 Manuel Albanese
Manuel Albanese Takanobu Tagawa
Takanobu Tagawa Wolfgang Hammerschmidt
Wolfgang Hammerschmidt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol. , 22 July 2022
Sec. Virology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.955603
Epstein-Barr virus (EBV) is a double-stranded DNA virus of the Herpesviridae family. This virus preferentially infects human primary B cells and persists in the human B cell compartment for a lifetime. Latent EBV infection can lead to the development of different types of lymphomas as well as carcinomas such as nasopharyngeal and gastric carcinoma in immunocompetent and immunocompromised patients. The early phase of viral infection is crucial for EBV to establish latency, but different viral components are sensed by cellular sensors called pattern recognition receptors (PRRs) as the first line of host defense. The efficacy of innate immunity, in particular the interferon-mediated response, is critical to control viral infection initially and to trigger a broad spectrum of specific adaptive immune responses against EBV later. Despite these restrictions, the virus has developed various strategies to evade the immune reaction of its host and to establish its lifelong latency. In its different phases of infection, EBV expresses up to 44 different viral miRNAs. Some act as viral immunoevasins because they have been shown to counteract innate as well as adaptive immune responses. Similarly, certain virally encoded proteins also control antiviral immunity. In this review, we discuss how the virus governs innate immune responses of its host and exploits them to its advantage.
Epstein-Barr virus (EBV), also referred to as human herpesvirus 4 (HHV-4) is a virus of the Gammaherpesvirinae subfamily with an encapsidated, linear, double-stranded DNA genome of approximately 160 kbs and an envelope of about 180 nm in diameter. More than 95% of the adult population worldwide is infected. Primary infection occurs mainly via saliva and is often asymptomatic in childhood, but is frequently associated with Infectious Mononucleosis (IM), if the first contact is delayed until adolescence or adulthood. IM patients have a higher risk of developing Multiple Sclerosis or malignant diseases later in life (Crawford, 2001; Ascherio and Munger, 2015). After infection, the virus establishes latency in mature B cells and persists in long-lived memory B cells. EBV is associated with certain B cell malignancies such as Hodgkin lymphoma, Burkitt’s lymphoma, and post-transplant lymphoproliferative disorder (PTLD) but the virus also causes nasopharyngeal carcinoma and gastric carcinoma in epithelial cells, which EBV can also infect. In immunocompromised individuals, EBV causes PTLD and the majority of acquired immunodeficiency syndrome (AIDS)-defining non-Hodgkin’s lymphoma (Yarchoan and Uldrick, 2018). EBV is a critical human tumor virus and a class 1 carcinogen (International Agency for Research on Cancer [IARC], 1997).
Epstein-Barr virus supports its lifelong persistence in us by minimizing the expression of viral proteins to limit viral detection and to escape from antiviral immunity. During EBV’s life cycle, three different latent phases are characterized by distinct viral gene expression profiles: latency 0/I [no viral proteins or only EBV nuclear antigen 1 (EBNA1)]; latency III [all latent viral proteins (6 EBNAs, 3 latent member proteins; LMPs)]; and latency II (a subset of latent viral proteins). During latency, up to 44 microRNAs (miRNAs) and two non-coding RNAs (EBERs) are also expressed. In in vitro infected human B cells, various lytic viral genes are expressed initially together with latent EBV genes in the so-called pre-latent phase of infection (Kalla et al., 2010; Kalla and Hammerschmidt, 2012), but lytic gene expression ceases when the incoming EBV DNA becomes epigenetically repressed within a few days.
During latent infection in resting cells, several copies of the EBV genome are stably maintained as fully chromatinized extrachromosomal plasmids which, in proliferating cells, replicate in synchrony with host cell DNA. To allow virus to spread to neighboring cells, reactivation occurs in B cells when they encounter their cognate antigen in vivo (Laichalk and Thorley-Lawson, 2005), but sporadic entry into EBV’s productive, lytic phase also occurs spontaneously in latently EBV infected cell lines in vitro. Lytic reactivation is initiated by the viral protein BZLF1, a member of the activator protein 1 (AP-1) family of transcription factors, which induces a massive synthesis of all viral proteins needed for the assembly and egress of virions (Buschle and Hammerschmidt, 2020).
The innate immune system is the frontline defense to protect the host from viral infection (Li and Wu, 2021). B cells and epithelial cells that EBV infects but also monocytes-derived macrophages (MDMs), dendritic cells (DCs), and plasmacytoid dendritic cells (pDCs) can sense virion components. These immune cells express a plethora of pattern recognition receptors, PRRs. Different PRRs recognize different viral components: Toll-like receptor 2 (TLR2) senses the viral dUTPAse in monocytes (Gaudreault et al., 2007; Ariza et al., 2009); in monocytes and pDCs and TLR9 senses the unmethylated CpG motifs of virion DNA (Fiola et al., 2010; Bouvet et al., 2021). The EBERs are recognized by TLR3, TLR7, and Retinoic acid-inducible gene I (RIG-I) in different types of established cell lines (Samanta et al., 2006; Iwakiri et al., 2009; Li et al., 2019) but not when EBV infects primary B cells (Bouvet et al., 2021), which is an unexpected finding. EBV is sensed by interferon gamma inducible protein 16 (IFI16) (Ansari et al., 2013) in epithelial cells in which EBV induces TRIM29, a negative regulator of STING (Xing et al., 2017) (stimulator of interferon genes) and an adaptor protein of cytosolic DNA sensors. A STING inhibitor suppressed B cell transformation by EBV (Miyagi et al., 2021) although B cells barely express STING (Mrozek-Gorska et al., 2019). Further research is needed to clarify its role in EBV infection.
Despite the known innate immune responses EBV establishes long-term latency in most of us, suggesting that it has evolved to escape from human immune recognition. EBV uses several viral gene products with immune evasive functions in different phases of the viral life cycle since many viral components are antigens and are recognized by the host. We summarize recent advancements in understanding the diverse classes of molecules that EBV uses to evade innate immune responses.
BPLF1 is a large (3149 aa) EBV protein expressed during EBV’s lytic phase and contained in the tegument of virions. BPLF1 stabilizes RAD18 ubiquitin complexes and acts as a deneddylase (removing the ubiquitin-like protein NEDD8) important for lytic viral DNA replication and production of infectious virions (Gastaldello et al., 2010; Kumar et al., 2014). Infection of humanized mice with a BPLF1-knockout virus showed reduced infectivity and delayed B cell transformation suggesting that BPLF1 contributes to but is not essential for the virus to spread in vivo (Whitehurst et al., 2015). The N-terminal region of the protein contains a deubiquitinating (DUB) domain, which interacts with proliferating cell nuclear antigen (PCNA) to reduce the cellular DNA damage response after lytic reactivation (Whitehurst et al., 2012). The DUB domain also reduces immune recognition during lytic activation by removing polyubiquitin chains from various components of TLR signaling, including NEMO, IKKα, and TRAF6 (Saito et al., 2013; van Gent et al., 2014). BPLF1 was further reported to interact with the 14-3-3 protein and the E3 ligase tripartite motif-containing protein 25 (TRIM25) mitigating type I interferon (IFN)-mediated responses by sequestering TRIM25 into a trimeric complex. Since RIG-I’s functions depend on ubiquitination by TRIM25, its inhibition causes the loss of the ubiquitin scaffold on RIG-I and affects successive downstream signaling cascades (Gupta et al., 2019). The decreased NF-κB activation in response to EBV reactivation reduces pro-inflammatory cytokine secretion levels. BPLF1 may also have an immune suppressive role immediately after B cell infection since BPLF1 contained in EBV’s infectious particles can control TLR signaling (van Gent et al., 2014) and BPLF1 is also transiently expressed during EBV’s pre-latent phase (Mrozek-Gorska et al., 2019).
EBV nuclear antigen 1 is a homo-dimeric protein expressed in all types of viral latency but latency 0. EBNA1 has crucial functions to maintain EBV genomes stably, to replicate viral DNA, and to partition the newly replicated EBV genomes to daughter cells. EBNA1 binds to the EBV origin of plasmid replication (oriP), which is critical for maintaining and replicating the viral genome and for its partitioning during mitosis (Chiu and Sugden, 2018). EBNA1’s amino-terminus binds cellular chromatin to localize EBV genomes to perichromatin and contains a glycine alanine repeat (GAr) domain with immune evasive function reducing presentation of EBNA1 epitopes to CD8+ cytotoxic T cells (Blake et al., 1997; Yin et al., 2003).
EBNA1’s role in regulating innate immunity is less clear. It interferes with functional NF-κB activation in different carcinoma cell lines (Valentine et al., 2010) and it can contribute to the development of EBV-associated tumors and their progression by activating AP-1 and signal transducer and activator of transcription 1 (STAT1) (O’Neil et al., 2008) and inhibiting TGF-β1 (Wood et al., 2007; Flavell et al., 2008). This is counterintuitive because STAT1 activation typically induces strong anti-viral IFN responses. EBV suppresses these “adverse effects” together with other immunoevasins such as EBNA2 or viral miRNAs. EBNA1 also induces CXCL12 secretion to recruit regulatory T cells and to create an immunosuppressive microenvironment (Huo et al., 2020). Finally, EBNA1 also regulates the expression of NKG2D ligands which reduces the recognition of EBV infected cells by NK cells shortly after infection (Westhoff Smith et al., 2021).
EBNA2 acts as the main transcriptional transactivator (Kempkes and Ling, 2015) and is essential for the reprogramming and transformation of primary B cells infected by EBV in vitro (Hammerschmidt and Sugden, 1989). EBNA2 induces a plethora of cellular genes including MYC (Kaiser et al., 1999) and genes coding for cell surface and adhesion molecules such as CD21 and CD23, but EBNA2 also activates multiple viral proteins including EBNA1, LMP1, and LMP2 (Kempkes and Ling, 2015). Regarding EBNA2’s role in innate immunity, it induces the expression of the IL-18 receptor (IL-18R) in primary B cells (Pagès et al., 2005). IL-18R may function differently depending on its ligands, the pro-inflammatory and anti-inflammatory cytokines IL-18 and IL-37, respectively. With IL-37, IL-18R can inhibit innate immune responses in general (Nold-Petry et al., 2015) but the contribution of the IL-18R pathway to EBV’s strategy of immune evasion needs to be assessed. EBNA2 was also shown to upregulate TNF-α and LT-α to support cell proliferation (Spender et al., 2001). EBNA2 also regulates host miRNAs hsa-miR-21 and miR-146a that act as negative feedback of type I IFN responses in the RIG-I pathway (Rosato et al., 2012). Already in the 1990s, EBNA2 was described to render the infected cells insensitive to IFN (Aman and von Gabain, 1990) and to suppress interferon stimulated genes (ISGs) but EBNA2 also induces low levels of IFN-β suggesting that chronic low levels of IFN may desensitize cells and dampen proper anti-viral responses (Kanda et al., 1992, 1999). In 293T and HeLa cells, EBNA2 activates STAT3, which could provide a growth advantage to EBV infected cells (Muromoto et al., 2009).
Epstein-Barr virus also manipulates innate immunity using viral IL-10 (vIL-10) encoded by the BCRF1 gene of EBV, a viral homolog of human IL-10 (Sin and Dittmer, 2012). vIL-10 is an anti-inflammatory cytokine that suppresses secretion of pro-inflammatory cytokines such as IFN-γ and IL-2 from EBV infected PBMCs (Jochum et al., 2012a). NK cells can sense and lyse EBV infected cells particularly during EBV’s lytic phase. CD4+ T cells can enhance NK cell activity, but vIL-10 suppresses this adjuvant T cell effect reducing the cytotoxic activity of NK cells (Jochum et al., 2012a). vIL-10 also controls adaptive immunity by suppressing TAP1, class I HLA, and co-stimulatory molecules such as ICAM-1 (Zeidler et al., 1997; Salek-Ardakani et al., 2002). Unlike hIL-10, vIL-10 does not induce class II MHC (Go et al., 1990) or promotes thymocyte proliferation (MacNeil et al., 1990) probably due to its lower receptor affinity compared to hIL-10 (Liu et al., 1997). In a recent study, vIL-10 reduced phosphorylation of STAT3 and vIL-10-treated monocytes showed weaker phagocytosis of apoptotic cells compared to hIL-10-treated cells (Jog et al., 2018). The impaired activity of monocytes to clear dying cells may lead to reduced antigen presentation indirectly supporting EBV infection in vivo. vIL-10 is not only a lytic gene but is markedly expressed in the pre-latent phase of B cell infection (Jochum et al., 2012a; Sin and Dittmer, 2012). Mice infected with a strain of murine gammaherpesvirus 76 engineered to encode EBV’s vIL-10 showed higher virus titers in the lung compared to wild-type virus but vIL-10 had no effect on virus reactivation or the number of latently infected splenic cells (Lindquester et al., 2014). Thus, vIL-10 can also contribute to a pro-viral microenvironment early after infection in this mouse model.
Latent membrane protein 1 contains 6 transmembrane domains and a C-terminal signaling domain and is located mainly in intracellular membranes (Lam and Sugden, 2003). LMP1 mimics a constitutive, i.e., ligand-independent CD40 receptor and its downstream signaling cascade (Kilger et al., 1998; Kieser and Sterz, 2015) contributing to survival and proliferation of EBV infected B cells (Wang et al., 1985; Zimber-Strobl et al., 1996; Kilger et al., 1998). LMP1 drives an innate anti-viral response but it also evokes an anti-viral state in infected cells. LMP1 induces interferon regulatory factor 7 (IRF7) via TRAF6 and RIP mediated ubiquitination (Zhang and Pagano, 2000; Huye et al., 2007; Ning et al., 2008) and activates the JAK/STAT pathway as well as both canonical and non-canonical NF-κB signaling pathways (Gires et al., 1999; Zhang L. et al., 2004; Huye et al., 2007). As a consequence, EBV infected cells release type I and II interferons leading to an upregulation of ISGs including ISG15, STAT1, and 2′,5′-oligoadenylate synthetase (OAS) (Richardson et al., 2003; Zhang J. et al., 2004; Najjar et al., 2005). Using LMP1 mutants, STAT1 regulation was shown to depend on both signaling domains of LMP1, CTAR1, and CTAR2 (Zhang L. et al., 2004). The authors suggested that this anti-viral response can be advantageous for the virus because it also supported latent infection in their experimental model (Zhang J. et al., 2004; Zhang L. et al., 2004). Of note, most of these reports rely on latently EBV infected, established B cell lines.
Recent reports identified additional immune evasive functions of LMP1. LMP1 downregulated TLR9 and reduced the recognition of non-CpG methylated viral DNA (Fathallah et al., 2010). Later, newly EBV infected B cells were found to be rather inert to sensing viral DNA (Bouvet et al., 2021), but plasmacytoid dendritic cells (pDCs) released high levels of type I IFN upon encounter with viral DNA and its detection by TLR9 (Bouvet et al., 2021). In 293T cells, LMP1 expression promoted RIG-I degradation (Xu et al., 2018), which might also dampen antiviral responses.
The viral LMP2 gene encodes two proteins, LMP2A and LMP2B, which differ in their N-terminal domains. Both LMP2A and LMP2B are integral transmembrane proteins embedded in the plasma membrane with their 12 transmembrane spanning domains. LMP2A’s N-terminal domain is cytosolic and contains two proline-rich PY domains as well as an immunoreceptor tyrosine-based activation motif (ITAM). The ITAM motif interacts with signal mediators of the B-cell receptor (BCR), mimics an active BCR (Engels et al., 2001a,b) and maintains a “tonic” B cell receptor-like signal (Mancao et al., 2005) and references therein. Concomitantly, LMP2A contributes to B cell survival as its expression supports viral latency and prevents acute BCR triggering (Brielmeier et al., 1996; Merchant et al., 2000; Mancao and Hammerschmidt, 2007). LMP2B has a short, 19 amino acid long N-terminal domain, only, which is incapable of signaling and was shown to form heterocomplexes with LMP2A regulating LMP2A’s activity (Lynch et al., 2002). LMP2A also controls adaptive immunity by downregulating CIITA, the master regulator of HLA class II alleles, reducing epitope presentation via HLA class II (Lin et al., 2015). LMP2A not only reduces immune recognition by CD8+ T cells (Rancan et al., 2015) but also innate immune responses in epithelial cell lines. IL-6 expression and secretion from carcinoma cell lines was suppressed by wild-type EBV but not by an LMP2A-deleted mutant EBV indicating that LMP2A shuts off this inflammatory cytokine (Stewart et al., 2004). LMP2A also downregulated LMP1, suggesting a fine-tuning of LMP1’s activities in EBV infected cells (Stewart et al., 2004) probably mediated by TRAF2 (Guasparri et al., 2008). Because LMP2A can induce the degradation of the receptors IFNAR1 and IFNGR1, LMP2A affects expression of ISGs and thus reduces type I and type II interferon responses in epithelial cells (Shah et al., 2009). Interestingly, LMP2B was also found to contribute to IFN receptor degradation suggesting that the N-terminal cytosolic tail of LMP2A, which LMP2B lacks is not important for this ubiquitin-dependent mechanism (Shah et al., 2009).
Epstein-Barr virus encodes 44 mature miRNAs which are important for cell survival and proliferation in the pre-latent phase of B cell infection (Seto et al., 2010; Feederle et al., 2011). Recently, many viral miRNAs were also found to govern innate and adaptive immune responses in vitro (Albanese et al., 2016; Tagawa et al., 2016; Bouvet et al., 2021) and in humanized mice in vivo (Murer et al., 2019). miRNAs regulate important proteins in different pathways of innate immunity. EBV miR-BART15 targets the NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome and EBV mir-BHRF1-2 was shown to regulate the IL-1 receptor preventing IL-1β release and downstream activation (Haneklaus et al., 2012; Skinner et al., 2017). EBV miR-BART2 targets MICB facilitating immune escape from Natural Killer (NK) cells (Nachmani et al., 2009). EBV miRNAs in primary B cells target key components of the interferon pathway reducing production and release of IFN from infected cells as well as autocrine and paracrine IFN mediated responses affecting ISG-driven transcription (Bouvet et al., 2021). Additional targets involved in reducing type I IFN response are CBP (CREB-binding protein) (Hooykaas et al., 2017) and RIG-I (Lu et al., 2017). Interestingly, pDCs, although they are only abortively infected, sense EBV’s genomic DNA in a TLR9 dependent manner but EBV’s miRNAs seem to counteract this innate response. The mode of action is not completely clear but could be mediated by miRNAs contained in virions (Jochum et al., 2012b; Bouvet et al., 2021). miRNAs are particularly interesting as immunoevasins. Viral proteins including those with anti-inflammatory functions are immunogenic and thus can stimulate adaptive immunity but viral miRNAs escape from immune recognition entirely.
In latently infected cells certain viral genes can control innate immunity, but lytic genes also undermine immune functions. IRF7 was shown to be regulated by the immediate early genes BZLF1 (Hahn et al., 2005) and BRLF1 (Bentz et al., 2010). Similarly, several lytic proteins, BGLF4 (Wang et al., 2009), BRLF1 (Bentz et al., 2010), BHRF1 (Vilmen et al., 2021), and BFRF1 (Wang et al., 2020) target IRF3. It and IRF7 are critical for type I IFN production and downstream ISG signaling, suggesting that the orchestrated suppression of both IRF3 and IRF7 is important in EBV’s lytic, productive phase. BGLF2 was identified to control all three types of IFN (α, β, ε) as it interferes with STAT1/STAT2 (Jangra et al., 2021) and tyrosine kinase 2 (TYK2) functions (Liu et al., 2020). Viral lytic genes also target PRRs. The BGLF5 exonuclease downregulates TLR2 (van Gent et al., 2015) and the dsDNA sensor TLR9 (van Gent et al., 2011). BLRF1 not only inhibits IRF3 but also suppresses transcription of RNA polymerase III-dependent immunogenic small RNAs dampening the RIG-I pathway (Long et al., 2021). Secreted BARF1 is a decoy receptor of macrophage colony stimulating factor (M-CSF) interfering with macrophage activation (Hoebe et al., 2012). Immune evasion appears to be particularly important during latent infection as compared to EBV’s lytic phase, when viral host shut-off mechanisms compromise cellular protein synthesis and thus immune functions (Rowe et al., 2007; Buschle et al., 2021). But, as in the case of BPLF1, viral genes that have been previously deemed lytic genes are now found to be expressed during EBV’s pre-latent phase of infection as well. These lytic immunoevasins are, therefore, potentially functional upon de novo infection.
The data discussed above and summarized in Table 1 and Figure 1 show that EBV has evolved to encode different classes of immune evasins, which contribute to successful infection and stable latency in its human host. Many factors have been found to play various but likely crucial roles in evading innate immunity and probably more factors and new principles will be discovered. Moreover, it appears difficult to assess the importance and contribution of each individual immunoevasin detected or claimed in the literature. Few findings have been independently confirmed or falsified. This uncertainty is partly due to the many different experimental systems and models used by different groups working in this field of virology and antiviral immunity.
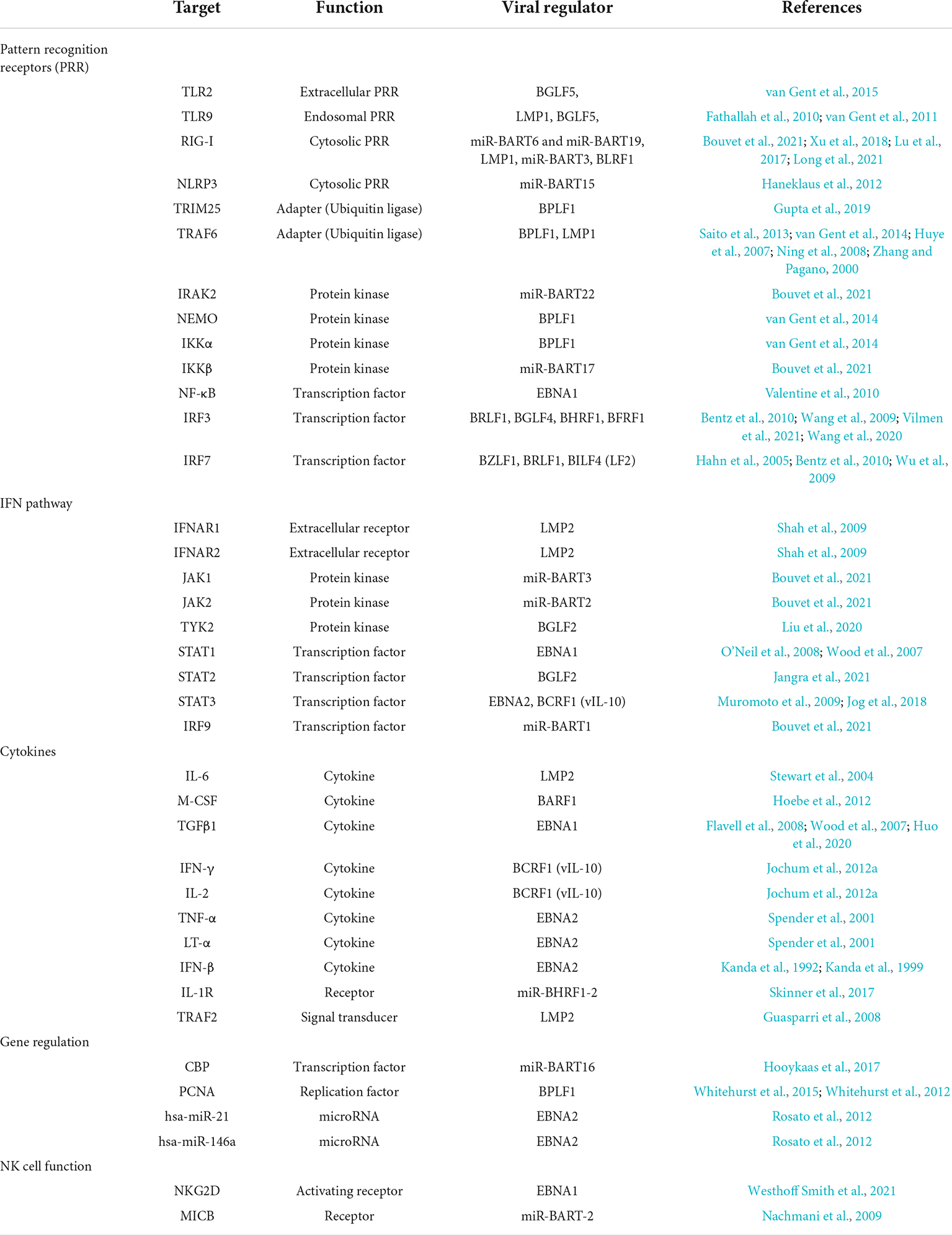
Table 1. Overview of cellular functions regulated by EBV proteins and miRNAs.
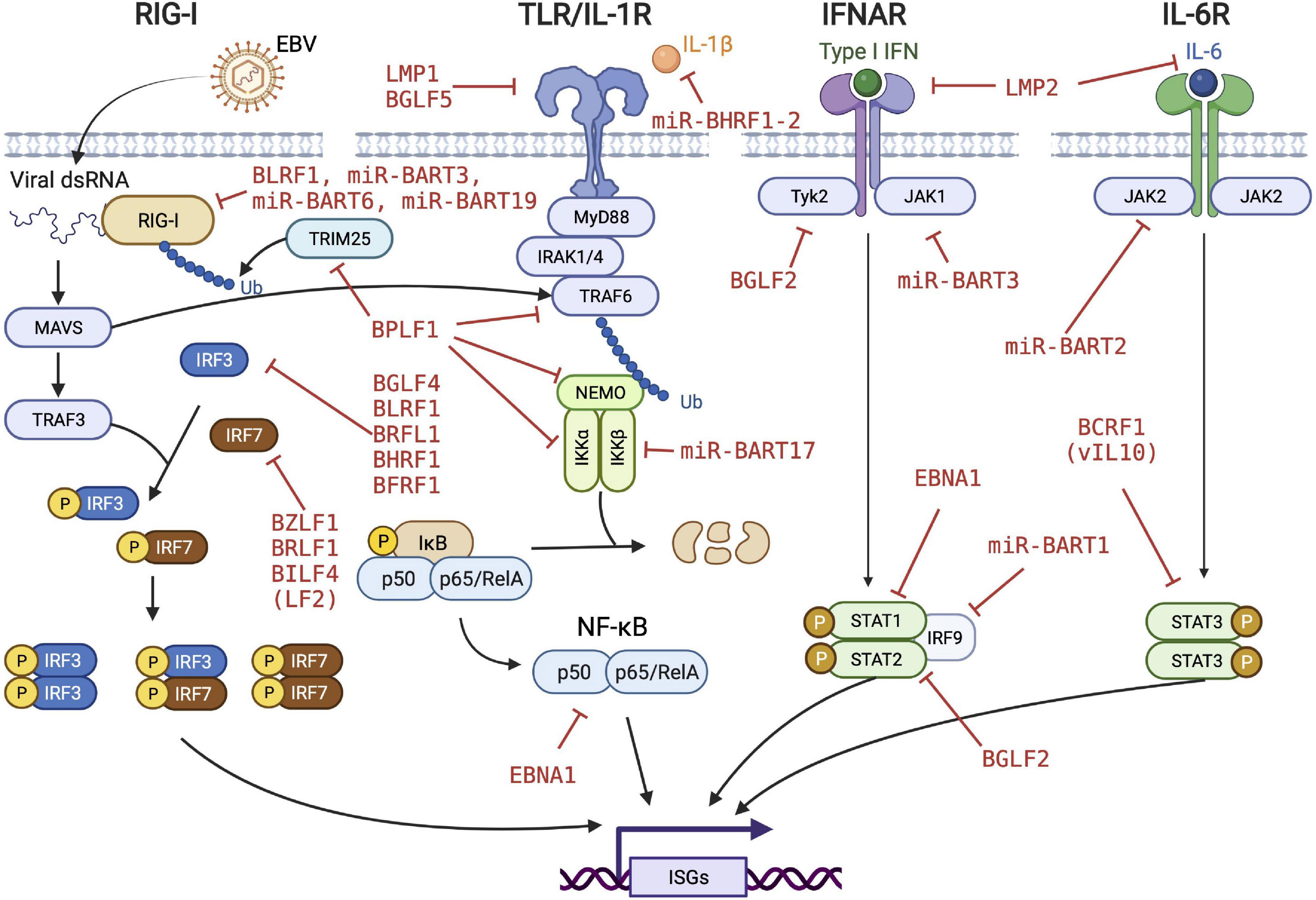
Figure 1. EBV’s strategy to evade innate immune responses. EBV evolved to encode many viral proteins and microRNAs to fend off different levels of innate immune responses of the infected cells. Viral immunoevasins and their regulatory functions are depicted in red. RIG-I, retinoic acid-inducible gene I; TLR, toll-like receptor; IFNAR, interferon-α/β receptor; MAVS, mitochondrial antiviral signaling protein; TRAF, TNF receptor associated factor; IRF, interferon regulatory factor; TRIM, tripartite motif containing; IRAK, interleukin receptor associated kinase; NEMO, nuclear factor-kappa B essential modulator; JAK, janus kinase; STAT, signal transducer and activator of transcription protein; ISG, interferon-stimulated gene. We acknowledge Biorender.com for graphical elements we used to compose the figure.
Use of models that closely mimic in vivo infection is a potential solution to this problem. We recently showed that primary human B cells constitute a valid model to assess the importance of candidates for immune evasion in the different phases of EBV infection. Using genetic EBV mutants that differ only in single genes are also supportive. For example, we found that the EBV gene BILF4 (LF2), which was shown to play a role in restricting innate immune response (Wu et al., 2009), did not show any reduction in IFN response in freshly infected B cells (Bouvet et al., 2021). This result suggested that data obtained from phenotypic studies in established cell lines are not necessarily relevant in models recapitulating critical steps in infection of EBV’s genuine target cells. A better understanding of the virus and its crucial factors that are responsible for successful infection will be important to identify potential therapeutic targets to prevent or cure EBV-associated diseases.
MA, TT, and WH conceptualized the topic. MA and TT researched and analyzed the literature, and drafted the manuscript including interpretations. WH analyzed the background literature, and wrote and finalized the manuscript together with MA and TT. All authors approved the final version of the manuscript, ensured the accuracy and integrity of the work, and agreed to be accountable for all aspects of the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank our colleagues in the EBV community and acknowledge their many publications which have been a valuable source while writing this mini review. We attempted to be as inclusive as we could but for size limitations we could not include all pertinent sources. We also apologize to authors whose contributions we have inadvertently missed.
Albanese, M., Tagawa, T., Bouvet, M., Maliqi, L., Lutter, D., Hoser, J., et al. (2016). Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. U. S. A. 113, E6467–E6475. doi: 10.1073/pnas.1605884113
Aman, P., and von Gabain, A. (1990). An Epstein-Barr virus immortalization associated gene segment interferes specifically with the IFN-induced anti-proliferative response in human B-lymphoid cell lines. EMBO J. 9, 147–152. doi: 10.1002/j.1460-2075.1990.tb08090.x
Ansari, M. A., Singh, V. V., Dutta, S., Veettil, M. V., Dutta, D., Chikoti, L., et al. (2013). Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I. II, and III in B and epithelial cells. J. Virol. 87, 8606–8623.
Ariza, M. E., Glaser, R., Kaumaya, P. T., Jones, C., and Williams, M. V. (2009). The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182, 851–859. doi: 10.4049/jimmunol.182.2.851
Ascherio, A., and Munger, K. L. (2015). EBV and autoimmunity. Curr. Top. Microbiol. Immunol. 390, 365–385.
Bentz, G. L., Liu, R., Hahn, A. M., Shackelford, J., and Pagano, J. S. (2010). Epstein-Barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-beta. Virology 402, 121–128. doi: 10.1016/j.virol.2010.03.014
Blake, N., Lee, S., Redchenko, I., Thomas, W., Steven, N., Leese, A., et al. (1997). Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity 7, 791–802. doi: 10.1016/s1074-7613(00)80397-0
Bouvet, M., Voigt, S., Tagawa, T., Albanese, M., Chen, Y. A., Chen, Y., et al. (2021). Multiple viral microRNAs regulate interferon release and signaling early during infection with Epstein-Barr virus. mBio 12:e3440. doi: 10.1128/mBio.03440-20
Brielmeier, M., Mautner, J., Laux, G., and Hammerschmidt, W. (1996). The latent membrane protein 2 gene of Epstein-Barr virus is important for efficient B cell immortalization. J. Gen. Virol. 77, 2807–2818.
Buschle, A., and Hammerschmidt, W. (2020). Epigenetic lifestyle of Epstein-Barr virus. Semin. Immunopathol. 42, 131–142.
Buschle, A., Mrozek-Gorska, P., Cernilogar, F. M., Ettinger, A., Pich, D., Krebs, S., et al. (2021). Epstein-Barr virus inactivates the transcriptome and disrupts the chromatin architecture of its host cell in the first phase of lytic reactivation. Nucleic Acids Res. 49, 3217–3241. doi: 10.1093/nar/gkab099
Chiu, Y. F., and Sugden, B. (2018). Plasmid partitioning by human tumor viruses. J. Virol. 92, e02170–17.
Crawford, D. H. (2001). Biology and disease associations of Epstein-Barr virus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 461–473.
Engels, N., Merchant, M., Pappu, R., Chan, A. C., Longnecker, R., and Wienands, J. (2001a). Epstein-Barr virus latent membrane protein 2A (LMP2A) employs the SLP-65 signaling module. J. Exp. Med. 194, 255–264. doi: 10.1084/jem.194.3.255
Engels, N., Wollscheid, B., and Wienands, J. (2001b). Association of SLP-65/BLNK with the B cell antigen receptor through a non-ITAM tyrosine of Ig-alpha. Eur. J. Immunol. 31, 2126–2134. doi: 10.1002/1521-4141(200107)31:7<2126::aid-immu2126>3.0.co;2-o
Fathallah, I., Parroche, P., Gruffat, H., Zannetti, C., Johansson, H., Yue, J., et al. (2010). EBV latent membrane protein 1 is a negative regulator of TLR9. J. Immunol. 185, 6439–6447. doi: 10.4049/jimmunol.0903459
Feederle, R., Linnstaedt, S. D., Bannert, H., Lips, H., Bencun, M., Cullen, B. R., et al. (2011). A viral microRNA cluster strongly potentiates the transforming properties of a human herpesvirus. PLoS Pathog. 7:e1001294. doi: 10.1371/journal.ppat.1001294
Fiola, S., Gosselin, D., Takada, K., and Gosselin, J. (2010). TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 185, 3620–3631. doi: 10.4049/jimmunol.0903736
Flavell, J. R., Baumforth, K. R., Wood, V. H., Davies, G. L., Wei, W., Reynolds, G. M., et al. (2008). Down-regulation of the TGF-beta target gene. PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood 111, 292–301. doi: 10.1182/blood-2006-11-059881
Gastaldello, S., Hildebrand, S., Faridani, O., Callegari, S., Palmkvist, M., Di Guglielmo, C., et al. (2010). A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat. Cell Biol. 12, 351–361.
Gaudreault, E., Fiola, S., Olivier, M., and Gosselin, J. (2007). Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 81, 8016–8024.
Gires, O., Kohlhuber, F., Kilger, E., Baumann, M., Kieser, A., Kaiser, C., et al. (1999). Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 18, 3064–3073.
Go, N. F., Castle, B. E., Barrett, R., Kastelein, R., Dang, W., Mosmann, T. R., et al. (1990). Interleukin 10, a novel B cell stimulatory factor: unresponsiveness of X chromosome-linked immunodeficiency B cells. J. Exp. Med. 172, 1625–1631. doi: 10.1084/jem.172.6.1625
Guasparri, I., Bubman, D., and Cesarman, E. E. B. V. (2008). LMP2A affects LMP1-mediated NF-kappaB signaling and survival of lymphoma cells by regulating TRAF2 expression. Blood 111, 3813–3820. doi: 10.1182/blood-2007-03-080309
Gupta, S., Ylä-Anttila, P., Sandalova, T., Sun, R., Achour, A., and Masucci, M. G. (2019). 14-3-3 scaffold proteins mediate the inactivation of trim25 and inhibition of the type I interferon response by herpesvirus deconjugases. PLoS Pathog. 15:e1008146. doi: 10.1371/journal.ppat.1008146
Hahn, A. M., Huye, L. E., Ning, S., Webster-Cyriaque, J., and Pagano, J. S. (2005). Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene. BZLF-1. J. Virol. 79, 10040–10052. doi: 10.1128/JVI.79.15.10040-10052.2005
Hammerschmidt, W., and Sugden, B. (1989). Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 340, 393–397. doi: 10.1038/340393a0
Haneklaus, M., Gerlic, M., Kurowska-Stolarska, M., Rainey, A. A., Pich, D., McInnes, I. B., et al. (2012). Cutting Edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J. Immunol. 189, 3795–3799. doi: 10.4049/jimmunol.1200312
Hoebe, E. K., Le Large, T. Y., Tarbouriech, N., Oosterhoff, D., De Gruijl, T. D., Middeldorp, J. M., et al. (2012). Epstein-Barr virus-encoded BARF1 protein is a decoy receptor for macrophage colony stimulating factor and interferes with macrophage differentiation and activation. Viral Immunol. 25, 461–470. doi: 10.1089/vim.2012.0034
Hooykaas, M. J. G., van Gent, M., Soppe, J. A., Kruse, E., Boer, I. G. J., van Leenen, D., et al. (2017). EBV microRNA BART16 suppresses type I IFN signaling. J. Immunol. 198, 4062–4073. doi: 10.4049/jimmunol.1501605
Huo, S., Luo, Y., Deng, R., Liu, X., Wang, J., Wang, L., et al. (2020). EBNA1 constructs an immunosuppressive microenvironment for nasopharyngeal carcinoma by promoting the chemoattraction of Treg cells. J. Immunother. Cancer 8:e001588. doi: 10.1136/jitc-2020-001588
Huye, L. E., Ning, S., Kelliher, M., and Pagano, J. S. (2007). Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol. Cell Biol. 27, 2910–2918. doi: 10.1128/MCB.02256-06
International Agency for Research on Cancer [IARC] (1997). Epstein-Barr Virus; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Lyon: IARC.
Iwakiri, D., Zhou, L., Samanta, M., Matsumoto, M., Ebihara, T., Seya, T., et al. (2009). Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 206, 2091–2099. doi: 10.1084/jem.20081761
Jangra, S., Bharti, A., Lui, W. Y., Chaudhary, V., Botelho, M. G., Yuen, K. S., et al. (2021). Suppression of JAK-STAT signaling by Epstein-Barr virus tegument protein BGLF2 through recruitment of SHP1 phosphatase and promotion of STAT2 degradation. J. Virol. 95:e0102721. doi: 10.1128/JVI.01027-21
Jochum, S., Moosmann, A., Lang, S., Hammerschmidt, W., and Zeidler, R. (2012a). The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 8:e1002704. doi: 10.1371/journal.ppat.1002704
Jochum, S., Ruiss, R., Moosmann, A., Hammerschmidt, W., and Zeidler, R. (2012b). RNAs in Epstein-Barr virions control early steps of infection. Proc. Natl. Acad. Sci. U. S. A. 109, E1396–E1404. doi: 10.1073/pnas.1115906109
Jog, N. R., Chakravarty, E. F., Guthridge, J. M., and James, J. A. (2018). Epstein-Barr virus interleukin 10 suppresses anti-inflammatory phenotype in human monocytes. Front. Immunol. 9:2198. doi: 10.3389/fimmu.2018.02198
Kaiser, C., Laux, G., Eick, D., Jochner, N., Bornkamm, G. W., and Kempkes, B. (1999). The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol. 73, 4481–4484. doi: 10.1128/JVI.73.5.4481-4484.1999
Kalla, M., and Hammerschmidt, W. (2012). Human B cells on their route to latent infection–early but transient expression of lytic genes of Epstein-Barr virus. Eur. J. Cell Biol. 91, 65–69. doi: 10.1016/j.ejcb.2011.01.014
Kalla, M., Schmeinck, A., Bergbauer, M., Pich, D., and Hammerschmidt, W. (2010). AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. U. S. A. 107, 850–855. doi: 10.1073/pnas.0911948107
Kanda, K., Decker, T., Aman, P., Wahlström, M., von Gabain, A., and Kallin, B. (1992). The EBNA2-related resistance towards alpha interferon (IFN-alpha) in Burkitt’s lymphoma cells effects induction of IFN-induced genes but not the activation of transcription factor ISGF-3. Mol. Cell Biol. 12, 4930–4936. doi: 10.1128/mcb.12.11.4930-4936.1992
Kanda, K., Kempkes, B., Bornkamm, G. W., von Gabain, A., and Decker, T. (1999). The Epstein-Barr virus nuclear antigen 2 (EBNA2), a protein required for B lymphocyte immortalization, induces the synthesis of type I interferon in Burkitt’s lymphoma cell lines. Biol. Chem. 380, 213–221. doi: 10.1515/BC.1999.029
Kempkes, B., and Ling, P. D. (2015). EBNA2 and Its coactivator EBNA-LP. Curr. Top. Microbiol. Immunol. 391, 35–59.
Kieser, A., and Sterz, K. R. (2015). The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 391, 119–149.
Kilger, E., Kieser, A., Baumann, M., and Hammerschmidt, W. (1998). Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 17, 1700–1709. doi: 10.1093/emboj/17.6.1700
Kumar, R., Whitehurst, C. B., and Pagano, J. S. (2014). The Rad6/18 ubiquitin complex interacts with the Epstein-Barr virus deubiquitinating enzyme. BPLF1, and contributes to virus infectivity. J. Virol. 88, 6411–6422. doi: 10.1128/JVI.00536-14
Laichalk, L. L., and Thorley-Lawson, D. A. (2005). Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79, 1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005
Lam, N., and Sugden, B. (2003). CD40 and its viral mimic. LMP1: similar means to different ends. Cell Signal. 15, 9–16. doi: 10.1016/s0898-6568(02)00083-9
Li, D., and Wu, M. (2021). Pattern recognition receptors in health and diseases. Signal. Transduct. Target Ther. 6:291.
Li, Z., Tsai, M. H., Shumilov, A., Baccianti, F., Tsao, S. W., Poirey, R., et al. (2019). Epstein-Barr virus ncRNA from a nasopharyngeal carcinoma induces an inflammatory response that promotes virus production. Nat. Microbiol. 4, 2475–2486. doi: 10.1038/s41564-019-0546-y
Lin, J. H., Lin, J. Y., Chou, Y. C., Chen, M. R., Yeh, T. H., Lin, C. W., et al. (2015). Epstein-Barr virus LMP2A suppresses MHC class II expression by regulating the B-cell transcription factors E47 and PU.1. Blood 125, 2228–2238. doi: 10.1182/blood-2014-08-594689
Lindquester, G. J., Greer, K. A., Stewart, J. P., and Sample, J. T. (2014). Epstein-Barr virus IL-10 gene expression by a recombinant murine gammaherpesvirus in vivo enhances acute pathogenicity but does not affect latency or reactivation. Herpesviridae 5:1.
Liu, X., Sadaoka, T., Krogmann, T., and Cohen, J. I. (2020). Epstein-Barr Virus (EBV) tegument protein BGLF2 suppresses type I interferon signaling to promote EBV reactivation. J. Virol. 94, e00258–20. doi: 10.1128/JVI.00258-20
Liu, Y., de Waal Malefyt, R., Briere, F., Parham, C., Bridon, J. M., Banchereau, J., et al. (1997). The EBV IL-10 homologue is a selective agonist with impaired binding to the IL-10 receptor. J. Immunol. 158, 604–613.
Long, X., Yang, J., Zhang, X., Yang, Z., Li, Y., Wang, F., et al. (2021). BRLF1 suppresses RNA Pol III-mediated RIG-I inflammasome activation in the early EBV lytic lifecycle. EMBO Rep. 22:e50714. doi: 10.15252/embr.202050714
Lu, Y., Qin, Z., Wang, J., Zheng, X., Lu, J., Zhang, X., et al. (2017). Epstein-Barr virus miR-BART6-3p inhibits the RIG-I pathway. J. Innate Immun. 9, 574–586. doi: 10.1159/000479749
Lynch, D. T., Zimmerman, J. S., and Rowe, D. T. (2002). Epstein-Barr virus latent membrane protein 2B (LMP2B) co-localizes with LMP2A in perinuclear regions in transiently transfected cells. J. Gen. Virol. 83, 1025–1035. doi: 10.1099/0022-1317-83-5-1025
MacNeil, I. A., Suda, T., Moore, K. W., Mosmann, T. R., and Zlotnik, A. (1990). IL-10, a novel growth cofactor for mature and immature T cells. J. Immunol. 145, 4167–4173.
Mancao, C., Altmann, M., Jungnickel, B., and Hammerschmidt, W. (2005). Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 106, 4339–4344.
Mancao, C., and Hammerschmidt, W. (2007). Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 110, 3715–3721. doi: 10.1182/blood-2007-05-090142
Merchant, M., Caldwell, R. G., and Longnecker, R. (2000). The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. J. Virol. 74, 9115–9124. doi: 10.1128/jvi.74.19.9115-9124.2000
Miyagi, S., Watanabe, T., Hara, Y., Arata, M., Uddin, M. K., Mantoku, K., et al. (2021). A sting inhibitor suppresses EBV-induced B cell transformation and lymphomagenesis. Cancer Sci. 112, 5088–5099. doi: 10.1111/cas.15152
Mrozek-Gorska, P., Buschle, A., Pich, D., Schwarzmayr, T., Fechtner, R., Scialdone, A., et al. (2019). Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. U. S. A. 116, 16046–16055. doi: 10.1073/pnas.1901314116
Murer, A., Rühl, J., Zbinden, A., Capaul, R., Hammerschmidt, W., Chijioke, O., et al. (2019). MicroRNAs of Epstein-Barr virus attenuate T-cell-mediated immune control In vivo. mBio 10, e01941–18.
Muromoto, R., Ikeda, O., Okabe, K., Togi, S., Kamitani, S., Fujimuro, M., et al. (2009). Epstein-Barr virus-derived EBNA2 regulates STAT3 activation. Biochem. Biophys. Res. Commun. 378, 439–443.
Nachmani, D., Stern-Ginossar, N., Sarid, R., and Mandelboim, O. (2009). Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5, 376–385. doi: 10.1016/j.chom.2009.03.003
Najjar, I., Baran-Marszak, F., Le Clorennec, C., Laguillier, C., Schischmanoff, O., Youlyouz-Marfak, I., et al. (2005). Latent membrane protein 1 regulates STAT1 through NF-kappaB-dependent interferon secretion in Epstein-Barr virus-immortalized B cells. J. Virol. 79, 4936–4943. doi: 10.1128/JVI.79.8.4936-4943.2005
Ning, S., Campos, A. D., Darnay, B. G., Bentz, G. L., and Pagano, J. S. (2008). TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell Biol. 28, 6536–6546. doi: 10.1128/MCB.00785-08
Nold-Petry, C. A., Lo, C. Y., Rudloff, I., Elgass, K. D., Li, S., Gantier, M. P., et al. (2015). IL-37 requires the receptors IL-18Rα and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 16, 354–365. doi: 10.1038/ni.3103
O’Neil, J. D., Owen, T. J., Wood, V. H. J., Date, K. L., Valentine, R., Chukwuma, M. B., et al. (2008). Epstein-Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J. Gen. Virol. 89, 2833–2842. doi: 10.1099/vir.0.2008/003392-0
Pagès, F., Galon, J., Karaschuk, G., Dudziak, D., Camus, M., Lazar, V., et al. (2005). Epstein-Barr virus nuclear antigen 2 induces interleukin-18 receptor expression in B cells. Blood 105, 1632–1639. doi: 10.1182/blood-2004-08-3196
Rancan, C., Schirrmann, L., Hüls, C., Zeidler, R., and Moosmann, A. (2015). Latent membrane protein LMP2A impairs recognition of EBV-infected cells by CD8+ T cells. PLoS Pathog. 11:e1004906. doi: 10.1371/journal.ppat.1004906
Richardson, C., Fielding, C., Rowe, M., and Brennan, P. (2003). Epstein-Barr virus regulates STAT1 through latent membrane protein 1. J. Virol. 77, 4439–4443.
Rosato, P., Anastasiadou, E., Garg, N., Lenze, D., Boccellato, F., Vincenti, S., et al. (2012). Differential regulation of miR-21 and miR-146a by Epstein-Barr virus-encoded EBNA2. Leukemia 26, 2343–2352. doi: 10.1038/leu.2012.108
Rowe, M., Glaunsinger, B., van Leeuwen, D., Zuo, J., Sweetman, D., Ganem, D., et al. (2007). Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. U. S. A. 104, 3366–3371. doi: 10.1073/pnas.0611128104
Saito, S., Murata, T., Kanda, T., Isomura, H., Narita, Y., Sugimoto, A., et al. (2013). Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-κB signaling during productive replication. J. Virol. 87, 4060–4070. doi: 10.1128/JVI.02020-12
Salek-Ardakani, S., Arrand, J. R., and Mackett, M. (2002). Epstein-Barr virus encoded interleukin-10 inhibits HLA-class I. ICAM-1, and B7 expression on human monocytes: implications for immune evasion by EBV. Virology 304, 342–351. doi: 10.1006/viro.2002.1716
Samanta, M., Iwakiri, D., Kanda, T., Imaizumi, T., and Takada, K. (2006). EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 25, 4207–4214. doi: 10.1038/sj.emboj.7601314
Seto, E., Moosmann, A., Gromminger, S., Walz, N., Grundhoff, A., and Hammerschmidt, W. (2010). Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 6:e1001063. doi: 10.1371/journal.ppat.1001063
Shah, K. M., Stewart, S. E., Wei, W., Woodman, C. B., O’Neil, J. D., Dawson, C. W., et al. (2009). The EBV-encoded latent membrane proteins. LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 28, 3903–3914. doi: 10.1038/onc.2009.249
Sin, S. H., and Dittmer, D. P. (2012). Cytokine homologs of human gammaherpesviruses. J. Interferon Cytokine Res. 32, 53–59.
Skinner, C. M., Ivanov, N. S., Barr, S. A., Chen, Y., and Skalsky, R. L. (2017). An Epstein-Barr virus microRNA blocks interleukin-1 (IL-1) signaling by targeting IL-1 receptor 1. J. Virol. 91, e00530–17. doi: 10.1128/JVI.00530-17
Spender, L. C., Cornish, G. H., Rowland, B., Kempkes, B., and Farrell, P. J. (2001). Direct and indirect regulation of cytokine and cell cycle proteins by EBNA-2 during Epstein-Barr virus infection. J. Virol. 75, 3537–3546. doi: 10.1128/JVI.75.8.3537-3546.2001
Stewart, S., Dawson, C. W., Takada, K., Curnow, J., Moody, C. A., Sixbey, J. W., et al. (2004). Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proc. Natl. Acad. Sci. U. S. A. 101, 15730–15735. doi: 10.1073/pnas.0402135101
Tagawa, T., Albanese, M., Bouvet, M., Moosmann, A., Mautner, J., Heissmeyer, V., et al. (2016). Epstein-Barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J. Exp. Med. 213, 2065–2080. doi: 10.1084/jem.20160248
Valentine, R., Dawson, C. W., Hu, C., Shah, K. M., Owen, T. J., Date, K. L., et al. (2010). Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol. Cancer 9:1. doi: 10.1186/1476-4598-9-1
van Gent, M., Braem, S. G., de Jong, A., Delagic, N., Peeters, J. G., Boer, I. G., et al. (2014). Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 10:e1003960. doi: 10.1371/journal.ppat.1003960
van Gent, M., Gram, A. M., Boer, I. G. J., Geerdink, R. J., Lindenbergh, M. F. S., Lebbink, R. J., et al. (2015). Silencing the shutoff protein of Epstein-Barr virus in productively infected B cells points to (innate) targets for immune evasion. J. Gen. Virol. 96, 858–865. doi: 10.1099/jgv.0.000021
van Gent, M., Griffin, B. D., Berkhoff, E. G., van Leeuwen, D., Boer, I. G., Buisson, M., et al. (2011). EBV lytic-phase protein BGLF5 contributes to TLR9 downregulation during productive infection. J. Immunol. 186, 1694–1702. doi: 10.4049/jimmunol.0903120
Vilmen, G., Glon, D., Siracusano, G., Lussignol, M., Shao, Z., Hernandez, E., et al. (2021). BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy 17, 1296–1315. doi: 10.1080/15548627.2020.1758416
Wang, D., Liebowitz, D., and Kieff, E. (1985). An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 43, 831–840.
Wang, J. T., Doong, S. L., Teng, S. C., Lee, C. P., Tsai, C. H., and Chen, M. R. (2009). Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 83, 1856–1869. doi: 10.1128/JVI.01099-08
Wang, P., Deng, Y., Guo, Y., Xu, Z., Li, Y., Ou, X., et al. (2020). Epstein-Barr virus early protein BFRF1 suppresses IFN-β activity by inhibiting the activation of IRF3. Front. Immunol. 11:513383. doi: 10.3389/fimmu.2020.513383
Westhoff Smith, D., Chakravorty, A., Hayes, M., Hammerschmidt, W., and Sugden, B. (2021). The Epstein-Barr virus oncogene EBNA1 suppresses Natural Killer cell responses and apoptosis early after infection of peripheral B cells. mBio 12:e0224321. doi: 10.1128/mBio.02243-21
Whitehurst, C. B., Li, G., Montgomery, S. A., Montgomery, N. D., Su, L., and Pagano, J. S. (2015). Knockout of Epstein-Barr virus BPLF1 retards B-cell transformation and lymphoma formation in humanized mice. mBio 6:e01574–15. doi: 10.1128/mBio.01574-15
Whitehurst, C. B., Vaziri, C., Shackelford, J., and Pagano, J. S. (2012). Epstein-Barr virus BPLF1 deubiquitinates PCNA and attenuates polymerase η recruitment to DNA damage sites. J. Virol. 86, 8097–8106. doi: 10.1128/JVI.00588-12
Wood, V. H., O’Neil, J. D., Wei, W., Stewart, S. E., Dawson, C. W., and Young, L. S. (2007). Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFbeta signaling pathways. Oncogene 26, 4135–4147. doi: 10.1038/sj.onc.1210496
Wu, L., Fossum, E., Joo, C. H., Inn, K. S., Shin, Y. C., Johannsen, E., et al. (2009). Epstein-Barr virus LF2: an antagonist to type I interferon. J. Virol. 83, 1140–1146. doi: 10.1128/JVI.00602-08
Xing, J., Zhang, A., Zhang, H., Wang, J., Li, X. C., Zeng, M. S., et al. (2017). TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 8:945. doi: 10.1038/s41467-017-00101-w
Xu, C., Sun, L., Liu, W., and Duan, Z. (2018). Latent Membrane Protein 1 of Epstein-Barr virus promotes RIG-I degradation mediated by proteasome pathway. Front. Immunol. 9:1446. doi: 10.3389/fimmu.2018.01446
Yarchoan, R., and Uldrick, T. S. (2018). HIV-associated cancers and related diseases. N. Engl. J. Med. 378, 1029–1041.
Yin, Y., Manoury, B., and Fåhraeus, R. (2003). Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 301, 1371–1374.
Zeidler, R., Eissner, G., Meissner, P., Uebel, S., Tampe, R., Lazis, S., et al. (1997). Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 90, 2390–2397.
Zhang, J., Das, S. C., Kotalik, C., Pattnaik, A. K., and Zhang, L. (2004). The latent membrane protein 1 of Epstein-Barr virus establishes an antiviral state via induction of interferon-stimulated genes. J. Biol. Chem. 279, 46335–46342. doi: 10.1074/jbc.M403966200
Zhang, L., and Pagano, J. S. (2000). Interferon regulatory factor 7 is induced by Epstein-Barr virus latent membrane protein 1. J. Virol. 74, 1061–1068.
Zhang, L., Hong, K., Zhang, J., and Pagano, J. S. (2004). Multiple signal transducers and activators of transcription are induced by EBV LMP-1. Virology 323, 141–152.
Keywords: herpesvirus, interferon, microRNA, immune evasion, natural killer cell, antiviral response, T cell, B cell
Citation: Albanese M, Tagawa T and Hammerschmidt W (2022) Strategies of Epstein-Barr virus to evade innate antiviral immunity of its human host. Front. Microbiol. 13:955603. doi: 10.3389/fmicb.2022.955603
Received: 28 May 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Mei-Ru Chen, National Taiwan University, TaiwanReviewed by:
Paul M. Lieberman, Wistar Institute, United StatesCopyright © 2022 Albanese, Tagawa and Hammerschmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Hammerschmidt, aGFtbWVyc2NobWlkdEBoZWxtaG9sdHotbXVlbmNoZW4uZGU=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.