 Hongbo Jiang
Hongbo Jiang Jie Bao
Jie Bao Yuenan Xing
Yuenan Xing Qijun Chen
Qijun Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 June 2022
Sec. Systems Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.939141
This article is part of the Research Topic Omics-Originated Exploration of Pathogenic Patterns and Molecular Mechanisms in Human and Animal Fungal Pathogens View all 7 articles
Metschnikowia bicuspidata is a globally distributed pathogenic yeast with a wide range of aquatic hosts. A new strain, M. bicuspidata LNES0119, isolated from the Chinese mitten crab Eriocheir sinensis, has caused a serious reduction in production and marked economic loss for the aquaculture industry in China. Therefore, the whole-genome sequence of M. bicuspidata LNES0119 was sequenced using Illumina and Oxford Nanopore technology; whole-genome annotation and comparative genomic analyses of this pathogen were performed as well. A high-quality genome of M. bicuspidata LNES0119 was 16.13 Mb in size, with six scaffolds and six contigs, and encoded 5,567 putative predicted genes. Of these, 1,467 genes shared substantial homology with genes in the pathogen–host interactions database. Comparative genomic analyses of three M. bicuspidata strains and one non-pathogenic yeast, M. aff. pulcherrima, showed 331 unique genes in M. bicuspidata LNES0119, 30 of which were putatively related to pathogenicity. Overall, we identified several meaningful characteristics related to pathogenicity and virulence that may play essential roles in the infection and pathogenicity of M. bicuspidata LNES0119. Our study will aid in identifying potential targets for further exploration of the molecular basis of the pathogenicity of M. bicuspidata as well as the therapeutic intervention of M. bicuspidata infection.
Metschnikowia bicuspidata (Metschnikoff) Kamienski (1899), which belongs to Fungi; Ascomycota; Saccharomycetales: Metschnikowiaceae: Metschnikowia, was first isolated from infected Daphnia magna by Metchnikoff (1884). Three strains are recognized according to their metabolic profile, biogeography, and habitat: M. bicuspidata var. bicuspidata, M. bicuspidata var. californica, and M. bicuspidata var. chathamia (Miller and Phaff, 1998).
M. bicuspidata is a globally distributed pathogenic fungus with a wide range of aquatic hosts. It exists in freshwater and marine environments worldwide, including in France, Romania, Russia, China, the United States, Canada, and even Antarctic waters (Bao et al., 2021). The hosts of M. bicuspidata include Daphnia, Artemia, snails, and the economically important freshwater prawn Macrobrachium rosenbergii, Chinese swimming crab Portunus trituberculatus, Chinese mitten crab Eriocheir sinensis, Chinese, grass shrimp (Palaemonetes sinensis) and chinook salmon (Moore and Strom, 2003; Wang et al., 2007; Bao et al., 2021; Cao et al., 2022). Among these hosts, the Daphnia-M. bicuspidata system has long been considered a model system in ecology and evolutionary biology and has been used as a model organism in the exploration of host-parasite theory. For example, M. bicuspidata is considered to be highly virulent and has been suggested to have affected the evolution of D. dentifera populations in Bristol Lake, United States (Duffy et al., 2008). In aquaculture, many economically important animals have been infected by M. bicuspidata, resulting in a decline in production and marked economic loss. For example, an outbreak of M. bicuspidata in Taiwan from May 2001 to December 2003 resulted in cumulative mortality rates of 20–95% in M. rosenbergii (see Chen et al., 2003, 2007), 40–60% in P. trituberculatus in Zhoushan, Zhejiang Province, China, from 2002 to 2006 (Shi et al., 2008), over 20% in E. sinensis in Panjin, Liaoning Province, China, from 2018 to 2019 (Bao et al., 2021), and 34.5% in larval chinook salmon fed on infected Artemia in California, United States (Moore and Strom, 2003). The aquatic hosts were infected directly by ingesting M. bicuspidata spores or indirectly by consuming diseased individuals, after which the spores in the body cavity began producing hyphae and adhered to nearby surfaces, producing conidia that rapidly increased in abundance within the host (Merrill and Cáceres, 2018; Jiang et al., 2022; Sun et al., 2022).
In 2019, we isolated the M. bicuspidata LNES0119 strain from infected E. sinensis from ponds in Panjin city, Liaoning Province, and using the D1/D2 domain of the 26S rDNA, we identified it as the pathogen causing milky disease in Chinese mitten crab (Bao et al., 2021), which is an economically important freshwater crustacean in China. This epidemic showed the characteristic symptoms of milky hemolymph and death due to organ failure. A milky disease epidemic was also detected in Xinjiang, Heilongjiang, and Jilin provinces, and the mortality rates in 2021 were even higher than that in 2020, which had seriously affected the Chinese mitten crab industry (Sun et al., 2022). Therefore, in this study, the whole-genome sequence of the strain M. bicuspidata LNES0119 was sequenced using Illumina and Oxford Nanopore technology. The whole-genome annotation and comparative genomic analyses with two M. bicuspidata stains and one non-pathogenic M. aff. pulcherrima stain were carried out to explore the genes or gene families associated with pathogenicity, our analyses will provide genomic resources for future exploration of the pathogenesis and molecular basis of the pathogenicity of M. bicuspidata.
The M. bicuspidata strain LNES0119 was isolated from a diseased Chinese mitten crab in Panjin city. The strain was stored at −80°C in 20% glycerol (v/v), incubated on Rose Bengal agar medium, cultured for 48 h at 28°C, and transferred onto a new medium, followed by further culturing for 48 h before collecting the yeast. Genomic DNA was extracted using the Qiagen Genomic-Tip 100/G Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA quality was assessed via 0.35% agarose gel electrophoresis and quantified using NanoDrop and Qubit Fluorometer 3.0 (Thermo Fisher Scientific, Waltham, MA, United States).
The M. bicuspidata strain LNES0119 was first surveyed using Illumina NovaSeq 6000 platform TruSeq libraries (150 bp paired end reads, insert size of 350 bp) and then sequenced using the long reads Oxford Nanopore sequencing platforms at Beijing Biomarker Technologies (Beijing, China). Large fragments of DNA (DNA fragments > 20 kb) were first collected using the BluePippin Size-Selection system (Sage Science, Beverly, MA, United States). Next, the DNA sequencing library was constructed, including DNA repair, end-prep, and adapter ligation, and clean-up steps were performed with the ligation kit SQK-LSK109 (Oxford Nanopore Technologies, Oxford, United Kingdom) according to the manufacturer’s instructions. The final product was quantified using a Qubit fluorometer (Thermo Fisher Scientific, Waltham, United States) and loaded into the PromethION flow cell, and real-time single-molecule sequencing was performed according to the manufacturer’s instructions (Oxford Nanopore Technologies, Oxford, United Kingdom). The Nanopore reads were base-called from the raw Fast5 files using Albacore (Oxford Nanopore Technologies). Fastq reads were filtered with a quality value of Q > 7. The filtered subreads were first corrected and assembled using Canu version 1.5 (Koren et al., 2017) and Wtdbg version 2.2 (Ruan and Li, 2020). Pilon version 1.22 (Walker et al., 2014) was further applied to correct any sequencing errors caused by using second-generation results, and a genome sequence with a higher accuracy was obtained. BUSCO v.2.0, Simão et al. (2015) was used to assess the completeness of the assembled genome based on the Benchmarking Universal Single-Copy Orthologs (BUSCOs) for Fungi (fungi_odb9) dataset.
For prediction of repeat sequences, LTR_FINDER v1.05 (Xu and Wang, 2007), MITE-Hunter (Han and Wessler, 2010), RepeatScout v1.0.5 (Price et al., 2005) and PILER-DF v2.4 (Edgar and Myers, 2005) software were used to construct a de novo repeats library of the M. bicuspidata LNES0119 genome using a combination of homology-based and de novo approaches. PASTEClassifier (Wicker et al., 2007) was used to classify the database, and then combined with the Repbase database (Jurka et al., 2005) to obtain the final repeat library, which was annotated using RepeatMasker V4.0.6 software (Chen, 2004). Transfer RNAs (tRNAs) were predicted by tRNAscan-SE 2.0 (Lowe and Eddy, 1997), whereas ribosomal RNAs (rRNAs) and other non-coding RNAs (ncRNAs) were predicted using Infernal 1.1 (Nawrocki and Eddy, 2013) based on Rfam database (Nawrocki et al., 2015).
Protein-coding genes in the M. bicuspidata LNES0119 genome were predicted using a combination of ab initio prediction, homology-based protein prediction, and transcriptome-based prediction. For ab initio prediction, Genscan (Burge and Karlin, 1997), Augustus V2.4 (Stanke and Waack, 2003), GlimmerHMM V3.0.4 (Majoros et al., 2004), GeneID V1.4 (Blanco et al., 2007), and SNAP (version 2006-07-28) (Korf, 2004) were used. For homologous protein-based prediction, protein sequences from M. bicuspidata were downloaded from the NCBI database and then used for gene annotation by aligning against the M. bicuspidata LNES0119 genome using GeMoMa V1.3.1 (Keilwagen et al., 2016). For transcriptome-based prediction, HISAT v2.0.4 and StringTie v1.2.3 (Pertea et al., 2016) were used to assemble and map our referenced RNA-seq data. TransDecoder v2.0,1 GeneMarkS-T v5.1 (Tang et al., 2015) and PASA v2.0.2 (Campbell et al., 2006) were used to predict assembled UniGene sequences. Finally, EVM V1.1.1 (Campbell et al., 2006) was used to integrate the above three prediction results, and PASA v2.0.2 was employed for modification.
Functional annotations of all predicted gene models were compared with the following databases: Eukaryotic Orthologous Groups (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG), NCBI non-redundant protein sequences (Nr), Swiss-Prot, and TrEMBL using BLASTP with an e-value cutoff of 1e-5. Blast 2GO (Conesa et al., 2005) was used for gene annotation based on gene ontology (GO). Potential pathogenicity-related proteins were identified using the pathogen–host interactions (PHI) database (Winnenburg et al., 2006). HMMER (Eddy, 1998) was used to detect transporters based on the Transporter Classification Database (TCDB) (Saier et al., 2006) and to annotate the predicted protein sequences using the Pfam database (Finn et al., 2016). Carbohydrate-active enzymes (CAZymes) were actualized using the CAZy database as well as the dbCAN CAZyme HMM database (Yin et al., 2012). Secreted proteins were predicted by Signal P 4.0 (Petersen et al., 2011) and TMHMM (Krogh et al., 2001), and the proteins containing signal peptides and without a putative transmembrane structure were regarded as secreted proteins. Potential effector proteins were predicted using EffectorP (Sperschneider et al., 2016) with a probability threshold of 0.5.
Two pathogenic strains of M. bicuspidata Baker 2002 (PRJNA421585) and M. bicuspidata NRRL YB-4993 (PRJNA207846), and one non-pathogenic M. aff. pulcherrima APC 1.2 (PRJNA508581) species were selected for comparison of their genomic data with the genomic data of M. bicuspidata LNES0119 (Table 1). Collinearity analysis was performed using the MCScanX software (Wang et al., 2012). Gene orthology analysis was conducted using OrthoMCL (Li et al., 2003), and the gene families were analyzed, including the gene families specific to the strains, the gene families common to all species, and the gene families with a single copy of each strain. Functional annotation of gene families was performed using the Pfam database (Finn et al., 2016). For comparative analysis of the CAZymes, data from M. bicuspidata LNES0119 and 12 other yeast strains were processed using the dbCAN CAZyme HMMs database.
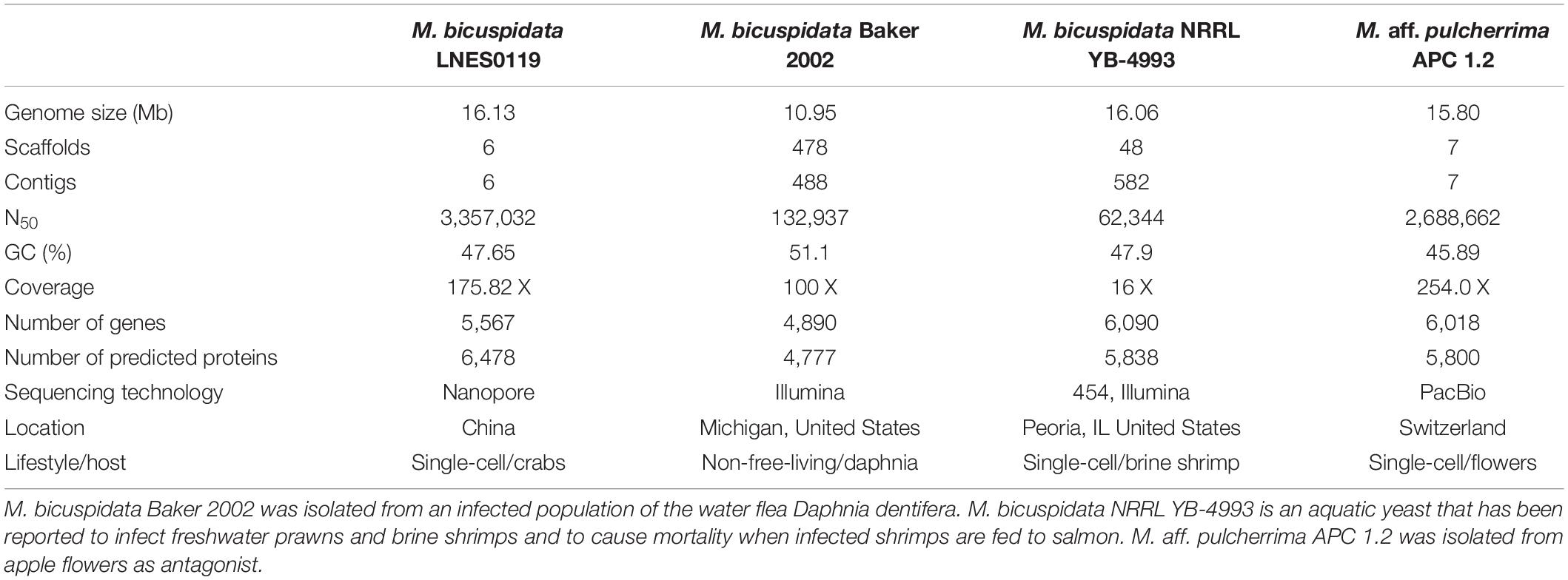
Table 1. Genome characteristics of four Metschnikowia species strains.
A total of 16.13 Mb of sequence data (175.82 X coverage), with six scaffolds and six contigs, were obtained from the whole genome of the M. bicuspidata strain LNES0119 (Table 2). Analysis of the length distribution of long and high-quality reads yielded a high-quality library (Supplementary Figure 1). The scaffold lengths of N50 and N90 were 3,357,032 and 2,548,385 bp, respectively, and the maximum length was 3,791,280 bp (Table 2). The average GC content was 47.65%. The number and average length of the predicted coding genes were 5,567 and 9,595,085 bp, respectively. The total length of the repeat sequences was 475,761 bp, accounting for 2.95% of the genomic length. These contained Class II transposable elements, including four Helitrons and 26 terminal inverted repeats, Class I or the retroelements including 359 LINE, 60 LTR/Copia, 153 LTR/Gypsy, and 426 non-LTR/LINE (Supplementary Table 1). With respect to RNAs, 88 rRNAs, 306 tRNAs, and 89 other ncRNAs were predicted (Supplementary Table 2). The genome was estimated to be 99.3% complete with 288 complete BUSCOs, one fragmented BUSCO, and one missing BUSCO from a total of 290 BUSCO groups. Overall, these results indicate that the genome of our M. bicuspidata strain was characterized as high-quality, complete, and accurate.

Table 2. Genome characteristics of the Metschnikowia bicuspidata strain LNES0119.
To annotate the function of the predicted genes in the M. bicuspidata LNES0119 genome build, 5,567 predicted genes were annotated using multiple public databases: Nr, GO, KEGG, KOG, TCDB, Pfam, TrEMBL, and Swiss-Prot databases are shown in Supplementary Table 3. According to the GO database, 3,175 predicted proteins that accounted for 57.03% of the entire genome were primarily distributed in three categories: cellular components, molecular function, and biological process (Supplementary Figure 2 and Supplementary Table 3). NCBI KOG mapping predicted that 3,590 genes (64.49%) were assigned to KOG categories (Figure 1). The most enriched KOG category is “General functional prediction only” (530), followed by “Posttranslational modification, protein turnover, chaperones” (386), “Translation, ribosomal structure and biogenesis” (305), “Intracellular trafficking, secretion, and vesicular transport” (282), “Function unknown” (262), and “Signal transduction mechanisms” (253). A total of 2,869 genes were annotated in the KEGG database and were separated into three specific categories: genetic information processing, environmental information processing, metabolism, and cellular processes (Supplementary Figure 3 and Supplementary Table 3). Among them, “Ribosome” (113) and “Biosynthesis of amino acids” (104) contained the largest number of genes.
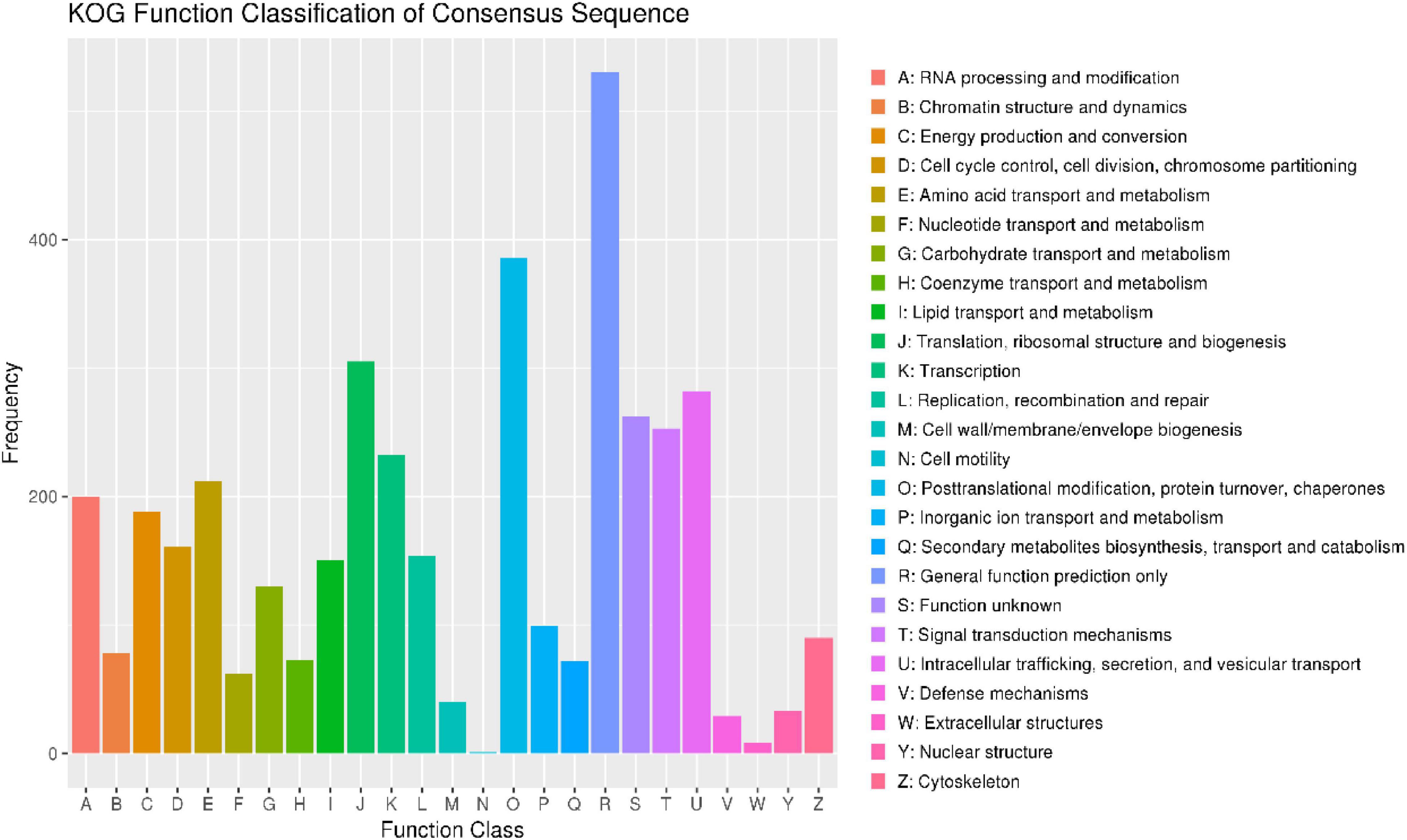
Figure 1. Clusters of orthologous groups of proteins (KOG) function classification of proteins in Metschnikowia bicuspidata LNES0119.
Moreover, 1,467 genes were annotated in the PHI database into different categories, which predicted protein function during host infection (Figure 2A and Supplementary Table 4). Of these genes, 653 genes were annotated to reduced virulence, 432 genes were annotated as unaffected pathogenicity, and there were 163 genes annotated as mixed outcomes. A total of 117 and 79 genes were annotated as pathogenic loss and lethal factors, respectively, whereas seven and 15 genes were annotated as chemistry targets and effectors (plant avirulence determinant), respectively. Only 10 genes were annotated as having enhanced virulence.
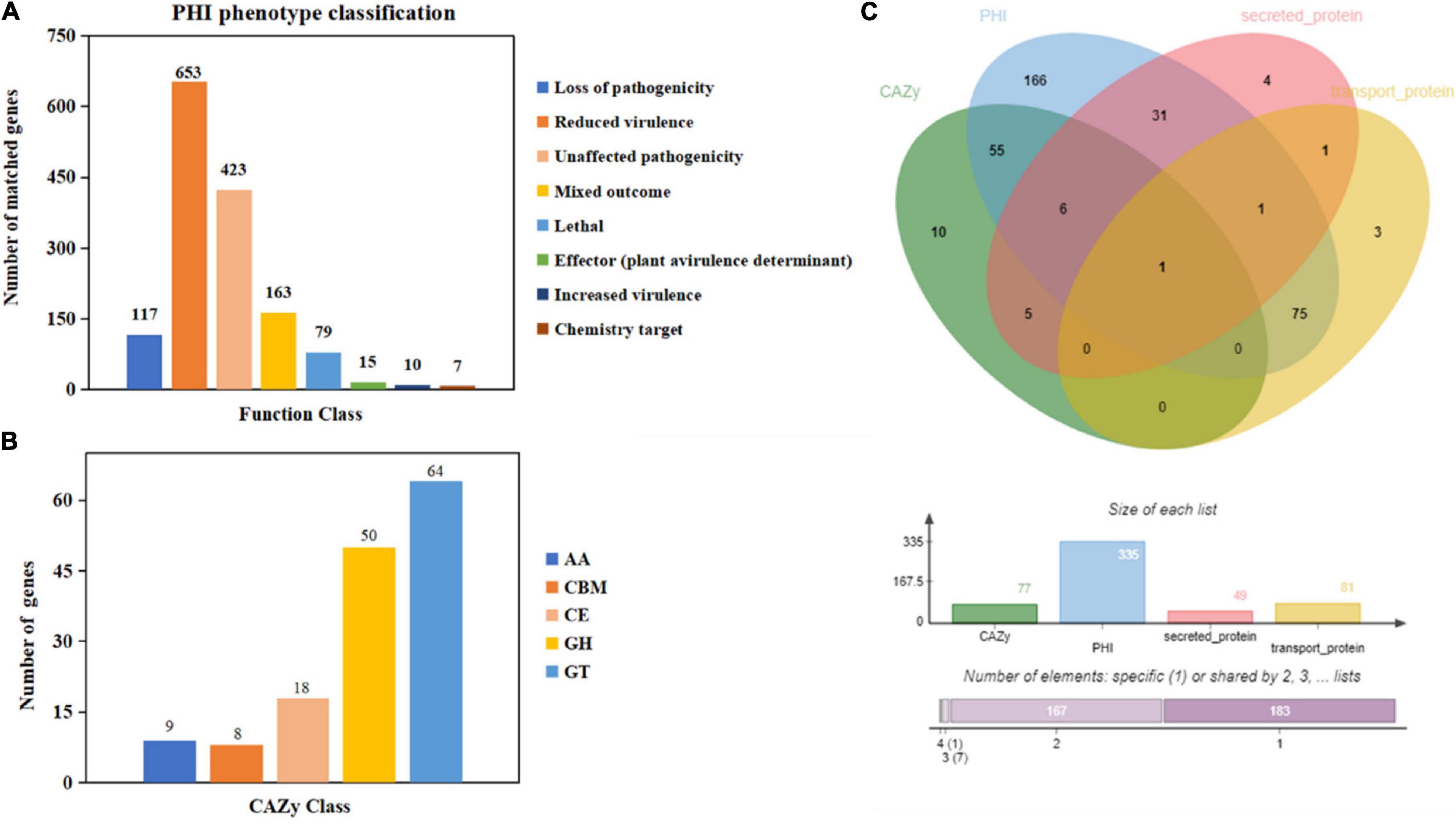
Figure 2. Gene annotation and gene prediction of M. bicuspidata LNES0119. (A) Genes were annotated and classified in the PHI database. Bars in different colors represent different PHI function classes, and lengths represent the number of genes. (B) Genes were annotated and classified in the CAZy database. Bars in different colors represent different CAZy categories, and lengths represent the number of genes. GH, glycoside hydrolase; GT, glycosyltransferase; CE, carbohydrate esterase; AA, auxiliary activity; CBM, carbohydrate-binding module. (C) Venn diagram showing the overlap of PHI-homologs and secretory proteins with transport protein and CAZymes.
There were 149 genes annotated in the CAZy database, which were divided into five categories, of which 64 genes (42.95%) annotated as glycosyl transferases (GTs), 50 genes (33.55%) annotated as glycoside hydrolases (GHs), 18 genes (12.08%) annotated as carbohydrate esterases (CEs), nine genes (6.04%) annotated as auxiliary activities enzymes (AAs), and eight genes (5.36%) annotated as carbohydrate-binding modules (CBMs) (Figure 2B).
The Venn map of M. bicuspidata LNES0119 was obtained according to the annotation results of CAZy, PHI, transported protein, and secreted protein (Figure 2C). A total of 291 proteins with signal peptides and 1,066 with transmembrane structures were detected in the genome of M. bicuspidata LNES0119. Of these, 158 were identified as potential secreted proteins (including 18 with putative effectors), of which 41 were annotated using the PHI database, and these secreted proteins were primarily cell wall proteins and hydrolytic proteins (Table 3 and Supplementary Table 5). In addition, 62 CAZymes protein clusters (including 64 genes) were annotated to PHI (Figure 2C), which were enriched in protein O-linked glycosylation (GO:0006493), glucan catabolic process (GO:0009251), and protein mannosylation (GO:0035268). The Venn diagram showed that there was only one protein cluster (multicopper oxidases) that was annotated to CAZymes, PHI, transported protein, and secreted protein, which belonged to iron ion homeostasis (GO:0055072).
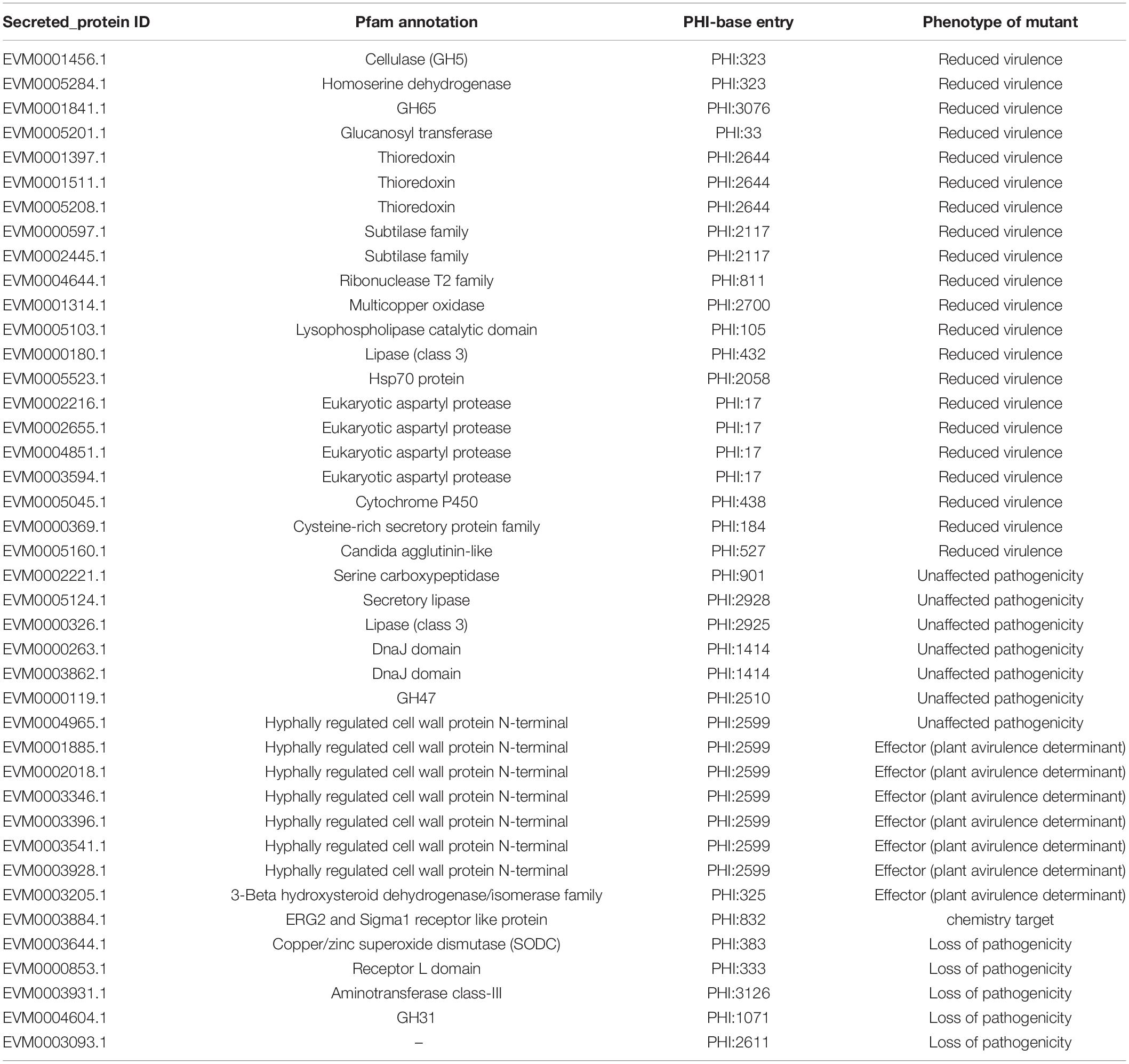
Table 3. Secretory proteins associated with pathogenicity.
The numbers of CAZymes in human/animal pathogenic and non-pathogenic yeasts varied between 117 and 173, all of which were without polysaccharide lyases (PLs). The lowest number was in M. bicuspidata Baker 2002 and highest in non-pathogen Debaryomyces hansenii (Figure 3). The number of CAZymes in M. bicuspidata LNES0119 was lower than that in the human pathogen C. albicans and non-pathogen Debaryomyces hansenii (Figure 3). CBM18, CBM21, CBM43, and CBM48 were present in each fungus, but CBM48 was absent in M. bicuspidata LNES0119 (Supplementary Table 6). In addition, M. bicuspidata LNES0119 had 18 CE, which was the highest among these yeast genomes. CBM20, CBM23, CE1 (8), CE8, CE10 (6), CE12, CE14, AA3, and AA7 are unique to the M. bicuspidata LNES0119 genome (Supplementary Table 6).
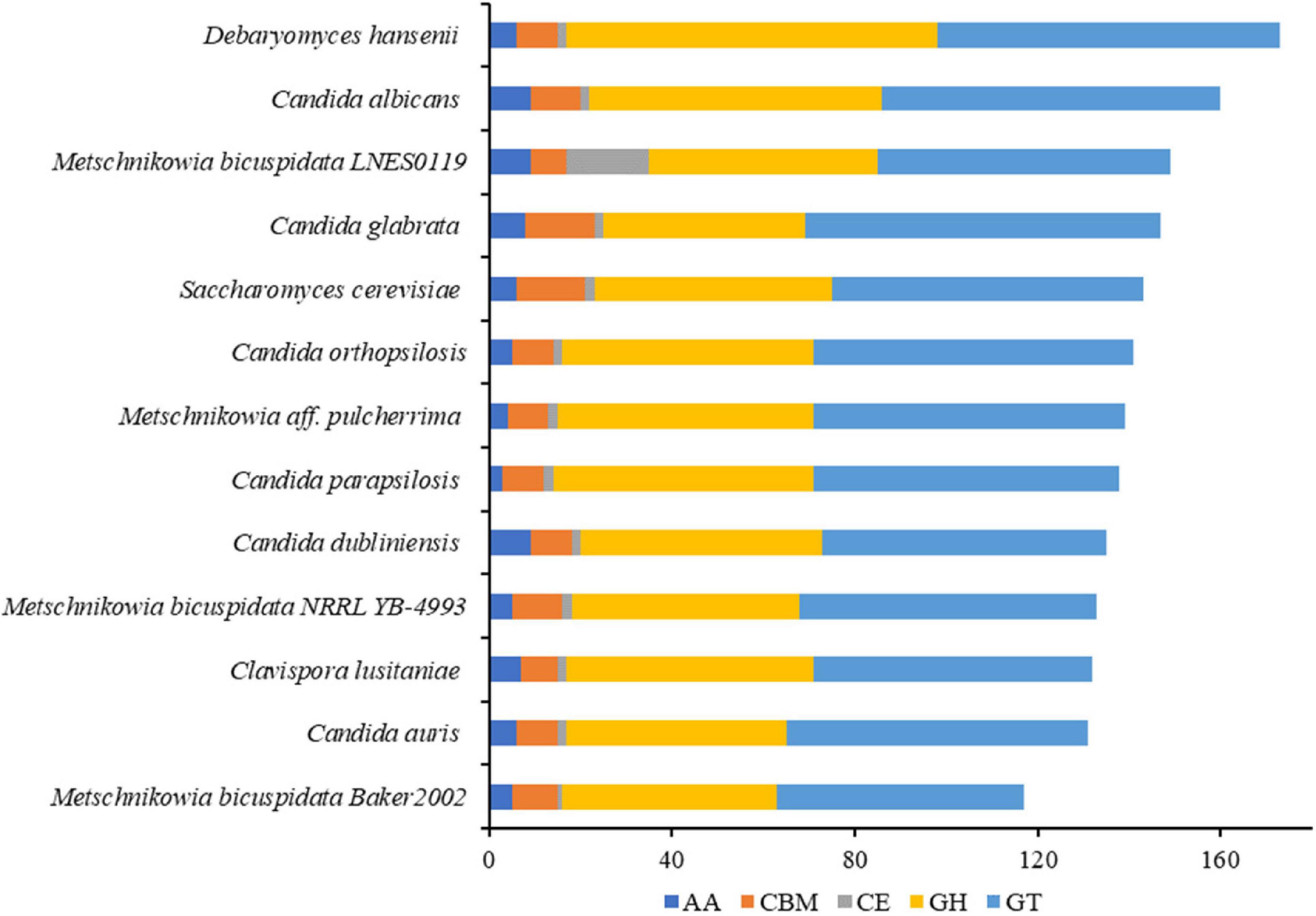
Figure 3. Number of CAZymes genes in M. bicuspidata LNES0119 and the other 12 yeasts. GH: glycoside hydrolase, GT, glycosyltransferase; CE, carbohydrate esterase; AA, auxiliary activity; CBM, carbohydrate-binding module.
As shown in Table 1, the genome size was similar between LNES0119 (16.13 Mb) and NRRL YB-4993 (16.06 Mb); meanwhile, the phylogenetic tree revealed that these two M. bicuspidata strains evolved closely, as determined according to their single copy homologous genes (Figure 4). In addition, there were 10,522 (92.52%) collinear genes between M. bicuspidata LNES0119 and NRRL YB-4993 strains, whereas there were 6,809 (65.83%) collinear genes between M. bicuspidata LNES0119 and Baker 2002 strains (Figures 4, 5). We found that the M. bicuspidata LNES0119 and NRRL YB-4993 strains were largely linearly syntenic, but there was a total of six evident inversions between the two assemblies (Figure 5).
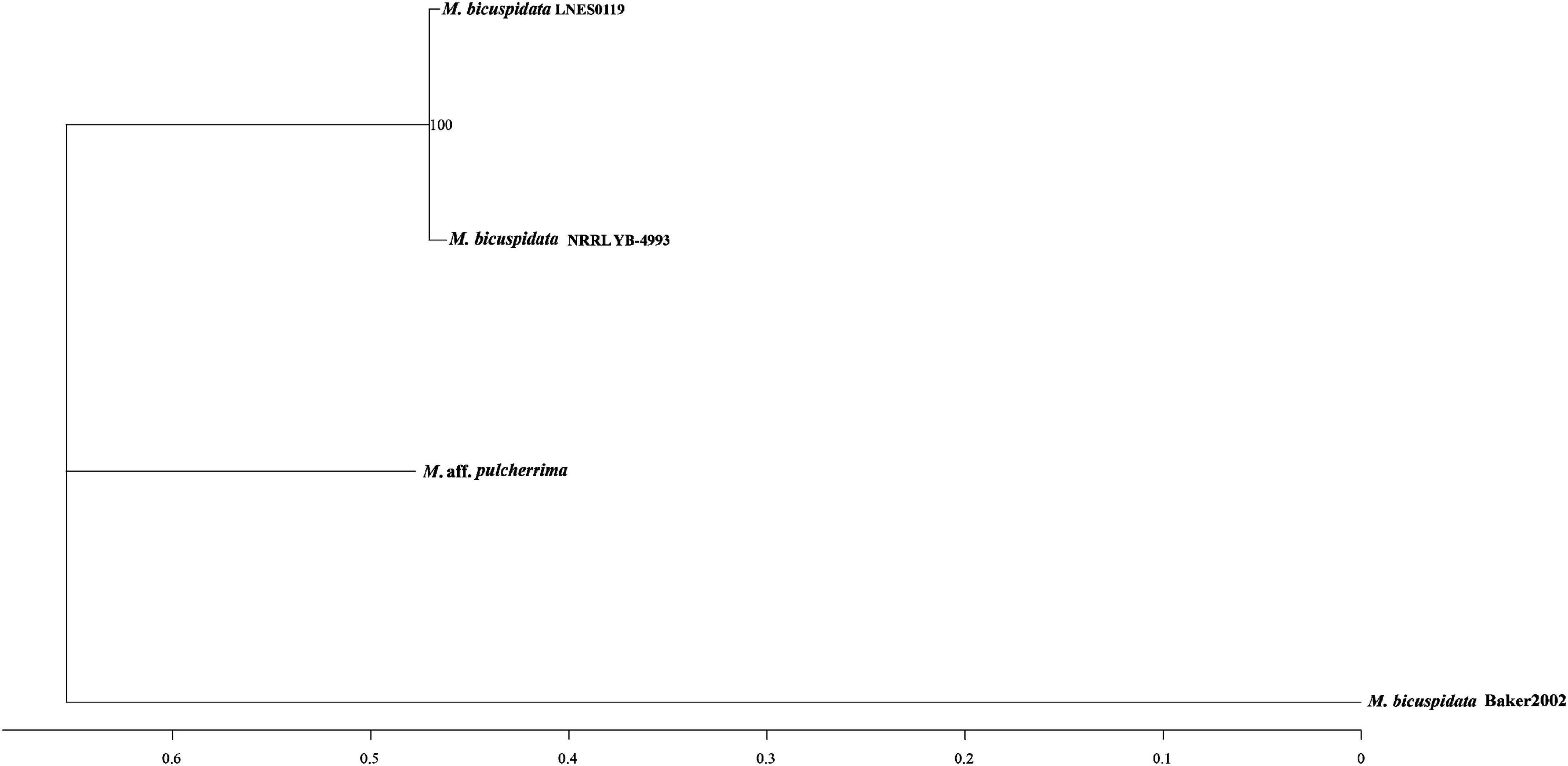
Figure 4. Interspecific phylogenic tree of three M. bicuspidata strains and M. aff. pulcherrima.

Figure 5. Parallel collinearity comparison between three M. bicuspidata strains LNES0119, NRRL YB-4993, and Baker 2002.
Comparative analyses of gene families were conducted among four Metschnikowia strains, including three M. bicuspidata strains and one non-pathogen, M. aff. Pulcherrima APC 1.2. We identified 5,411 putative gene families, of which 2,948 homologous families were shared among four Metschnikowia species, including 2,785 single-copy genes. There were 82 common family genes present in three pathogenic strains of M. bicuspidata that were absent in the non-pathogenic species M. aff. pulcherrima APC 1.2 (Table 4 and Supplementary Table 7). A total of 331 unique genes were found in M. bicuspidata LNES0119, 30 of which were putatively related to pathogenicity, and they were primarily cell wall genes, including eight Hyr/Iff-like genes (Pfam annotation to hyphally regulated N-terminal) as well as one agglutinin-like protein 5 and secreted aspartyl protease (Table 4 and Supplementary Table 8). In addition, only 12 unique gene families were observed in M. bicuspidata LNES0119, of which two gene families (GF3117 and GF3118) represented by six genes belonged to the Pfam domain (PF11765.3, hyphally regulated cell wall protein N-terminal) and were putatively associated with pathogenicity (Table 4 and Supplementary Table 9).

Table 4. Statistics of the gene families of four Metschnikowia strains.
The genome of M. bicuspidata LNES0119 strain was successively sequenced using Illumina and Oxford Nanopore platforms in combination, and the assembly size of the genome was similar to that of M. bicuspidata NRRL YB-4993. The six contigs and 99.3% complete BUSCO estimation demonstrated that the genome was a high-integrity assembly. When compared with that of the three M. bicuspidata strains, the homology of M. bicuspidata LNES0119 and NRRL YB-4993 strains was found to be higher than that of the Baker 2002 strain in a phylogenetic tree analysis, and this result is consistent with the results of the synteny analysis. Ahrendt et al. (2018) considered that the Daphnia parasite M. bicuspidata Baker 2002 was unculturable and not conspecific to the brine shrimp parasite M. bicuspidata NRRL YB-4993. The two strains of M. bicuspidata LNES0119 and NRRL YB-4993 had good coverage with each other, but there were six large segment inversions, which revealed that the two genomes experienced a particular genome structure variation during the process of evolution, which might have led to changes in coding genes and even changes in functional proteins.
CAZymes are responsible for the breakdown, biosynthesis, or modification of glycoconjugates, oligo-, and polysaccharides, and are known to play important roles in host-pathogen interactions (Zhao et al., 2013). In our study, 64 CAZymes of M. bicuspidata LNES0119 annotated to PHI are involved in carbohydrate metabolic processes of protein O-linked glycosylation, the glucan catabolic process, and protein mannosylation. Previous studies have demonstrated that these proteins play vital roles in cell wall assembly and construction of human pathogenic yeasts against environmental stress conditions, act as virulence factors in pathogenicity, or cause a cell-mediated host immune response (Chaffin et al., 1998; Stubbs et al., 1999; Chaffin, 2008; Mora-Montes et al., 2009; Hall and Gow, 2013). This suggests that these CAZymes of M. bicuspidata LNES0119 might be involved in the composition and maintenance of the cell wall during infection. In addition, comparative analysis of the CAZymes genes of 13 species of human/animal and non-pathogenic yeast showed that they all lacked PLs and the number of CAZy genes varied from 117 to 173, which is far less than that found in the genomes of ascomycete fungi (Gazis et al., 2016; Peng et al., 2017). This distinction in the composition and size of CAZymes was closely related to host specificity and the lifestyle adaptations of the fungi. For example, the number of CAZymes in 97 species of saprophytic, pathogenic, and endophytic fungi varied from 215 to 1,938 (Wan, 2019). Compared to the 94 saprophytic, facultative parasitic, hemi-biotrophic, biotrophic, and symbiotic fungi, there were 12 saprophytic or facultative parasitic species such as yeasts and fungi in the genus Trichophyton that lacked PLs (Zhao et al., 2013). By comparing the genomes of pathogenic animal and plant fungi, it was found that specialization to different hosts drives a distinct CAZyme family repertoire (Gaulin et al., 2018). In our study, the lowest number of CAZymes shown in M. bicuspidata Baker 2002 might be closely related to its non-free-living lifestyle. It was found that M. bicuspidata Baker 2002 was unculturable in vitro because it lacked 15 CAZymes that were broadly linked to aspects of urea, sulfate, and thiamine metabolism when compared with that of the NRRL YB-4993 strain (Ahrendt et al., 2018). These results suggest that M. bicuspidata Baker 2002 might have a reduced number of CAZymes as a strategy to adapt to the host environment. In a similar way, the reduced number of 30 CAZymes observed in the endophytic fungus Xylona heveae may be an adaptation to enable intercellular growth in its rubber tree host (Gazis et al., 2016). However, the CE1 and CE10 gene families were unique in M. bicuspidata LNES0119 and the extended role of this gene family is unknown. The enrichment of CE1 and CE10 homologs observed in hemibiotrophic and necrotrophic Oomycetes suggests that they are used either in a species-specific manner during infection or only for pathogens with certain similar characteristics (Gaulin et al., 2018). Taken together, these results might provide insights into the close relationship of CAZymes with the lifestyle and pathogenicity of pathogenic yeasts, which highlights the need for further research.
Fungal lifestyle closely relies on proteins that are secreted extracellularly for growth within their hosts, and these secreted proteins play an important role in mediating interactions with hosts (McCotter et al., 2016). In the M. bicuspidata LNES0119 strain, 41 secretory proteins were found to be related to pathogenicity. Of these, cell wall proteins were the most abundant (Table 3). It is well known that the cell wall of pathogenic fungi maintains the integrity of the cell and interacts with the environment; it also plays a major role in the interaction with the host cells by adhering to their surfaces, invading tissues, and protecting the pathogen from host defense mechanisms. Interestingly, we found that cell wall proteins of hyphally regulated N-terminal domains, eukaryotic aspartyl protease domains, and the Candida agglutinin-like domain were unique to M. bicuspidata LNES0119 (Supplementary Table 8); however, these cell wall proteins were also found to be highly enriched in the strong human pathogens C. albicans, C. tropicalis, and C. parapsilosis (Butler et al., 2009). In these Candida spp., these proteins have been confirmed to be closely associated with pathogenicity and virulence by primarily participating in adhesion to host surfaces and invasion of host cells (Bailey et al., 1996; De Bernardis et al., 2001; Naglik et al., 2003; Butler et al., 2009; de Groot et al., 2013). Our results imply that the roles of unique cell wall proteins in M. bicuspidata LNES0119 are similar to those of human pathogens during infection. Furthermore, eight secreted proteins in M. bicuspidata LNES0119 were shown to be hydrolytic proteins, including lipases, proteases, and phospholipases (Table 3). Previous studies have demonstrated that fungal hydrolytic enzymes (lipases, proteases, and phospholipases) as virulence factors increase the pathogenicity to insects and humans (Hruskova-Heidingsfeldova, 2008; Gaillardin, 2010; Bentubo and Gompertz, 2014; Mondal et al., 2016; Petrisor and Stoian, 2017). For human Candida species, these hydrolytic enzymes can contribute to their invasion of the host tissue through digestion or destruction of the cell membrane, help the pathogen avoid the host defense immune system, and allow the microorganism to utilize host cell macromolecules as a source of nutrients (Schaller et al., 2005; Hruskova-Heidingsfeldova, 2008). For entomopathogenic fungi, hydrolytic enzymes are primarily involved in the initial stages of the adhesion and penetration of insect cuticles (Petrisor and Stoian, 2017). Thus, it was reasonable to infer that these hydrolytic proteins of M. bicuspidata LNES0119 are involved in adaptation and pathogenicity to their host. Finally, other secreted proteins, such as HSP70, multicopper oxidase, and thioredoxin might have taken part in the process of thermal adaptation, nutrient acquisition and stress tolerance, which have also been detected as virulence factors involved in the pathogenicity of fungi (Zhu and Williamson, 2004; Puri and Edgerton, 2013; Chakraborty et al., 2020; Wang et al., 2020). Further research should focus on these proteins because they might play a vital role in the regulation of crab–pathogen interactions.
In the present study, high-quality assembly and complete genome analysis were performed in M. bicuspidata LNES0119. Genomic and comparative analyses revealed that the genome of M. bicuspidata LNES0119 possesses a variety of putative pathogenic genes, which are primarily involved in cell wall assembly and construction and might play a vital role in adapting to the host environment or acting as virulence factors in pathogenicity or causing a cell-mediated host immune response. Therefore, these candidate factors provide a novel resource for further study of the pathogenic mechanisms in M. bicuspidata–associated diseases as well as for the identification of potential targets for further research and therapeutic intervention.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA803590.
The animal study was reviewed and approved by the Animal Experiments Ethics Committee of Shenyang Agricultural University.
HJ and QC conceived and designed the project. JB, HJ, and YX prepared the strain samples and conducted the bioinformatics analysis. HJ, JB, XL, and QC wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the China Agriculture Research System of MOF and MARA (CARS-48), Liaoning Province Department of Education fund item (LSNQN20 2002), Liaoning Province Key R&D Planning Project (2021JH1/10400040), and Shenyang Science and Technology Mission Project (21-116-3-38).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.939141/full#supplementary-material
Ahrendt, S. R., Quandt, C. A., Ciobanu, D., Clum, A., Salamov, A., Andreopoulos, B., et al. (2018). Leveraging single-cell genomics to expand the fungal tree of life. Nat. Microbiol. 3, 1417–1428. doi: 10.1038/s41564-018-0261-0
Bailey, D. A., Feldmann, P. J., Bovey, M., Gow, N. A., and Brown, A. J. (1996). The Candida albicans HYR1 gene, which is activated in response to hyphal development, belongs to a gene family encoding yeast cell wall proteins. J. Bacterial. 178, 5353–5360. doi: 10.1128/jb.178.18.5353-5360.1996
Bao, J., Jiang, H. B., Shen, H. B., Xing, Y. N., Feng, C. C., Li, X. D., et al. (2021). First description of milky disease in the Chinese mitten crab Eriocheir sinensis caused by the yeast Metschnikowia bicuspidata. Aquaculture 532:735984. doi: 10.1016/j.aquaculture.2020.735984
Bentubo, H. D. L., and Gompertz, O. F. (2014). Effects of temperature and incubation time on the in vitro expression of proteases, phospholipases, lipases and DNases by different species of Trichosporon. SpringerPlus 3:377. doi: 10.1186/2193-1801-3-377
Blanco, E., Parra, G., and Guigó, R. (2007). Using geneid to identify genes. Curr. Protoc. Bioinformatics 18, 4.3.1–4.3.28. doi: 10.1002/0471250953.bi0403s18
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Butler, G., Rasmussen, M. D., Lin, M. F., Santos, M. A., Sakthikumar, S., Munro, C. A., et al. (2009). Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662. doi: 10.1038/nature08064
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M., and Buell, C. R. (2006). Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7:327. doi: 10.1186/1471-2164-7-327
Cao, G., Bao, J., Feng, C., Li, X., Lang, Y., Xing, Y., et al. (2022). First report of Metschnikowia bicuspidata infection in Chinese grass shrimp (Palaemonetes sinensis) in China. Transbound. Emerg. Dis. 1–9. doi: 10.1111/tbed.14460 [Epub ahead of print].
Chaffin, W. L. (2008). Candida albicans cell wall proteins. Microbiol. Mol. Biol. Rev. 72, 495–544. doi: 10.1128/MMBR.00032-07
Chaffin, W. L., López-Ribot, J. L., Casanova, M., Gozalbo, D., and Martínez, J. P. (1998). Cell wall and secreted proteins of Candida albicans: identification, function, and expression. Microbiol. Mol. Biol. Rev. 62, 130–180. doi: 10.1128/MMBR.62.1.130-180.1998
Chakraborty, T., Tóth, R., Nosanchuk, J. D., and Gácser, A. (2020). Multicopper oxidases in Saccharomyces cerevisiae and human pathogenic fungi. J. Fungi. 6:56. doi: 10.3390/jof6020056
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4:Unit4.10. doi: 10.1002/0471250953.bi0410s05
Chen, S. C., Chen, T. H., Wang, P. C., Chen, Y. C., and Liaw, L. L. (2003). Metschnikowia bicuspidate and Enterococcus faecium coinfection in the giant freshwater prawn Macrobrachium rosenbergii. Dis. Aquat. Org. 55, 161–167. doi: 10.3354/dao055161
Chen, S. C., Chen, Y. C., Kwang, J., Manopo, I., Wang, P. C., Chaung, H. C., et al. (2007). Metschnikowia bicuspidata dominates in Taiwanese cold-weather yeast infections of Macrobrachium rosenbergii. Dis. Aquat. Org. 75, 191–199. doi: 10.3354/dao075191
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
De Bernardis, F., Sullivan, P. A., and Cassone, A. (2001). Aspartyl proteinases of Candida albicans and their role in pathogenicity. Med. Mycol. 39, 303–313. doi: 10.1080/mmy.39.4.303.313
de Groot, P. W., Bader, O., de Boer, A. D., Weig, M., and Chauhan, N. (2013). Adhesins in human fungal pathogens: glue with plenty of stick. Eukaryot. Cell 12, 470–481. doi: 10.1128/EC.00364-12
Duffy, M. A., Brassil, C. E., Hall, S. R., Tessier, A. J., Cáceres, C. E., and Conner, J. K. (2008). Parasite-mediated disruptive selection in a natural Daphnia population. BMC Evol. Biol. 8:80. doi: 10.1186/1471-2148-8-80
Eddy, S. R. (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. doi: 10.1093/bioinformatics/14.9.755
Edgar, R. C., and Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158. doi: 10.1093/bioinformatics/bti1003
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., and Mitchell, A. L. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Gaillardin, C. (2010). “Lipases as pathogenicity factors of fungi,” in Handbook of Hydrocarbon and Lipid Microbiology, ed. K. N. Timmis (Berlin: Springer). doi: 10.1007/978-3-540-77587-4_247
Gaulin, E., Pel, M. J. C., Camborde, L., San-Clemente, H., Courbier, S., Dupouy, M. A., et al. (2018). Genomics analysis of Aphanomyces spp. identifies a new class of oomycete effector associated with host adaptation. BMC Biol. 16:43. doi: 10.1186/s12915-018-0508-5
Gazis, R., Kuo, A., Riley, R., Butti, K., Lipzen, A., Lin, J. Y., et al. (2016). The genome of Xylona heveae provides a window into fungal endophytism. Fungal Biol. 120, 26–42. doi: 10.1016/j.funbio.2015.10.002
Hall, R. A., and Gow, N. A. R. (2013). Mannosylation in Candida albicans: role in cell wall function and immune recognition. Mol. Microbiol. 90, 1147–1161. doi: 10.1111/mmi.12426
Han, Y., and Wessler, S. R. (2010). MITE-hunter: a program for discovering miniature invertedrepeat transposable elements from genomic sequences. Nucleic Acids Res. 38:e199. doi: 10.1093/nar/gkq862
Hruskova-Heidingsfeldova, O. (2008). Secreted proteins of Candida albicans. Front. Biosci. 13:7227–7242. doi: 10.2741/3224
Jiang, H., Bao, J., Cao, G., Xing, Y., Feng, C., Hu, Q., et al. (2022). Experimental transmission of the yeast, Metschnikowia bicuspidata, in the Chinese Mitten Crab, Eriocheir sinensis. J. Fungi 8:210. doi: 10.3390/jof8020210
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Jan, G., and Frank, H. (2016). Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44:e89. doi: 10.1093/nar/gkw092
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
McCotter, S. W., Horianopoulos, L. C., and Kronstad, J. W. (2016). Regulation of the fungal secretome. Curr. Genet. 62, 533–545. doi: 10.1007/s00294-016-0578-2
Merrill, T., and Cáceres, C. E. (2018). Within-host complexity of a plankton-parasite interaction. Ecology 99, 2864–2867. doi: 10.1002/ecy.2483
Metchnikoff, E. (1884). Ueber eine sprosspilzkrankheit der daphnien. Beitrag Zur Lehre Über Den Kampf Der Phagozyten Gegen Krankheitserreger. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 96, 178–195. doi: 10.1055/s-0029-1209659
Miller, M. W., and Phaff, H. J. (1998). “Metschnikowia kamienski,” in The Yeasts, a Taxonomic Study, 4th Edn. eds C. P. Kurtzman and J. W. Fell (Amsterdam: Elsevier), 256–267.
Mondal, S., Baksi, S., Koris, A., and Vatai, G. (2016). Journey of enzymes in entomopathogenic fungi. Pac. Sci. Rev. A Nat. Sci. Eng. 18, 85–99. doi: 10.1016/j.psra.2016.10.001
Moore, M. M., and Strom, M. S. (2003). Infection and mortality by the yeast Metschnikowia bicuspidata var. bicuspidata in Chinook salmon fed live adult brine shrimp (Artemia franciscana). Aquaculture 220, 43–57. doi: 10.1016/S0044-8486(02)00271-5
Mora-Montes, H. M., Ponce-Noyola, P., Villagómez-Castro, J. C., Gow, N., and López-Romero, A. E. (2009). Protein glycosylation in Candida. Future Microbiol. 4, 1167–1183. doi: 10.2217/fmb.09.88
Naglik, J. R., Challacombe, S. J., and Hube, B. (2003). Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol. Mol. Biol. Rev. 67, 400–428. doi: 10.1128/MMBR.67.3.400-428.2003
Nawrocki, E. P., Burge, S. W., Bateman, A., Daub, J., Eberhardt, R. Y., Eddy, S. R., et al. (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137. doi: 10.1093/nar/gku1063
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Peng, M., Dilokpimol, A., Mäkelä, M. R., Hildén, K., Bervoets, S., Riley, R., et al. (2017). The draft genome sequence of the ascomycete fungus Penicillium subrubescens reveals a highly enriched content of plant biomass related Cazymes compared to related fungi. J. Biotechnol. 246, 1–3. doi: 10.1016/j.jbiotec.2017.02.012
Pertea, M., Kim, D., Pertea, G. M., Leek, J. K., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Petersen, T. N., Brunak, S., Von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Petrisor, C., and Stoian, G. (2017). The role of hydrolytic enzymes produced by entomopathogenic fungi in pathogenesis of insects mini review. Rom. J. Plant Prot. 10, 66–72.
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Puri, S., and Edgerton, M. (2013). “Candida albicans Ssa: an Hsp70 homologue and virulence factor,” in Moonlighting Cell Stress Proteins in Microbial Infections. Heat Shock Proteins, Vol. 7, ed. B. Henderson (Dordrecht: Springer). doi: 10.1007/978-94-007-6787-4_14
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. doi: 10.1038/s41592-019-0669-3
Saier, M. H., Tran, C. V., and Barabote, R. D. (2006). TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 34, D181–D186. doi: 10.1093/nar/gkj001
Schaller, M., Borelli, C., Korting, H. C., and Hube, B. (2005). Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 48, 365–377. doi: 10.1111/j.1439-0507.2005.01165.x
Shi, H., Xu, W. J., Xu, H. X., and Shi, H. D. (2008). The18S rRNA sequencing and analysis of a pathogenic yeast, a causative agent of emulsification disease in Portunus trituberculatus. Mar. Fish. Res. 4, 34–38.
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. Z. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Sperschneider, J., Gardiner, D. M., Dodds, P. N., Tini, F., Covarelli, L., Singh, K. B., et al. (2016). EffectorP: predicting fungal effector proteins from secretomes using machine learning. New Phytol. 210, 743–761. doi: 10.1111/nph.13794
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, 215–225. doi: 10.1093/bioinformatics/btg1080
Stubbs, H. J., Brasch, D. J., Emerson, G. W., and Sullivan, P. A. (1999). Hydrolase and transferase activities of the β−1, 3-exoglucanase of Candida albicans. Eur. J Biochem. 263, 889–895. doi: 10.1046/j.1432-1327.1999.00581.x
Sun, N., Bao, J., Liang, F., Liu, F., Jiang, H., and Li, X. (2022). Prevalence of ‘milky disease’ caused by Metschnikowia bicuspidata in Eriocheir sinensis in Panjin city, China. Aquac. Res. 53, 1136–1140. doi: 10.1111/are.15625
Tang, S., Lomsadze, A., and Borodovsky, M. (2015). Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43:e78. doi: 10.1093/nar/gkv227
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wan, R. P. (2019). Comparative Genomic Analysis of CAZymes in Plant Saprophytic, Pathogenic and Endophytic Fungi. Master’s thesis. Nanchang: Jiangxi Normal University.
Wang, J., Chen, J., Hu, Y., Ying, S. H., and Feng, M. G. (2020). Roles of six Hsp70 genes in virulence, cell wall integrity, antioxidant activity and multiple stress tolerance of Beauveria bassiana. Fungal Genet. Biol. 144:103437. doi: 10.1016/j.fgb.2020.103437
Wang, X., Chi, Z., Yue, L., Li, J., Li, M., and Wu, L. (2007). A marine killer yeast against the pathogenic yeast strain in crab (Portunus trituberculatus) and an optimization of the toxin production. Microbiol. Res. 162, 77–85. doi: 10.1016/j.micres.2006.09.002
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Winnenburg, R., Baldwin, T. K., Urban, M., Rawlings, C., Köhler, J., and Hammond-Kosack, K. E. (2006). PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 34, D459–D464. doi: 10.1093/nar/gkj047
Xu, Z., and Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yin, Y., Mao, X., Yang, J., Chen, X., Mao, F., and Xu, Y. (2012). dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451. doi: 10.1093/nar/gks479
Zhao, Z. T., Liu, H. Q., Wang, C. F., and Xu, J. R. (2013). Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 14:274. doi: 10.1186/1471-2164-14-274
Keywords: Metschnikowia bicuspidata, genome annotation, comparative genomics, pathogenicity, Eriocheir sinensis
Citation: Jiang H, Bao J, Xing Y, Li X and Chen Q (2022) Comparative Genomic Analyses Provide Insight Into the Pathogenicity of Metschnikowia bicuspidata LNES0119. Front. Microbiol. 13:939141. doi: 10.3389/fmicb.2022.939141
Received: 08 May 2022; Accepted: 24 May 2022;
Published: 13 June 2022.
Edited by:
Donglei Sun, Shanghai Jiao Tong University, ChinaReviewed by:
Yanli Chen, University of Maryland, College Park, United StatesCopyright © 2022 Jiang, Bao, Xing, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qijun Chen, cWlqdW5jaGVuNzU5QHN5YXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.