 David Gerlach1,2,3*†
David Gerlach1,2,3*† Raphael N. Sieber4
Raphael N. Sieber4 Jesper Larsen4
Jesper Larsen4 Janes Krusche1,2,3
Janes Krusche1,2,3 Cristina De Castro5
Cristina De Castro5 Juliane Baumann1,2,3
Juliane Baumann1,2,3 Antonio Molinaro5
Antonio Molinaro5 Andreas Peschel1,2,3*
Andreas Peschel1,2,3*- 1Interfaculty Institute of Microbiology and Infection Medicine, Infection Biology Section, University of Tübingen, Tübingen, Germany
- 2German Center for Infection Research (DZIF), Partner Site Tübingen, Tübingen, Germany
- 3Cluster of Excellence EXC2124 Controlling Microbes to Fight Infection, University of Tübingen, Tübingen, Germany
- 4Statens Serum Institut, Copenhagen, Denmark
- 5Department of Chemical Sciences, University of Naples, Naples, Italy
Methicillin-resistant Staphylococcus aureus (MRSA), a major human pathogen, uses the prophage-encoded tarP gene as an important immune evasion factor. TarP glycosylates wall teichoic acid (WTA) polymers, major S. aureus surface antigens, to impair WTA immunogenicity and impede host defence. However, tarP phages appear to be restricted to only a few MRSA clonal lineages, including clonal complexes (CC) 5 and 398, for unknown reasons. We demonstrate here that tarP-encoding prophages can be mobilized to lysogenize other S. aureus strains. However, transfer is largely restricted to closely related clones. Most of the non-transducible clones encode tarM, which generates a WTA glycosylation pattern distinct from that mediated by TarP. However, tarM does not interfere with infection by tarP phages. Clonal complex-specific Type I restriction-modification systems were the major reasons for resistance to tarP phage infection. Nevertheless, tarP phages were found also in unrelated S. aureus clones indicating that tarP has the potential to spread to distant clonal lineages and contribute to the evolution of new MRSA clones.
Introduction
In order to maintain and optimize its role as a major human pathogen, Staphylococcus aureus relies on horizontal gene transfer (HGT), by which virulence factors and resistance genes are exchanged to create new clonal lineages with increased virulence, resistance, or dissemination capacities (Lindsay, 2010) or with new host specificities (Oliveira et al., 2014; Sheppard et al., 2018). Bacteriophages, bacterial viruses, are the crucial vehicles of HGT in S. aureus (Xia and Wolz, 2014). Siphophages often form prophages, which can integrate into host bacterial chromosomes to contribute up to 20% of the bacterial genomic content. Some of these phages contain genes and gene clusters that interfere with host immune function, including the immune evasion cluster 1 (IEC-1, from now on referred to as “IEC”) that enables S. aureus to evade the human innate immune response (Richardson et al., 2018) and the tarP gene, which impairs the immunogenicity of wall teichoic acid (WTA), a major S. aureus surface antigen (Gerlach et al., 2018). Moreover, phages can mobilize resistance determinants such as the mecA gene of methicillin-resistant S. aureus (MRSA) (Maslanova et al., 2013; Scharn et al., 2013).
Phages use the species and clone-specific structure of WTA to bind to and infect appropriate bacterial host cells (Winstel et al., 2013). In most S. aureus, WTA is a surface glycopolymer consisting of polyribitol phosphate (poly-RboP) repeats that are linked covalently to the peptidoglycan via a linkage unit (Weidenmaier and Peschel, 2008; Brown et al., 2013). RboP-WTA is modified with N-acetylglucosamine (GlcNAc) residues, which can be linked in different conformations, thereby governing the susceptibility of S. aureus to specific phage groups and the immunogenicity of WTA (Winstel et al., 2014; Gerlach et al., 2018; Ingmer et al., 2019). Most S. aureus isolates encode the housekeeping GlcNAc transferase (GT) tarS, which confers a β-1,4-GlcNAc-RboP glycosylation pattern (Brown et al., 2012). Some S. aureus isolates express another genome-encoded GT, TarM, which attaches α-1,4-GlcNAc residues to RboP (Xia et al., 2010). Like tarS, tarM does not appear to be associated with either phages or other mobile genetic elements (MGEs) (Li et al., 2015). In contrast, the phage-encoded GT TarP uses the same substrate as TarS and TarM (UDP-GlcNAc) but attaches GlcNAc to RboP with a β-1,3 configuration (Gerlach et al., 2018). We previously demonstrated that tarP is prevalent in some of the major hospital-associated (HA), MRSA sequence types (STs), 5 and 225, both of which belong to clonal complex (CC) 5 and the major livestock-associated (LA) lineage CC398, contributing to the immune evasion capacities of these clones (Gerlach et al., 2018). It has remained unclear though why tarP is absent from many other MRSA clones and whether it could potentially spread to give rise to new pathogen lineages.
Horizontal transfer of phages and other MGEs is not only restricted by WTA modification, but also by Type I restriction-modification (RM) (Oliveira et al., 2014, 2016). The importance of Type I RM systems is reflected by their high abundance in almost all S. aureus genomes along with clone-specific conservation but high inter-clonal diversity of specific RM types (Sadykov, 2016). Three different host specificity determinants make up Type I RM systems: the nuclease HsdR, designated as SauI in S. aureus (Waldron and Lindsay, 2006); the DNA methyltransferase HsdM; and HsdS that confers specificity via two DNA target recognition domains to HsdR, and HsdM in a clonal complex-specific manner (Wilson and Murray, 1991).
Here, we comprehensively analyzed all genomes of S. aureus deposited to public databases for the presence of tarP and its association with tarS and tarM. We investigated genetic diversity of tarP phages and their transmission dynamics between different hosts and demonstrate that its horizontal transfer is not affected by tarM or tarS-mediated WTA modifications but by incompatibility between Type I RM systems of bacterial phage donor and acceptor clones.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains used for this study can be found in Supplementary Table S3. S. aureus was cultivated in tryptic soy broth (TSB) and Escherichia coli in Lysogeny broth (LB), in the presence of appropriate antibiotics (chloramphenicol 10 μg/ml, ampicillin 100 μg/ml) at 37°C. For the calculation of growth rates in TSB or RPMI media bacterial densities were monitored at 595 nm using an Epoch 2 (BioTek) device. The maximum slope was extracted as growth rate from the logarithmic growth curve after background correction.
Molecular biology
All oligonucleotide primers used for this study are listed in Supplementary Table S4. Modification of tarP phage genomes with antibiotic resistance genes or marker-less deletion of tarP and tarS were established by using the E. coli/S. aureus shuttle vector pBASE6 plasmid and procedures described previously (Geiger et al., 2012).
tarP was deleted in NCTC13132 using the previously published plasmid pIMAY_tarP (Gerlach et al., 2018). Mutagenesis was accomplished as described recently (Monk et al., 2015).
hsdR (SAUSA300_0196) was amplified from genomic DNA of either USA300 (intact hsdR) or RN4220 (non-functional hsdR) with primers containing restriction sites BamHI and SacI and ligated into shuttle vector pRB474. The resulting plasmids were subsequently used to transform the restriction-deficient strain RN4220 by electroporation.
Phage-biological methods
Induction of prophages
Bacterial strains harboring prophages were grown in TSB to an OD 595 nm of 0.4–0.5 and prophages were induced by addition of 1 μg/ml mitomycin C for 4 h at 30°C with slow agitation and eventually overnight incubation at ambient temperature without shaking. Phages were purified by removing bacteria and cell debris by centrifugation and filtration with a pore size of 0.22 μm. Phage titers of the obtained lysates were determined by adding appropriate dilutions in soft ager to agar plates with susceptible test strains and counting of phage plaques. For phage ΦN315, indicator strain S. aureus R5 was used, for phage ΦSa1int-tarP and ΦSebago_int-tarP S. aureus RN4220.
Phage transfer
Lysogenization by induced phages was analyzed by diluting overnight cultures of bacterial recipient strains to an OD595 nm of 0.1 and mixing 200 μl of bacterial suspension with 100 μl phage lysate followed by incubation for 15 min at 37°C with mild agitation. As an exception, for transfer assays with restored hsdR in RN4220 (depicted in Figure 5), 100 μl bacteria were mixed with 200 μl phage suspension. Phage lysates were then plated on TSB plates containing the appropriate antibiotic (either 3 μg/ml tetracycline for ΦN315 or 2.5 μg/ml erythromycin for ΦSa1int-tarP and ΦSebago_int-tarP) and incubated for 2 days at 37°C. Successful lysogenization by tarP phages was confirmed by molecular typing of the tarP gene or, in case of ΦN315, additionally by altered hemolysis phenotype on TSB blood plates, because this phage integrates into the beta-hemolysin gene.
Phage susceptibility assay
Susceptibility of bacterial strains to phages was determined by the previously described soft agar overlay method (Xia et al., 2010). Briefly, bacterial overnight cultures were diluted to an OD595 nm of 0.1 and 200 μl were mixed with 5 ml prewarmed soft ager (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 0.1% K2HPO4, 0.1% glucose, 0.4% agar) and poured in TSB plates. Phage suspensions (10 μl) were dropped onto the soft agar and incubated at 37°C overnight.
WTA isolation
Isolation of WTA was conducted as previously described (Gerlach et al., 2018). Briefly, WTA was isolated from stationary-phase bacteria grown in BM broth supplemented with extra glucose (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 0.1% K2HPO4, 0.25% glucose). After repeated washing of bacterial cells in AA buffer (20 mM, ammonium acetate pH 4.8) cells were disrupted in a cell mill (Euler biotechnologies), and cell lysates were treated with DNAse (40 U/ml) and RNAse (80 U/ml) at 37°C for at least 6 h. Subsequently cell lysates were supplement with 2% SDS, incubated at 65°C for 60 min, and ultrasonicated for 15 min. After removal of SDS by extensive washing of cell walls with AA buffer, WTA was released from the insoluble cell wall fraction by incubation with 5% trichloroacetic acid for 4 h at 60°C. The pH of the WTA solution was adjusted to pH 4–5 and WTA was dialyzed against distilled water using a Spectra/Por3 dialysis membrane (MWCO of 3.5 kDa; VWR International GmbH, Darmstadt).
Analysis of WTA
Phosphorus and GlcNAc content of WTA was determined by the method of Chen and Warner (1956) and Smith (1979), respectively.
NMR data collection
All NMR experiments were carried out as described previously (Speciale et al., 2022). Briefly, samples (about 7–10 mg) were solved in D2O (550 μl) and measured at 298 K with a Bruker DRX-600 spectrometer equipped with a cryo-probe by using standard Bruker software. Chemical shift of spectra was measured in ppm relative to internal acetone (2.225 and 31.45 ppm). The spectral width was set to 10 ppm and the frequency carrier placed at the residual HOD peak, suppressed by presaturation. Two-dimensional spectra (TOCSY with a mixing time of 100 ms, gHSQC, gHMBC, and HSQC-TOCSY with a mixing time of 100 ms) were measured for all samples. For NCTC13132 ΔtarP and RN4220 ΦN135, additional spectra were acquired. HSQC-TOCSY with a mixing time of 20 ms was recorded for both, while a T-ROESY (350 ms of mixing time) and a NOESY (200 ms of mixing time) spectra were acquired for RN4220 ΦN135 and NCTC13132 ΔtarP, respectively.
For all the experiments, 512 free induction decays (FIDs) of 2,048 complex data points were collected; for homonuclear spectra, 32 scans per FID were acquired; heteronuclear 1H-13C spectra were measured in the 1H-detected mode, gHSQC spectrum was acquired with 40 scans per FID, gHMBC and HSQC-TOCSY scans doubled, or tripled, those of the gHSQC spectrum, respectively. During processing, each data matrix was zero-filled in both dimensions to give a matrix 4 K × 2 K points and was resolution-enhanced in both dimensions by a cosine-bell function before Fourier transformation; data processing and analysis was performed with Bruker Topspin 3 program.
Additional information on the NMR analysis procedure can be found in the Supplementary material.
Bioinformatic analysis
General analysis of the datasets occurred using R (Version 4.02) or GraphPad Prism.
Analysis of restriction modification systems
Restriction modification patterns were analyzed using a custom R script to survey MGEs for described recognition sequences of Type-I restriction modification systems (Cooper et al., 2017).
Genomic analysis of gene presence-absence
All available S. aureus genomes were downloaded from the RefSeq Genome Database (accessed 10. December 2020) (O’Leary et al., 2016) and sequence-typed using the MLST software1 based on information stored on the PubMLST website (Jolley and Maiden, 2010). BLASTN searches (Altschul et al., 1990) were performed using reference sequences S. aureus COL (Genbank accession no. NC_002951) for mecA, tarS, and tarM, S. aureus N315 (Genbank accession no. NC_002745) for tarP, S. aureus LGA251 (Genebank accession no. NC_017349) for mecC, and Newman (Genbank accession no. NC_009641) for scn (an indicator of the IEC element) and the Sa3int. The genes were considered present when showing at least 90% length and 90% identity to the reference gene, except for tarP, where 80% identity was considered sufficient in order to account for a rare tarP variant with 82% identity to the reference.
tarP-associated integrase genes were identified by extraction and annotation of the 2-kb flanking regions in both directions for all tarP genes. Annotation was performed in Geneious Prime 2019 (Biomatters Ltd.) from an in-house collection of integrase genes based on Goerke et al. (2009).
Phylogenetic analysis of selected phyla
The contigs of selected CC’s or ST’s were used to call single nucleotide polymorphisms (SNPs) using NASP (Sahl et al., 2016). To remove SNPs falling into regions of putative recombination, Gubbins (Croucher et al., 2015) was used using default parameters. The maximum likelihood phylogenies were then established using IQTREE (Nguyen et al., 2015) using a GTR model of substitution and annotated using package ggtree (Yu et al., 2016) in R version 3.6. The respective phylogeny files are deposited as Supplementary material: Data sheet 1 – CC1; Data sheet 2 – CC45; Data sheet 3 – ST5.
Results
tarP is confined to distinct clonal lineages
tarS was present in 99% of the 11,984 S. aureus genomes present in the NCBI Reference Sequence Database (Figure 1A). In agreement with our previous findings, tarP was most frequently present in CC5 and CC398 (30 and 27% of genomes, respectively; Table 1, Figure 1B). tarP was also prevalent in CC45 and CC1 (27 and 12% of the clones, respectively). In addition, tarP was found in CC8, CC15, CC30, and CC97, albeit at low frequencies (range 0.2–3.2%). Finally, tarP was present in few clones with very small numbers of available genomes (Table 1).
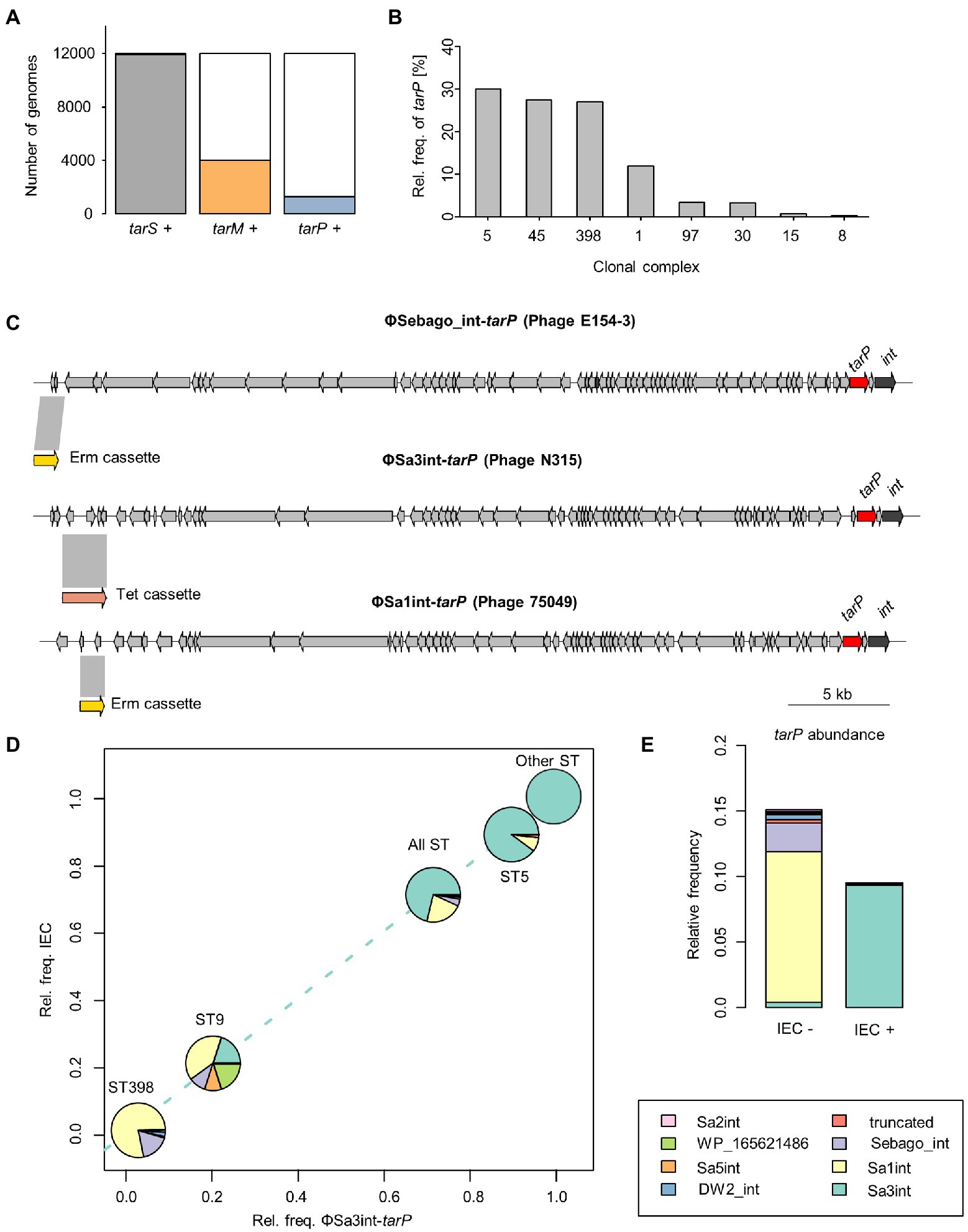
Figure 1. Distribution of tarP phages across Staphylococcus aureus isolates. (A) Absolute frequency of RboP WTA glycosyltransferases (GT). (B) Relative frequency in percent of tarP in clonal complexes. Clonal complexes were used to summarize ST. (C) Genetic structure of manipulated tarP phages, presented in this work, indicating location of tarP, integrase (red) and the respective, inserted antibiotic resistance marker (yellow/pink). (D) tarP phage distribution of main tarP-encoding ST (other STs comprise 45, 180, 188, 225, 228, and 3,410). Each ST is placed according to its relative frequencies of IEC and Sa3int-tarP phages. (E) Relative frequency of tarP in relation to IEC. tarP-associated phage integrases are indicated by the same colours as in panel D.
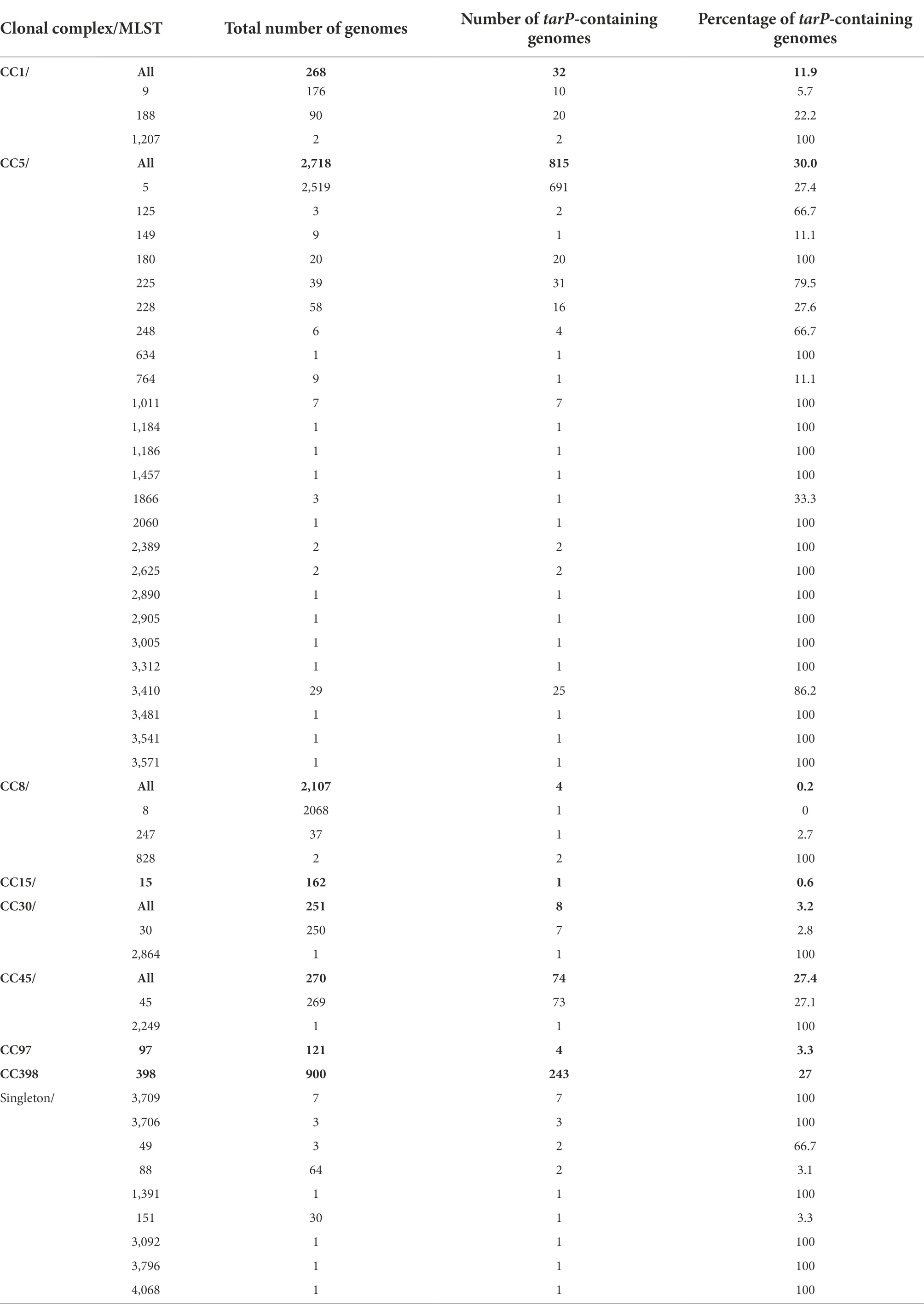
Table 1. Abundance of tarP-positive S. aureus sequence types (MLST) in the used BLAST dataset.
In order to assess the risk for the future transfer of tarP phages to other important MRSA clones we sought to understand the underlying principles of the discrete phylogenetic distribution of tarP phages. tarP has a conserved position in diverse phage genomes, close to the gene for the phage integrase (Figure 1C), which directs the insertion position of the prophage in the bacterial genome. Integrase gene types can be utilized to classify staphylococcal siphophages into more than 12 different families (Goerke et al., 2009). We sought to understand the diversity of tarP phages in the analyzed genomes by mapping the tarP-associated integrase type. Sa3int was the most frequent integrase associated with tarP, although we observed strong differences across different STs (Figure 1D). ΦSa3int also harbors the IEC element that enables S. aureus to evade the human innate immune response and is highly prevalent in human S. aureus clones, whereas it is virtually absent from S. aureus clones from non-human animals (Richardson et al., 2018). Accordingly, IEC was significantly enriched in S. aureus genomes of human origin (p < 10−15, Fisher’s exact test) (Supplementary Figure 1). tarP was usually located on ΦSa3int phages in S. aureus clones harboring the IEC element, whereas it was present on many different phages in S. aureus clones lacking the IEC element, including ΦSa1int, ΦSa2int, ΦSa5int, ΦSa7int, and the newly described ΦSebago (Klotz et al., 2019) that was previously misidentified as Sa9int (Figure 1D) (Gerlach et al., 2018). In addition, we observed that tarP is 40% more frequent in strains lacking the IEC element than in those with IEC (p < 10−13 by Fisher’s exact test), suggesting that tarP is more prevalent in S. aureus clones from non-human animals (Figure 1E).
To assess the functional mobility of tarP phages, we constructed phylogenies of ST5, CC1, and CC45 (Supplementary material). In CC45 tarP was confined to a deep-branching clade, located on ΦSa3int, which reflects a history of vertical spread with no horizontal transfer of tarP phages to isolates belonging to other clades. In CC1, tarP was associated with different phages and showed a more scattered distribution, supporting the notion that it has been introduced into CC1 on multiple occasions. In ST5, tarP was located on ΦSa3int phages but showed a more scattered distribution than in CC45, suggesting a history of multiple introductions and/or losses and, therefore, potentially dynamic WTA glycosylation alterations.
To study tarP phage dynamics in an experimental approach, we inserted antibiotic resistance markers into three dominant tarP prophages: ΦSa3int-tarP (ΦN315) in the genome of hospital-associated (HA)-MRSA N315 (CC5), ΦSa1int-tarP in LA-MRSA 75049 (ST398), and ΦSebago_int-tarP in LA-MRSA 70153 (ST398) (Figure 1C). The markers were placed by removing non-essential or non-coding genetic regions upstream of the phage holin/lysins genes to maintain phage activity. We attempted to mobilize the marked tarP-encoding phages ΦN315 and ΦSa1int-tarP by mitomycin C treatment and monitored lysogenization of the widely used phage-susceptible lab strain S. aureus RN4220. Both phage preparations could lysogenize RN4220 indicating that the two phages are indeed intact and can be mobilized (Figure 2). Next, we probed S. aureus isolates devoid of tarP prophages from different clonal complexes and backgrounds for susceptibility to the tar phages ΦSa3int-tarP (Figure 2A) and ΦSa1int-tarP (Figure 2B). In both cases we observed transfer only to acceptor strains that shared the same clonal background as the donor strains N315 (CC5) or 75,049 (CC398).

Figure 2. Transfer of tarP phages induced from different S. aureus backgrounds into different recipient strains. MLST, clonal complex, associated RM I system make-up, and successful transfer are indicated (NF, non-functional Type I RM system). RM systems that share identical make-up with the donor strain are marked by color. Transfer efficiency is depicted as Tru/PFU as mean + SD of 3 independent replicates. (A) Transfer of ΦN315 induced from a CC5 background. (B) Transfer of ΦSa1int-tarP induced from a CC398 background.
tarP phages require glycosylated WTA for binding
Next, we probed whether a lack of phage receptor compatibility, a major necessity for phage transduction (Winstel et al., 2013; Li et al., 2015; Gerlach et al., 2018), might interfere with spread of tarP phages to other clades. Recipients of phage transduction need to carry appropriate phage receptor structures (Winstel et al., 2013). To study if clonal lineages lacking tarP may have altered phage receptor, we probed the glycosyltransferes repertoire of those lineages. Doing so we observed a strong negative association of tarP with the second accessory WTA GT genes tarM among S. aureus genomes (p < 2.2 × 10−16, Fisher’s exact test) (Figure 3A) in a sequence type-dependent manner (Figure 3B). A decrease in lysogenization by ΦSa3int-tarP and ΦSa1int-tarP of a mutant lacking any WTA glycosylation (RN4220 ΔtarM/tarS) indicated a preference but no absolute dependence of tarP phages for binding glycosylated WTA (Figure 3C). Restoration of glycosylation by expression of either TarM, TarP, or TarS led to similar levels of transduction, which indicated that TarM glycosylation does not directly prevent adsorption of tarP phages and cannot explain the lineage-dependent presence or absence of tarP prophages.
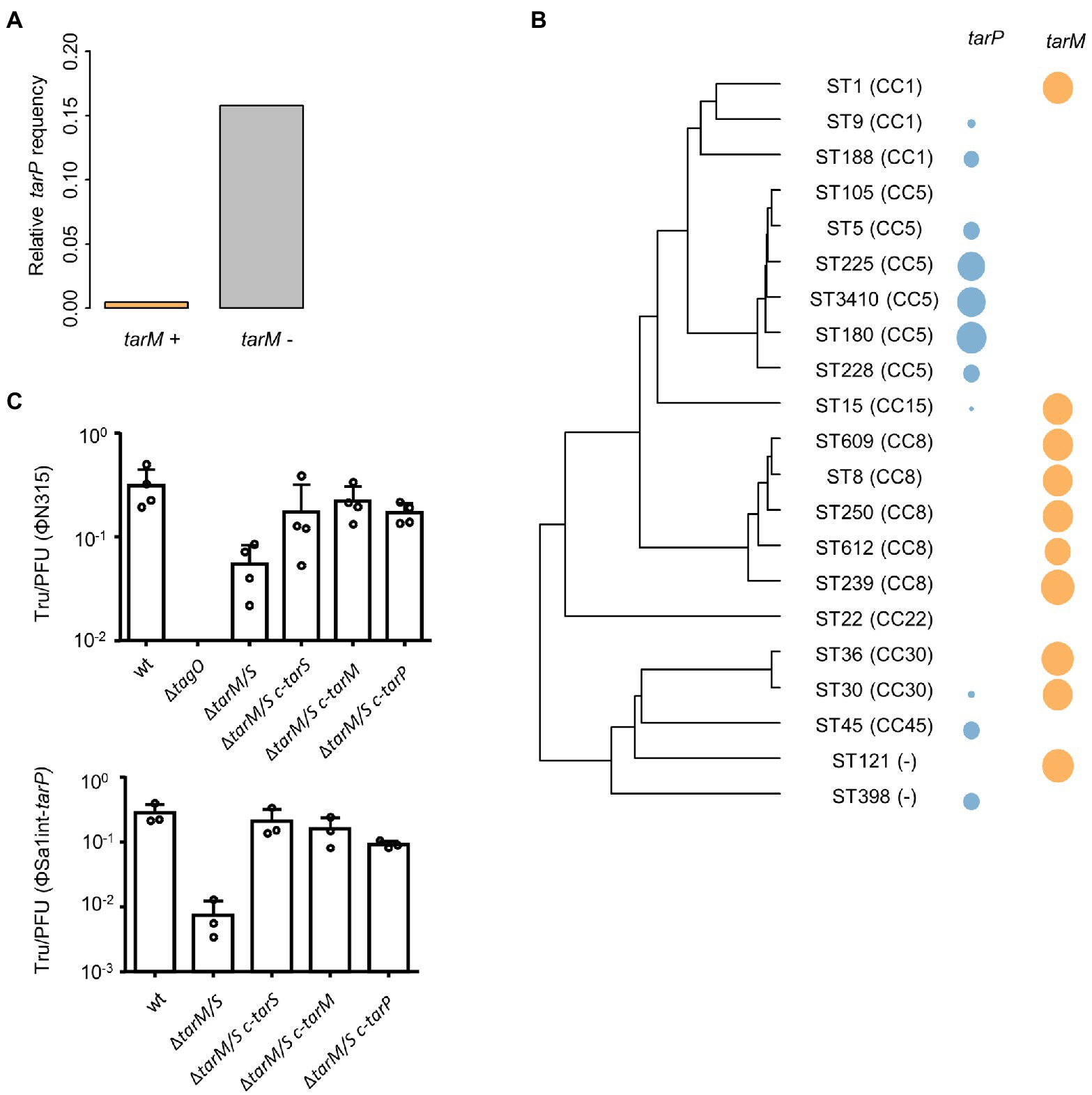
Figure 3. Influence of tarM on tarP phage prevalence. (A) Relative frequency of tarP presence in in dependence of tarM status. (B) Phylogeny based on concatenated MLST alleles for most prevalent STs. Relative frequency of tarP and tarM are indicated by colored circles. (C) Transfer rates of tarP phages into RN4420 WTA mutant panel. RN4220 WT, RN4220 WTA mutant (ΔtagO), and WTA glycosylation mutant RN4220 (ΔtarM/tarS) expressing different WTA GTs. Transfer rates are represented as obtained transductants per input PFU of the respective phage. Top, transfer of phage ΦN315 (mean + s.d. of 4 independent experiments). Bottom, transfer of phage ΦSa1int-tarP (mean + s.d. of 3 independent biological replicates).
The strong negative association of tarM and tarP was analyzed in more detail to understand, if the two GT might interfere with each other functionally. We searched for naturally occurring tarM-encoding, tarP phages lysogenic strains in public genomic datasets: of the 11,984 available S. aureus genomes we identified only 21, mostly from CC8 and CC30, encoding all three GTs, the accessory genes tarM, tarP, as well as the housekeeping GT gene tarS (Figure 4A). Next, we confirmed the proper glycosylation capability of the engineered tarP phages, by utilizing a special set of podoviridae, which depend exclusively on a TarS-conferred 1,4-beta-GlcNAc WTA pattern (Li et al., 2015). These podophages failed to infect the initially sensitive S. aureus isolates after lysogenization by the engineered tarP phages, indicating a successful conversion of the phage receptor by TarP (Supplementary Figure 2A). We used one of these phages, ΦN315, to lysogenize tarM-encoding RN4220 and compared the WTA structure with the native lysogen NCTC13132 (CC8) (Figure 4B) using nuclear magnetic resonance (NMR) spectroscopy. All NMR-analyzed strains showed similar growth behavior (Supplementary Figure 2B). TarM-mediated glycosylation was found to be dominant over that conferred by TarS in the tarP-negative strains RN4220 and NCTCT13132 ΔtarP (Figure 4B left and Table 2), which reflects our previous findings (Xia et al., 2010). In contrast, the two strains additionally expressing tarP from the ΦN315 prophage showed signals for both, α-1,4-GlcNAc-RboP and β-1,3-GlcNAc-RboP (conferred by TarM and TarP, respectively) with similar frequency (48 vs. 52% for RN4420 and 40 vs. 60% for NCTC13132) (Figure 4B; Table 2) indicating that TarP and TaM compete with similar efficacy for RboP glycosylation sites and do not exclude each other. To our knowledge this represents the first description of S. aureus isolates expressing a novel α-1,4-GlcNAc-RboP -β-1,3-GlcNAc-RboP-WTA mix type, whose biological impact could be of future interest.
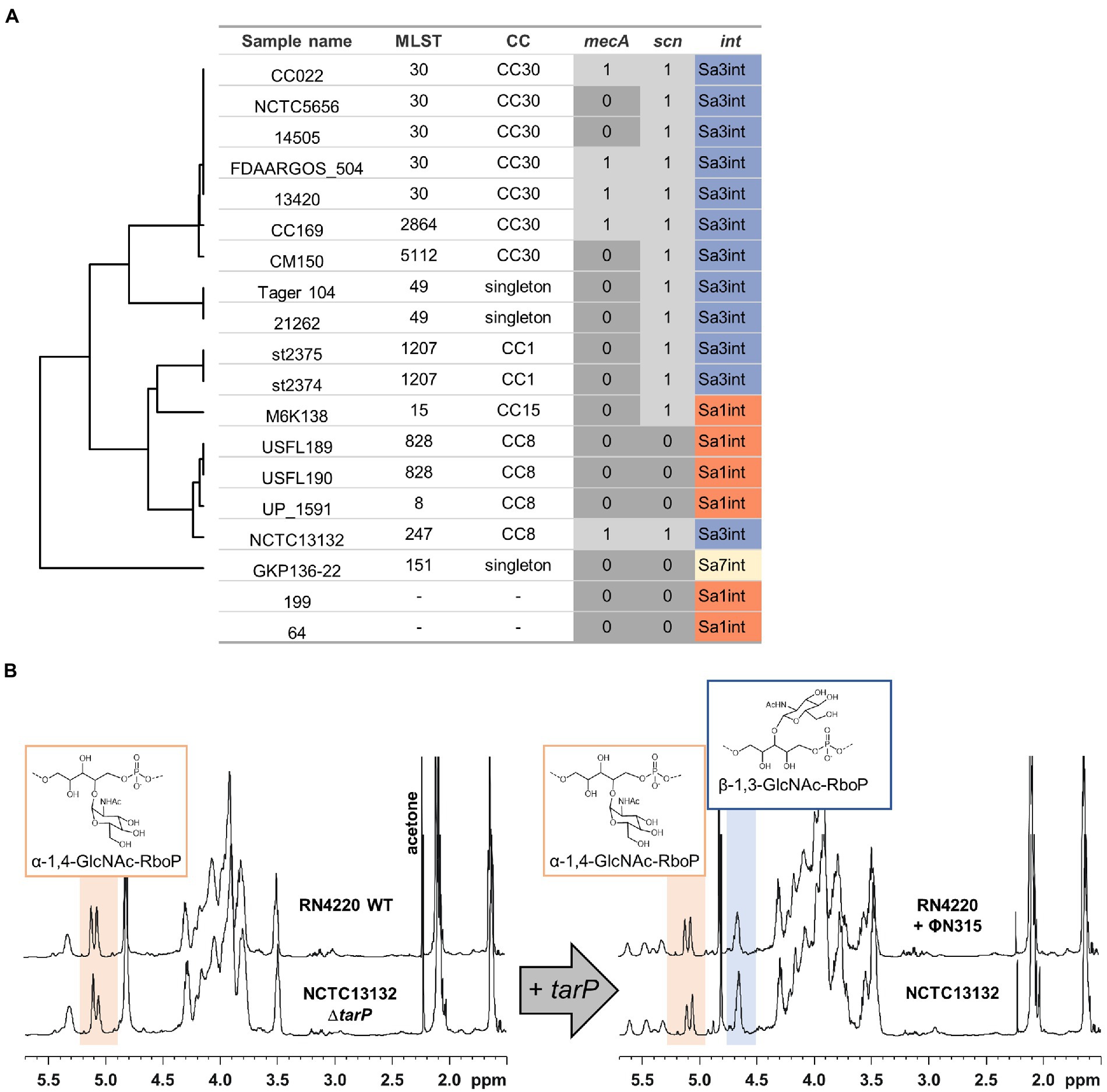
Figure 4. Identified S. aureus encoding tarP and tarM. MRSA determinant mecA, IEC, and tarP-associated integrase are indicated. (A) Isolates are grouped by MLST similarity as in Figure 3B (in 2 cases MLST could not be determined). (B) 1H-NMR spectra of tarM-positive S. aureus without (left) or with tarP (right), switching WTA glycosylation from a TarM-type (α-1,4-GlcNAc-RboP) to a TarM/TarP-type (α-1,4-GlcNAc-RboP + β-1,3-GlcNAc-RboP).
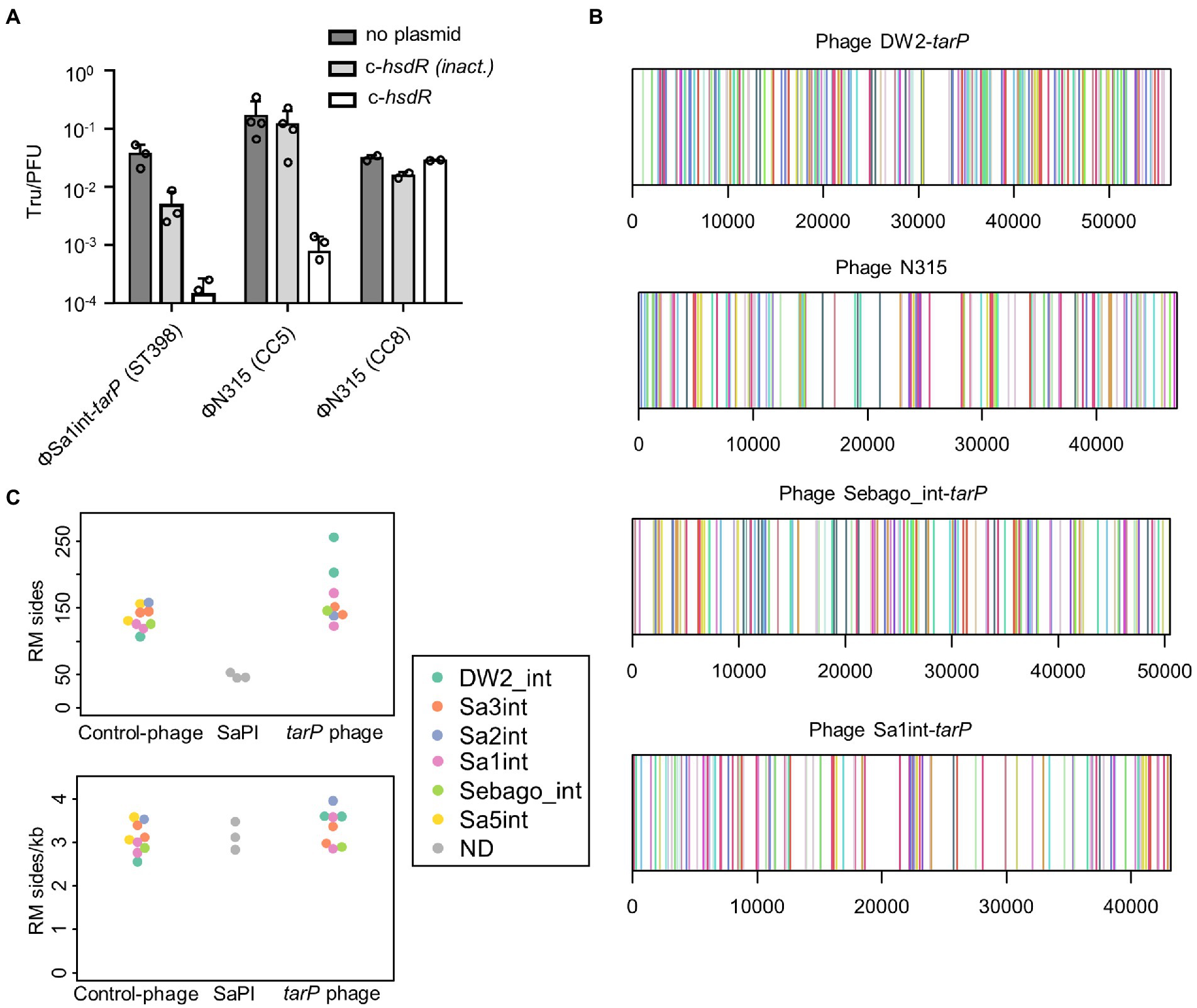
Figure 5. Influence of Type I RM systems on tarP phage mobility. (A) Transfer of ΦSa1int-tarP induced from ST398 background or ΦN315, induced from CC5 or CC8 background, into restriction deficient strain RN4220 (CC8) expressing no plasmid, a control plasmid with the native inactivating hsdR mutation (576G>A) of RN4220 or a functional hsdR variant from a CC8 isolate (mean Tru/PFU + s.d. of 2 or 3 independent replicates). (B) Genetic location of 21 different RM recognition sites (indicated by color) in 4 representative tarP phages of the most prevalent tarP phage int classes, including phages used for in-vitro studies. (C) Comparison of the sum of RM recognition sites between tarP phages, non-tarP phages and phage-like SaPIs. The int group for phages is indicated. Top, absolute number of recognition sides. Bottom relative frequency given as sites per 1 kb.
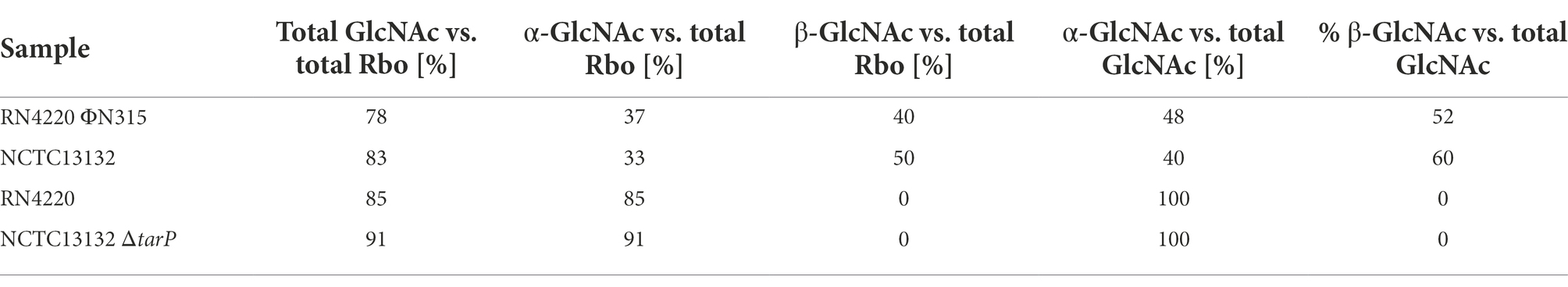
Table 2. Relative abundance of NMR motifs for analyzed tarM-encoding tarP phage lysogens (lab-made RN4220 ΦN315 and native NCTC13132) and the corresponding strains without tarP locus (RN4420 and NCTC13132 ΔtarP).
Clone-specific RM systems restrict the transfer of tarP phages
Staphylococcus aureus CCs usually share specific types of lineage-specific RM systems, which might explain the clonal distribution of tarP phages (Lee et al., 2019). S. aureus mutants lacking Type I RM systems were used to assess this hypothesis. ΦN315 could not lysogenize USA300 JE2 (CC8) but it could be transferred effectively to an isogenic mutant with inactivated hsdR locus (Figure 2A). This also demonstrates that tarP phages appear not to be affected by prophage immunity, since USA300 harbours a non-tarP Sa3int prophage. Overall recipients could be lysogenized by ΦN315 with similar efficacy when they shared the clonal background of the donor strain (Figures 2A,B), which is in agreement with a crucial role of RM systems in susceptibility to tarP phage lysogenization. In a second experiment, reminiscent of previous work by Waldron and Lindsay (Waldron and Lindsay, 2006), we reintroduced a functional copy of sauI (hsdR) from another CC8 isolate into RN4220 (CC8) to restore its Type I RM functionality, which resulted in a profound decrease of lysogenization efficiency for tarP phages Sa1int and Sa3int (ΦN315) (Figure 6A). Moreover, transferring ΦSa3int-tarP released from a donor strain (USA300) to RN4420 with the same CC8-specific DNA methylation pattern resulted in very similar transfer efficiencies (Figure 6A).
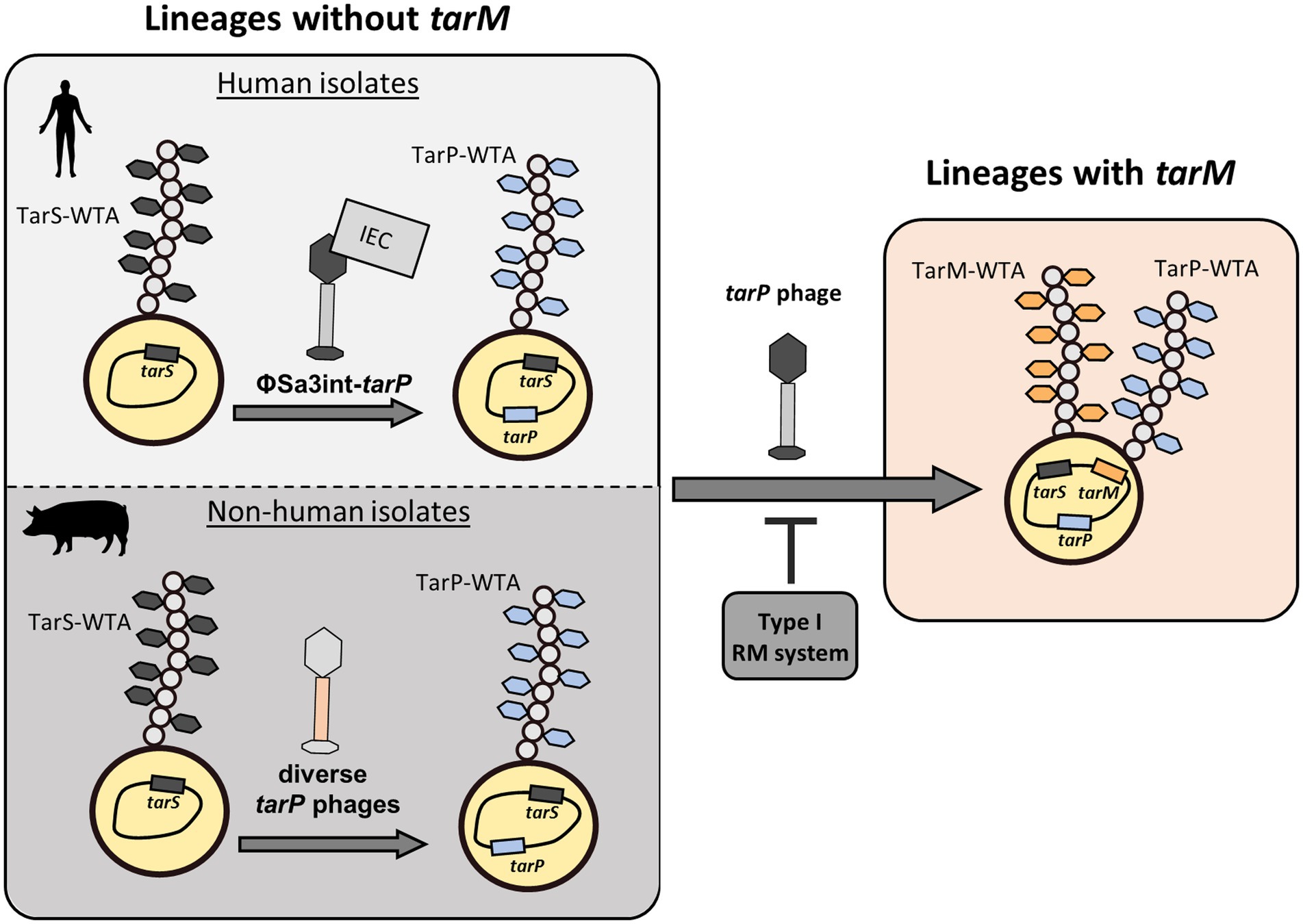
Figure 6. Summary of distribution of tar phages. tarP phages are prolific in clonal lineages of S. aureus that are devoid of the genome-encoded, alternative glycosyltransferase tarM. Here, TarP-glycosylation appears to be dominant over TarS-glycosylation. Furthermore, the make-up of tarP phages shows a stark discrepancy between Sa3int-phages with presumed IEC in human isolates and diverse non-Sa3int-phages in non-human isolates. Upon integration of tarP phages, lysogens express a dominant tarP-WTA. Transfer of tarP phages into tarM-encoding clonal lineages such as (CC1, CC8, and CC30) is mainly suppressed by type I RM systems. However rare bypassing of the host restriction barrier leads to expression of a TarM/TarP-hybrid-WTA.
To assess the potential of HsdR proteins to cleave prophage DNA we searched for selected recognition sequences of published SauI RM systems (Cooper et al., 2017) in tarP phage genomes. All probed tarP phages contained at least one target sequence of each of the 21 known HsdS types. The recognition sites were spread over the entire phage genomes (Figure 3A) implying potentially successful restriction and interception of the infection process by the various HdsS variants. tarP phages showed similar frequencies of S. aureus HdsS recognition sites compared to control siphophages or phage-related S. aureus pathogenicity islands (SaPI), which are devoid of the tarP gene (Figure 6C). Thus, distribution of tarP phages in other clonal lineages and their dissemination appears to be similarly limited by Type I RM systems as reported for other temperate phages (McCarthy and Lindsay, 2012).
tarP phages were abundant in all STs of CC5 besides ST105 (Table 1), which is in agreement with the shared RM systems in this CC (Lee et al., 2019). CC5 and CC8 both use the Newman_MS1 HsdS variant (Lee et al., 2019), which may be a reason why some CC8 isolates also harbor tarP phages (Table 1). However, the two clonal groups have different additional RM systems and CC8 encodes tarM in its core genome, which may explain why tarP has remained rare in CC8. tarP is prevalent in CC5, CC398, and CC45 (Table 1) although these clones have quite different RM systems (Lee et al., 2019), indicating that tarP phages can expand in different clonal backgrounds once they have traversed a RM barrier. Fortunately, such host clone jumps may have remained rare because tarP was not found in any of the genomes from several other major S. aureus clonal groups.
Discussion
Staphylococcus aureus is a quickly evolving pathogen, with new clonal lineages continuously emerging and outcompeting existing clones. It is a major concern that HGT will lead to new combinations of virulence, fitness, and resistance factors, creating new clones with even more dangerous properties, than the existing clones (Lee et al., 2018). The tarP gene is an S. aureus virulence factor of concern as it resides on an MGE and provides host bacteria with extraordinary immune evasion capacities (Gerlach et al., 2018). It has remained enigmatic, why it is found only in a narrow subset of S. aureus clonal lineages.
We provide evidence that tarP phages are indeed mobile and can lysogenize susceptible S. aureus host cells. Intriguingly, the presence of tarP was strongly associated with the absence of the alternative GT gene tarM, raising the possibility that tarM-mediated WTA modification might interfere with infection by tarP phages. However, we found that TarM-glycosylation has no impact on S. aureus susceptibility to tarP phages, while the major factors limiting the spread of tarP phages to distant clonal lineages turned out to be RM systems (Figure 6). In a species wide analysis we detected a low frequency of tarM-encoding isolates that show lysogenization by tarP phages, which led to the production of a novel TarM/TarP-hybrid WTA. The low frequency of tarP phages in tarM-encoding S. aureus lineages was corroborated also by a recent study (Xiong et al., 2020). Using PCR typing, the authors identified only 2 out of 555 S. aureus isolates encoding both, tarP and tarM.
The fact that tarP is also found in LA isolates, localized on non-Sa3int phages, points towards an important role of TarP glycosylation also in non-human hosts, as reported previously (Sieber et al., 2020). However, the obvious preference of tarP for Sa3int-phages, might be driven by complex coevolution events of tarP, IEC, and the phage itself.
TarP and TarM have similar impacts on many different physiological processes such as resistance to podophages (i.e., 44AHJD-like podophages) (Li et al., 2015; Gerlach et al., 2018) or reduced recognition by host IgGs (van Dalen et al., 2019, 2020). This indicates that tarP phages may be particularly successful in providing additional fitness to clonal lineages, which have lost the tarM locus and only produce TarS-glycosylated WTA. Here TarP glycosylation appears to become dominant over TarS glycosylation upon lysogenization by the respective prophage (Supplementary Figure 2A). tarP prophages would allow their hosts to modulate the immune and phage evasion capacities. A recent study pointed towards similar capabilities of TarS- and TarP-WTA to bind the skin dendritic cell receptor langerin. However, the consequences of TarP and TarS-mediated WTA modification are not identical in terms of immune activation, since tarP-expressing S. aureus showed increased levels of IL-8 and TNFα secretion by Langerhans cells compared to those with TarS-WTA (Hendriks et al., 2021).
tarP phages were present in two S. aureus lineages (ST5 and CC1) in a manner that appeared to result from horizontal spread among different phylogenetic backgrounds, while tarP distribution patterns in other lineages were in line with those expected under vertical inheritance. In support of a prominent role of restriction mechanisms in suppression of trans-lineage spread of prophages, we found similar abundances of Type-I RM targets in phages with or without tarP, supporting the notion that siphophages including tarP phages show a broad specificity for RboP WTA receptors with GlcNAc, irrespective of the glycosylation pattern, but are impeded strongly by host restriction (Figure 6). Once tarP phages form infectious particles, they are not hindered by WTA glycosylation configurations to lysogenize host strains.
Genotypic risk assessment strategies for S. aureus have previously focused strongly on the presence of antibiotic resistance or toxins genes associated with disease severity (Shopsin and Kreiswirth, 2001). Our increasing knowledge of prevalence, function, and phenotypes of WTA GTs may lead to additional suitable biomarkers for monitoring S. aureus immune evasion capacities. Understanding the capacities of resistance genes to spread among S. aureus strains will be important for estimating their potential contribution to the evolution of new clonal lineages. In this regard, the high mobility of tarP remains a matter of concern despite its partial restriction by RM systems.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
DG, JK, and JB performed phage experiments, WTA extractions, and molecular cloning. CC and AM analyzed WTA via NMR. RS, DG, and JL performed bioinformatic analysis of Staphylococcus aureus genomes. DG and AP analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financed by grants from the Deutsche Forschungsgemeinschaft, grants TRR34, SPP 2330, TRR261 project ID 398967434, and PE 805/7-1 to AP; and the German Center of Infection Research to AP. AP was supported by infrastructural funding from the Cluster of Excellence EXC 2124 “Controlling Microbes to Fight Infections” project ID 390838134.
Acknowledgments
The authors thank Petra Kühner for technical support, and Christiane Wolz and Carina Rohmer for helpful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.951333/full#supplementary-material
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Brown, S., Santa Maria, J. P. Jr., and Walker, S. (2013). Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 67, 313–336. doi: 10.1146/annurev-micro-092412-155620
Brown, S., Xia, G., Luhachack, L. G., Campbell, J., Meredith, T. C., Chen, C., et al. (2012). Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. U. S. A. 109, 18909–18914. doi: 10.1073/pnas.1209126109
Chen, P. S. T. Y., and Warner, H. (1956). Microdetermination of phosphorus. Anal. Chem. 28, 1756–1758. doi: 10.1021/ac60119a033
Cooper, L. P., Roberts, G. A., White, J. H., Luyten, Y. A., Bower, E. K. M., Morgan, R. D., et al. (2017). DNA target recognition domains in the type I restriction and modification systems of Staphylococcus aureus. Nucleic Acids Res. 45, 3395–3406. doi: 10.1093/nar/gkx067
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Geiger, T., Francois, P., Liebeke, M., Fraunholz, M., Goerke, C., Krismer, B., et al. (2012). The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 8:e1003016. doi: 10.1371/journal.ppat.1003016
Gerlach, D., Guo, Y., De Castro, C., Kim, S. H., Schlatterer, K., Xu, F. F., et al. (2018). Methicillin-resistant Staphylococcus aureus alters cell wall glycosylation to evade immunity. Nature 563, 705–709. doi: 10.1038/s41586-018-0730-x
Goerke, C., Pantucek, R., Holtfreter, S., Schulte, B., Zink, M., Grumann, D., et al. (2009). Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 191, 3462–3468. doi: 10.1128/JB.01804-08
Hendriks, A., van Dalen, R., Ali, S., Gerlach, D., van der Marel, G. A., Fuchsberger, F. F., et al. (2021). Impact of glycan linkage to Staphylococcus aureus wall teichoic acid on Langerin recognition and Langerhans cell activation. ACS Infect. Dis. 7, 624–635. doi: 10.1021/acsinfecdis.0c00822
Ingmer, H., Gerlach, D., and Wolz, C. (2019). Temperate phages of Staphylococcus aureus. Microbiol. Spectr 7. doi: 10.1128/microbiolspec.GPP3-0058-2018
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Klotz, K., Korn, A., Newkirk, H., Liu, M., Gill, J. J., and Ramsey, J. (2019). Complete genome sequence of Staphylococcus aureus Siphophage Sebago. Microbiol. Resour. Announc. 8. doi: 10.1128/MRA.00765-19
Lee, J. Y. H., Carter, G. P., Pidot, S. J., Guérillot, R., Seemann, T., Gonçalves da Silva, A., et al. (2019). Mining the methylome reveals extensive diversity in Staphylococcus epidermidis restriction modification. MBio 10. doi: 10.1128/mBio.02451-19
Lee, A. S., de Lencastre, H., Garau, J., Kluytmans, J., Malhotra-Kumar, S., Peschel, A., et al. (2018). Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers. 4:18033. doi: 10.1038/nrdp.2018.33
Li, X., Gerlach, D., Du, X., Larsen, J., Stegger, M., Kuhner, P., et al. (2015). An accessory wall teichoic acid glycosyltransferase protects Staphylococcus aureus from the lytic activity of podoviridae. Sci. Rep. 5:17219. doi: 10.1038/srep17219
Lindsay, J. A. (2010). Genomic variation and evolution of Staphylococcus aureus. Int. J. Med. Microbiol. 300, 98–103. doi: 10.1016/j.ijmm.2009.08.013
Maslanova, I., Doskar, J., Varga, M., Kuntova, L., Muzik, J., Maluskova, D., et al. (2013). Bacteriophages of Staphylococcus aureus efficiently package various bacterial genes and mobile genetic elements including SCCmec with different frequencies. Environ. Microbiol. Rep. 5, 66–73. doi: 10.1111/j.1758-2229.2012.00378.x
McCarthy, A. J., and Lindsay, J. A. (2012). The distribution of plasmids that carry virulence and resistance genes in Staphylococcus aureus is lineage associated. BMC Microbiol. 12:104. doi: 10.1186/1471-2180-12-104
Monk, I. R., Tree, J. J., Howden, B. P., Stinear, T. P., and Foster, T. J. (2015). Complete bypass of restriction Systems for Major Staphylococcus aureus lineages. MBio 6, e00308–e00315. doi: 10.1128/mBio.00308-15
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Oliveira, P. H., Touchon, M., and Rocha, E. P. (2014). The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 42, 10618–10631. doi: 10.1093/nar/gku734
Oliveira, P. H., Touchon, M., and Rocha, E. P. (2016). Regulation of genetic flux between bacteria by restriction-modification systems. Proc. Natl. Acad. Sci. U. S. A. 113, 5658–5663. doi: 10.1073/pnas.1603257113
Richardson, E. J., Bacigalupe, R., Harrison, E. M., Weinert, L. A., Lycett, S., Vrieling, M., et al. (2018). Gene exchange drives the ecological success of a multi-host bacterial pathogen. Nat. Ecol. Evol. 2, 1468–1478. doi: 10.1038/s41559-018-0617-0
Sadykov, M. R. (2016). Restriction-modification systems as a barrier for genetic manipulation of Staphylococcus aureus. Methods Mol. Biol. 1373, 9–23. doi: 10.1007/7651_2014_180
Sahl, J. W., Lemmer, D., Travis, J., Schupp, J. M., Gillece, J. D., Aziz, M., et al. (2016). NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb. Genom. 2:e000074. doi: 10.1099/mgen.0.000074
Scharn, C. R., Tenover, F. C., and Goering, R. V. (2013). Transduction of staphylococcal cassette chromosome mec elements between strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 57, 5233–5238. doi: 10.1128/AAC.01058-13
Sheppard, S. K., Guttman, D. S., and Fitzgerald, J. R. (2018). Population genomics of bacterial host adaptation. Nat. Rev. Genet. 19, 549–565. doi: 10.1038/s41576-018-0032-z
Shopsin, B., and Kreiswirth, B. N. (2001). Molecular epidemiology of methicillin-resistant Staphylococcus aureus. Emerg. Infect. Dis. 7, 323–326. doi: 10.3201/eid0702.010236
Sieber, R. N., Urth, T. R., Petersen, A., Moller, C. H., Price, L. B., Skov, R. L., et al. (2020). Phage-mediated immune evasion and transmission of livestock-associated methicillin-resistant Staphylococcus aureus in humans. Emerg. Infect. Dis. 26. doi: 10.3201/eid2611.201442
Smith, L. R. G. E. (1979). Quantitation of glycosaminoglycan hexosamine using 3-methyl-2-benzothiazolone hydrazone hydrochloride. Anal. Biochem. 98, 478–480. doi: 10.1016/0003-2697(79)90170-2
Speciale, I., Notaro, A., Garcia-Vello, P., Di Lorenzo, F., Armiento, S., Molinaro, A., et al. (2022). Liquid-state NMR spectroscopy for complex carbohydrate structural analysis: a hitchhiker's guide. Carbohydr. Polym. 277:118885. doi: 10.1016/j.carbpol.2021.118885
van Dalen, R., Molendijk, M. M., Ali, S., van Kessel, K. P. M., Aerts, P., van Strijp, J. A. G., et al. (2019). Do not discard Staphylococcus aureus WTA as a vaccine antigen. Nature 572, E1–E2. doi: 10.1038/s41586-019-1416-8
van Dalen, R., Peschel, A., and van Sorge, N. M. (2020). Wall teichoic acid in Staphylococcus aureus host interaction. Trends Microbiol. 28, 985–998. doi: 10.1016/j.tim.2020.05.017
Waldron, D. E., and Lindsay, J. A. (2006). Sau1: a novel lineage-specific type I restriction-modification system that blocks horizontal gene transfer into Staphylococcus aureus and between S. aureus isolates of different lineages. J. Bacteriol. 188, 5578–5585. doi: 10.1128/JB.00418-06
Weidenmaier, C., and Peschel, A. (2008). Teichoic acids and related cell-wall glycopolymers in gram-positive physiology and host interactions. Nat. Rev. Microbiol. 6, 276–287. doi: 10.1038/nrmicro1861
Wilson, G. G., and Murray, N. E. (1991). Restriction and modification systems. Annu. Rev. Genet. 25, 585–627. doi: 10.1146/annurev.ge.25.120191.003101
Winstel, V., Liang, C., Sanchez-Carballo, P., Steglich, M., Munar, M., Broker, B. M., et al. (2013). Wall teichoic acid structure governs horizontal gene transfer between major bacterial pathogens. Nat. Commun. 4:2345. doi: 10.1038/ncomms3345
Winstel, V., Xia, G., and Peschel, A. (2014). Pathways and roles of wall teichoic acid glycosylation in Staphylococcus aureus. Int. J. Med. Microbiol. 304, 215–221. doi: 10.1016/j.ijmm.2013.10.009
Xia, G., Maier, L., Sanchez-Carballo, P., Li, M., Otto, M., Holst, O., et al. (2010). Glycosylation of wall teichoic acid in Staphylococcus aureus by TarM. J. Biol. Chem. 285, 13405–13415. doi: 10.1074/jbc.M109.096172
Xia, G., and Wolz, C. (2014). Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 21, 593–601. doi: 10.1016/j.meegid.2013.04.022
Xiong, M., Zhao, J., Huang, T., Wang, W., Wang, L., Zhao, Z., et al. (2020). Molecular characteristics, virulence gene and Wall teichoic acid glycosyltransferase profiles of Staphylococcus aureus: a multicenter study in China. Front. Microbiol. 11:2013. doi: 10.3389/fmicb.2020.02013
Keywords: Staphylococcus aureus, bacteriophage, teichoic acids, bacterial genomics, horizontal gene transfer
Citation: Gerlach D, Sieber RN, Larsen J, Krusche J, De Castro C, Baumann J, Molinaro A and Peschel A (2022) Horizontal transfer and phylogenetic distribution of the immune evasion factor tarP. Front. Microbiol. 13:951333. doi: 10.3389/fmicb.2022.951333
Edited by:
Sophia Johler, University of Zurich, SwitzerlandReviewed by:
Tom Grunert, University of Veterinary Medicine Vienna, AustriaAlexandra Fetsch, Bundesanstalt für Risikobewertung (BfR), Germany
Copyright © 2022 Gerlach, Sieber, Larsen, Krusche, De Castro, Baumann, Molinaro and Peschel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Gerlach, ZGF2aWQuZ2VybGFjaEB1bmktd3VlcnpidXJnLmRl; Andreas Peschel, YW5kcmVhcy5wZXNjaGVsQHVuaS10dWViaW5nZW4uZGU=
†Present address: David Gerlach, Biocenter, Brochado Lab, Department of Microbiology, Würzburg University, Würzburg, Germany