode of action of butyrate in intestinal physiology and lipid metabolism. The research will contribute to the consideration of butyrate as an HDAC inhibitor in preventing and treating DKD kidney injury.
Recently, long non-coding RNAs (lncRNAs; Vinolo et al., 2011) and miRNAs (Du et al., 2020b) have been paid elaborate attention to decipher the molecular mechanisms of butyrate-mediated renal protection. miRNAs are small non-coding RNAs that regulate gene expression through the 3′ untranslated region of messenger RNAs (mRNAs). In DKD, extensive alterations in the expression of miRNAs such as miR-184 (Zanchi et al., 2017), miR-192 (Kato et al., 2013), and miR-21 (Kölling et al., 2017) was noted. Butyrate has been reported to influence miRNAs expression in oncogenic signalling pathways such as miR-92a, miR-22, miR-3,935, miR-574-3p, and miR-106B (Hu et al., 2015; Pant et al., 2017; Xiao et al., 2018). Du et al. (2020b) demonstrated that miR-7a-5p was prominently reduced in the HG-induced SV40-MES-13 cells and the kidneys of db/db mouse, while butyrate induced the upregulation of Mir-7a-5P. Later, the Mir-7a-5p inhibitors were administered to block the antifibrotic effects of butyrate.
Yang et al. (2021) also confirmed that sodium butyrate improved renal dysfunction in DKD model mice induced by db/db mice. Further RNA-seq results (Yang et al., 2021) showed that certain lncRNAs and mRNAs in the DKD+ sodium butyrate group had reverse changes compared with the DKD groups, and subsequent bioinformatics analysis also suggested that these changes might affect nephritis. These cumulative results indicated that sodium butyrate could protect DKD by altering the expression of lncRNA in mouse kidneys.
Autophagy is a highly conserved process and maintains cellular homeostasis (Liu et al., 2017). The expression of autophagy-related protein 7 in renal tubular epithelial cells was increased by SCFAs, suggesting that SCFAs regulate autophagy in acute kidney injury (Andrade-Oliveira et al., 2015). In colorectal and bladder cells, sodium butyrate also stimulated autophagy and inhibited tumor growth (Zhang et al., 2016; Luo et al., 2019; Wang et al., 2020). These results suggest a correlation between SCFAs and DKD via autophagy signaling.
AMPK is activated and downregulates the mTOR pathway under hunger or increased energy needs, which may induce autophagy to maintain homeostasis (Alers et al., 2012). AMPK phosphorylation was activated by butyrate in CKD rats, reducing renal injury (Gonzalez et al., 2019). Also, sodium butyrate causes autophagy-mediated cell death and reactivates the tumor suppressor gene DIRAS1 in the UOK146 renal cell carcinoma cell line (Verma et al., 2018). By increasing intestinal barrier function and activating the free fatty acid receptor 2-mediated PI3K/AKT/mTOR pathway, butyrate protects against DKD-induced muscle atrophy (Tang et al., 2022). Cai et al. (2022) first revealed that sodium butyrate boosted the phosphorylation of AMPK (Thr172), inhibited the phosphorylation of mTOR (Ser2448), and initiated autophagy in DKD rats. However, extensive research is needed to evaluate the mechanisms and interactions involved in gene knockout or overexpression in vitro and in vivo.
We previously described that butyrate improves DKD by inhibiting HDAC (Khan and Jena, 2014; Dong et al., 2017; Du et al., 2020a). In the recent years, histone butyrylation (Chen et al., 2007), crotonylation (Tan et al., 2011), β-hydroxybutyrylation (Xie et al., 2016), and other novel histone post-translational modifications have been found. Pelletier N reported these mechanisms to cooperate or antagonize with histone acetylation and methylation, which plays a crucial role in gene expression regulation and cell fate decision (Pelletier et al., 2017). In 2007, Zhao Y’s team found that butyrate metabolite-butyryl-CoA, catalyzed by transcriptional coactivator P300 with acetyltransferase activity, can transfer the butyl group to the histone lysine side chain, and produce a novel modification-histone butyrylation (Chen et al., 2007). Yang et al. (2021) reported that butyrylation of histone H3K9 is negatively regulated by high-fat stress in mouse heart tissue. Also, Ishikawa-Kobayashi E and Gaikwad AB reported that the acetylation of H3K9 is involved in obesity and diabetic heart disease (Gaikwad et al., 2010; Ishikawa-Kobayashi et al., 2012). The β-hydroxybutyrylation of H3K9 up-regulated matrix metalloproteinase-2 and improved DKD (Luo et al., 2020). Nie et al. (2017) reported that the butyrylation of H3K18 in hepatocytes of high-fat induced obesity mice model significantly decreased, and the acetylation was up-regulated. The above studies thus suggest that the crosstalk between these modifications at the same lysine site may have synergistic or antagonistic effects on related gene expression. Since butyrate may induce butyrylation of histone, the role of this modification in the pathogenesis and prevention of DKD deserves further attention.
In a limited number of investigations conducted to date, butyrate or sodium butyrate has been used therapeutically in DKD in vivo and in vitro studies. Research conducted on the application of butyrate in DKD is summarized in Table 1.
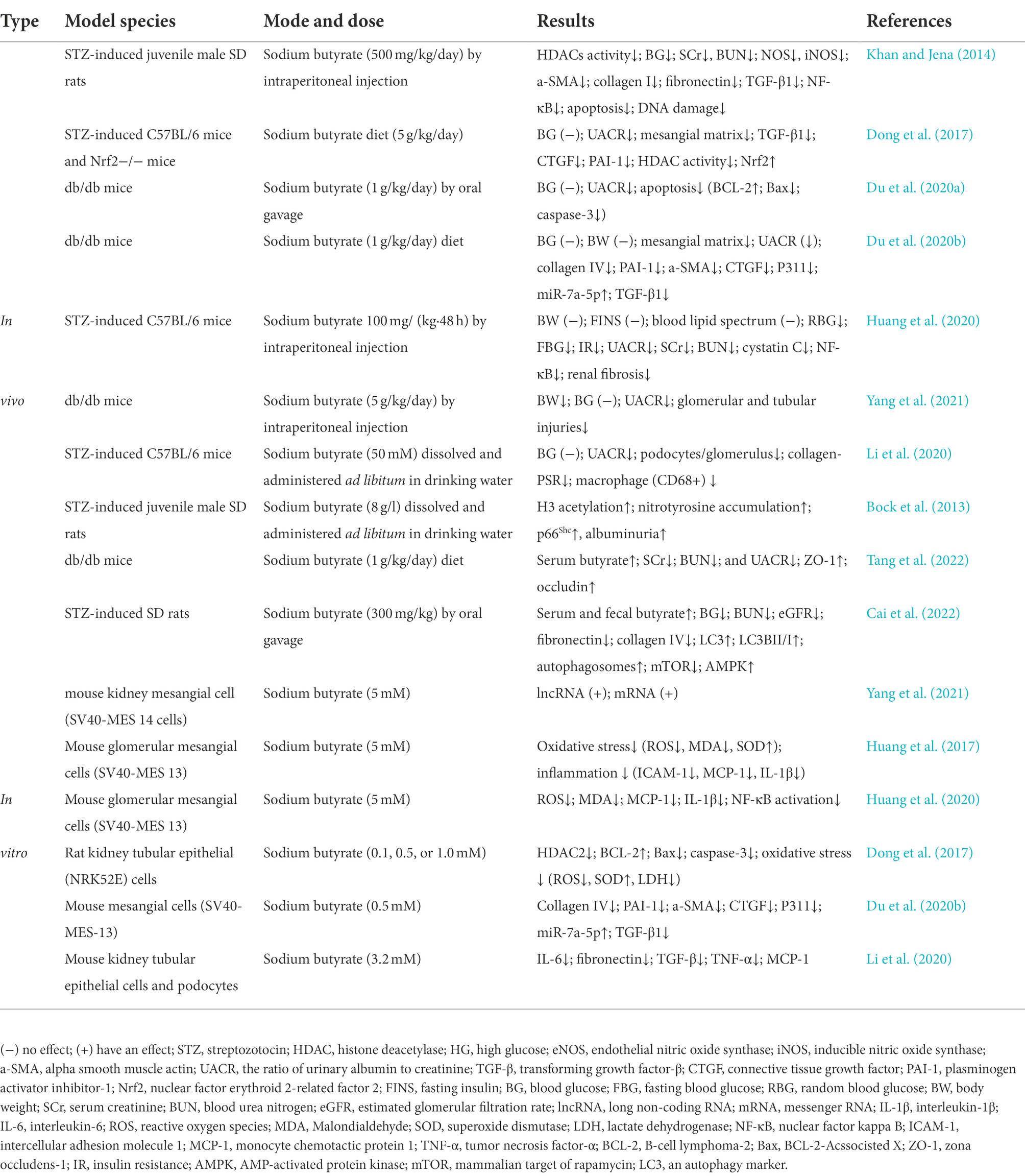
Table 1. Application of butyrate in vivo and in vitro model of DKD.
In a study by Khan S (Khan and Jena, 2014), sodium butyrate reduced plasma glucose, creatinine, urea, histological alterations, and descent of the HDACs activity. It also curbed the expression of endothelial nitric oxide synthase, inducible nitric oxide synthase, alpha-smooth muscle actin, collagen I, fibronectin, and transforming growth factor-β (TGF-β), and NF-κB in the kidneys of STZ-induced diabetic mice (Khan and Jena, 2014). However, Du Y’s study (Du et al., 2020a) showed that sodium butyrate as an HDAC2 inhibitor had no significance on the blood glucose levels of diabetic mice, though it significantly improved the ratio of urinary albumin to creatinine (UACR). Further, Dong et al. (2017) found that sodium butyrate did not alter blood glucose levels in STZ-induced diabetic C57BL/6 Nrf2 knockout and the WT mice, but significantly reduced UACR.
Surprisingly, Bock et al. (2013) reported that supplementing the drinking water with 8 g/l sodium butyrate eliminated the protective effect of activated protein C in diabetic mice. It further increased renal H3 acetylation, PAS-positive staining, nitrotyrosine accumulation, and urine albumin without affecting blood glucose levels or albuminuria in non-diabetic control animals. Chen et al. (2014) showed that in sodium butyrate-treated HBZY-1 cells (the rat mesangial cells), mRNA levels of monocyte chemotactic protein 1, intercellular adhesion molecule 1, and vascular cell adhesion molecule 1 significantly increased (1.7, 1.3 and 3.1 times). Apelin13, the most active member of the adipokine apelin group, inhibited sodium butyrate-induced inflammation and HDAC1 reduction in HBZY-1 cells by regulating histone acetylation (Chen et al., 2014). Therefore, a new mechanism was proposed to determine whether butyrate mediated inhibition of HDAC and contributed to the improvement of DKD. However, more studies are necessary to characterize and explain the controversial effects of endogenous butyrate.
In addition to protecting against DKD by inhibiting the HDAC pathway, butyrate also acts as an agonist of GPCRs to ameliorate DKD. DKD is a chronic low-grade inflammatory disease, and SCFAs have proven to positively affect inflammation and kidney injury (Huang et al., 2017; Xu et al., 2018). Previous studies by Huang et al. (2017) showed that butyrate and GPR43 agonists reversed the HG and LPS-induced mesangial cell proliferation, reduced reactive oxygen species and malondialdehyde generation, and controlled the inflammatory cytokine release. Ensuing research (Huang et al., 2020) evaluated the effects of butyrate in mice fed with a high-fat diet, with STZ-induced type 2 diabetes, DKD, and on HG-induced mouse glomerular mesangial cells. The results showed that exogenous butyrate improved blood glucose and insulin resistance and avoided UACR, mesangial matrix accumulation, and renal fibrosis in mice. Butyrate is responsible for the aforementioned effects via GPR43-mediated suppression of oxidative stress and NF-κB signalling.
Snelson et al. (2020) first studied the effects of GPR109A deficiency on DKD through analysis of GPR109A signalling deficiency on the development of DKD renal injury by GPR109A gene deletion. The findings of the long-term study (24 weeks) suggested that GPR109A deficiency does not play a vital role in the occurrence and development of DKD and gastrointestinal homeostasis. On the contrary, Li et al. (2020) explored the protective effects of dietary fiber on experimental DKD in STZ-induced diabetic mice or GPR43/GPR109A knockout mice. The diabetic mice fed on a high-fat diet had lower rates of albuminuria, glomerular hypertrophy, podocyte injury, and interstitial fibrosis compared with diabetic controls. Intake of fiber beneficially increased faecal and systemic SCFA concentrations and decreased the expression of inflammatory factors, chemokines, and fibrosis-promoting proteins in diabetic kidneys.
Since butyrate is recognized by the GPR109A, GPR41, and GPR43 receptors (Miyamoto et al., 2016), it has been argued that single-gene knockout models are insufficient to effectively illustrate the function of these receptors. This demands the use of double or triple knockout models (Tan et al., 2017). So far, the association between GPR41 and kidney injury has not been verified, and extensive studies are needed to investigate and further unravel the relationship between GPCRs and DKD.
Recently, Cai et al. (2022) explored the protective effects and mechanism of sodium butyrate on STZ-induced DKD rats through activation of autophagy. They reported that oral sodium butyrate improved blood glucose, serum nitrogen levels, fibronectin, and collagen IV expression. They also confirmed that the beneficial effects of sodium butyrate on the said DKD phenotype were due to the activation of the AMPK/mTOR signalling pathway, which induces autophagy.
Studies by Tang et al. (2022) also showed that sodium butyrate lowered serum creatinine, blood urea nitrogen, and UACR levels in DKD models replicated in db/db mice. They further evaluated the protective effects of butyrate against DKD-induced muscle atrophy. Butyrate provides a protective effect in db/db mice and HG/LPS-induced C2C12 myoblasts by suppressing autophagy and oxidative stress and activating the PI3K/AKT/mTOR pathway. Since the relationship between autophagy and DKD is not entirely deciphered yet, butyrate’s role in the regulation of autophagy and the prevention and treatment of DKD needs further evaluation.
We previously described the epigenetic control by HDAC inhibition mediated by butyrate that leads to amelioration of DKD, which recent studies have corroborated. Du et al. (2020b) found that butyrate alleviated renal dysfunction, serum creatinine and UACR levels, and mesangial matrix expansion in db/db mice by modulating the miR-7a-5p/P311/TGF-β1 pathway. However, no apparent differences in blood glucose levels and body weight were noted. As reviewed by Yang et al. (2021), sodium butyrate significantly improved body weight, urinary microalbumin, and urinary creatinine in diabetic mice by altering lncRNA expression, with no significant improvement in blood glucose levels.
Studies have found that butyrylation, similar to histone acetylation, can competitively inhibit gene transcription (Nie et al., 2017). Also, exogenous crotonate-mediated histone crotonylation inhibits the transcriptional activation of renal inflammatory genes and improves renal function in mice with acute AKI models (Ruiz-Andres et al., 2016). There is also evidence that histone β-hydroxybutyrylation is potentially beneficial for DKD (Luo et al., 2020). However, whether butyrate or sodium butyrate improves DKD renal injury through the histone butyrylation pathway has not been deciphered. Therefore, exploring the role of novel epigenetic mechanisms in DKD gene transcription regulation can provide great insights into the development of novel therapeutics against this disease.
All in all, endogenous butyrate or exogenous sodium butyrate supplementation improves body weight, glucose, and lipid metabolism, benefits a large population of type 2 diabetes, and protects against DKD, suggesting a new therapeutic reagent for DKD (Hong et al., 2016; Bridgeman et al., 2020). However, in vivo experiments have not clarified whether butyrate’s protective effect on DKD is independent or dependent on improving glycolipid metabolism, and the in vitro studies have reported that butyrate within a certain concentration range improves HG-induced renal intrinsic cells injury (Huang et al., 2017; Gu et al., 2019), but the harmful effects of butyrate have also been reported when the intervention concentration or time exceeds a certain level (Bock et al., 2013), furthermore, it is unclear whether the beneficial results observed in animal studies can be extrapolated to humans.
As shown in Figure 2, the molecular mechanism of butyrate improving DKD is extremely complex. As GPR41, GPR43, or GPR109A agonist, butyrate may exert anti-inflammatory effect through these receptors signalling pathways. At the same time, butyrate also acts as epigenetic regulators in response to the environment or therapeutic modulation by inhibiting HDAC, up-regulation of miR-7a-5p, or induction of the histone butyrylation and autophagy processes. It is worth mentioning that butyrate-induced histone butyrylation, as a novel histone post-translational modification, has demonstrated its renal protective effects on diverse nephropathies, which provides a novel perspective for elucidating the pharmacological mechanisms of butyrate. Therefore, further basic experiments and well-designed clinical studies are necessary to explore the pharmacological effects and molecular mechanism of butyrate in DKD prevention and treatment.
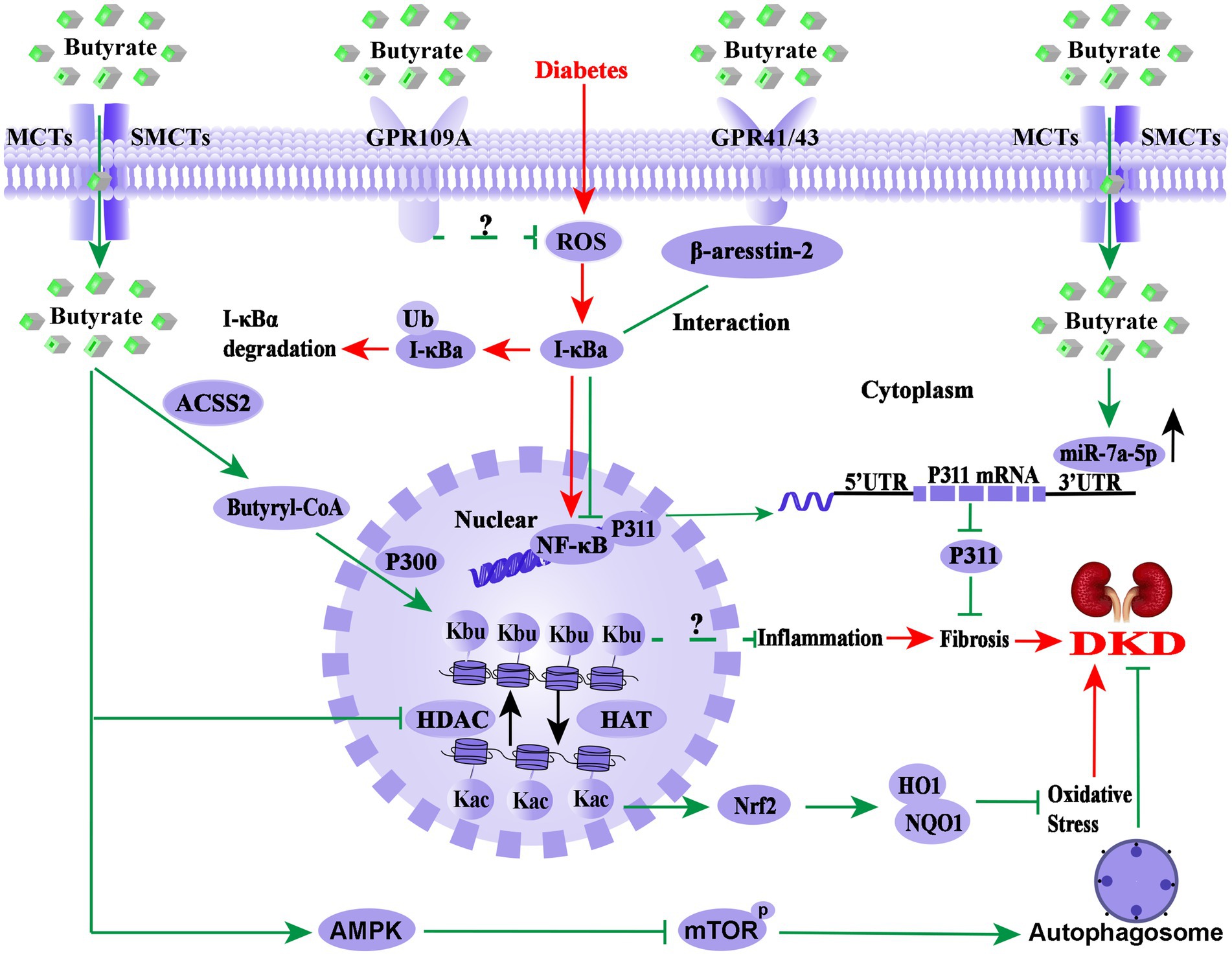
Figure 2. Overview of the molecular mechanism of butyrate in the prevention and treatment of DKD. The pathological process of DKD involves persistent HG-induced oxidative stress, immune system disorders, and inflammation (red arrows). Endogenous or exogenous butyrate (green arrows) inhibits the activity of HDAC, opens the structure of chromatin, and facilitates the expression of the Nrf2 gene, which may enter the nucleus and upregulate the downstream targets HO1 and NQO1 and then inhibits oxidative stress and inflammation in DKD. Meanwhile, GPR43 and GPR109A are important receptors of butyrate for renal protection, and the interaction between β-arrestin-2 and I-κBα is induced by butyrate via GPR43, suggesting that butyrate-mediated GPR43-β-arrestin-2 signaling may be a novel and promising target for DKD (green arrows). Moreover, it has been found that butyrate reverses HG-induced the downregulation of miR-7a-5p and inhibits the expression of P311, followed by the inhabitation of the kidney fibrosis of DKD (green arrows) and activated autophagy via the AMPK/mTOR pathway to delay the DKD progression. Notably, butyl-CoA, a metabolite of butyrate, is the substrate of histone butyrylation modification, irrespective of whether butyrate or sodium butyrate improves DKD renal injury through histone butyrylation pathway or the cross-talk of the histone post-translational modifications has not been reported. Nrf2, Nuclear factor erythroid 2-related factor 2; HO1, heme oxygenase 1; NQO1, NAD(P)H dehydrogenase quinone 1; HDAC, histone deacetylase; HAT, histone acetyltransferase; UTR, untranslated region; NF-κB, nuclear factor kappa B; Kbu, histone lysine butyrylation; Kac, histone lysine acetylation; ACSS2, acetyl-CoA synthetase 2; p300, a histone acetylation transferase that mediates butyrylation; P311, an RNA-binding protein, which could stimulate fibrosis; AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin.
WH contributed to the conception and design of the research. XC and TZ contributed to the design of the research. YoX contributed to the acquisition and analysis of the data. YuX and YH contributed to the analysis of the data. WH contributed to the acquisition, analysis, and interpretation of the data. All authors drafted the manuscript, critically revised the manuscript, agree to be fully accountable for ensuring the integrity and accuracy of the work, and read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81800741 and 82170834), the Program of Sichuan Provincial Science and Technology Department (No. 2019YJ0697 and 2018JY0059), and Office of Science Technology and Talent Work of Luzhou (No. 2020LZXNYDP02 and 2021LZXNYD-G01).
We would like to thank the Clinical Center Laboratory (Affiliated Hospital of Southwest Medical University) for technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
DKD, Diabetic kidney disease; SCFAs, Short-chain fatty acids; UACR, The ratio of urinary albumin to creatinine; LPS, Lipopolysaccharide; AMPK, AMP-activated protein kinase; β-arrs, β-arrestins; CKD, Chronic kidney disease; STZ, Streptozotocin; GPCRs, G-protein coupled receptors; CoA, Coenzyme A; NF-κB, Nuclear factor-kappa B; mRNA, Messenger RNA; Nrf2, Nuclear factor erythroid 2-related factor 2; mTOR, Mammalian target of rapamycin; miRNA, microRNAs; lncRNAs, Long non-coding RNAs; HDAC, Histone deacetylase; HG, High glucose.
Alers, S., Löffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11. doi: 10.1128/mcb.06159-11
Andrade-Oliveira, V., Amano, M. T., Correa-Costa, M., Castoldi, A., Felizardo, R. J., de Almeida, D. C., et al. (2015). Gut bacteria products prevent AKI induced by ischemia-reperfusion. J. Am. Soc. Nephrol. 26, 1877–1888. doi: 10.1681/asn.2014030288
Bergman, E. N. (1990). Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 70, 567–590. doi: 10.1152/physrev.1990.70.2.567
Bock, F., Shahzad, K., Wang, H., Stoyanov, S., Wolter, J., Dong, W., et al. (2013). Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc. Natl. Acad. Sci. U. S. A. 110, 648–653. doi: 10.1073/pnas.1218667110
Bose, P., Dai, Y., and Grant, S. (2014). Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol. Ther. 143, 323–336. doi: 10.1016/j.pharmthera.2014.04.004
Bridgeman, S., Ellison, G., Newsholme, P., and Mamotte, C. (2021). The HDAC inhibitor butyrate impairs β cell function and activates the disallowed gene hexokinase I. Int. J. Mol. Sci. 22. doi: 10.3390/ijms222413330
Bridgeman, S. C., Northrop, W., Melton, P. E., Ellison, G. C., Newsholme, P., and Mamotte, C. D. S. (2020). Butyrate generated by gut microbiota and its therapeutic role in metabolic syndrome. Pharmacol. Res. 160:105174. doi: 10.1016/j.phrs.2020.105174
Brown, A. J., Goldsworthy, S. M., Barnes, A. A., Eilert, M. M., Tcheang, L., Daniels, D., et al. (2003). The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 278, 11312–11319. doi: 10.1074/jbc.M211609200
Cai, K., Ma, Y., Cai, F., Huang, X., Xiao, L., Zhong, C., et al. (2022). Changes of gut microbiota in diabetic nephropathy and its effect on the progression of kidney injury. Endocrine 76, 294–303. doi: 10.1007/s12020-022-03002-1
Cai, T. T., Ye, X. L., Li, R. R., Chen, H., Wang, Y. Y., Yong, H. J., et al. (2020). Resveratrol modulates the gut microbiota and inflammation to protect against diabetic nephropathy in mice. Front. Pharmacol. 11:1249. doi: 10.3389/fphar.2020.01249
Castillo-Rodriguez, E., Fernandez-Prado, R., Esteras, R., Perez-Gomez, M. V., Gracia-Iguacel, C., Fernandez-Fernandez, B., et al. (2018). Impact of altered intestinal microbiota on chronic kidney disease progression. Toxins 10:300. doi: 10.3390/toxins10070300
Chen, H., Li, J., Jiao, L., Petersen, R. B., Li, J., Peng, A., et al. (2014). Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in Akita mouse. J. Physiol. 592, 505–521. doi: 10.1113/jphysiol.2013.266411
Chen, Y., Sprung, R., Tang, Y., Ball, H., Sangras, B., Kim, S. C., et al. (2007). Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteom. 6, 812–819. doi: 10.1074/mcp.M700021-MCP200
Corfe, B. M. (2012). Hypothesis: butyrate is not an HDAC inhibitor, but a product inhibitor of deacetylation. Mol. BioSyst. 8, 1609–1612. doi: 10.1039/c2mb25028d
Counillon, L., Bouret, Y., Marchiq, I., and Pouysségur, J. (2016). Na(+)/H(+) antiporter (NHE1) and lactate/H(+) symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta 1863, 2465–2480. doi: 10.1016/j.bbamcr.2016.02.018
Cruz-Bravo, R. K., Guevara-González, R. G., Ramos-Gómez, M., Oomah, B. D., Wiersma, P., Campos-Vega, R., et al. (2014). The fermented non-digestible fraction of common bean (Phaseolus vulgaris L.) triggers cell cycle arrest and apoptosis in human colon adenocarcinoma cells. Genes Nutr. 9:359. doi: 10.1007/s12263-013-0359-1
Cummings, J. H., Pomare, E. W., Branch, W. J., Naylor, C. P., and Macfarlane, G. T. (1987). Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221–1227. doi: 10.1136/gut.28.10.1221
Dong, W., Jia, Y., Liu, X., Zhang, H., Li, T., Huang, W., et al. (2017). Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J. Endocrinol. 232, 71–83. doi: 10.1530/joe-16-0322
Dou, X., Yan, D., Ma, Z., Gao, N., and Shan, A. (2022). Sodium butyrate alleviates LPS-induced kidney injury via inhibiting TLR2/4 to regulate rBD2 expression. J. Food Biochem. 46:e14126. doi: 10.1111/jfbc.14126
Du, Y., Tang, G., and Yuan, W. (2020a). Suppression of HDAC2 by sodium butyrate alleviates apoptosis of kidney cells in db/db mice and HG-induced NRK-52E cells. Int. J. Mol. Med. 45, 210–222. doi: 10.3892/ijmm.2019.4397
Du, Y., Yang, Y. T., Tang, G., Jia, J. S., Zhu, N., and Yuan, W. J. (2020b). Butyrate alleviates diabetic kidney disease by mediating the miR-7a-5p/P311/TGF-β1 pathway. FASEB J. 34, 10462–10475. doi: 10.1096/fj.202000431R
Eichel, K., Jullié, D., Barsi-Rhyne, B., Latorraca, N. R., Masureel, M., Sibarita, J. B., et al. (2018). Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386. doi: 10.1038/s41586-018-0079-1
Elgamal, D. A., Abou-Elghait, A. T., Ali, A. Y., Ali, M., and Bakr, M. H. (2020). Ultrastructure characterization of pancreatic β-cells is accompanied by modulatory effects of the HDAC inhibitor sodium butyrate on the PI3/AKT insulin signaling pathway in juvenile diabetic rats. Mol. Cell. Endocrinol. 503:110700. doi: 10.1016/j.mce.2019.110700
Evenepoel, P., Poesen, R., and Meijers, B. (2017). The gut-kidney axis. Pediatr nephrology (Berlin, Germany). 32, 2005–2014. doi: 10.1007/s00467-016-3527-x
Felizardo, R. J. F., de Almeida, D. C., Pereira, R. L., Watanabe, I. K. M., Doimo, N. T. S., Ribeiro, W. R., et al. (2019). Gut microbial metabolite butyrate protects against proteinuric kidney disease through epigenetic-and GPR109a-mediated mechanisms. FASEB J. 33, 11894–11908. doi: 10.1096/fj.201901080R
Feng, W., Wu, Y., Chen, G., Fu, S., Li, B., Huang, B., et al. (2018). Sodium butyrate attenuates diarrhea in weaned piglets and promotes tight junction protein expression in colon in a GPR109A-dependent manner. Cell. Physiol. Biochem. 47, 1617–1629. doi: 10.1159/000490981
Gaikwad, A. B., Sayyed, S. G., Lichtnekert, J., Tikoo, K., and Anders, H. J. (2010). Renal failure increases cardiac histone h3 acetylation, dimethylation, and phosphorylation and the induction of cardiomyopathy-related genes in type 2 diabetes. Am. J. Pathol. 176, 1079–1083. doi: 10.2353/ajpath.2010.090528
Gao, Z., Yin, J., Zhang, J., Ward, R. E., Martin, R. J., Lefevre, M., et al. (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517. doi: 10.2337/db08-1637
Gonzalez, A., Krieg, R., Massey, H. D., Carl, D., Ghosh, S., Gehr, T. W. B., et al. (2019). Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol. Dia. Transplant. 34, 783–794. doi: 10.1093/ndt/gfy238
Goudarzi, A., Zhang, D., Huang, H., Barral, S., Kwon, O. K., Qi, S., et al. (2016). Dynamic competing histone H4 K5K8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol. Cell 62, 169–180. doi: 10.1016/j.molcel.2016.03.014
Gu, J., Huang, W., Zhang, W., Zhao, T., Gao, C., Gan, W., et al. (2019). Sodium butyrate alleviates high-glucose-induced renal glomerular endothelial cells damage via inhibiting pyroptosis. Int. Immunopharmacol. 75:105832. doi: 10.1016/j.intimp.2019.105832
Guilloteau, P., Martin, L., Eeckhaut, V., Ducatelle, R., Zabielski, R., and Van Immerseel, F. (2010). From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr. Res. Rev. 23, 366–384. doi: 10.1017/s0954422410000247
Haenen, D., Zhang, J., Souza da Silva, C., Bosch, G., van der Meer, I. M., van Arkel, J., et al. (2013). A diet high in resistant starch modulates microbiota composition, SCFA concentrations, and gene expression in pig intestine. J. Nutr. 143, 274–283. doi: 10.3945/jn.112.169672
Hong, J., Jia, Y., Pan, S., Jia, L., Li, H., Han, Z., et al. (2016). Butyrate alleviates high fat diet-induced obesity through activation of adiponectin-mediated pathway and stimulation of mitochondrial function in the skeletal muscle of mice. Oncotarget 7, 56071–56082. doi: 10.18632/oncotarget.11267
Hu, S., Liu, L., Chang, E. B., Wang, J. Y., and Raufman, J. P. (2015). Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol. Cancer 14:180. doi: 10.1186/s12943-015-0450-x
Hu, Y., Liu, J., Yuan, Y., Chen, J., Cheng, S., Wang, H., et al. (2018). Sodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ. Toxicol. Pharmacol. 64, 112–121. doi: 10.1016/j.etap.2018.09.002
Hu, X., Ouyang, S., Xie, Y., Gong, Z., and Du, J. (2020). Characterizing the gut microbiota in patients with chronic kidney disease. Postgrad. Med. 132, 495–505. doi: 10.1080/00325481.2020.1744335
Huang, W., Guo, H. L., Deng, X., Zhu, T. T., Xiong, J. F., Xu, Y. H., et al. (2017). Short-chain fatty acids inhibit oxidative stress and inflammation in mesangial cells induced by high glucose and lipopolysaccharide. Exp. Clin. Endocrinol. Diabet. 125, 98–105. doi: 10.1055/s-0042-121493
Huang, W., Man, Y., Gao, C., Zhou, L., Gu, J., Xu, H., et al. (2020). Short-chain fatty acids ameliorate diabetic nephropathy via GPR43-mediated inhibition of oxidative stress and NF-κB signaling. Oxidative Med. Cell. Longev. 2020, 4074832–4074821. doi: 10.1155/2020/4074832
Huang, W., Zhou, L., Guo, H., Xu, Y., and Xu, Y. (2017). The role of short-chain fatty acids in kidney injury induced by gut-derived inflammatory response. Metab. Clin. Exp. 68, 20–30. doi: 10.1016/j.metabol.2016.11.006
Ishikawa-Kobayashi, E., Ushijima, K., Ando, H., Maekawa, T., Takuma, M., Furukawa, Y., et al. (2012). Reduced histone H3K9 acetylation of clock genes and abnormal glucose metabolism in ob/ob mice. Chronobiol. Int. 29, 982–993. doi: 10.3109/07420528.2012.706765
Jiang, S., Xie, S., Lv, D., Zhang, Y., Deng, J., Zeng, L., et al. (2016). A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Van Leeuwenhoek 109, 1389–1396. doi: 10.1007/s10482-016-0737-y
Jin, T., and Weng, J. (2016). Hepatic functions of GLP-1 and its based drugs: current disputes and perspectives. Am. J. Physiol. Endocrinol. Metab. 311, E620–E627. doi: 10.1152/ajpendo.00069.2016
Kato, M., Dang, V., Wang, M., Park, J. T., Deshpande, S., Kadam, S., et al. (2013). TGF-β induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci. Signal. 6:ra43. doi: 10.1126/scisignal.2003389
Kawanami, D., Matoba, K., and Utsunomiya, K. (2016). Signaling pathways in diabetic nephropathy. Histol. Histopathol. 31, 1059–1067. doi: 10.14670/hh-11-777
Keshari, K. R., Wilson, D. M., Sai, V., Bok, R., Jen, K. Y., Larson, P., et al. (2015). Noninvasive in vivo imaging of diabetes-induced renal oxidative stress and response to therapy using hyperpolarized 13C dehydroascorbate magnetic resonance. Diabetes 64, 344–352. doi: 10.2337/db13-1829
Khan, S., and Jena, G. (2014). Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 73, 127–139. doi: 10.1016/j.fct.2014.08.010
Khan, S., and Jena, G. (2016). Sodium butyrate reduces insulin-resistance, fat accumulation and dyslipidemia in type-2 diabetic rat: a comparative study with metformin. Chem. Biol. Interact. 254, 124–134. doi: 10.1016/j.cbi.2016.06.007
Khan, S., Jena, G., and Tikoo, K. (2015). Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the inhibition of histone deacetylases in diabetic rat. Exp. Mol. Pathol. 98, 230–239. doi: 10.1016/j.yexmp.2015.01.003
Kölling, M., Kaucsar, T., Schauerte, C., Hübner, A., Dettling, A., Park, J. K., et al. (2017). Therapeutic miR-21 silencing ameliorates diabetic kidney disease in mice. Mol. Ther. 25, 165–180. doi: 10.1016/j.ymthe.2016.08.001
Kumar, A., Alrefai, W. A., Borthakur, A., and Dudeja, P. K. (2015). Lactobacillus acidophilus counteracts enteropathogenic E. coli-induced inhibition of butyrate uptake in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G602–G607. doi: 10.1152/ajpgi.00186.2015
Lee, S. U., In, H. J., Kwon, M. S., Park, B. O., Jo, M., Kim, M. O., et al. (2013). β-Arrestin 2 mediates G protein-coupled receptor 43 signals to nuclear factor-κB. Biol. Pharm. Bull. 36, 1754–1759. doi: 10.1248/bpb.b13-00312
Leonel, A. J., and Alvarez-Leite, J. I. (2012). Butyrate: implications for intestinal function. Curr. Opin. Clin. Nutr. Metab. Care 15, 474–479. doi: 10.1097/MCO.0b013e32835665fa
Li, Y. J., Chen, X., Kwan, T. K., Loh, Y. W., Singer, J., Liu, Y., et al. (2020). Dietary fiber protects against diabetic nephropathy through short-chain fatty acid-mediated activation of G protein-coupled receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 31, 1267–1281. doi: 10.1681/asn.2019101029
Li, J., Moturi, K. R., Wang, L., Zhang, K., and Yu, C. (2019). Gut derived-endotoxin contributes to inflammation in severe ischemic acute kidney injury. BMC Nephrol. 20:16. doi: 10.1186/s12882-018-1199-4
Li, Y., Qin, G. Q., Wang, W. Y., Liu, X., Gao, X. Q., Liu, J. H., et al. (2022). Short chain fatty acids for the risk of diabetic nephropathy in type 2 diabetes patients. Acta Diabetol. 59, 901–909. doi: 10.1007/s00592-022-01870-7
Lin, M. Y., de Zoete, M. R., van Putten, J. P., and Strijbis, K. (2015). Redirection of epithelial immune responses by short-chain fatty acids through inhibition of histone deacetylases. Front. Immunol. 6:554. doi: 10.3389/fimmu.2015.00554
Liu, N., Xu, L., Shi, Y., and Zhuang, S. (2017). Podocyte autophagy: a potential therapeutic target to prevent the progression of diabetic nephropathy. J. Diabetes Res. 2017, 3560238–3560236. doi: 10.1155/2017/3560238
Louis, P., and Flint, H. J. (2017). Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41. doi: 10.1111/1462-2920.13589
Lu, Y., Fan, C., Li, P., Lu, Y., Chang, X., and Qi, K. (2016). Short chain fatty acids prevent high-fat-diet-induced obesity in mice by regulating G protein-coupled receptors and gut microbiota. Sci. Rep. 6:37589. doi: 10.1038/srep37589
Luo, S., Li, Z., Mao, L., Chen, S., and Sun, S. (2019). Sodium butyrate induces autophagy in colorectal cancer cells through LKB1/AMPK signaling. J. Physiol. Biochem. 75, 53–63. doi: 10.1007/s13105-018-0651-z
Luo, W., Yu, Y., Wang, H., Liu, K., Wang, Y., Huang, M., et al. (2020). Up-regulation of MMP-2 by histone H3K9 β-hydroxybutyrylation to antagonize glomerulosclerosis in diabetic rat. Acta Diabetol. 57, 1501–1509. doi: 10.1007/s00592-020-01552-2
Machado, R. A., Constantino Lde, S., Tomasi, C. D., Rojas, H. A., Vuolo, F. S., Vitto, M. F., et al. (2012). Sodium butyrate decreases the activation of NF-κB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast-induced nephropathy. Nephrol. Dial. Transplant. 27, 3136–3140. doi: 10.1093/ndt/gfr807
Macia, L., Tan, J., Vieira, A. T., Leach, K., Stanley, D., Luong, S., et al. (2015). Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 6:6734. doi: 10.1038/ncomms7734
Madhusudhan, T., Ghosh, S., Wang, H., Dong, W., Gupta, D., Elwakiel, A., et al. (2020). Podocyte integrin-β (3) and activated protein C coordinately restrict RhoA signaling and ameliorate diabetic nephropathy. J. Am. Soc. Nephrol. 31, 1762–1780. doi: 10.1681/asn.2019111163
Manson, S. R., Song, J. B., Hruska, K. A., and Austin, P. F. (2014). HDAC dependent transcriptional repression of Bmp-7 potentiates TGF-β mediated renal fibrosis in obstructive uropathy. J. Urol. 191, 242–252. doi: 10.1016/j.juro.2013.06.110
Marshall, C. B. (2016). Rethinking glomerular basement membrane thickening in diabetic nephropathy: adaptive or pathogenic? Am. J. Physiol. Renal Physiol. 311, F831–F843. doi: 10.1152/ajprenal.00313.2016
Miyamoto, J., Hasegawa, S., Kasubuchi, M., Ichimura, A., Nakajima, A., and Kimura, I. (2016). Nutritional signaling via free fatty acid receptors. Int. J. Mol. Sci. 17:450. doi: 10.3390/ijms17040450
Mokhtari, Z., Gibson, D. L., and Hekmatdoost, A. (2017). Nonalcoholic fatty liver disease, the gut microbiome, and diet. Adv. Nutr. 8, 240–252. doi: 10.3945/an.116.013151
Mollica, M. P., Mattace Raso, G., Cavaliere, G., Trinchese, G., De Filippo, C., Aceto, S., et al. (2017). Butyrate regulates liver mitochondrial function, efficiency, and dynamics in insulin-resistant obese mice. Diabetes 66, 1405–1418. doi: 10.2337/db16-0924
Nie, L., Shuai, L., Zhu, M., Liu, P., Xie, Z. F., Jiang, S., et al. (2017). The landscape of histone modifications in a high-fat diet-induced obese (DIO) mouse model. Mol. Cell. Proteom. 16, 1324–1334. doi: 10.1074/mcp.M117.067553
Noureldein, M. H., Bitar, S., Youssef, N., Azar, S., and Eid, A. A. (2020). Butyrate modulates diabetes-linked gut dysbiosis: epigenetic and mechanistic modifications. J. Mol. Endocrinol. 64, 29–42. doi: 10.1530/jme-19-0132
Pant, K., Yadav, A. K., Gupta, P., Islam, R., Saraya, A., and Venugopal, S. K. (2017). Butyrate induces ROS-mediated apoptosis by modulating miR-22/SIRT-1 pathway in hepatic cancer cells. Redox Biol. 12, 340–349. doi: 10.1016/j.redox.2017.03.006
Pelletier, N., Grégoire, S., and Yang, X. J. (2017). Assays for acetylation and other acylations of lysine residues. Curr. Protoc. Protein Sci. 87, 14.11.1–14.11.18. doi: 10.1002/cpps.26
Rodríguez-Carlos, A., Trujillo, V., Gonzalez-Curiel, I., Marin-Luevano, S., Torres-Juarez, F., Santos-Mena, A., et al. (2020). Host defense peptide RNase 7 is down-regulated in the skin of diabetic patients with or without chronic ulcers, and its expression is altered with metformin. Arch. Med. Res. 51, 327–335. doi: 10.1016/j.arcmed.2020.03.006
Ruiz-Andres, O., Sanchez-Niño, M. D., Cannata-Ortiz, P., Ruiz-Ortega, M., Egido, J., Ortiz, A., et al. (2016). Histone lysine crotonylation during acute kidney injury in mice. Dis. Model. Mech. 9, 633–645. doi: 10.1242/dmm.024455
Sanna, S., van Zuydam, N. R., Mahajan, A., Kurilshikov, A., Vich Vila, A., Võsa, U., et al. (2019). Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 51, 600–605. doi: 10.1038/s41588-019-0350-x
Si, X., Shang, W., Zhou, Z., Strappe, P., Wang, B., Bird, A., et al. (2018). Gut microbiome-induced shift of acetate to butyrate positively manages dysbiosis in high fat diet. Mol. Nutr. Food Res. 62:1700670. doi: 10.1002/mnfr.201700670
Simpson, H. L., and Campbell, B. J. (2015). Review article: dietary fibre-microbiota interactions. Aliment. Pharmacol. Ther. 42, 158–179. doi: 10.1111/apt.13248
Snelson, M., Clarke, R. E., Nguyen, T. V., Penfold, S. A., Forbes, J. M., Tan, S. M., et al. (2021). Long term high protein diet feeding alters the microbiome and increases intestinal permeability, systemic inflammation and kidney injury in mice. Mol. Nutr. Food Res. 65:e2000851. doi: 10.1002/mnfr.202000851
Snelson, M., Tan, S. M., Higgins, G. C., Lindblom, R. S. J., and Coughlan, M. T. (2020). Exploring the role of the metabolite-sensing receptor GPR109a in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 318, F835–F842. doi: 10.1152/ajprenal.00505.2019
Sun, X., Zhang, B., Hong, X., Zhang, X., and Kong, X. (2013). Histone deacetylase inhibitor, sodium butyrate, attenuates gentamicin-induced nephrotoxicity by increasing prohibitin protein expression in rats. Eur. J. Pharmacol. 707, 147–154. doi: 10.1016/j.ejphar.2013.03.018
Sun, Y., Zhou, C., Chen, Y., He, X., Gao, F., and Xue, D. (2022). Quantitative increase in short-chain fatty acids, especially butyrate protects kidney from ischemia/reperfusion injury. J. Invest. Med. 70, 29–35. doi: 10.1136/jim-2020-001715
Tan, M., Luo, H., Lee, S., Jin, F., Yang, J. S., Montellier, E., et al. (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028. doi: 10.1016/j.cell.2011.08.008
Tan, J. K., McKenzie, C., Mariño, E., Macia, L., and Mackay, C. R. (2017). Metabolite-sensing G protein-coupled receptors-facilitators of diet-related immune regulation. Annu. Rev. Immunol. 35, 371–402. doi: 10.1146/annurev-immunol-051116-052235
Tang, G., Du, Y., Guan, H., Jia, J., Zhu, N., Shi, Y., et al. (2022). Butyrate ameliorates skeletal muscle atrophy in diabetic nephropathy by enhancing gut barrier function and FFA2-mediated PI3K/Akt/mTOR signals. Br. J. Pharmacol. 179, 159–178. doi: 10.1111/bph.15693
Terpstra, M. L., Sinnige, M. J., Hugenholtz, F., Peters-Sengers, H., Remmerswaal, E. B., Geerlings, S. E., et al. (2019). Butyrate production in patients with end-stage renal disease. Int. J. Nephrol. Renov. Dis. 12, 87–101. doi: 10.2147/ijnrd.S200297
Trachsel, J., Bayles, D. O., Looft, T., Levine, U. Y., and Allen, H. K. (2016). Function and phylogeny of bacterial butyryl coenzyme A: acetate transferases and their diversity in the proximal colon of swine. Appl. Environ. Microbiol. 82, 6788–6798. doi: 10.1128/aem.02307-16
Verma, S. P., Agarwal, A., and Das, P. (2018). Sodium butyrate induces cell death by autophagy and reactivates a tumor suppressor gene DIRAS1 in renal cell carcinoma cell line UOK146. In Vitro Cell. Dev. Biol. Anim. 54, 295–303. doi: 10.1007/s11626-018-0239-5
Vinolo, M. A., Rodrigues, H. G., Hatanaka, E., Sato, F. T., Sampaio, S. C., and Curi, R. (2011). Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J. Nutr. Biochem. 22, 849–855. doi: 10.1016/j.jnutbio.2010.07.009
Walters, R. W., Shukla, A. K., Kovacs, J. J., Violin, J. D., DeWire, S. M., Lam, C. M., et al. (2009). Beta-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J. Clin. Invest. 119, 1312–1321. doi: 10.1172/jci36806
Wang, J., and Friedman, E. (2000). Downregulation of p53 by sustained JNK activation during apoptosis. Mol. Carcinog. 29, 179–188. doi: 10.1002/1098-2744(200011)29:3<179::aid-mc7>3.0.co;2-k
Wang, S., Lv, D., Jiang, S., Jiang, J., Liang, M., Hou, F., et al. (2019). Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin. Sci. 133, 1857–1870. doi: 10.1042/cs20190171
Wang, F., Wu, H., Fan, M., Yu, R., Zhang, Y., Liu, J., et al. (2020). Sodium butyrate inhibits migration and induces AMPK-mTOR pathway-dependent autophagy and ROS-mediated apoptosis via the miR-139-5p/Bmi-1 axis in human bladder cancer cells. FASEB J. 34, 4266–4282. doi: 10.1096/fj.201902626R
Wu, C., Chen, Z., Zhang, L., Zhu, Y., Deng, M., Huang, C., et al. (2021). Sodium butyrate ameliorates deoxycorticosterone acetate/salt-induced hypertension and renal damage by inhibiting the MR/SGK1 pathway. Hypertens. Res. 44, 168–178. doi: 10.1038/s41440-020-00548-3
Wu, J., Jiang, Z., Zhang, H., Liang, W., Huang, W., Zhang, H., et al. (2018). Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic. Biol. Med. 124, 454–465. doi: 10.1016/j.freeradbiomed.2018.06.034
Xiao, X., Cao, Y., and Chen, H. (2018). Profiling and characterization of microRNAs responding to sodium butyrate treatment in A549 cells. J. Cell. Biochem. 119, 3563–3573. doi: 10.1002/jcb.26547
Xie, Z., Zhang, D., Chung, D., Tang, Z., Huang, H., Dai, L., et al. (2016). Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell 62, 194–206. doi: 10.1016/j.molcel.2016.03.036
Xu, Y. H., Gao, C. L., Guo, H. L., Zhang, W. Q., Huang, W., Tang, S. S., et al. (2018). Sodium butyrate supplementation ameliorates diabetic inflammation in db/db mice. J. Endocrinol. 238, 231–244. doi: 10.1530/joe-18-0137
Yaku, K., Enami, Y., Kurajyo, C., Matsui-Yuasa, I., Konishi, Y., and Kojima-Yuasa, A. (2012). The enhancement of phase 2 enzyme activities by sodium butyrate in normal intestinal epithelial cells is associated with Nrf2 and p53. Mol. Cell. Biochem. 370, 7–14. doi: 10.1007/s11010-012-1392-x
Yang, Z., He, M., Austin, J., Pfleger, J., and Abdellatif, M. (2021). Histone H3K9 butyrylation is regulated by dietary fat and stress via an acyl-CoA dehydrogenase short chain-dependent mechanism. Mol. Metab. 53:101249. doi: 10.1016/j.molmet.2021.101249
Yang, J., Lim, S. Y., Ko, Y. S., Lee, H. Y., Oh, S. W., Kim, M. G., et al. (2019). Intestinal barrier disruption and dysregulated mucosal immunity contribute to kidney fibrosis in chronic kidney disease. Nephrol. Dial. Transplant. 34, 419–428. doi: 10.1093/ndt/gfy172
Yang, H., Zhang, Z., Peng, R., Zhang, L., Liu, H., Wang, X., et al. (2021). RNA-Seq analysis reveals critical transcriptome changes caused by sodium butyrate in DN mouse models. Biosci. Rep. 41:BSR20203005. doi: 10.1042/bsr20203005
Zanchi, C., Macconi, D., Trionfini, P., Tomasoni, S., Rottoli, D., Locatelli, M., et al. (2017). MicroRNA-184 is a downstream effector of albuminuria driving renal fibrosis in rats with diabetic nephropathy. Diabetologia 60, 1114–1125. doi: 10.1007/s00125-017-4248-9
Zhang, M. Z., Yao, B., Yang, S., Yang, H., Wang, S., Fan, X., et al. (2012). Intrarenal dopamine inhibits progression of diabetic nephropathy. Diabetes 61, 2575–2584. doi: 10.2337/db12-0046
Zhang, J., Yi, M., Zha, L., Chen, S., Li, Z., Li, C., et al. (2016). Sodium butyrate induces endoplasmic reticulum stress and autophagy in colorectal cells: implications for apoptosis. PLoS One 11:e0147218. doi: 10.1371/journal.pone.0147218
Keywords: butyrate, diabetic kidney disease, immune, inflammation, epigenetics
Citation: Cheng X, Zhou T, He Y, Xie Y, Xu Y and Huang W (2022) The role and mechanism of butyrate in the prevention and treatment of diabetic kidney disease. Front. Microbiol. 13:961536. doi: 10.3389/fmicb.2022.961536
Edited by:
Mingzhi Zhu, Hunan Agricultural University, ChinaReviewed by:
Biao Yuan, China Pharmaceutical University, ChinaCopyright © 2022 Cheng, Zhou, He, Xie, Xu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Xu, eHl3eWxsQGFsaXl1bi5jb20=; Wei Huang, aHVhbmd3ZWkxMjEyNTIwQDE2My5jb20=; Tingting Zhou, emhvdXRpbmd0aW5nMzYxQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.