 Shiqin Zhang
Shiqin Zhang Zhiye Bai
Zhiye Bai Zichen Wang1
Zichen Wang1 Wen Wang
Wen Wang Qingli Dong
Qingli Dong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 04 August 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.950065
This article is part of the Research TopicNew Knowledge of Food Microbiology in Asia, Volume IIView all 15 articles
Shiga toxin-producing Escherichia coli (STEC) is an important food-borne pathogen, which can cause diseases such as diarrhea, hemorrhagic enteritis, and hemolytic uremic syndrome in humans. Twelve STEC isolates were collected from beeves and feces of commercial animals in China between 2019 and 2020 for this study. In addition to the determination of serotype and Shiga toxin subtype, whole-genome sequencing (WGS) was used for determining phylogenetic relationships, antimicrobial resistance (AMR), virulence genes, and sequence type (ST) of isolates. A total of 27 AMR genes were detected, and each STEC isolate carried more than 10 AMR genes. Eight STEC isolates from ground beef and four STEC isolated from feces were screened. A total of seven serotypes were identified, and one isolate ONT:H10 was undetermined by SeroTypeFinder. Three O157:H7 strains were confirmed and the remaining five serogroups were confirmed as O26:H11, O81:H31, O105:H8, O178:H19, and O136:H12. The phylogenetic analysis showed that STEC isolates of the same serotype or ST were clustered together based on cgMLST. The comparison of the genomes of 157 STEC reference isolates worldwide with our local STEC isolates showed that STEC isolates screened in China represented various collections and could not form a separate cluster but were interspersed among the STEC reference collection, which suggested that several STEC isolates shared a common ancestor irrespective of STEC serotype isolates. cgMLST revealed that isolates of the same O serotype clustered irrespective of their H type. Further investigation is required to determine the pathogenic potential of other serotypes of STEC, particularly in regard to these rare serotypes.
Shiga toxin-producing Escherichia coli (STEC) is an important food-borne pathogen causing zoonotic diseases. As a subpopulation of diarrheagenic E. coli (DEC), STEC can result in severe cases of disease such as hemorrhagic colitis (HC) and hemolytic uremic syndrome (HUS). The pathogenicity of STEC is associated with the production of Shiga-like toxins (Stx) encoding either one or both of the stx1 and stx2 genes (Gonzalez and Cerqueira, 2020). Since STEC was first described in 1982, more than 400 different serotypes of STEC have been identified, and research hotspots have focused on O157:H7 (Karmali et al., 2003). Moreover, several non-O157 serotypes have been associated with sporadic cases and outbreaks, such as O26, O45, O103, O111, O121, and O145, which were considered as the top six non-STEC serogroups (Mathusa et al., 2010; Amézquita-López et al., 2018).
In China, the outbreak of STEC was traced back in Xuzhou, Jiangsu province in 1999, which led to 195 hospitalized HUS patients and 177 deaths (Wang et al., 2011). The traceability analysis revealed that the food was contaminated by the fecal shedding of courtyard animals carrying STEC O157:H7 (Xiong et al., 2012). Although the main reservoir of STEC was considered to be cattle, there has been an increase in non-bovine food-related outbreaks worldwide (Smith et al., 2014). Some researches show that the STEC was transmitted mainly through foods, such as raw poultry, fresh vegetables, fruits, water, ground beef, and dairy products, to humans (Smith et al., 2014). Various transmission routes of STEC have been proposed, and the primary one is presumably via consumption of contaminated food or water (Parsons et al., 2016). However, some transmissions are not clear. For epidemiologic purposes, several genetic fingerprinting methods have been used to classify, trace, and prevent the dissemination of STEC (Heir et al., 2000). Pulsed-field gel electrophoresis (PFGE) and multilocus variable-number of tandem-repeat (MLVA) have mostly been applied in analyzing the molecular epidemiology of STEC and proved to be reliable among the sequence-based methods. However, low discriminatory power is exposed in the methods of complex workflows, expensive reagents, and time-consuming process.
The advances in next-generation sequencing in the past decade have made it true to perform WGS of organisms, including STEC at affordable costs (Joensen et al., 2014, 2015), and there was an ever-increasing evidence demonstrating that WGS was not only epidemiologic typing to detect and support outbreak investigations but also to define transmission pathways of pathogens and to reveal laboratory cross-contamination (Keoser et al., 2012; Chattaway et al., 2016; Baha et al., 2019). WGS data have the potential to provide powerful, high-level phylogenetic analysis and to show insight into the evolutionary background of the outbreak strains by using quantifiable genetic differences (Baltasar et al., 2014; Mikhail et al., 2018). For example, the comparison of 62 STEC local isolates from Chile with STEC isolated from the rest of the world by core genome multilocus sequence typing (cgMLST) typing method showed Chile STEC did not cluster with genomes of the rest of the world, suggesting local STEC isolates and STEC isolated from worldwide were not phylogenetical (Smith et al., 2014; Gutierrez et al., 2021) and indicating STEC phylogeny was affected by the origin of geographical isolation.
An earlier study described the diversity of Chinese E. coli O157 obtained from different sources such as mutton, beef, chicken, pork, and vegetable salad. Twenty-two pulsotypes by PFGE and 23 types by MLVA were found, and this study demonstrated the diversity among 30 STEC O157 isolates (Li et al., 2015). However, other STEC serotypes have not been studied in China. In this study, non-O157 STEC and O157 STEC were mainly isolated from cattle feces and ground beef in China, and serotype, sequence type (ST), and virulence genes of these strains were characterized. Furthermore, based on WGS data, cgMLST analysis from STEC isolates screened by our lab and National Center for Biotechnology Information (NCBI) confirmed phylogenetic relationships among different STEC serotypes found and suggested possibly high-risk foods causing STEC. The comparison of these characteristics with those of foodborne and clinical isolates around the worldwide could provide some information for food safety risk assessment.
Between 2019 and 2020, the STEC isolates used in this study were isolated from animal food and feces in Shanghai. All samples were collected in sterile sample bags and transported in ice as soon as possible to the laboratory for immediate processing. The enrichment method was modified from the GB4789.6-2016 food microbiological examination of Escherichia coli (National Food Safety Standards of China). Briefly, 225 ml of sterile Tryptone Soya Broth was added to a sterile sample bag with 25 g of each sample. Then, incubated at 37°C for 16–22 h on a shaking platform (200 rpm). Enrichment solutions were inoculated into CHROMagar™ STEC plates (CHEOMagar, Pairs, France). Discrete, strongly mauve colonies were picked and streaked out on MacConkey agar (MAC) and sorbitol MacConkey agar (CT-SMAC; Hopebio, Qingdao, China) for 18–22 h (John et al., 2018). STEC was identified by using PCR for the targeted genes wzxO157, stx1, and stx2 screening, as shown in Table 1. All positive samples were further processed to obtain pure stx-positive isolates (Boer et al., 2015).

Table 1. Sequences of all primers and annealing temperature used in this work.
Genomic DNA of STEC isolates was extracted from overnight cultures using boiling. Briefly, 0.5 g of feces and 25 g of food samples were added together into 225 ml of lysogeny broth (LB) and incubated at 37°C for 18–24 h. The enrichment broth (1 ml) was centrifuged at 4,000 rpm for 2 min, then centrifuged at 12,000 rpm for 5 min, and supernatant was removed. Finally, 200-μl sterile deionized water was added and boiled at 100°C for 15 min.
Whole-genome sequencing (WGS) using a 400-bp paired end was performed on the STEC genomic DNA on an Illumina Novaseq according to the manufacturer's instructions. The genomic DNA libraries were prepared using the TIANamp Bacteria DNA Kit (Tiangen Biotech Beijing Co., Ltd., China). The raw data were assembled using SPAdesv3.11.1 software to obtain scaffolds sequences with default parameters, and raw data were cleaned by Fastp (v0.19.4; number of bases to average across: 4, average quality required: 20, fold coverage was required to be >30 for the cleaned data) (Bolger et al., 2014; Shifu et al., 2018).
The serotype of each STEC isolate was determined by uploading the assembled genomes to the SerotypeFinder 2.0 (https://cge.cbs.dtu.dk/services/SerotypeFinder/) of the Center for Genomic Epidemiology (CGE) website: the threshold of identity was set to 85% and the minimum length was set to 60%, VirulenceFinder 2.0 web-based tool (https://cge.cbs.dtu.dk/services/VirulenceFinder/) was used to determine virulence genes of each STEC isolates in this study with the following parameters: the 90% threshold identity and 60% minimum length. ResFinder 4.1 web-based tool (https://cge.food.dtu.dk/services/ResFinder/)was used to determine AMR genes for each STEC isolate with default parameters. The sequence types (STs) were identified by uploading assembled genomes to the MLST Finder (https://cge.cbs.dtu.dk/services/MLST/) (Jaureguy et al., 2008).
To determine the phylogenetic relationship of the isolates, a gene-by-gene approach was performed by SeqSphere (v3.1.1-rc04, Ridom) and BLAST (v2.2.12) (Michaela et al., 2021). The key parameters identity was 90% and aligned was 100%. Briefly, a core genome MLST scheme was developed using the genome of E. coli O157:H7 strain Sakai (accession no. NC_002695; https://www.cgmlst.org/ncs/schema/8896773/) as a reference genome and an additional eleven E. coli as query genomes to extract open reading frames (ORFs) from the genome of each isolate using MLST+ (v2.11.0+) of SeqSphere (v3.1.1-rc04, Ridom). The genes shared by all isolates analyzed were defined as the core genome for phylogenetic analysis. Loci were detected by chewBBACA (https://github.com/B-UMMI/chewBBACA), BLAST Score Ratio threshold was 0.8, and the number of loci present in genomes was 95% (Jagadesan et al., 2019). By default, a minimum spanning tree (MST) was calculated based on loci, which were carried out using SeqSphere (v3.1.1-rc04, Ridom).
The phylogenetic relationship of the STEC isolates of this study with isolates of the STEC reference collection (n = 157; Supplementary File 1) from NCBI was determined by cgMLST method. The download criteria for raw data of reference collection were as follows: (1) sequence depth cannot be lower than 200×, and (2) sequence length in the range of 4.7–5.5 M.
As shown in Table 2, 289 samples from cattle feces, lettuce, and ground beef were tested during 2019–2020, 4.41% (9/204) of cattle feces, 1.81% (1/55) of lettuce, and 0.00% (0/30) of ground beef were stx1 and stx2-positive. Further, 4 STEC strains were isolated from nine STEC-positive samples of cattle feces, whereas no STEC strains were found in any samples of lettuce that tested positive for STEC. The prevalence of STEC in cattle feces was higher than the other samples, with a PCR-positive rate of 4.41% and an isolation rate of 1.96%. Eight STEC strains were isolated from ground beef earlier, and 12 STEC strains were collected for this study. STEC has emerged as an important food-borne pathogen, several reports indicated that STEC cases have been transmitted to humans via food (Smith et al., 2014; Jenkins et al., 2019; Mohammad et al., 2021). In this study, STEC isolates were collected from cattle feces and ground beef in Shanghai, China. The findings in this study supported that cattle carry various serotypes (Monaghan et al., 2012).

Table 2. Prevalence of STEC isolates in cattle feces, lettuce, and ground beef samples.
The serotypes of 12 STEC isolates were identified by Serotypefinder software. In addition to the common serotypes such as O157:H7 and O26:H11, other serotypes such as O81:H31, O136:H12, and O105:H8 were isolated. However, an unknown STEC serotype ONT:H10 was found, which may be due to the incomplete coverage of the area involved in O-antigen determination, resulting in the generation of unknown serotype STEC. As a result, three O157:H7 strains were confirmed and the remaining five serogroups were O26:H11, O81:H31, O105:H8, O178:H19, and O136:H12. In this study, seven housekeeping (adk, fumC, gyrB, icd, mdh, purA, and recA) genes were used for MLST analysis, and seven STs were identified, as shown in Table 3. Three O157:H7 were ST 11, four O136:H12 belong to ST 329. O26:H11 clustered in ST 21, ONT:H10 clustered in ST 441, O178:H19 clustered in ST 192, O81:H31 clustered in ST101, and O105:H8 clustered in ST 13.
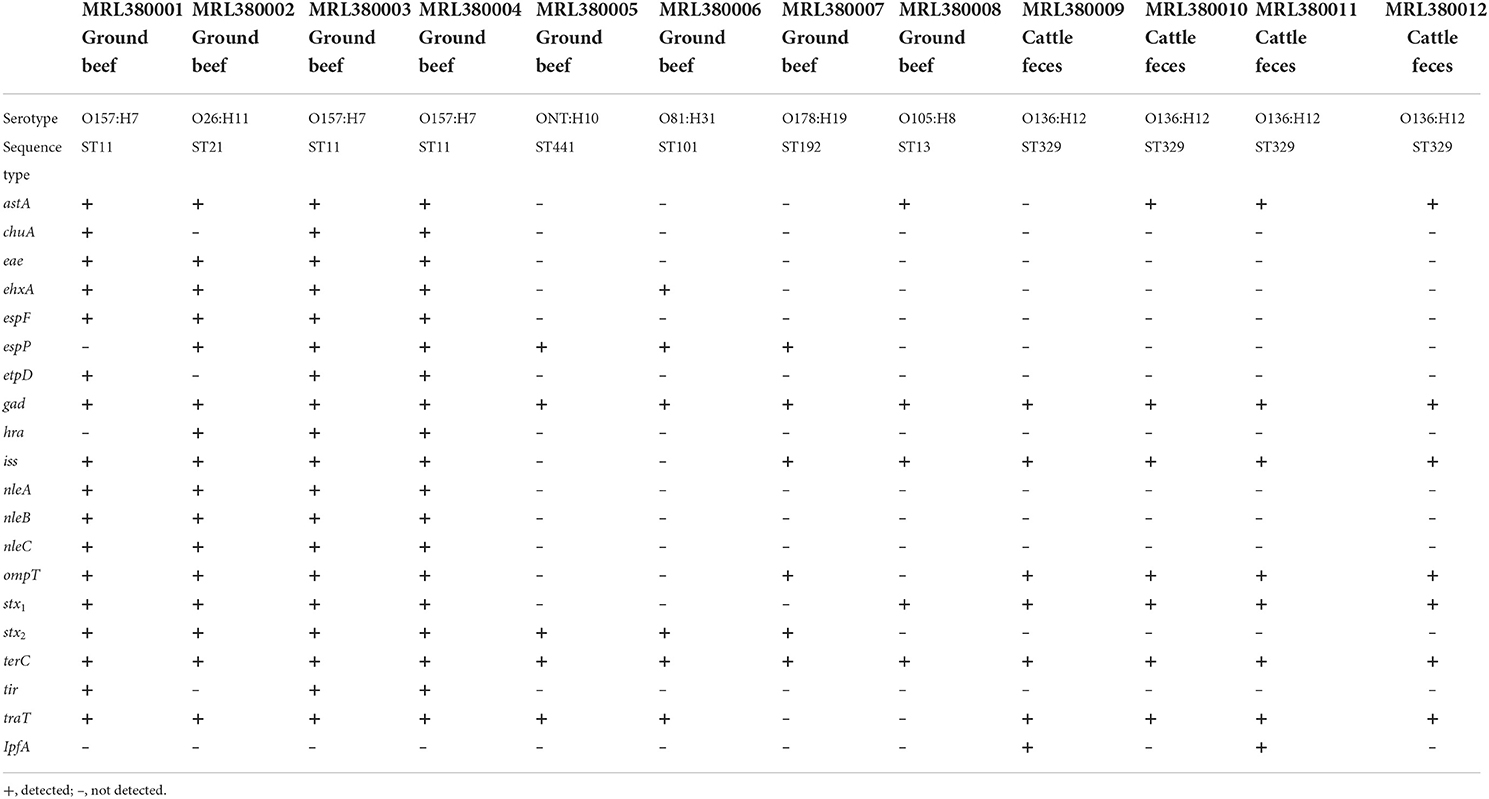
Table 3. Sequence types and virulence genes detected by WGS in 12 STEC strains in this study.
A total of 20 virulence genes were detected among the 12 STEC strains. The distribution of virulence genes among strains of different serogroups is shown in Table 3. Glutamate decarboxylase-encoding gad gene and tellurium ion resistance protein-encoding terC were the most widespread gene and were detected in all strains. Other well-known virulence genes found were as follows, with their distribution noted in parentheses: eae which encodes intimin (33.3%, n = 4), astA which encodes heat-stable enterotoxin (66.7%, n = 8), and ompT which encodes outer membrane protease (75%, n = 9). Four isolates with the combination of stx1 and stx2 as the most frequently detected type were found. Three isolates O157:H7 and one O26:H11 with stx1 and stx2 virulence genes were acquired, and the non-O157 serotype STEC isolated from ground beef with single stx2 was found. This result was consistent with previous studies in China, which reported that stx1 and stx2 were common in retail meat or slaughterhouses (Leung et al., 2001; Li et al., 2015, 2016). All the STEC obtained from 204 cattle feces were O136:H12 serotype and only contained stx1 virulence gene. WGS characterization of the isolates revealed that 12 isolates possessed AMR genes that can confer resistance to at least six classes of antimicrobials. As shown in Table 4, 27 AMR genes were detected, and each STEC isolate carried more than 10 AMR genes, suggesting that 12 STEC isolates may be multidrug-resistant. According to the health outcome of reported confirmed human STEC cases, serogroups O81:H31, ONT:H10 were classified to group E indicating non-human only disease, serogroups O178:H19, O105:H8 serotype were classified to group A/B/C indicating a HUS-associated serotype (Hazards, 2013). All those STEC isolates distributions indicated a higher diversity of serotypes in cattle and revealed a potential threat to consumers.
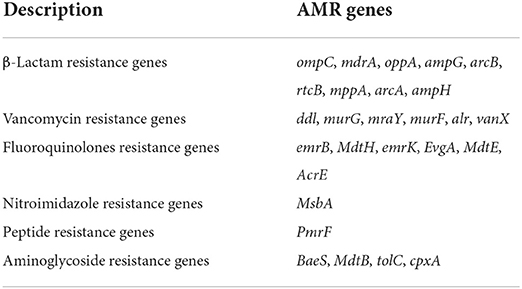
Table 4. Summary of resistance genes carried by STEC isolates.
The cgMLST scheme including 3,152 cgMLST targets, 1,485 accessory targets (Supplementary File 2), and 567 low-quality genes were filtered out. According to the cgMLST typing scheme, based on genome typing with PanGen.py in the chewBBACA tool, which uses Prodigal annotation, a total of 2,514 loci were selected as cgMLST targets shared by 12 STEC and 2,586 loci were selected as cgMLST targets shared by 169 STEC (Supplementary Files 3, 4). The MST was constructed based on the loci for the analysis of phylogenetic relationship with default.
The number of locus differences in core genome MLST minimum spanning tree ranged from 1 to 2,437 between 12 STEC isolates (Figure 1). Isolates of the same serotype are clustered together, four O136:H12 (MRL380009, MRL380010, MRL380011, and MRL380012) can be considered homologous because of their small allelic differences. Figure 2 shows a minimum spanning tree representing 12 STEC isolates from our lab and 157 STEC reference collections (Supplementary File 1) by cgMLST. In this study, STEC isolates representing various serotypes collections and without forming a separate cluster suggested that STEC isolates were phylogenetically related to STEC reference collections, on the one hand. On the other hand, epidemiologically related strains grouped together or were even part of a clonal cluster as shown for strains concurrently isolated from the farm. The top six non-STEC serotypes O145:H25 and four local isolated STEC strains O136:H12 clustered together, suggesting O136:H12 may be pathogenic. In order to determine the relatedness between different serotype STEC strains, the present study calculated the allelic distances between strains ranging from 1 to 2532. The greatest distances were observed between the O136:H12 and O145:H25. In addition, the same somatic antigen (O antigen) and the different flagella antigen (H antigen) STEC were divided into same lineages, such as O121:HNT, O121:H19 and O111:NM, O111:H8.
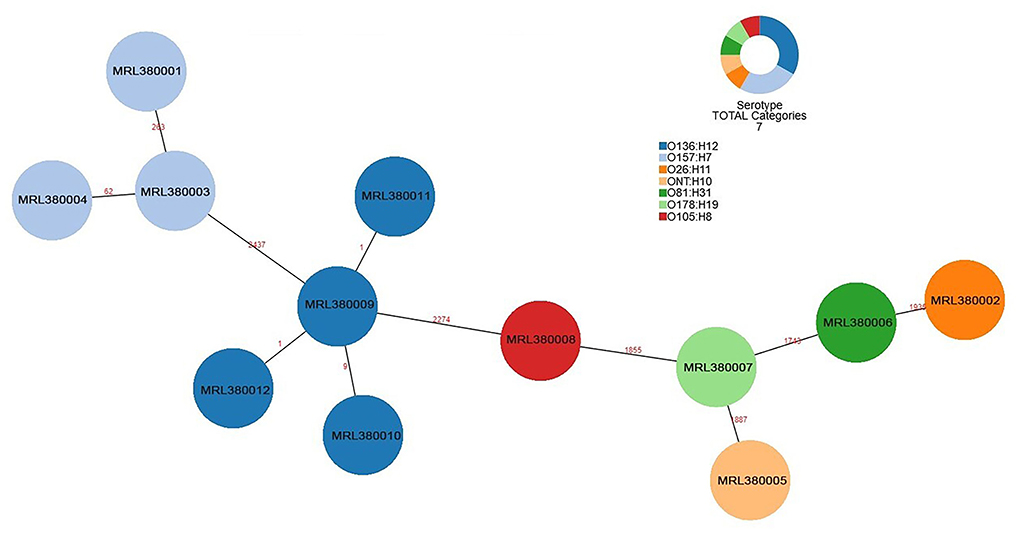
Figure 1. Minimum spanning tree for 12 STEC isolates from 289 samples based on the 2,514 loci, a total of 7 serotypes, different colors represent different serotypes.
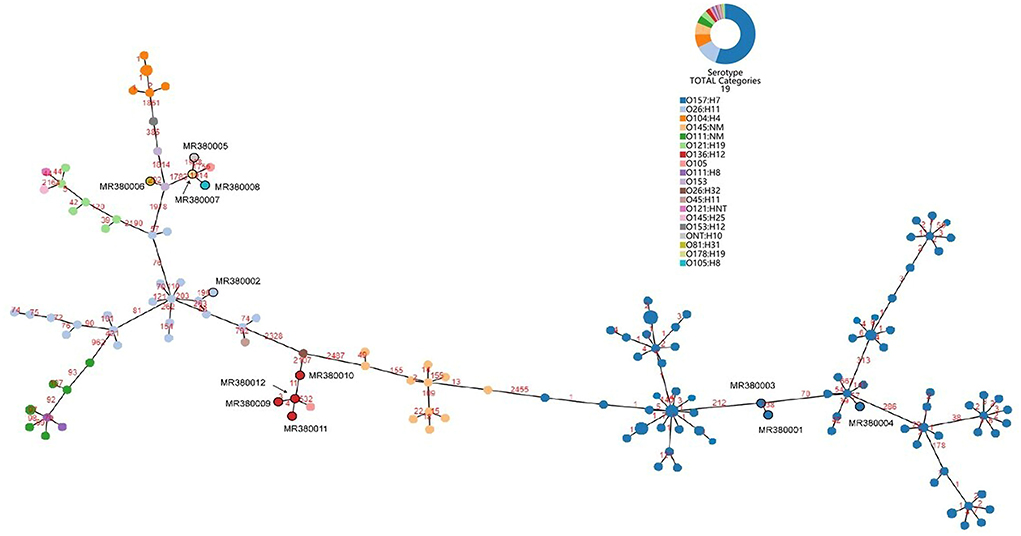
Figure 2. Minimum spanning tree for 12 STEC isolates from 289 samples and 157 STEC strains from NCBI based on the 2,586 loci, a total of 19 serotypes, different colors represent different serotypes.
Based on sequencing data and in silico analysis, STEC isolates from our lab and STEC reference collection strains revealed that the STEC isolates represented a heterogeneous group. A minimum spanning tree was constructed by all 169 STEC strains based on cgMLST method illustrated that STEC of the same serotype or ST were clustered together. However, STEC strains of the same O serogroup were located in the same phylogenetic clusters, isolates of the same H type, irrespective of their O serotype may not share a common ancestor. This finding was not consistent with the results from cgMLST typing analysis revealing H serogroups were described as monophyletic, while O serogroups were described as polyphyletic (Ju et al., 2012; Steyert et al., 2012; Ferdous et al., 2016).
Several limitations existed in this study. Firstly, STEC isolation and identification methods should be further improved, because parasites in cow manure can cause false positives during the initial screening process (Ferdous et al., 2016). STEC strain false positives can be decreased but not entirely eliminated by choosing CHROMagar™ STEC plates, MacConkey agar, and sorbitol MacConkey agar. The limited local STEC isolates and lacking clinical isolates might have influenced the reported diversity.
In conclusion, the results analyzing 12 isolates from food sources and feces of commercial animals, indicated a low prevalence of STEC in Shanghai, China. The capability of WGS providing rapid data for identification, serotyping, sequence typing, and virulence genes of STEC strains compared with traditional methods was further confirmed. STEC strains of the same O serogroup were located in the same phylogenetic clusters, isolates of the same H type, irrespective of their O serotype may not share a common ancestor by cgMLST. The study revealed that cgMLST typing method could be useful for outbreak investigations of STEC strains. In addition, the data stemmed from wide-ranging molecular characteristics with WGS resolution could be used as an effective approach for comparing with similar human, food, or animal isolates at the international level.
The data presented in the study are deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2017) in National Genomics Data Center (Nucleic Acids Res 2021), accession number CRA007022.
QD and HL conceived of the study and modified the first draft of the article. SZ is responsible for the experimental work, article writing, and data analysis. ZB and ZW isolated laboratory strains. XW and WW provided help with research ideas. All authors reviewed and approved the final manuscript.
This study was supported by foundation 2010DS700124-KF2001 from State Key Laboratory for Managing Biotic and Chemical Threats to the Quality and Safety of Agro-products, China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.950065/full#supplementary-material
Amézquita-López, B. A., Soto-Beltrán, B. G. L., and Jaszemyn, C. (2018). Isolation, genotyping and antimicrobial resistance of Shiga toxin-producing Escherichia coli. J. Microbiol. Immunol. 51, 425–434. doi: 10.1016/j.jmii.2017.07.004
Baha, M., Bouska, A. C. B., Zhang, W., and Brianna, L. S. H. (2019). Whole genome sequencing to characterize shiga toxin-producing Escherichia coli O26 in a public health setting. J. Infect. Public Health 12, 884–889. doi: 10.1016/j.jiph.2019.06.008
Baltasar, M., Caio, T. C. C., Raquel, S. P., and Susana, D. (2014). Impact of next generation sequencing techniques in food microbiology. Curr. Genomics. 15, 293–309. doi: 10.2174/1389202915666140616233211
Boer, R. F., Ferdous, M., Ott, A., and Scheper, H. R. (2015). Assessing the public health risk of Shiga toxin-producing Escherichia coli by use of a rapid diagnosticscreening algorithm. J. Clin. Microbiol. 53, 1588–1598. doi: 10.1128/JCM.03590-14
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chattaway, M. A., Dallman, T. J., Gentle, A., Michael, J. W., Sophie, E. L., and P. M. A.. (2016). Whole genome sequencing for public health surveillance of shiga toxin-producing Escherichia coli other than serogroup O157. Front. Microbiol. 7, 251–264. doi: 10.3389/fmicb.2016.00258
Ferdous, M, Friedrich, AW, Grundmann, H, de Boer, RF, Croughs, PD, and Islam, MA. (2016). Molecular characterization and phylogeny of Shiga toxin–producing Escherichia coli isolates obtained from two Dutch regions using whole genome sequencing. Clin. Microbiol. Infec. 22, 1.e1–1.e9. doi: 10.1016/j.cmi.2016.03.028
Gonzalez, A. G. M., and Cerqueira, A. M. F. (2020). Shiga toxin-producing Escherichia coli in the animal reservoir and food in Brazil. J. Appl. Microbiol. 128, 1268–1282. doi: 10.1111/jam.14500
Gutierrez, S., Díaz, L., Reyes-Jara, A., Yang, X., Meng, J., González-Escalona, N., et al. (2021). Whole-genome phylogenetic analysis reveals a wide diversity of non-O157 STEC isolated from ground beef and cattle feces. Front. Microbiol. 11, 622663. doi: 10.3389/fmicb.2020.622663
Hazards, E. (2013). Scientific opinion on VTEC-seropathotype and scientific criteria regarding pathogenicity assessment. EFSA J. 11, 3138. doi: 10.2903/j.efsa.2013.3138
Heir, E., Lindstedt, B. A., Vardund, T., Wasteson, Y., and Kapperud, G. (2000). Genomic finger printing of shiga toxin-producing Escherichia coli (STEC) strains: comparison of Pulsed-field gel electrophoresis (PFGE) and fluorescent amplified-fragment-length polymorphism (FAFLP). Epidemiol. Infect. 125, 537–548. doi: 10.1017/S0950268800004908
Jagadesan, S., Udayakumar, S. V., Paramasamy, G., and Jeyaprakash, R. (2019). Development and evaluation of a core genome multilocus sequence typing (cgMLST) scheme for Brucella spp. Infect. Genet. Evol. 67, 38–43. doi: 10.1016/j.meegid.2018.10.021
Jaureguy, F., Landraud, L., Passet, V., Diancourt, L., Frapy, E., Guigon, G., et al. (2008). Phylogenetic and genomic divers-ity of human bacteremic Escherichia coli straints. BMC Genomic. 9, 550–564. doi: 10.1186/1471-2164-9-560
Jenkins, C., Dallman, T. J., and Grant, K. A. (2019). Impact of whole genome sequencing on the investigation of food-borne outbreaks of Shiga toxin-producing Escherichia coli serogroup O157:H7, England, 2013 to 2017. Euro Surveill. 24:1800346. doi: 10.2807/1560-7917.ES.2019.24.4.1800346
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Nielsen, E. M., and Aaretrup, F. M. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Joensen, K. G., Tetzschner, A. M., Iguchi, A., Aarestrup, F. M., and Scheutz, F. (2015). Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. doi: 10.1128/JCM.00008-15
John, B. K., Suereta, F., Bridget, C., and Lourens, R. (2018). Proteomic comparison of three clinical diarrhoeagenic drug-resistant Escherichia coli isolates grown on CHROMagarTM STEC media. J. Proteomics. 180, 25–35. doi: 10.1016/j.jprot.2017.09.003
Ju, W. T., Cao, G., Timme, R., Errol, S., Yan, L., and Meng, J. L. (2012). Phylogenetic analysis of non-O157 Shiga toxin-producing Escherichia coli strains by whole-genome sequencing. J. Clin. Microbiol. 50, 4123–4127. doi: 10.1128/JCM.02262-12
Karmali, M. A., Mascarenhas, M., Shen, S., Ziebell, K., Clarl, C., Rahn, K., et al. (2003). Association of genomic O island 122 of Escherichia coli EDL 933 with verocytotoxin-producing Escherichiacoli seropathotypes that are linked to epidemic and/or serious disease. J. Clin. Microbiol. 41, 4930–4940. doi: 10.1128/JCM.41.11.4930-4940.2003
Keoser, C. U., Ellington, M. J., Gillespie, S. H., Brown, N. M., and Farrington, M. (2012). Routine use of microbial whole genome sequencing in diagnostic and public he-alth microbiology. PLoS Pathog. 8, e1002824. doi: 10.1371/journal.ppat.1002824
Leung, P. H., Yam, W. C., and Ng, W. W. (2001). The prevalence and characterization of verotoxin-producing Escherichia coli isolated from cattle and pigs in an abattoir in Hong Kong. Epidemiol. Infect. 126, 173–179. doi: 10.1017/S0950268801005210
Li, B., Yunchang, G., Ruiting, L., Yingping, D., Wei, W., Yujie, H., et al. (2015). Genotypic characterization of Shiga toxin-producing Escherichia coli O157:H7 isolates in food products from China between 2005 and 2010. Food Control 50, 209–214. doi: 10.1016/j.foodcont.2014.08.045
Li, R., Xiao, T., Xiao, J., Hongxun, W., Zhiguo, L., Min, Z., et al. (2016). Molecular screening and characterization of Shiga toxin-producing Escherichia coli in retail foods. Food Control 60, 180–188. doi: 10.1016/j.foodcont.2015.07.045
Mathusa, E. C., Chen, Y., Enache, E., and Lloyd, H. (2010). Non-O157 Shiga toxin-producing Escherichia coli in foods. J. Food Protect. 73, 1721–1736. doi: 10.4315/0362-028X-73.9.1721
Michaela, P., Marina, C. L., Petra, G., Andr,é, G., Sandra, C., Lorenz, W., et al. (2021). Genetic diversity and pathogenic potential of Shiga toxin-producing Escherichia coli (STEC) derived from German flour. Int. J. Food Microbiol. 347, 109197. doi: 10.1016/j.ijfoodmicro.2021.109197
Mikhail, A. F. W., Jenkins, C., Dallman, T. J., Inns, T., Douglas, A., Martin, A. I. C., et al. (2018). An outbreak of Shiga toxin-producing Escherichia coli O157:H7 associated with contaminated salad leaves: epidemiological, genomic and food trace back investigations. Epidemiol. Infect. 146, 87–196. doi: 10.1017/S0950268817002874
Mohammad, M., Masoumeh, D., Abbas, R. F., and Rahimeh, S. (2021). Molecular characterization and prevalence of virulence factor genes of Shiga toxin-producing Escherichia coli (STEC) isolated from diarrheic children. Gene Rep. 25, 101379. doi: 10.1016/j.genrep.2021.101379
Monaghan, Á., Byrne, B., Fanning, S., Sweeney, T., McDowell, D., and Bolton, D. J. (2012). Serotypes and virulotypes of non-O157 Shiga-toxin producing Escherichia coli (STEC) on bovine hides and carcasses. Food Microbiol. 32, 223–229. doi: 10.1016/j.fm.2012.06.002
Parsons, B., Zelyas, N., Berenger, B., and Chui, L. (2016). Detection, characterization, and typing of Shiga toxin-producing Escherichia coli. Front. Microbiol. 07, 478–490. doi: 10.3389/fmicb.2016.00478
Shifu, C., Yanqing, Z., Jia, G., and Yaru, C. (2018). fastp: an ultra-fast all-in-one FASTQ pre-processor. Bioinformatics 34, 884–890. doi: 10.1093/bioinformatics/bty560
Smith, J. L., Fratamico, P. M., and Gunther, N. (2014). Shiga toxin-producing Escherichia coli. Adv. Appl. Microbiol. 10, 145–197. doi: 10.1016/B978-0-12-800262-9.00003-2
Steyert, S. R., Sahl, J. W., Fraser, C. M., Fraser, C. M., Teel, L. D., Scheutz, F., et al. (2012). Comparative genomics and stx phage characterization of LEE-negative Shiga toxin-producing Escherichia coli. Front. Cell. Infect. Microbiol. 2, 133. doi: 10.3389/fcimb.2012.00133
Wang, P., Xiong, Y., Lan, R. T., Changyun, Y., Hua, W., Ren, J., et al. (2011). pO157_Sal, a novel conjugative plasmid detected in outbreak isolates of Escherichia coli O157: H7. J. Clin. Microbiol. 49, 1594–1597. doi: 10.1128/JCM.02530-10
Keywords: STEC, food, cgMLST, serogroups, phylogenetic relationships
Citation: Zhang S, Bai Z, Wang Z, Wang X, Wang W, Li H and Dong Q (2022) Molecular characterization and phylogeny of Shiga toxin-producing Escherichia coli derived from cattle farm. Front. Microbiol. 13:950065. doi: 10.3389/fmicb.2022.950065
Received: 22 May 2022; Accepted: 07 July 2022;
Published: 04 August 2022.
Edited by:
Lin Lin, Jiangsu University, ChinaReviewed by:
Alexandre Leclercq, Institut Pasteur, FranceCopyright © 2022 Zhang, Bai, Wang, Wang, Wang, Li and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei Li, c3VubnlzYW5kQDEyNi5jb20=; Qingli Dong, cWRvbmdAdXNzdC5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.