
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 09 September 2022
Sec. Microbiotechnology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.928307
 Tian Luan1†Lu Wang2†Jiyu Zhao3†Hui Luan1
Tian Luan1†Lu Wang2†Jiyu Zhao3†Hui Luan1 Yueling Zhang1Chunlai Wang1
Yueling Zhang1Chunlai Wang1 Paul R. Langford4Siguo Liu1
Paul R. Langford4Siguo Liu1 Wanjiang Zhang1*
Wanjiang Zhang1* Gang Li1*
Gang Li1*Actinobacillus pleuropneumoniae is an important respiratory pig pathogen that causes substantial losses in the worldwide swine industry. Chronic or subclinical infection with no apparent clinical symptoms poses a challenge for preventing transmission between herds. Rapid diagnostics is important for the control of epidemic diseases. In this study, we formulated an A. pleuropneumoniae species-specific apxIVA-based CRISPR/Cas12a-assisted rapid detection platform (Card) that combines recombinase polymerase amplification (RPA) of target DNA and subsequent Cas12a ssDNase activation. Card has a detection limit of 10 CFUs of A. pleuropneumoniae, and there is no cross-reactivity with other common swine pathogens. The detection process can be completed in 1 h, and there was 100% agreement between the conventional apxIVA-based PCR and Card in detecting A. pleuropneumoniae in lung samples. Microplate fluorescence readout enables high-throughput use in diagnostic laboratories, and naked eye and lateral flow test readouts enable use at the point of care. We conclude that Card is a versatile, rapid, accurate molecular diagnostic platform suitable for use in both laboratory and low-resource settings.
The bacterium Actinobacillus pleuropneumoniae is a member of the Pasteurellaceae family and is the cause of porcine pleuropneumonia, which is characterized by hemorrhagic, fibrinous, and necrotic lung lesions (Frey, 1995). A. pleuropneumoniae is responsible for substantial morbidity, mortality, and economic losses in the worldwide swine industry (Losinger, 2005). Nineteen serovars have been identified based on capsule loci (Stringer et al., 2021a), and the isolates are assigned as biovar 1 and 2 if they are NAD-dependent or -independent, respectively (Serrano-Rubio et al., 2008). Vaccination is considered the best approach to control epidemic diseases, but there are currently no commercially available vaccines that provide cross-protection against all serovars (Loera-Muro and Angulo, 2018).
Therefore, to prevent and control the epidemic of porcine pleuropneumonia, rapid diagnostics play a vital role (Stringer et al., 2021b). Serology, e.g., ApxI-IV ELISAs, can be used to monitor the presence of clinical infections (Nielsen et al., 2000; Andresen et al., 2002; Wei et al., 2012; Gimenez-Lirola et al., 2014), but it is less suitable for the detection of carrier pigs in tonsils or nasal cavities (Sidibe et al., 1993; Chiers et al., 2002). Typically, the identification of A. pleuropneumoniae is established by selective bacterial examination followed by phenotypic and/or molecular tests, e.g., based on the presence of the species-specific apxIVA gene (Schaller et al., 2001). This can take days to get the results back. So, there is a need for more a rapid, especially point-of-care, testing to enable early antibiotic treatment and/or changes in herd management to control disease (Stringer et al., 2021b). Now, there has been a move toward molecular diagnostics, such as PCR (Gram et al., 1996; Klein et al., 2003; Hricinova et al., 2010) and quantitative real-time PCR (qPCR) (Tobias et al., 2012; Watt et al., 2020). These techniques are well developed and are typically based on the apxIVA gene (Schaller et al., 2001). However, these methods still have some limitations. For example, they require expensive thermal cycling instruments and skilled personnel, which have limited their application at the point of care and in low-resource settings. Recently, isothermal amplification techniques, such as recombinase polymerase amplification (RPA) (Li et al., 2019) and loop-mediated isothermal amplification (LAMP) (Yang et al., 2009), have been used to detect A. pleuropneumoniae and have potential use for rapid point-of-care diagnostics. Here, we describe a rapid A. pleuropneumoniae detection platform based on a combination of RPA with elements of the adaptive clustered regularly interspaced short palindromic repeats (CRISPR)/Cas immune system.
Bacteria and archaea have evolved CRISPR and their associated proteins (Cas), collectively referred to as the CRISPR-Cas immune system, to specifically target exogenous viral infections (Koonin and Makarova, 2019). The CRISPR-Cas systems have precise double-stranded DNA (dsDNA) cleavage activity due to their nucleotide sequence-specific recognition capabilities (Zetsche et al., 2015). The CRISPR-Cas9 system is widely used for genome editing in various organisms, such as bacteria, plants, and animals (Jiang et al., 2015; Burgio, 2018; Lu and Tian, 2022). However, recently discovered Cas12a and Cas13a systems have features that are different from the Cas9 system (Zetsche et al., 2015). For example, they can be complexed with a CRISPR RNA (crRNA) containing a programmable spacer sequence to form a nuclease-inactive ribonucleoprotein (RNP) complex. When the RNP binds to complementary target DNA/RNA, their collateral cleavage activity is activated and then precisely cleaves the surrounding single-stranded DNA (ssDNA) (Chen et al., 2018) or RNA (ssRNA) substrates (Fozouni et al., 2021). The substrate can be linked by a fluorophore-quencher pair, and the cleavage activity is detected by measuring the fluorescence (East-Seletsky et al., 2016). This has permitted the development of nucleic acid detection platforms for different pathogens that combine the CRISPR-Cas12a and Cas13a systems and gene amplification, such as DETECTR (Chen et al., 2018), SHERLOCK (Gootenberg et al., 2018), and HOLMES (Li et al., 2018), which have high sensitivities (at the single-molecule level) and specificities (single base resolution). CRISPR/Cas-based diagnostic technology had been successfully applied to detect many common pathogens, e.g., Mycobacterium tuberculosis (Xiao et al., 2020), African swine fever virus (ASFV) (Lu et al., 2020), SARS-CoV-2 (Cao et al., 2021), Zika virus (ZIKV), Dengue virus (DENV) (Gootenberg et al., 2018), and human papillomavirus (HPV) (Chen et al., 2018). The Cas13-based SHERLOCK system requires the target DNA to be reverse-transcribed to RNA for detection by Cas13a (Gootenberg et al., 2018). For bacterial genome targets, the use of the DNA-sensing Cas12 avoids the need for transcribing RNA to DNA (Chen et al., 2018). Therefore, the Cas12a-based system using a single-stranded DNA probe is more suitable for the detection of bacterial pathogens.
To our knowledge, no CRISPR-Cas12a-based platform has been developed to detect A. pleuropneumoniae. Here, we report the development of a rapid CRISPR-Cas12a-based platform for direct detection of A. pleuropneumoniae based on the species-specific gene apxIVA, which we have named the Cas12a/crRNA-Assisted Rapid Detection (Card) platform.
Since the apxIVA gene was reported to be a suitable locus for the identification of A. pleuropneumoniae (Schaller et al., 2001; da Costa et al., 2004; Tobias et al., 2012), we chose it as the target sequence for the design of crRNA probes. Eight species-specific crRNA probes were designed based on a conserved region of the apxIVA gene (Park et al., 2015). The DNA template for crRNA transcription was synthesized by Comate Biotech with a T7 promoter sequence added at its 3′ end. In vitro transcription was performed using the HiScribe T7 Quick High Yield RNA Synthesis kit (New England Biolabs, United States) following the manufacturer’s recommendations. The final translation products were treated with the addition of DNase I to remove template DNA and subsequently purified using the RNA Clean and Concentrator kit (Zymo Research, United States) following the manufacturer’s protocol. crRNA concentration was quantified in a Nanodrop spectrophotometer (IMPLEN, United States). The sequences of all the oligonucleotides synthesized (Comate Biotech, China) in this study are shown in Table 1.

Table 1. The crRNAs and primers used for amplification.
Genomic DNA from A. pleuropneumoniae of different serovar reference strains and other common bacterial pathogens used in the specificity assay were extracted with the Genomic DNA Extraction kit (TIANGEN, China). Viral genomes were extracted with the AxyPrep Body Fluid Viral DNA/RNA Miniprep kit (Axygen Scientific, United States). The DNA from lung samples was extracted with the Tissue and Blood DNA extraction kit (TIANGEN, China) according to the manufacturer’s instructions.
The RPA primer sequences for apxIVA were designed by the Primer 5.0 software by setting the product length to 100–200 bp, with a size preference of 30–35 nt, and with a GC content between 30 and 70%. Standard RPA reactions were performed according to the manufacturer’s instructions of the TwistAmp Basic kit (TwistDx Ltd, United Kingdom) with minor modifications. Every single reaction contained 14.75 μL of rehydration buffer, 0.48 μM forward and reverse primers, an appropriate amount of genomic DNA, 1.25 μL of magnesium acetate (MgAc), and sterile water up to 20 μL. The mixture was incubated at 37°C for 15 min.
LbCas12a-crRNA complexes were preassembled by incubating 1 μM LbCas12a (New England Biolabs, United States) and 1.25 μM crRNA at room temperature for 10 min. DNA targets of various concentrations were dissolved in 1 × Binding Buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5 mM MgCl2, 0.1 mg/mL bovine serum albumin [BSA]) and mixed with LbCas12a-crRNA complexes and 500 nM ssDNA reporter probe in 20 μL reaction volume. In the assay, the final concentrations of LbCas12a and crRNA were 50 and 62.5 nM, respectively. The mixture was incubated in a constant temperature incubator at 37°C for 15 min.
The detection mechanism is based on the non-specific ssDNA probe cleavage by Cas12a-crRNA complexes. To report the presence of A. pleuropneumoniae genomic DNA from different samples, ssDNA probes with different label methods were used. Three different readouts were used. An ssDNA probe (5′-FAM-TTATT-TAMRA-3′) linked with fluorescent and quencher groups was used for fluorescence-based detection either by the naked eye under blue light (Sangon Biotech, China), or by a fluorescent microplate reader (PerkinElmer EnSpire, United States). An ssDNA probe (5′-FAM-TTATT-Biotin-3′) with fluorescein on one end and biotin on the other was used for detection colorimetrically in lateral flow strips (Milenia Biotec GmbH, United Kingdom). On the lateral flow strips, the antibodies labeled with gold nanoparticles (NPs) were embedded in the sample area of the strip and could bind the ssDNA reporter to form a dark purple color line. When no genomic DNA of A. pleuropneumoniae appears in the reaction system, all the uncleaved ssDNA reporter would be captured by the streptavidin present in the control line, and only one purple line on the strip would be present. In contrast, a positive result arises from the reporter being cleaved and the gold NP–labeled antibodies continuously flowing toward the test line covered with anti-FAM antibodies and forming another dark purple color line, which indicates the presence of the target in the system. All the colorimetric processes were completed in less than 5 min.
To evaluate the specificity of this detection platform, apart from the A. pleuropneumoniae serovar 7 strain S8 (Li et al., 2012), several other common pathogens and closely related species were also tested. The Glaesserella parasuis serovar 5 reference strain Nagasaki (Frandoloso et al., 2012) and Streptococcus suis serovar 2 strain 05ZYH33 (Feng et al., 2014) were stored in our laboratory. The Pasteurella multocida CVCC438 and Salmonella enterica serovar choleraesuis CVCC 79500 were purchased from the China Veterinary Culture Collection Center (CVCC). The genome of porcine circovirus type 2, porcine pseudorabies virus, and porcine epidemic diarrhea virus were kindly provided by other research groups of the Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, China. The specificity of the Card platform for these pathogens was assessed by all the readout formats. Additionally, the species from the Pasteurellaceae family (Actinobacillus suis, Actinobacillus equuli, Actinobacillus lignieresii, Actinobacillus indolicus, Actinobacillus porcitonsillarum, Actinobacillus porcinus, Actinobacillus pneumotropica, and Actinobacillus minor) and all the reference strains (serovar 1, strain 4074; serovar 2, S1536; serovar 3, S1421; serovar 4, M62; serovar 5a, K17; serovar 6, Fem∅; serovar 7, WF83; serovar 8, 405; serovar 9, CVJ13261; serovar 10, D13039; serovar 11, strain 56153; serovar 12, strain 1096; serovar 13, strain N273; serovar 14, strain 3906; serovar 15, strain HS143; serovar 16, strain A-85/14; serovar 17, strain 16287-1; serovar 18, strain 7311555; serovar 19, strain 7213384-1) were also tested. Another 10 field isolates [serovar 1 (isolates 2 and 5), serovar 2 (isolates 1, 3, and 8), serovar 7 (isolates 4, 6, 7, 9, and 10)], which had been isolated from the lungs of suspected A. pleuropneumoniae infections in different farms of China and identified by PCR (Stringer et al., 2021a), were also tested. The specificity for these species was assessed in a fluorescent microplate reader.
To evaluate the sensitivity of the Card platform, we first determined the sensitivity using serial dilutions of the plasmid template. The fragment containing the chosen conserved region of apxIVA was amplified by PCR with primer pairs P1 and P2. Then, the PCR products were purified by using a gel extraction kit following the manufacturer’s protocol. The final purified fragment was ligated to the pUT18 plasmid (Euromedex, United States). The plasmid concentration was then quantified by Nanodrop spectrophotometry, and the initial copy number was calculated according to plasmid length and concentration. The serially diluted plasmid was used as a template in the RPA reaction system.
Additionally, serial dilutions of pure culture of an A. pleuropneumoniae strain were also used to determine the sensitivity. The A. pleuropneumoniae serovar 7 strain S8 was cultured overnight, serial dilutions were plated on TSA medium, and the colony forming units (CFUs) were counted. Subsequently, 1 mL cultures of different dilutions (106 to 100 CFUs/mL) were centrifuged at 5,000 × g for 5 min, resuspended in 100 μL PBS buffer, boiled for 10 min, and samples were centrifuged at 12,000 × g for 10 min. The resulting supernatants were used as templates in the RPA reaction system. All the tests were done in triplicate. Meanwhile, to compare the LOD of the Card platform, the same template was also subjected to the apxIVA-based PCR (Schaller et al., 2001) and qPCR (Tobias et al., 2012). The qPCR analysis was done using an Applied Biosystems QuantStudio 5 thermocycler with primer pair ApxIVA-q-F and ApxIVA-q-R.
To further validate utility, genomic DNA isolated from pig lung tissues was tested in the Card platform. These lung samples were obtained from experimental infection studies in pigs challenged with the A. pleuropneumoniae serovar 7 strain S8 that had been carried out in our laboratory previously (Xie et al., 2017). The lung tissues (20–50 mg) were mixed with PBS buffer, homogenized for 10 min, and centrifuged at 3,000 × g for 5 min, and a 50-μL supernatant was spread on the TSA medium at 37°C for 12 h. Suspected A. pleuropneumoniae colonies were identified by PCR with the primer pair ApxIV-F and ApxIV-R. Genomic DNA from homogenized lung tissues was extracted according to the manufacturer’s instructions. Confirmation of A. pleuropneumoniae for genomic DNA was carried out by PCR with the same primer pair. The CFUs from these samples were also assessed by qPCR.
The GraphPad Prism v.8.1.2 software was used for data analysis and graphic design. The data in the figures were expressed as mean ± standard deviation.
A schematic overview of the Card platform for the identification of A. pleuropneumoniae is shown in Figure 1. First, the nucleic acid is extracted from the clinical sample by a commercial genome extraction kit, and the target DNA is amplified by RPA for 15 min. The amplified product is mixed with crRNA, Cas12a, and a quenched fluorescence reporter, ssDNA (total time = 15 min). When the amplified target DNA is recognized by the specific crRNA probe, Cas12a is activated, and the complex cleaves the free ssDNA reporter in the reaction mixture. Fluorescence can be directly detected in an appropriate microplate reader or colorimetrically in lateral flow strips or tubes. The entire detection process can be completed within 1 h.
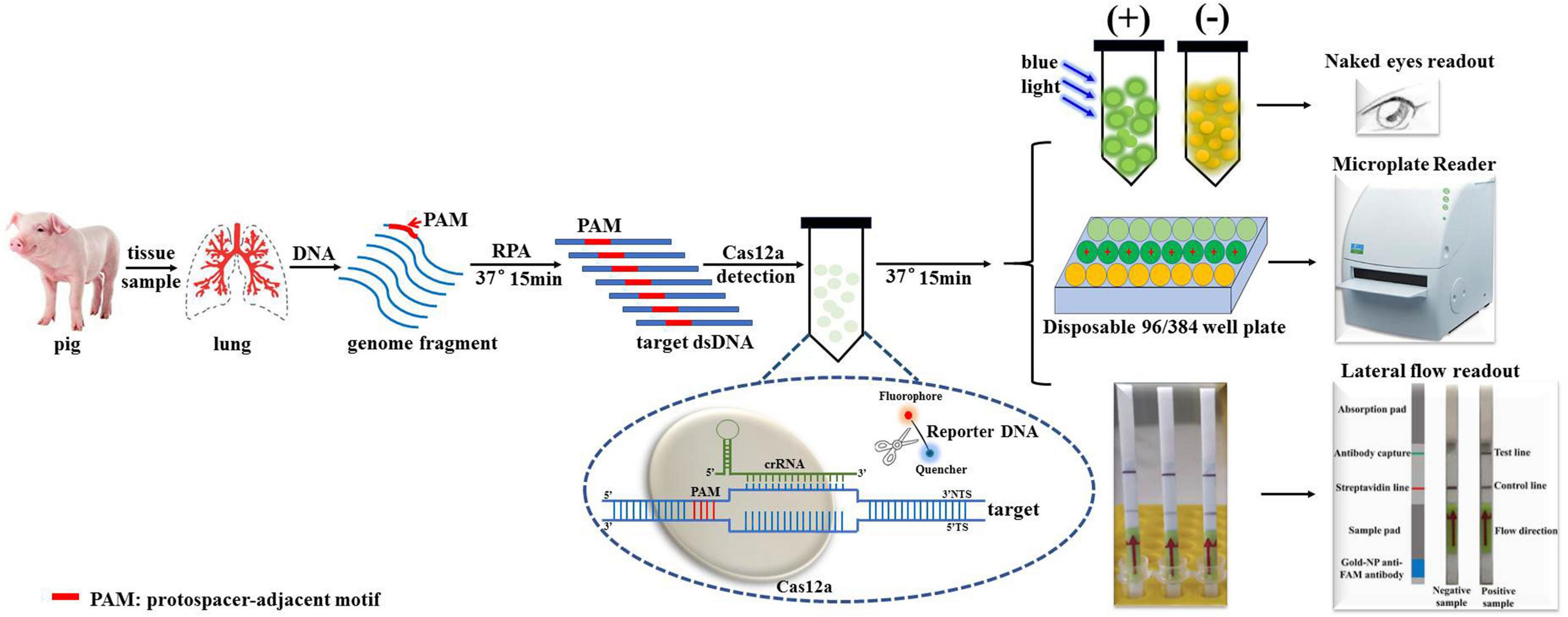
Figure 1. Schematic diagram of Cas12a/crRNA-assisted rapid detection (Card) platform for identification of A. pleuropneumoniae. Gene targets are specifically pre-amplified from genome fragments isolated from clinical samples by RPA. The specific crRNA probes were designed to target amplified fragments, and mixed with Cas12a/crRNA/ssDNA-FQ. Once the target DNA is recognized by the complex formed by Cas12a and crRNA, Cas12a exhibits non-specific endonuclease activity and cleaves the fluorescently labeled reporter (fluorophore [F] is quenched by a quencher [Q]) resulting in fluorescence, which is detected in a microplate reader, or observed by the naked eye under blue light.
Initially, we designed eight crRNA probes (Table 1) based on the species-specific apxIVA gene of A. pleuropneumoniae (Schaller et al., 2001). Every Cas12a-crRNA RNP recognized a single 20-nucleotide region of the apxIVA gene (Figure 2), and each crRNA was initially individually tested in a microplate detection assay. The assay used LbCas12a and a quenched fluorescence reporter, ssDNA, together with in vitro transcribed crRNA, which had been designed to correspond to a conserved region within the apxIVA gene. Initial testing was carried out with plasmid pUT18, which harbored a conserved region of the apxIVA gene. At a target DNA concentration of 1.0 × 105 copies/μL, eight crRNAs with reactivity above the RNP alone control (RNP and probe but no target DNA) were identified. Similar trends in specific crRNA performance were found (Figure 3). For further studies, crRNA 7 was selected since it generated the highest fluorescence value in the shortest reaction time.
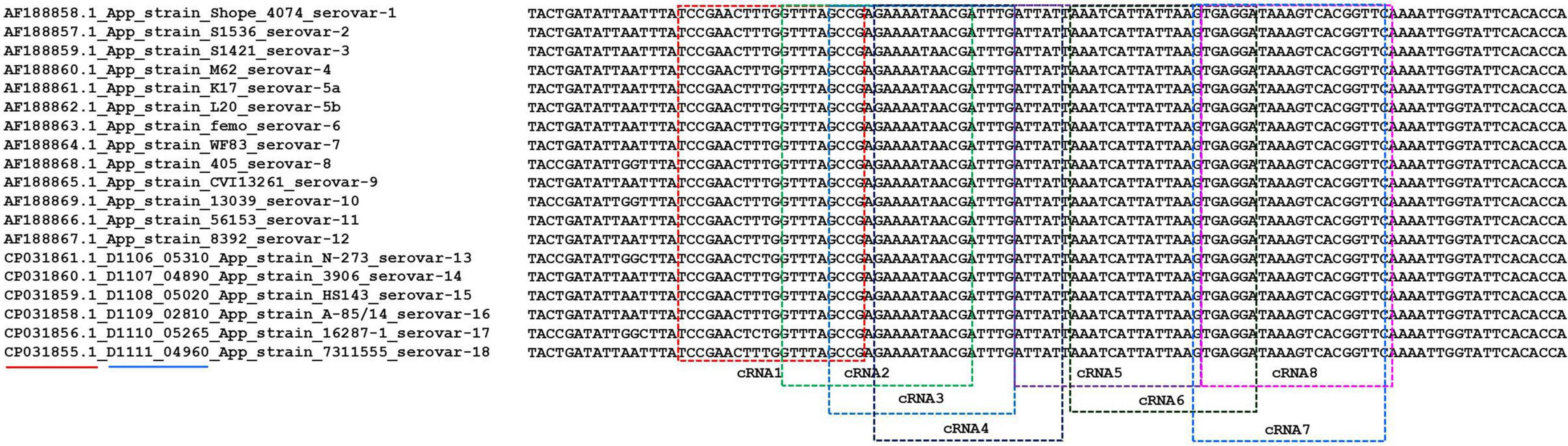
Figure 2. The alignment of partial coding sequence of apxIVA of A. pleuropneumoniae serovars 1–18 downloaded from the NCBI database. Red underline indicates the accession number of the sequence in the NCBI database. Blue underline indicates the locus tag of the gene in the genomic sequence. The different colored frames indicate the recognized region of different crRNAs.
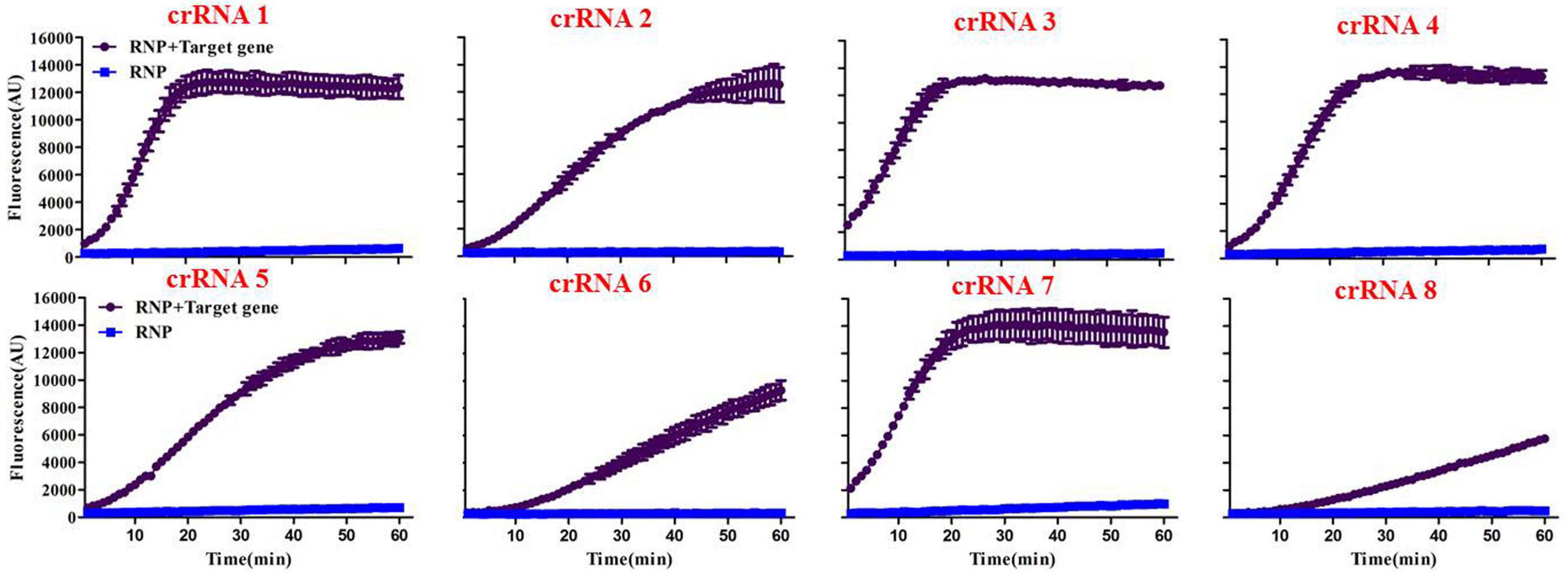
Figure 3. Screening of specific crRNAs by the Card platform. The performance of crRNA 1 to crRNA 8 was determined by testing 50 nM of each RNP individually against 105 copies/μL of plasmid including a conserved region of the A. pleuropneumoniae-specific apxIVA gene. RNP = no target DNA in the reaction system. Data are represented as mean ± standard deviation.
A major advantage of CRISPR diagnostics is their high specificity. To confirm the specificity of the crRNA 7, we tested it against other pathogens, including G. parasuis, Streptococcus suis, S. choleraesuis, P. multocida, porcine circovirus, pseudorabies virus, and porcine epidemic diarrhea virus, which are common pathogens that infect pigs and cause severe economic losses. The specificity of the crRNA 7 probe was evaluated preliminarily using extracted genomic or reverse-transcribed DNA from these pathogens as appropriate. High-emitted fluorescence was found when using a microplate reader when the genome of A. pleuropneumoniae was used as a template, and there was no cross-reaction with the other pathogens evaluated (Figure 4A). Similarly, green fluorescence was observable under blue light for A. pleuropneumoniae but not for other pathogens (Figure 4B). In the lateral flow format, there was a clear purple control line with all samples but an additional purple line with A. pleuropneumoniae (Figure 4C). Meanwhile, the specificity for genomic DNA from pure cultures of reference serovars, closely related species, and clinical isolates was also evaluated. There was high fluorescence in the microplate format from all of the A. pleuropneumoniae serovar reference strains (Figure 4D), related species (Figure 4E), and the isolates with various serovars (Figure 4F). These results indicated that the crRNA 7 probe had high specificity for A. pleuropneumoniae and could be applied to the Card platform for use with multiple readout formats.
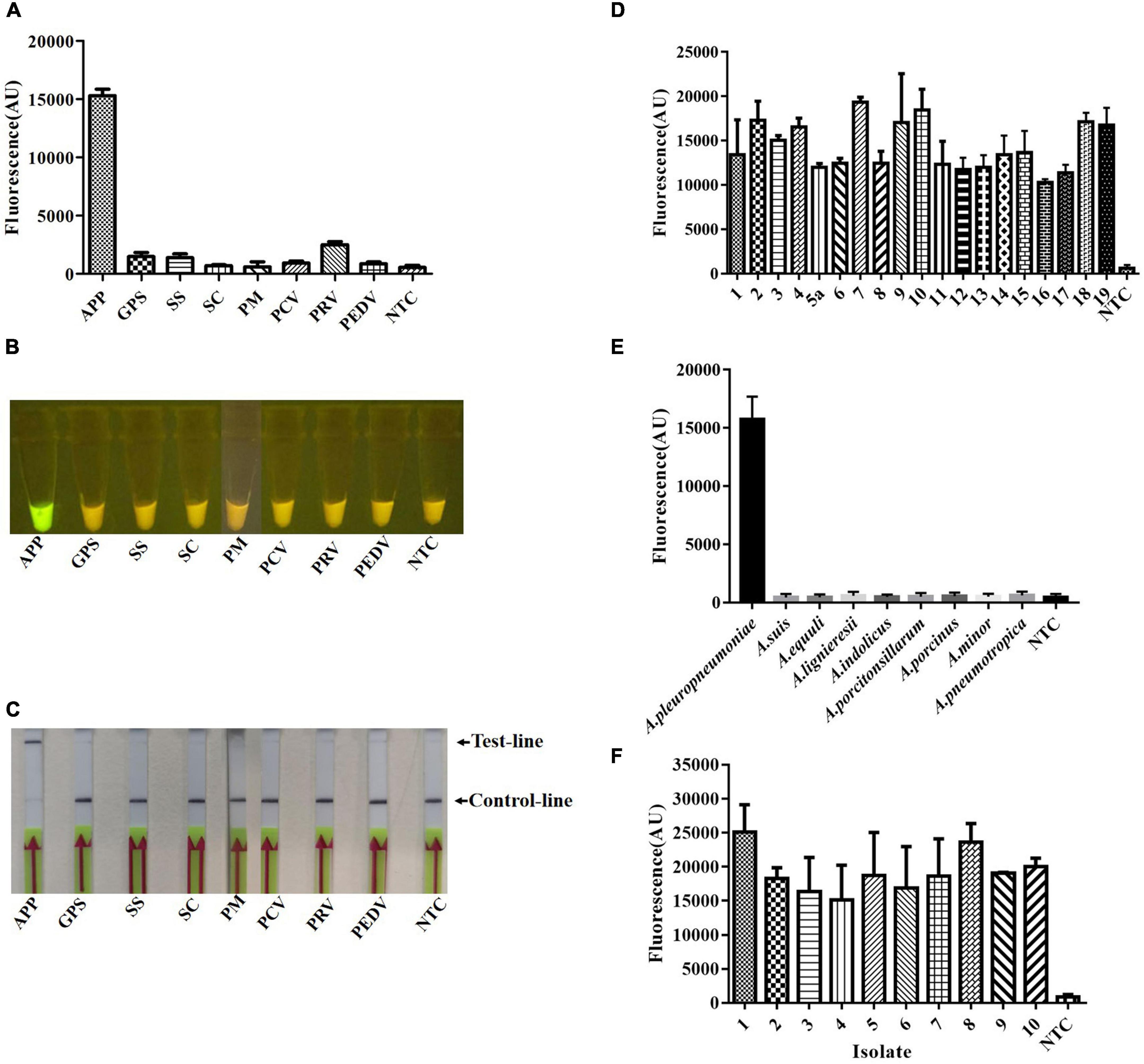
Figure 4. Specificity of the crRNA 7 probe for A. pleuropneumoniae, and detection in multiple formats. The specificity of Card was determined by using genomic or reverse-transcribed DNA extracted from the different pathogens associated with diseases in pigs. (A) Endpoint fluorescence of RPA amplification products detected by a fluorescence microplate reader. (B) Visual detection of RPA amplification products under blue light: A. pleuropneumoniae appears green and negative samples appear yellow. (C) Detection of A. pleuropneumoniae RPA amplification products in a lateral flow strip format. A purple control line is present in all lateral flow strips, but an additional line is present when A. pleuropneumoniae genomic DNA was used as the template. (D) Endpoint fluorescence (microplate reader) for genomic DNA from A. pleuropneumoniae serovars 1–19. (E) Endpoint fluorescence (microplate reader) for genomic DNA from related species to A. pleuropneumoniae. (F) Endpoint fluorescence (microplate reader) for genomic DNA from ten field isolates of A. pleuropneumoniae serovar 1 (isolate 2, 5), serovar 2 (isolate 1, 3, 8), serovar 7 (isolates 4, 6, 7, 9, and 10). In all cases, 50 ng genomic DNA or cDNA were used in the reaction system, and three replicates were conducted for each test, with one representative being shown. A. pleuropneumoniae (APP), Glaesserella parasuis (GPS), Streptococcus suis (SS), Salmonella enterica subsp. enterica serovar choleraesuis (SC), Pasteurella multocida (PM), Porcine circovirus (PCV), Pseudorabies virus (PRV), and Porcine Epidemic Diarrhea Virus (PEDV), non-template control (NTC). Error bars represent the standard deviations of the mean of the three replicates (n = 3).
To determine the sensitivity of the Card platform, we first carried out serial dilutions of the plasmid template to determine the limit of detection for this platform. The plasmid DNA was prepared by ligated amplified apxIVA gene fragment to pUT18 vector, and the extracted plasmid was 10-fold diluted from a starting concentration of 109 copies/μL. The results showed that, if more than 101 copies (≈1 fg) of the plasmid are present in the reaction system, it can be successfully detected as positive (Figures 5A–C).
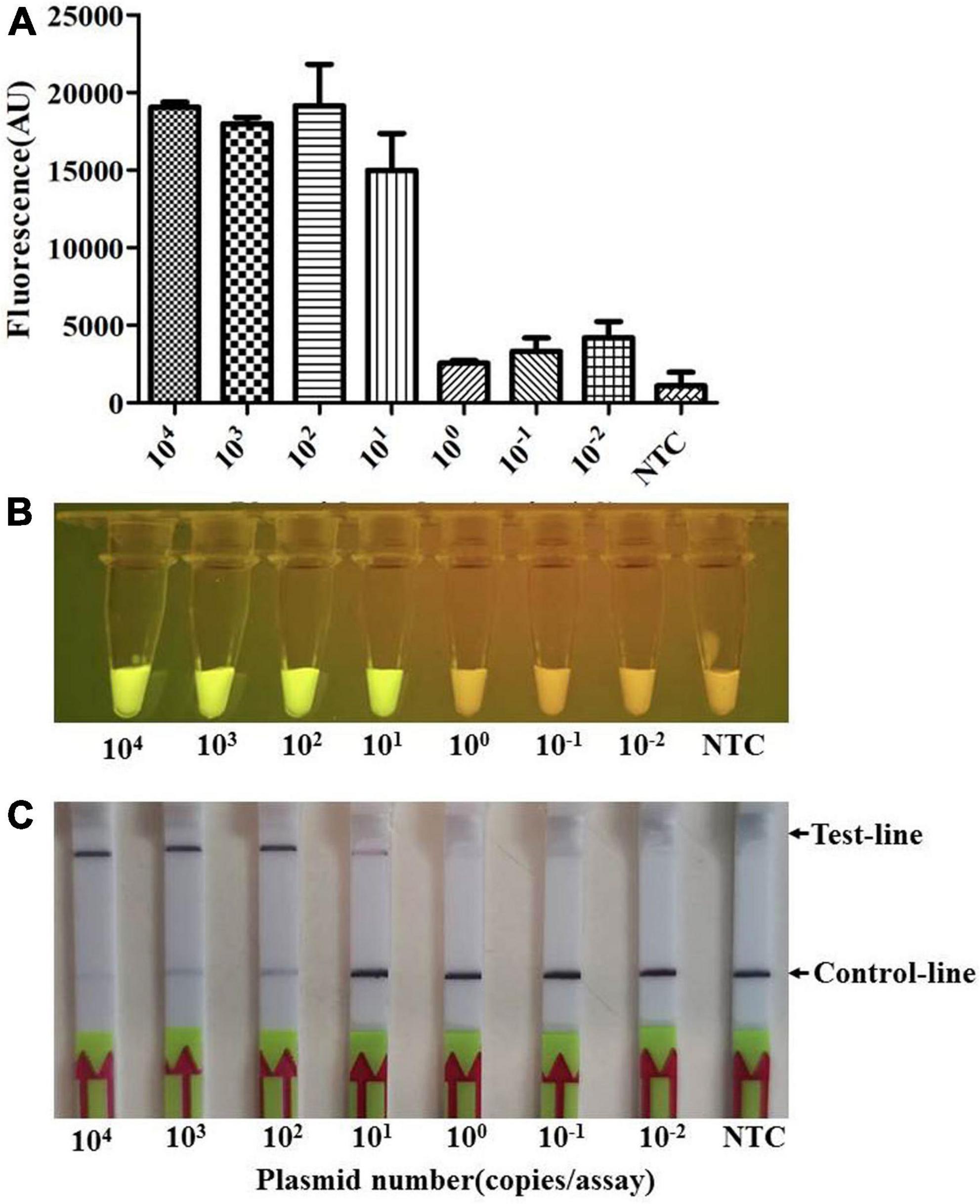
Figure 5. The limit of detection (LOD) of the Card platform for the plasmid sample. The LOD for the plasmid sample was determined by using the diluted plasmid containing apxIVA DNA insert as a template. (A) Endpoint fluorescence of RPA amplification products detected in a fluorescence microplate reader. Error bars represent the standard deviations of the mean of three replicates (n = 3). (B) Visual detection of RPA amplification products under blue light: positive samples appear green and negative samples appear yellow. (C) Detection of RPA amplification products in a lateral flow strip format. A purple control line is present in all lateral flow strips, but an additional line is present when the plasmid DNA was detected by the Card platform. NTC, non-template control. In all cases, three replicates were conducted for each test, with one representative being shown.
In addition, the genomic DNA of serial dilutions of pure culture from A. pleuropneumoniae serovar 7 strain S8 in PBS buffer, equivalent to DNA from 104 to 10–2 CFUs per reaction, was evaluated in the reaction system. The results indicated that the analytical sensitivity could reach 101 CFUs (≈100 fg) per reaction (Figures 6A–C). This is consistent with qPCR (Figure 6E) and 100 times higher than PCR, where a positive band was only found when more than 103 CFUs were included in the reaction system (Figure 6D).
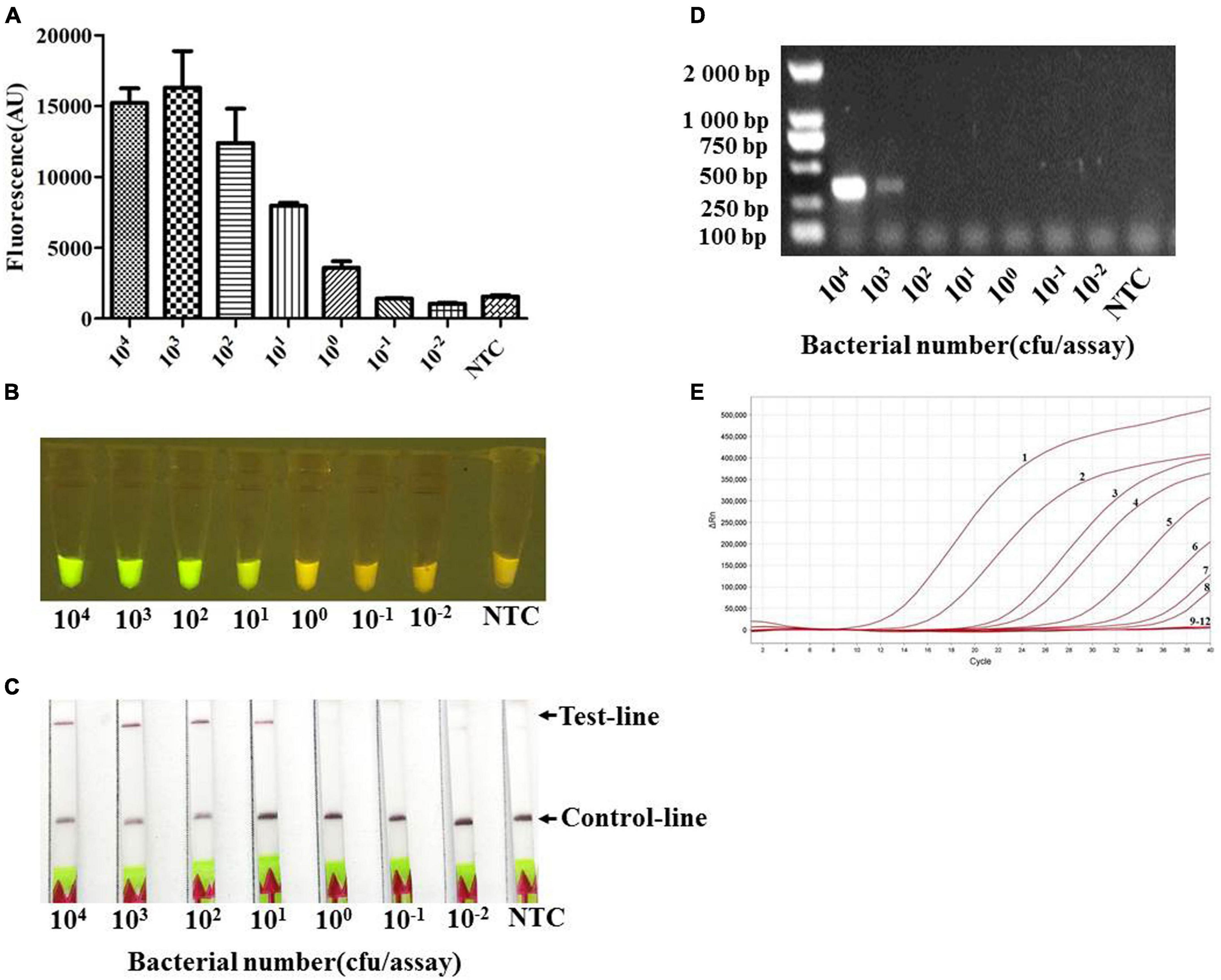
Figure 6. The limit of detection (LOD) of the Card platform for a pure culture of App S8 serovar 7. The LOD for different CFUs was determined by using the genomic DNA from serial dilutions of pure culture as a template. (A) Endpoint fluorescence of RPA amplification products detected in a fluorescence microplate reader. Error bars represent the standard deviations of the mean of the three replicates (n = 3). (B) Visual detection of RPA amplification products under blue light: positive samples appear green and negative samples appear yellow. (C) Detection of RPA amplification products in a lateral flow strip format. A purple control line is present in all lateral flow strips, but an additional line is present when the genomic DNA was detected by the Card platform. (D) The genomic DNA from serial dilutions of pure culture was detected by the PCR. (E) The genomic DNA from serial dilutions of pure culture was detected by quantitative real-time PCR. The numbers 1–8 represent the CFUs from 108 to 101 per assay, numbers 9–12 represent the CFUs from 100 to 10–2, and NTC (non-template control) per assay.
We examined whether the Card Platform could be used for the detection of A. pleuropneumoniae in lung samples. The genomic DNA from these samples was identified by PCR amplification of the apxIVA gene. The homogenized lung tissues were also spread on a selective medium and identified by colony PCR. A total of 30 positive and 5 negative samples were confirmed by both PCR methods. The bacterial load of positive samples was determined by qPCR with the genomic DNA from positive samples, and the results showed that the CFUs of positive samples ranged from 4.8 × 103 to 3.6 × 106 CFUs/g. All the genomic DNA were examined by the Card platform and observed with the naked eyes. The results showed that all apxIVA PCR positive samples produced green fluorescence, while all negative samples emitted yellow light. There was a 100% correlation between the Card platform and the apxIVA PCR method.
There is a need for rapid on-site diagnostics that can be used to detect the major pig pathogen A. pleuropneumoniae. In the present study, we combined RPA with CRISPR-Cas12a to formulate the Card platform to detect A. pleuropneumoniae. Initially, eight specific crRNAs were designed for binding to a conversed region of the A. pleuropneumoniae specific apxIVA gene. Although the aligned sequences were not from all of the 19 known A. pleuropneumoniae serovars, the region is high conserved between the A. pleuropneumoniae 1–18 reference strains. These designed crRNAs showed different specificity in target dsDNA recognition, and we chose the crRNA 7 to take forward as it showed the highest fluorescence. There was no cross-reaction with nucleic acid samples extracted from other common bacterial or viral pathogens of pigs. Such specificity is important for the successful application of such tests to the field (Alon et al., 2021; Gong et al., 2021; Liu et al., 2021; Ma et al., 2021).
To validate the sensitivity of the Card platform, DNA samples from different origins were used in the reaction system. The LOD of the Card platform could reach 10 copies of plasmid or CFUs of A. pleuropneumoniae. It was similar to that of qPCR (Tobias et al., 2012) and more sensitive than PCR, where the LOD was 103 copies. When Card was used on lung samples, there was a 100% correlation with a conventional apxIVA-based PCR. This means that it had good agreement with the most widely used methods (Schaller et al., 2001; Fittipaldi et al., 2003; Klein et al., 2003). Due to the unavailability of samples, we did not assess whether Card can be used on the nasal or tonsillar material, which will be the subject of a future study. We speculate that, being a nucleic-based test, it will be effective for use on such samples, but its clinical utility remains to be determined. Now, all the tests were done using the genomic DNA extracted from pure cultures and lung tissues in a commercial kit. There were some simplified extraction protocols for sample preparation that had been developed to minimize the number of steps in the assay (Myhrvold et al., 2018; Joung et al., 2020), which would bring greater convenience to on-site detection.
The versatile readout styles of the Card platform make it not only suitable for high-throughput professional veterinary diagnostic laboratories (microplate reader format) but also for on-site point-of-care (naked eyes and lateral flow formats) diagnosis of A. pleuropneumoniae. In particular, Card is rapid (results in less than 1 h), which facilitates its use at the point of care, and does not require the use of more expensive instruments that are required for conventional PCR and qPCR. However, the reagent cost per test of the Card platform is higher than the commonly used PCR and qPCR (Rauch et al., 2021), two of the least expensive testing modalities currently available. This bottleneck may be gradually solved with the increasing popularity.
We conclude that the Card platform shows great promise as a rapid, point-of-care diagnostic for A. pleuropneumoniae.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
WZ and GL designed the experiments. TL, LW, JZ, WZ, and GL wrote the manuscript. TL, LW, and JZ performed the experiments. TL, LW, JZ, HL, YZ, and CW collected and processed materials. TL, LW, JZ, HL, and YZ analyzed the data. PL and SL revised the manuscript. All authors read and approved the submitted version.
This work was funded by the National Natural Science Foundation of China (Nos. 31672575 and 32172853) and the Central Public-interest Scientific Institution Basal Research Fund (No. 1610302022001). PL was supported by the UK Biotechnology and Biological Sciences Research Council (BB/S019901/1 and BB/K020765/1).
We thank Shenye Yu for their technical assistance. The genomic DNA of closely related species and reference strains of serovars 11–19 were a kind gift from Yanwen Li of Imperial College London, Tao He of the Veterinary Research Institute, Jiangsu Academy of Agricultural Sciences, and Lu Li of Huazhong Agricultural University, China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alon, D. M., Hak, H., Bornstein, M., Pines, G., and Spiegelman, Z. (2021). Differential Detection of the Tobamoviruses Tomato Mosaic Virus (ToMV) and Tomato Brown Rugose Fruit Virus (ToBRFV) Using CRISPR-Cas12a. Plants 10:1256. doi: 10.3390/plants10061256
Andresen, L. O., Klausen, J., Barfod, K., and Sorensen, V. (2002). Detection of antibodies to Actinobacillus pleuropneumoniae serotype 12 in pig serum using a blocking enzyme-linked immunosorbent assay. Vet. Microbiol. 89, 61–67. doi: 10.1016/s0378-1135(02)00156-6
Burgio, G. (2018). Redefining mouse transgenesis with CRISPR/Cas9 genome editing technology. Genome Biol. 19:27. doi: 10.1186/s13059-018-1409-1
Cao, Y., Wu, J., Pang, B., Zhang, H., and Le, X. C. (2021). CRISPR/Cas12a-mediated gold nanoparticle aggregation for colorimetric detection of SARS-CoV-2. Chem. Commun. 57, 6871–6874. doi: 10.1039/d1cc02546e
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. doi: 10.1126/science.aar6245
Chiers, K., Donne, E., Van Overbeke, I., Ducatelle, R., and Haesebrouck, F. (2002). Evaluation of serology, bacteriological isolation and polymerase chain reaction for the detection of pigs carrying Actinobacillus pleuropneumoniae in the upper respiratory tract after experimental infection. Vet. Microbiol. 88, 385–392. doi: 10.1016/s0378-1135(02)00150-5
da Costa, M. M., Klein, C. S., Balestrin, R., Schrank, A., Piffer, I. A., da Silva, S. C., et al. (2004). Evaluation of PCR based on gene apxIVA associated with 16S rDNA sequencing for the identification of Actinobacillus pleuropneumoniae and related species. Curr. Microbiol. 48, 189–195. doi: 10.1007/s00284-003-4162-x
East-Seletsky, A., O’Connell, M. R., Knight, S. C., Burstein, D., Cate, J. H., Tjian, R., et al. (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. doi: 10.1038/nature19802
Feng, Y., Zhang, H., Wu, Z., Wang, S., Cao, M., Hu, D., et al. (2014). Streptococcus suis infection: An emerging/reemerging challenge of bacterial infectious diseases? Virulence 5, 477–497. doi: 10.4161/viru.28595
Fittipaldi, N., Broes, A., Harel, J., Kobisch, M., and Gottschalk, M. (2003). Evaluation and field validation of PCR tests for detection of Actinobacillus pleuropneumoniae in subclinically infected pigs. J. Clin. Microbiol. 41, 5085–5093. doi: 10.1128/JCM.41.11.5085-5093.2003
Fozouni, P., Son, S., Diaz de Leon Derby, M., Knott, G. J., Gray, C. N., D’Ambrosio, M. V., et al. (2021). Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184, 323–333.e9. doi: 10.1016/j.cell.2020.12.001
Frandoloso, R., Martinez-Martinez, S., Gutierrez-Martin, C. B., and Rodriguez-Ferri, E. F. (2012). Haemophilus parasuis serovar 5 Nagasaki strain adheres and invades PK-15 cells. Vet. Microbiol. 154, 347–352. doi: 10.1016/j.vetmic.2011.07.022
Frey, J. (1995). Virulence in Actinobacillus pleuropneumoniae and RTX toxins. Trends Microbiol. 3, 257–261.
Gimenez-Lirola, L. G., Jiang, Y. H., Sun, D., Hoang, H., Yoon, K. J., Halbur, P. G., et al. (2014). Simultaneous detection of antibodies against Apx toxins ApxI, ApxII, ApxIII, and ApxIV in pigs with known and unknown Actinobacillus pleuropneumoniae exposure using a multiplexing liquid array platform. Clin. Vaccine Immunol. 21, 85–95. doi: 10.1128/CVI.00451-13
Gong, J., Zhang, G., Wang, W., Liang, L., Li, Q., Liu, M., et al. (2021). A simple and rapid diagnostic method for 13 types of high-risk human papillomavirus (HR-HPV) detection using CRISPR-Cas12a technology. Sci. Rep. 11:12800. doi: 10.1038/s41598-021-92329-2
Gootenberg, J. S., Abudayyeh, O. O., Kellner, M. J., Joung, J., Collins, J. J., and Zhang, F. (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. doi: 10.1126/science.aaq0179
Gram, T., Ahrens, P., and Nielsen, J. P. (1996). Evaluation of a PCR for detection of Actinobacillus pleuropneumoniae in mixed bacterial cultures from tonsils. Vet. Microbiol. 51, 95–104. doi: 10.1016/0378-1135(96)00013-2
Hricinova, M., Holoda, E., Mudronova, D., and Ondrasovicova, S. (2010). Multiplex PCR assay for detection of Actinobacillus pleuropneumoniae, Pasteurella multocida and Haemophilus parasuis in lungs of pigs from a slaughterhouse. Folia Microbiol. 55, 635–640. doi: 10.1007/s12223-010-0103-9
Jiang, Y., Chen, B., Duan, C., Sun, B., Yang, J., and Yang, S. (2015). Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514. doi: 10.1128/AEM.04023-14
Joung, J., Ladha, A., Saito, M., Kim, N. G., Woolley, A. E., Segel, M., et al. (2020). Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N. Engl. J. Med. 383, 1492–1494. doi: 10.1056/NEJMc2026172
Klein, C. S., Piffer, I. A., Ceroni da Silva, S., Schrank, A., Favero, M. B., and Schrank, I. S. (2003). Detection of Actinobacillus pleuropneumoniae by PCR on field strains from healthy and diseased pigs. Curr. Microbiol. 46, 443–447. doi: 10.1007/s00284-002-3890-7
Koonin, E. V., and Makarova, K. S. (2019). Origins and evolution of CRISPR-Cas systems. Philos. Trans R. Soc. Lond. B Biol. Sci. 374:20180087. doi: 10.1098/rstb.2018.0087
Li, G., Xie, F., Zhang, Y., and Wang, C. (2012). Draft genome sequence of Actinobacillus pleuropneumoniae serotype 7 strain S-8. J. Bacteriol. 194, 6606–6607. doi: 10.1128/JB.01650-12
Li, R., Wang, J., Liu, L., Zhang, R., Hao, X., Han, Q., et al. (2019). Direct detection of Actinobacillus pleuropneumoniae in swine lungs and tonsils by real-time recombinase polymerase amplification assay. Mol. Cell. Probes 45, 14–18. doi: 10.1016/j.mcp.2019.03.007
Li, S. Y., Cheng, Q. X., Wang, J. M., Li, X. Y., Zhang, Z. L., Gao, S., et al. (2018). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4:20. doi: 10.1038/s41421-018-0028-z
Liu, S., Huang, M., Xu, Y., Kang, J., Ye, S., Liu, S., et al. (2021). CRISPR/Cas12a technology combined with RT-ERA for rapid and portable SARS-CoV-2 detection. Virol. Sin. 36, 1083–1087. doi: 10.1007/s12250-021-00406-7
Loera-Muro, A., and Angulo, C. (2018). New trends in innovative vaccine development against Actinobacillus pleuropneumoniae. Vet. Microbiol. 217, 66–75. doi: 10.1016/j.vetmic.2018.02.028
Losinger, W. C. (2005). Economic impacts of reduced pork production associated with the diagnosis of Actinobacillus pleuropneumoniae on grower/finisher swine operations in the United States. Prev. Vet. Med. 68, 181–193. doi: 10.1016/j.prevetmed.2004.12.004
Lu, Q. S. M., and Tian, L. (2022). An efficient and specific CRISPR-Cas9 genome editing system targeting soybean phytoene desaturase genes. BMC Biotechnol. 22:7. doi: 10.1186/s12896-022-00737-7
Lu, S., Li, F., Chen, Q., Wu, J., Duan, J., Lei, X., et al. (2020). Rapid detection of African swine fever virus using Cas12a-based portable paper diagnostics. Cell Discov. 6:18. doi: 10.1038/s41421-020-0151-5
Ma, L., Peng, L., Yin, L., Liu, G., and Man, S. (2021). CRISPR-Cas12a-Powered Dual-Mode Biosensor for Ultrasensitive and Cross-validating Detection of Pathogenic Bacteria. ACS Sens 6, 2920–2927. doi: 10.1021/acssensors.1c00686
Myhrvold, C., Freije, C. A., Gootenberg, J. S., Abudayyeh, O. O., Metsky, H. C., Durbin, A. F., et al. (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448. doi: 10.1126/science.aas8836
Nielsen, R., van den Bosch, J. F., Plambeck, T., Sorensen, V., and Nielsen, J. P. (2000). Evaluation of an indirect enzyme-linked immunosorbent assay (ELISA) for detection of antibodies to the Apx toxins of Actinobacillus pleuropneumoniae. Vet. Microbiol. 71, 81–87. doi: 10.1016/s0378-1135(99)00157-1
Park, J., Bae, S., and Kim, J. S. (2015). Cas-Designer: A web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31, 4014–4016. doi: 10.1093/bioinformatics/btv537
Rauch, J. N., Valois, E., Solley, S. C., Braig, F., Lach, R. S., Audouard, M., et al. (2021). A Scalable, Easy-to-Deploy Protocol for Cas13-Based Detection of SARS-CoV-2 Genetic Material. J. Clin. Microbiol. 59:e02402–20. doi: 10.1128/JCM.02402-20
Schaller, A., Djordjevic, S. P., Eamens, G. J., Forbes, W. A., Kuhn, R., Kuhnert, P., et al. (2001). Identification and detection of Actinobacillus pleuropneumoniae by PCR based on the gene apxIVA. Vet. Microbiol. 79, 47–62. doi: 10.1016/s0378-1135(00)00345-x
Serrano-Rubio, L. E., Tenorio-Gutierrez, V., Suarez-Guemes, F., Reyes-Cortes, R., Rodriguez-Mendiola, M., Arias-Castro, C., et al. (2008). Identification of Actinobacillus pleuropneumoniae biovars 1 and 2 in pigs using a PCR assay. Mol. Cell. Probes 22, 305–312. doi: 10.1016/j.mcp.2008.09.001
Sidibe, M., Messier, S., Lariviere, S., Gottschalk, M., and Mittal, K. R. (1993). Detection of Actinobacillus pleuropneumoniae in the porcine upper respiratory tract as a complement to serological tests. Can. J. Vet. Res. 57, 204–208.
Stringer, O. W., Bossé, J. T., Lacouture, S., Gottschalk, M., Fodor, L., Angen, O., et al. (2021a). Proposal of Actinobacillus pleuropneumoniae serovar 19, and reformulation of previous multiplex PCRs for capsule-specific typing of all known serovars. Vet. Microbiol. 255:109021. doi: 10.1016/j.vetmic.2021.109021
Stringer, O. W., Bossé, J. T., Lacouture, S., Gottschalk, M., Fodor, L., Angen, O., et al. (2021b). Rapid Detection and Typing of Actinobacillus pleuropneumoniae Serovars Directly From Clinical Samples: Combining FTA® Card Technology With Multiplex PCR. Front. Vet. Sci. 8:728660. doi: 10.3389/fvets.2021.728660
Tobias, T. J., Bouma, A., Klinkenberg, D., Daemen, A. J., Stegeman, J. A., Wagenaar, J. A., et al. (2012). Detection of Actinobacillus pleuropneumoniae in pigs by real-time quantitative PCR for the apxIVA gene. Vet. J. 193, 557–560. doi: 10.1016/j.tvjl.2012.02.004
Watt, A. E., Browning, G. F., Markham, P. F., and Marenda, M. S. (2020). Detection of naturally aerosolized Actinobacillus pleuropneumoniae on pig farms by cyclonic air sampling and qPCR. Vet. Microbiol. 250:108856. doi: 10.1016/j.vetmic.2020.108856
Wei, B., Li, F., Yang, H., Yu, L., Zhao, K., Zhou, R., et al. (2012). Magnetic beads-based enzymatic spectrofluorometric assay for rapid and sensitive detection of antibody against ApxIVA of Actinobacillus pleuropneumoniae. Biosens. Bioelectron. 35, 390–393. doi: 10.1016/j.bios.2012.03.027
Xiao, G., He, X., Zhang, S., Liu, Y., Liang, Z., Liu, H., et al. (2020). Cas12a/Guide RNA-Based Platform for Rapid and Accurate Identification of Major Mycobacterium Species. J. Clin. Microbiol. 58:e01368–19. doi: 10.1128/JCM.01368-19
Xie, F., Li, G., Zhou, L., Zhang, Y., Cui, N., Liu, S., et al. (2017). Attenuated Actinobacillus pleuropneumoniae double-deletion mutant S-8 △clpP/apxIIC confers protection against homologous or heterologous strain challenge. BMC Vet. Res. 13:14. doi: 10.1186/s12917-016-0928-9
Yang, W., Pin, C., Haibing, G., Yang, C., Hui, L., and Qigai, H. (2009). Loop-mediated isothermal amplification targeting the apxIVA gene for detection of Actinobacillus pleuropneumoniae. FEMS Microbiol. Lett. 300, 83–89. doi: 10.1111/j.1574-6968.2009.01779.x
Keywords: CRISPR, Cas12a, crRNA, Actinobacillus pleuropneumoniae, diagnostics
Citation: Luan T, Wang L, Zhao J, Luan H, Zhang Y, Wang C, Langford PR, Liu S, Zhang W and Li G (2022) A CRISPR/Cas12a-assisted rapid detection platform by biosensing the apxIVA of Actinobacillus pleuropneumoniae. Front. Microbiol. 13:928307. doi: 10.3389/fmicb.2022.928307
Received: 25 April 2022; Accepted: 19 August 2022;
Published: 09 September 2022.
Edited by:
Weiwen Zhang, Tianjin University, ChinaReviewed by:
Bo Yu, Institute of Microbiology (CAS), ChinaCopyright © 2022 Luan, Wang, Zhao, Luan, Zhang, Wang, Langford, Liu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Li, bGlnYW5nQGNhYXMuY24=; Wanjiang Zhang, emhhbmd3YW5qaWFuZ0BjYWFzLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.