
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 25 October 2022
Sec. Infectious Agents and Disease
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.949328
This article is part of the Research Topic Genomics-based Strategies for Advanced Drug Resistance and Epidemiological Surveillance of Mycobacterium tuberculosis and other Non-Tuberculous Mycobacteria View all 8 articles
 Nienke A. Kok1†
Nienke A. Kok1† Nilay Peker1
Nilay Peker1 Leonard Schuele1
Leonard Schuele1 Jessica L. de Beer1,2
Jessica L. de Beer1,2 John W. A. Rossen1,3,4‡
John W. A. Rossen1,3,4‡ Bhanu Sinha1‡
Bhanu Sinha1‡ Natacha Couto1,5*‡
Natacha Couto1,5*‡Identification and phenotypic drug-susceptibility testing for mycobacteria are time-consuming and challenging but essential for managing mycobacterial infections. Next-generation sequencing (NGS) technologies can increase diagnostic speed and quality, but standardization is still lacking for many aspects (e.g., unbiased extraction, host depletion, bioinformatic analysis). Targeted PCR approaches directly on sample material are limited by the number of targets that can be included. Unbiased shotgun metagenomics on direct material is hampered by the massive amount of host DNA, which should be removed to improve the microbial detection sensitivity. For this reason, we developed a method for NGS-based diagnosis of mycobacteria directly from patient material. As a model, we used the non-tuberculous mycobacterium (NTM) Mycobacterium abscessus. We first compared the efficiency of three different DNA extraction kits for isolating DNA (quality and concentration). The two most efficient kits were then used in a follow-up study using artificial sputum. Finally, one extraction kit was selected and further evaluated for DNA isolation from a patients’ sputum mixture spiked with M. abscessus at three concentrations (final concentrations 108, 107, 106 CFU/ml). The spiked sputum samples were processed with and without saponin treatment (ST) in combination with DNAse treatment prior to bacterial DNA extraction to evaluate the recovery of bacteria and depletion of host DNA by PCR and Illumina sequencing.
While Ct values of the qPCR targeting mycobacterial ITS DNA remained rather stable, Ct values in the qPCR targeting the human β-actin gene increased by five Ct values in ST samples. In subsequent Illumina sequencing, a decrease of 89% of reads mapped to the human genome was observed in ST samples. The percentage of reads mapped to M. abscessus (108 CFU/ml) increased by 89%, and the sequencing depth increased two times when undergoing ST.
In conclusion, the sensitivity of M. abscessus detection in artificial sputum was increased using a saponin pre-treatment step. The saponin followed by the DNase I treatment approach could be efficiently applied to detect and characterize mycobacterial infections, including tuberculosis, directly from sputum.
Mycobacteria causing infections are classified as tuberculosis-causing and non-tuberculosis mycobacteria (NTM) (Johansen et al., 2020). Tuberculosis (TB) caused by Mycobacterium tuberculosis is one of the leading causes of death, with over 1 million deaths worldwide yearly (World Health Organization, 2019). Although the M. tuberculosis mutation rate is relatively low under optimized treatment conditions, multidrug-resistant (MDR) and extensively drug-resistant (XDR) M. tuberculosis are advancing rapidly (World Health Organization, 2018). A prompt and accurate characterization of the complete resistance profile of TB, which currently takes 2–6 weeks or longer in routine diagnostic laboratories, is critical to optimize appropriate antimicrobial therapy, thereby reducing antimicrobial selection pressure (World Health Organization, 2021). To implement a faster method to detect and characterize TB and NTM, broad genotypic testing of direct material using molecular techniques such as polymerase chain reaction (PCR) is already applied in many laboratories. These sensitive molecular techniques can provide results within hours and have been successfully implemented even in low resource settings (World Health Organization, 2018). Next-generation sequencing (NGS), on the other hand, is being used for comprehensive genotypic drug susceptibility testing (DST) and is usually performed on culture isolates as direct testing of clinical samples by shotgun metagenomics (SMg) results in the sequencing of vast amounts of human DNA (World Health Organization, 2021). This hampers the sensitivity of detection and the genome coverage necessary for predicting genotypic drug susceptibility and inferring genetic relatedness.
Enrichment of bacterial DNA by human DNA depletion has improved pathogen detection by NGS (Hasan et al., 2016). In this study, human DNA was depleted using a saponin pre-treatment (ST) step. Saponin is a secondary plant compound that permeabilizes cell membranes by targeting cholesterol. The formation of a saponin-cholesterol complex in the cell membrane causes pores in the cell wall of eukaryotic cells resulting in membrane instability (Böttger and Melzig, 2013). Because bacteria do not have cholesterol, they are not affected by this treatment. The ST protocol used in this study is based on a recently published ST protocol optimized for nanopore sequencing of lower respiratory infections, but that did not include mycobacterial species (Charalampous et al., 2019). Since saponin has been reported to have a negative effect on the DNA recovery of some bacterial species (Street et al., 2020), we wanted to test the saponin protocol in mycobacteria-containing samples. Additionally, the efficiency of three different DNA extraction kits was studied. Mycobacterium abscessus is a rapidly growing mycobacterium associated with pulmonary infections (Brown-Elliott and Philley, 2017) that does not need to be processed under Biosafety Level-3 (BSL 3) laboratory conditions. Additionally, M. abscessus has been shown to share conserved strategies for host adaptation and persistence with M. tuberculosis (Low et al., 2017). Therefore, we developed an NGS-based method for detecting the presence and resistance patterns of M. abscessus (ATCC 19977) as a model for mycobacteria. Overall, we aimed to develop an optimized SMg protocol that can be applied to detect and characterize mycobacterial infections.
Mycobacterium abscessus ATCC 19977 (ATCC, London, United Kingdom) was initially grown on blood agar plates (product and manufacturer) at 37°C for 3–5 days until bacterial growth was visible. A stock suspension of M. abscessus (1.21 × 109 CFU/ml) was prepared in 1 × PBS (Thermo Scientific, Waltham, MA, United States), and 10-fold serial dilutions were prepared in duplicate, ranging from a concentration of 1.21 × 107 to 1.21 × 109 CFU/ml. Bacterial concentrations were confirmed by plating eight serial dilutions on blood agar plates (BD, Franklin Lakes, United States; 100 μl/plate) and counting plates that showed 30–300 colonies.
DNA extraction from M. abscessus isolates was performed using the (i) DNeasy UltraClean Microbial kit (Qiagen Benelux, Venlo, Netherlands; DNeasy), (ii) QIAamp DNA Mini kit (Qiagen; DNA Mini), and (iii) AllPrep PowerFecal DNA/RNA kit (Qiagen; AllPrep). All kits were used following the respective manufacturer’s recommended protocol with the following minor modifications (Qiagen, 2016, 2017, 2018a): for all kits, the starting volume was 200 μl, and the DNA was eluted twice to a total elution volume of 30 μl of the respective elution buffer provided by the kits; the AllPrep protocol was adjusted using 2 ml PowerBead Pro tubes (cat. No. 19301; Qiagen) instead of the provided Microbial Lysis Tubes; centrifugation steps were adjusted to a maximum of 10,000 ×g, and the centrifugation time was doubled if a higher centrifugation force was required (Merck KGaA, 2018). The purity of DNA was analyzed using the NanoDrop™ 2000 spectrophotometer (Thermo Scientific), and DNA concentrations were measured using the Qubit™ 2.0 and the dsDNA High Sensitivity Assay Kit (Thermo Scientific). Afterwards, a quantitative PCR (qPCR) targeting the 16S rRNA gene was performed using the Applied Biosystems 7500 Real-Time PCR System (Thermo Scientific) with the primers listed in Supplementary Table S1.
Spike-in experiments were initially conducted using artificial sputum, consisting of mucin, salmon sperm, egg yolk and salts (the protocol is available at: https://protocols.scienceexchange.com/protocols/artificial-sputum-medium) (Sriramulu et al., 2005). After preparation, the artificial sputum was aliquoted and stored at –80°C. Artificial sputum samples (250 μl) were spiked in triplicate with 50 μl of the M. abscessus bacterial stock suspension at two initial dilutions (1.21 × 109 and 1.21 × 108 CFU/ml; final concentrations of 2.02 × 108 and 2.02 × 107 CFU/ml, respectively). Prior to DNA extraction, 300 μl of spiked samples were treated with 200 μl of a 2.5% saponin solution (Merck; diluted in 1 × PBS and sterile filtered). Samples were vortexed thoroughly and incubated at room temperature for 10 min to promote host cell lysis. Subsequently, 350 μl of nuclease-free water (Merck) was added, and samples were incubated for 30 s. For the osmotic shock, 12 μl of 5 M NaCl (Merck) were added to all samples. Samples were centrifuged at 6,000 × g for 5 min, and pellets were resuspended in 100 μl 1 × PBS (Thermo Scientific). Next, 15 μl DNase I [20,000 Units (50 to 375 U/μl); Thermo Scientific] and 11 μl of 10 × DNase buffer (Thermo Scientific) were added to digest free DNA. Samples were incubated in a thermomixer (Eppendorf, Hamburg, Germany) with shaking at 800 rpm at 37°C for 30 min. After incubation, samples were centrifuged at 6,000 × g for 3 min, and pellets were washed twice with 800 μl 1 × PBS (Sriramulu et al., 2005). Finally, pellets were resuspended in the lysis buffer of the AllPrep and DNeasy kits, following the respective manufacturer’s instructions. Extracted DNA was eluted twice with 15 μl of elution buffer and centrifuged at 6,000 × g for 1 min. Concentration and purity of DNA were measured using the Qubit™ 2.0 with the dsDNA High Sensitivity Assay Kit (Thermo Scientific) and the NanoDrop™ 2000 spectrophotometer (Thermo Scientific), respectively. Fragment length of ST and saponin not-treated (NT) samples spiked with 1.21 × 109 CFU/ml M. abscessus was measured using the Agilent 2,200 TapeStation (Agilent, Santa Clara, CA, United States). Then, qPCR targeting the human β-actin gene and 16S rRNA gene (Supplementary Table S1) was performed using the LightCycler 480 Instrument (Roche Diagnostics).
The workflow is illustrated in Figure 1. Mycobacterium-negative sputum samples screened for using an internal transcribed spacer high resolution melting curve (ITS-HRM) PCR, using a mycobacterial specific primer set (Akkerman et al., 2013) from different patients analyzed for general microbiological diagnostics in Labmicta (Hengelo, Netherlands) were pooled until a total sputum volume of approximately 15 ml was obtained. To promote mucolysis, approximately 1 gram, depending on the viscosity of the sputum, of N-Acetyl-L-Cysteine (NALC; Merck, Darmstadt, Germany) was added and vortexed until liquefied. Samples were processed as described above (50 μl of bacterial stock was spiked into 250 μl sputum), an additional dilution was used (1.21 × 107 CFU/ml), and experiments were performed in duplicate. Only the AllPrep kit was tested further using this protocol based on prior validation experiments. Additionally, the ITS-HRM PCR was performed using a primer/probe set (Supplementary Table S1) that specifically detects mycobacterial DNA using the Applied Biosystems 7,500 Real-Time PCR System (Thermo Scientific).
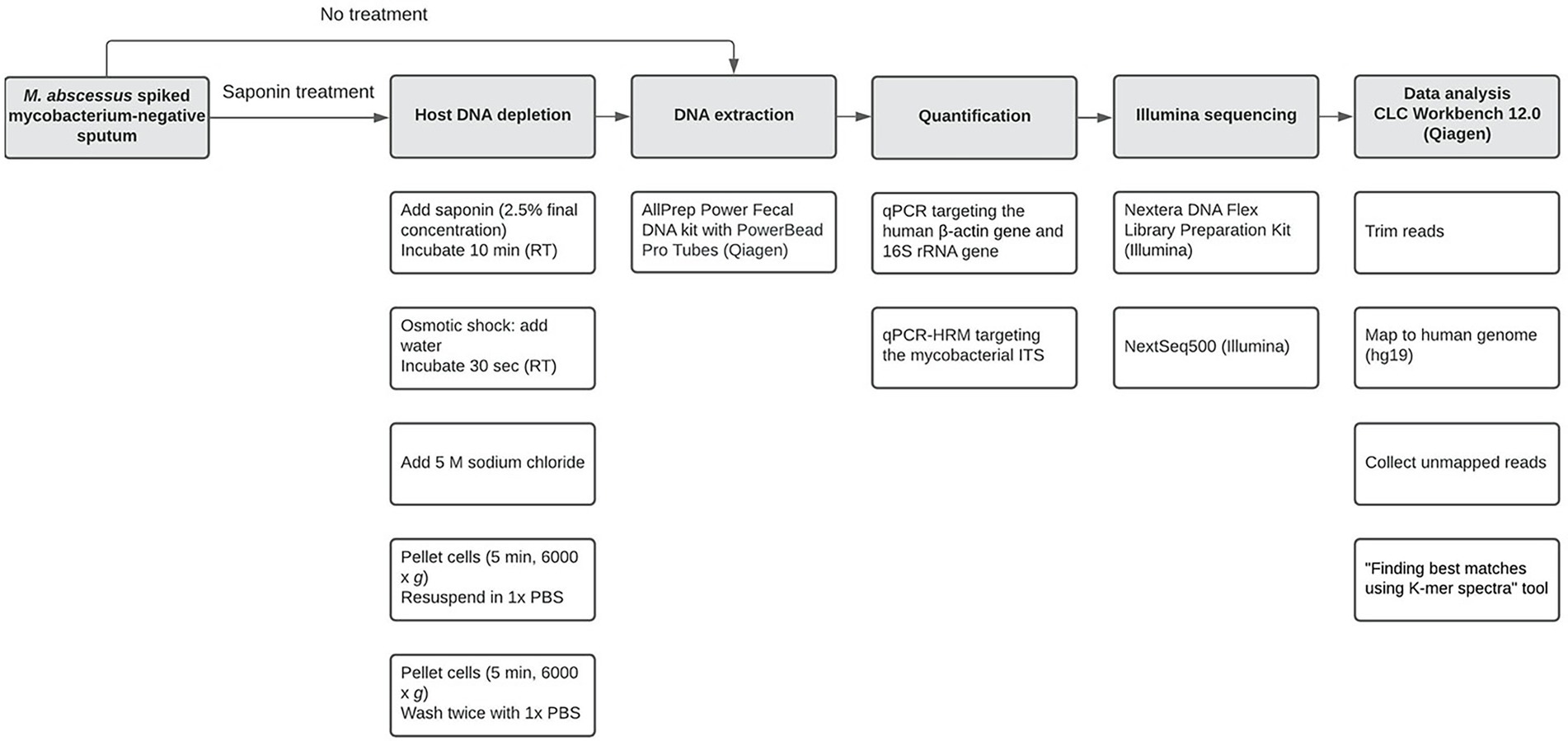
Figure 1. Workflow used in the current study from spiking Mycobacterium-negative sputum until data analysis. Open boxes in a column represent specific subsequent sub-steps (from top to bottom) for major steps (shaded boxes).
DNA pellets of duplicate samples were pooled and purified with 1.8 volumes of AMPure XP Beads (Beckman Coulter, Indianapolis, United States) according to the manufacturer’s instructions and eluted in 20 μl water (Coulter, 2016). Sequencing libraries were prepared using the Nextera DNA Flex Library Prep Kit according to the manufacturer’s protocol (Illumina, San Diego, CA, United States) (Illumina, 2019) and sequenced using the Illumina NextSeq 500 (Illumina) to generate 2 × 150 bp paired-end reads. The M. abscessus average sequencing depth was 40.4 (maximum 85.5 and minimum 3.4; Supplementary Table S2).
Sequencing reads (fastq files) were analyzed using the CLC Genomics Workbench v21.0.5 (Qiagen) (Qiagen, 2018b) with default settings and parameters unless mentioned otherwise. First, the paired-end reads were quality and adapter trimmed. Then they were mapped (Map Reads to Reference v1.8) to the reference human genome (hg19), and unmapped reads were analyzed using the “Find Best Matches using K-mer Spectra” tool v1.4 (CLC Microbial Genomics Module 21.1) and an in-house microbial database (2018-10-18) build-in CLC Genomics Workbench. The relative abundance of M. abscessus was compared between ST and NT sputum spiked samples. For microbial characterization, reads were also uploaded to the One Codex Platform (One Codex, San Francisco, CA, United States; Minot et al., 2015).
Statistical analysis was performed using Prism 9 for macOS v9.3.0 (GraphPad Software, San Diego, CA, United States). Results were considered significant at p < 0.05. For numerical variables, the parametric paired t-test (following Shapiro–Wilk normality test) or nonparametric Mann–Whitney or Wilcoxon signed rank tests were used to compare groups.
Sputum samples not used for routine diagnostics were collected with approval from the Medical Ethics Review Board of UMCG (METc number 2018/393). The study was performed in adherence to the guidelines of the Declaration of Helsinki and local regulations, and all patient data were treated pseudo-anonymously.
Bias is a pervasive problem when performing DNA extraction (McCarthy et al., 2015). An important factor is the difference in lysis efficiencies of different bacterial populations, which vary depending on the extraction protocol (McCarthy et al., 2015). Therefore, we tested three different extraction kits on M. abscessus suspensions. The quality and concentration of DNA using the three different extraction kits were compared. DNA concentrations were highest when using the DNeasy and AllPrep kits (Table 1).
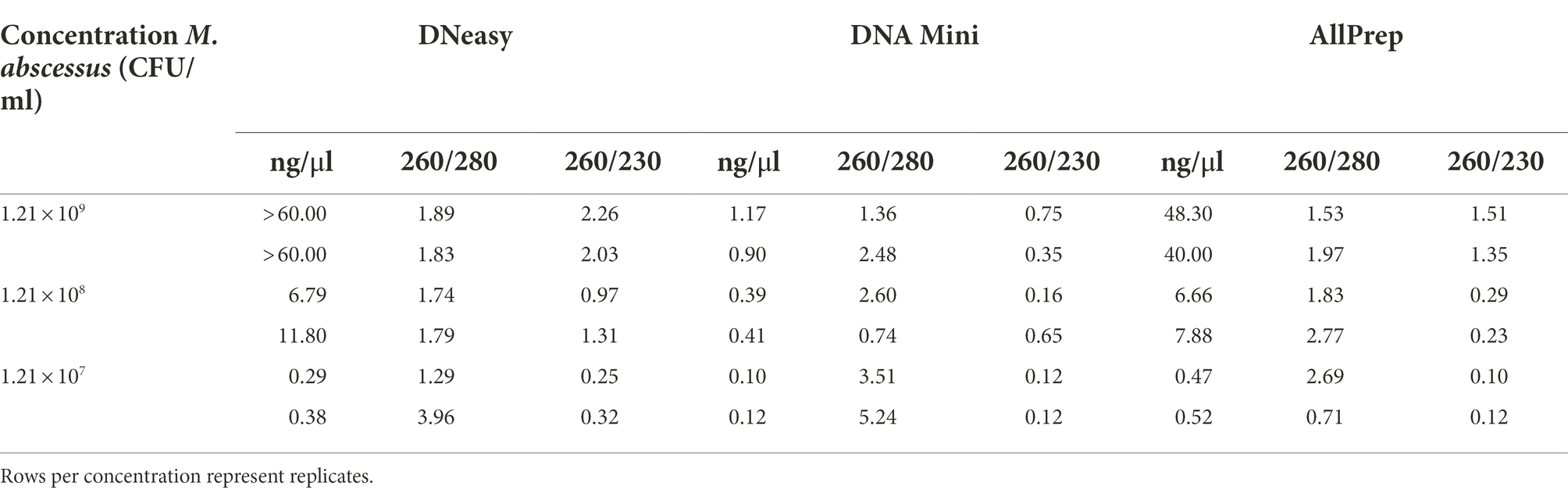
Table 1. Quantification and quality of DNA extracted through three commercially available kits (DNeasy, DNA Mini, and AllPrep).
Additionally, quantification of the 16S rRNA gene by qPCR showed that the DNeasy and AllPrep kits both yielded more bacterial DNA than the Mini kit (Figure 2).
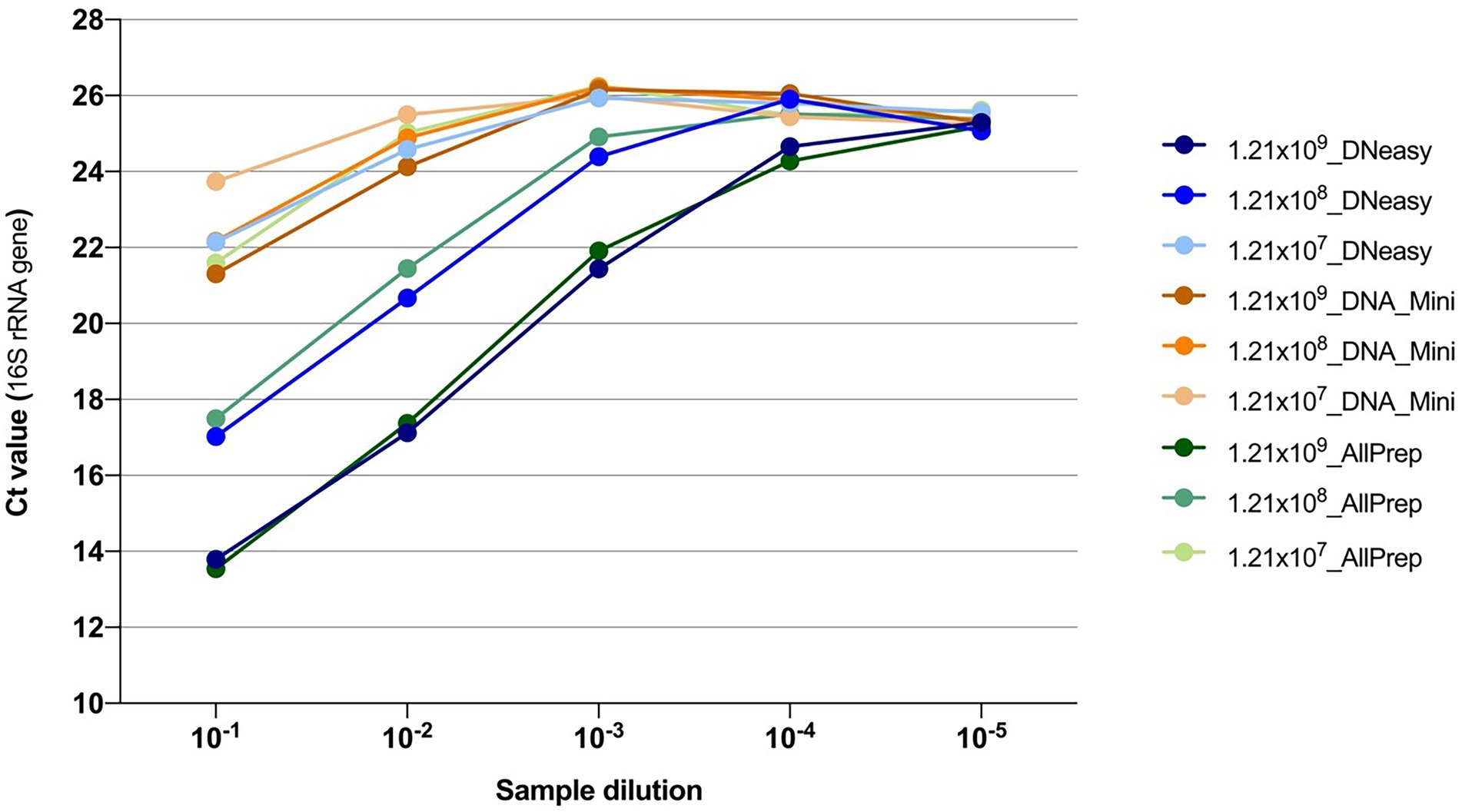
Figure 2. Quantification of the 16S rRNA gene using qPCR from DNA extractions. Colors indicate the combination of the concentration of the bacteria and the kit used for extracting the DNA. After extraction, DNA samples were 10-fold serially diluted, and five dilutions were subsequently tested.
Interestingly, in a recent study comparing DNA extraction strategies for livestock fecal microbiota, the Allprep PowerFecal DNA/RNA Kit (Qiagen) yielded the lowest concentrations of DNA and the lowest quality (260/280 values were all outside the acceptable range of 1.8–2.0) (Wegl et al., 2021). This is discordant with the results obtained in the present study. However, it should be noted that the sample matrix and complexity can differ substantially between studies. Moreover, we substituted the Microbial Lysis Tubes in the Allprep kit with the PowerBead Pro tubes to increase DNA extraction efficiency, improving DNA concentration and quality. Since the DNeasy and AllPrep kits showed the most promising DNA yield and quality results, they were used in the follow-up study using artificial sputum.
Host depletion or target enrichment is an important step in a metagenomics workflow to increase the sensitivity of the method (Schuele et al., 2021). Saponin treatment effectively depletes host DNA in respiratory samples with a maximum of 104-fold depletion (median 600-fold) of human DNA (Charalampous et al., 2019). However, saponin has been reported to have a negative impact on the DNA recovery of certain bacterial species (Street et al., 2020). In this study, we opted for a host depletion step because the method developed here could potentially be applied for the detection and characterization of multiple respiratory pathogens (not only mycobacteria) and even for microbiome studies.
We first tested our saponin protocol using artificial sputum to understand the effect of saponin treatment on yields of mycobacterial DNA. As shown in Figure 3A and Supplementary Table S3, ST resulted in slightly lower DNA concentrations, independent of the extraction kit used, although differences were not statistically significant (p > 0.05). Additionally, this did not result in significantly (p > 0.05) different 16S rRNA gene quantifications (Figure 3B; Supplementary Table S3), suggesting that although higher DNA concentrations were obtained in NT samples, this was not reflected in higher mycobacterial DNA concentrations and probably reflects the extraction of salmon DNA, which is digested in the ST method by the addition of DNase I. When comparing ST and NT samples, the fragment lengths of extracted DNA were similar (μ = 15,567 bp compared to μ = 15,945 bp, respectively; Supplementary Figure S1).
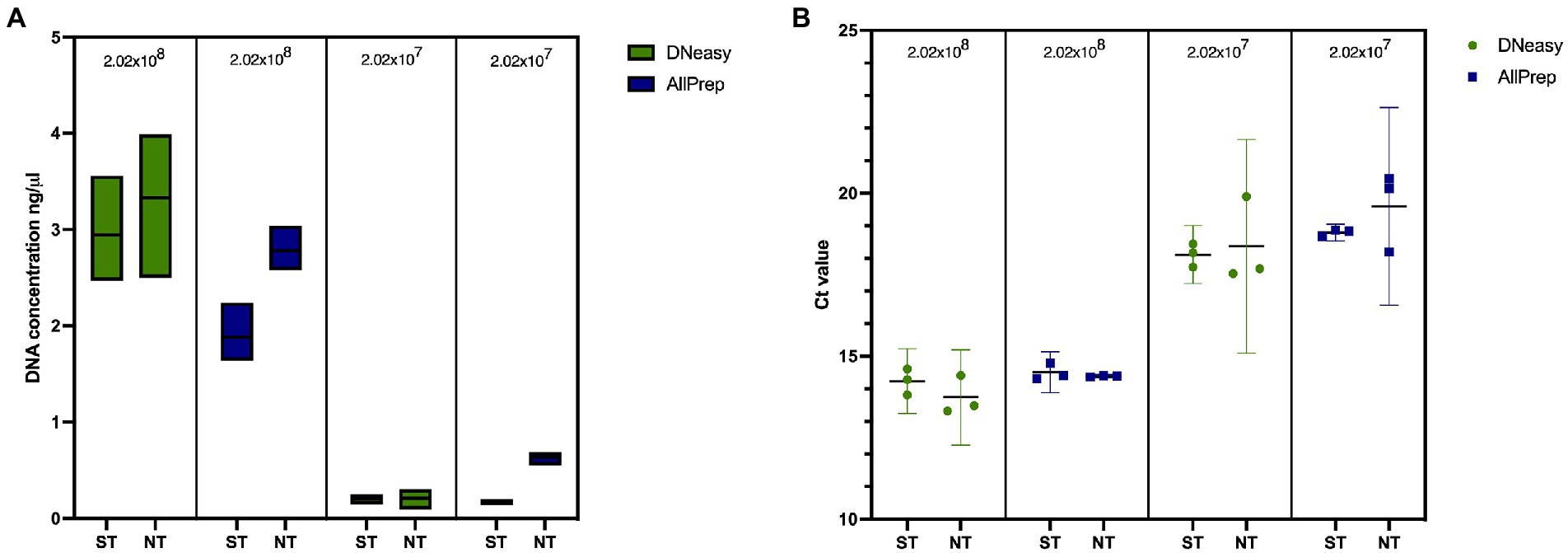
Figure 3. Effect of saponin and two DNA extraction kits on artificial sputum samples in terms of (A) DNA concentration measured with QubitTM 2.0 and (B) qPCR targeting the 16S rRNA gene in spiked artificial sputum. ST, saponin treated; NT, not treated.
Lower DNA concentrations were obtained when extracting samples with the AllPrep kit than with the DNeasy kit. However, these differences were less prominent after quantifying the 16S rRNA gene (Figure 3B; Supplementary Table S3). Fragment lengths were longer when using the AllPrep kit (μ = 17,603 bp) than with the DNeasy kit (μ = 13,912 bp), although this difference was not statistically significant (p = 0.3333; Supplementary Figure S1). Contrary to the DNeasy kit, the AllPrep kit allows for differential nucleic acid extraction of DNA and RNA in separate eluates, which allows the detection of RNA viruses or look at indicators of host response (if no depletion step is used). The results provided by this kit were more repeatable (with less variation between replicates). Therefore, we decided to use the AllPrep kit for experiments with human sputum.
After evaluating the effect of saponin treatment on the recovery of M. abscessus DNA, we tested the effect of saponin treatment on the differential lysis of human cells and subsequently depletion of human DNA. To comply with ethical standards and for practical reasons, we pooled mycobacterial-negative sputum samples from different individuals. Higher DNA concentrations were obtained in the NT samples (Figure 4A). The human β-actin gene was detectable five cycles earlier in the NT conditions indicating a higher amount of human DNA in these samples (Figure 4B). This difference was statistically significant (p = 0.0008; paired t-test) and suggested that saponin had a significant lytic effect on human cells, which then resulted in lysis of the human DNA by the DNase I treatment. The effect of the ST on M. abscessus was negligible looking at the Ct values of the qPCR targeting the mycobacterial ITS, which hardly changed in ST compared to NT samples (Figure 4C), indicating it had no negative impact on M. abscessus DNA recovery.
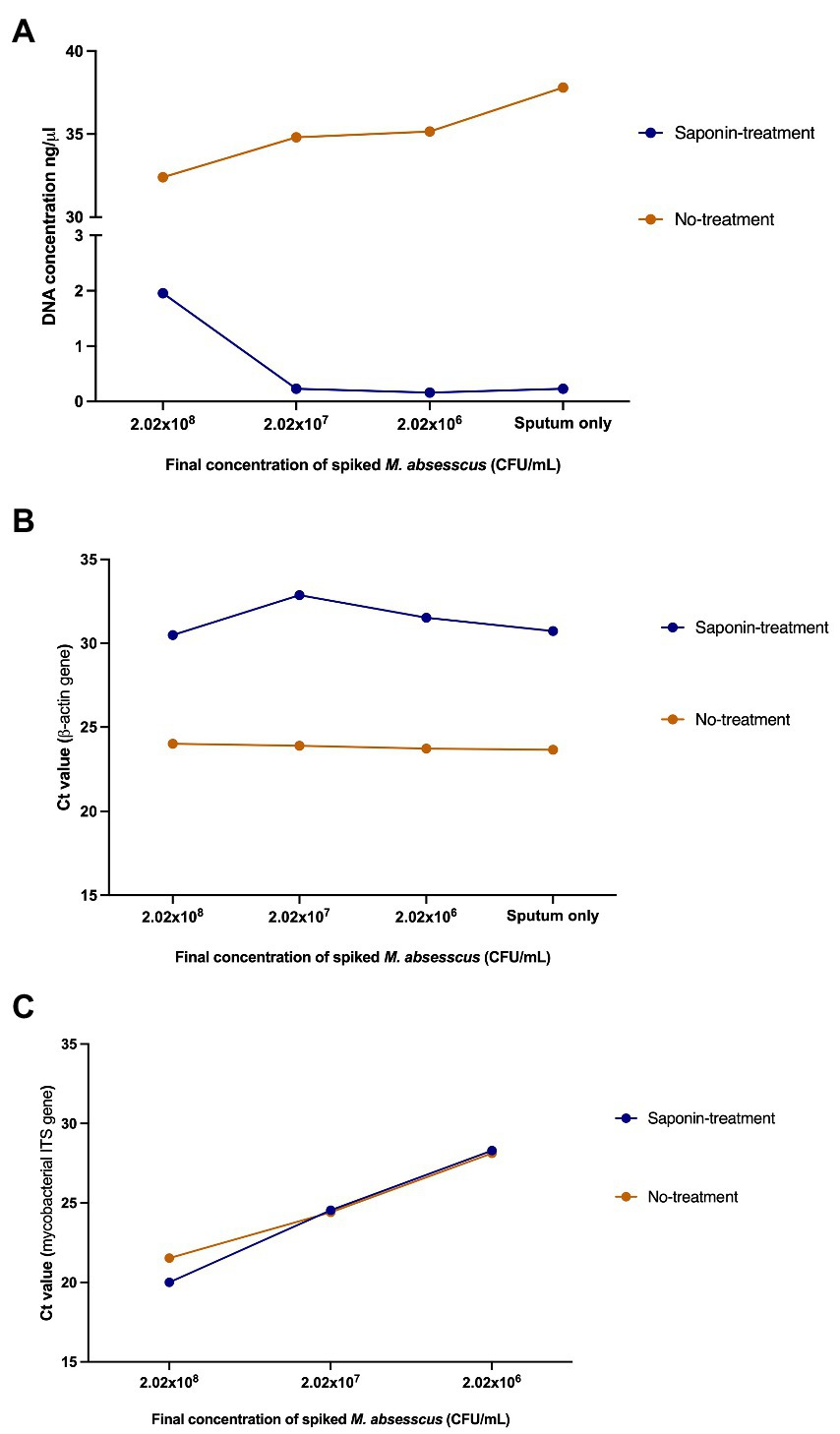
Figure 4. Effect of saponin treatment on human and M. abscessus DNA. (A) DNA quantification using QubitTM v2.0, (B) qPCR targeting the human β-actin gene, and (C) qPCR targeting the mycobacterial internal transcribed spacer (ITS) on spiked human sputum (sputum only: non-spiked sputum sample). Concentrations and Ct values shown in this figure represent the average of the duplicates.
To confirm the previously obtained qPCR results, we sequenced the samples and assessed the efficiency of saponin in depleting host DNA by comparing the proportion of M. abscessus and human reads between ST and NT samples. In ST samples with a final M. abscessus concentration of 2.02 × 108 CFU/ml, 2.02 × 107 CFU/ml and 2.02 × 106 CFU/ml, approximately 1.5, 35.8 and 82.9% of the reads mapped to the human genome, respectively, compared to 90.5, 97.1 and 97.1% in the NT samples, respectively (Figure 5; Supplementary Table S2).
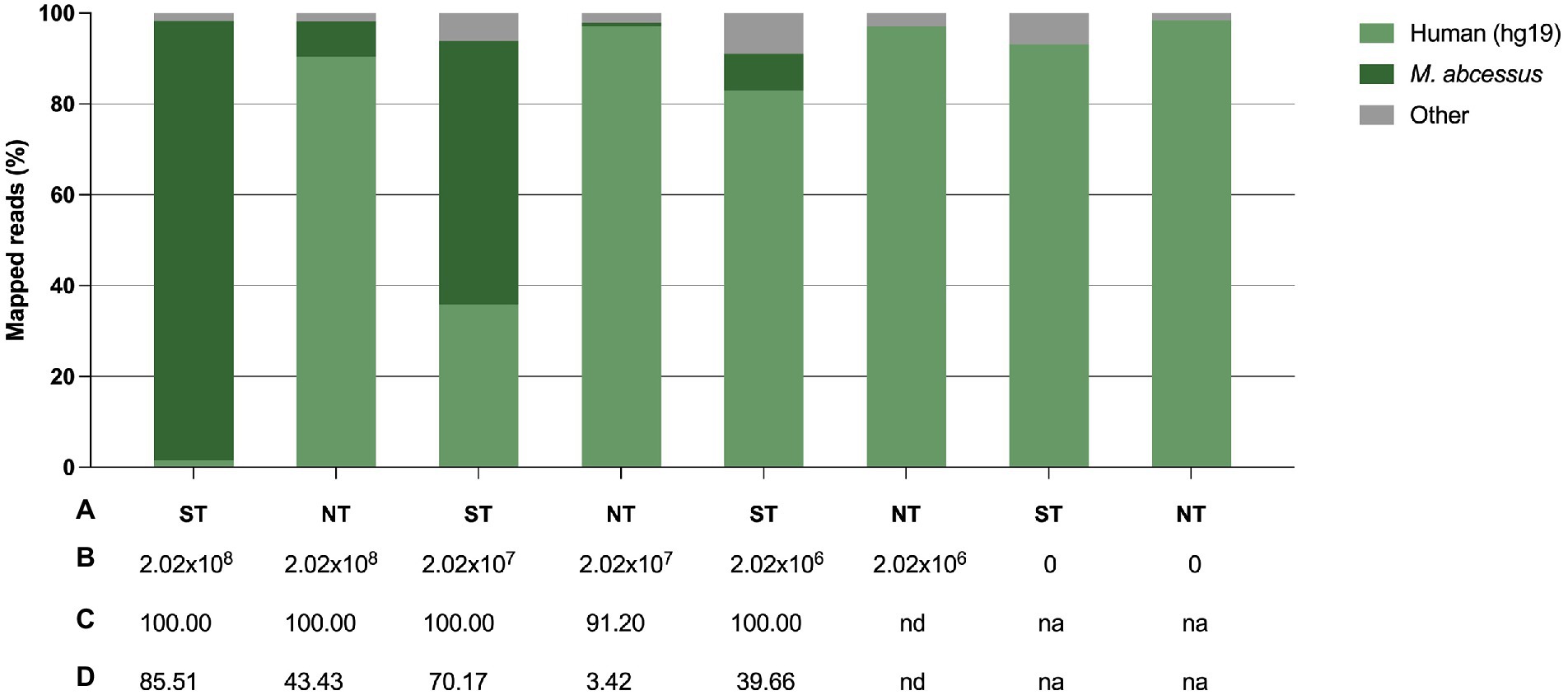
Figure 5. Effect of saponin treatment (depleting host DNA) on sputum spiked with M. abscessus at three different concentrations. (A) Condition, i.e., saponin treated (ST) or non-treated (NT), (B) Final concentration of spiked M. abscessus (CFU/ml), (C) M. abscessus genome coverage (%), (D) M. abscessus mean sequencing depth (average number of reads per sites). 0 concentration refers to non-spiked sputum samples. na, not applicable; nd, not detected.
A difference in the proportion of reads mapped to the human genome, although smaller, was also noted in treated and non-treated non-spiked control samples (sputum only). Overall, there was a 60-fold, 2.7-fold and 1.2-fold decrease in the proportion of human reads in ST samples with a final M. abscessus concentration of 2.02 × 108 CFU/ml, 2.02 × 107 CFU/ml and 2.02 × 106 CFU/ml, respectively. There was a 12.5-fold, 74.5-fold increase in the proportion of M. abscessus reads in ST samples with a final M. abscessus concentration of 2.02 × 108 CFU/ml and 2.02 × 107 CFU/ml, respectively. In samples with a final M. abscessus concentration of 2.02 × 106 CFU/ml, 8.1% of the reads were classified for M. abscessus in ST samples, whereas M. abscessus could not be detected in NT samples. At lower concentrations of M. abscessus (2.02 × 107 and 2.02 × 106 CFU/ml), full genome coverage was only obtained after ST. ST also resulted in a higher sequencing depth for M. abscessus. High genome coverage is essential to identify bacteria to the species level, gene sequences associated with antimicrobial resistance, and for accurate variant (or SNP) calling for outbreak management.
Other methods of human DNA depletion have been tested in M. tuberculosis-positive samples. One was based on osmotic lysis by adding a large volume of sterile water to a sputum sample to increase osmotic pressure and cause human cells to burst while leaving the more robust M. tuberculosis cells intact (Doughty et al., 2014). A DNase enzyme is then used to degrade the released human DNA prior to M. tuberculosis DNA extraction. However, this approach is severely limited for acquiring sufficient (due to the high host to bacteria cell ratio) bacterial DNA from low load samples without enrichment (Doughty et al., 2014) and some co-infection causing pathogens of interest, such as Gram-negative bacteria, are also susceptible to osmotic pressure (Bachmann et al., 2018). Votintseva et al. (2017) used a commercial kit that selectively lyses human and other eukaryotic cells in M. tuberculosis-positive respiratory samples. This kit is based on a chaotropic buffer that selectively lyses eukaryotic cells while keeping the bacterial cells intact, however, the identity of the chaotropic buffer is not disclosed by the manufacturer (Bachmann et al., 2018). They were able to identify M. tuberculosis in all samples from which DNA was successfully extracted, and sufficient data for antimicrobial susceptibility prediction was obtained from 62% of the samples (Votintseva et al., 2017). The sample composition affected the number of M. tuberculosis reads and depended on the amount of human and bacterial DNA in the sample (Votintseva et al., 2017). Although, the use of certain commercial extraction kits can reduce the turnaround time and are easier to standardize and validate (Votintseva et al., 2017), they are usually more costly and might not be suitable for low- and middle-income settings where there is unavailability or inaccessibility of certain kits/reagents (Boro and Stoll, 2022).
To understand the broader impact of human DNA depletion, we evaluated the effect of saponin treatment followed by DNase I digestion on other microbial species in the sputum samples. Using the One Codex platform (Minot et al., 2015), we determined the presence of other bacterial species (Figure 6) and viruses (Supplementary Figure S3) in the sputum samples used for the spiking experiments.
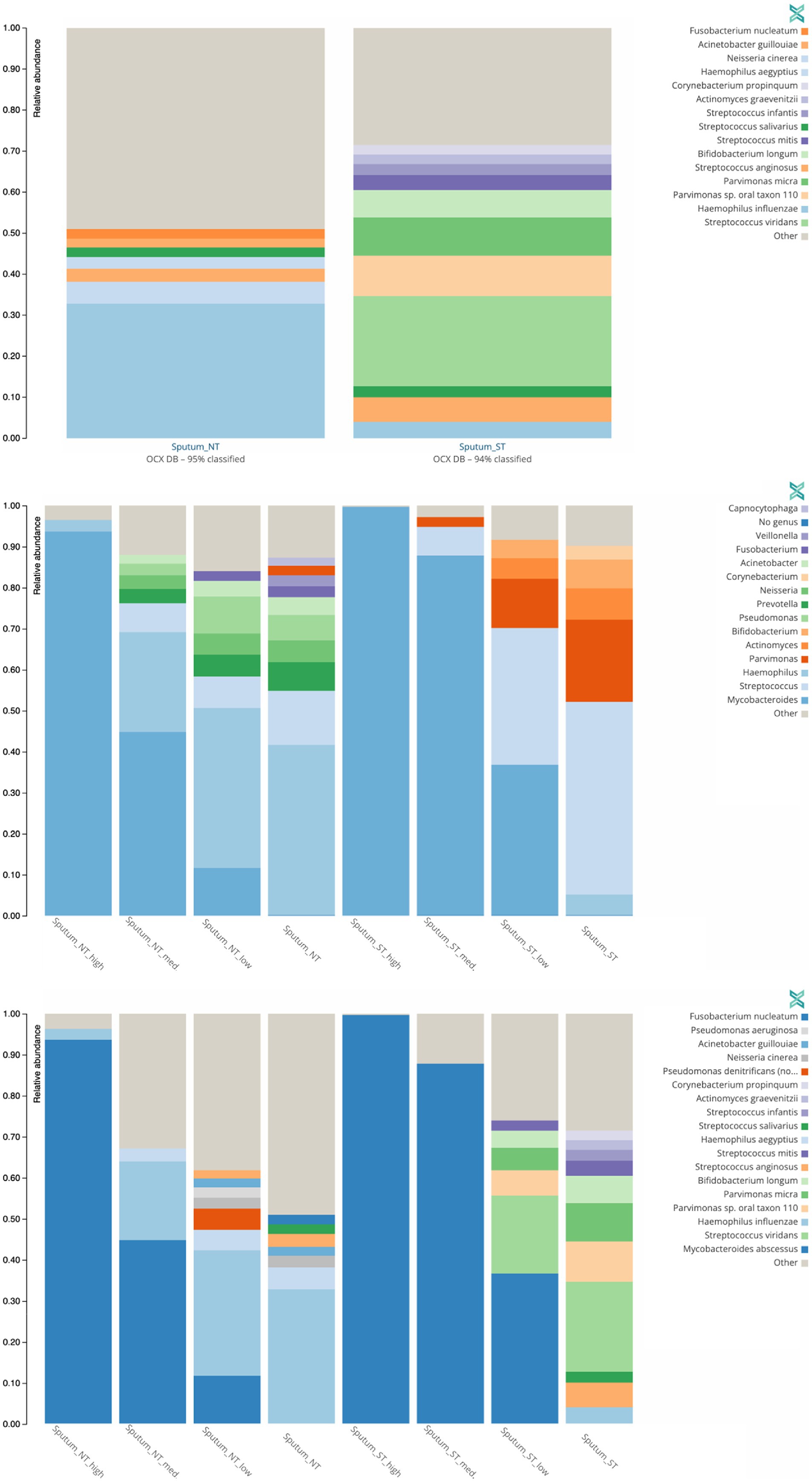
Figure 6. Effect of saponin treatment (ST) on microbiota present in patient sputa. Non-spiked sputa (A) and sputa spiked (B,C) with different concentrations of M. abscessus (high: 2.02 × 108 CFU/ml, med: 2.02 × 107 CFU/ml, low: 2.02 × 106 CFU/ml). Subsequently, sputa were incubated (ST) or not (NT) with saponin followed by DNAse treatment. Relative abundance of bacterial genera (A,B) or species (C) with at least 2% reads are shown. Other refers to low abundant species.
The two microorganisms predominantly found in non-spiked ST and NT sputa were Streptococcus viridans (22%) and Haemophilus influenzae (33%), respectively (Figure 6A). Similarly, S. viridians and H. influenzae were mostly found in M. abscessus spiked ST and NT sputum, respectively (Figures 6B,C). These results suggest that saponin followed by DNase I treatment depletes Gram-negative bacteria. Saponin treatment combined with a DNase I step is an efficient option to deplete host DNA (Bachmann et al., 2018; Charalampous et al., 2019; Boro and Stoll, 2022), however, the effect on different bacterial species varies depending on the study. In a study optimizing culture-independent genome sequencing, saponin treatment resulted in almost undetectable levels of human DNA with minimal loss of Bordetella pertussis DNA (Fong et al., 2020). In another study, however, depletion of human DNA with saponin treatment increased Neisseria gonorrhoeae yields in simulated infections but decreased yields in clinical samples (Osei-Wusu et al., 2021). Both species are Gram-negative bacteria. Hence, variations in the effect of saponin treatment cannot be solely explained by differences in the cell wall structure.
Furthermore, another study examining the effect of saponin followed by DNase I treatment also showed an increase in the proportion of reads detected for DNA viruses (Hasan et al., 2016). Although we did not mainly focus on viruses in our study, we also observed an increase in the proportion of reads detected for viruses in ST samples (Supplementary Figure S3). However, our protocol also included osmotic lysis and centrifugation, which might have degraded viruses susceptible to osmotic stress. A balance between effective host depletion and minimum negative impact on microbial cells (and therefore microbial DNA) must be achieved while designing a saponin-DNase I treatment protocol. Our study shows that the protocol used here did not affect the overall mycobacterial DNA extraction, while significantly reducing the human DNA load, which resulted in higher mycobacterial read counts and higher sensitivity. Our protocol, nonetheless, might not be applicable to other bacterial species such as H. influenzae.
A limitation of this study is the restricted number of bacterial dilutions of M. abscessus tested. Mycobacteria can be present in clinical samples at low concentrations (101–103 CFU/ml) (Walker et al., 2015), which is substantially lower than the dilutions tested using this protocol. However, in this study, we aimed to quantify the effect of saponin treatment on human cells and M. abscessus, which would have been challenging in samples with unquantifiable DNA concentrations. It is worth to mention, however, that in the study conducted by Charalampous et al. (2019) using a similar extraction method, the limit of detection (LoD) of the method ranged from 103 to 105 CFU/ml for E. coli and S. aureus and depended on different levels of background commensal/human DNA. Accordingly, and in another study using SMg, researchers were able to identify and characterize M. tuberculosis in samples with low AFB scores (+1; approx. 5,000 bacilli per milliliter of specimen) and no growth in MGIT culture (Votintseva et al., 2017). Taking this into consideration and assuming that similar LoDs would be identified for mycobacteria, the LoD is probably the main drawback of our current SMg protocol. However, we believe the method proposed here is of substantial diagnostic/therapeutic value because it has the potential to complement more sensitive diagnostic tests (if there is a strong suspicion of TB or NTM infection even though culture/microscopy is negative) and speed up the turnaround time for DST and/or phylogenetic inference in case of a suspected resistant strain that would lead to therapeutic failure (e.g., recurrent mycobacterial infection) or in case an outbreak is suspected, respectively.
The second limitation of this study was the lack of clinical Mycobacterium-positive sputum samples to test our protocol. We attempted to overcome this limitation by spiking sputum from patients with mycobacteria, but we acknowledge that this may not entirely reflect the clinical situation.
The present study aimed to develop an optimized extraction protocol to complement available TB/NTM-diagnostic tests for faster susceptibility testing and outbreak management of mycobacterial infections using NGS. We showed that saponin combined with DNase I treatment was an effective method for host depletion, by decreasing the availability of human DNA for PCR amplification during library preparation, and with minimal impact on the recovery of mycobacterial DNA as opposed to previous observations for other pathogens. SMg can complement other diagnostic tests (e.g., culture, microscopy, POC tests, PCR), by decreasing the turnaround time for a comprehensive drug susceptibility testing and by combining detailed and comprehensive diagnostic information (Peker et al., 2021), including typing of isolates, in a single test. It has the potential to provide increased sensitivity, potential for automation, and further decreased time to result in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA823663.
NC, JR, BS, and NP conceived and supervised the study. NK, NP, and LS adjusted the host depletion pre-treatment protocol and performed the sequencing experiments. NK, NP, and JB performed the host depletion and DNA extraction. NK, NP, LS, and NC analyzed the data. JB contributed with sputum samples. NK, NP, LS, and NC wrote the draft manuscript. All authors contributed to the article and approved the submitted version.
This study was funded in part by the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement 713660 (MSCA-COFUND-2015-DP “Pronkjewail”) and by the Stichting Beatrixoord Noord-Nederland.
We would like to thank Erwin C. Raangs for his excellent technical assistance.
NK is currently employed by Becton, Dickinson and Company. JR and NC consulted for IDbyDNA. JR consults for ARES-Genetics. NC consults for the University of Oxford. However, this did not influence the writing of the manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.949328/full#supplementary-material
ITS, internal transcribed spacer; HRM, high resolution melting curve; MDR, multidrug-resistant; NALC, N-acetyl-L-cysteine; NGS, next-generation sequencing; NT, not-treated; NTM, non-tuberculous mycobacteria; qPCR, quantitative PCR; SMg, Shotgun metagenomics; ST, saponin treated; TB, tuberculosis; WHO, World Health Organization; XDR, extensively-drug resistant.
Akkerman, O. W., van der Werf, T. S., de Boer, M., de Beer, J. L., Rahim, Z., Rossen, J. W. A., et al. (2013). Comparison of 14 molecular assays for detection of mycobacterium tuberculosis complex in bronchoalveolar lavage fluid. J. Clin. Microbiol. 51, 3505–3511. doi: 10.1128/JCM.00843-13
Bachmann, N. L., Rockett, R. J., Timms, V. J., and Sintchenko, V. (2018). Advances in clinical sample preparation for identification and characterization of bacterial pathogens using metagenomics. Front. Public Health 6:363. doi: 10.3389/fpubh.2018.00363
Boro, E., and Stoll, B. (2022). Barriers to COVID-19 health products in Low-and middle-income countries during the COVID-19 pandemic: a rapid systematic review and evidence synthesis. Front. Public Health 10:928065. doi: 10.3389/fpubh.2022.928065
Böttger, S., and Melzig, M. F. (2013). The influence of saponins on cell membrane cholesterol. Bioorg. Med. Chem. 21, 7118–7124. doi: 10.1016/j.bmc.2013.09.008
Brown-Elliott, B. A., and Philley, J. V. (2017). Rapidly growing mycobacteria. Microbiol. Spectr. 5:TNMI7-0027-2016. doi: 10.1128/microbiolspec.TNMI7-0027-2016
Charalampous, T., Kay, G. L., Richardson, H., Aydin, A., Baldan, R., Jeanes, C., et al. (2019). Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 37, 783–792. doi: 10.1038/s41587-019-0156-5
Coulter, B. (2016). Agencourt AMPure XP instructions for use. [Online]. Available at: https://www.beckmancoulter.com/wsrportal/techdocs?docname=B37419 (Accessed February 2019).
Doughty, E. L., Sergeant, M. J., Adetifa, I., Antonio, M., and Pallen, M. J. (2014). Culture-independent detection and characterisation of mycobacterium tuberculosis and M. africanum in sputum samples using shotgun metagenomics on a benchtop sequencer. PeerJ. 2:e585. doi: 10.7717/peerj.585
Fong, W., Rockett, R., Timms, V., and Sintchenko, V. (2020). Optimization of sample preparation for culture-independent sequencing of Bordetella pertussis. Microb. Genom. 6:e000332. doi: 10.1099/mgen.0.000332
Hasan, M. R., Rawat, A., Tang, P., Jithesh, P. V., Thomas, E., Tan, R., et al. (2016). Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J. Clin. Microbiol. 54, 919–927. doi: 10.1128/JCM.03050-15
Illumina (2019). Nextera DNA flex library prep reference guide. [Online]. Available at: https://emea.support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/samplepreps_nextera/nextera-xt/nextera-xt-library-prep-reference-guide-15031942-05.pdf (Accessed February 2019).
Johansen, M. D., Herrmann, J. L., and Kremer, L. (2020). Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 18, 392–407. doi: 10.1038/s41579-020-0331-1
Low, J. L., Wu, M.-L., Aziz, D. B., Laleu, B., and Dick, T. (2017). Screening of TB actives for activity against Nontuberculous mycobacteria delivers high hit rates. Front. Microbiol. 8:1539. doi: 10.3389/fmicb.2017.01539
McCarthy, A., Chiang, E., Schmidt, M. L., and Denef, V. J. (2015). RNA preservation agents and nucleic acid extraction method bias perceived bacterial community composition. PLoS One 10:e0121659. doi: 10.1371/journal.pone.0121659
Merck KGaA (2018). Mark Frei. Centrifugation basics. [Online]. Available at: https://www.sigmaaldrich.com/NL/en/technical-documents/technical-article/protein-biology/protein-pulldown/centrifugation-basics
Minot, S. S., Krumm, N., and Greenfield, N. B. (2015). One codex: a sensitive and accurate data platform for genomic microbial identification. bioRxiv [Epub ahead of preprint]. doi: 10.1101/027607
Osei-Wusu, S., Morgan, P., Asare, P., Adams, G., Musah, A. B., Siam, I. M., et al. (2021). Bacterial load comparison of the three Main lineages of Mycobacterium tuberculosis complex in West Africa. Front. Microbiol. 12:3176. doi: 10.3389/fmicb.2021.719531
Peker, N., Schuele, L., Kok, N., Terrazos, M., Neuenschwander, S. M., de Beer, J., et al. (2021). Evaluation of whole-genome sequence data analysis approaches for short- and long-read sequencing of Mycobacterium tuberculosis. Microb. Genom. 7:000695. doi: 10.1099/mgen.0.000695
Qiagen (2016). QIAamp DNA mini and blood mini kit handbook [Online]. Available at: https://www.qiagen.com/us/shop/sample-technologies/dna/genomic-dna/qiaamp-dna-mini-kit/#resources (Accessed February 2019).
Qiagen (2017). DNeasy UltraClean microbial kit handbook [Online]. Available at: https://www.qiagen.com/us/shop/sample-technologies/dna/genomic-dna/dneasy-ultraclean-microbial-kit/#resources (Accessed February 2019).
Qiagen (2018a). AllPrep PowerFecal DNA/RNA kit handbook [Online]. Available at: https://www.qiagen.com/us/shop/sample-technologies/combined-sample-technologies/preparation/allprep-powerfecal-dna-rna-kit/#orderinginformation
Qiagen (2018b). CLC genomics workbench. [online]. Available: https://www.qiagenbioinformatics.com/products/clc-genomics-workbench/.
Schuele, L., Cassidy, H., Peker, N., Rossen, J. W. A., and Couto, N. (2021). Future potential of metagenomics in microbiology laboratories. Expert. Rev. Mol. Diagn. 21, 1273–1285. doi: 10.1080/14737159.2021.2001329
Sriramulu, D. D., Lünsdorf, H., Lam, J. S., and Römling, U. (2005). Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J. Med. Microbiol. 54, 667–676. doi: 10.1099/jmm.0.45969-0
Street, T. L., Barker, L., Sanderson, N. D., Kavanagh, J., Hoosdally, S., Cole, K., et al. (2020). Optimizing DNA extraction methods for nanopore sequencing of Neisseria gonorrhoeae directly from urine samples. J. Clin. Microbiol. 58, e01822–e01819. doi: 10.1128/JCM.01822-19
Votintseva, A. A., Bradley, P., Pankhurst, L., Del Ojo Elias, C., Loose, M., Nilgiriwala, K., et al. (2017). Same-day diagnostic and surveillance data for tuberculosis via whole-genome sequencing of direct respiratory samples. J. Clin. Microbiol. 55, 1285–1298. doi: 10.1128/JCM.02483-16
Walker, T. M., Kohl, T. A., Omar, S. V., Hedge, J., Del Ojo Elias, C., Bradley, P., et al. (2015). Whole-genome sequencing for prediction of mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect. Dis. 15, 1193–1202. doi: 10.1016/S1473-3099(15)00062-6
Wegl, G., Grabner, N., Köstelbauer, A., Klose, V., and Ghanbari, M. (2021). Toward best practice in livestock microbiota research: a comprehensive comparison of sample storage and DNA extraction strategies. Front. Microbiol. 12:322. doi: 10.3389/fmicb.2021.627539
World Health Organization (2018). Tuberculosis: multidrug-resistant tuberculosis (MDR-TB). Available at: https://www.who.int/news-room/questions-and-answers/item/tuberculosis-multidrug-resistant-tuberculosis-(mdr-tb) (Accessed February 2019).
World Health Organization (2019). Global Tuberculosis Report 2019. Geneva: World Health Organization.
Keywords: internal transcribed spacer, N-acetyl-L-cysteine, next-generation sequencing, shotgun metagenomics, saponin treatment, tuberculosis, host DNA depletion, drug-resistant
Citation: Kok NA, Peker N, Schuele L, de Beer JL, Rossen JWA, Sinha B and Couto N (2022) Host DNA depletion can increase the sensitivity of Mycobacterium spp. detection through shotgun metagenomics in sputum. Front. Microbiol. 13:949328. doi: 10.3389/fmicb.2022.949328
Edited by:
Sivakumar Shanmugam, National Institute of Research in Tuberculosis (ICMR), IndiaReviewed by:
Radha Gopalaswamy, National Institute of Research in Tuberculosis (ICMR), IndiaCopyright © 2022 Kok, Peker, Schuele, de Beer, Rossen, Sinha and Couto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natacha Couto, bi5tb25nZS5nb21lcy5kby5jb3V0b0B1bWNnLm5s
†Present address: Nienke A. Kok, Becton, Dickinson and Company, R&D Department, Drachten, Netherlands
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.