 Christoph Jans
Christoph Jans Joseph Wambui
Joseph Wambui Marc J. A. Stevens
Marc J. A. Stevens Taurai Tasara
Taurai Tasara
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 15 August 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.923080
This article is part of the Research Topic Rising Stars in Food Microbiology: 2021 View all 5 articles
Staphylococcus aureus infection is considered to be a neglected tropical disease with huge impact on human and animal health alike. Dairy production in sub-Saharan Africa (SSA) relies heavily on various animals such as cows, goats, and camels, depending on the region. S. aureus causes mastitis and exhibits high prevalence in raw milk. The population structure including genotypic and phenotypic traits of dairy S. aureus in relation to animal and human isolates is, however, unknown for SSA. In this work, 20 S. aureus dairy isolates from East and West Africa were selected for comparative genomics and phenotypic analysis. Comparing their population structure revealed a large diversity of different origins suggesting milk to be a reservoir for human and animal strains alike. Furthermore, a novel putative siderophore was detected in multiple strains in a distinct animal-clade with strains of global origin. This putative siderophore shares a high genetic identity with that from Streptococcus equi suggesting possible horizontal gene transfer. These findings combined with the virulence genes harbored by these dairy-derived strains such as pvl, human evasion factor scn, various enterotoxin, leucocidin and antibiotic resistance genes, stresses the need for an integrative One Health approach to tackle the problem of S. aureus infections in animals and humans in sub-Saharan Africa.
Staphylococcus aureus is an important pathogen responsible for a broad range of human and animal diseases and toxin-mediated illnesses such as staphylococcal food poisoning and toxic shock syndrome (Tong et al., 2015; Zeaki et al., 2019). S. aureus pathogenic success is attributed to the possession of various virulence factors that include adhesins, immune evasion factors, enterotoxins, hemolysins, exfoliative toxins, Panton-Valentine leukocidin (PVL) and toxic shock syndrome toxin (TSST-1; Malachowa and DeLeo, 2010; Tong et al., 2015; Tam and Torres, 2019; Zeaki et al., 2019). In addition, S. aureus is able to acquire and develop resistance against various antimicrobial compounds including antibiotics frequently used in treatments such as beta-lactams. This is an attribute that limits therapeutic options and increases human and animal healthcare costs due to S. aureus infections (Malachowa and DeLeo, 2010; Nelson et al., 2018; Turner et al., 2019). Especially methicillin-resistant S. aureus (MRSA) strains are notorious and are routinely detected in healthcare (HA-MRSA), community (CA-MRSA) and livestock (LA-MRSA)-associated staphylococcal infections (Enright et al., 2002; Monecke et al., 2011; Lakhundi and Zhang, 2018).
In Africa, S. aureus infection is still considered to be one of the neglected diseases despite the acknowledged high burden of S. aureus infections within communities, healthcare settings and livestock production systems (Reddy et al., 2010; Herrmann et al., 2013; Schaumburg et al., 2014). Paucity of knowledge currently exists with respect to population structure, prevalence, characteristics and health burdens due to different S. aureus genotypes especially with respect to livestock and livestock-derived food products, particularly in Africa (Schaumburg et al., 2014; Lozano et al., 2016). Raw and spontaneously fermented milk products constitute an important nutrient source that is widely consumed by communities in Africa, particularly in rural and pastoral or agro-pastoral communities. These products also pose significant public health risks as potential vehicles for transmission of S. aureus and its toxins to human populations in general (Njage et al., 2013; Jans et al., 2017; Paudyal et al., 2017; Wambui et al., 2019; Owusu-Kwarteng et al., 2020). Also MRSA was reported in over 50% of strains isolated from bulk can milk and raw milk products and over 90% of strains carried at least one enterotoxin genes (Asiimwe et al., 2017) These public health risks are further exacerbated due to poor sanitation and milking hygiene practices, suboptimal milk handling and distribution infrastructures, as well as the existing high prevalence of human and intramammary dairy livestock colonization and infections with S. aureus (Owusu-Kwarteng et al., 2020; Seligsohn et al., 2020).
DNA sequence-based bacterial strain typing tools such as multilocus sequence typing (MLST) and whole genome sequence (WGS) are valuable tools in the interrogation of population structure, molecular epidemiology, dissemination routes and associated human and animal health risks of the different S. aureus genotypes (Enright et al., 2000; Harris et al., 2010). MLST assigns S. aureus strains to sequence types and clonal complexes (CC) based on the partial DNA sequence of the alleles of seven house-keeping gene loci. WGS in contrast uses the entire sequenced genome information for a more precise and high-resolution molecular typing of S. aureus strains (Enright et al., 2000; Harris et al., 2010). MLST-based interrogation of S. aureus population structures has previously revealed the clonal nature of S. aureus and identification of virulent MRSA and methicillin-sensitive S. aureus (MSSA) lineages as well as CC associated with the HA-MRSA, CA-MRSA and LA-MRSA strains (Enright et al., 2000, 2002; David and Daum, 2010; Tong et al., 2015).
We previously found that raw and fermented milk products derived from cows, goats and camels, products which are widely consumed by pastoral and sedentary communities in SSA, are also potential reservoirs for HA and LA S. aureus strains (Jans et al., 2017). DNA microarray-based analysis of such strains established that they were genetically diverse harboring various virulence and antibiotic resistance genes. Therefore, these strains may pose health risks for human consumption. We, however, currently lack high-resolution insights into the population structure and detailed genetic makeup of these strains. Generation of such information would be crucial in determining the public health risks posed by locally predominant S. aureus genotypes and their reservoirs. Furthermore, such information would contribute to the design of public health risk mitigation strategies in these widely consumed food products. In the present study, we aimed to utilize WGS to characterize this SSA milk-associated S. aureus strain collection and to provide information on their population structure and genetic composition and thus extrapolate on associated public health risks.
African S. aureus strains (ILS03 to ILS79, n = 20, ILS = Institut für Lebensmittelsicherheit und –hygiene = Institute of Food Safety and Hygiene) were isolated from raw cow, camel and goat artisanal milk and their fermented products previously collected in Côte d’Ivoire, Kenya and Somalia between 2007 and 2014 (Jans et al., 2017; Table 1). Details on isolation source and milk sample types were previously published (Jans et al., 2012, 2013; Njage et al., 2013; Jans et al., 2017). Strains examined here were selected from the set of previously described strains based on representation of different product types and geographical origin. Reference strains for all laboratory work included S. aureus HG003, S. aureus Newman and S. aureus USA300. USA300 is a CA-MRSA (Diep et al., 2006) whereas HG003 is a NCTC8325 derivative with restored rsbU and tcaR genes (Herbert et al., 2010). Strains were routinely cultivated in Brain Heart Infusion (BHI) broth (Biolife, Milan, Italy) at 37°C under aerobic conditions. All materials were autoclaved at 121°C for 15 min.
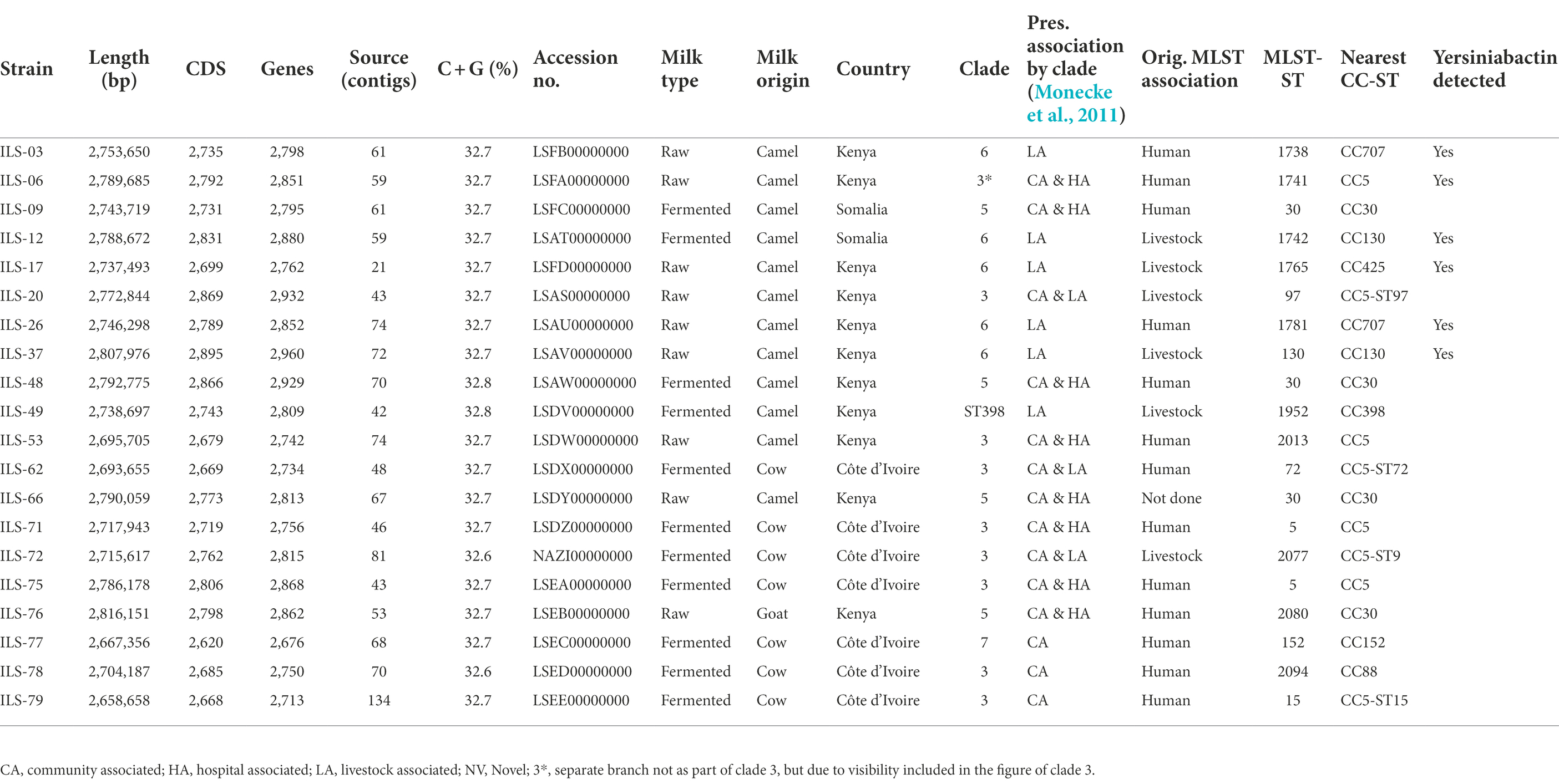
Table 1. Genome statistics of Staphylococcus aureus strains of African dairy sources obtained and characterized in this study (genome sequences available under BioProject PRJNA310553 on GenBank).
DNA for whole genome sequencing of the 20 S. aureus ILS strains was isolated from an overnight culture inoculated with material from a single colony. The extraction was performed as previously described using the DNAeasy Blood and Tissue Kit (Qiagen, Hilden, Germany; Jans et al., 2017).
The genomes of all 20 S. aureus ILS isolates were sequenced at the Functional Genome Centre of ETH Zurich and the University of Zurich (Zurich, Switzerland). Libraries were prepared using the Nextera XT kit (Illumina Inc., San Diego, United States). MiSeq 2 × 150 bp platform and chemistry (Miseq V2 5–0-4) was used for genome sequencing (Illumina Inc.).
Genome assembly was performed using CLC Genomics Workbench de novo assembly algorithm (v11, Qiagen, Aarhus, Denmark) in multiple runs. Assemblies were performed after initial adapter trimming and read quality trimming using default settings. The option “map reads back to contigs” and combined with “update contig = yes” was activated for all assemblies. Assembly quality was checked in CLC and assembly stats.1 Annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline version 3.1. All annotated genomes were submitted to GenBank and are available under Bioproject ID PRJNA, BioSample ID range SAMN04452702-SAMN04452721 and corresponding GenBank accession No. as indicated in Table 1.
Phylogenetic analysis was performed in several steps and using different analysis methods. Primarily, WGSA.net/pathogen.watch2 was used to obtain an overview of the phylogenetic relationships of the African S. aureus ILS strains in a global collection of S. aureus strains. To do so, 936 genomes out of >4,000 genomes in the Pathogen.watch database were selected based on representation of at least one member of each sequence type, each country and inclusion of all reference genomes. Individual strains per criteria were then selected at random whereas all strains identifiable as being of African origin were included. Together with the 20 ILS genomes of this study, this yielded a collection of 956 genomes (Supplementary Tables S1, S2) to calculate the core genome MLST (cgMLST) of S. aureus.
Basic sequences statistics were performed using CLC Genomics Workbench (v11, Qiagen). MLST-ST confirmation was performed on MLST 2.0 (Larsen et al., 2012). Singleton calculations on strain and group levels were performed on EDGAR v2.3 using default settings and the automated homology cutoff calculations as described in the original publications (Blom et al., 2009, 2016). A reduced set of 79 strains was used for the initial comparison (Supplementary Tables S1, S3). The set was selected based on the two criteria “reference strain” and the origin of the strains being “Africa” to facilitate faster initial comparison calculations on presumably unique CDS. Those CDS identified as presumably unique based on this small strain set, were subsequently blasted against the available S. aureus genomes on NCBI Genbank to provide a comprehensive coverage.
Genomes were analyzed for virulence factors using all S. aureus genes archived within the virulence factor database VFDB (Chen et al., 2005). A bidirectional best hit approach was applied using blastp and an identity cutoff of 70%. Mobtyper (Robertson and Nash, 2018) and ResFinder (v4.1) were used to extract information on virulence factor genes (VFGs), plasmid replicons and antimicrobial resistance genes (ARGs) from the genome assemblies uploaded to the Center for Genomic Epidemiology3 (Zankari et al., 2012; Cosentino et al., 2013; Joensen et al., 2014). These online tools were applied using the default parameters for query coverage and percentage identity.
The sensitivity of the 20 S. aureus ILS strains against a panel of six selected antibiotics was determined using the disk diffusion method. The assays were performed in duplicate on two separate occasions following the Clinical Laboratory Standards Institute (CLSI) guidelines (CLSI, 2017). The panel of tested antibiotics included bacitracin (0.04 units), ciprofloxacin (5 μg), cefoxitin (30 μg), penicillin (10 IU), teicoplanin (30 μg), fosfomycin (200 μg) and tetracycline (30 μg). The S. aureus USA 300 strain was used as a quality control to achieve the minimum standards dictated by the CLSI (2017).
Genomes of 928 S. aureus strains were screened for the presence of yersiniabactin operon structures. For this, the full operon DNA sequence was added to a blast-database in CLC Genomics workbench. This blast-database was then used to query the genomes of the 928 S. aureus strains using default settings. Hits of >1,000 bp and higher 90% sequence identity were visually confirmed for each strain prior to listing a strain as a carrier of the putative yersiniabactin siderophore operon.
For primer design and PCR development, genomes of strains harboring yersiniabactin-enconding genes were selected. The DNA sequence of the operon structure extracted from S. aureus strain ILS-03 in comparison with that of strain BSAR58 was used to identify conserved sequences. Four primer pairs were designed to target the four main genes comprised within the putative yersiniabactin siderophore-encoding operon (Table 2), namely the genes encoding for amp ligase (ILS-03 locus tag AXE88_09125), non-ribosomal peptide synthetase (AXE88_09130), polyketide synthase (AXE88_09135) and polyketide synthetase (AXE88_09145). Further genes upstream and downstream of the yersiniabactin siderophore-encoding operon were selected based on their conserved nature across yersiniabactin carrier and non-carrier strains. Genes encoding for the AraC family transcriptional regulator (AXE88_09195) and ATPase (AXE88_04705) were determined as conserved genes for this purpose and targeted using two additional primer pairs. All primers were designed in CLC Genomics Workbench with a universal nearest neighbor melting temperature of 58–60°C. All PCR assays were operated with 1 μM of each primer, 50 ng of DNA template and PCR master mix to a final volume of 25 μl. The PCR protocol encompassed an initial denaturation of 2 min at 95°C followed by 35 cycles of 30 s denaturation at 95°C, 30 s annealing at 56°C and 30 s replication at 72°C. PCR products were resolved on a 1.5% agarose gel and visualized under UV light after ethidium bromide staining.
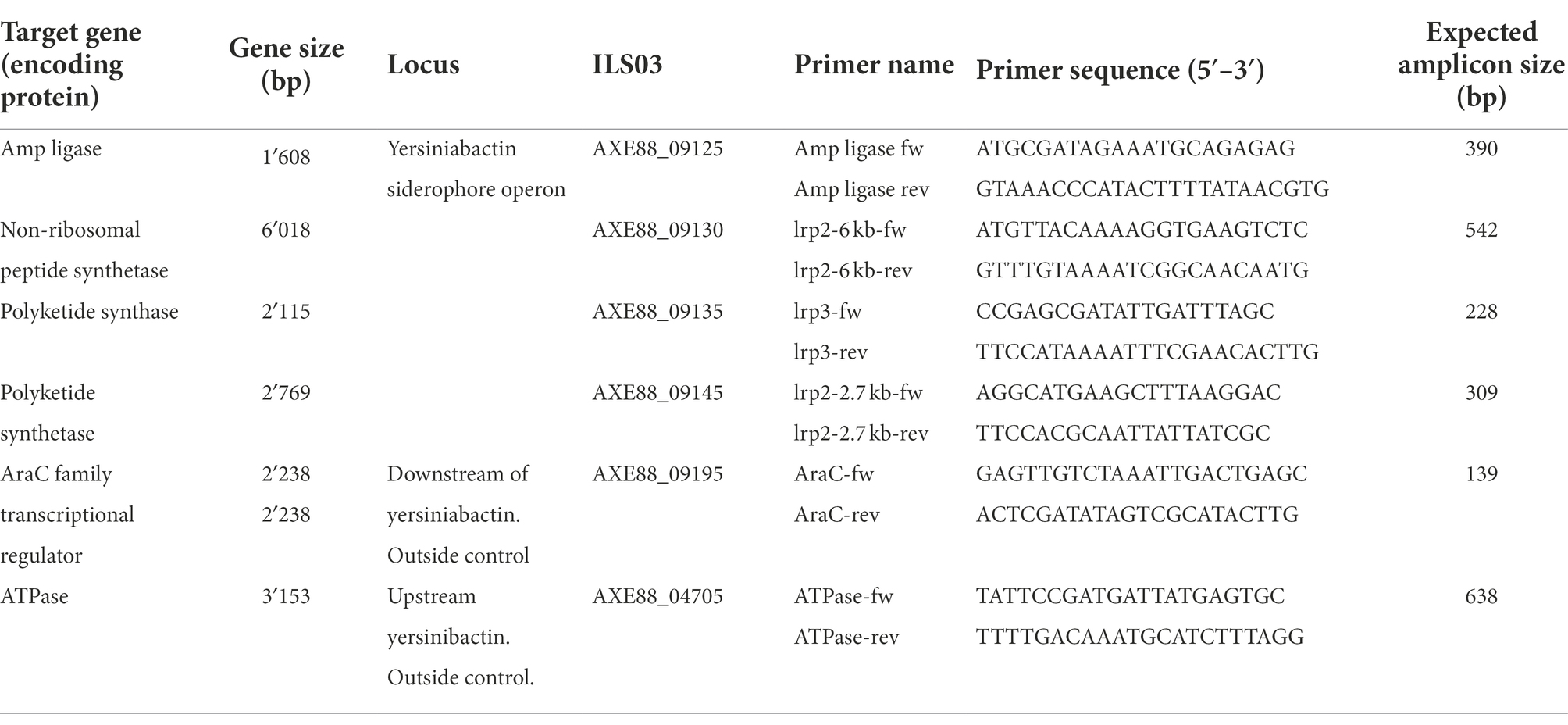
Table 2. Primers for yersiniabactin locus detection in S. aureus.
Iron-limited growth conditions were generated as previously described (Ledala et al., 2014) by chelating divalent cations in TSB with 5% Chelex-100 (BioRad Laboratories, Hercules, California, United States) for 20 h at 4°C while stirring. Afterward, the Chelex was removed by filtrating through a 0.2-μm filter membrane (Thermo Fisher Scientific, Waltham, Massachusetts, United States). Defferated TSB was supplemented with 25 μM ZnSO4, 25 μM MnSO4, 1 mM MgSO4, 100 μM CaCl2, and 5 μM ferrous Iron (DTSBfe; Sigma-Aldrich Co., Missouri, United States). To compare growth, overnight cultures (16 h at 37°C and 150 rpm) of yersiniabactin siderophore operon-positive (n = 11) and negative (n = 18) S. aureus strains cultivated in normal TSB were harvested by centrifugation and washed in 10 ml PBS (Life Technologies Ltd., Paisely, United Kingdom) at 8,000 × g for 5 min. A 20-μl culture suspension in PBS standardized to an optical density 600 nm (OD600) of 1.0 for each strain was inoculated in 180 μl DTSB and TSB in 96-well microtiter plates (Corning Incorporated, New York, United States). Cultures were grown at 37°C with continuous medium shaking. Growth was monitored by measuring OD600 at 30-min intervals for 24 h in an Eon microtiter plate reader (BioTek, Lucerne, Switzerland). Each strain was assessed in three independent biological experiments that were performed in duplicates. Growth parameters, namely, lag phase (LPD), maximum growth rate (MGR), and area under the curve (AUC), in TSB and DTSB were determined from the OD-based growth curve data using the R package ‘opm’ as previously described (Wambui et al., 2020). Statistical analyses were performed using the program GraphPad Prism (version 9.3.1). DTSB growth parameters were all standardized to account for growth variability between strains in normal TSB without limited iron stress. To this end DTSB growth parameters of each strain were expressed relative to those determined in normal TSB. The Mann–Whitney U rank-sum test for nonparametric data was used to analyze statistical significant differences in standardized DTSB growth parameters (LPD, MGR, and AUC) between the groups of yersiniabactin operon positive and negative strains.
The 20 genomes of S. aureus ILS strains obtained from camel, cow, and goat milk products in Côte d’Ivoire, Kenya, and Somalia featured a genome size of 2.66–2.82 Mb with a G + C content of 32.6%–32.7% and encoding for 2,676–2,960 genes (Table 1). Genome de novo assembly yielded 21–134 contigs (Table 1). Genome assembly statistics are provided in Supplementary Table S4.
Analysis of the 20 ILS S. aureus strains and 936 S. aureus genomes selected from the pathogen.watch cgMLST identified seven clades of which five clades incorporated the majority of strains. These clades were represented by S. aureus reference strains USA300 FPR3757, COL, Newman and NCTC8325 (clade 1, mainly CC8, 186 isolates), TW20 and JKD6008 (clade 2, mainly CC239, 161 isolates), Mu50, N315, and JH1 (clade 3, mainly CC5, 213 isolates), HO50960412 (clade 4, mainly CC22, 208 isolates), and MRSA252 (clade 5, mainly CC30, 111 isolates; Figure 1). S. aureus strains derived from human sources in Africa (n = 33) were distributed among all clades except clade 1. The majority of the African human isolates clustered in clades 6 and 7 (n = 12), in or near clade 3 (n = 11), clade 4 (n = 6), clade 5 (n = 3), and clade 2 (n = 1).
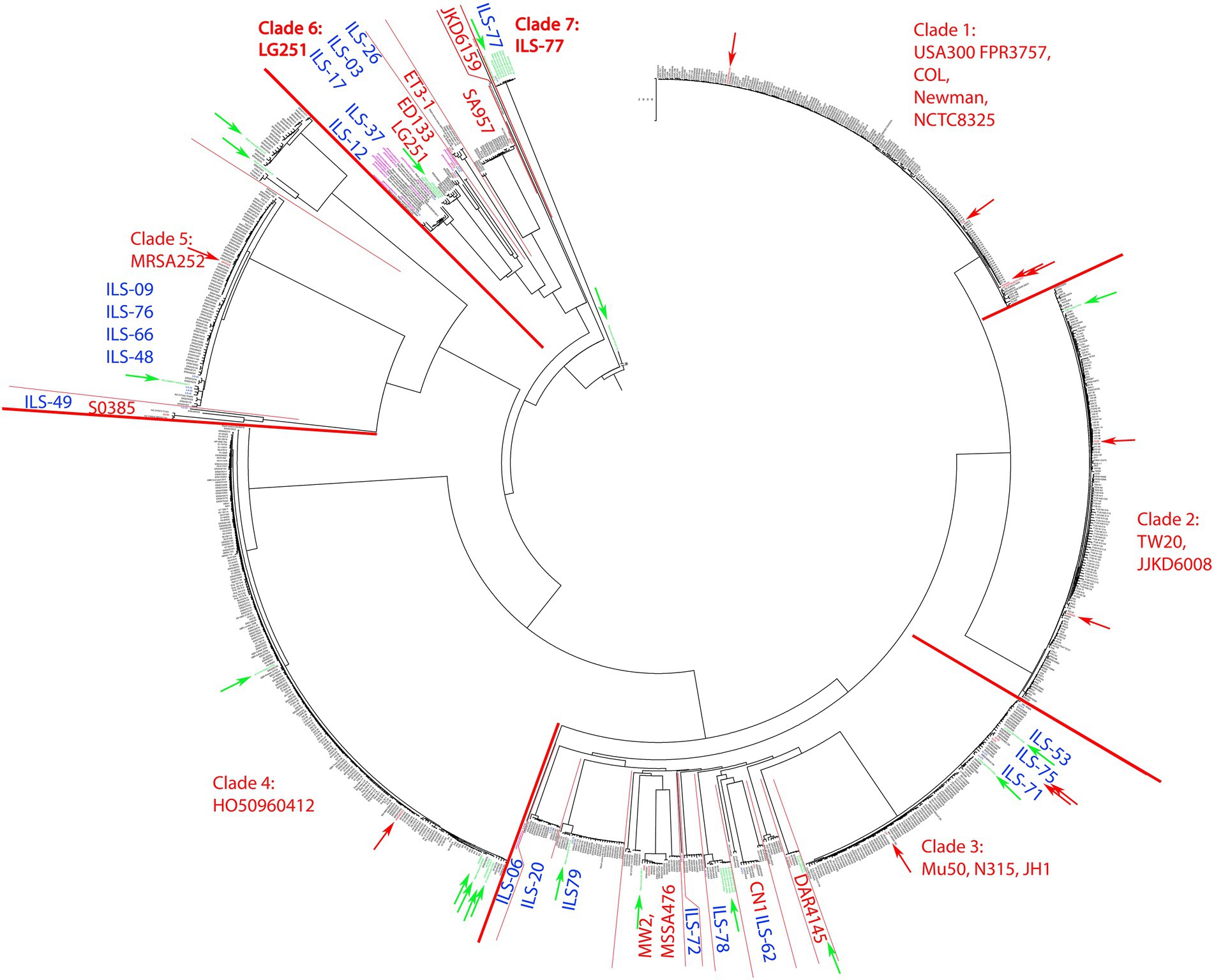
Figure 1. cgMLST tree of 956 S. aureus strains calculated on pathogen.watch using various reference S. aureus strains highlighted in each clade. For better visualization of the main phylogenetic tree structure, the branch length connecting the base of the tree (S. aureus BU G1201 t13 as the most distant strain) was cut from 854′621 units to 200 units and indicated accordingly. Blue indicates ILS strains reported in this study. Red boundaries and arrows indicate clade boundaries and tree location of all main reference strains, respectively. Green arrows indicate strains of African origin not originating from this study.
Overall the 20 S. aureus ILS strains derived from African dairy products featured a selective distribution among several distinct clades whereas some strains formed novel clades. A large number of African isolates were directly part of clade 3 or clustered in close relationship to clade 3 together with human-derived African strains (Supplementary Figure S1). This included ILS-53, ILS-75, and ILS-71, which clustered within clade 3. ILS-06, ILS-20, ILS-79, ILS-72, ILS-78, and ILS-62 formed small diverging clades within clade 3 and in connection to reference strains MW2, MSSA476, CN1 and DAR4145. Members of clade 3 and adjacent clades were mainly assigned to CC5. Other ILS isolates (ILS-09, ILS-76, ILS-66, ILS-48, and ILS-49) clustered in close relationship to clade 5 and reference strain MRSA252 (Supplementary Figure S2). Clade 5 featured predominantly strains of CC30 (regularly associated with MRSA), but none of the ILS strains were found to be methicillin-resistant.
Furthermore, ILS-49, a member of the LA CC398, formed a separate clade adjacent to clade 5 defined by S0385. S0385 is a globally present LA-MRSA of sequence type (ST) 398 (Schijffelen et al., 2010; Supplementary Figure S2). ILS-49 was determined not to be methicillin-resistant.
Near the base of the tree, the structure showed a diversification into multiple smaller clades summarized as clade 6 and comprised of reference strains LG251, ED133, ET3-1, SA957, and JKD6159 (Figure 2). ILS-03, ILS-26 (both CC707) clustered closest to ED133. ILS-17 (CC425) clustered closest to LG251 whereas ILS-37 and ILS-12 (both CC130) clustered in a separate clade comprised of European human, animal and dairy isolates. ILS-77 (CC152) together with eight African human isolates formed clade 7 comprised of only African-derived isolates. S. aureus BU G1201 t13 as the most distant isolate formed the base of the tree. For better visualization, its branch length was corrected from 854′621 units to 200 units. In this context, the branch length within isolates associated with LG251 as well as neighboring other clades suggest less homogeneity and more distant relationships among these isolates in contrast to the rather flat hierarchy observed in clades 1, 2, 4, and 5 as well as parts of clade 3. Clades neighboring LG251 seem to be in a region of the cgMLST tree that is still less represented by the selected sample collection and available genomes.
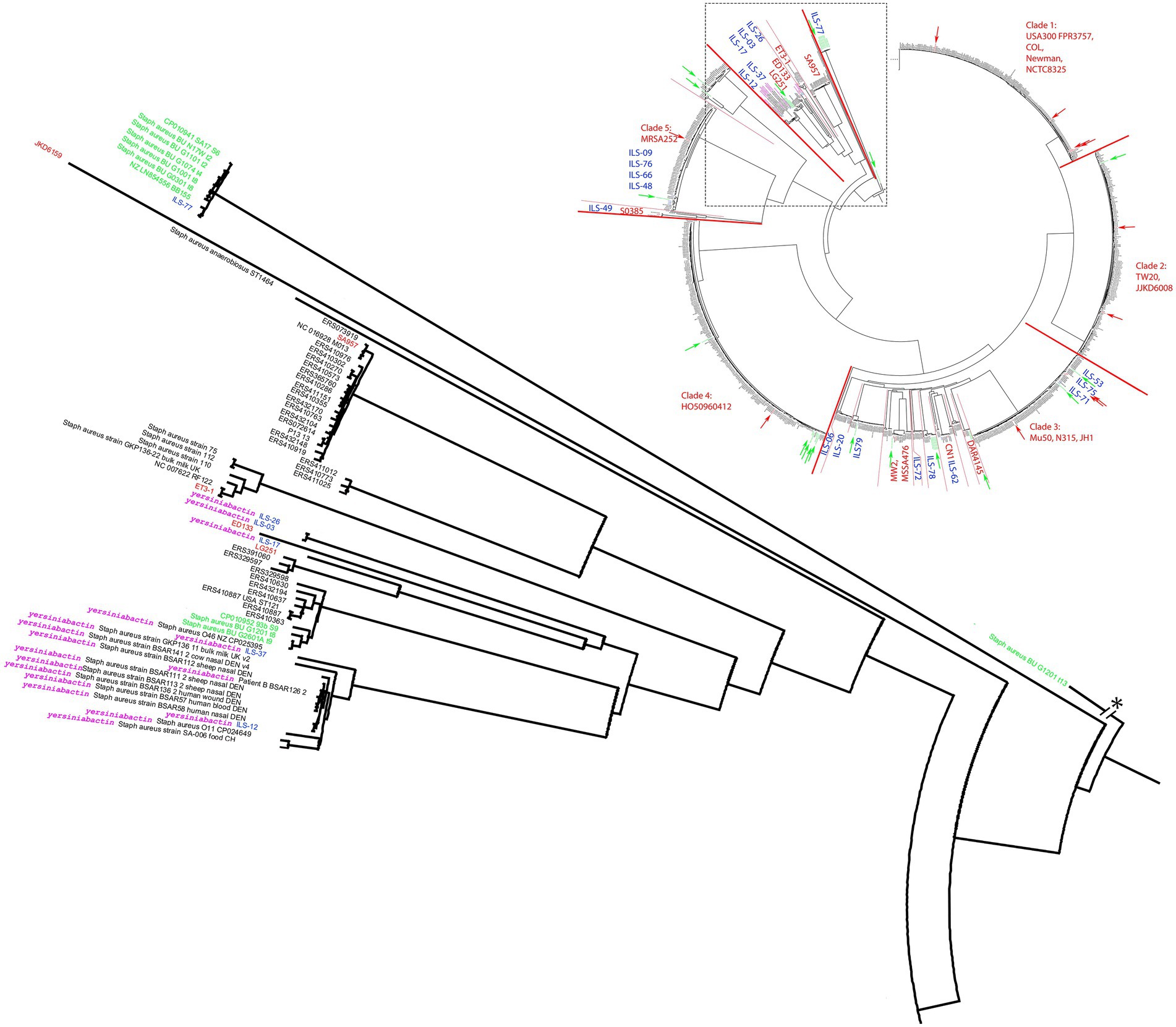
Figure 2. Detailed view of the S. aureus cgMLST tree clade comprised of strains carrying the putative yersiniabactin operon. The clade is extracted near the root of the cgMLST tree of 956 S. aureus strains (Figure 1). All putative yersiniabactin carriers are highlighted in purple. Strains derived from this study are highlighted with blue ILS-tags. Strains of African origin are highlighted in green. Reference strains are highlighted in red.
The 20 ILS strains represent a unique insight into dairy S. aureus of East and West Africa. In order to identify genes that might be shared among these ILS strains but that are unique among other S. aureus genomes, we performed a group-wise comparison. Using this approach, 225 genes (147 of which encoding for hypothetical proteins) were found to be unique in at least one ILS strain in comparison with the 79 other strains (Table 3). When expanding the grouping to merge ILS strains with all other African strains in comparison with strains outside of Africa, 431 CDS (227 encoding for hypothetical proteins) were found to be unique (Table 3). Subsequent NCBI Blast analysis of all genes presumptively identified to be unique among the limited strain pool of 20 ILS and 79 other S. aureus strains returned no major undescribed unique CDS. However, future research is needed to provide clarity into the multiple CDS still described as encoding for hypothetical proteins.
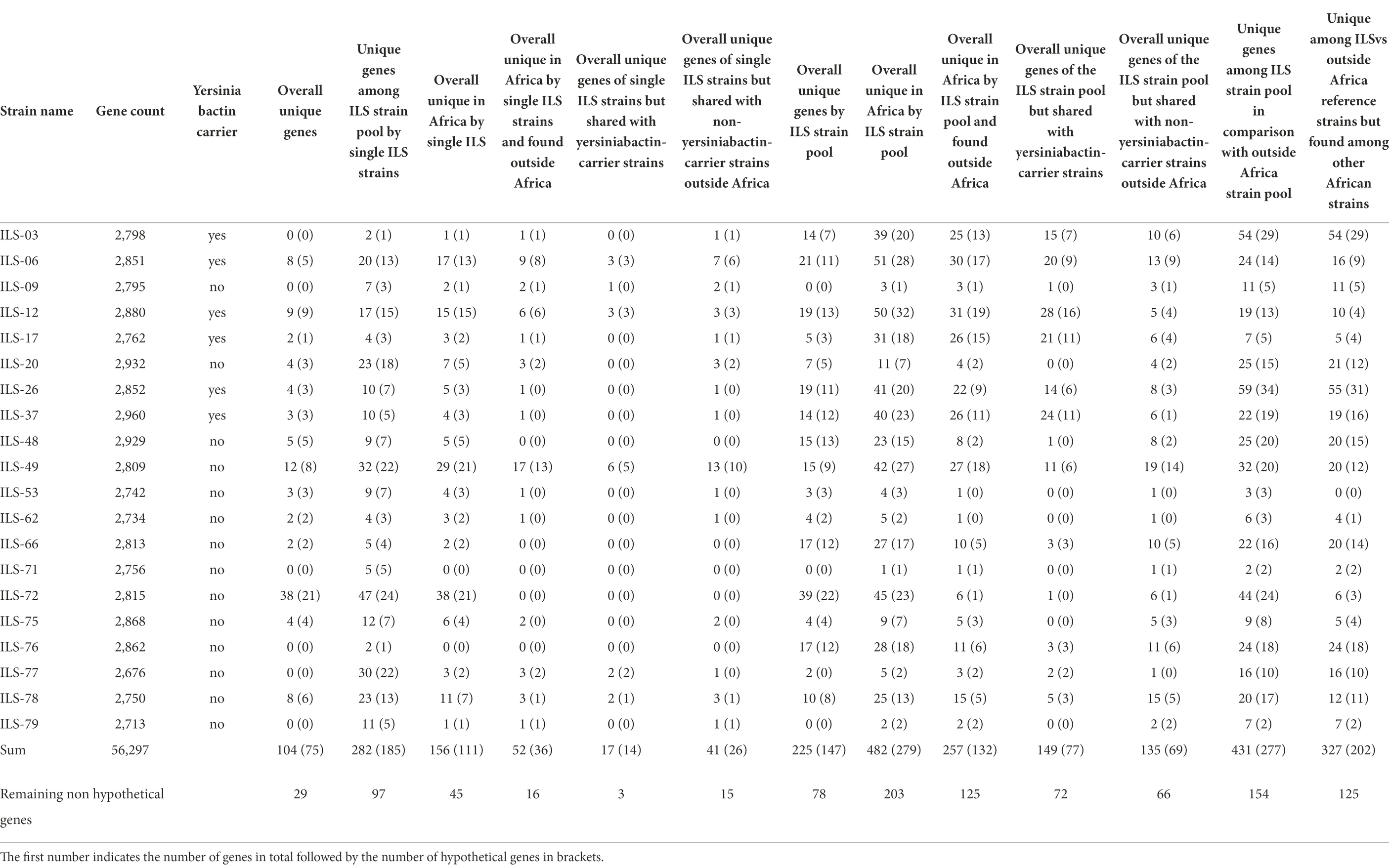
Table 3. Comparative genomics of 20 ILS S. aureus strains to 79 other S. aureus isolates selected according to their cgMLST relationship.
All 20 ILS isolates and 79 other S. aureus strains (Supplementary Table S1) selected from the different cgMLST clades were used for comparative genomics to perform a first screening for potentially unique CDS and patterns in African isolates. Among these 20 ILS and 79 other strains, ILS-06, ILS-49, ILS-72, and ILS-78 were the strains with the highest number of unique CDS overall. Only 104 CDS from these 20 ILS strains were fully unique, meaning that they appeared only once in the 20 ILS and 79 other S. aureus strains. Out of these 104 CDS, 75 CDS were identified as encoding for hypothetical proteins (Table 3). Only 29 CDS encoded proteins with annotated functions (Supplementary Table S3). These 29 CDS were further analyzed in comparison with GenBank, which found matching CDS at >95% sequence identity to CDS in other S. aureus strains for 12 CDS. Therefore, only a final set of 17 CDS originating from five ILS strains (ILS-06, ILS-17, ILS-49, ILS-72, and ILS-78) were found to be truly unique. Of these 17 CDS, 11 originated from ILS-72, seven of which were related to a putative Staphylococcus-plasmid encoding among others for type VI secretion proteins, plasmid functions and DNA binding. In ILS-06, the three unique CDS were all located adjacent to each other on the same contig. All three CDS were associated with transporter function. In ILS-17, one unique CDS was identified as encoding for a toxin-antitoxin system, whereas single unique CDS in ILS-49 and ILS-78 were identified as encoding for transcriptional regulators.
Interestingly, the in-depth analysis of seemingly unique CDS led to the discovery of a lineage of strains defined by the presence of a set of CDS termed to be involved in yersiniabactin synthesis. Yersiniabactin according to its annotated function is supposedly encoding for a siderophore involved in iron scavenging. The ILS strains identified as yersiniabactin carriers were: ILS-03, ILS-06, ILS-12, ILS-17, ILS-26, and ILS-37, all of which originated from Kenya or Somalia.
In order to investigate the presence of this putative siderophore, we analyzed 928 S. aureus genomes. Among the 928 strains queried in our cgMLST analysis, yersiniabactin CDS were limited to 18 strains, six of which being the ILS strains described above (Figure 2). The other strains identified as yersiniabactin carriers were BSAR57, BSAR58, BSAR111_2, BSAR112, BSAR113_2, BSAR136_2, BSAR141_2, GKP136-11, O11, O46, and SA-006 (Figure 2). Interestingly, the majority of strains carrying yersiniabactin were LA or food-associated strains (n = 14) with the exception of only three strains being associated with humans (BSAR57, BSAR58, and BSAR136_2). Furthermore, yersiniabactin seems to be present in two subclades within this main clade, but absent in two other subclades (Figure 2). Interestingly, ILS-06 seems to have a nearly identical yersiniabactin locus, but overall, the strain forms a very distinct separate new lineage adjacent to clade 3 and not clade 6. A reason for this distribution is so far not known.
A general comparison among several S. aureus strains (Figure 3) clustering in the same phylogenetic clade as ILS-03 revealed that this general organization of the yersiniabactin-encoding region is highly conserved among African (ILS-03, ILS-06, ILS-12, ILS-17, ILS-26, and ILS-37) and European S. aureus strains (BSAR57, BSAR58, BSAR111_2, BSAR112, BSAR113_2, BSAR136_2, BSAR141_2, GKP136-11, O11, O46, and SA-006).
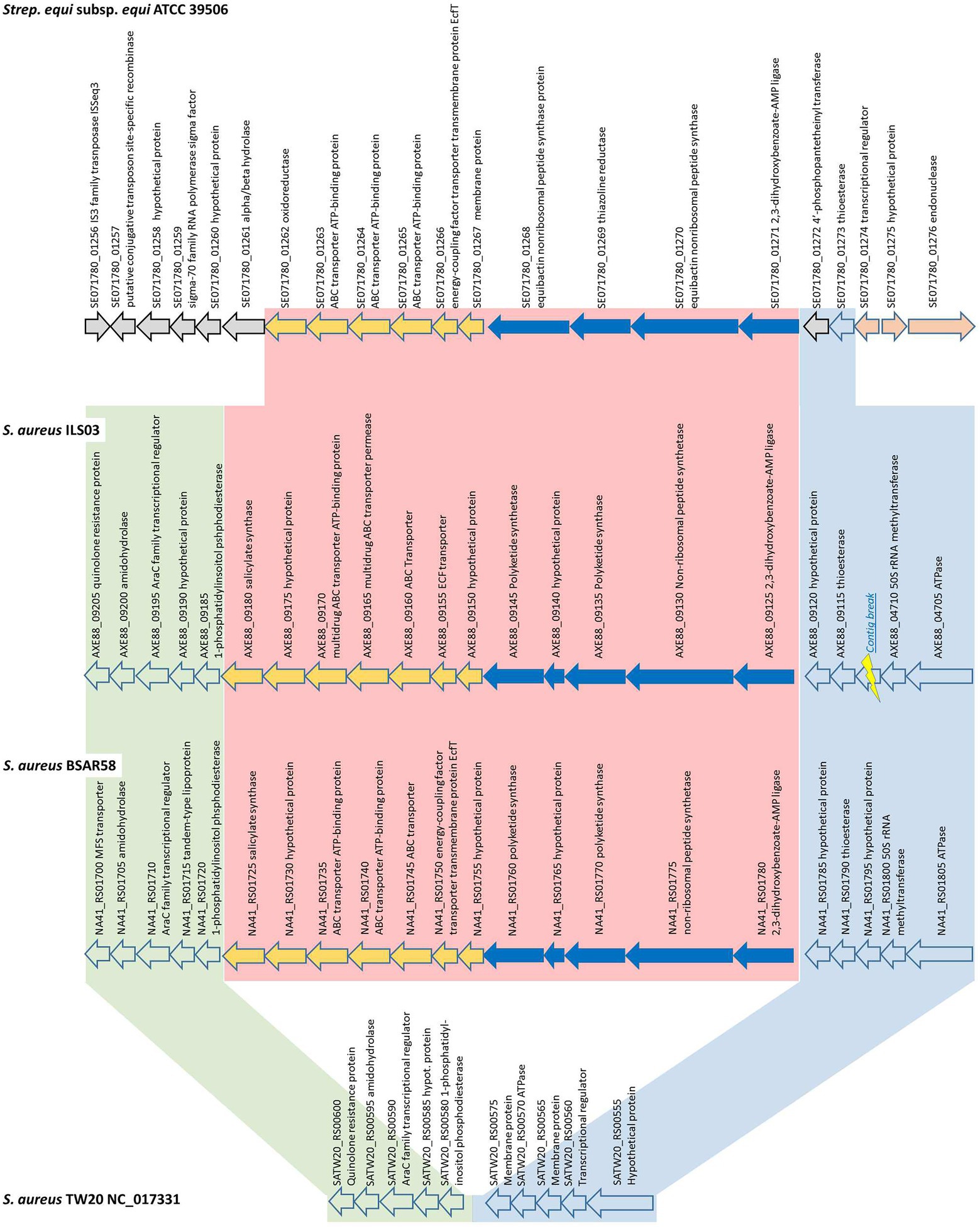
Figure 3. Detailed schematic overview of the putative yersiniabactin-encoding operon and short flanking regions shared among yersiniabactin-carriers (ILS-03 and BSAR58) and non-carrier (TW20) S. aureus strains as well as S. equi subsp. equi ATCC 39506 as closest other match.
In detail, the putative yersiniabactin-encoding genome region is comprised of 12 CDS from AXE88_09125 to AXE88_09180 using ILS-03 as a model strain (Figure 3). The first five CDS (AXE88_09125 to AXE88_09145) are the main CDS of the putative yersiniabactin production machinery and encode for putative AMP ligase, peptide synthetase, polyketide synthases, and hypothetical proteins. This is followed by seven CDS (AXE88_09150 to AXE88_09175) encoding mainly for different transporters, followed by AXE88_09180 encoding for a salicylate synthase.
To further analyze this genomic region, S. aureus TW20 (NC_017331) served as an interesting comparison in that it features only the conserved flanking regions but not the 12 yersiniabactin-related CDS (Figure 3; Supplementary Figure S3). A blast comparison of the genome section SATW20_RS00600-SATW20_RS00555 among multiple reference strains distributed among all major branches in our phylogenetic cgMLST analysis revealed that the genomic organization of TW20 is shared as well by strains US300_FPR3757, Mu50, JKD6159, CN1, MW2, MRSA252, and COL, featuring 94.6–99.9% sequence identity. This finding could in part support a hypothesis for this yersiniabactin region to be an insertion.
Following this idea of insertion, this prompted the search for other organisms sharing similar CDSs. A general blastp search on RAST suggested Streptococcus equi subsp. equi ATCC 39506 as a potential match with CDS identities and positive scores ranging between 26–56% and 49–77%, respectively (Supplementary Table S5). Subsequent bidirectional best hit comparison between ATCC 39506 and ILS-03 in CLC Genomics Workbench confirmed these matches for AXE88_01975 to AXE88_09125 including also two CDS (AXE88_09120 and AXE88_09115) directly adjacent in the upstream flanking region (Figure 3). AXE88_09115 encodes for a putative thioesterase linked possibly also to iron scavenging abilities. This shared region outside the actual region under investigation could contradict the above hypothesis of an insertion and would also support the hypothesis of a common ancestor with similar genetic organization and the subsequent deletion of the yersiniabactin-encoding region in all other strains.
Due to presumptive livestock-association of yersiniabactin carriage, we decided to screen our in-house African and Swiss S. aureus strain collections for the presence of several genes of the putative yersiniabactin operon. First, an in silico analysis was conducted revealing the presence of the yersiniabactin siderophore operon in six (ILS-03, ILS-06, ILS-12, ILS-17, ILS-26, and ILS-37) of the 20 African S. aureus genomes. To assess the distribution of this operon among the S. aureus isolates a multiplex PCR assay system targeting the genes encoding for ATPase, AMP ligase, lrp3, and AraC was developed and applied (Figure 4). This PCR assay correctly detected yersiniabactin operon presence in the six strains found to harbor this operon from in silico genome analysis (Supplementary Table S1). Its application to 51 S. aureus isolates from African fermented milk collection with yet un-sequenced genomes led to the detection of further 24 (24/51; 47%) more yersiniabactin siderophore operon carrying ILS strains (Supplementary Table S6). On the other hand, only one isolate (2.5%) tested positive for this operon when a collection of 43 Swiss mastitis milk S. aureus isolates available from the ILS strain collection was tested as an outgroup (data not shown).
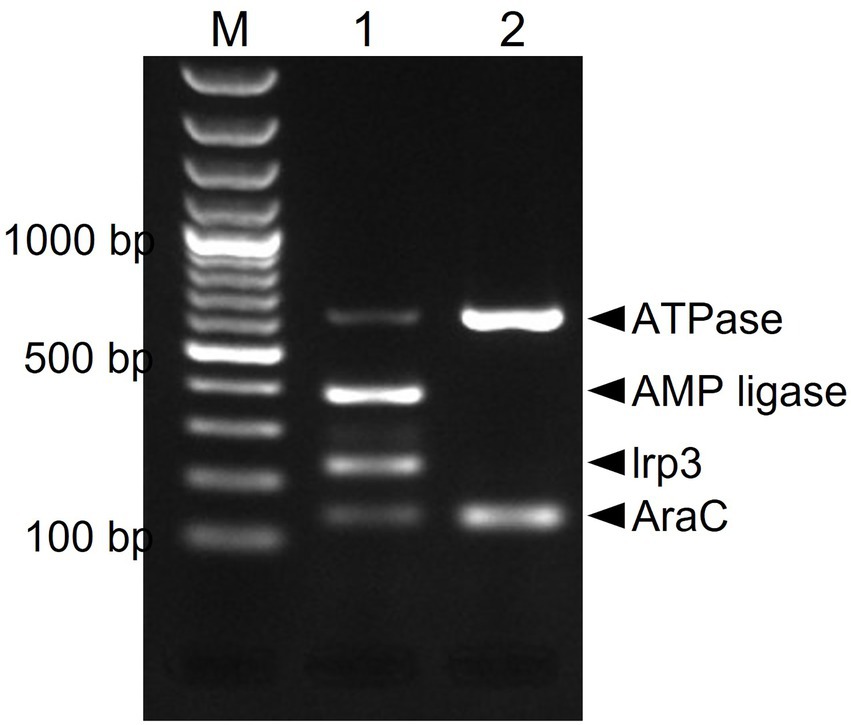
Figure 4. Multiplex PCR amplification and detection of the putative yersiniabactin operon genes in S. aureus. Primers targeting the AraC [139 bp], Lrp3 [228 bp], AMP ligase [390 bp], and ATPase [638 bp] genes were used. PCR amplicons were separated and visualized using agarose gel electrophoresis. M-100 bp DNA ladder, 1-ILS03 (positive control), and 2-ILS 09 (negative control).
To assess the possible siderophore function for the discovered putative yersiniabactin operon we compared growth under limited iron availability conditions between a group of yersiniabactin operon carrying S. aureus strains (n = 11) and a control group comprising strains (n = 18) without this operon. Growth curve parameters total area under growth curves (AUC) capturing overall growth dynamics, lag phased duration (LPD), maximum growth rate (MGR), and final maximum cell density (MD), determined for each strain in deferrated TSB media (limited iron availability conditions), were normalized to correct for strain specific growth differences in non-deferrated control TSB broth. Comparing the normalized parameters between the two strain groups revealed that although the yersiniabactin operon positive strains as a group displayed slightly higher AUC and shorter LPD medians than the yersiniabactin negative control strain group, such differences were not statistically significant (Figure 5). On the other hand comparing MGR revealed that the yersiniabactin operon strain group had significantly higher (p < 0.05; Mann–Whitney U test) MGRs than the control strain group without this operon during growth under limited iron availability conditions (Figure 5) This observation thus suggests that yersiniabactin operon carrying strains possess a growth fitness advantage than strains without the operon under the limited iron availability conditions applied in our studies. The discovered putative yersiniabactin operon might thus be beneficial for growth of S. aureus strains possessing this operon under limited iron availability.
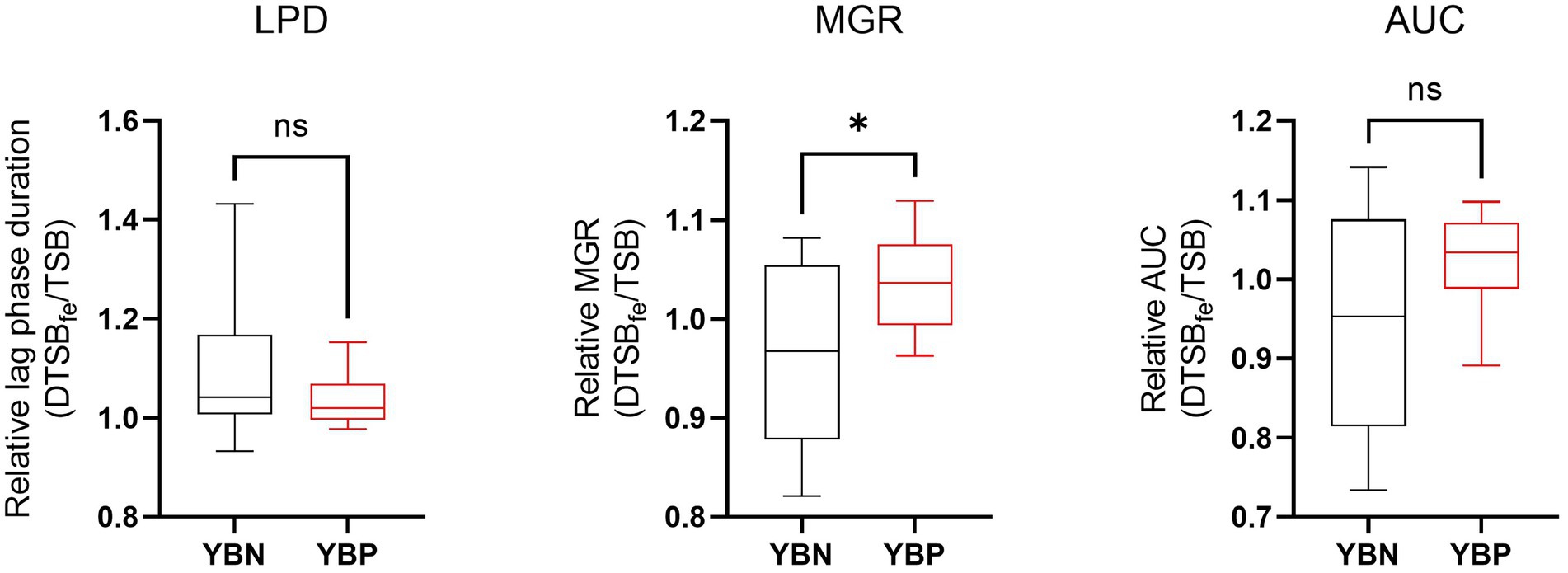
Figure 5. Yersiniabactin operon possession enhances growth rate under iron limited conditions in deferrated TSB media supplemented with 5 μM ferrous Iron (DTSBfe). Box plots comparison of relative growth parameters; area under growth curve (AUC), lag phase duration (LPD), and maximum growth rate (MGR) determined under limited iron conditions (5 μM Fe) in DTSB for the yersiniabactin negative (YBN; n = 18) and positive (YBP; n = 11) ILS S. aureus strain groups. Growth curve parameters determined under limited iron conditions (DTSBfe) were normalized to growth efficiency in normal TSB for each strain to correct for strain-dependent growth efficiency variation observed in this media. *Statistically significant differences (P < 0.05) between the YBP and YBN groups identified using the Mann–Whitney U test.
VirulenceFinder analysis of the 20 African S. aureus genomes revealed that ILS strains harbored between 5 and 21 of the known virulence factor genes (VFGs; Table 4; Supplementary Tables S6, S7). Besides a core set of 5 VFGs possessed by all the 20 strains, there were 16 VFGs, which were variably distributed among the strains including enterotoxin, toxic shock syndrome, leukotoxin, and epidermal cell differentiation inhibitor genes. Panton-Valentine leucocidin genes (lukS/F-PV) were present in three (ILS-09, ILS-77, and ILS-79) strains, whereas the leukotoxin DE genes (lukD/E) were harbored in 12 strains. One (ILS-53) and four (ILS-12, ILS-37, ILS-72, and ILS-77) strains, respectively, carried the toxic shock syndrome (tst1) and epidermal cell differentiation inhibitor B (edinB) genes. The staphylococcal complement inhibitor (scn) gene was detected in seven (ILS-09, ILS-20, ILS-53, ILS-62, ILS-71, ILS-75, and ILS-79) strains, whereas eight strains, most (7/8) of which had the egc enterotoxin gene cluster (seg, sei, sem, sen, seo, and seu) carried enterotoxin genes. The enterotoxin A (sea) gene was detected in five (ILS-48, ILS-53, ILS-66, ILS-75, and IL-S79) strains. SED (sed), SEJ (sej) and SER (ser) genes were detected in a single strain ILS-75, which also harbored sea and the egc cluster genes. PlasmidFinder detected seven plasmid replicon types among the 20 African S. aureus strain genomes with 15 (75%) genomes containing between one and three plasmid replicon types, whereas five genomes had none (Table 4; Supplementary Table S6). ARG screening using ResFinder revealed that 9 of the 20 genomes contained between one and eight resistance genes targeting aminoglycosides (aad and aa6; n = 3), tetracyclines (tetK; n = 8), beta-lactams (blaZ; n = 5), quinolones (grlA and gyrA; n = 1), and sulphonamides (dfrG; n = 1). Meanwhile examining the sequence environment associated with the tetracycline (tetK) and beta-lactam resistance (blaZ) ARGs showed that they were located on four plasmid types based on contig size and sequence composition. Most of the tetK genes were located on a 4-kb contig harboring plasmid replicon type 7a but in one strain (ILS-75) the gene was located on large 36-kb contig harboring a rep 7a type plasmid replicon, β-lactam, bleomycin and cadmium resistance genes as well as sed and sej enterotoxin genes. The β-lactam resistance genes in three strains ILS-62, ILS-20, and ILS-09 was located on contigs carrying plasmid replicon types rep7c, 7a, and 20 origins, respectively, as well as cadmium resistance and bacteriocin transporter genes. One strain (ILS-79) had β-lactam resistance genes located together with bacteriocin transporter genes on 20-kb contig carrying a plasmid replicon type rep 7a origin. Phenotypic resistance testing against selected antibiotics showed that all strains were sensitive to fosfomycin and teicoplanin but resistant to bacitracin (Supplementary Table S6). There were eight, five and one strain, respectively, that were resistant to tetracycline, penicillin and ciprofloxacin, which harbored corresponding ARGs in agreement with the genomic resistance profiles inferred from ResFinder genome analysis (Table 4).
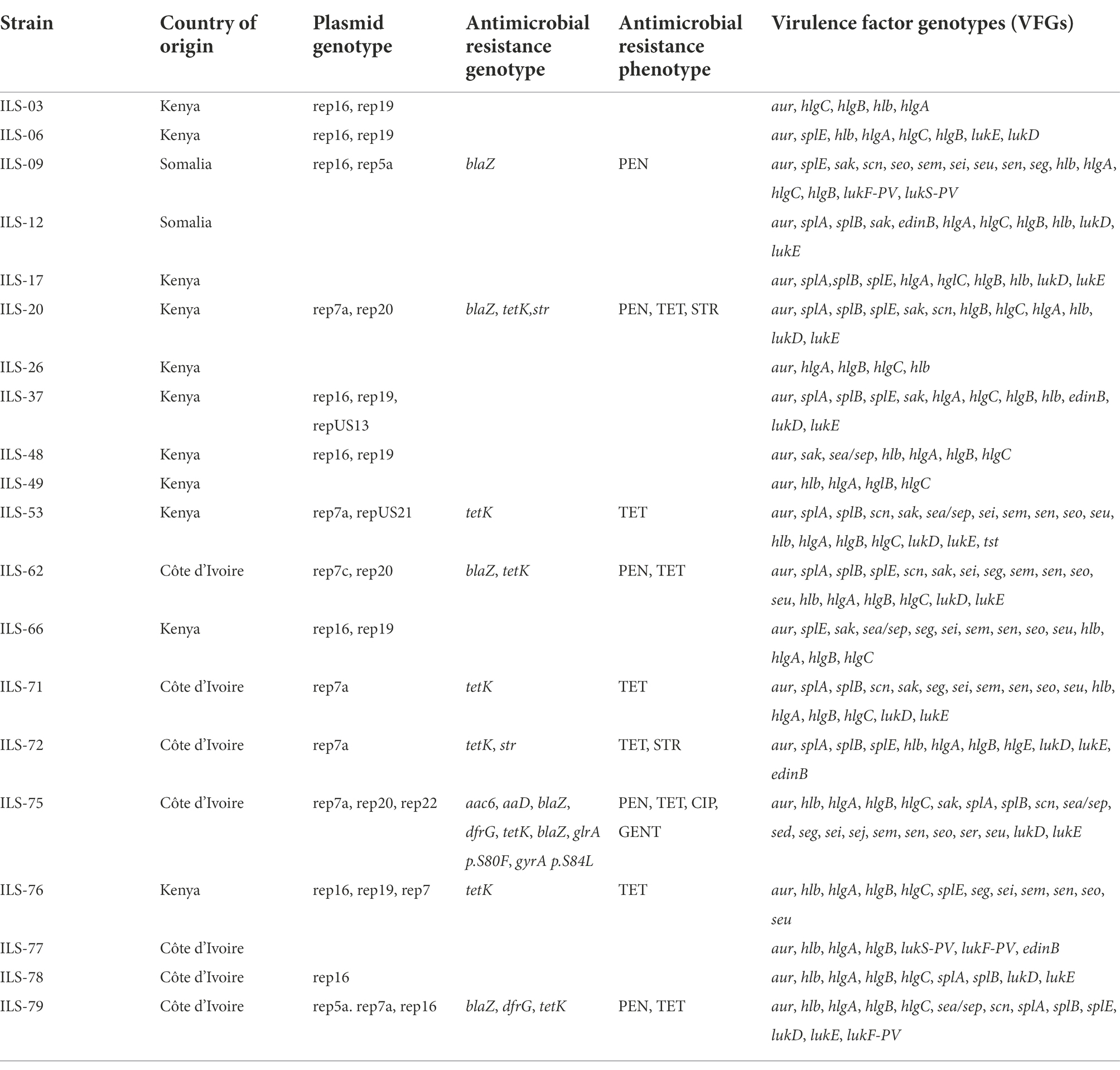
Table 4. ILS S. aureus strains and their genotype and phenotype details on plasmids, antimicrobial resistance, and virulence factors.
Our study aimed at presenting the first insight into genetic makeup of African food-borne S. aureus strains and their population structure through WGS and comparative genomics. Initial characterization on these strains was previously performed through classic MLST and single gene analysis tools (Jans et al., 2017). With the growing wide availability of WGS, we wanted to use this technology for a much more in-depth look at the phylogeny and genetic makeup of these food-borne isolates in comparison with other HA and LA strains of African and global origin. This is also due to the importance of S. aureus as a human and animal pathogen as well as a causative agent for a neglected tropical disease (Herrmann et al., 2013).
In this context, it was important to elucidate the phylogenetic relationship of food-borne S. aureus and whether food-borne isolates are more likely to be related to CA, HA, or LA S. aureus lineages in the African setting. Given the current selection of isolates, phylogenetic analysis allowed for no clear trend to be identified toward any of these three reservoirs as nearly equal numbers of isolates relating to putative CA, HA, and LA lineages were observed. This observation based on WGS data confirms our previous observations on these strains using spa-typing and MLST (Jans et al., 2017).
Importantly, this observation supports previous hypotheses on the role of raw milk in the distribution of S. aureus between human and animal sources as well as within the human or animal reservoir or ecosystem (Tomao et al., 2020; van den Brom et al., 2020). Interestingly, these reports mainly observed the issue of LA lineages being present in milk (Tomao et al., 2020) or the limited development of animal-derived S. aureus infections in humans (Boss et al., 2016). However, the presence not only of LA strains including ST-398 but also CA and HA strains in East and West African milk products suggests a concern for animal and human health alike. Furthermore, it supports the need to classify S. aureus infection as important but so far a neglected tropical disease (Herrmann et al., 2013). Good manufacturing practices during milk production, processing as well as animal welfare would therefore be of great importance for animal and human health alike (van den Brom et al., 2020).
Our population structure analysis confirms and expands previous findings on S. aureus population structure observed in Africa (Ruffing et al., 2017). Our study also indicates that the majority of African isolates are assigned to CCs 5, 15, and 30. These CCs relate to typical S. aureus isolates of Europe and Africa (Ruffing et al., 2017). Interestingly, however, despite that the largest subgroup of ILS isolates belong to CC5 (8 out of 20 isolates), which represents also the predominant CC in the African healthcare setting and a main reservoir for MRSA (Abdulgader et al., 2015), none of the ILS isolates were MRSA. This suggest that at least in the environment and food setting in the countries studied in this work and in contrast to Uganda (Asiimwe et al., 2017), MRSA might not be as prevalent as elsewhere. On the other hand, ILS-77 and ILS-78 were classified as members of CCs 152 and 88, respectively, two CCs reported to be typical for African S. aureus (Abdulgader et al., 2015; Ruffing et al., 2017; Kyany'a et al., 2019). In case of CC152, eight other African isolates formed a distinct African clade with ILS-77, all of which belong to ST152. CC152 is significantly associated with HA and in part also CA strains in Africa (Ruffing et al., 2017) supporting again the potential role of milk as reservoir for various S. aureus lineages. In addition, isolates belonging to previously undescribed CCs 130, 398 and 425 for the African ecosystem were isolated from milk. Particularly the detection of a ST-398 strain, which is related to a highly transmissible clone (Uhlemann et al., 2012), might be concerning regarding its distribution in the community via milk. On a positive note, at least within our strain set, no USA300/ST8 isolates were found, although they are known to have established in the communities in SSA (Strauss et al., 2017). Although our isolate set was small and probably not representative of the entire SSA S. aureus population, these findings showed some clear distinctions from the otherwise expected typical strain portfolio of African community-sourced isolates. Our findings suggest that the population structure of S. aureus in SSA is still in many ways unknown and likely diverse across SSA countries as observed for example in milk products from some regions of Uganda, where a much higher MRSA-prevalence was detected (Asiimwe et al., 2017). Our findings of milk as a reservoir for CA, HA and LA strains furthermore highlight the need for an integrative approach such as that under the One Health concept in order to reduce the burden of S. aureus on animals and humans.
Interestingly, the overall genetic makeup of the 20 ILS S. aureus strains was not significantly different from the publicly available other S. aureus genomes globally. This was reflected in the small number of CDS identified as unique to these strains most of which do not yet possess an annotated function. Therefore, the ILS strains still contributed to the open pan genome of S. aureus (Bosi et al., 2016) even though the rate of added new genes per strains seems to be minor.
Despite the small number of unidentified CDS, by utilizing clade-specific markers, we observed differences that were shared mainly within a clade but so far not further described. One such feature was the detection of a putative siderophore operon annotated as yersiniabactin and the identification of a main “Yersiniabactin clade.” This clade is mainly comprised of LA-isolates of African and a few European sources. The isolates were identified as members of CCs 707, 130, 425 with the exception of ILS-06 being assigned to CC5. Siderophores have been described in S. aureus in the past such as staphyloferrin A and B (Conroy et al., 2019) which was identified, e.g., in ILS-03. However, yersiniabactin was previously not described in S. aureus and no other blast hits could be found. The closest link was established by sequence identity and genetic organization to S. equi. It remains, however, unclear, whether this was an insertion or deletion event. Given the complete absence of any remaining pseudogenes or genetic leftovers in other S. aureus strains, we hypothesize this yersiniabactin to be an insertion, for which not sufficient numbers of isolates are yet available to reconstruct the evolutionary tree. Our newly developed PCR-test identified at least one other LA-isolate of Swiss origin and could be helpful in finding answers. In support of its annotated role in iron acquisition, we showed by comparing strain growth dynamics in vitro that strains possessing this operon might have a growth fitness advantage compared to those without as they displayed a higher growth rate under limited iron availability conditions. We thus hypothesize that possession of this operon might provide growth advantage to S. aureus strains within infected hosts when iron availability is limited. The hypothesized role of this operon in iron acquisition would, however, need to be examined through further experimental work including confirmation experiments conducted using knock-out (isogenic deletion) and complemented mutants of S. aureus strains. The hypothesis of a growth advantage under iron limited conditions was, however, previously already suggested in connection with Bifidobacterium strains isolated from iron deficient Kenyan infants (Vazquez-Gutierrez et al., 2015).
In addition to providing a high-resolution genetic population structure of these SSA milk-associated S. aureus strains, WGS further confirmed previously described public health risks of such strains as many of them possess virulence factors and ARGs. Although the strain set analyzed was comprised entirely of MSSA strains, the examined strains also harbored resistance genes against other antibiotics commonly used in human and veterinary medicine such as penicillin and tetracycline. Although some differences in distribution and genetic sequences of some virulence factors and ARGs were observed, there were no obvious genetic features identified that are unique to the examined SSA S. aureus strain collection. Typical virulence factors identified included scn, a marker gene encoding for an immune evasion cluster (IEC) frequently associated with human origin S. aureus, as well as genes encoding for classical food poisoning-associated enterotoxins and the toxic shock syndrome (TSST-1). Moreover, three strains harbored pvl showing that significant populations of methicillin-sensitive, pvl-positive S. aureus strains that were reported for African healthcare settings may have extended their reservoir beyond the clinical setting (Abdulgader et al., 2015). In combination with the genotypically identified antimicrobial resistance, which in many cases were phenotypically confirmed, these findings support our previous conclusions and further stresses the need to further investigate non-clinical S. aureus in order to tackle S. aureus infections as a neglected tropical disease (Herrmann et al., 2013). It is, however, also important to point out that our evaluation of virulence and antibiotic resistance genes within the examined set of SSA S. aureus strain genomes is by no means exhaustive. Our genetic comparison is limited to the extent of the current information that is available within the publicly available databases that were used in our study. It is thus also expected that, besides variation in the genetic elements identified here, the SSA S. aureus genomes examined also possess further genetic variation in other putative virulence and ARGs that are not captured within the databases queried in our study.
In conclusion, the population structure, genotypic makeup, presence of virulence factors and antimicrobial resistance supports the theory of milk as a reservoir for S. aureus in SSA as many of the features reported to be typical for African clinical S. aureus isolates could be observed in milk isolates. Given the role of S. aureus infections as neglected tropical disease, the impact of S. aureus on human and animal health alike and as such also on the livestock economy, an integrative One Health approach is required for future success. Comparative genomics in this case again helped to identify novel traits such as the putative yersiniabactin siderophore and to develope detection methods. Despite the vast number of available isolates and whole genome sequences, detailed characterization on genome sequence level still proves to be highly beneficial and supportive toward a better understanding of S. aureus, even if such isolates were previously already analyzed using MLST or other typing schemes.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
CJ and TT wrote the grant application and the manuscript draft, designed the experiments, supervised lab work, and performed analyses and interpretation of data for comparative genomics, phylogeny, genetic composition and link to phenotype data. JW performed and interpreted phenotypic and genotypic antimicrobial resistance and siderophore experiments. MS performed the analysis on plasmids, ARGs, and virulence genes and provided subsequent interpretations. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was funded by the World Food System Center of ETH Zurich, Switzerland and the Institute of Food Safety and Hygiene from the University of Zurich. This study was based on the reanalysis of milk samples previously collected during the frame of projects funded by the North–South Centre of ETH Zurich and the UBS Optimus Foundation in Côte d’Ivoire, Kenya and Somalia.
The authors greatly acknowledge the original sample collection team of the Laboratory of Food Biotechnology, ETH Zurich (Sabine Tanner and Leo Meile), Switzerland; Centre Suisse de Recherches Scientifiques en Côte d’Ivoire (Sylvie Kouamé Sina and Bassirou Bonfoh); the University of Nairobi in Kenya (Dasel Wambua Mulwa Kaindi, John Wangoh, and Patrick Murigu Kamau Njage); and Vétérinaires Sans Frontières Germany in Kenya (Mario Younan and Illona Glücks).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.923080/full#supplementary-material
1. ^https://github.com/sanger-pathogens/assembly-stats (Accessed July 20, 2022).
2. ^https://pathogen.watch/ (Accessed July 20, 2022).
3. ^http://www.genomicepidemiology.org/ (Accessed July 20, 2022).
Abdulgader, S. M., Shittu, A. O., Nicol, M. P., and Kaba, M. (2015). Molecular epidemiology of methicillin-resistant Staphylococcus aureus in Africa: a systematic review. Front. Microbiol. 6:348. doi: 10.3389/fmicb.2015.00348
Asiimwe, B. B., Baldan, R., Trovato, A., and Cirillo, D. M. (2017). Prevalence and molecular characteristics of Staphylococcus aureus, including methicillin resistant strains, isolated from bulk can milk and raw milk products in pastoral communities of south-West Uganda. BMC Infect. Dis. 17:422. doi: 10.1186/s12879-017-2524-4
Blom, J., Albaum, S. P., Doppmeier, D., Puhler, A., Vorholter, F. J., Zakrzewski, M., et al. (2009). EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics 10:154. doi: 10.1186/1471-2105-10-154
Blom, J., Kreis, J., Spanig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Bosi, E., Monk, J. M., Aziz, R. K., Fondi, M., Nizet, V., and Palsson, B. O. (2016). Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 113, E3801–E3809. doi: 10.1073/pnas.1523199113
Boss, R., Cosandey, A., Luini, M., Artursson, K., Bardiau, M., Breitenwieser, F., et al. (2016). Bovine Staphylococcus aureus: subtyping, evolution, and zoonotic transfer. J. Dairy Sci. 99, 515–528. doi: 10.3168/jds.2015-9589
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
CLSI (2017). Performance Standards for Antimicrobial Susceptibility Testing. CLSI supplement m100. 27th Edn. Wayne, PA: Clinical and Laboratory Standards Institute.
Conroy, B. S., Grigg, J. C., Kolesnikov, M., Morales, L. D., and Murphy, M. E. P. (2019). Staphylococcus aureus heme and siderophore-iron acquisition pathways. Biometals 32, 409–424. doi: 10.1007/s10534-019-00188-2
Cosentino, S., Voldby Larsen, M., Aarestrup, F. M., and Lund, O. (2013). PathogenFinder: distinguishing friend from foe using bacterial whole genome sequence data. PLoS One 8:e77302. doi: 10.1371/journal.pone.0077302
David, M. Z., and Daum, R. S. (2010). Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616. doi: 10.1128/cmr.00081-09
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739. doi: 10.1016/S0140-6736(06)68231-7
Enright, M. C., Day, N. P., Davies, C. E., Peacock, S. J., and Spratt, B. G. (2000). Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38, 1008–1015.
Enright, M. C., Robinson, D. A., Randle, G., Feil, E. J., Grundmann, H., and Spratt, B. G. (2002). The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. U. S. A. 99, 7687–7692. doi: 10.1073/pnas.122108599
Harris, S. R., Feil, E. J., Holden, M. T., Quail, M. A., Nickerson, E. K., Chantratita, N., et al. (2010). Evolution of MRSA during hospital transmission and intercontinental spread. Science 327, 469–474. doi: 10.1126/science.1182395
Herbert, S., Ziebandt, A. K., Ohlsen, K., Schäfer, T., Hecker, M., Albrecht, D., et al. (2010). Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78, 2877–2889. doi: 10.1128/IAI.00088-10
Herrmann, M., Abdullah, S., Alabi, A., Alonso, P., Friedrich, A. W., Fuhr, G., et al. (2013). Staphylococcal disease in Africa: another neglected 'tropical' disease. Future Microbiol. 8, 17–26. doi: 10.2217/fmb.12.126
Jans, C., Bugnard, J., Njage, P. M. K., Lacroix, C., and Meile, L. (2012). Lactic acid bacteria diversity of African raw and fermented camel milk products reveals a highly competitive, potentially health-threatening predominant microflora. LWT-Food Sci. Technol. 47, 371–379. doi: 10.1016/j.lwt.2012.01.034
Jans, C., Kaindi, D. W. M., Böck, D., Njage, P. M. K., Kouamé-Sina, S. M., Bonfoh, B., et al. (2013). Prevalence and comparison of Streptococcus infantarius subsp. infantarius and Streptococcus gallolyticus subsp. macedonicus in raw and fermented dairy products from East and West Africa. Int. J. Food Microbiol. 167, 186–195. doi: 10.1016/j.ijfoodmicro.2013.09.008
Jans, C., Merz, A., Johler, S., Younan, M., Tanner, S. A., Kaindi, D. W. M., et al. (2017). East and West African milk products are reservoirs for human and livestock-associated Staphylococcus aureus. Food Microbiol. 65, 64–73. doi: 10.1016/j.fm.2017.01.017
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Kyany'a, C., Nyasinga, J., Matano, D., Oundo, V., Wacira, S., Sang, W., et al. (2019). Phenotypic and genotypic characterization of clinical Staphylococcus aureus isolates from Kenya. BMC Microbiol. 19:245. doi: 10.1186/s12866-019-1597-1
Lakhundi, S., and Zhang, K. (2018). Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin. Microbiol. Rev. 31:e00020-18. doi: 10.1128/CMR.00020-18
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/jcm.06094-11
Ledala, N., Zhang, B., Seravalli, J., Powers, R., and Somerville, G. A. (2014). Influence of iron and aeration on Staphylococcus aureus growth, metabolism, and transcription. J. Bacteriol. 196, 2178–2189. doi: 10.1128/JB.01475-14
Lozano, C., Gharsa, H., Ben Slama, K., Zarazaga, M., and Torres, C. (2016). Staphylococcus aureus in animals and food: methicillin resistance, prevalence and population structure. A review in the African continent. Microorganisms 4:12. doi: 10.3390/microorganisms4010012
Malachowa, N., and DeLeo, F. R. (2010). Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 67, 3057–3071. doi: 10.1007/s00018-010-0389-4
Monecke, S., Coombs, G., Shore, A. C., Coleman, D. C., Akpaka, P., Borg, M., et al. (2011). A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One 6:e17936. doi: 10.1371/journal.pone.0017936
Nelson, R. E., Jones, M., Liu, C. F., Samore, M. H., Evans, M. E., Stevens, V. W., et al. (2018). The impact of healthcare-associated methicillin-resistant Staphylococcus aureus infections on postdischarge health care costs and utilization across multiple health care systems. Health Serv. Res. 53, 5419–5437. doi: 10.1111/1475-6773.13063
Njage, P. M. K., Dolci, S., Jans, C., Wangoh, J., Lacroix, C., and Meile, L. (2013). Biodiversity and enterotoxigenic potential of staphylococci isolated from raw and spontaneously fermented camel milk. Br. Microbiol. Res. J. 3, 128–138. doi: 10.9734/BMRJ/2013/2896
Owusu-Kwarteng, J., Akabanda, F., Agyei, D., and Jespersen, L. (2020). Microbial safety of milk production and fermented dairy products in Africa. Microorganisms 8:752. doi: 10.3390/microorganisms8050752
Paudyal, N., Anihouvi, V., Hounhouigan, J., Matsheka, M. I., Sekwati-Monang, B., Amoa-Awua, W., et al. (2017). Prevalence of foodborne pathogens in food from selected African countries: a meta-analysis. Int. J. Food Microbiol. 249, 35–43. doi: 10.1016/j.ijfoodmicro.2017.03.002
Reddy, E. A., Shaw, A. V., and Crump, J. A. (2010). Community-acquired bloodstream infections in Africa: a systematic review and meta-analysis. Lancet Infect. Dis. 10, 417–432. doi: 10.1016/S1473-3099(10)70072-4
Robertson, J., and Nash, J. H. E. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 4:e000206. doi: 10.1099/mgen.0.000206
Ruffing, U., Alabi, A., Kazimoto, T., Vubil, D. C., Akulenko, R., Abdulla, S., et al. (2017). Community-associated Staphylococcus aureus from sub-Saharan Africa and Germany: a cross-sectional geographic correlation study. Sci. Rep. 7:154. doi: 10.1038/s41598-017-00214-8
Schaumburg, F., Alabi, A. S., Peters, G., and Becker, K. (2014). New epidemiology of Staphylococcus aureus infection in Africa. Clin. Microbiol. Infect. 20, 589–596. doi: 10.1111/1469-0691.12690
Schijffelen, M. J., Boel, C. H., van Strijp, J. A., and Fluit, A. C. (2010). Whole genome analysis of a livestock-associated methicillin-resistant Staphylococcus aureus ST398 isolate from a case of human endocarditis. BMC Genomics 11:376. doi: 10.1186/1471-2164-11-376
Seligsohn, D., Nyman, A. K., Younan, M., Sake, W., Persson, Y., Bornstein, S., et al. (2020). Subclinical mastitis in pastoralist dairy camel herds in Isiolo, Kenya: prevalence, risk factors, and antimicrobial susceptibility. J. Dairy Sci. 103, 4717–4731. doi: 10.3168/jds.2019-17701
Strauss, L., Stegger, M., Akpaka, P. E., Alabi, A., Breurec, S., Coombs, G., et al. (2017). Origin, evolution, and global transmission of community-acquired Staphylococcus aureus ST8. Proc. Natl. Acad. Sci. U. S. A. 114, E10596–E10604. doi: 10.1073/pnas.1702472114
Tam, K., and Torres, V. J. (2019). Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol. Spectr. 7:2018. doi: 10.1128/microbiolspec.GPP3-0039-2018
Tomao, P., Pirolo, M., Agnoletti, F., Pantosti, A., Battisti, A., Di Martino, G., et al. (2020). Molecular epidemiology of methicillin-resistant Staphylococcus aureus from dairy farms in North-Eastern Italy. Int. J. Food Microbiol. 332:108817. doi: 10.1016/j.ijfoodmicro.2020.108817
Tong, S. Y., Davis, J. S., Eichenberger, E., Holland, T. L., and Fowler, V. G. Jr. (2015). Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28, 603–661. doi: 10.1128/CMR.00134-14
Turner, N. A., Sharma-Kuinkel, B. K., Maskarinec, S. A., Eichenberger, E. M., Shah, P. P., Carugati, M., et al. (2019). Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat. Rev. Microbiol. 17, 203–218. doi: 10.1038/s41579-018-0147-4
Uhlemann, A. C., Porcella, S. F., Trivedi, S., Sullivan, S. B., Hafer, C., Kennedy, A. D., et al. (2012). Identification of a highly transmissible animal-independent Staphylococcus aureus ST398 clone with distinct genomic and cell adhesion properties. MBio 3:e00027. doi: 10.1128/mBio.00027-12
van den Brom, R., de Jong, A., van Engelen, E., Heuvelink, A., and Vellema, P. (2020). Zoonotic risks of pathogens from sheep and their milk borne transmission. Small Rumin. Res. 189:106123. doi: 10.1016/j.smallrumres.2020.106123
Vazquez-Gutierrez, P., Lacroix, C., Jaeggi, T., Zeder, C., Zimmerman, M. B., and Chassard, C. (2015). Bifidobacteria strains isolated from stools of iron deficient infants can efficiently sequester iron. BMC Microbiol. 15:3. doi: 10.1186/s12866-014-0334-z
Wambui, J., Eshwar, A. K., Aalto-Araneda, M., Pontinen, A., Stevens, M. J. A., Njage, P. M. K., et al. (2020). The analysis of field strains isolated from food, animal and clinical sources uncovers natural mutations in Listeria monocytogenes nisin resistance genes. Front. Microbiol. 11:549531. doi: 10.3389/fmicb.2020.549531
Wambui, J., Njage, P. M. K., Tasara, T., and Buys, E. M. (2019). Meta-analysis and meta-regression indicate dynamic prevalence and moderators of foodborne pathogens in African indigenous fermented milk. Microorganisms 7:563. doi: 10.3390/microorganisms7110563
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Staphylococcus aureus, siderophore, iron scavenger, population structure, African dairy products, One Health, neglected tropical disease
Citation: Jans C, Wambui J, Stevens MJA and Tasara T (2022) Comparative genomics of dairy-associated Staphylococcus aureus from selected sub-Saharan African regions reveals milk as reservoir for human-and animal-derived strains and identifies a putative animal-related clade with presumptive novel siderophore. Front. Microbiol. 13:923080. doi: 10.3389/fmicb.2022.923080
Edited by:
Vincenzina Fusco, Italian National Research Council, ItalyReviewed by:
Timothy J. Foster, Trinity College Dublin, Ireland Mingsong Kang, Canadian Food Inspection Agency, CanadaCopyright © 2022 Jans, Wambui, Stevens and Tasara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taurai Tasara, dGFzYXJhdEBmc2FmZXR5LnV6aC5jaA==
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.