 Madjid Morsli
Madjid Morsli Jean Philippe Lavigne
Jean Philippe Lavigne Michel Drancourt
Michel Drancourt
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 05 July 2022
Sec. Infectious Agents and Disease
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.926240
This article is part of the Research Topic Advances and Challenges in the Detection and Treatment of Pathogenic Microorganisms in Infectious Disease Control View all 25 articles
Current routine diagnosis of community-acquired meningitis (CAM) by multiplex real-time polymerase chain reaction (RT-PCR) is limited in the number of tested pathogens and their full characterisation, requiring additional in vitro investigations to disclose genotype and antimicrobial susceptibility. We reviewed 51 studies published through December 2021 reporting metagenomic next generation sequencing (mNGS) directly applied to the cerebrospinal fluid (CSF). This approach, potentially circumventing the above-mentioned limitations, indicated 1,248 investigated patients, and 617 patients dually investigated by routine diagnosis and mNGS, in whom 116 microbes were detected, including 50 by mNGS only, nine by routine methods only, and 57 by both routine methods and mNGS. Of 217 discordant CSF findings, 103 CSF samples were documented by mNGS only, 87 CSF samples by routine methods only, and 27 CSF samples in which the pathogen identified by mNGS was different than that found using routine methods. Overall, mNGS allowed for diagnosis and genomic surveillance of CAM causative pathogens in real-time, with a cost which is competitive with current routine multiplex RT-PCR. mNGS could be implemented at point-of-care (POC) laboratories as a part of routine investigations to improve the diagnosis and molecular epidemiology of CAM, particularly in the event of failure of routine assays.
Community-acquired meningitis (CAM), a life-threatening condition with 40% mortality (Beaman, 2018), warrants a rapid diagnosis, possibly within 2 h at a point-of-care (POC) laboratory, providing identification of the causative pathogen among a repertoire of fewer than 20 pathogens (Leber et al., 2016; Launes et al., 2017; Soucek et al., 2019; Vincent et al., 2020), those most frequently encountered in the general population of patients, and supporting the initial medical decisions for hospitalisation and antibiotic treatment (Leber et al., 2016; Soucek et al., 2019; Tansarli and Chapin, 2020). Such a POC diagnosis, which is routinely made using nested or semi-nested polymerase chain reactions (PCRs), is prone to cross-contamination resulting in false-positive diagnoses (Boudet et al., 2019; Vincent et al., 2020). Current POC diagnosis also relies on the detection of a limited number of pathogens, and may thus miss microorganisms acting as life-threatening pathogens in selected populations, such as Cryptococcus in HIV-infected patients, in whom Cryptococcus most frequently causes fungal meningitis, with 223,000 new cases each year and 81% mortality (Ahn et al., 2017; Xing et al., 2019) and amoebas, such as Acanthamoeba (Greninger et al., 2015; Behera et al., 2016). The precise characterisation of such detected pathogens, including genotyping and anti-infectious susceptibility profiling, are pieces of medical information which are relevant for the immediate medical management of patients and contacts, and source tracing.
Metagenomic next generation sequencing (mNGS) directly applied to cerebrospinal fluid (CSF) emerged less than 10 years ago, in the specific context of healthcare-associated meningitis (Naccache et al., 2015), as an alternative to the laboratory diagnosis of CAM, potentially surpassing above-mentioned limitations of the current multiplex PCR approach, as discussed below (Table 1). However, no review papers have been published including recommendations and the routine diagnostic implications of this approach. To update existing knowledge about the direct diagnosis of CAM by metagenomics, we conducted a literature search for studies applying mNGS directly to CSF for the diagnosis of CAM.
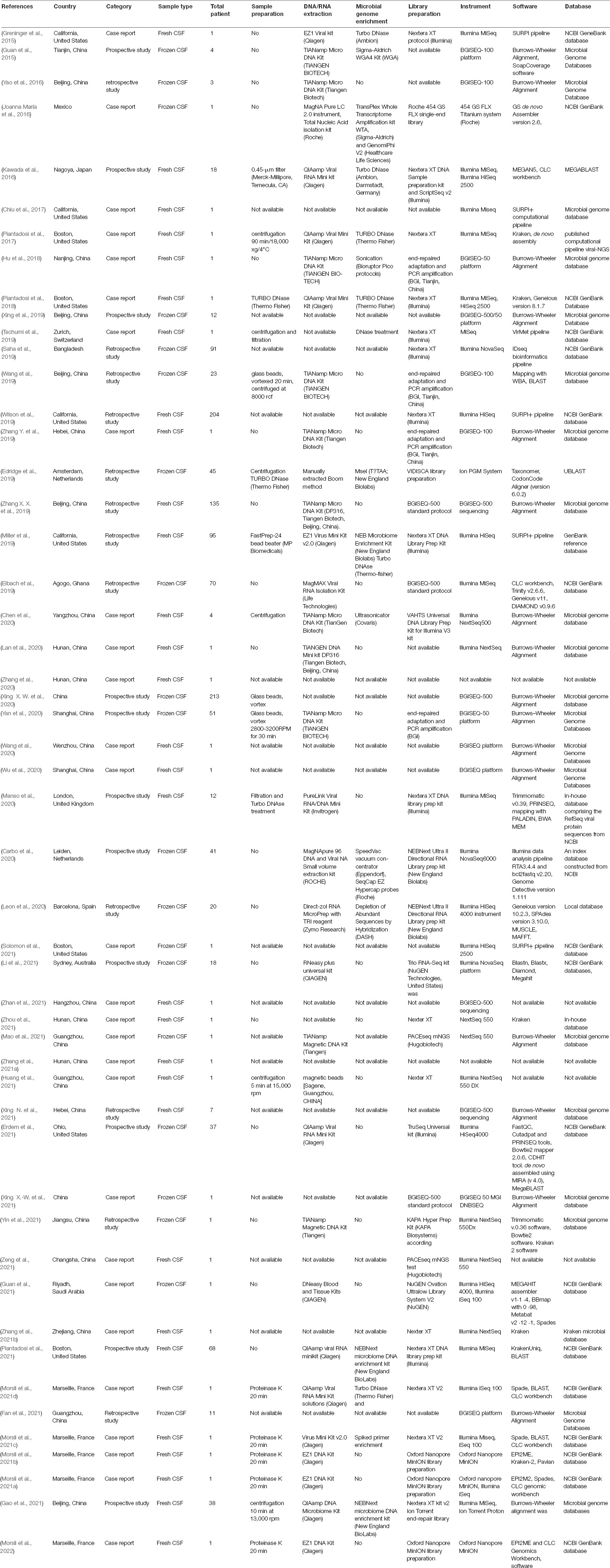
Table 1. Metagenomics pipelines.
This study was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (Moher et al., 2015). A systematic bibliography search was conducted on PubMed, Google Scholar, Web of Science, Microsoft Academic, Crossref and Semantic Scholar databases for studies published in English between January 1, 2015 and December 31, 2021 and related to the diagnosis of CAM by metagenomic NGS. Duplicate studies were removed in a first screening and the remaining papers were further screened based on title and abstract, according to the eligibility criteria. After full-text screening, only studies that met the inclusion criteria were included in this review, using the keywords: “metagenomic,” “diagnosis,” “meningitis,” “encephalitis,” “next generation sequencing,” and “cerebrospinal fluid.” These keywords were used in combination to perform an exhaustive search, as presented in Figure 1.
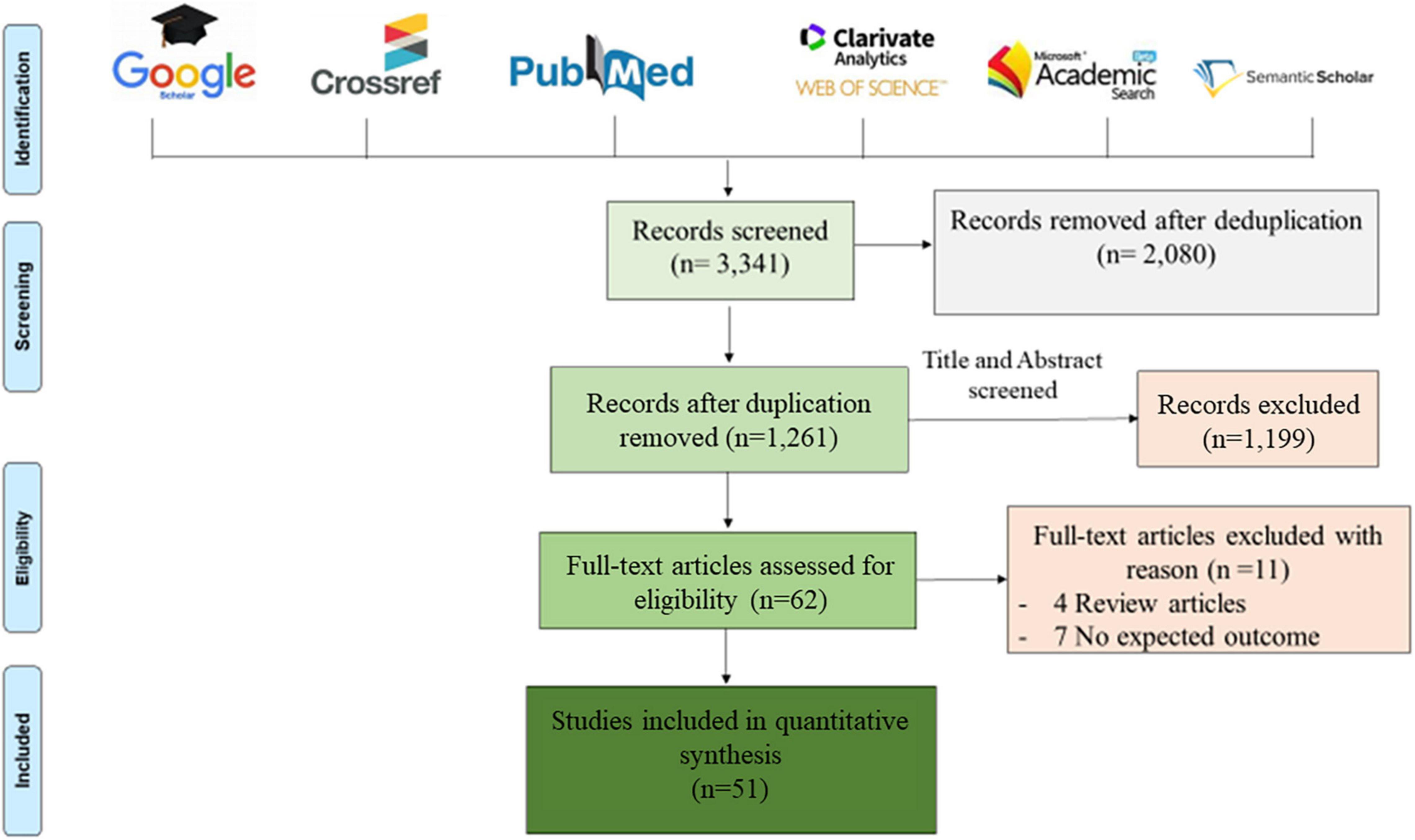
Figure 1. Prisma diagram of eligible articles recorded after removing duplications. Six bibliographic databases were reviewed using the following key words: “metagenomic,” “diagnosis,” “meningitis,” “encephalitis,” “next generation sequencing,” and “cerebrospinal fluid,” used alone and/or in combination. Only 51 articles were eligible for this review, after screening according to the inclusion criteria described above.
Studies with the following criteria were included in this review: Studies using mNGS-CSF for: (1) case reports; (2) prospective series; (3) retrospective series for non-routinely detected pathogens” (Table 1 and Supplementary Data). Review articles, studies performed on animals, non-clinical studies, and benchmarking studies were not included. Data extracted from selected studies included: first author name and year of publication, authors’ country, nucleic acid extraction method including commercially available kits, mNGS platform and sequencer instrument, pipeline data analysis and software, and reference database used for pathogen genome detection of identified microorganisms. Included were the total number and discordance/agreement of mNGS data with data yielded by routinely used methods, including molecular tests and culture. These data were reviewed and extracted after validation of the inclusion criteria by the authors.
Bibliographic searches identified 3,341 articles and, following the Prisma diagram (Figure 1), 2,080 articles were removed after deduplication and 1,199 additional articles were removed by title and abstract screening. From the 62 remaining articles, 11 articles were removed after full-text reading, including four review articles and seven non-relevant articles, resulting in 51 articles analysed in this review.
In 2015, the first reported application of mNGS directly on CSF (and brain biopsy) enabled the post-mortem diagnosis of Astrovirus-related encephalitis in an immunocompromised patient (Naccache et al., 2015). A 7-year review of publications indicated only nine studies published between 2015 and 2018, ten in 2019 and 42/51 (82%) publications between 2019 and 2021, including 22 (43%) publications in 2021 (Figure 2). As for the geographic origin of published studies, 28 (56%) were published in Asia, including 25 (49%) from Chinese laboratories (Table 1), one each from Saudi Arabia (Guan et al., 2021), Bangladesh (Saha et al., 2019), and Japan (Kawada et al., 2016) (6.1%); nine in the United States (Table 1); one in Mexico (Joanna María et al., 2016); five in France (10%) (Tabel 1); two in Netherlands (Edridge et al., 2019; Carbo et al., 2020); and one each in Switzerland, the United Kingdom, and Spain (Tschumi et al., 2019; Leon et al., 2020; Manso et al., 2020). One study was also published in Guinea and another in Australia (Eibach et al., 2019; Li et al., 2021; Figure 2).
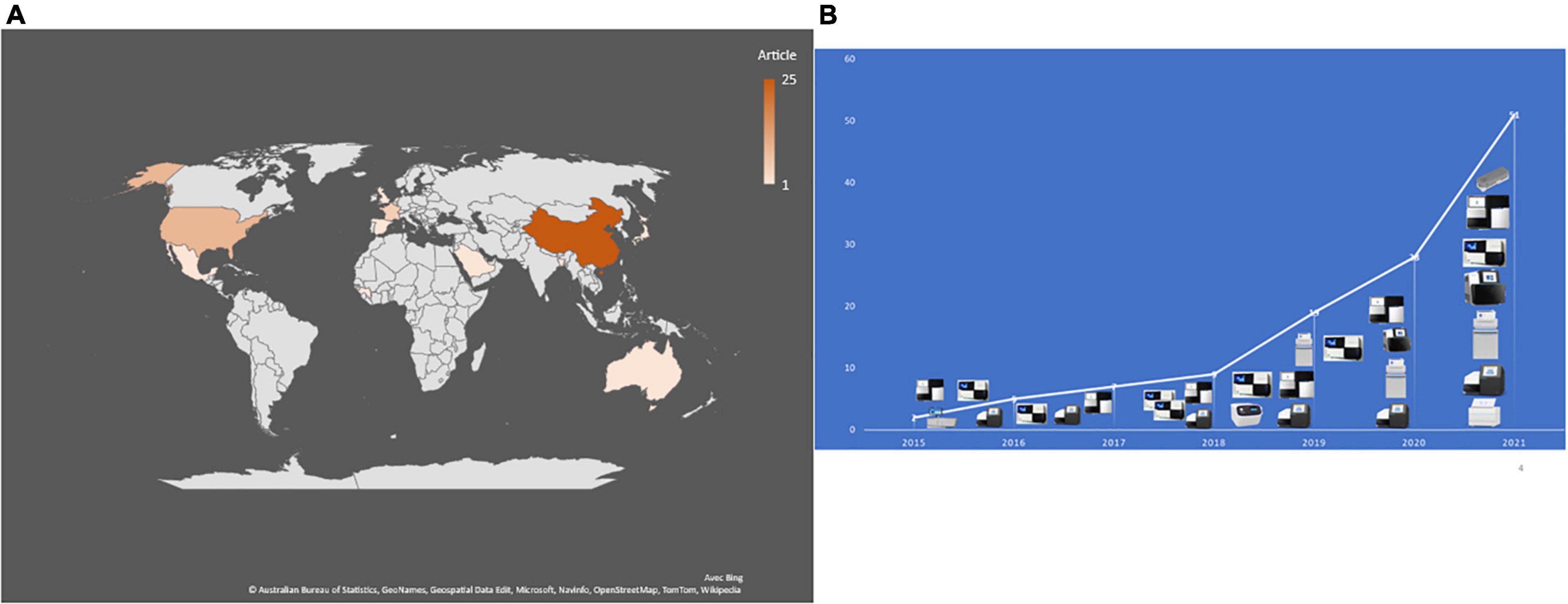
Figure 2. (A) Number and geographical distribution of publication records after duplication removed. (B) Publication chronology according to the used sequencing technologies.
Only CSF investigations [apart from one post-mortem brain biopsy in the founding study (Naccache et al., 2015)] were reviewed in this study. Depending on the prospective or retrospective nature of the investigations, fresh CSF was used in 63% of studies and frozen CFS in 37% of studies. DNA and RNA were manually extracted in 44% of studies, automatic extraction was performed in 20%, and 36% of studies did not report on the nucleic acid extraction method. The sample pre-treatment reported in 18/51 (35%) studies included centrifugation (∼15,000 g), filtration and vortexing in 12 studies, DNase treatment in four studies (Piantadosi et al., 2018; Edridge et al., 2019; Manso et al., 2020; Morsli et al., 2021d) and proteinase K treatment in five studies (Table 1; Guan et al., 2015; Miller et al., 2019; Carbo et al., 2020; Manso et al., 2020; Morsli et al., 2021a,c; Piantadosi et al., 2021), while no pre-treatment was performed in 15 studies and no such information was provided in 16 studies (Table 1). Extracted nucleic acids (retro-transcribed RNA and DNA) were always quantified using either a Qubit fluorometer and a Qubit DNA and RNA High Sensitivity Assay Kit (Life Technology, United States) (Table 1; Guan et al., 2015; Miller et al., 2019; Carbo et al., 2020; Manso et al., 2020; Morsli et al., 2021a,c; Piantadosi et al., 2021), or NanoDrop spectrophotometer (Thermo Fisher Scientific, United States) (Li et al., 2021). Optimisation in the human-to-microbial ratio of DNA was achieved by intermediate microbial genome enrichment or human genome depletion (Table 1). DNase treatment was used to reduce human DNA in extracted RNA for mNGS investigations of RNA-viruses (Table 1; Guan et al., 2015; Miller et al., 2019; Carbo et al., 2020; Manso et al., 2020; Morsli et al., 2021a,c; Piantadosi et al., 2021), and bead-based capture kits were also used to remove human DNA (Miller et al., 2019; Leon et al., 2020; Gao et al., 2021; Piantadosi et al., 2021). Further, microbial genomes could be enriched by specific primer amplification for whole genome investigations (Table 1). cDNA and double-stranded synthesis were needed prior to library preparation when RNA was investigated (Table 1), and in the case of RNA genome enrichment, an RT-one-step protocol was followed by reverse-transcription and pathogen genome amplification (Morsli et al., 2021c). In two studies, DNA was mechanically broken prior to library construction (Hu et al., 2018; Zhang X. X. et al., 2019).
Three main protocols were used for mNGS library preparation (Table 1 and Figure 3). Illumina pair-end protocols and reagents were used for mNGS library preparation in 19/51 (37%) studies, including Nextera XT DNA Library Prep Kit in 16/51 (31%) and one each with the TruSeq Universal kit (Erdem et al., 2021) and VAHTS Universal DNA Library Prep Kit (Chen et al., 2020). Standard BGISEQ pair-end protocols were used in 6/51 studies. Two studies used the PACEseq mNGS test (Hugobiotech, Beijing, China) (Mao et al., 2021; Zeng et al., 2021), two studies used VIDISCA and Ion Torrent end-repair library (Edridge et al., 2019; Gao et al., 2021), and one study each used the NEBNext Ultra II Directional RNA Library prep kit (New England Biolabs, Ipswich, MA, United States) (Carbo et al., 2020; Leon et al., 2020), the KAPA Hyper Prep Kit (Kapa Biosystem, Potters Bar, United Kingdom) (Yin et al., 2021), the NuGEN Ovation Ultralow Library System V2 kit (NuGEN Technologies, United States) (Guan et al., 2015) the Trio RNA-Seq kit (NuGEN Technologies) (Li et al., 2021), and the Roche 454 GS FLX single-end library (Roche Diagnostics, Branford, CT, United States) (Joanna María et al., 2016). In three studies, the mNGS library was constructed following the single-end Oxford Nanopore library preparation protocol (Morsli et al., 2021a,b, 2022). Finally, the library preparation protocol and kits were not reported in 14 studies (Table 1 and Figure 3). mNGS libraries were sequenced using Illumina sequencers in 30/51 (59%) studies, MiSeq platform in 11/30, HiSeq platform in 8/30, Nextseq platform in 6/30, iSeq 100 platform in 4/30, and 3/30 studies used the NovaSeq platform. BGISEQ platforms were used in 12/51 (24%) of studies (Table 1), including BGISEQ-50, 100, and 500 platforms, while three studies used the Roche 454 GS FLX Titanium system platform, Ion PGM System platforms and Ion Torrent Proton Sequencer (Life Technologies, United States), respectively (Joanna María et al., 2016; Edridge et al., 2019; Gao et al., 2021). Real-time sequencing with the Oxford Nanopore MinION sequencer was used in three studies (Morsli et al., 2021a,b, 2022) and no information about the sequencer was reported in five studies (Table 1). The use of Nanopore technologies, i.e., single-long-read sequencing platforms combined with direct read blasting against the NCBI GenBank database using EPI2ME online software1 or against an internal database using in-house pipelines, allowed for pathogen identification within the first minutes of sequencing (Morsli et al., 2021a). Furthermore, all additional pieces of information such as bacteria profiling and genotyping, was obtained in less than 6 h (Morsli et al., 2021a,b, 2022).
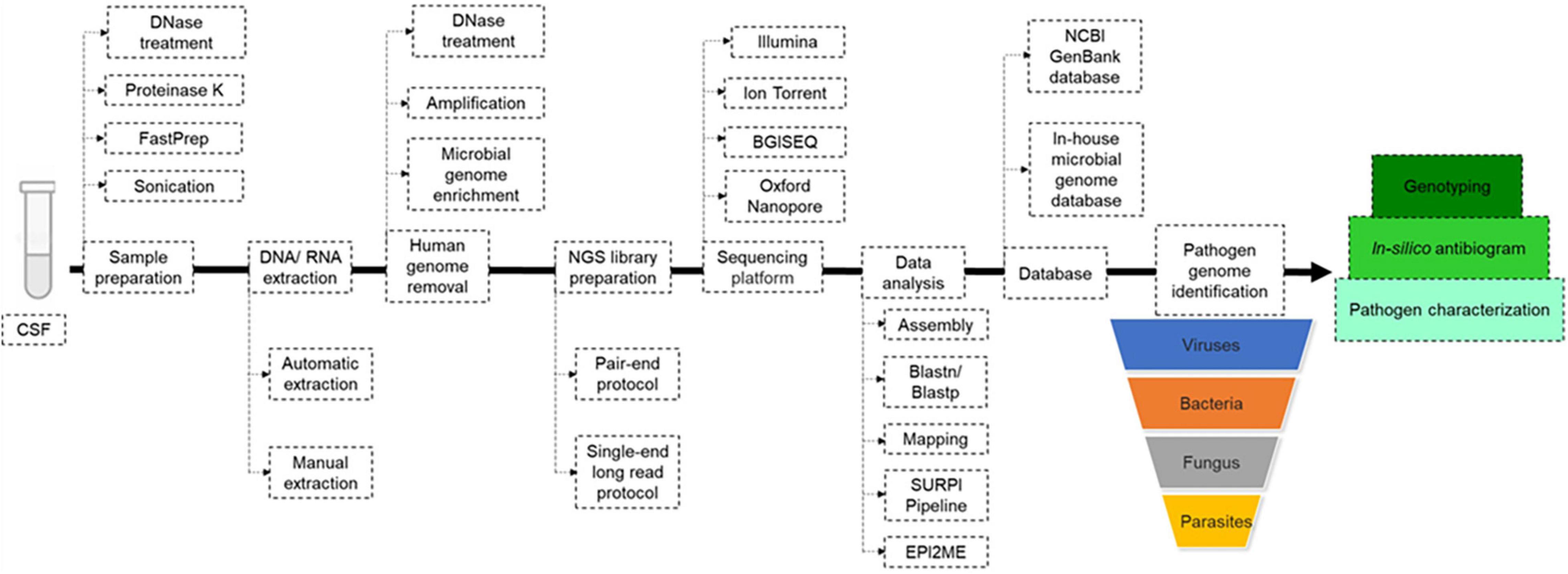
Figure 3. Metagenomic next generation sequencing (mNGS) workflow. Upon reception of the cerebrospinal fluid (CSF), mechanical and enzymatic sample pre-treatment was performed before manual or automatic DNA/RNA extraction. Before mNGS library preparation, the human genome was removed with or without microbial genome enrichment, then mNGS libraries were prepared following pair-end or single-end long read protocol, depending on the sequencing platform. Pathogen genome identified directly by blast against NCBI GenBank and/or local microbial genome database downloaded from GenBank, using in-house pipelines. In-silico antibiogram and genotyping were performed depending on the nature of the identified pathogenic agent.
Next generation sequencing data quality was controlled using FastQC (Erdem et al., 2021; Yin et al., 2021), and the human genome could be removed by mNGS reads mapping to the Homo sapiens reference genome (hg19) using Burrows-Wheeler Alignment (BWA) in 40% of studies. Non-mapped reads could be assembled using Spades and de novo assembly or directly against the reference genome using the BWA and CLC Genomic workbench (Table 1). For an exhaustive pathogen genome investigation, mNGS data were blasted [BLAST: Basic Local Alignment Search Tool (nih.gov)] against the NCBI GenBank database or against an in-house microbial genome database, regardless of the sequencing technology, yet with different timing as for Illumina and Nanopore technologies, as mentioned above (Table 1). The sequence-based ultra-rapid pathogen identification computational pipeline (SURPI) was a powerful method for pathogen identification, combining blast and mapping of both pathogen genome and protein encoding sequences directly from mNGS outputs (Greninger et al., 2015; Miller et al., 2019). When only one specific aetiology was investigated, a reference database constructed with such specific pathogen genomes and proteins could be used for its accurate identification (Piantadosi et al., 2017; Manso et al., 2020). Depending on the previous pathogen identification, a specific pathogen genome was extracted directly as consensus sequences in FASTA files, after mapping to the reference genome directly from CLC and BWA software for further analysis (Table 1 and Figure 3). Virus genotyping based on hit-blast identified strains could be confirmed by phylogenetic analysis based on sequence similarity level, while bacteria genotyping could be predicted online by Multi-Locus Sequence Typing online platform (Figure 3). Also, the in silico antibiotic resistance pattern could be predicted by aligning bacterial genome sequences against online databases of antibiotic-resistance encoding genes (Table 1; Guan et al., 2015; Miller et al., 2019; Carbo et al., 2020; Manso et al., 2020; Morsli et al., 2021a,c; Piantadosi et al., 2021), mostly represented by ResFinder on the Centre for Genomic Epidemiology server (Bortolaia et al., 2020; Morsli et al., 2021a,b, 2022).
Of the 51 studies analysed, a total of 1,248 CSF samples collected from CAM patients were investigated in parallel by mNGS reference and routine molecular diagnosis and culture. Routine methods and mNGS yielded concordant results in 1,031 (82.6%) CSF samples, including no pathogen documentation in 566 CSF samples and concordant identification in 465 CSF samples. Discordant results were observed in 217 (17.4%) CSF samples, including 87 CSF samples documented by routine methods only, 103 CSF samples documented by mNGS only, and 27 CSF samples in which documented microorganisms differed from routine methods and mNGS. Altogether, the 116 detected microorganisms included 63 bacteria, 38 viruses, 10 fungi, and five parasites. mNGS detected 106/116 (91.4%) different microorganisms, including 50 (43.1%) detected by mNGS only, whereas 56 microorganisms were detected by both routine methods and mNGS. In addition, five microorganisms were detected by routine molecular diagnosis only, four microorganisms by culture only and one Enterococcus gallinarum strain was diagnosed by routine molecular diagnosis and culture (Figure 4). Further, there was concordant documentation of 56 different pathogens, including 30 bacterial pathogens, 19 viral pathogens, six fungi and one Balamuthia mandrillaris (B. mandrillaris) as the only amoeba detected in a CSF sample (Table 1 and Figure 4). Of the 217 CSF samples in which mNGS investigation yielded results discordant with the routine method, mNGS conclusively found a pathogen in 38/103 CSF samples in which routine investigations found no pathogen, as reported in 25/51 studies: mNGS over-detected RNA viruses, including Human rhinovirus and Human coronavirus (Li et al., 2021), Coxsackievirus 9 and Mumps virus (Kawada et al., 2016), Saint-Louis Encephalitis virus (Chiu et al., 2017), Powassan Virus (Piantadosi et al., 2018), Toscana virus (Tschumi et al., 2019), Jamestown Canyon virus (Solomon et al., 2021), Enterovirus A71 (Leon et al., 2020), and Hepatitis E virus (Carbo et al., 2020), while only HSV-1, HHV-6, and EBV were identified as DNA viruses (Zhang Y. et al., 2019; Carbo et al., 2020). Bacterial pathogens documented in 11% of these cases included Listeria monocytogenes in four cases (Yao et al., 2016; Lan et al., 2020), Ureaplasma parvum in three cases (Wang et al., 2020; Xing X.-W. et al., 2021; Zhan et al., 2021) and one case each was detected of Klebsiella pneumoniae (Zeng et al., 2021), Pasteurella multocida (Morsli et al., 2022), Enterococcus faecalis (Zhang et al., 2021a), Nocardia farcinica (Zhang et al., 2021b), Streptococcus suis (Zhang et al., 2020), and Psychrobacter sp. (Joanna María et al., 2016). Toxoplasma gondii, B. mandrillaris, and Naegleria fowleri parasite species were identified in four patients (N. fowleri in two patients) in whom routine diagnostic methods completely missed N. fowleri (Hu et al., 2018; Wu et al., 2020; Guan et al., 2021; Huang et al., 2021), while Coccidioides posadasii was the single fungal infection exclusively detected by mNGS in one patient with no previously documented infectious meningitis (Mao et al., 2021; Supplementary Data; Figure 4).
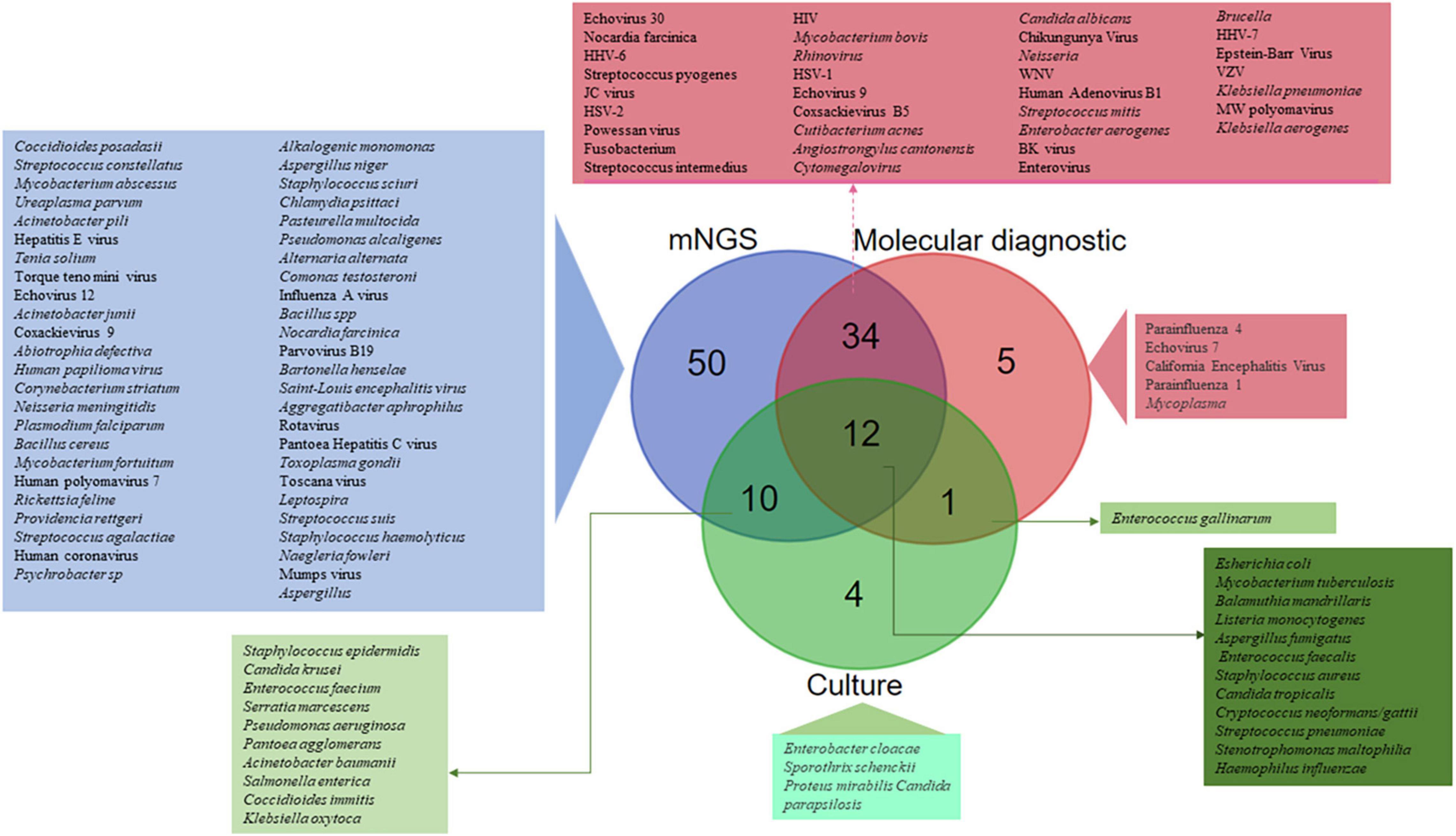
Figure 4. SAM pathogens identified by routine molecular diagnosis, culture, and metagenomic next generation sequencing (mNGS). A total of 116 different microorganisms were identified, including 50 (43.1%) only by mNGS, 43 by both routine molecular diagnosis and mNGS, 12 by culture, mNGS and routine molecular diagnosis, 10 by culture and mNGS, five only by routine molecular diagnosis, four only by culture, and one only by culture and routine molecular diagnosis.
A review of published data indicated that in 4/217 cases, mNGS failed to detect a pathogen when the pathogen had been detected by real-time polymerase chain reaction (RT-PCR) with a Ct > 37, mainly due to a low pathogen inoculum (Miller et al., 2019), as well as for a pathogen detected by immunodetection assay (Xing X.-W. et al., 2021). These limitations could be due to:
1) Storage conditions: storage of the CSF at temperature above -80°C may cause nucleic acid molecule damage. Moreover, lack of proper storage in the presence of RNase inhibitors, will likely result in RNA viruses undergoing some degradation.
2) Limited CSF volume affecting the DNA quantity used for NGS library preparation (Miller et al., 2019), especially for the Oxford Nanopore libraries, which require 1 μg of DNA (Morsli et al., 2021a,b).
3) A CSF leukocyte count (>103/mL) increasing the ratio of human genome, which required microbial genome enrichment (Guan et al., 2015; Miller et al., 2019; Carbo et al., 2020; Manso et al., 2020; Morsli et al., 2021a,c; Piantadosi et al., 2021), and human genome depletion (Table 1). DNase treatment usually used to reduce human DNA in the extracted CSF can, therefore, generate false negative results, due to the degradation of the DNA pathogens (Edridge et al., 2019). Alternatively, one limitation is related to the elimination of human DNA prior tosequencing, such as CSF pathogens whose genome is partially or totally integrated into the human genome, such as the case of HHV-6 (Edridge et al., 2019). Library preparation and cross-contamination removal during sample preparation is essential to avoid any confusion of detection and interpretation, especially when working on several samples in the same run (Table 1; Guan et al., 2015; Miller et al., 2019; Carbo et al., 2020; Manso et al., 2020; Morsli et al., 2021a,c; Piantadosi et al., 2021). Depth, coverage, and sequence quality may be influenced by the sequencing platforms, as well as the kit used for library preparation, which is relative to the initial pathogen load in the CSF (Table 1; Guan et al., 2015; Miller et al., 2019; Carbo et al., 2020; Manso et al., 2020; Morsli et al., 2021a,c; Piantadosi et al., 2021), depending on the number of sequencing cycles for paired-end sequencing and the read size for single-end long read sequencing. Assembly and data analysis mainly depend on the quantity and quality of reads generated by sequencers (Morsli et al., 2021a,c,d). Pathogen identification is mainly based on in-house databases constructed with a limited number of pathogen genomes and species downloaded from the NCBI GenBank (Table 1) used for blast and mapping, which needs regular updating for the exhaustive identification of all possible microbial genomes detected in the CSF sample (Table 1). Performance analysis requires powerful software and pipelines to blast all mNGS data against the GenBank database, which is limited by cost and accessibility, such as the EPI2ME software used for real-time analysis of sequencing data generated by Nanopore Platforms (Morsli et al., 2021a,b, 2022).
Direct mNGS appears to be a relevant alternative diagnostic approach to simplex and multiplex RT-PCRs for the POC diagnosis of CAM. mNGS adds one-shot pieces of medically relevant information, including genotyping and antimicrobial susceptibility profile, in comparison to current PCR-based methods (Morsli et al., 2021a,b; Zhou et al., 2021). The suitability of this new approach should be evaluated in all laboratories already providing molecular detection of pathogen-genome sequences in the CSF of patients with meningitis, and as a new technique in new POC laboratories bypassing previous multiplex RT-PCR (Boudet et al., 2019; Vincent et al., 2020; Morsli et al., 2022). The basis for such an evaluation includes the flow of samples and cost of mNGS relative to multiplex PCR. Indeed, mNGS also provides an antimicrobial susceptibility profile to guide medical management of the patient (Morsli et al., 2021a,b, 2022) and genotyping to detect outbreaks and to guide source tracing compared to multiplex PCRs (Broberg et al., 2018). Such genotyping information may be medically relevant and associated with a particular prognosis of CAM (case of a few Enteroviruses) and certainly relevant to source tracing in the context of clustered cases and outbreaks. Compared to the limited number of the pathogens investigated by routine multiplex RT-PCR, mNGS identified 106 different microorganisms in CSFs from patients with meningitis, i.e., covering 91.4% of total microorganisms identified here. This allowed the detection of pathogen genomes in 38 non-routinely documented CSFs. In addition, mNGS can operate within infinitely greater open databases than those currently supporting available multiplex RT-PCR assays, which rely on fewer than 30 such entries, whereas genome sequence databases such as NCBI consist of more than 83,124 complete microbial genomes (including bacteria, viruses, fungi, and parasites), offering opportunities for the diagnosis of rare pathogens, as reviewed in this study.
Finally, a few current limitations of the mNGS approach are opportunities for improvements from the perspective of routine use. Most enzymes used in the above-described sequencing protocols were issued from cloning in competent bacteria such as Escherichia coli (E. coli) (Morsli et al., 2021a), meaning potential contamination by E. coli DNA and a risk of a false-positive diagnosis of E. coli meningitis. As an illustration, Nanopore sequencing results regularly consisted of E. coli DNA resulting from the recommended internal library control, and Shigella and T4 phage reads issued from repair and ligation enzymes (Morsli et al., 2021a,b, 2022). This limit could be overcome by replacing the E. coli control and using the human genome as an internal control, in line with our own practice. The quantity of data generated by sequencing was sometimes insufficient to support an accurate interpretation, requiring additional enrichment and human genome depletion steps to improve the sensitivity and specificity of detection of CAM causative agents (Miller et al., 2019; Carbo et al., 2020; Gao et al., 2021; Morsli et al., 2021c; Piantadosi et al., 2021). Moreover, sequence variation linked to high mutational levels, as observed in RNA viruses including Enterovirus (Piantadosi et al., 2017; Morsli et al., 2021d,c), with more than 300 genotypes (Isaacs et al., 2018), and obscuring the identification capability of the mNGS approach, further indicates the necessity for pathogen genome enrichment to achieve appropriate sequencing depth. This is usually limited by the CSF volume which makes it not possible to extract the DNA and RNA separately (Li et al., 2021). The sensitivity of mNGS depended on the nature of the causative pathogen, being lower than routine techniques for bacterial and fungal meningitis, at 73.3% and <85%, respectively (Xing X. W. et al., 2020; 2021). In contrast, mNGS appeared to be highly sensitive (>90%) in cases of mycobacterial and viral meningitis (Kawada et al., 2016; Miller et al., 2019; Xing X. W. et al., 2020; Zhang et al., 2021b). However, human genome depletion by DNase treatment can produce false-negative results in the case of RNA investigation, by degrading the pathogen DNA (Edridge et al., 2019; Wilson et al., 2019).
The mNGS approach is not currently standardised and remains in its infancy. Microbiologists may, therefore, wish to develop different strategies to investigate RNA and/or DNA pathogens, relying on available mNGS materials and reagents as well as mNGS platform, and in-house pipelines using local epidemiology-driven, specific pathogen genome databases (Figure 3). Finally, because this identification is database-dependent, attention should be given to curating and updating local databases (Ji et al., 2020; Carbo et al., 2021; Xing X.-W. et al., 2021). In contrast to routine tests based on nested and semi-nested PCRs generating up to 25% false-positive results (Boudet et al., 2019), the mNGS approach does not involve any prior amplification and no specific pathogen target, thus limiting the opportunity for such false-positives. Additional information about genotyping and in silico antibiotic susceptibility testing was added, even in cases of uncultured bacteria.
As a time and cost-effective approach, real-time mNGS could be implemented in POC laboratories for the diagnosis of CAM, especially with regards to undocumented meningeal disease (Morsli et al., 2022).
MM: literature search, data collection, data cleaning, data interpretation, validation, and writing of the manuscript. JL and MD: design of the study, data interpretation, validation, funding, critical review of the manuscript, coordination, and direction of the work. The data presented here were extracted by MM and validated by JL and MD after review. All authors have read and approved the manuscript.
MM was a Ph.D. student supported by the Fondation Méditerranée Infection. This work was supported by the French Government under the Investissements d’Avenir (Investments in the Future) programme managed by the ANR [Agence Nationale de la Recherche (National Agency for Research)] (reference: Méditerranée Infection 10-IAHU-03).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the support provided by the Fondation Méditerranée Infection, IHU Méditerranée Infection, Marseille, France. MM received a Ph.D. grant from the Fondation Méditerranée Infection.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.926240/full#supplementary-material
Ahn, H., Weaver, M., Lyon, D., Choi, E., and R. N R. B. F. (2017). Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis. 176, 139–148. doi: 10.1016/S1473-3099(17)30243-8
Beaman, M. H. (2018). Encephalitis: a narrative review. Med. J. Aust. 209, 449–454. doi: 10.5694/mja17.01073
Behera, H. S., Satpathy, G., and Tripathi, M. (2016). Isolation and genotyping of Acanthamoeba spp. from Acanthamoeba meningitis/ meningoencephalitis (AME) patients in India. Parasit. Vectors 9:442. doi: 10.1186/s13071-016-1729-5
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). Mette pinholt 16, muna f. anjum 18, nicholas a. duggett 18. J. Antimicrob. Chemother. 16, 3491–3500. doi: 10.1093/jac/dkaa345
Boudet, A., Pantel, A., Carles, M. J., Boclé, H., Charachon, S., Enault, C., et al. (2019). A review of a 13-month period of filmarray meningitis/encephalitis panel implementation as a first-line diagnosis tool at a university hospital. PLoS One 14:e0223887. doi: 10.1371/journal.pone.0223887
Broberg, E. K., Simone, B., Jansa, J., Prochazka, B., Wyndham-Thomas, C., Van Ranst, M., et al. (2018). Upsurge in echovirus 30 detections in five EU/EEA countries, April to September, 2018. Eurosurveillance 23:1800537. doi: 10.2807/1560-7917.ES.2018.23.44.1800537
Carbo, E. C., Blankenspoor, I., Goeman, J. J., Kroes, A. C. M., Claas, E. C. J., and De Vries, J. J. C. (2021). Viral metagenomic sequencing in the diagnosis of meningoencephalitis: a review of technical advances and diagnostic yield. Expert Rev. Mol. Diagn. 21, 1139–1146. doi: 10.1080/14737159.2021.1985467
Carbo, E. C., Buddingh, E. P., Karelioti, E., Sidorov, I. A., Feltkamp, M. C. W., Borne, P. A. V. D., et al. (2020). Improved diagnosis of viral encephalitis in adult and pediatric hematological patients using viral metagenomics. J. Clin. Virol. 130:104566. doi: 10.1016/j.jcv.2020.104566
Chen, L., Xu, Y., Liu, C., Huang, H., Zhong, X., Ma, C., et al. (2020). Clinical features of aseptic meningitis with varicella zoster virus infection diagnosed by next-generation sequencing: case reports. BMC Infect. Dis. 20:435. doi: 10.1186/s12879-020-05155-8
Chiu, C. Y., Coffey, L. L., Murkey, J., Symmes, K., Sample, H. A., Wilson, M. R., et al. (2017). Diagnosis of fatal human case of st. louis encephalitis virus infection by metagenomic sequencing, california, 2016. Emerg. Infect. Dis. 23, 1694–1698. doi: 10.3201/eid2310.161986
Edridge, A. W. D., Deijs, M., Van Zeggeren, I. E., Kinsella, C. M., Jebbink, M. F., Bakker, M., et al. (2019). Viral metagenomics on cerebrospinal fluid. Genes (Basel) 10:332. doi: 10.3390/genes10050332
Eibach, D., Hogan, B., Sarpong, N., Winter, D., Struck, N. S., Adu-Sarkodie, Y., et al. (2019). Viral metagenomics revealed novel betatorquevirus species in pediatric inpatients with encephalitis/meningoencephalitis from Ghana. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-38975-z
Erdem, G., Kaptsan, I., Sharma, H., Kumar, A., Aylward, S. C., Kapoor, A., et al. (2021). Cerebrospinal fluid analysis for viruses by metagenomic next-generation sequencing in pediatric encephalitis: not yet ready for prime time? J. Child Neurol. 36, 350–356. doi: 10.1177/0883073820972232
Fan, X., Wang, Q., Li, P., Ai, B., Song, Y., Peng, Q., et al. (2021). The diagnostic value of metagenomic next-generation sequencing in Angiostrongylus cantonensis encephalitis/meningitis. J. Behav. Brain Sci. 11, 216–226. doi: 10.4236/jbbs.2021.119017
Gao, D., Hu, Y., Jiang, X., Pu, H., Guo, Z., and Zhang, Y. (2021). Applying the pathogen-targeted next-generation sequencing method to pathogen identification in cerebrospinal fluid. Ann. Transl. Med. 9, 1675–1675. doi: 10.21037/atm-21-5488
Greninger, A. L., Messacar, K., Dunnebacke, T., Naccache, S. N., Federman, S., Bouquet, J., et al. (2015). Clinical metagenomic identification of Balamuthia mandrillaris encephalitis and assembly of the draft genome: the continuing case for reference genome sequencing. Genome Med. 7, 1–14. doi: 10.1186/s13073-015-0235-2
Guan, H., Shen, A., Lv, X., Yang, X., Ren, H., Zhao, Y., et al. (2015). Detection of virus in CSF from the cases with meningoencephalitis by next-generation sequencing. J. Neurovirol. 22, 240–245. doi: 10.1007/s13365-015-0390-7
Guan, Q., Alhuthali, B., Mfarrej, S., Halim, M. A., Almaghrabi, R. S., and Pain, A. (2021). Metagenomics-driven rapid diagnosis of an imported fatal case of rare amoebic meningoencephalitis. J. Travel Med. Taab 172, 1–3. doi: 10.1093/jtm/taab172
Hu, Z., Weng, X., Xu, C., Lin, Y., Cheng, C., Wei, H., et al. (2018). Metagenomic next-generation sequencing as a diagnostic tool for toxoplasmic encephalitis. Ann. Clin. Microbiol. Antimicrob. 17, 4–9. doi: 10.1186/s12941-018-0298-1
Huang, S., Liang, X., Han, Y., Zhang, Y., Li, X., and Yang, Z. (2021). A pediatric case of primary amoebic meningoencephalitis due to Naegleria fowleri diagnosed by next-generation sequencing of cerebrospinal fluid and blood samples. BMC Infect. Dis. 21:1251. doi: 10.1186/s12879-021-06932-9
Isaacs, S. R., Kim, K. W., Cheng, J. X., Bull, R. A., Stelzer-Braid, S., Luciani, F., et al. (2018). Amplification and next generation sequencing of near full-length human enteroviruses for identification and characterisation from clinical samples. Sci. Rep. 8, 1–9. doi: 10.1038/s41598-018-30322-y
Ji, X. C., Zhou, L. F., Li, C. Y., Shi, Y. J., Wu, M. L., Zhang, Y., et al. (2020). Reduction of human DNA contamination in clinical cerebrospinal fluid specimens improves the sensitivity of metagenomic next-generation sequencing. J. Mol. Neurosci. 70, 659–666. doi: 10.1007/s12031-019-01472-z
Joanna María, O. A., José Miguel, S. C., Fabiola, G. A., Elizabeth, G. D., Araceli, R. C., Patricia, A. P., et al. (2016). Fatal Psychrobacter sp. infection in a pediatric patient with meningitis identified by metagenomic next-generation sequencing in cerebrospinal fluid. Arch. Microbiol. 198, 129–135. doi: 10.1007/s00203-015-1168-2
Kawada, J. I., Okuno, Y., Torii, Y., Okada, R., Hayano, S., Ando, S., et al. (2016). Identification of viruses in cases of pediatric acute encephalitis and encephalopathy using next-generation sequencing. Sci. Rep. 6, 1–8. doi: 10.1038/srep33452
Lan, Z. W., Xiao, M. J., Guan, Y. L., Zhan, Y. J., and Tang, X. Q. (2020). Detection of Listeria monocytogenes in a patient with meningoencephalitis using next-generation sequencing: a case report. BMC Infect. Dis. 20:721. doi: 10.1186/s12879-020-05447-z
Launes, C., Casas-Alba, D., Fortuny, C., Valero-Rello, A., Cabrerizo, M., and Muñoz-Almagro, C. (2017). Utility of FilmArray meningitis/encephalitis panel during outbreak of brainstem encephalitis caused by enterovirus in catalonia in 2016. J. Clin. Microbiol. 55, 336–338. doi: 10.1128/JCM.01931-16
Leber, A. L., Everhart, K., Balada-Llasat, J. M., Cullison, J., Daly, J., Holt, S., et al. (2016). Multicenter evaluation of biofire filmarray meningitis/encephalitis panel for detection of bacteria, viruses, and yeast in cerebrospinal fluid specimens. J. Clin. Microbiol. 54, 2251–2261. doi: 10.1128/JCM.00730-16
Leon, K. E., Schubert, R. D., Casas-Alba, D., Hawes, I. A., Ramachandran, P. S., Ramesh, A., et al. (2020). Genomic and serologic characterization of enterovirus A71 brainstem encephalitis. Neurol. Neuroimmunol. Neuroinflammation 7:21523. doi: 10.1212/NXI.0000000000000703
Li, C., Burrell, R., Dale, R. C., and Kesson, A. (2021). Diagnosis and analysis of unexplained cases of childhood encephalitis in Australia using metagenomic next- generation sequencing. bioRxiv [Preprint] doi: 10.1101/2021.05.10.443367
Manso, C. F., Bibby, D. F., Mohamed, H., Brown, D. W. G., Zuckerman, M., and Mbisa, J. L. (2020). Enhanced detection of DNA viruses in the cerebrospinal fluid of encephalitis patients using metagenomic next-generation sequencing. Front. Microbiol. 11:1879. doi: 10.3389/fmicb.2020.01879
Mao, Y., Li, X., Lou, H., Shang, X., Mai, Y., Yang, L., et al. (2021). Detection of Coccidioides posadasii in a patient with meningitis using metagenomic next-generation sequencing: a case report. BMC Infect. Dis. 21:968. doi: 10.1186/s12879-021-06661-z
Miller, S., Naccache, S. N., Samayoa, E., Messacar, K., Arevalo, S., Federman, S., et al. (2019). Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 29, 831–842. doi: 10.1101/gr.238170.118
Moher, D., Shamseer, L., Clarke, M., Ghersi, D., Liberatî, A., Petticrew, M., et al. (2015). Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 4:1.
Morsli, M., Bechah, Y., Coulibaly, O., Toro, A., Fournier, P., Houhamdi, L., et al. (2022). Direct diagnosis of Pasteurella multocida meningitis using next-generation sequencing. Lancet Microbe 3:e6. doi: 10.1016/s2666-5247(21)00277-9
Morsli, M., Zandotti, C., Morand, A., Colson, P., and Drancourt, M. (2021d). Direct diagnosis of echovirus 12 meningitis using metagenomic next generation sequencing. Pathogens 10, 3–7. doi: 10.3390/pathogens10050610
Morsli, M., Kerharo, Q., Amrane, S., Parola, P., Fournier, P. E., and Drancourt, M. (2021a). Real-time whole genome sequencing direct diagnosis of Streptococcus pneumoniae meningitis: a case report. J. Infect. 10, 14–16. doi: 10.1016/j.jinf.2021.10.002
Morsli, M., Vincent, J. J., Milliere, L., Colson, P., and Drancourt, M. (2021c). Direct next-generation sequencing diagnosis of echovirus 9 meningitis. France. Eur. J. Clin. Microbiol. Infect. Dis. 40, 2037–2039. doi: 10.1007/s10096-021-04205-6
Morsli, M., Kerharo, Q., Delerce, J., Roche, P., Troude, L., and Drancourt, M. (2021b). Haemophilus influenzae meningitis direct diagnosis by metagenomic next-generation sequencing: a case report. Pathogens 10:461. doi: 10.3390/pathogens10040461
Naccache, S. N., Peggs, K. S., Mattes, F. M., Phadke, R., Garson, J. A., Grant, P., et al. (2015). Diagnosis of neuroinvasive astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin. Infect. Dis. 60, 919–923. doi: 10.1093/cid/ciu912
Piantadosi, A., Kanjilal, S., Ganesh, V., Khanna, A., Hyle, E. P., Rosand, J., et al. (2018). Rapid detection of powassan virus in a patient with encephalitis by metagenomic sequencing. Clin. Infect. Dis. 66, 789–792. doi: 10.1093/cid/cix792
Piantadosi, A., Mukerji, S. S., Chitneni, P., Cho, T. A., Cosimi, L. A., Hung, D. T., et al. (2017). Metagenomic sequencing of an echovirus 30 genome from cerebrospinal fluid of a patient with aseptic meningitis and orchitis. Open Forum Infect. Dis. 4, 30–32. doi: 10.1093/ofid/ofx138
Piantadosi, A., Mukerji, S. S., Ye, S., Leone, M. J., Freimark, L. M., Park, D., et al. (2021). Enhanced virus detection and metagenomic sequencing in patients with meningitis and encephalitis. mBio 12:e01143–21. doi: 10.1128/mBio.01143-21
Saha, S., Ramesh, A., Kalantar, K., Malaker, R., Hasanuzzaman, M., Khan, L. M., et al. (2019). Unbiased metagenomic sequencing for pediatric meningitis in bangladesh reveals neuroinvasive chikungunya virus outbreak and other unrealized pathogens. mBio 10: 02877–19. doi: 10.1128/mBio.02877-19
Solomon, I. H., Ganesh, V. S., Yu, G., Deng, X. D., Wilson, M. R., Miller, S., et al. (2021). Fatal case of chronic jamestown canyon virus encephalitis diagnosed by metagenomic sequencing in patient receiving rituximab. Emerg. Infect. Dis. 27, 238–242. doi: 10.3201/EID2701.203448
Soucek, D. K., Dumkow, L. E., VanLangen, K. M., and Jameson, A. P. (2019). Cost justification of the biofire filmarray meningitis/encephalitis panel versus standard of care for diagnosing meningitis in a community hospital. J. Pharm. Pract. 32, 36–40. doi: 10.1177/0897190017737697
Tansarli, G. S., and Chapin, K. C. (2020). Diagnostic test accuracy of the BioFire§FilmArray§meningitis/encephalitis panel: a systematic review and meta-analysis. Clin. Microbiol. Infect. 26, 281–290. doi: 10.1016/j.cmi.2019.11.016
Tschumi, F., Schmutz, S., Kufner, V., Heider, M., Pigny, F., Schreiner, B., et al. (2019). Meningitis and epididymitis caused by toscana virus infection imported to switzerland diagnosed by metagenomic sequencing: a case report. BMC Infect. Dis. 19:591. doi: 10.1186/s12879-019-4231-9
Vincent, J. J., Zandotti, C., Baron, S., Kandil, C., Levy, P. Y., Drancourt, M., et al. (2020). Point-of-care multiplexed diagnosis of meningitis using the FilmArray§ME panel technology. Eur. J. Clin. Microbiol. Infect. Dis. 39, 1573–1580. doi: 10.1007/s10096-020-03859-y
Wang, Q., Wang, K., Zhang, Y., Lu, C., Yan, Y., Huang, X., et al. (2020). Neonatal Ureaplasma parvum meningitis: a case report and literature review. Transl. Pediatr. 9, 174–179. doi: 10.21037/TP.2020.02.04
Wang, S., Chen, Y., Wang, D., Wu, Y., Zhao, D., Zhang, J., et al. (2019). The feasibility of metagenomic next-generation sequencing to identify pathogens causing tuberculous meningitis in cerebrospinal fluid. Front. Microbiol. 10:1993. doi: 10.3389/fmicb.2019.01993
Wilson, M. R., Sample, H. A., Zorn, K. C., Arevalo, S., Yu, G., Neuhaus, J., et al. (2019). Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N. Engl. J. Med. 380, 2327–2340.
Wu, X., Yan, G., Han, S., Ye, Y., Cheng, X., Gong, H., et al. (2020). Diagnosing Balamuthia mandrillaris encephalitis via next-generation sequencing in a 13-year-old girl. Emerg. Microbes Infect. 9, 1379–1387. doi: 10.1080/22221751.2020.1775130
Xing, N., Zhao, Z., Li, Q., Dong, Y., Li, J., and Zhang, S. (2021). Ureaplasma parvum meningitis following atypical choroid plexus papilloma resection in an adult patient: a case report and literature review. BMC Infect. Dis. 21:1276. doi: 10.1186/s12879-021-06975-y
Xing, X. W., Zhang, J. T., Ma, Y. B., He, M. W., Yao, G. E., Wang, W., et al. (2020). Metagenomic next-generation sequencing for diagnosis of infectious encephalitis and meningitis: a large, prospective case series of 213 patients. Front. Cell. Infect. Microbiol. 10:88. doi: 10.3389/fcimb.2020.00088
Xing, X. W., Zhang, J. T., Ma, Y. B., Zheng, N., Yang, F., and Yu, S. Y. (2019). Apparent performance of metagenomic next-generation sequencing in the diagnosis of cryptococcal meningitis: a descriptive study. J. Med. Microbiol. 68, 1204–1210. doi: 10.1099/jmm.0.000994
Xing, X.-W., Yu, S.-F., Zhang, J.-T., Tan, R.-S., Ma, Y.-B., Tian, X., et al. (2021). Metagenomic next-generation sequencing of cerebrospinal fluid for the diagnosis of cerebral aspergillosis. Front. Microbiol. 12:787863. doi: 10.3389/fmicb.2021.787863
Yan, L., Sun, W., Lu, Z., and Fan, L. (2020). Metagenomic next-generation sequencing (mNGS) in cerebrospinal fluid for rapid diagnosis of Tuberculosis meningitis in HIV-negative population. Int. J. Infect. Dis. 96, 270–275. doi: 10.1016/j.ijid.2020.04.048
Yao, M., Zhou, J., Zhu, Y., Zhang, Y., Lv, X., Sun, R., et al. (2016). Detection of Listeria monocytogenes in CSF from three patients with meningoencephalitis by next-generation sequencing. J. Clin. Neurol. 12, 736–745. doi: 10.3988/jcn.2016.12.4.446
Yin, X. W., Mao, Z. D., Zhang, Q., Ou, Q. X., Liu, J., Shao, Y., et al. (2021). Retrospective Study clinical metagenomic sequencing for rapid diagnosis of pneumonia and meningitis caused by Chlamydia psittaci. World J. Clin. Cases 9, 7693–7703. doi: 10.12998/wjcc.v9.i26.7693
Zeng, S., Yan, W. Q., Wu, X. M., and Zhang, H. N. (2021). Case report: diagnosis of Klebsiella pneumoniae invasive liver abscess syndrome with purulent meningitis in a patient from pathogen to lesions. Front. Med. 8:714916. doi: 10.3389/fmed.2021.714916
Zhang, X., Wu, Z., and Wang, K. (2020). Diagnosis of Streptococcus suis meningoencephalitis with metagenomic next-generation sequencing of the cerebrospinal fluid: a case report with literature review. BMC Infect. Dis. 20:884. doi: 10.1186/s12879-020-05621-3
Zhan, C., Chen, L., and Hu, L. (2021). Neonatal Ureaplasma parvum meningitis complicated with subdural hematoma: a case report and literature review. BMC Infect. Dis. 21:268. doi: 10.1186/s12879-021-05968-1
Zhang, X. X., Guo, L. Y., Liu, L. L., Shen, A., Feng, W. Y., Huang, W. H., et al. (2019). The diagnostic value of metagenomic next-generation sequencing for identifying Streptococcus pneumoniae in paediatric bacterial meningitis. BMC Infect. Dis. 19:495. doi: 10.1186/s12879-019-4132-y
Zhang, X., Jiang, C., and Zhou, C. (2021a). Diagnosis of Enterococcus faecalis meningitis associated with long-term cerebrospinal fluid rhinorrhoea using metagenomics next-generation sequencing: a case report. BMC Infect. Dis. 21:1105. doi: 10.1186/s12879-021-06797-y
Zhang, X., Wang, R., Luo, J., Xia, D., and Zhou, C. (2021b). Detection of meningoencephalitis caused by Listeria monocytogenes with ischemic stroke-like onset using metagenomics next-generation sequencing: a case report. Medicine (Baltimore) 100:e26802. doi: 10.1097/MD.0000000000026802
Zhang, Y., Hong, K., Zou, Y., and Bu, H. (2019). Rapid detection of human herpes virus by next-generation sequencing in a patient with encephalitis. Virol. J. 16, 1–6. doi: 10.1186/s12985-019-1205-x
Keywords: metagenomic next generation sequencing, diagnosis, point-of-care laboratory, multiplex RT-PCR, culture, pathogen genome, antibiotic resistance, genotyping
Citation: Morsli M, Lavigne JP and Drancourt M (2022) Direct Metagenomic Diagnosis of Community-Acquired Meningitis: State of the Art. Front. Microbiol. 13:926240. doi: 10.3389/fmicb.2022.926240
Received: 22 April 2022; Accepted: 06 June 2022;
Published: 05 July 2022.
Edited by:
Tianmu Chen, Xiamen University, ChinaReviewed by:
Peter Michael Keller, University of Bern, SwitzerlandCopyright © 2022 Morsli, Lavigne and Drancourt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel Drancourt, bWljaGVsLmRyYW5jb3VydEB1bml2LWFtdS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.