
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 12 July 2022
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.944416
This article is part of the Research Topic The Potential Role of Gut Microbiome in Animal Gut-Linked Diseases View all 16 articles
 Zhuoen He1,2†Yunong Zeng2†Shuyu Li2†Lizhen Lin2†Ruisi Zhou2Fangzhao Wang2
Zhuoen He1,2†Yunong Zeng2†Shuyu Li2†Lizhen Lin2†Ruisi Zhou2Fangzhao Wang2 Wenjiao Yang3Yuhao Wu2Junhao Yang4Ali Chen5
Wenjiao Yang3Yuhao Wu2Junhao Yang4Ali Chen5 Zhang Wang4
Zhang Wang4 Hong Yang1
Hong Yang1 Xiaoshan Zhao2
Xiaoshan Zhao2 Wei Xiao2,6*
Wei Xiao2,6* Lei Li7*
Lei Li7* Shenhai Gong1,2*
Shenhai Gong1,2*Background and Aims: Drug-induced liver injury (DILI) is a common cause of acute liver failure and represents a significant global public health problem. When discussing the gut-liver axis, although a great deal of research has focused on the role of gut microbiota in regulating the progression of DILI, the gut commensal fungal component has not yet been functionally identified.
Methods: Mice were pretreated with fluconazole (FC) to deplete the gut commensal fungi and were then subject to acetaminophen (APAP) gavage. In addition, transcriptome sequencing was performed to identify differentially expressed genes (DEGs) between control and fluconazole-pretreated groups of the mice challenged with APAP.
Results: Gut commensal fungi ablation through fluconazole pretreatment predisposed mice to APAP-induced hepatotoxicity, characterized by elevated serum liver enzyme levels and more severe centrilobular necrosis, which appears to be caused by robust inflammation and oxidative stress. The 16S rDNA sequencing results indicated that Akkermansia muciniphila abundance had significantly decreased in gut fungi-depleted mice, whereas increased abundance of Helicobacter rodentium was observed. The gene interaction network between DEGs identified by the transcriptome sequencing highlighted a significant enrichment of Cyp2a5 in the liver of APAP-treated mice that were preadministrated with fluconazole. Pharmacological inhibition of Cyp2a5 by 8-methoxypsoralen (8-MOP) could significantly attenuate hepatic inflammation and oxidative stress in mice, thereby conferring resistance to acute liver injury caused by APAP administration.
Conclusion: Our data highlighted the significance of gut commensal fungi in hepatic inflammation and oxidative stress of APAP mice, shedding light on promising therapeutic strategies targeting Cyp2a5 for DILI treatment.
Drug-induced liver injury (DILI) is becoming an increasingly severe public health problem worldwide as it is one of the leading causes of acute liver failure (ALF; Bernal and Wendon, 2013). A large body of evidence has revealed that acetaminophen (APAP) is extensively used to treat pain and fever, although hepatotoxicity caused by an APAP overdose is the major cause of DILI in many European and North American countries (Larson et al., 2005; Bernal et al., 2013; Stravitz and Lee, 2019). APAP-induced ALF is a progressive disease that is characterized by extensive hepatocellular necrosis and has unacceptably high levels of mortality (Larson et al., 2005; Fernandez-Checa et al., 2021). Although substantial efforts have been made to improve medical management of the condition, APAP-induced liver failure nevertheless causes more than 500 deaths annually in the US (Li et al., 2020). The pathogenesis of APAP hepatotoxicity is linked to the intracellular depletion of hepatic reduced glutathione (GSH), with the consequence of predisposing hepatocytes to mitochondrial reactive oxygen species (ROS; Yuan and Kaplowitz, 2013). A normal dose of APAP can be mostly metabolized in the liver by glucuronidation and sulfation into non-toxic substances, thereby promoting the excretion of APAP via the kidneys (Watkins and Seeff, 2006). However, APAP is metabolized to a large number of N-acetyl-p-benzoquinoneimine (NAPQI) through cytochrome P450 enzymes when both glucuronidation and sulfation pathways become saturated following an APAP overdose (Krenkel et al., 2014). After hepatic GSH exhaustion, the excessive NAPQI bonds covalently with hepatocellular macromolecules to form APAP-protein adducts, which leads to hepatocyte necrosis or ferroptosis through mitochondrial dysfunction (Jaeschke et al., 2019; Shojaie et al., 2020).
The gut microbiota is an enormous microscopic community consisting of bacteria, fungi, viruses, and microeukaryotes (Norman et al., 2014). During the dynamic development of the gut microbiome in early life, bacteria-fungi crosstalk is important in maintaining microecological homeostasis and can define the trajectory of the health of the host (Iliev and Leonardi, 2017; Schei et al., 2017). In recent years, the ecological balance in the gut has been shown to regulate the pathogenesis of liver disease via the gut-liver axis (Albillos et al., 2020). Prior studies from our laboratory have revealed that the oscillation of gut microbiota mediated the diurnal variation of acetaminophen-induced acute liver injury through the generation of the gut microbial metabolite 1-phenyl-1,2-propanedione (PPD) in mice (Gong et al., 2018). With the increased understanding of intestinal microecology, research is gradually focusing on the potential of the gut fungal component in treating liver disease. For example, the yeast Saccharomyces boulardii seems to hinder the growth of pathogenic bacteria, revealing the potential therapeutic implications of certain fungi against infectious disease (Chen et al., 2020). In addition, gut commensal fungal dysbiosis induced by fluconazole (FC) could aggravate allergic airway disease in a house dust mite challenge mode by increasing the infiltration of gut-resident mononuclear phagocytes (MNPs) that express the fractalkine receptor CX3CR1 (Li et al., 2018). However, exactly what role the gut commensal fungi play in preventing the development and progression of drug-induced ALF remains unknown. In the present study, we revealed that gut commensal fungi ablation by fluconazole resulted in hepatic Cyp2a5 overexpression, thereby increasing susceptibility to APAP hepatotoxicity due to enhanced inflammatory responses and oxidative stress.
A number of 6–8-week-old male specific-pathogen-free C57BL/6 J mice were housed in standard laboratory conditions under a cycle of 12 h light/12 h dark at room temperature, with food and water ad libitum.
The mice were initially treated with fluconazole dissolved in distilled drinking water at a concentration of 0.5 g/l for 14 days (with the solution replaced every 2 days) in order to deplete the gut commensal fungi (Iliev et al., 2012). The mice in the control group were given distilled water. After fluconazole pretreatment, mice were given a single oral dose of 300 mg/kg APAP dissolved in phosphate-buffered saline (PBS) before being sacrificed for tissues collection 24 h later. In addition, mice were treated with a dose of 20 mg/kg 8-methoxypsoralen (8-MOP) dissolved in corn oil to investigate the role of Cyp2a5 expression toward inhibiting inflammatory responses and oxidative stress in hepatotoxicity caused by APAP.
All experimental procedures were performed in accordance with the National Institutes of Health guidelines and were approved by the local Animal Care and Use Committee of Southern Medical University, Guangzhou, China.
The levels of alanine transaminase (ALT) and aspartate transaminase (AST) in the serum were quantitated using commercial assay kits (Jiancheng, Nanjing, China) according to the manufacturer’s instructions. Malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione (GSH) activity in the liver tissues were determined using a corresponding commercial assay kit (Jiancheng, Nanjing, China). TNF-α, IL-6, and MCP-1 concentrations in the plasma were measured by the ELISA assay kits (Neobioscience, Shenzhen, China), and the serum MCP-3 level was detected with an ELISA assay kit (CUSABIO, Wuhan, China).
Liver tissues were fixed in 4% paraformaldehyde (PFA) for 24 h at room temperature, before being embedded in paraffin and sectioned. Hematoxylin and eosin (HE) staining were performed to assess pathological changes in the liver. At least 6–8 randomly selected fields per sample were used to calculate the area of liver necrosis over the whole field using ImageJ software (National Institute of Health).
The paraffin-embedded slides were dewaxed, rehydrated, and incubated in 3% H2O2 for 20 min and calf serum for 15 min, respectively. Next, the slides were incubated with anti-CD11b antibody (Servicebio, Wuhan, China) at 4°C overnight, before being incubated with horseradish peroxidase (HRP)-labeled secondary antibody (Gene Tech, Shanghai, China). After washing with PBS, the slides were stained with DAB and hematoxylin, respectively. Finally, eight fields per slide were randomly photographed in order to assess and quantify CD11b-positive cells using ImageJ software.
The paraffin-embedded liver sections were subjected to terminal-deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) staining using a commercial assay kit (KeyGEN BioTECH, Nanjing, China) according to the manufacturer’s instructions. At least 6–7 fields per slide were randomly selected in order to determine the number of TUNEL-positive cells.
To detect the hepatic ROS levels, the frozen liver sections were incubated with dihydroethidium (DHE, Thermo Scientific, MA, United States) at a final concentration of 2 μM at 37°C for 30 min. Between four and eight fields per slide were then selected at random and the mean fluorescence intensity (MFI) was analyzed using ImageJ software.
Feces from mice were collected on day 14 after fluconazole treatment and were immediately stored at-80°C. The fecal samples were mashed in PBS containing 0.5% Tween20 solution and were then subjected to repeated freezing at-80°C for 10 min and thawing at 60°C for 5 min. The next steps involved extracting and purifying the DNA using the phenol-chloroform isoamyl alcohol method and a commercial reagent (Solarbio, Beijing, China). Next, the concentration and purity of total DNA were determined by using a NanoDrop spectrophotometer (Thermo Scientific, MA, United States). The hypervariable region 4 (V4) of the bacterial 16S rRNA was amplified by a polymerase chain reaction (PCR) using primers V4-F (5′-GTGTGYCAGCMGCCGCGGTAA-3′) and V4-R (5′-CCG GACTACNVGGGTWTCTAAT-3′). DNA sequencing was performed on an Illumina Hiseq PE250, and the raw data of 16S rRNA gene sequencing were analyzed with the QIIME2 platform (v2020.2).
RNA was extracted from the liver tissues using TRIzol reagent (Thermo Scientific, MA, United States) and separated by chloroform. cDNA was obtained by reverse transcription with ReverTra Ace qPCR RT Kit (Toyobo, Shanghai, China). Quantitative real-time PCR (qRT-PCR) analysis were performed on a 7,500 Real-Time PCR System (Applied Biosystems, CA, United States) and 18S ribosomal RNA was used for normalization and quantification of the target gene expression levels using the comparative CT method. All primers for qRT-PCR analysis were listed in Table 1.
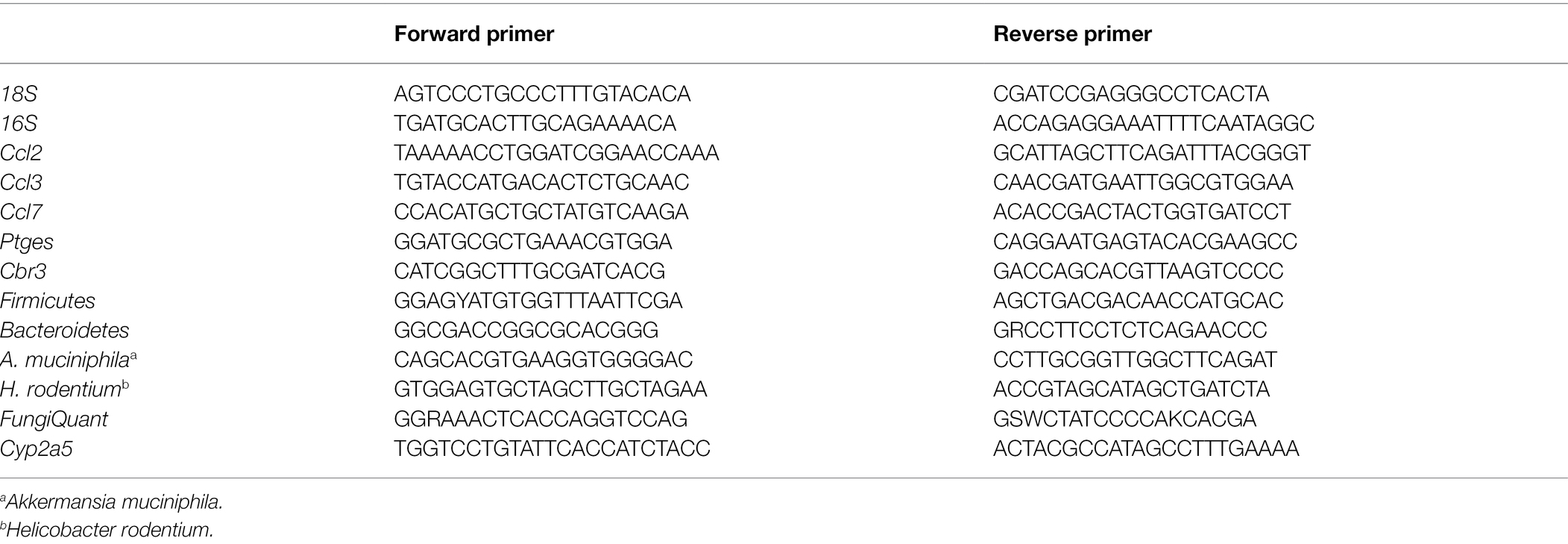
Table 1. Primers for qPCR.
Liver tissues were homogenized in RIPA lysis buffer (Beyotime, Shanghai, China) on ice to prepare for total protein extraction. Total protein extractions were denatured with SDS loading buffer by heating at 95°C for 5 min. After blocking with 5% non-fat milk in tris-buffered saline tween (TBST), the membranes were incubated separately with total JNK (Cell Signaling Technology, MA, United States), p-JNK (Cell Signaling Technology, MA, United States), total p65 (Cell Signaling Technology, MA, United States), p-p65 (Cell Signaling Technology, MA, United States), total p38 (Cell Signaling Technology, MA, United States), p-p38 (Cell Signaling Technology, MA, United States), total ERK (Cell Signaling Technology, MA, United States), and p-ERK (Cell Signaling Technology, MA, United States) antibodies overnight at 4°C. Finally, the protein bands were incubated with secondary antibodies (Proteintech, Wuhan, China) for 1 h at room temperature and were then visualized using an enhanced chemiluminescence (ECL) detection kit (Vazyme, Wuhan, China).
To conduct the transcriptomic sequencing analysis, the total RNA from the livers of the APAP-treated mice with or without fluconazole pretreatment was extracted using TRIzol reagent (Thermo Scientific, MA, United States). Purified total RNA was performed to construct libraries, followed by subjection to sequence through Illumina NovaSeq 6,000 (Novogene Co., Ltd., Beijing, China). Statistical analysis was performed with the DESeq2 R package. The genes with |log2 (fold change)| > 1 and adjusted p < 0.05 were identified as differentially expressed genes (DEGs).
The transcriptome and 16S rDNA data have been uploaded to the China Nucleotide Sequence Archive (CNSA, https://db.cngb.org/cnsa/) under accession codes CNP0002810 and CNP0002825, respectively.
All data were expressed as mean ± standard error of the mean (SEM) and were evaluated by an unpaired two-tailed Student’s t-test. A value of p lower than 0.05 was considered significant (*p < 0.05).
To explore the role of gut commensal fungi in APAP-induced hepatotoxicity, the mice were orally pretreated with fluconazole (FC) for 14 days to deplete the gut commensal fungi, followed by subjection to APAP administration (Figure 1A). First, we determined whether the gut commensal fungi depletion through fluconazole administration affected energy intake. As shown in Figures 1B,C, we found that food consumption and body weight were not altered by the presence of fluconazole in mice. Serum biochemistry results showed that ALT levels were comparable in control and fluconazole-treated mice (Figure 1D), which was further confirmed by the hepatic expression of inflammatory chemokine including Ccl2, Ccl3 and Ccl7 (Figure 1E). The above data suggested that the dose of fluconazole used in our study did not directly cause hepatotoxicity. Consistent with previous study (Qiu et al., 2015), the qRT-PCR result showed that the fluconazole-treated mice had a lesser abundance of gut fungi in comparison with the control group (Figure 1F). Interestingly, gut fungi ablation caused by the fluconazole pretreatment resulted in a significant increase in plasma ALT and AST levels in response to APAP administration (Figure 1G). Histopathological examination displayed that gut commensal fungi depletion significantly elevated the area of necrosis in the liver tissues of APAP-treated mice (Figure 1H). This result was further supported by the TUNEL staining, which measured the level of cell death including necrosis, apoptosis and necroptosis (Figure 1I; Grasl-Kraupp et al., 1995). All of the above-mentioned results indicated that gut commensal fungi depletion predisposed mice to APAP-induced hepatotoxicity.
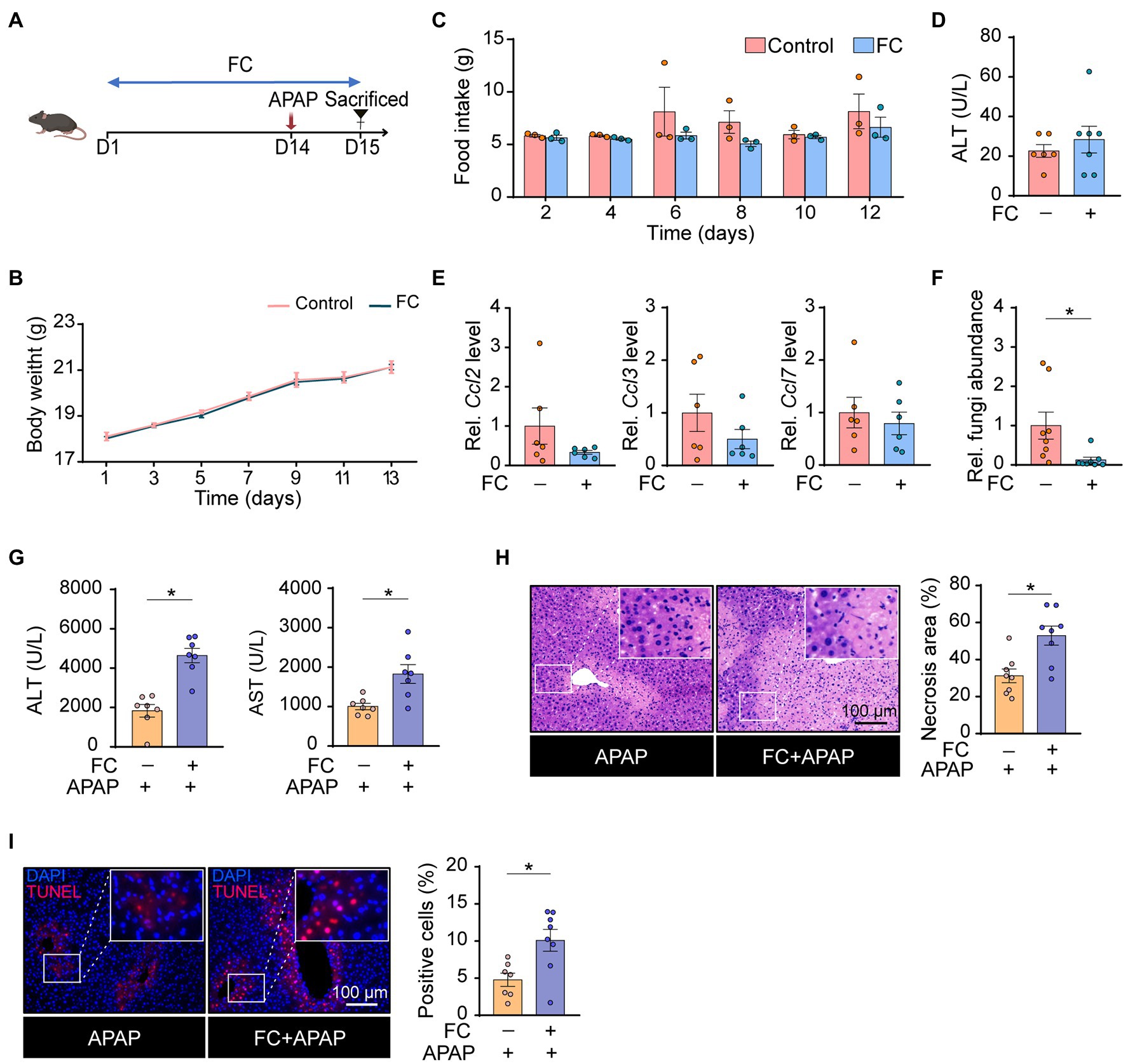
Figure 1. Gut commensal fungi protect mice against APAP-induced acute liver failure. (A) Study design. C57BL/6 mice were pretreated with fluconazole for 14 days to deplete the gut commensal fungi, followed by subjection to APAP administration for 24 h. Tissues were collected after mice were euthanized. (B) Body weight change in the control and fluconazole-only group. (C) Food intake of the control and fluconazole-only group. (D) Effect of fluconazole administration alone on serum ALT levels. (E) Relative mRNA levels of Ccl2, Ccl3, and Ccl7 in the liver of control and fluconazole-only group were detected by qRT-PCR. (F) Relative abundance of fungi in the gut of the control and fluconazole-only group. (G) Plasma ALT and AST levels in APAP-treated mice with or without preadministration of fluconazole. (H) Representative H&E staining images and quantification of necrotic areas in the livers of APAP-treated mice with or without preadministration of fluconazole. (I) TUNEL staining of the liver from APAP-treated mice with or without preadministration of fluconazole and quantification of dead cells. *p < 0.05. Data were expressed as mean ± SEM and were evaluated by a two-tailed unpaired Student’s t-test. n = 3–8. Scale bar = 100 μm. FC, fluconazole; APAP, acetaminophen; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Rel, Relative; H&E, hematoxylin and eosin; and TUNEL, terminal-deoxynucleotidyl transferase-mediated nick end labeling.
A previous study had shown that the interactions between gut commensal fungi and bacteria are important for maintaining intestinal health and improving disease resistance (Sam et al., 2017). For this reason, we next performed 16S rDNA sequencing to determine the alterations in gut bacterial community structure in response to fluconazole treatment. The qRT-PCR result indicated a significant decrease in the Firmicutes/Bacteroidetes ratio, reflecting the composition of gut microbes in mice subjected to fluconazole treatment for 14 days (Figure 2A). There were significant changes in the alpha diversity of gut bacteria as shown by Faith’s Phylogenetic Distance (PD) and Observed OTUs, although not in the Shannon diversity index (Figure 2B). Consistently, significant intergroup differences among control and fluconazole-treated mice were assessed using the Bray-Curtis metric distance according to the principal coordinates analysis (Figure 2C). Moreover, gut fungi ablation through fluconazole treatment induced a distinct clustering of microbiota composition at the phylum level in mice (Figure 2D).
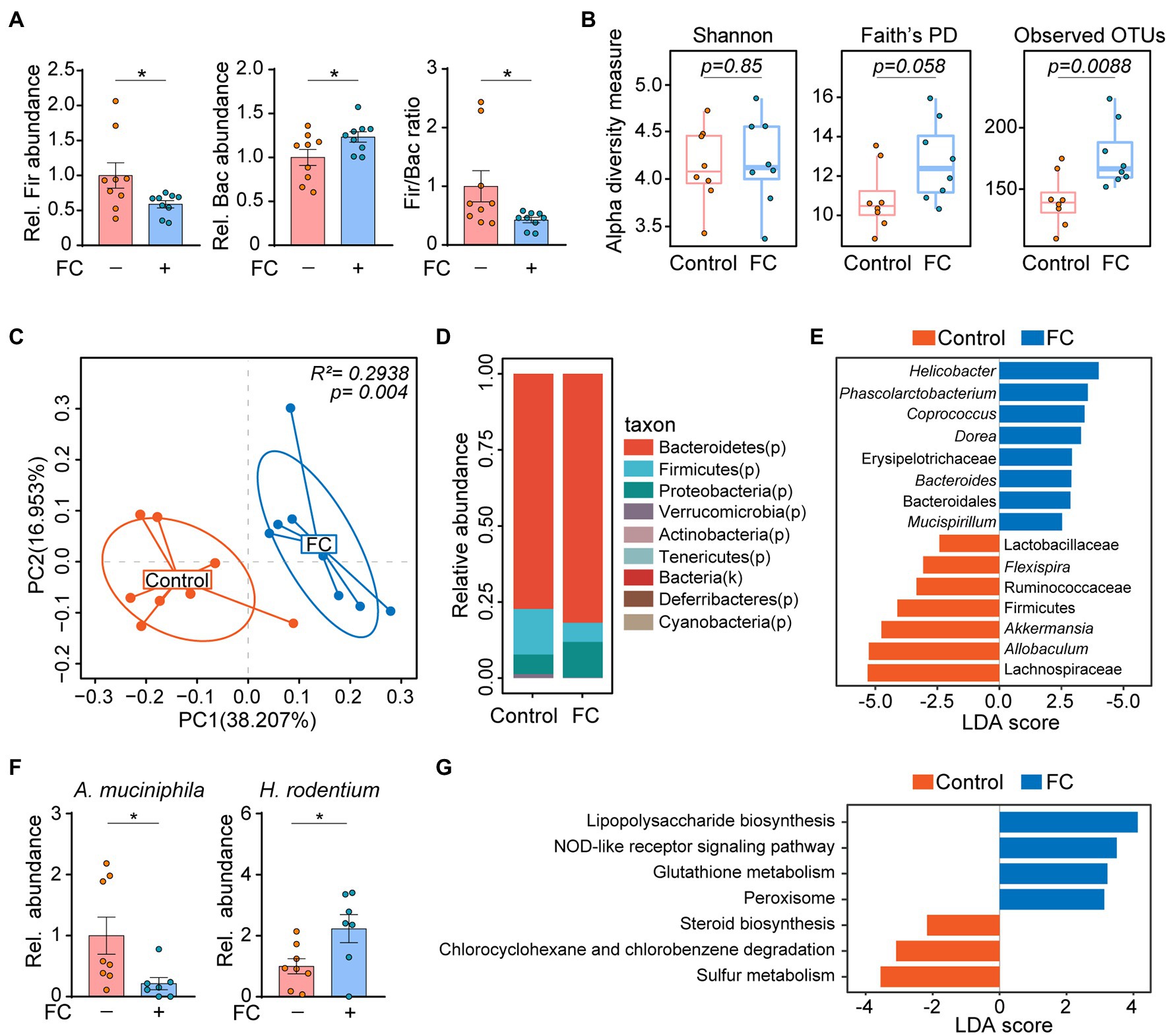
Figure 2. Gut commensal fungi depletion alters bacterial community structure in mice. (A) C57BL/6 mice were treated with fluconazole to deplete the gut commensal fungi. Feces were collected after 14 days of fluconazole treatment. Relative abundance of Firmicutes and Bacteroidetes as well as the ratio of Firmicutes/Bacteroidetes in the feces of control and fluconazole-only group. (B) Alpha diversity based on Shannon Index, Faith’s PD, and Observed OTUs of gut microbiota in the control and fluconazole-only group. These data were evaluated by Wilcoxon rank-sum test. (C) Principal coordinate analysis (PCoA) using Bray–Curtis distance of microbial composition in control and fluconazole-treated mice. These data were evaluated by the Adonis test. (D) Relative abundance of gut microbiota at phylum level from control and fluconazole-treated mice. (E) The differences in specific microbiota taxa between the control and fluconazole-treated mice were identified by linear discriminant analysis effect size (LEfSe) analysis. (F) Relative abundance of Akkermansia muciniphila and Helicobacter rodentium in control and fluconazole-treated mice. (G) Predicated functional KEGG pathways were inferred from OTUs by PICRUSt analysis in control and fluconazole-treated mice. *p < 0.05. All bar graph data were expressed as mean ± SEM and were evaluated by a two-tailed unpaired Student’s t-test. n = 7–9. Rel, Relative; FC, fluconazole; Fir, Firmicutes; Bac, Bacteroidetes; A. muciniphila, Akkermansia muciniphila; H. rodentium, Helicobacter rodentium; LEfSe, linear discriminant analysis effect size; and LDA, linear discriminant analysis.
Specially, the linear discriminant analysis (LDA) effect size (LEfSe) statistical analysis indicated that mice subjected to fluconazole treatment were enriched for members of genus Helicobacter and Dorea, whereas genus Akkermansia and Allobaculum were enriched in control group (Figure 2E). To validate these results, we performed qRT-PCR to determine the changes in gut microbiota in response to fluconazole treatment and found that the abundance of Akkermansia muciniphila (A. muciniphila) was decreased, however, Helicobacter rodentium (H. rodentium) abundance exhibited an opposite trend in fluconazole-treated mice compared with the control group (Figure 2F). PICRUSt analysis on the OTU derived from the 16S rDNA sequence was also conducted to predict potential metabolic functions of gut microbiota in fluconazole-treated mice. In company with increased abundance of Helicobacter, which is defined as pathogens associated with generation of lipopolysaccharides (Hynes et al., 2004), the genomic abundance of some pathways, which included lipopolysaccharide biosynthesis and NOD-like receptor signaling pathway were significantly enhanced in fluconazole-treated mice compared with the control group (Figure 2G). In contrast, we also found that some pathways including sulfur metabolism and steroid biosynthesis were enriched in the control group (Figure 2G). All of the above data demonstrated that the crosstalk of bacteria and fungi is important in maintaining microecological homeostasis.
To explore how gut commensal fungi protected mice against APAP-induced acute liver injury, we performed transcriptome analysis of the liver tissues collected from the APAP-treated mice subjected to fluconazole preadministration. Notably, the KEGG pathway enrichment analysis of DEGs showed that the pathway of arachidonic acid metabolism was enhanced in fluconazole-pretreated mice compared with the control group in the presence APAP administration (Figure 3A), which was also confirmed by PCA analysis (Figure 3B). Concomitantly, volcano plots displayed that mice subjected to fluconazole pretreatment exhibited an increase in the hepatic mRNA levels of the Ptges and Cbr3 genes responsible for inflammatory mediator prostaglandin E2 biosynthesis and metabolism in the presence of APAP (Figure 3C). The DEGs identified by the transcriptome analysis were also convincingly supported by real-time quantitative PCR analysis (Figure 3D).
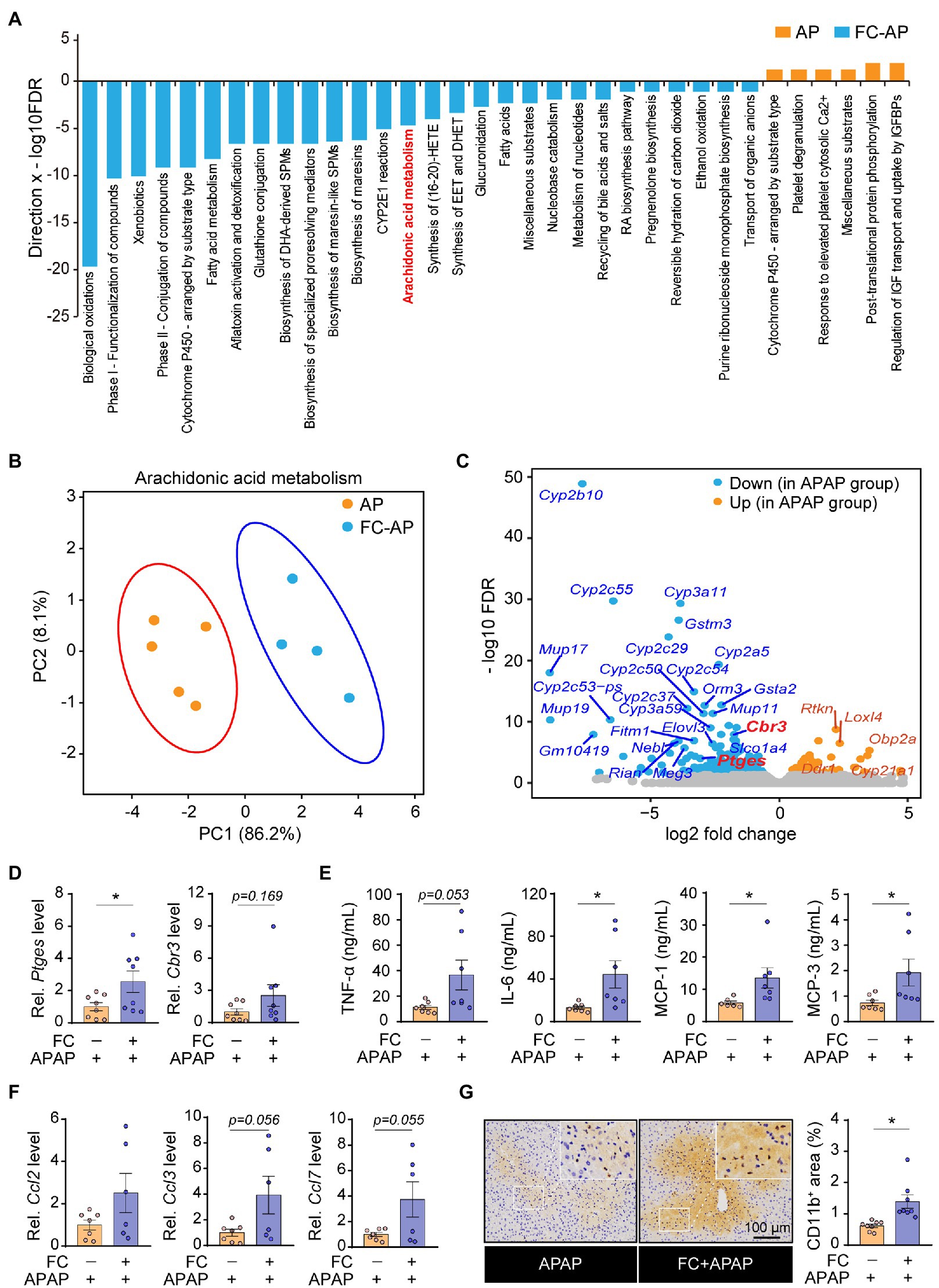
Figure 3. Gut commensal fungi ablation promotes the inflammatory response associated with aberrant arachidonic acid metabolism in APAP-treated mice. (A) Hypergeometric analysis showing pathway enrichment analysis of the liver tissues from APAP-treated mice with or without preadministration of fluconazole. (B) The principal component analysis (PCA) of arachidonic acid metabolism is based on transcriptomic analysis in APAP-treated mice with or without preadministration of fluconazole. (C) Volcano plot of DEGs in the control group compared with fluconazole-pretreated mice in presence of APAP administration. (D) Relative mRNA levels of Ptges and Cbr3 in liver tissues of APAP-treated mice with or without preadministration of fluconazole. (E) Quantification of pro-inflammatory cytokines including TNF-α, IL-6, MCP-1 and MCP-3 in the blood from APAP-treated mice with or without preadministration of fluconazole. (F) Relative mRNA levels of Ccl2, Ccl3, and Ccl7 in the liver of APAP-treated mice with or without preadministration of fluconazole were detected by qRT-PCR. (G) Immunohistochemical staining of CD11b and quantification of positive cells in the liver tissues of APAP-treated mice with or without preadministration of fluconazole. *p < 0.05. All bar graph data were expressed as mean ± SEM and were evaluated by a two-tailed unpaired Student’s t-test. n = 4–8. Scale bar = 100 μm. APAP, acetaminophen; FC, fluconazole; and Rel, Relative.
Emerging evidence indicated that the aberrant metabolism of arachidonic acid profoundly triggers an inflammatory response in the host, thus resulting in an acceleration of the disease process (Harizi et al., 2008). Therefore, it was reasonable to speculate that gut commensal fungi ablation through fluconazole treatment predisposed mice to hepatotoxicity caused by APAP overdose, which appeared to be due to vigorous proinflammatory cytokine production. Indeed, compared with control mice, fluconazole pretreatment resulted in a larger increase in the concentrations of cytokine and chemokine including TNF-α, IL-6, MCP-1 and MCP-3 in the blood of APAP-treated mice (Figure 3E). In addition, we also observed that gut commensal fungi depletion exacerbated hepatic inflammatory responses in response to APAP administration, as indicated by the increase in the mRNA levels of chemokines (Figure 3F). Furthermore, immunohistochemical (IHC) staining displayed that the number of infiltrating CD11b-positive cells, defined as inflammatory cells (Solovjov et al., 2005), increased in the liver tissues of APAP-treated mice who had been pretreated with fluconazole (Figure 3G). In all, the above results suggested that the gut fungi ablation by fluconazole pretreatment have enhanced hepatic inflammation in mice upon APAP exposure.
A previous study demonstrated that the depletion of hepatic reduced glutathione and the subsequent oxidative stress is involved in the pathogenesis of APAP-induced acute liver injury (Goldring et al., 2004). In order to further understand the underlying mechanism by which gut fungi ablation enhanced hepatotoxicity induced by APAP in mice, we thus determined the level of oxidative stress in the liver from APAP-treated mice subjected to fluconazole pretreatment. We first found that hepatic malondialdehyde (MDA) levels in APAP-treated mice subjected to fluconazole preadministration were significantly higher than in the APAP-only group (Figure 4A). Correspondingly, the markers of antioxidant capacity, SOD activity and GSH levels were significantly repressed in the livers of APAP-treated mice subjected to fluconazole preadministration (Figures 4B,C). In addition, DHE staining showed that fluconazole preadministration significantly increased ROS accumulation in the liver of APAP-treated mice (Figure 4D). We next focused on the down signaling pathway which was affected by fluconazole preadministration in APAP-treated mice. As shown in Figure 4E, the phosphorylation level of JNK was significantly increased in the livers of APAP-treated mice subjected to fluconazole preadministration compared with the APAP-only group, whereas other MAPK and NF-κB signaling pathways remained comparable in the two groups. All of these data indicated that fluconazole-induced oxidative stress and sustained JNK activation predisposed mice to APAP-induced hepatotoxicity.
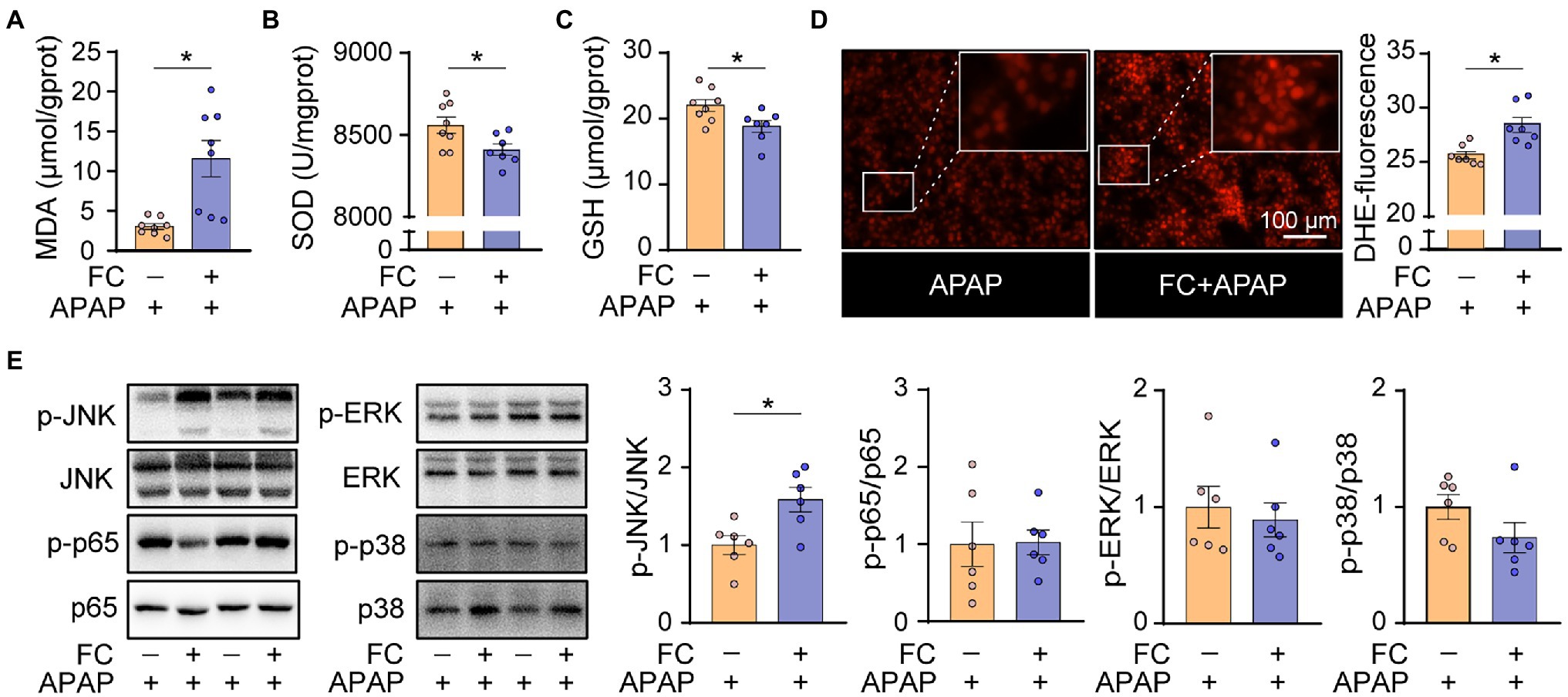
Figure 4. Gut commensal fungi ablation exacerbates hepatic oxidative stress in APAP-treated mice. (A–C) C57BL/6 mice were pretreated with fluconazole for 14 days to deplete the gut commensal fungi and were then subjected to APAP administration for 1 h. Hepatic MDA (A), SOD (B) and total GSH (C) levels in APAP-treated mice with or without preadministration of fluconazole. (D) Representative images of DHE immunofluorescence staining and quantification of hepatic ROS production in APAP-treated mice with or without preadministration of fluconazole. (E) Western blot was performed with specific antibodies to quantify the phosphorylated JNK, ERK, p38 and p65 in the liver tissues from APAP-treated mice with or without preadministration of fluconazole. *p < 0.05. Data were expressed as mean ± SEM and were evaluated by a two-tailed unpaired Student’s t-test. n = 6–8. Scale bar = 100 μm. APAP, acetaminophen; FC, fluconazole; MDA, malonaldehyde; SOD, superoxide dismutase; GSH, glutathione; and DHE, dihydroethidium.
To further illustrate the underlying molecular mechanism of fluconazole pretreatment responsible for excessive inflammatory response and oxidative stress in APAP-treated mice, we performed transcriptome analysis and selected the top twenty DEGs ranked by value of p for interaction network analysis. Figures 5A,B show that the expression of Cyp2a5, identified as the hub gene and highlighted in red, was increased in liver tissues of APAP-treated mice subjected to fluconazole pretreatment. On the basis of these observations, we considered that gut commensal fungi ablation through fluconazole treatment could promote hepatic Cyp2a5 overexpression, thereby resulting in increasing the susceptibility to APAP-induced hepatotoxicity in mice.
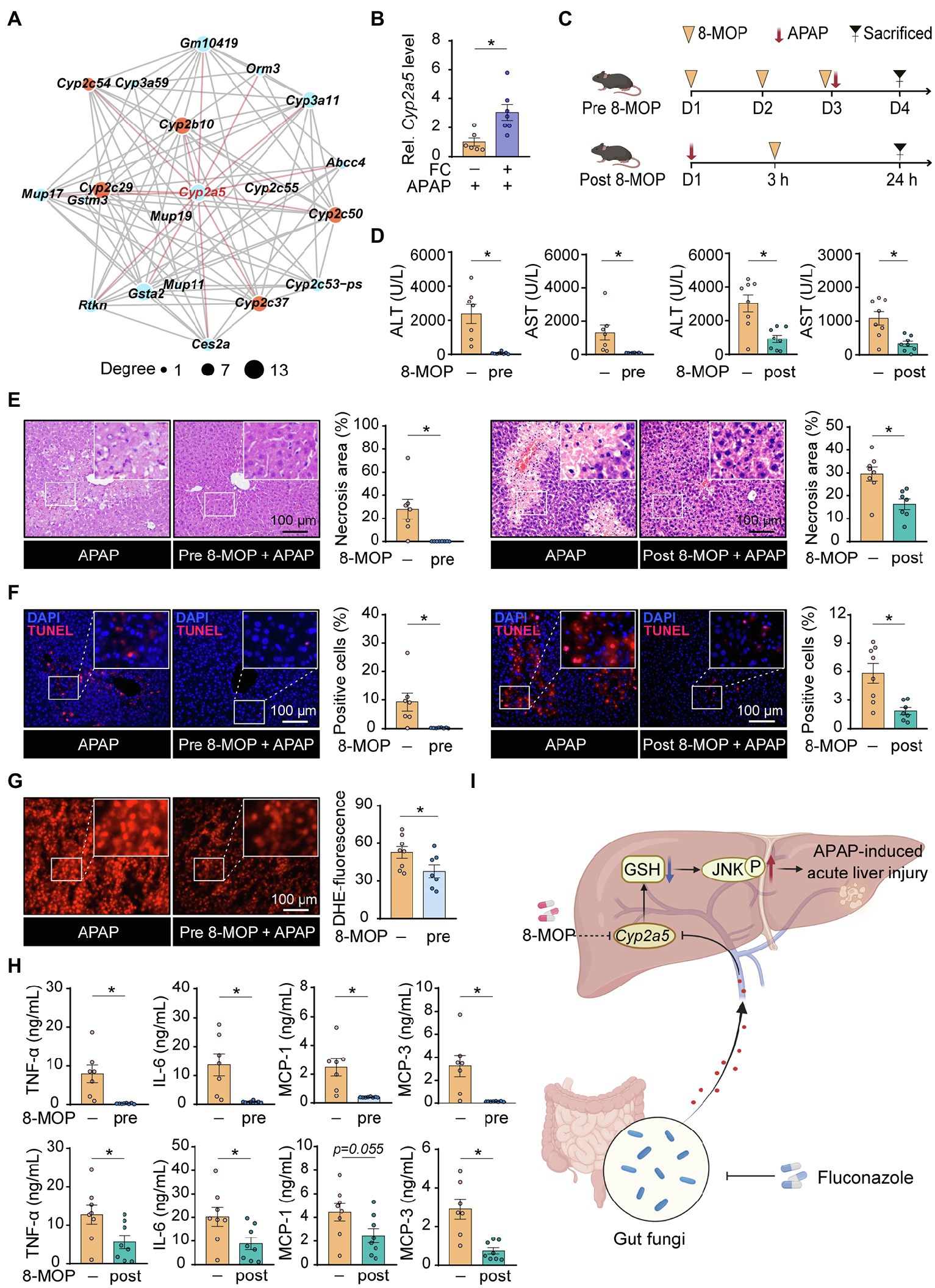
Figure 5. Gut commensal fungi ablation enhances the susceptibility to APAP-induced hepatotoxicity by upregulation of hepatic Cyp2a5 level. (A) The interaction network of the top twenty DEGs in the control group compared with fluconazole-pretreated mice in presence of APAP administration. C57BL/6 mice were pretreated with fluconazole for 14 days to deplete the gut commensal fungi, followed by subjection to APAP administration for 24 h. Liver tissues were collected for transcriptome sequencing analysis. (B) Relative mRNA levels of Cyp2a5 in the liver tissues from APAP-treated mice with or without preadministration of fluconazole. (C) Study design. Mice were pre- and post-treated with the Cyp2a5 inhibitor, 8-MOP, in presence of APAP administration. After 24 h of APAP treatment, the mice were sacrificed, and tissues were collected for further analysis. (D) Effect of 8-MOP on ALT and AST levels in the blood of APAP-treated mice. (E) Representative H&E staining images and quantification of necrotic areas in the liver from APAP-treated mice with or without administration of 8-MOP. (F) TUNEL staining of the liver from APAP-treated mice with or without administration of 8-MOP and quantification of dead cells. (G) Representative images of DHE immunofluorescence staining and quantification of hepatic ROS production in APAP-treated mice with or without preadministration of 8-MOP. (H) Quantification of pro-inflammatory cytokines including TNF-α, IL-6, MCP-1 and MCP-3 in the blood of APAP-treated mice with or without 8-MOP administration. (I) Working model: Mice with gut fungi depletion by fluconazole treatment showed more sensitivity to APAP-induced acute liver injury through the promotion of hepatic Cyp2a5 overexpression (Created with BioRender.com). *p < 0.05. Data were expressed as mean ± SEM and were evaluated by a two-tailed unpaired Student’s t-test. n = 6–8. Scale bar = 100 μm. APAP, acetaminophen; FC, fluconazole; Rel, Relative; 8-MOP, 8-methoxypsoralen; ALT, alanine aminotransferase; AST, aspartate aminotransferase; H&E, hematoxylin and eosin; TUNEL, terminal-deoxynucleotidyl transferase-mediated nick end labeling; and DHE, dihydroethidium.
To verify this hypothesis, mice were pre- and post-treated with 8-methoxypsoralen (8-MOP, known as a Cyp2a5 inhibitor) in the presence of APAP administration, respectively (Figure 5C). Interestingly, pharmacologic inhibition of Cyp2a5, by administration of 8-MOP to control mice, fully reversed the hepatotoxicity induced by APAP administration, as indicated by the result of serum biochemistry (Figure 5D). Furthermore, histopathological examination displayed that the percentage of cell death apparently decreased in the liver tissues of APAP-treated mice subjected to 8-MOP treatment compared with the APAP-only group (Figures 5E,F). In line with the above results, pharmacologic inhibition of Cyp2a5 significantly suppressed the accumulation of hepatic ROS as well as cytokine and chemokine release in APAP-treated mice (Figures 5G,H). Taken together, these discoveries suggested that inhibiting hepatic Cyp2a5 overexpression may hold promise as a novel pharmacological strategy for treating APAP-induced acute liver injury.
DILI is a common acute liver disease characterized by high mortality and is mainly caused by an overdose of APAP in Western countries (Andrade et al., 2019). The pathogenesis of APAP-induced hepatotoxicity is associated with the persistent depletion of hepatic reduced glutathione (GSH), leading to predispose hepatocytes to sustained oxidative stress-induced necroptosis and ferroptosis (Matsumaru et al., 2003; Yamada et al., 2020). The incidence of DILI was progressively increased worldwide that accompany with the action of abuse drugs (Haque et al., 2016). Therefore, novel targeted strategies for the treatment of drug-induced hepatotoxicity are essential.
Although maintaining the ecological homeostasis in the gut has recently been shown to increase resistance to ALF, the biological functions of gut commensal fungi in regulation of the sustained oxidative stress during acute liver injury induced by APAP overdose remain largely undefined (Chopyk and Grakoui, 2020). Our data clearly revealed that fluconazole pretreatment predisposed mice to accelerated hepatotoxicity induced by APAP. We next explored whether the effects of fluconazole in the progression of APAP-induced liver injury were in gut fungi-dependent manner. It is well known that glutathione biosynthesis is dependent on the uptake of amino acids during food processing and digestion (Minich and Brown, 2019). We first monitored food consumption and body weight periodically until the end of the fluconazole treatment period to exclude the possibility that any difference in energy metabolism between the fluconazole-treated mice and the control group influenced the process of glutathione biosynthesis in the liver. Additionally, we also found that fluconazole treatment alone did not cause liver injury in our study, as confirmed by the serum level of ALT, and the expression of chemokines in control and fluconazole-treated mice, whereas we found that the gut fungi load was significantly reduced in fluconazole-treated mice compared with the control group, a result consistent with previous literature (Li et al., 2018). These above results indicated that gut commensal fungi could protect mice against APAP-induced liver injury, which suggested that maintaining the gut commensal fungi balance could be a promising therapeutic strategy for preventing DILI. Further preclinical and clinical research will be necessary to identify the specific intestinal-resident fungi responsible for protecting against drug-induced hepatotoxicity.
Since commensal bacteria coexist with fungi in the intestine, maintaining microecological homeostasis can increase resistance to disease through the modulation of host immunity (Clemente et al., 2012). This notion is supported by the other observation that antibiotic-mediated depletion of intestinal-resident bacteria may shift the organizational structure of gut commensal fungi, leading to result in an overabundance of Saccharomycetes fungi in particular (Jiang et al., 2017). However, less is known about whether fluconazole-induced fungi depletion leads to persistent changes in the composition of gut bacteria. In the present study, we noted that gut commensal fungi ablation through fluconazole treatment could increase the diversity of intestinal bacteria communities, as shown by Faith’s Phylogenetic Distance (PD) and the Observed OTUs, which appears to be due to a strong competition between enteric bacterial and fungal components for commonly available nutrients in a shared environment. In addition, our data showed that the relative ratio of Firmicutes and Bacteroidetes was decreased in fluconazole-treated mice compared with the control group, indicating the occurrence of a gut microbiota disorder. In particular, we found that gut fungi ablation leads to an overabundance of the enteric bacterial pathogen H. rodentium, while the level of probiotic A. muciniphila was decreased. Therefore, our study has provided new insight into the significance of enteric bacteria-fungi interactions, although the functions of the bacteria or its derived metabolites modulated by fungal components in the gut need further clarification.
A key finding of the current study was the highlighting of inhibition of hepatic Cyp2a5 expression as a novel therapeutic strategy for treating drug-induced liver failure. As a member of the CYP2A subfamily, murine Cyp2a5, and its human ortholog, Cyp2a6, have been identified as important xenobiotic-metabolizing enzymes in the liver (Kim et al., 2005; Kirby et al., 2011). Recent research indicated that exposure to chemicals including ethanol, thioacetamide, and cadmium could promote upregulations of hepatic Cyp2a5 expression in mice, thereby modulating disease susceptibility by affecting oxidative stress (Lu et al., 2011; Hong et al., 2016). However, the pathogenic role of Cyp2a5 in regulating acute liver injury induced by APAP overdose remains unclear. In the present study, we found that the expression of Cyp2a5 was increased in APAP-treated mice subjected to fluconazole pretreatment, suggesting that Cyp2a5 appears to be a dominant-positive regulator of APAP hepatotoxicity. To verify this hypothesis, we applied a known Cyp2a5 inhibitor, 8-methoxypsoralen (8-MOP), to APAP-treated mice and found that the pharmacologic inhibition of Cyp2a5 fully reversed the hepatotoxicity induced by APAP administration.
Given the possibility that the protective role of 8-MOP on APAP-induced hepatotoxicity was mediated through the blocking of APAP metabolic bioactivation, mice were treated with 8-MOP after 3 h of APAP administration, by which time the processes of intrahepatic APAP metabolic is end (Souza et al., 2022). Serum biochemical analysis showed that administering 8-MOP post-treatment also reduced APAP-induced hepatotoxicity in mice, a conclusion which was further confirmed by histopathological examination. The pharmacologic inhibition of Cyp2a5, therefore, could play a role in preventing drug-induced liver failure, although the potential therapeutic effects of 8-MOP will require further clinical study.
In summary, our findings identified the protective role of gut commensal fungi against acute APAP-induced liver injury through an attenuated inflammatory response and oxidative stress in mice and have shed light on the significance of downregulating Cyp2a5 by 8-MOP as a novel pharmacological strategy for treating DILI.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Southern Medical University, Guangzhou, China.
SG, LL, WX, XZ, and HY were responsible for the study design, supervision, and manuscript preparation. ZH, YZ, SL, LL, RZ, FW, WY, and YW were responsible for the experiment. JY, AC, and ZW were responsible for sequencing analysis. All authors contributed to the article and approved the submitted version.
This work was financially supported by the National Natural Science Foundation of China (81973804), Shenzhen Science and Technology Program (RCBS20210706092252059), the Key Project of National Natural Science Foundation of China (81830117), and the National Natural Science Foundation of China (82104382).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Jiaqi Li for the excellent technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.944416/full#supplementary-material
DILI, drug-induced liver injury; ALF, acute liver failure; APAP, acetaminophen; GSH, glutathione; ROS, reactive oxygen species; NAPQI, n-acetyl-p-benzoquinoneimine; PPD, 1-phenyl-1,2-propanedione; MNPs, mononuclear phagocytes; FC, fluconazole; PBS, phosphate-buffered saline; 8-MOP, 8-methoxypsoralen; ALT, alanine transaminase; AST, aspartate transaminase; MDA, malondialdehyde; SOD, superoxide dismutase; PFA, paraformaldehyde; HE, hematoxylin and eosin; HRP, horseradish peroxidase; TUNEL, terminal-deoxynucleotidyl transferase-mediated nick end labeling; DHE, dihydroethidium; MFI, mean fluorescence intensity; PCR, polymerase chain reaction; qRT-PCR, quantitative real-time polymerase chain reaction; TBST, tris-buffered saline tween; ECL, enhanced chemiluminescence; DEGs, differentially expressed genes; SEM, standard error of the mean; PD, phylogenetic distance; LDA, linear discriminant analysis; LEfSe, linear discriminant analysis effect size; A. muciniphila, Akkermansia muciniphila; H. rodentium, Helicobacter rodentium; IHC, immunohistochemical; PCoA, principal coordinate analysis; PCA, principal component analysis.
Albillos, A., De Gottardi, A., and Rescigno, M. (2020). The gut-liver axis in liver disease: pathophysiological basis for therapy. J. Hepatol. 72, 558–577. doi: 10.1016/j.jhep.2019.10.003
Andrade, R. J., Chalasani, N., Björnsson, E. S., Suzuki, A., Kullak-Ublick, G. A., Watkins, P. B., et al. (2019). Drug-induced liver injury. Nat. Rev. Dis. Primers. 5:58. doi: 10.1038/s41572-019-0105-0
Bernal, W., Hyyrylainen, A., Gera, A., Audimoolam, V. K., McPhail, M. J., Auzinger, G., et al. (2013). Lessons from look-back in acute liver failure? A single Centre experience of 3300 patients. J. Hepatol. 59, 74–80. doi: 10.1016/j.jhep.2013.02.010
Bernal, W., and Wendon, J. (2013). Acute liver failure. N. Engl. J. Med. 369, 2525–2534. doi: 10.1056/NEJMra1208937
Chen, K., Zhu, Y., Zhang, Y., Hamza, T., Yu, H., Saint Fleur, A., et al. (2020). A probiotic yeast-based immunotherapy against Clostridioides difficile infection. Sci. Transl. Med. 12:eaax4905. doi: 10.1126/scitranslmed.aax4905
Chopyk, D. M., and Grakoui, A. (2020). Contribution of the intestinal microbiome and gut barrier to hepatic disorders. Gastroenterology 159, 849–863. doi: 10.1053/j.gastro.2020.04.077
Clemente, J. C., Ursell, L. K., Parfrey, L. W., and Knight, R. (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. doi: 10.1016/j.cell.2012.01.035
Fernandez-Checa, J. C., Bagnaninchi, P., Ye, H., Sancho-Bru, P., Falcon-Perez, J. M., Royo, F., et al. (2021). Advanced preclinical models for evaluation of drug-induced liver injury–consensus statement by the European drug-induced liver injury network [PRO-EURO-DILI-NET]. J. Hepatol. 75, 935–959. doi: 10.1016/j.jhep.2021.06.021
Goldring, C. E., Kitteringham, N. R., Elsby, R., Randle, L. E., Clement, Y. N., Williams, D. P., et al. (2004). Activation of hepatic Nrf2 in vivo by acetaminophen in CD-1 mice. Hepatology 39, 1267–1276. doi: 10.1002/hep.20183
Gong, S., Lan, T., Zeng, L., Luo, H., Yang, X., Li, N., et al. (2018). Gut microbiota mediates diurnal variation of acetaminophen induced acute liver injury in mice. J. Hepatol. 69, 51–59. doi: 10.1016/j.jhep.2018.02.024
Grasl-Kraupp, B., Ruttkay-Nedecky, B., Koudelka, H., Bukowska, K., Bursch, W., and Schulte-Hermann, R. (1995). In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21, 1465–1468. doi: 10.1002/hep.1840210534
Haque, T., Sasatomi, E., and Hayashi, P. H. (2016). Drug-induced liver injury: pattern recognition and future directions. Gut. Liver 10, 27–36. doi: 10.5009/gnl15114
Harizi, H., Corcuff, J.-B., and Gualde, N. (2008). Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol. Med. 14, 461–469. doi: 10.1016/j.molmed.2008.08.005
Hong, F., Si, C., Gao, P., Cederbaum, A. I., Xiong, H., and Lu, Y. (2016). The role of CYP2A5 in liver injury and fibrosis: chemical-specific difference. Naunyn. Schmiedebergs Arch. Pharmacol. 389, 33–43. doi: 10.1007/s00210-015-1172-8
Hynes, S. O., Ferris, J. A., Szponar, B., Wadström, T., Fox, J. G., O’Rourke, J., et al. (2004). Comparative chemical and biological characterization of the lipopolysaccharides of gastric and enterohepatic helicobacters. Helicobacter 9, 313–323. doi: 10.1111/j.1083-4389.2004.00237.x
Iliev, I. D., Funari, V. A., Taylor, K. D., Nguyen, Q., Reyes, C. N., Strom, S. P., et al. (2012). Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317. doi: 10.1126/science.1221789
Iliev, I. D., and Leonardi, I. (2017). Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 17, 635–646. doi: 10.1038/nri.2017.55
Jaeschke, H., Ramachandran, A., Chao, X., and Ding, W.-X. (2019). Emerging and established modes of cell death during acetaminophen-induced liver injury. Arch. Toxicol. 93, 3491–3502. doi: 10.1007/s00204-019-02597-1
Jiang, T. T., Shao, T.-Y., Ang, W. G., Kinder, J. M., Turner, L. H., Pham, G., et al. (2017). Commensal fungi recapitulate the protective benefits of intestinal bacteria. Cell Host Microbe 22, 809.e4–816.e4. doi: 10.1016/j.chom.2017.10.013
Kim, D., Wu, Z.-L., and Guengerich, F. P. (2005). Analysis of coumarin 7-hydroxylation activity of cytochrome P450 2A6 using random mutagenesis. J. Biol. Chem. 280, 40319–40327. doi: 10.1074/jbc.M508171200
Kirby, G. M., Nichols, K. D., and Antenos, M. (2011). CYP2A5 induction and hepatocellular stress: an adaptive response to perturbations of heme homeostasis. Curr. Drug Metab. 12, 186–197. doi: 10.2174/138920011795016845
Krenkel, O., Mossanen, J. C., and Tacke, F. (2014). Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 3, 331–343. doi: 10.3978/j.issn.2304-3881.2014.11.01
Larson, A. M., Polson, J., Fontana, R. J., Davern, T. J., Lalani, E., Hynan, L. S., et al. (2005). Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42, 1364–1372. doi: 10.1002/hep.20948
Li, X., Leonardi, I., Semon, A., Doron, I., Gao, I. H., Putzel, G. G., et al. (2018). Response to fungal dysbiosis by gut-resident CX3CR1+ mononuclear phagocytes aggravates allergic airway disease. Cell Host Microbe 24, 847–856.e4. doi: 10.1016/j.chom.2018.11.003
Li, L., Wang, H., Zhang, J., Sha, Y., Wu, F., Wen, S., et al. (2020). SPHK1 deficiency protects mice from acetaminophen-induced ER stress and mitochondrial permeability transition. Cell Death Differ. 27, 1924–1937. doi: 10.1038/s41418-019-0471-x
Lu, Y., Zhuge, J., Wu, D., and Cederbaum, A. I. (2011). Ethanol induction of CYP2A5: permissive role for CYP2E1. Drug Metab. Dispos. 39, 330–336. doi: 10.1124/dmd.110.035691
Matsumaru, K., Ji, C., and Kaplowitz, N. (2003). Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology 37, 1425–1434. doi: 10.1053/jhep.2003.50230
Minich, D. M., and Brown, B. I. (2019). A review of dietary (phyto) nutrients for glutathione support. Nutrients 11:2073. doi: 10.3390/nu11092073
Norman, J. M., Handley, S. A., and Virgin, H. W. (2014). Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology 146, 1459–1469. doi: 10.1053/j.gastro.2014.02.001
Qiu, X., Zhang, F., Yang, X., Wu, N., Jiang, W., Li, X., et al. (2015). Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Sci. Rep. 5:10416. doi: 10.1038/srep10416
Sam, Q. H., Chang, M. W., and Chai, L. Y. A. (2017). The fungal mycobiome and its interaction with gut bacteria in the host. Int. J. Mol. Sci. 18:330. doi: 10.3390/ijms18020330
Schei, K., Avershina, E., Øien, T., Rudi, K., Follestad, T., Salamati, S., et al. (2017). Early gut mycobiota and mother-offspring transfer. Microbiome 5:107. doi: 10.1186/s40168-017-0319-x
Shojaie, L., Iorga, A., and Dara, L. (2020). Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 21:9682. doi: 10.3390/ijms21249682
Solovjov, D. A., Pluskota, E., and Plow, E. F. (2005). Distinct roles for the α and β subunits in the functions of integrin αMβ2. J. Biol. Chem. 280, 1336–1345. doi: 10.1074/jbc.M406968200
Souza, V. D., Shetty, M., Badanthadka, M., Mamatha, B., and Vijayanarayana, K. (2022). The effect of nutritional status on the pharmacokinetic profile of acetaminophen. Toxicol. Appl. Pharmacol. 438:115888. doi: 10.1016/j.taap.2022.115888
Stravitz, R. T., and Lee, W. M. (2019). Acute liver failure. Lancet 394, 869–881. doi: 10.1016/s0140-6736(19)31894-x
Watkins, P. B., and Seeff, L. B. (2006). Drug-induced liver injury: summary of a single topic clinical research conference. Hepatology 43, 618–631. doi: 10.1002/hep.21095
Yamada, N., Karasawa, T., Kimura, H., Watanabe, S., Komada, T., Kamata, R., et al. (2020). Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 11:144. doi: 10.1038/s41419-020-2334-2
Keywords: acetaminophen, gut fungi, Cyp2a5, acute liver injury, inflammation, oxidative stress
Citation: He Z, Zeng Y, Li S, Lin L, Zhou R, Wang F, Yang W, Wu Y, Yang J, Chen A, Wang Z, Yang H, Zhao X, Xiao W, Li L and Gong S (2022) Gut Commensal Fungi Protect Against Acetaminophen-Induced Hepatotoxicity by Reducing Cyp2a5 Expression in Mice. Front. Microbiol. 13:944416. doi: 10.3389/fmicb.2022.944416
Edited by:
Hui Zhang, South China Agricultural University, ChinaReviewed by:
Dayou Shi, South China Agricultural University, ChinaCopyright © 2022 He, Zeng, Li, Lin, Zhou, Wang, Yang, Wu, Yang, Chen, Wang, Yang, Zhao, Xiao, Li and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenhai Gong, Z3NoMDUyNkBzbXUuZWR1LmNu; Lei Li, MjY5NzM2MzcxNEBxcS5jb20=; Wei Xiao, eHc3Njg4QHNtdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.