 Xiaoge Hou
Xiaoge Hou Ming Hui1
Ming Hui1 Xin Shi
Xin Shi Ruifang Li
Ruifang Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 July 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.934696
This article is part of the Research Topic Innovative Microbial Technologies for Future and Sustainable Food Science View all 12 articles
Medium-temperature Daqu (MT-Daqu), a saccharification-fermentation agent and aroma-producing agent, is used to produce Chinese strong-flavor Baijiu. Many related studies have been published; however, less is known about microbial community and quality properties inside and outside the MT-Daqu from fermentation to storage. Here, along with determining the physicochemical index, the microbial community of MT-Daqu was investigated using both culture-dependent and culture-independent methods during 31 days of fermentation and 4 months of storage. Volatile compounds of mature MT-Daqu were analyzed using headspace solid-phase microextraction (HS-SPME) combined with gas chromatography-mass spectrometry (GC–MS). The results indicated obvious variation in the microbial community due to the changes in environmental conditions, and the physicochemical indices shifted from fluctuations in the fermentation period to relative stability after storage for 3 months. Moreover, the microbial counts and physicochemical indices of the inner layers of MT-Daqu differed from those of the outer layers. The dominant communities, including the bacterial phyla Firmicutes, Proteobacteria, and Actinobacteria and the fungal phyla Ascomycota and Mucoromycota, showed different abundances in the two parts of the mature MT-Daqu, and different microbial communities were enriched in both parts. Additionally, pyrazines and alcohols were the most abundant volatile aroma compounds in the mature MT-Daqu.
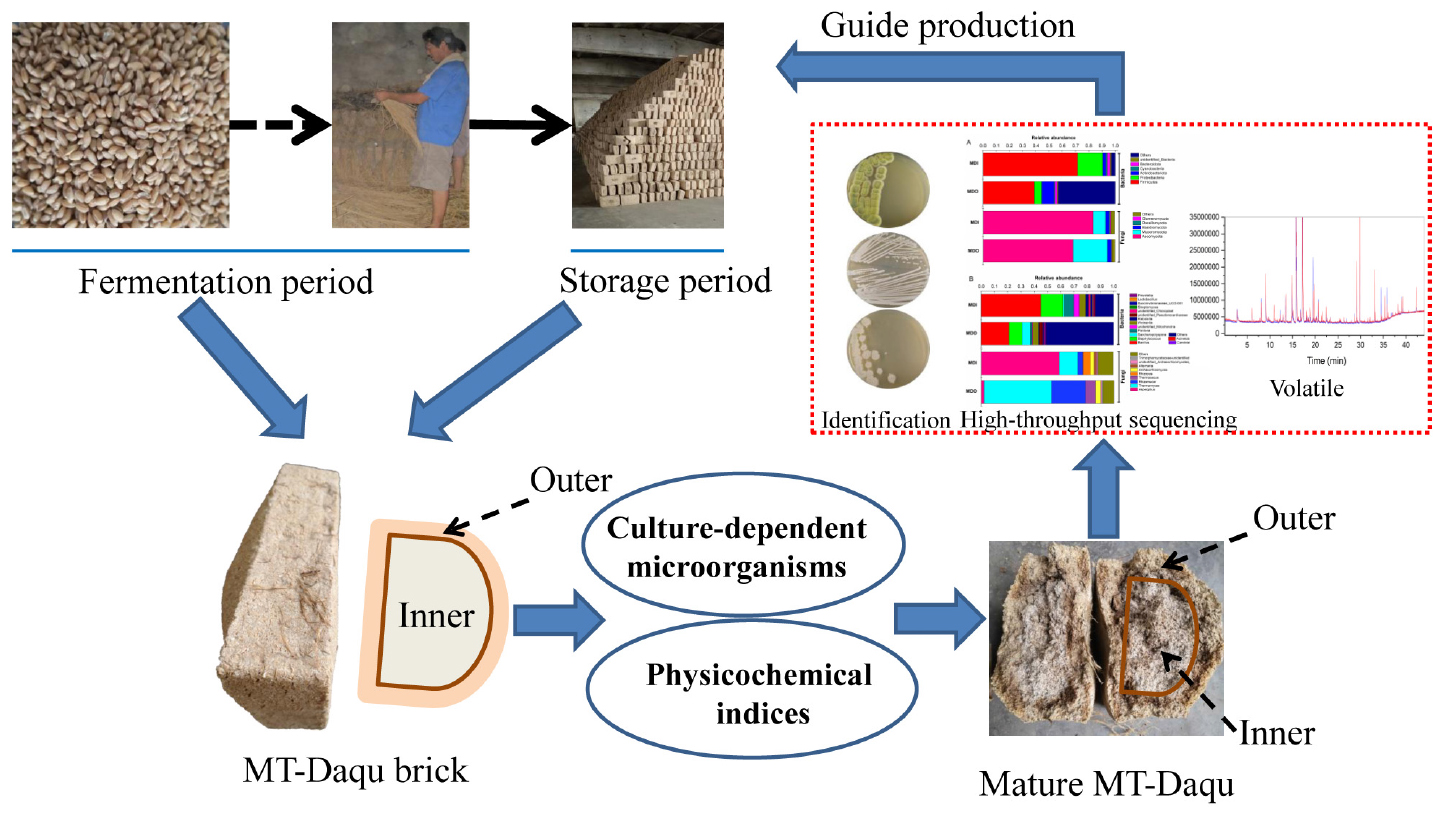
GRAPHICAL ABSTRACT.
Baijiu is one of the six well-known distillates worldwide (Jin et al., 2017; He et al., 2019), and Daqu was used as a saccharification and fermentation agent and aroma-producing agent for Baijiu production (Wang et al., 2017). Thus, Daqu constituents exert a direct effect on the final distilled Baijiu. Due to the abundant accumulation of microorganisms, enzymes and flavor compounds are formed during an open spontaneous solid-state fermentation process (Cai et al., 2021; Deng et al., 2021). According to the highest fermenting temperature, Daqu is mainly classified into three types: high-temperature Daqu (60–70°C), medium-temperature Daqu (50–60°C), and low-temperature Daqu (40–50°C) (Deng et al., 2020; Cai et al., 2021; Hu et al., 2021; Wang et al., 2021). These three types of Daqu have been employed to produce Jiang-flavor, strong-flavor, and light-flavor Baijiu, which are the three most popular Baijiu products on the Chinese market (Hu et al., 2021; Zhang H. et al., 2021).
Strong-flavor Baijiu, with ethyl caproate as the main aroma, has the characteristics of a fragrant flavor, soft mouthfeel, and long-lasting aftertaste and is the most famous type of Baijiu, with an annual output of more than 70% of all Chinese liquor (Xu et al., 2017; Zou et al., 2018; Deng et al., 2021). The generation of the flavor of strong-flavor Baijiu is closely linked to MT-Daqu. As a saccharifying starter for 1000 of years and as raw material, MT-Daqu is combined with approximately 20% raw grains for strong-flavor Baijiu fermentation (Xu et al., 2017; Zou et al., 2018). MT-Daqu is produced by solid fermentation using a wheat or wheat mixture as raw materials, and the process involves three phases: material mixing and shaping, spontaneous solid-state fermenting, and maturing (Supplementary Figure 1; Zou et al., 2018). During MT-Daqu production, the fermentation phase is crucial and proceeds through five stages, namely, the Anqu, Peijun, Dinghuo, Huanluo, and Dalong phases (Yan et al., 2019). As a result, numerous microorganisms, such as Bacillus, Acetobacter, Pseudomonas, Weissella, Alternaria, Aspergillus, Mucor, Saccharomyces, and Hansenula, inhabited the fermentation broth, and enzymes, such as liquefaction enzymes, saccharifying enzymes, proteases, and esterifying enzymes, accumulated through microbial metabolism. Flavor compounds, such as organic acids, alcohols, esters, aldehydes, and ketones, contributing to the generation of flavor of Baijiu are also enriched in this period (Yan et al., 2013a,2019; Liu et al., 2018; Deng et al., 2021). Additionally, fresh Daqu must mature for 3–6 months to exclude undesirable microorganisms and balance metabolic compounds (Fan et al., 2019; Yang et al., 2021). Only mature Daqu can be used for Baijiu brewing. Therefore, understanding the rule of microbial succession and metabolism in the MT-Daqu production process might contribute to the stability and quality of Daqu.
However, the microbial composition and physicochemical indices in Daqu undergo continuous succession during production due to environmental factors (Fu et al., 2021). Meanwhile, different microbiotas and physicochemical properties have been observed in different parts of the same Daqu brick due to the difficulty in transferring heat, water, and oxygen (Jin et al., 2019; Chen et al., 2020; Zhang Y. et al., 2021). The aforementioned results all affect the quality of Daqu. At present, the criteria that are accepted by Baijiu industry have clearly specified the organoleptic requirement and predominant physicochemical index requirement for a fermentation MT-Daqu that is mature (People’s Republic of China Light Industry Professional Standard., 2011b), QB/T 4259-2011, 2011; (Shangdong Province Local Standard, 2009, in China, DB37/T 1231-2009, 2009). The microorganism and the physicochemical property indices inside and outside the strong flavor Daqu are not included in the industrial criteria. Therefore, analyzing the microbiotas and physicochemical indices inside and outside the Daqu during production might provide a scientific theory for establishing a relatively comprehensive method to evaluate the quality and control the process of Daqu. Some studies on these have been performed (Jin et al., 2019; Chen et al., 2020; Zhang Y. et al., 2021). Jin et al. (2019) revealed that the dominant microbes and volatile flavors on the surface and the core of soy-flavor Daqu had different distribution characteristics and found that microbial interactions were stronger in the core than on the surface (Jin et al., 2019). Thereafter, Zhang Y. et al. (2021) explored the microbiota, enzymes, and metabolites in different parts of fresh and mature soy-flavor Daqu, and the dominant enzymes varied significantly and identified the predominant microbial community migrated between the two parts of Daqu during production (Zhang Y. et al., 2021). In addition to Jiang-flavor Daqu, studies on differences in microbiota and physicochemical indices between surface and central parts of special-flavor Baijiu Daqu were reported, and the microbial distribution and physicochemical indices in the two parts were significantly different (Chen et al., 2020). These findings potentially provide a theoretical basis for microbial resources and quality control of Daqu (Wang et al., 2021). However, Chinese flavor Baijiu brewing enterprises are widely distributed over South China and North China and have their own unique ecological environments and diverse regulations for the production of Daqu. Therefore, a comprehensive understanding of the mechanisms underlying the composition of Daqu might contribute to better control of the Daqu process and standardize local Baijiu production (Xu et al., 2017; Liu et al., 2018). Currently, studies that examine different parts or different types of Daqu mainly focus on high-temperature or low-temperature Daqu (Chen et al., 2020; Cai et al., 2021; Wang et al., 2021; Zhang Y. et al., 2021), and a comprehensive study of the microbial compositions and the quality characteristics of different parts of MT-Daqu have not yet been conducted.
In this study, the bacterial and fungal communities in different parts of strong-flavor Daqu from the fermentation period to the mature period were investigated using a culture-dependent method, and the changes in the physicochemical properties throughout the production process were also explored. Based on these results, the maturity period of MT-Daqu was evaluated, and the analysis of microbial composition, physicochemical indices, and volatile compounds of different parts of mature MT-Daqu using the Illumina MiSeq platform and headspace solid-phase microextraction (HS-SPME)-GC–MS was the main focus. This study provides guidance for the determination of the mature stage, for the process control of MT-Daqu, and for improving the quality of strong-flavor Baijiu.
Medium-temperature-Daqu samples were obtained from Songhe Co., Ltd., in Henan, China (longitude 115.48°E and latitude 33.86°N), a typical distillery that produces strong-flavor Baijiu in Henan Province. MT-Daqu was produced by traditional solid-state fermentation in summer (Supplementary Figure 1). The same batch of MT-Daqu bricks was removed from the fermentation room on days 1, 5, 9, 13, 17, 21, 26, and 31 and from the storage room at 0.5, 1, 1.5, 2, 3, and 4 months. The Daqu samples were collected as described in the previous reports (Yan et al., 2019). A total of three Daqu bricks were randomly selected from the top, middle, and bottom of stacked layers. Subsequently, each Daqu brick was divided into two parts based on the fire-circle strategy (Wang et al., 2020): the 0.5–2-cm thick outer layer (named MDO) and the inner layer of the remaining part (named MDI). Then, the same parts (equal weight) of each Daqu brick were mixed into one sample and separated into two parts for subsequent testing. Samples were collected during two parallel batches of MT-Daqu production, and four biological replicates of each sample were obtained. Approximately 50 g of the sample was immediately brought to the laboratory to analyze the microbial community, 100 g of sample was stored at –20°C for the analysis of physicochemical indices and volatile compounds, and 50 g of sample was sealed in sterile bags and stored at –80°C for Illumina MiSeq sequencing.
During the MT-Daqu fermentation period, the temperature and humidity in the fermentation room and the interior of Daqu bricks were measured using thermometers and hygrometers, respectively (Li et al., 2016). The physicochemical indices were determined according to the industrial general standard methods with a slight modification (People’s Republic of China Light Industry Professional Standard., 2011a), QB/T 4257-2011, 2011; Shangdong Province Local Standard in China, DB37/T (1231-2009). Moisture (g/g,%) was measured as the weight loss of the samplers after drying at 105°C for 3–5 h until constant weight. The total acidity (mmol/10 g) was determined by titrating with 0.1 M NaOH to pH 8.2. Liquefying activity (g/g● h) was determined by the reaction time (minutes) with a mixture of iodine solution and starch solution. Saccharifying activity (mg/g●h) was measured using Fehling’s solution titration method (Xiao et al., 2017). The esterifying activity (mg/g●100 h) was determined using the saponification method and reported as the amount of ethyl caproate synthesized from caproic acid and ethanol per 100 h by 1 g of dried Daqu. Fermenting activity (gCO2/g●72 h) was determined by the weight loss of sorghum saccharifying solution incubated with 0.5 g of Daqu, and it was reported as the amount of CO2 released in 72 h by 1 g dried Daqu.
Microbial enumeration using culture-dependent methods has described in the previous study (Hu et al., 2017).
About 10 g of Daqu sample was mixed with 90 ml of sterile saline (0.85% w/v sodium chloride) in a-250 ml sterile conical flask and shaken at 25°C for 30 min at 120 rpm to obtain homogenized samples. After allowing the mixture to stand for a few minutes, the supernatant was diluted 10-folds. Next, 0.2 ml of the diluted suspension was spread in triplicate on the surface of plate count agar medium (Oxoid CM0325) and cultured at 37°C for 24 h to enumerate total aerobic bacteria and spread on potato dextrose agar medium (Oxoid CM0139) and Czapek Dox medium containing 0.1 g/L ampicillin and cultured at 30°C for 3 days to enumerate yeasts and molds (Wang et al., 2018). For the estimation of spore-forming bacteria, a 10% (w/v) sample suspension was heated at 80°C for 10 min and then spread on plate count agar medium, followed by incubation at 55°C for 72 h (Zheng et al., 2012). Colony-forming units (CFUs) per gram of fresh weight sample were calculated based on their morphological features.
Considering the application of the mature MT-Daqu for Baijiu production, the strains from the mature MT-Daqu were isolated and identified. A total of five to ten colonies with the square root of the total number of colonies were picked randomly from each plate for purification using the successive streak plate method (Yang et al., 2022). Genomic DNA was used as a template for PCR to amplify the bacterial 16S rDNA using the universal primer pairs 27F and 1492R, the mold 18S rDNA using the primers NS1(5′-GTAGTCATATGCTTGTCTC-3′) NS6 (5′-GCATCACAGACCTGTTATTGCCTC-3′), and the yeast internal transcribed spacer (ITS) regions using the universal primers ITS1/ITS4 (Zhang H. et al., 2021). Then, purified PCR products were sequenced by Sangon Biotech Co., Ltd. (Shanghai, China), and the sequences were submitted to GenBank for comparison with those in the NCBI GenBank database using the BLAST search tool for the identification of the strains.
Mature MT-Daqu samples were removed for Illumina sequencing. Metagenomic DNA was extracted using the CTAB/SDS method, and DNA concentration and purity were monitored on 1% agarose gels. According to the concentration, DNA was diluted to 1 ng/μl using sterile water (Xiao et al., 2017). Variable regions V4 of the bacterial 16S rRNA gene were amplified with degenerate PCR primers: 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). ITS1 of the ITS region was amplified with degenerate PCR primers: ITS1F (CTTGGTCATTTAGAGGAAGTAA) and ITS2R (GCTGCGTTCTTCATCGATGC). The PCR products were purified with a Qiagen Gel Extraction Kit (Qiagen, Germany), sent to Novogene Co., Ltd. (Beijing, China), and sequenced using the Illumina NovaSeq platform. The raw 16S rRNA and ITS gene sequencing reads were merged using FLASH version 1.2.7, quality-filtered using the QIIME (v1.9.1) quality control process, and the effective tags were finally obtained by removing chimera sequences using the UCHIME algorithm (UCHIME Algorithm). The sequence analysis was performed using UPARSE v7.0.1001, and sequences with ≥97% similarity were assigned to the same OTUs. Rarefaction curves were obtained by comparing the richness and evenness of OTUs among samples. A representative sequence for each OTU was screened for annotation using the Silva Database (Release 138) for bacteria and the Unite Database (Release 8.0) for fungi. The species diversity, including alpha diversity and beta diversity, was analyzed using QIIME (1.9.1) software and displayed with R software (version 2.15.3). The original sequence was deposited in the NCBI Sequence Read Archive (SRA, PRJNA815814).
Volatile compounds in mature MT-Daqu were analyzed using HS-SPME-GC–MS. Using a 50/30 μm DVB/CAR/PDMS fiber (Supelco, Bellefonte, PA, United States) preconditioned in the GC injection port at 250°C for 30 min, volatile extraction was performed using a published method with a minor modification (He et al., 2019). Briefly, 2.0 g of Daqu powder and 5 μl internal standard (2-octanol, 0.0722 g/L, Sigma-Aldrich, St. Louis, MO, United States) were placed into a 20-ml headspace vial containing 5 ml of saturated sodium chloride solution. Then, the vial was placed in a thermostatic block stirrer to equilibrate for 10 min at 50°C and subsequently extracted for 45 min with stirring at 500 r/min. After extraction, the SPME fiber was withdrawn into the needle and immediately inserted into the injection port of the Thermo Fisher Scientific GC–MS system coupled with a trace TR-WAX fused silica capillary column (30 m × 0.25 mm i.d., 0.25-μm film thickness, Thermo Fisher Scientific, United States). Ultrahigh-purity helium was used as the carrier gas at a constant flow velocity of 1 ml/min, and the temperature of the injector and detector was maintained at 250°C. Split injection (10:1) was used. The oven temperature was programmed as follows: 35°C for 2 min, 5°C/min ramp to 110°C for 5 min, and 7°C/min ramp to 230°C for 7 min. The temperatures of the interface and ion source were 250 and 240°C, respectively. The electron impact (EI) ionization energy was 70 eV. EI spectra were recorded in the 30–450 amu range in full scan acquisition mode. Volatile compounds were identified by comparison with the MS spectral database library of the National Institute for Standards and Technology (NIST 2.3, 2017) and were required to fit logically with respect to the retention time in the chromatograms. Semiquantitative determination of the compounds was conducted using a reported method (Tang H. L. et al., 2019).
The biomass of every group of microbes was determined by counting viable colonies on agar in plates with appropriately diluted samples and calculated with the formula LogCFU/g using EXCEL2016. The statistical significance of differences between the samples was determined using SPSS Statistics 16.0 software with one-way ANOVA followed by Duncan’s test, and significant differences were defined as p < 0.05 or p < 0.01. The linear relationship between predominant physiochemical indices was analyzed by calculating the Pearson correlation coefficient using SPSS 16.0 software. Principal coordinates analysis (PCoA) was applied to explore the possible differences between the samples based on Bray–Curtis distances via ANOSIM using R (version 3.3.1). The linear discriminant analysis (LDA) effect size (LEfSe) method was used to identify significant variations among different parts of MT-Daqu samples (LDA > 3.5, p < 0.05).
The core temperature of MT-Daqu reached 60°C after 7 days, maintained 60°C for 7 days, and then decreased gradually until 24 days (Supplementary Figure 2). The relative humidity of fermentation room was close to saturation (100%) after 5 days. As shown in Table 1, the moisture of MDO significantly decreased to 14.18% in the first 9 days (p < 0.05), while that of MDI began to decrease from 34.20%, and the moisture in MDI was higher than that in MDO from 5 days to 75 days; then, the moisture content of both sections reached a plateau (<13%) after storage for 2 months. Similarly, the acidity of MDI was higher than that of MDO before 3 months, whereas the acidity of MDO changed slightly, but that of MDI increased sharply in the first 5 days (p < 0.05), peaked (2.10 mmol/10 g) on the 5th day, and then gradually decreased to a relatively stable value (1.0 mmol/10 g). In contrast, the liquefying activity and saccharifying activity of MDO were both higher than those of MDI after fermentation for 5 days (p < 0.05). The saccharifying activity in MDI decreased substantially from 722.50 mg/g●h to 22.50 mg/g●h in the early stage of fermentation, then increased slightly, and finally exhibited a relatively stable range from 222 to 450 mg/g●h. The saccharifying activity and liquefying activity in MDO increased to relatively stable ranges of 852 to 925 mg/g●h and 3.10 to 3.92 g/g●h after 5 days, respectively. The esterifying activity in the two parts of MT-Daqu exhibited a relatively high value during storage and tended to be consistent after 75 days (> 12.0 mg/g●100 h). The fermenting activity in both MDI and MDO underwent similar changes: increasing within 1–5 days, decreasing gradually within 5–31 days, and then plateauing at approximately 1.50 gCO2/g●72 h until the end of storage.
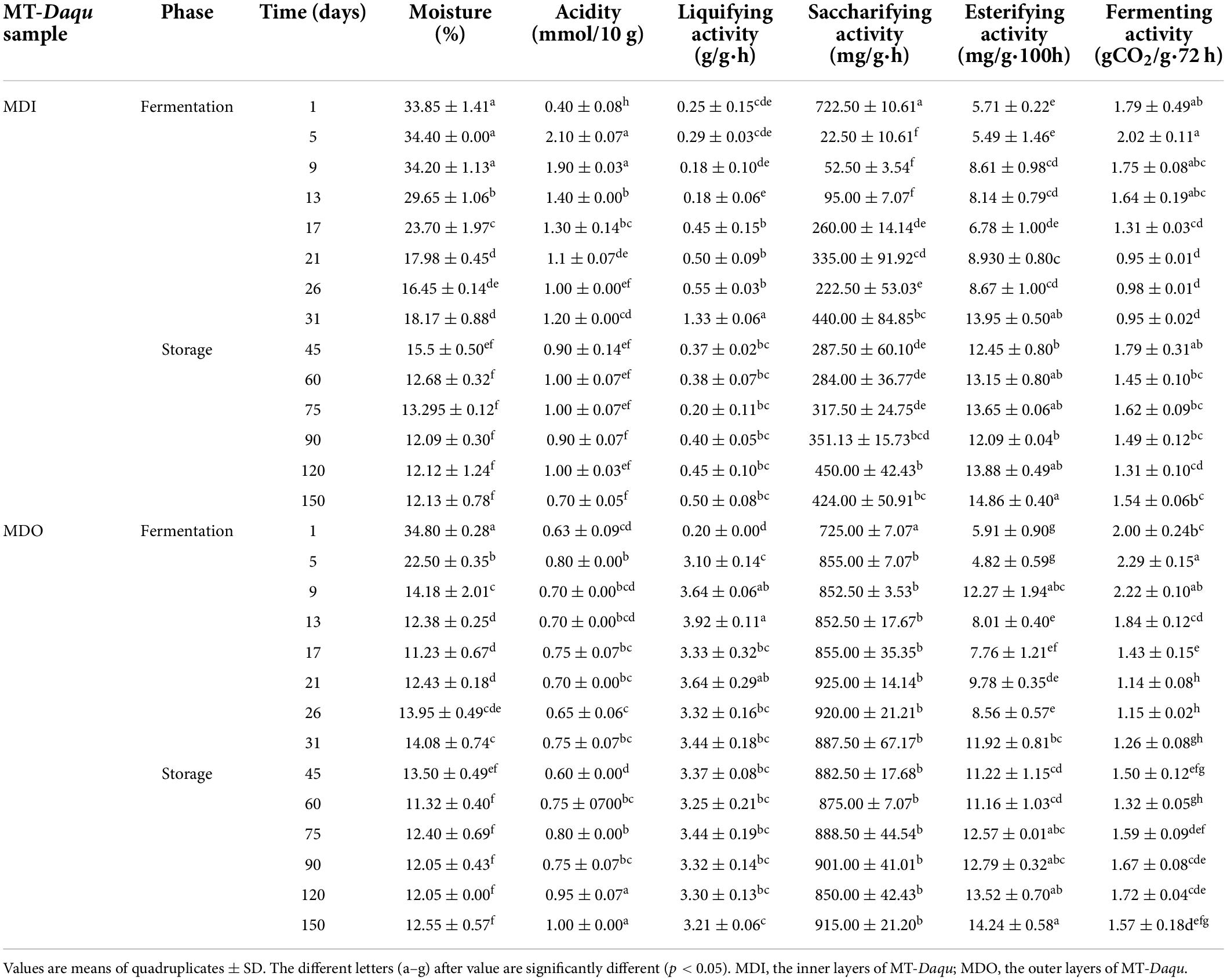
Table 1. Differences in physicochemical indices of samples during MT-Daqu production.
The aerobic bacteria (Figure 1A, the upper y-coordinate) and yeast counts (Figure 1B, the upper y-coordinate) in MDI showed a similar pattern in the 31-day fermentation: reaching the minimum value on the 17th day. The aerobic bacteria in MDO (Figure 1A, the bottom of the y-axis) decreased rapidly in the first 9 days, then increased slowly to reach the maximum at 45 days, and subsequently decreased and stabilized at 90 days. The total yeast in MDO (Figure 1B, the bottom y-coordinate) decreased rapidly to reach the minimum value on the 17th day, then was restored to its original counts on the 45th day, and continued to decrease during storage. The total spore-forming bacteria counts in both parts of Daqu showed no obvious difference and similarly increased during the fermentation period and remained stable in the storage period (Figure 1A). In the fermentation period, the changes in mold in MDI (Figure 1B, the upper y-coordinate) were similar to those in spore-forming bacteria in MDI, whereas the mold in MDO increased rapidly in the first 5 days and remained stable thereafter (Figure 1B, the bottom of the y-axis).
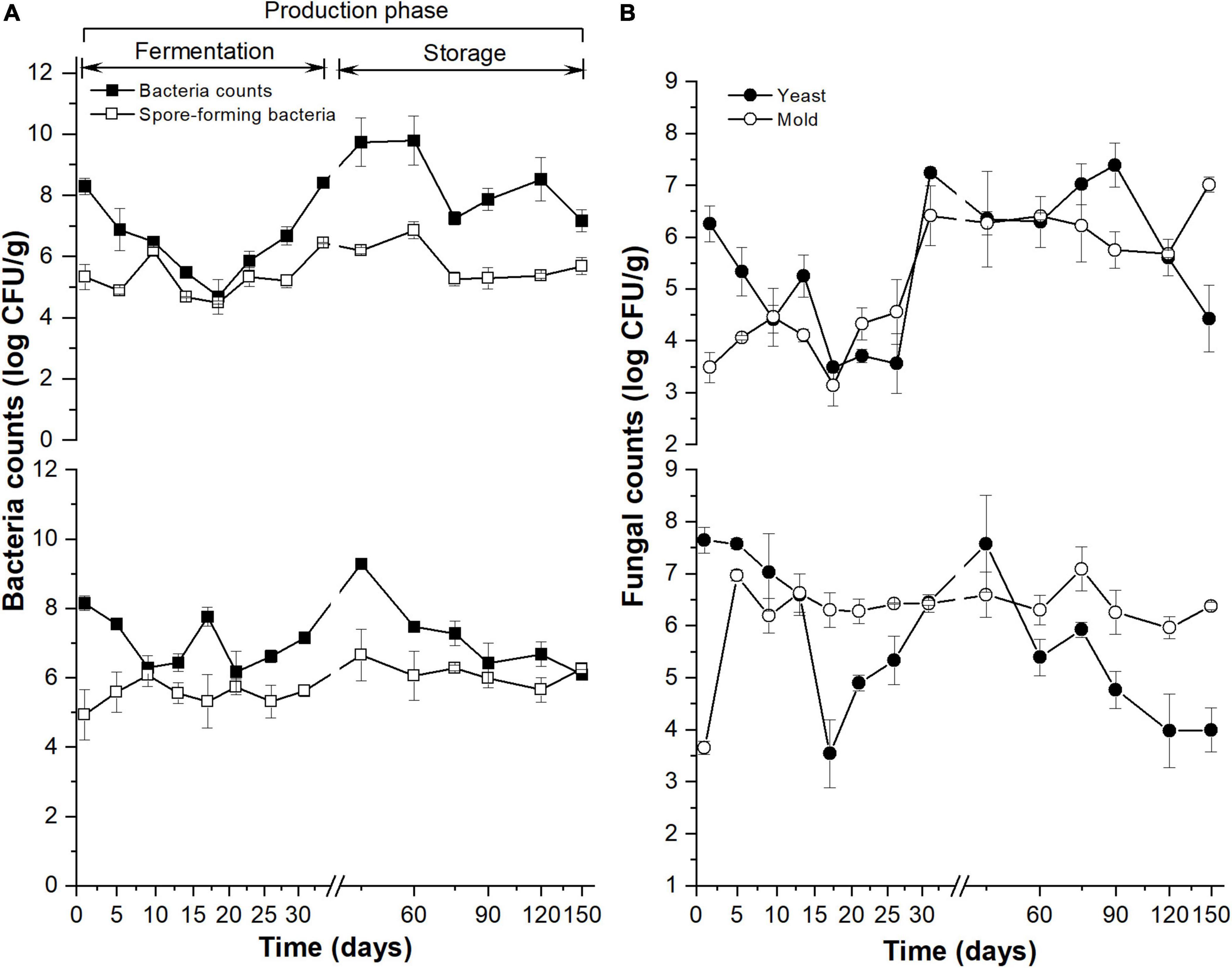
Figure 1. Dynamics of culture-dependent bacteria (A) and fungi (B) in MT-Daqu during entire production, including 31-day fermentation period and 4 months storage. The upper and lower y-axis indicate the microbial counts in MDI (the inner layers of MT-Daqu) and MDO (the outer layers of MT-Daqu), respectively.
As shown in Figure 2, during storage, the microbial counts, except those of molds, in the MDI were significantly higher than those in the MDO (p < 0.05) in the first 2 months, whereas counts of the four groups of microbial in the MDI were close to those of MDO after 3 months of storage. Furthermore, all microbial counts in the MDO remained relatively constant after 1 month of storage.
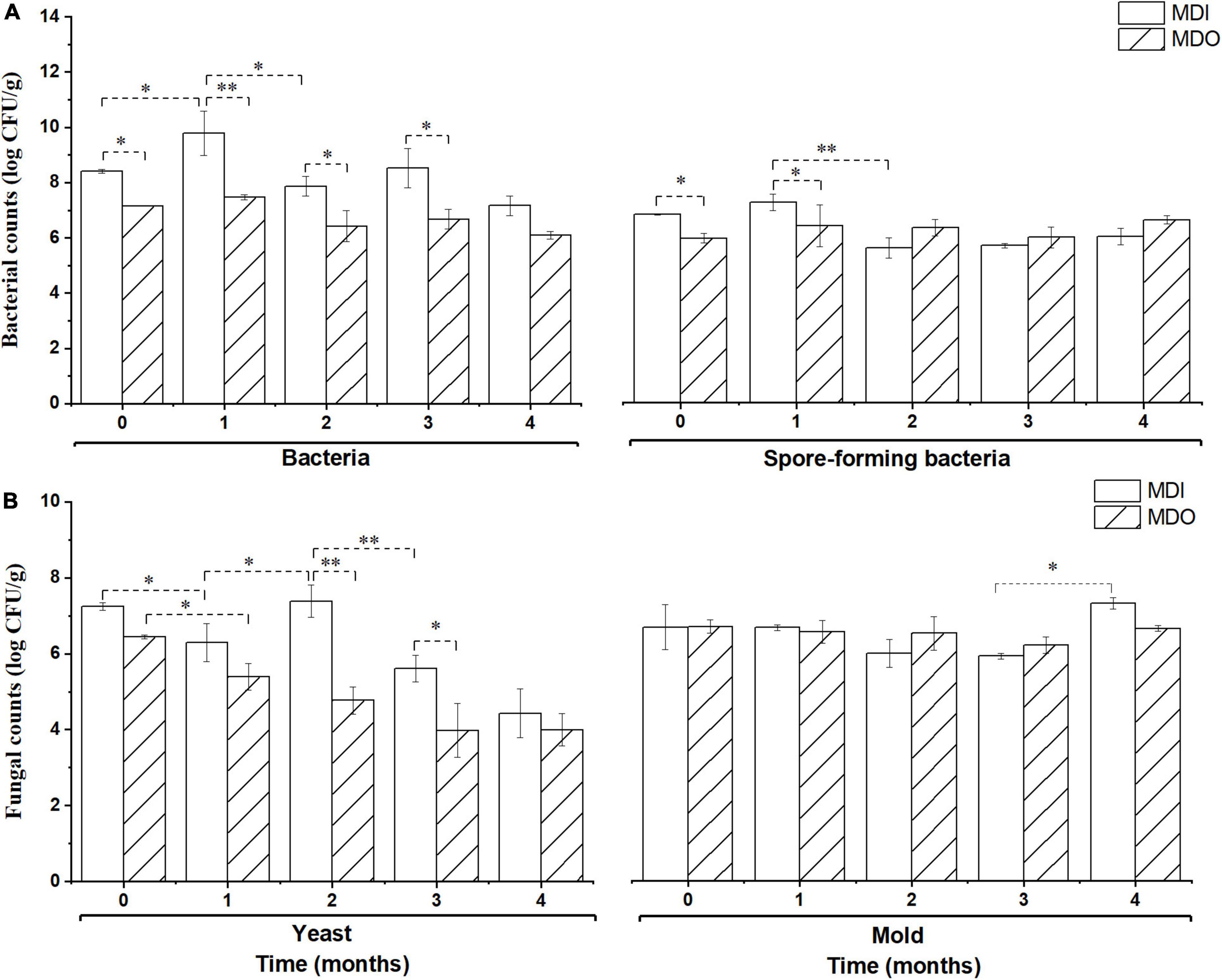
Figure 2. Comparison of the bacteria (A) and fungi (B) in the MDI and MDO samples during storage. Significant differences are noted by * (p < 0.05) and ** (p < 0.01).
A total of eighty-two bacterial isolates were obtained from the mature samples. Based on the analysis of the 16S rRNA gene sequence, 6 bacterial genera (15 species) were identified (Supplementary Table 1). Of these, Bacillus and Staphylococcus accounted for 70.7 and 13.4% of the bacterial community, respectively, at the genus level. A total of fifty fungal isolates were selected for identification by ITS region sequencing and 18S rRNA gene sequence analysis. A total of nine fungal species were identified, including Wickerhamomyces anomalus (12 strains), Pichia jadinii (8 strains), Aspergillus awamori (13 strains), Aspergillus ustus (4 strains), Penicillium purpurogenum (7 strains), Penicillium sp. (1 strain), Alternaria alternata (4 strains), and Lichtheimia corymbifera (1 strain) (Supplementary Table 2). W. anomalus and A. awamori mainly appeared in MDI, whereas other fungal species exhibited a similar distribution in both parts of MT-Daqu.
Illumina sequencing of the bacterial 16S rRNA genes and fungal ITS gene generated a total of 283,896 and 520,350 effective sequences, respectively. After clustering all the sequences, 597-632 and 625-773 OTUs of observed species in the outer layer and inner layer of MT-Daqu were obtained, respectively. The community diversity of either bacteria or fungi in the MDI was greater than that in the MDO, whereas the number of bacterial OTUs (observed species) was less than that of fungal OTUs (Supplementary Table 3). At the phylum level (Figure 3A), the three dominant bacterial phyla were Firmicutes (75.68%), Proteobacteria (17.27%), and Actinobacteria (2.72%) in MDI. In MDO, Firmicutes and Proteobacteria were still dominant, but at lower percentages, e.g., only 39.26 and 5.74%, respectively. Moreover, the two dominant fungal phyla were Ascomycota and Mucoromycota, and the former was the predominant type, which accounted for 82.20 and 65.79% of the total sequences in MDI and MDO, respectively. At the genus level (Figure 3B), bacteria were dominated by Bacillus, Staphylococcus, Pantoea, Weissella, Saccharopolyspora, Klebsiella, and Prevotella. Bacillus and Staphylococcus were highly abundant (>9.83%) in the MDI and MDO samples, whereas Bacillus was relatively more abundant in MDI (47.02%) than in MDO (21.07%), and Saccharopolyspora was much less abundant in MDI (0.79%) than in MDO (6.11%). For fungi, Thermomyces, Aspergillus, Rhizomucor, Thermoascus, Rhizopus, Archaeorhizomyces, and Alternaria were the predominant genera in both parts of MT-Daqu. The abundances of Aspergillus and Rhizopus in MDI (58.88 and 5.34%, respectively) were higher than in MDO (20.05 and 0.14%). However, the abundances of Thermomyces, Rhizomucor, and Thermoascus in the MDI (13.56, 3.82, and 0.60%, respectively) were less than in MDO (50.67, 25.72, and 7.51%, respectively).
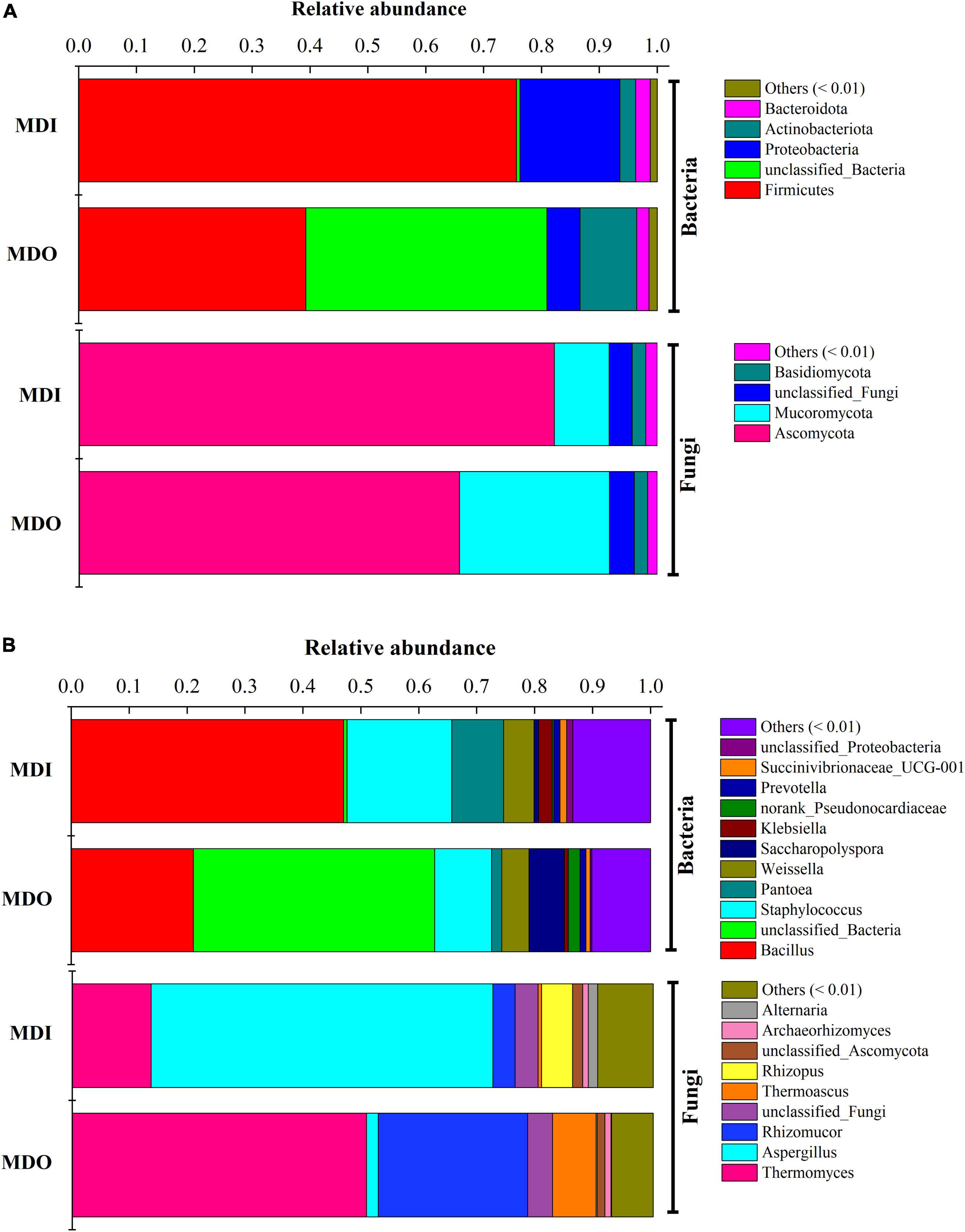
Figure 3. Microbial composition in different parts of the mature MT-Daqu. (A) Bacterial and fungal composition at the phylum level (more than 0.5%); (B) bacterial and fungal composition at the genus level (more than 0.5%). MDI, inner layers of MT-Daqu; MDO, outer layers of MT-Daqu.
Principal coordinates analysis showed that the cumulative contribution of the first two principal coordinates was 89.16% for bacterial communities (Figure 4A) and 89.92% for fungal communities (Figure 4B). Moreover, the two PCoA plots showed that the samples of the same part of MT-Daqu clustered together, and PERMANOVA revealed that the differences were significant (p = 0.034 < 0.05). LEfSe indicated that as many as 30 bacteria (Figure 4C) and 29 fungi (Figure 4D) exhibited significant differences (p < 0.05) in different parts of MT-Daqu samples. Bacillus, Klebsiella, Cronobacter, and Pantoea were the most significantly enriched bacterial genera in MDI, whereas Saccharopolyspora, unclassified or no rank Pseudonocardiaceae, and unclassified Bacteria were enriched in MDO. A total of eighteen taxa and 11 taxa exerted significant effects (p < 0.05) on fungal community structure in the MDI and MDO samples, respectively. Furthermore, Aspergillus, Alternaria, and Rhizopus were enriched fungal genera in the MDI but Thermomyces, Thermoascus, and Rhizomucor were enriched in the MDO. At the genus level, the different types of samples contained a higher proportion of type-specific microorganisms.
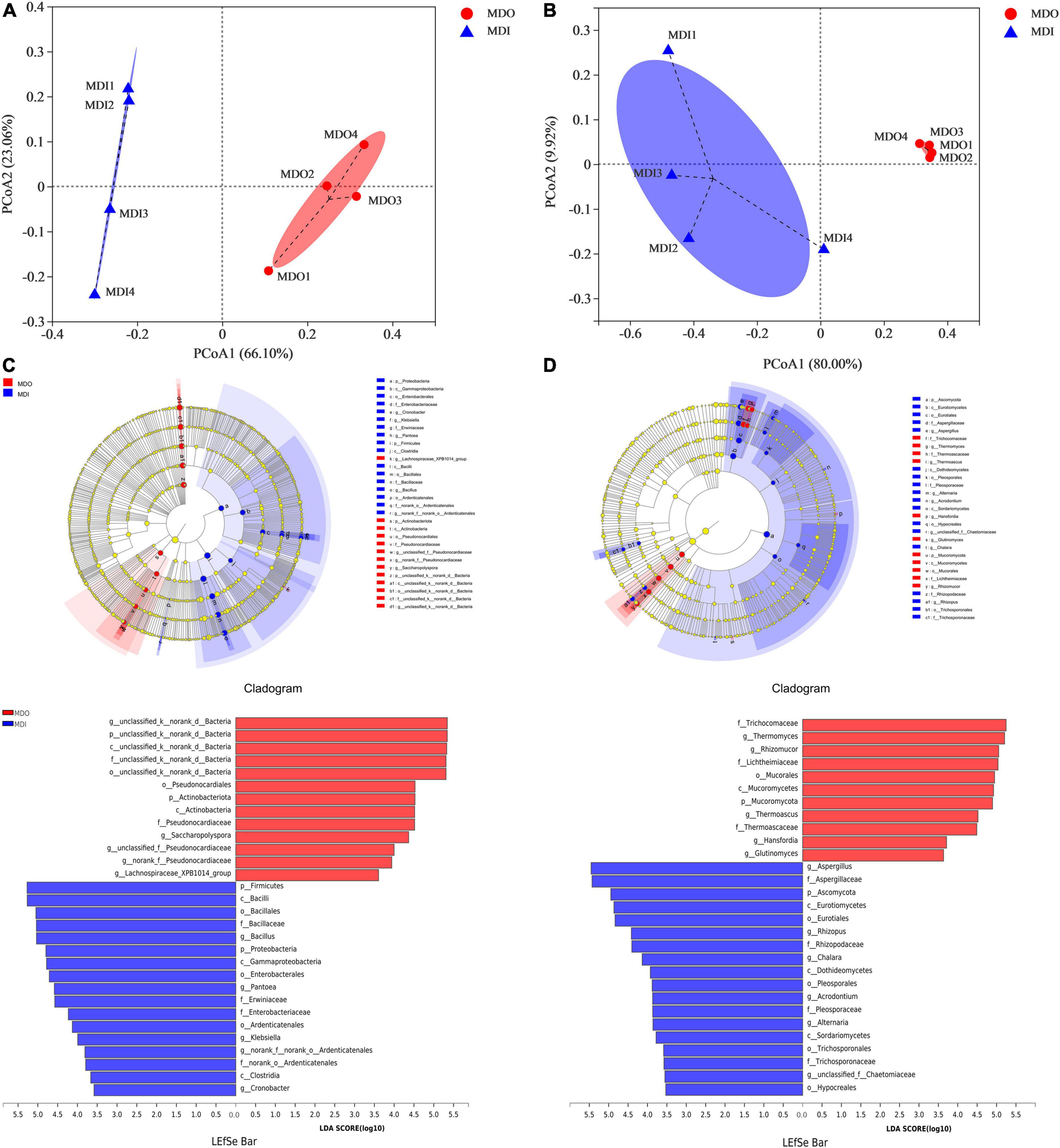
Figure 4. Principal coordinates analysis score plots of microbial β-diversity based on Bray–Curtis distance (p < 0.05) and LEfSe analysis of the MDI and MDO samples (LDA > 3.5, p < 0.05). Bacterial (A,C) and fungal (B,D) taxa showed significantly different abundances.
A total of sixty-five volatile compounds, including 17 alcohols, 10 esters, 13 aldehydes, 7 ketones, 10 pyrazines, and 8 other compounds, were identified and semiquantified (Supplementary Table 4). Of these, isoamyl alcohol, hexanol, 2,3-butanediol, benzyl alcohol, phenylethyl alcohol, ethylhexanoate, ethylpalmitate, ethyllinoleate hexanal, benzaldehyde, geranylacetone, and tetramethylpyrazine were dominant in mature MT-Daqu. The contents of six species of volatile compounds are shown in Figure 5. The contents of alcohols and pyrazines were the highest in all samples, and only the alcohol content in the MDI (699.05 μg/kg) was significantly higher than that in the MDO (348.88 μg/kg). In the MDI sample, no significant difference was observed between the contents of alcohols and pyrazines, whereas in the MDO sample, the content of pyrazines at 590.96 μg/kg was the highest, which was 25 times higher than the lowest content of ketones (23.57 μg/kg).
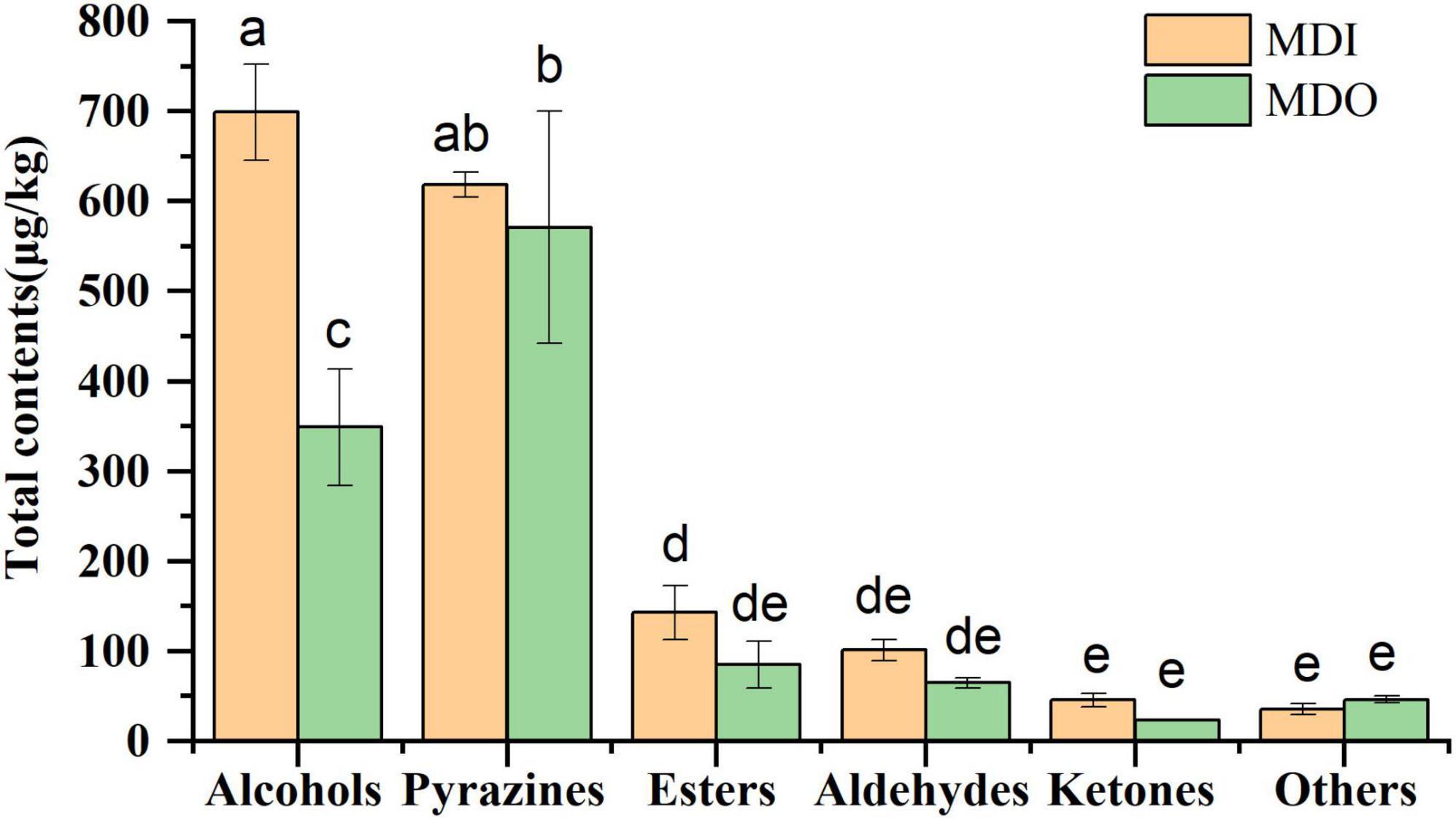
Figure 5. Relative abundance of six categories of volatile compounds in MDI and MDO samples of the mature MT-Daqu. Different letters indicate significant differences (p < 0.05).
The investigation of the culture-dependent microbial abundance and physicochemical properties of Daqu provided the first insight into the microbial composition and metabolic changes, which provides a basis for further omics analyses (Huang et al., 2020) and might be used to initially evaluate the maturity of Daqu. Despite the high workload and incomplete coverage, culture-dependent methods isolate cultivable strains that can be used as functional starters with contributions to the quality of ferment liquor and foods (Zheng et al., 2012; Lv et al., 2015; Yang et al., 2022). Considering bacteria, yeasts and molds as the three major types of microbiota in Daqu (Cai et al., 2021) and spore-forming bacteria, especially Bacillus as producers of enzymes and aroma compounds that contribute to Daqu flavor and quality (Yan et al., 2013b; He et al., 2019), two groups of bacteria, namely, aerobic bacteria and spore-forming bacteria and two groups of fungi, namely, yeast and mold, were analyzed using culture-dependent methods during MT-Daqu production. In the fermentation period, the counts of aerobic bacteria in MDI and yeast in both parts of MT-Daqu rapidly decreased to the minimum values at 17 days (Figures 1A,B) as the temperature increased to 60°C (Supplementary Figure 2) and then increased until the end of fermentation, consistent with previous findings (Li et al., 2016; Yan et al., 2019). This result might be attributed to the decrease in non-thermotolerant bacterial genera, such as lactic acid bacteria (LAB), and fungal genera, such as Pichia, which was restrained when the temperature was higher than 40°C (Li P. et al., 2015). The counts of spore-forming bacteria and molds in both parts of MT-Daqu increased rapidly in the first 9 days during the Peijun stage and then decreased slowly from 9 days to 17 days at the Dinghuo stage, thereafter increasing gradually or remaining stable until the end of fermentation. In the Peijun and Dinghuo stages, thermotolerant spore-forming bacteria, such as Bacillus, and filamentous fungi, such as Aspergillus, Rhizopus, Rhizomucor, and Amylomyces, survived and were commonly involved in solid-state fermentation with increasing temperature and decreasing moisture and increasing environmental relative humidity (Supplementary Figure 2; Li P. et al., 2015; Shanqimuge et al., 2015; Yan et al., 2019). This finding was confirmed by the changes in the total aerobic bacteria in MDI. The counts of total aerobic bacteria were essentially coincident with those of spore-forming bacteria from 9 days to 17 days, showing that the spore-forming bacteria are capable of producing endospores that are highly resistant to unfavorable environmental conditions of heat and desiccation, and thus, they were predominant among the total aerobic bacteria in this stage (Shanqimuge et al., 2015). Additionally, the cell counts of the four microbial groups in MDO were higher than those in MDI from 5 days to 21 days, which is fairly consistent with the results of previous studies (Zheng et al., 2012). The explanation for this observation may be that less access to water and air and a higher temperature MDI were not suitable for the growth of bacteria, except spore-forming bacteria, whereas the interface containing abundant air and nutrient substances easily formed in MDO, leading to the promotion of microbial growth (Li et al., 2013; Zhang Y. et al., 2021).
Consistent with previous studies, the cell counts of aerobic bacteria and yeasts in MDI and MDO samples were significantly different before and after the samples were stored for 3 months, and a similar difference was observed in the counts of spore-forming bacteria before 2 months (Li et al., 2013; Jin et al., 2019). These results supported the hypothesis that these environmental conditions promote three groups of microorganisms to propagate further in the inner layers. After at least 3 months of storage, the microbial counts in both parts of MT-Daqu were not significantly different (p > 0.05) because the changes in the environmental conditions, including temperature, moisture, and acidity, during storage, and drove the microorganisms in both parts of MT-Daqu to become similar and eventually form a stable system (Zhang Y. et al., 2021). Overall, culture-dependent microorganisms are stabilized after 3 months of storage.
The acidity of both parts quickly peaked in the early stage of fermentation, and the value of MDI was higher than that of MDO before 3 months (Table 1). Acidity was regarded as the most significant factor correlated with the composition of the microbial communities in the early stage of Daqu fermentation, and some Lactobacillus species were detected at higher percentages in the central part (Li et al., 2013), leading to the continuous synthesis of numerous organic acids (Shanqimuge et al., 2015). The liquefying activity and the saccharifying activity of Daqu are the two major contributors to liquefaction and saccharification in Baijiu production. Therefore, the two activities in different parts of Daqu showed similar changes throughout production (Table 1), and the two indices in MDI after fermenting 5 days and in MDO during production exhibited significantly positive correlations (Pearson correlation coefficient ρ = 0.58, p < 0.05 and ρ = 0.81, p < 0.01). The results might be attributed to the thermal stability of amylase and glucoamylase at high incubation temperatures of approximately 60°C (Li P. et al., 2015). Moreover, changes in both activities in the MDI and the MDO samples were similar to the counts of mold and spore-forming bacteria, respectively. A few studies have reported that some mold species, such as Thermoascus, Aspergillus, and Rhizomucor, and spore-forming bacterial species, such as B. licheniformis, B. subtilis, and B. cereus, which were identified as the core genera in Daqu production, produce various hydrolytic enzymes for starch liquefication and saccharification (Li Z. M. et al., 2015; Wang et al., 2018; Yan et al., 2019). Esterifying activity mainly contributes to the synthesis of esters that are the prominent flavor of Baijiu (Guan et al., 2021). In this study, along with the decrease in yeast counts, esterifying activity remained at a lower range until complete fermentation. Similar to acidity, the fermenting activity, especially in MDO, increased in the early fermentation period but did not change significantly in storage, mainly due to the higher counts of yeasts in the early phase (Fan et al., 2020).
Consistent with previous reports (Fan et al., 2020; Guan et al., 2021), we showed that storage for at least 3 months is necessary for MT-Daqu to reach a stable state in Figure 2. Moreover, we found that five main bacterial phyla, Firmicutes, Proteobacteria, Actinobacteria, Cyanobacteria, and Bacteroidetes, and two dominant fungal phyla, Ascomycota and Mucoromycota, were identified in both parts of MT-Daqu. These dominant microbial communities in MT-Daqu have been reported in other studies (Guan et al., 2021), and the same discovery was documented in both low-temperature and high-temperature Daqu (Li et al., 2013; Jin et al., 2019). Bacillus, Klebsiella, and Pantoea might represent potential biomarkers for MDI and Saccharopolyspora for MDO. The result that Saccharopolyspora were dominant outside of MT-Daqu is consistent with a previous study, and the study indicated that they were derived from the environment or raw materials to provide the initial microbiota (Zhang Y. et al., 2021). In addition, large amounts of Bacillus, Staphylococcus, and Weissella emerged in both samples. This finding was consistent with the analysis in terms of the predominance of Bacillus and Staphylococcus isolated and identified using culture-dependent methods (Supplementary Table 1). This study also showed that fungal communities in MT-Daqu were mainly comprised of Aspergillus, Thermomyces, Rhizomucor, Thermoascus, and Rhizopus, and Aspergillus and Rhizopus were dominant in MDI, whereas Thermomyces, Rhizomucor, and Thermoascus were predominant in MDO. The same composition of molds at the genus level in MT-Daqu was observed in liquor industries of South China, such as Sichuan and Jiangsu, which are the dominant regions producing strong-flavor Baijiu (Guan et al., 2021). Additionally, LEfSe revealed that the fungal genera Aspergillus, Alternaria, and Rhizopus were enriched in the MDI, whereas Thermomyces, Thermoascus, and Rhizomucor were enriched in the MDO. Unlike their abundances in high-temperature Daqu, Byssochlamys, and Fusarium were predominant in the inner and outer parts of high-temperature Daqu, respectively, and Thermoascus possessed an absolutely dominant position in both positions (Zhang Y. et al., 2021). In MT-Daqu, we did not detect Byssochlamys and only detected a low abundance of Fusarium (0.013%). These results may be due to the different production processes for different types of Daqu or different regional environments (Cai et al., 2021). Therefore, MT-Daqu has a specific fungal community structure. Aspergillus, Alternaria, and Rhizopus might be considered as biomarkers for MDI, whereas Thermomyces, Thermoascus, and Rhizomucor may be considered as biomarkers for MDO of MT-Daqu (Hu et al., 2021). The dominant fungal community comprised of molds was discovered in high-temperature and MT-Daqu (Hu et al., 2017; Wang et al., 2018), whereas the fungal composition with a high proportion of yeasts, such as Wickerhamomyces, Saccharomycopsis, and Pichia, was detected in low-temperature Daqu because of its fermentation at a lower temperature (Cai et al., 2021; Hu et al., 2021). Therefore, the special fungal composition of MT-Daqu is different from that of low-temperature Daqu but close to that of high-temperature Daqu, which potentially explains why the strong-flavor Daqu has its own unique flavor and physiological–biochemical characteristics.
Flavors in Daqu play an important role in the flavor classification of Chinese Baijiu (Zhang et al., 2011; Wang et al., 2020). In this study, isoamyl alcohol, hexanol, 2,3- butanediol, benzyl alcohol, phenylethyl alcohol, ethylhexanoate, ethylpalmitate, ethyllinoleate, hexanal, benzaldehyde, geranylacetone, and tetramethylpyrazine were detected as the predominant flavors in mature MT-Daqu (Supplementary Table 4), consistent with the findings of Luzhou-Daqu (Sichuan, China) (Zhang et al., 2011; He et al., 2019) and Yingjia-Daqu (Anhui, China) (Yan et al., 2019). Of these compounds, tetramethylpyrazine was present the highest relative abundance in both parts of MT-Daqu, followed by phenylethyl alcohol and benzaldehyde. Pyrazines, which are nitrogen-containing compounds, are the important volatile compounds of Chinese Baijiu that provide baked, roasted, and nutty notes (Fan and Qian, 2006) and are reported to be the key active compounds of Jiang-flavor Baijiu (Jin et al., 2019). A total of ten pyrazines were also identified in our samples, five of which were detected in “Wuliangye” and “Jiannanchun” strong-flavored Baijiu (Fan and Qian, 2006). He et al. (2019) reported that 10 pyrazines were identified in Luzhoulaojiao-Daqu, and the relatively high content of tetramethylpyrazine was consistent with the high abundance of Bacillus in fortified MT-Daqu inoculation with Bacillus velezensis and Bacillus subtilis (He et al., 2019). According to Jin et al. (2019), Bacillales, Thermoactinomyces, and Aspergillus are the principal contributors to pyrazines (Jin et al., 2019). We found that alcohols were also the most dominant flavor compounds and total alcohols were detected at the highest abundance in MDI (Figure 5). Alcohols, as important precursors of acids and esters, comprise the largest group of compounds, and most of them have been detected in Chinese liquor, which often impart fruity, floral, green, and alcoholic odors and contribute to the flavor of Chinese liquor (Zhang et al., 2011). Phenethyl alcohol, imparting floral components, such as rose and fruity notes, was the main constituent in alcohols in MT-Daqu (Yang et al., 2021) and was the second most abundant compound in this study. In addition to phenethyl alcohol, isoamyl alcohol, hexanol, and S-2,3-butanediol were present at high contents in all samples, and their contents in MDI were higher than those in MDO. These higher alcohols might be formed from amino acid catabolism via the Ehrlich metabolic pathway or from lipid oxidation by some microbial strains, such as Enterobacter, Acinetobacter, Aspergillus, Rhizopus, and Alternaria (Zhang et al., 2011; Tang Q. X. et al., 2019; Yang et al., 2022). In addition, geranylacetone, which was present at an intermediate content, was first identified in MT-Daqu, and its function and source in Daqu are still unknown and require further exploration.
This study provided a comprehensive understanding of the microbial community and physicochemical properties inside and outside the MT-Daqu used for strong-flavor Baijiu production from the fermentation period to the mature period. Both culture-dependent and culture-independent methods provided a view of changes in the microbial community during production and discrepancies in the different parts of the MT-Daqu. The physicochemical property indices in both parts of MT-Daqu shifted from the fermentation period to a relatively stable range after storage for 3 months, and a difference between the inner layers and the outer layers was also observed. The findings provide theoretical evidence for industrial process control and evaluating the mature period of MT-Daqu. This study also investigated the predominant bacterial and fungal composition and volatile compounds inside and outside the mature MT-Daqu. The results will assist in quality judgments of the mature MT-Daqu and identifying the functional microbial sources to apply in Baijiu brewing.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
XH conducted all the experiments and wrote the initial manuscript. RL and MH conceived and designed the experiments. ZS and JW revised and proofread the manuscript. XS and RX analyzed the experimental data. XL and CP contacted the liquor manufacturer and participated in the partial production experiment. All authors read and approved the manuscript.
This work was supported by the Major Special Subproject of Henan Province (181100211400-8), the Key Project of Scientific and Technological Research in Henan Province (182102110008), and the Innovative Funds Plan of Henan University of Technology (2020ZKCJ23).
We thank Fuqiang Li, Zihong Liu, and Peixue Yan at the Henan Songhe Distillery Co., Ltd., for their contribution to the sampling of MT-Daqu.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.934696/full#supplementary-material
Cai, W., Wang, Y., Ni, H., Liu, Z., Liu, J., Zhong, J., et al. (2021). Diversity of microbiota, microbial functions, and flavor in different types of low-temperature Daqu. Food Res. Int. 150:110734. doi: 10.1016/j.foodres.2021.110734
Chen, Y., Li, K., Liu, T., Li, R., Fu, G., Wan, Y., et al. (2020). Analysis of difference in microbial community and physicochemical indices between surface and central parts of Chinese special-Flavor Baijiu Daqu. Front. Microbiol. 11:592421. doi: 10.3389/fmicb.2020.592421
Deng, L., Mao, X., Liu, D., Ning, X. Q., Shen, Y., Chen, B., et al. (2020). Comparative analysis of physicochemical properties and microbial composition in High-Temperature Daqu with different colors. Front. Microbiol. 11:588117. doi: 10.3389/fmicb.2020.588117
Deng, Y., Huang, D., Han, B., Ning, X., Yu, D., Guo, H., et al. (2021). Correlation: between autochthonous microbial diversity and volatile metabolites during the fermentation of Nongxiang Daqu. Front. Microbiol. 12:688981. doi: 10.3389/fmicb.2021.688981
Fan, G. S., Fu, Z. L., Sun, B. G., Zhang, Y. H., Wang, X. L., Xia, Y. Q., et al. (2019). Roles of aging in the production of light-flavored Daqu. J. Biosci. Bioeng. 127, 309–317. doi: 10.1016/j.jbiosc.2018.08.005
Fan, G. S., Fu, Z. L., Teng, C., Liu, P. X., Wu, Q. H., Rahman, M. K. R., et al. (2020). Effects of aging on the quality of roasted sesame-like flavor Daqu. BMC Microbiol. 20:67. doi: 10.1186/s12866-020-01745-3
Fan, W. L., and Qian, M. C. (2006). Characterization of aroma components of Chinese “Wuliangye” and “Jiannanchuan” liquors by aroma extracr dilution analysis. J. Agric. Food Chem. 54, 2695–2704. doi: 10.1021/jf052635t
Fu, G. M., Deng, M. F., Chen, K. D., Chen, Y. R., Cai, W. Q., Wu, C. F., et al. (2021). Peak-temperature effects of starter culture (Daqu) on microbial community succession and volatile substances in solid-state fermentation (Jiupei) during traditional Chinese special-flavour Baijiu production. Lwt 152:112132. doi: 10.1016/j.lwt.2021.112132
Guan, T. W., Yang, H., Ou, M. Y., and Zhang, J. X. (2021). Storage period affecting dynamic succession of microbiota and quality changes of strong-flavor Baijiu Daqu. Lwt 139:110544. doi: 10.1016/j.lwt.2020.110544
He, G. Q., Dong, Y., Huang, J., Wang, X. J., Zhang, S. Y., Wu, C. D., et al. (2019). Alteration of microbial community for improving flavor character of Daqu by inoculation with Bacillus velezensis and Bacillus subtilis. Lwt 111, 1–8. doi: 10.1016/j.lwt.2019.04.098
Hu, Y. L., Dun, Y. H., Li, S. A., Fu, B., Xiong, X. M., Peng, N., et al. (2017). Changes in microbial community during fermentation of high-temperature Daqu used in the production of Chinese ‘Baiyunbian’ liquor. J. Ins. Brew. 123, 594–599. doi: 10.1002/jib.455
Hu, Y. N., Huang, X. N., Yang, B., Zhang, X., Han, Y., Chen, X. X., et al. (2021). Contrasting the microbial community and metabolic profile of three types of light-flavor Daqu. Food Biosci. 44:101395. doi: 10.1016/j.fbio.2021.101395
Huang, X., Fan, Y., Lu, T., Kang, J., Pang, X., Han, B., et al. (2020). Composition and metabolic functions of the microbiome in fermented grain during light-flavor Baijiu fermentation. Microorganisms 8:1281. doi: 10.3390/microorganisms8091281
Jin, G. Y., Zhu, Y., and Xu, Y. (2017). Mystery behind Chinese liquor fermentation. Trends Food Sci. Technol. 63, 18–28. doi: 10.1016/j.tifs.2017.02.016
Jin, Y., Li, D., Ai, M., Tang, Q., Huang, J., Ding, X., et al. (2019). Correlation between volatile profiles and microbial communities: a metabonomic approach to study Jiang-flavor liquor Daqu. Food Res. Int. 121, 422–432. doi: 10.1016/j.foodres.2019.03.021
Li, P., Liang, H., Lin, W. T., Feng, F., and Luo, L. (2015). Microbiota Dynamics Associated with Environmental Conditions and Potential Roles of Cellulolytic Communities in Traditional Chinese Cereal Starter Solid-State Fermentation. Appl. Environ. Microbiol. 81, 5144–5156. doi: 10.1128/AEM.01325-15
Li, Z. M., Chen, L., Bai, Z. H., Wang, D. L., Gao, L. P., and Hui, B. D. (2015). Cultivable bacterial diversity and amylase production in two typical light-flavor Daqus of Chinese spirits. Front. Life Sci. 8, 264–270. doi: 10.1080/21553769.2015.1041188
Li, P., Lin, W., Liu, X., Wang, X., and Luo, L. (2016). Environmental factors affecting microbiota dynamics during traditional solid-state fermentation of Chinese Daqu starter. Front. Microbiol. 7:1237. doi: 10.3389/fmicb.2016.01237
Li, X. R., Ma, E. B., Yan, L., Meng, Z. H., Du, X. W., and Quan, Z. X. (2013). Bacterial and fungal diversity in the starter production process of Fen liquor, a traditional Chinese liquor. J. Microbiol. 51, 430–438. doi: 10.1007/s12275-013-2640-9
Liu, J., Chen, J., Fan, Y., Huang, X., and Han, B. (2018). Biochemical characterisation and dominance of different hydrolases in different types of Daqu – a Chinese industrial fermentation starter. J. Sci. Food Agric. 98, 113–121. doi: 10.1002/jsfa.8445
Lv, X. C., Chen, Z. C., Jia, R. B., Liu, Z. B., Zhang, W., Chen, S. J., et al. (2015). Microbial community structure and dynamics during the traditional brewing of Fuzhou Hong Qu glutinous rice wine as determined by culture-dependent and culture-independent techniques. Food Control 57, 216–224. doi: 10.1016/j.foodcont.2015.03.054
People’s Republic of China Light Industry Professional Standard. (2011b). Strong Flavour Daqu (QB/T 4259-2011), Ministry of Industry and Information Technology of the People’s Republic of China. Beijing: China Light Industry Press, 1–4.
People’s Republic of China Light Industry Professional Standard. (2011a). General Methods of Analysis for Daqu (QB/T 4257-2011), Ministry of Industry and Information Technology of the People’s Republic of China. Beijing: China Light Industry Press, 1–3.
Shangdong Province Local Standard. (2009). General Technical Conditions of Daqu (DB37/T 1231-2009). China: Shangdong Province Bureau of Quality and Technical Supervision, 1–15.
Shanqimuge Liang, H., Zhang, C., Shao, C., Peng, X., Liang, L., et al. (2015). A DGGE marker-mediated mast monitoring of bacterial diversity and comprehensive identification of high-temperature Daqu starter. J. Food Sci. 80:M1519–M1525. doi: 10.1111/1750-3841.12903
Tang, H. L., Liang, H. B., Song, J. K., Lin, W. F., and Luo, L. X. (2019). Comparison of microbial community and metabolites in spontaneous fermentation of two types Daqu starter for traditional Chinese vinegar production. J. Biosci. Bioeng. 128, 307–315. doi: 10.1016/j.jbiosc.2019.03.011
Tang, Q. X., He, G. Q., Huang, J., Wu, C. D., Jin, Y., and Zhou, R. Q. (2019). Characterizing relationship of microbial diversity and metabolite in Sichuan Xiaoqu. Front. Microbiol. 10:696. doi: 10.3389/fmicb.2019.00696
Wang, P., Wu, Q., Jiang, X. J., Wang, Z. Q., Tang, J. L., and Xu, Y. (2017). Bacillus licheniformis affects the microbial community and metabolic profile in the spontaneous fermentation of Daqu starter for Chinese liquor making. Int. J. Food Microbiol. 250, 59–67. doi: 10.1016/j.ijfoodmicro.2017.03.010
Wang, X. D., Ban, S. D., and Qiu, S. Y. (2018). Analysis of the mould microbiome and exogenous enzyme production in Moutai-flavor Daqu. J. Ins. Brew. 124, 91–99. doi: 10.1002/jib.467
Wang, Y., Cai, W., Wang, W., Shu, N., Zhang, Z., Hou, Q., et al. (2021). Analysis of microbial diversity and functional differences in different types of high-temperature Daqu. Food Sci. Nutr. 9, 1003–1016. doi: 10.1002/fsn3.2068
Wang, Z. M., Wang, C. T., Shen, C. H., Wang, S. T., Mao, J. Q., Li, Z., et al. (2020). Microbiota stratification and succession of amylase-producing Bacillus in traditional Chinese Jiuqu (fermentation starters). J. Sci. Food Agric. 100, 3544–3553. doi: 10.1002/jsfa.10405
Xiao, C., Lu, Z. M., Zhang, X. J., Wang, S. T., Ao, L., Shen, C. H., et al. (2017). Bio-Heat Is a Key Environmental Driver Shaping the Microbial Community of Medium-Temperature Daqu. Appl. Environ. Microbiol. 83:e01550–17. doi: 10.1128/AEM.01550-17
Xu, Y. Q., Sun, B. G., Fan, G. S., Teng, C., Xiong, K., Zhu, Y. P., et al. (2017). The brewing process and microbial diversity of strong flavour Chinese spirits: a review. J. Ins. Brew. 123, 5–12. doi: 10.1002/jib.404
Yan, S. B., Tong, Q. Q., and Guang, J. Q. (2019). Yeast dynamics and changes in volatile compounds during the fermentation of the traditional Chinese strong-flavor Daqu. Lwt 106, 57–63. doi: 10.1016/j.lwt.2019.02.058
Yan, Z., Zheng, X. W., Chen, J. Y., Han, J. S., and Han, B. Z. (2013a). Effect of different bacillus strains on the profile of organic acids in a liquid culture of Daqu. J. Ins. Brew. 119, 78–83. doi: 10.1002/jib.58
Yan, Z., Zheng, X. W., Han, B. Z., Han, J. S., Nout, M. J., and Chen, J. Y. (2013b). Monitoring the ecology of Bacillus during Daqu incubation, a fermentation starter, using culture-dependent and culture-independent methods. J. Microbiol. Biotechnol. 23, 614–622. doi: 10.4014/jmb.1211.11065
Yang, H. Y., Peng, Q., Zhang, H. B., Sun, J. Q., Shen, C., and Han, X. Y. (2022). The volatile profiles and microbiota structures of the wheat Qus used as traditional fermentation starters of Chinese rice wine from Shaoxing region. Lwt 154:112649. doi: 10.1016/j.lwt.2021.112649
Yang, Y., Wang, S. T., Lu, Z. M., Zhang, X. J., Chai, L. J., Shen, C. H., et al. (2021). Metagenomics unveils microbial roles involved in metabolic network of flavor development in medium-temperature Daqu starter. Food Res. Int. 140:110037. doi: 10.1016/j.foodres.2020.110037
Zhang, C. L., Ao, Z. H., Chui, W. Q., Shen, C. H., Tao, W. Y., and Zhang, S. Y. (2011). Characterization of the aroma-active compounds in Daqu: a tradition Chinese liquor starter. Eur. Food Res. Technol. 234, 69–76. doi: 10.1007/s00217-011-1616-4
Zhang, H., Wang, L., Tan, Y., Wang, H., Yang, F., Chen, L., et al. (2021). Effect of Pichia on shaping the fermentation microbial community of sauce-flavor Baijiu. Int. J. Food Microbiol. 336:108898. doi: 10.1016/j.ijfoodmicro.2020.108898
Zhang, Y., Shen, Y., Cheng, W., Wang, X., Xue, Y., Chen, X., et al. (2021). Understanding the shifts of microbial community and metabolite profile from wheat to mature Daqu. Front. Microbiol. 12:714726. doi: 10.3389/fmicb.2021.714726
Zheng, X. W., Yan, Z., Han, B. Z., Zwietering, M. H., Samson, R. A., Boekhout, T., et al. (2012). Complex microbiota of a Chinese “Fen” liquor fermentation starter (Fen-Daqu), revealed by culture-dependent and culture-independent methods. Food. Microbiol. 31, 293–300. doi: 10.1016/j.fm.2012.03.008
Keywords: MT-Daqu, microbial community, physicochemical indices, volatile compounds, fermentation, mature
Citation: Hou X, Hui M, Sun Z, Li X, Shi X, Xiao R, Wang J, Pan C and Li R (2022) Comparative analysis of the microbiotas and physicochemical properties inside and outside medium-temperature Daqu during the fermentation and storage. Front. Microbiol. 13:934696. doi: 10.3389/fmicb.2022.934696
Received: 02 May 2022; Accepted: 30 June 2022;
Published: 27 July 2022.
Edited by:
Yu Xia, Jiangnan University, ChinaReviewed by:
Beizhong Han, China Agricultural University, ChinaCopyright © 2022 Hou, Hui, Sun, Li, Shi, Xiao, Wang, Pan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruifang Li, bHJmQGhhdXQuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.