 Xiancui Zhang
Xiancui Zhang Huihui Feng1
Huihui Feng1 Abrar Muhammad
Abrar Muhammad Fan Zhang
Fan Zhang- 1College of Animal Sciences, Institute of Sericulture and Apiculture, Zhejiang University, Hangzhou, China
- 2Key Laboratory of Animal Resistance Biology of Shandong Province, College of Life Science, Shandong Normal University, Jinan, China
The complex gut microbiome is a malleable microbial community that can undergo remodeling in response to many factors, including the gut environment and microbial properties. Enterococcus has emerged as one of the predominant gut commensal bacterial and plays a fundamental role in the host physiology and health of the major economic agricultural insect, Bombyx mori. Although extensive research on gut structure and microbiome diversity has been carried out, how these microbial consortia are established in multifarious niches within the gut has not been well characterized to date. Here, an Enterococcus species that was stably associated with its host, the model organism B. mori, was identified in the larval gut. GFP–tagged E. faecalis LX10 was constructed as a model bacterium to track the colonization mechanism in the intestine of B. mori. The results revealed that the minimum and optimum colonization results were obtained by feeding at doses of 105 CFU/silkworm and 107 CFU/silkworm, respectively, as confirmed by bioassays and fluorescence-activated cell sorting analyses (FACS). Furthermore, a comprehensive genome-wide exploration of signal sequences provided insight into the relevant colonization properties of E. faecalis LX10. E. faecalis LX10 grew well under alkaline conditions and stably reduced the intestinal pH through lactic acid production. Additionally, the genomic features responsible for lactic acid fermentation were characterized. We further expressed and purified E. faecalis bacteriocin and found that it was particularly effective against other gut bacteria, including Enterococcus casselifavus, Enterococcus mundtii, Serratia marcescens, Bacillus amyloliquefaciens, and Escherichia coli. In addition, the successful colonization of E. faecalis LX10 led to drastically increased expression of all adhesion genes (znuA, lepB, hssA, adhE, EbpA, and Lap), defense genes (cspp, tagF, and esp), regulation gene (BfmRS), secretion gene (prkC) and immune evasion genes (patA and patB), while the expression of iron acquisition genes (ddpD and metN) was largely unchanged or decreased. This work establishes an unprecedented conceptual model for understanding B. mori–gut microbiota interactions in an ecological context. Moreover, these results shed light on the molecular mechanisms of gut microbiota proliferation and colonization in the intestinal tract of this insect.
Introduction
The insect gut is colonized by a complex bacterial community that governs a wide range of functions contributing to the host’s physiology, pathogen defense, development, and immune protection (Engel and Moran, 2013; Xiao et al., 2017). Thus, the gut microbiota may be recognized as a virtual “bacterial organ” that is integrated into the host’s biological system and fundamental to its health (Aksoy, 2018; Schretter, 2020). For instance, a comparison between aposymbiotic and symbiotic insects showed that the Burkholderia symbiont dramatically increases the fecundity, growth rate, and body size of stinkbugs by providing essential metabolites, and the insect host can also develop resistance against the insecticide fenitrothion (Kikuchi et al., 2007; Kikuchi and Fukatsu, 2014). The phytophagous leafminer Phyllonorycter blancardella (Lepidoptera) co-opts the bacterial endosymbiont Wolbachia to improve its nutritional and physiological environment (Kaiser et al., 2010). Furthermore, associations with specific microorganisms may improve environmental fitness or provide new opportunities for exploring new niches (Robinson et al., 2018). Although myriad microorganisms inhabit the guts of insects, many gut community members are transient, and it is sometimes difficult to create a resident microbiota due to various intrinsic factors (developmental stage, sex, genetics, gut pH, digestive enzymes, etc.) (Engel and Moran, 2013; Hammer et al., 2017).
The silkworm, Bombyx mori, is a powerful model organism for studying the bacterial colonization of the invertebrate intestine and for deciphering the interactions between the host and its gut microbiota (Chen et al., 2018, 2020). The midguts of silkworm larvae exhibit extremely high alkalinity, with pH levels of 10–11, and their digestive enzymes are adapted to the alkaline environment (Liang et al., 2018). Recent extensive surveys have revealed that these adverse conditions may support the growth of alkaline-resistant and facultative anaerobic microorganisms, such as Firmicutes, Planctomycetes, and Clostridium (Bignell et al., 2011; Yeruva et al., 2020). For example, Enterococcus (Firmicutes), which plays a crucial role in metabolic adaptability against pathogenic or plant toxins and anti-herbivore defense, was found to be one of the predominant gut microorganism of lepidopteran insects, including B. mori, Helicoverpa zea, and Porthetria dispar (Paniagua Voirol et al., 2018; Zhang et al., 2022). Some other insects harbor similar bacterial lineages in their alkaline guts (Egert et al., 2003; Broderick et al., 2004). Several physiological and biochemical characteristics of the gut environment influence the colonization of such symbionts, including the constant expulsion of intestinal contents via peristalsis, pH levels, reactive oxygen species, nutrient availability, digestive enzymes, and possibly antimicrobial agents (Siegel and Weiser, 2015). Thus, it appears likely that bacteria stably inhabiting the gut would have developed sophisticated mechanisms to resist these challenges.
The green fluorescent protein (GFP) of the jellyfish Aequorea victoria, first described in the 1970s, is a bright, stable molecular marker used in live-cell imaging to facilitate bacterial localization, isolation, and tracking in biological studies of several host species (Rizzo et al., 2009; Viswanathan et al., 2015). Genetically modified bacteria labeled with GFP or other fluorescent proteins have provided a unique and powerful tool for understanding the spatiotemporal dynamics and ecological interactions of the gut microbiota (Wang S. et al., 2017). Recently, similar research has been performed in Pseudomonas-zebrafish, Photorhabdus-nematode, Burkholderia-bean bug, and other model symbiotic systems (Rawls et al., 2007; Ciche et al., 2008; Kikuchi and Fukatsu, 2014). For example, the Pseudomonas aeruginosa: GFP transcriptional reporter strain is detectable in the Caenorhabditis elegans head and intestine regions using fluorescence microscopy (Sellegounder et al., 2019). Despite the recognized importance of gut-associated bacteria to B. mori, how and where its gut microbiota achieves colonization and the genetic and molecular bases of colonization are still open questions. In the current study, we constructed a genetically engineered strain of E. faecalis LX10 expressing GFP to visualize how the test organism adapted to the gut environment of silkworms. E. faecalis LX10 grew well under alkaline conditions and showed efficient lactic acid production. We further analyzed bacteriocin production and bacterial competition based on whole-genome sequencing. In addition, we quantified colonization-related genes following the growth of the bacterium in the gut using quantitative real-time PCR (RT–qPCR). The present study enhances our understanding of gut colonization by an indigenous bacterium and may promote the application of probiotic targeting regulation in agricultural and economic insects or for pest control of lepidopterans.
Materials and Methods
Sample Collection and Rearing
The B. mori Haoyue × Jingsong strain was hatched from eggs provided by the Silkworm Germplasm Bank at the College of Animal Sciences, Zhejiang University, China. Larvae were raised on fresh mulberry leaves at 25 ± 1°C under 70 ± 5% relative humidity and an alternating 14:10 h light/dark photoperiod.
Survey and Characteristics of Enterococcus From the Gut of Bombyx mori
Enterococcus bacteria were isolated from the gut of normal B. mori larvae using Enterococcus-selective agar with 1% 2,3,5-triphenyl tetrazolium chloride (TTC) in the appropriate medium (Hopebio, Qingdao, China) (Supplementary Table 1). Random samples of bacterial colonies were picked from agar plates and subcultured at least three times before identification. Molecular analysis was performed by isolating genomic DNA and amplifying bacterial 16S rRNA genes using the 27 F/1492 R universal primers. The synthesis and sequencing of PCR products were performed by Sangon Biotech (Shanghai, China). The phylogenetic tree was constructed via the multiple sequence alignment of 16S rRNA genes performed with ClustalW and the neighbor-joining method in MEGA7 (7.0.14) software.
After anesthetizing the B. mori larvae on ice, the gut tissue was dissected from larvae of each developmental stage (1st–5th-instar larvae), transferred to a 2 mL sterile centrifuge tube containing 500 μL sterile PBS and then thoroughly homogenized. The number of gut Enterococcus bacteria in each sample was quantified by standard colony-forming unit (CFU) enumeration and absolute quantification via quantitative real-time PCR (qPCR). To determine CFU counts, each sample was serially diluted from 1 × 10–1 to 1 × 10–6 and plated, and the plates were incubated for 48 h at 30°C until colonies formed. Colonies were then manually counted (n = 10). For absolute quantification, total DNA was extracted from 1st–5th-instar larvae using a DNA kit (Epicenter, Madison, United States) following the manufacturer’s protocol. The final DNA concentration and purity were measured on a 2100 Bioanalyzer (Agilent, Palo Alto, United States), and DNA quality was then checked by 1% agarose gel electrophoresis. The absolute quantification of Enterococcus species, including E. faecalis, E. mundtii, and E. casseliflavus, was conducted by qPCR analysis (n = 10). The primers used in the qPCR assays are shown in Supplementary Table 2. The thermocycler conditions were as follows: 95°C for 5 min and 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 20 s, followed by a final melting curve step (from 65 to 92°C, 0.5°C/s). A standard curve was generated from a mixed sample based on a series of 10–fold dilutions with the same reaction parameters, and the total 16S rRNA gene copies in each sample were calculated according to the standard curve.
For scanning electron microscopy (SEM) analysis, a E. faecalis LX10 culture was directly transferred to a fixative solution containing 2.5% glutaraldehyde (pH 7.4). Gut samples were then postfixed in 1% osmium tetroxide (OsO4) for 2 h, followed by dehydration in a graded ethanol series (70–100%) for 15 min. After washing with PBS, samples were dehydrated and critical point dried using carbon dioxide. Specimens were mounted on adhesive carbon films, coated with platinum (1 nm), and examined via SEM (Hitachi SU8000, Tokyo, Japan).
For fluorescence in situ hybridization (FISH) analysis, OCT-embedded gut tissue was frozen in a dry ice bath and sectioned at a 5 μm thickness on a freezing microtome (CryoStar, NX70, France). FISH was performed with a Cy3–labeled (GAAAGCGCCTTTCACTCTTATGC) E. faecalis–specific probe (red), and DNA was counterstained with DAPI (blue) in hybridization buffer (900 mM NaCl, 20% formamide, 20 mM Tris–HCl, 1% SDS) (Wellinghausen et al., 2007). Hybridization was achieved under previously described conditions (Koga et al., 2009).
Construction of Green Fluorescent Protein–Enterococcus faecalis LX10
The bacterial strains and plasmids used in this study are listed in Supplementary Table 1. The GFP-containing plasmids were maintained in the Escherichia coli DH5α strain (TaKaRa, Dalian, China). E. coli strains were grown at 30°C in lysogenic broth (LB) medium. An enterococcal erythromycin ribosomal methylase (ermB) promoter drives green fluorescent protein 5 (GFP5) in the pTRKH3–ermGFP construct (Swinfield et al., 1990; Lizier et al., 2010) (Addgene plasmid 27169). pTRKH3–ermGFP was electroporated into E. faecalis LX10 competent cells using an Eppendorf Electroporator 2510 (Eppendorf, Hamburg, Germany) at 1.5 kV, 600 Ω, and 10 μF, with a pulse length of 3.6 ms. The electroporated cells were immediately mixed with 900 μL Todd Hewitt broth (THB), incubated for 1 h at 30°C, and plated on THB-agar containing erythromycin (5 μg/mL). Total DNA was extracted from GFP–tagged bacteria using a MasterPure™ complete DNA and RNA Purification Kit (Epicenter, Madison, United States) following the manufacturer’s protocol. The GFP gene (735 bp, GFP F/R) was amplified by PCR in a final volume of 25 μL using Taq DNA polymerase according to the manufacturer’s instructions (TaKaRa, Dalian, China). Images were captured with a confocal laser scanning microscope (CLSM) (LSM780, ZEISS, Germany). Growth curve analyses of E. faecalis LX10 and GFP–E. faecalis LX10 were conducted in 96-well honeycomb plates (Corning, United States), measuring the absorption at 600 nm for a total of 40 h in 4 h intervals.
Analysis of Green Fluorescent Protein–Enterococcus faecalis Stability and Proliferation in the Bombyx mori Gut
Bacterial colonization challenges were performed with GFP–E. faecalis LX10. Each silkworm was fed on mulberry leaves (5 cm × 5 cm) that were previously treated with a GFP–E. faecalis LX10 suspension (1 × 103, 1 × 105, 1 × 107, or 1 × 109 CFU/mL) for 4 h. The same volume of sterilized PBS solution was used in the control groups (CK). The gut stability protocol conducted in a 90 mm petri dish was conducted with a single silkworm per dish, and feces were removed every 6 h. The gut tissue from each single gut was homogenized after 0, 1, 3, 5, or 7 days as described above.
Total gut GFP–E. faecalis counts were quantified by CFU enumeration, qPCR, and FACS. The CFUs were counted on each plate after a 48-h incubation period at 30°C (n = 10). Absolute quantification of the amount of GFP–E. faecalis 16S rRNA gene copies was conducted with the GFP F/R primer pair (Supplementary Table 2) (n = 10). For FACS, fluorescent E. faecalis bacteria were separated from the intestinal homogenate by filtration via 40 μm sterile cell strainers (Corning, United States) and diluted to the appropriate concentration. FACS analyses were performed using a BD FACSAria™ Fusion Cell Sorter (Becton Dickinson, Heidelberg, Germany). The filtrates were then separated into aliquots of 1 mL for each sample. GFP fluorescence was detected using a 488–nm blue laser line for signal excitation, and the wavelength was set to 493–578 nm. Intestinal bacteria were subjected to FACS to sort the GFP–positive and GFP–negative fractions. The corresponding control samples included a positive control (GFP–E. faecalis) and a negative control (intestinal homogenate without GFP) (n = 3). In addition, GFP fluorescence in the silkworm intestinal tract was visualized under an LSM780 confocal microscope (Zeiss) with oil immersion objectives after the silkworms had fed on GFP–E. faecalis for 5 days. Notably, the peritrophic membrane (PM) and intestinal gut digestive juice were mounted on slides and stored at 4°C until use. To determine if intestinal juice affects E. faecalis morphology, gut tissues were homogenized in a Precellys–24 homogenizer (Bertin Technologies, Aixen, France) at 5,500 × g for 20 s, centrifuged briefly at 8,000 g, and filtered through a 0.22 μm filter. E. faecalis was grown statically at 30°C in the presence of the extracted sterile intestinal juice added to THB liquid media at 1:1.
Larvae were weighed 7 d after inoculation with GFP–E. faecalis LX10. Pupation and adult emergence rates, whole cocoon weight, and cocoon shell weight were recorded after cocooning (n = 10).
Growth Test at Different pH Levels and Measurement of Gut pH and Lactic Acid Production
To determine the growth rate of E. faecalis LX10 at different pH levels (6–11), the THB medium was dissolved in solutions with different pH levels in 100 mM NaHCO3/Na2CO3 buffer. E. faecalis LX10 cells were then immediately subjected to lactic acid measurement under different initial pH levels (n = 5, pH = 6–11) and different bacterial concentrations (n = 5, pH = 11, 103, 105, 107, or 109 CFU/mL) after 24 h of incubation using a Lactic Acid (LA) Content Assay Kit (D799099–0100, Sangon Biotech, Shanghai, China) (n = 5). A pH microelectrode (Unisense, Aarhus, Denmark) with a 20–30 μm tip diameter was used to measure the pH levels of fresh gut extracts obtained after silkworms had fed on a GFP–E. faecalis LX10 suspension (1 × 103, 1 × 105, 1 × 107, or 1 × 109 CFU/mL) for 5 d as previously described (Liu et al., 2016), with healthy germ-free B. mori larvae serving as a negative control (CK) (n = 10).
Genome Sequencing, Assembly, and Annotation
The whole genome of E. faecalis LX10 was sequenced on the PacBio RSII platform at Majorbio Bio–pharm Technology Co., Ltd. (Shanghai, China). A total of 1,605 Mbp of high–quality reads were generated and filtered to remove sequences whose 5’ ends contained non–A, T, G, and C bases, sequences < 25 bp in length, sequences containing >10% Ns, and low-quality reads (≤ Q20). Short Oligonucleotide Analysis Package (SOAP) de novo V version 1.05 was used to generate contigs and scaffolds using different kmer sizes, and gaps were filled via the PCR amplification method (Liang et al., 2018; Xie et al., 2019). The raw dataset including both single and paired-end reads (average read length of 14,796 bp), is deposited at GenBank under BioProject and accession number ID: PRJNA810349 and CP092784-CP092785. Genes of strain LX10 were predicted by using Glimmer V3.02 software.1
The obtained genome component predictions included the prediction of protein-coding genes, tandem repetitive sequences, interspersed repeats, transposons, non-coding RNAs, prophages, and clustered regularly interspaced short palindromic repeat (CRISPR) sequences. The applied steps were as follows. The GeneMarkS program (Version 4.17) was used to identify related protein-coding genes (Besemer et al., 2001). Interspersed repetitive sequences were identified from genomic sequences using RepeatMasker,2 and tandem repeats were identified using Tandem Repeats Finder (TRF) (Benson, 1999; Saha et al., 2008). Virulence factors and antibiotic resistance genes were annotated based on the Virulence Factors Database (VFDB) (Liu et al., 2022) and the Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020). PHAST was used for prophage prediction3 (Zhou et al., 2011). Transfer RNA (tRNA) and ribosomal RNA (rRNA) genes were detected with tRNAscan-SE V1.3.14 and Barrnap V0.4.2,5 respectively (Lowe and Eddy, 1997). Small nuclear RNAs (snRNAs) were predicted by BLAST searches against the Rfam database (Nawrocki et al., 2015). The CRISPRFinder online tool6 was used for CRISPR identification (Grissa et al., 2007), and the genomic circle map was visualized by using Circos V0.64.7 Furthermore, the predicted gene functions were compared against the non-redundant protein NCBI non-redundant protein(NR), Clusters of Orthologous Groups (COG), Pfam, Swiss–Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO) databases using BLASTp (BLAST 2.2.28 +) with an E-value cut off of 1e-5 (Ashburner et al., 2000).
Overexpression and Purification of Bacteriocin and Measurement of Antimicrobial Activity
Total genomic DNA from E. faecalis LX10 was obtained and used as target DNA for PCR amplification of the bacteriocin gene (372 bp). The coding regions of bacteriocin (residues 22–126) were further cloned into the pET28a vector and transformed into E. coli BL21 (DE3) cells (TaKaRa, Dalian, China) by homologous recombination using the ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China). DE3 strains were cultured in LB medium, and expression was induced at an OD600 = 0.6 with 1 mM IPTG at 30°C for 6 h. DE3 cells were lysed in lysis buffer (10% glycerol, 25 mM Tris⋅HCl pH 8.0, 1% IGEPAL® CA–630, 0.1 M sodium chloride, and 1 mM PMSF) by sonication for 15 min in an ice bath, followed by centrifugation at 10,000 × g for 30 min at 4°C. The hexahistidine-tagged bacteriocin protein was purified by using Ni–NTA agarose following the recommendations of the column manufacturer (QIAGEN, Germany). The bacteriocin protein was eluted in elution buffer (100 mM sodium chloride, 50 mM sodium phosphate, and 150 mM imidazole) in a small chromatography column (Sangon, Shanghai, China), and the elution fractions were collected. The collected fractions were then subjected to 15% (w/v) sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
The E. faecalis LX10 strain was cultured in THB (Hopebio, Qingdao, China) medium at 30°C for 36 h. The supernatant of E. faecalis LX10 was collected after centrifugation at 12,000 × g for 5 min and filtered through a 0.22 μm PVDF membrane (Millipore Corp., Billerica, MA) to obtain the cell-free fermentation liquid. After incubation at 4°C overnight, the crude proteins were precipitated with 60% saturated ammonium sulfate [(NH4)2SO4] and dialyzed through regenerated cellulose membrane tubing (3.5 kDa, Spectrum) in distilled water. Then, the crude proteins were concentrated via ultrafiltration with Amicon Ultra 3 kDa (Millipore Corp., Billerica, MA).
Antimicrobial activity of E. faecalis was evaluated using the agar well diffusion method (Zhang et al., 2017). For the determination of inhibitory spectra, bacteria strains, including the isolated E. mundtii, E. casseliflavus, S. marcescens, and B. amyloliquefaciens, E. coli, Pseudomonas fulva, Staphylococcus sciuri, and Methylobacterium populi type strains, were grown in the BHI media recommended by culture collections. Holes (10 mm) were cut from inoculated agar plates with sterile Pasteur pipettes and then filled with 100 μL of the tested culture filtrates. A 100 μL sample of the bacteriocin protein (2 mg/mL) or crude proteins (2 mg/mL) of E. faecalis was added to each well, and THB broth was used as a negative control. The inhibition zones were observed after 24–48 h of incubation at 30°C.
Quantitative Real-Time PCR Validation of Genes
After feeding the silkworms GFP–E. faecalis LX10 (107 CFU/mL), the expression levels of colonization-related genes [zinc ABC transporter substrate-binding protein (znuA), response regulator transcription factor (BfmRS), ABC transporter A (patA), ABC transporter B (patB), signal peptidase I (lepB), glycosyltransferase family 2 protein (hssA), bifunctional acetaldehyde-CoA/alcohol dehydrogenase (adhE), endocarditis and biofilm-associated pilus tip protein (EbpA), acetaldehyde dehydrogenase (Lap), cell surface protein precursor (cspp), CDP-glycerol glycerophosphotransferase family protein (tagF), ABC transporter ATP-binding protein (ddpD), serine/threonine protein kinase IreK (prkC), ATP-binding cassette domain-containing protein (metN) and cell surface protein (csp)] were determined at 0, 1, 3, 5, and 7 days using RT–qPCR (n = 5). Total RNA was isolated from the silkworm gut using a Promega Eastep® Total RNA Super Extraction Kit (Beijing Biotec. Co., Ltd.) according to the supplier’s protocol. The RNA quality and concentration were measured with a NanoDrop spectrophotometer (Biodropsis BD–2000) and by gel electrophoresis (1% agarose gel). RNA of the desired quality (1 μg) was reverse transcribed into cDNA using HiScript® II Q RT SuperMix R223–01 (Vazyme Biotech Co., Ltd.). RT–qPCR was performed according to a previously described of this study. All the results were normalized by using the 23 SrRNA housekeeping gene. The gene-specific primers used for transcript quantification are listed in the Supplementary Table 2. The 2– ΔΔCt method was used to calculate the relative transcript abundance of the genes of interest.
Statistical Analysis
Statistical analyses were carried out using GraphPad Prism (version 9.0). The appropriate test (parametric/non-parametric) was chosen after the data were checked for normality and homogeneity of variance. Analysis of variance (one–way ANOVA, Tukey’s post-hoc test) or Student’s t-test was performed to compare the results regarding gut bacterial quantification, Enterococcus quantification, and GFP–E. faecalis colonization, body mass, pupation and adult emergence rates, whole cocoon weight, cocoon shell weight, gut pH levels, lactic acid production, and gene expression changes across the experimental and control groups. The P-value for distinguishing the significance of the data was set at <0.05. All assays were repeated at least three times.
Results
Characteristics of Enterococcus Isolated From Bombyx mori
Before a silkworm metamorphoses into an adult, it goes through five larval instars (L1–L5), among which the 5th instar has the longest growth period (6–8 days). The gut population analysis revealed that Enterococcus bacteria were present from the first instar to the fifth instar, and the number of Enterococcus progressively increased with B. mori age (Figure 1A). Based on phenotypic characteristics and 16S rRNA gene sequence analysis, three Enterococcus species were identified from the silkworm gut, including E. faecalis LX10, E. mundtii LX11, and E. casseliflavus LX12. E. faecalis LX10 was the most abundant of these strains on average relative to E. mundtii LX11 and E. casseliflavus LX12 isolated from silkworm samples of increasing age. The results for E. faecalis LX10 ranged from 1.30 × 103 to 1.38 × 108 16S copies per larva, and this strain was consistently recovered from different batches of samples (Figure 1B). The obtained 16S rRNA gene sequences of E. faecalis LX10, E. mundtii LX11, and E. casseliflavus LX12 showed 100, 92, and 94% similarity, respectively, with available published sequences in GenBank. Additionally, the phylogenetic analysis revealed that these isolates from the silkworm gut clustered within the Enterococcus clade (Supplementary Figure 1). One representative strain, designated E. faecalis LX10 (as the dominant strain), was selected for further analysis. The cells of this bacterium are 0.5–1.0 μm in diameter and occur in pairs (Figure 1C). FISH with a Cy3-labeled E. faecalis-specific probe (red) showed large amounts of E. faecalis adhesioning to the PM of the B. mori gut epithelium (blue), where the bacterium formed a structural barrier (Figure 1D).
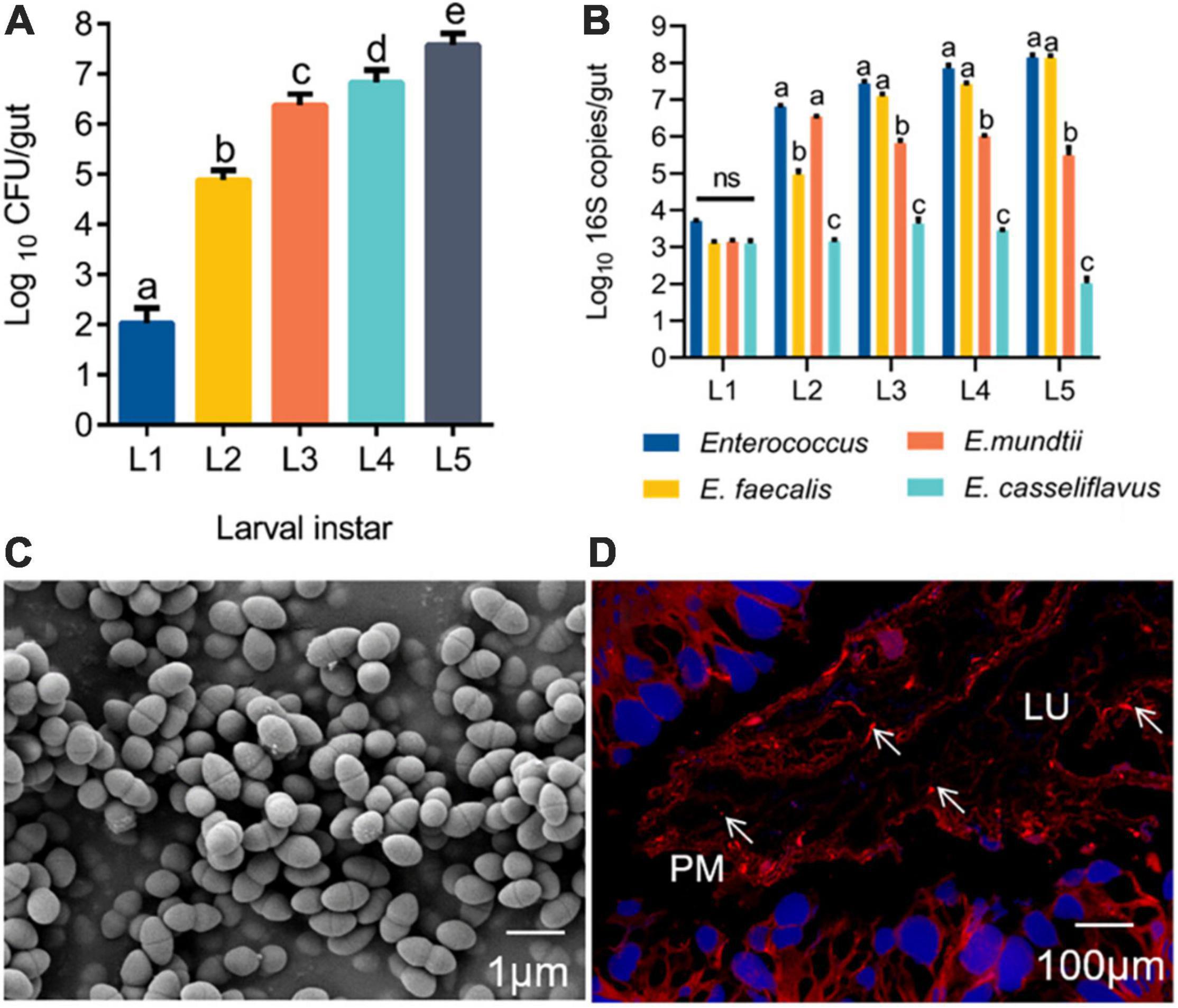
Figure 1. Enterococcus is widely found in the gut of silkworms. (A) Change in the Enterococcus population in the gut microbiota of B. mori larvae across different instars (L1, first instar; L2, second instar; L3, third instar; L4, fourth instar; L5, fifth instar). Proportional data are modeled using the mean number of CFUs counted (n = 10). (B) Absolute quantification (qPCR) of the total Enterococcus, Enterococcus faecalis, Enterococcus mundtii, and Enterococcus casselifavus 16S gene numbers in gut samples (L1-L5; mean with 95% confidence intervals). (C) Scanning electron microscopy (SEM) images of E. faecalis. Scale bars are 1 μm. (D) FISH with a Cy3–labeled E. faecalis-specific probe (red) shows a high density of bacterial cells adhered to the peritrophic membrane. lu, gut lumen; pm, peritrophic membrane; epithelium (blue, nuclei stained with DAPI). Statistically significant changes were determined by ANOVA and the post hoc Tukey honestly significant difference (HSD) test. Different letters represent statistically significant differences (p < 0.05, n = 10).
GFP–Enterococcus faecalis Proliferates in the Gut of Bombyx mori
The fluorescence intensity E. faecalis transformed with wild-type and pTRKH3–erm GFP E. faecalis was detected (Figures 2A,B). Interestingly, GFP–E. faecalis could switch from diplococcus form to occurring as chains in the presence of gut digestive juice (Figure 2C). In the intermediate and advanced stages (3–7 days) of ingestion, the fluorescent bacteria could be seen scattered among the gut contents of the 5th-instar larvae (Figure 2D). GFP bacteria accumulated around the PM, suggesting that there may be specific, stable cell attachment sites, and the bacteria seemed to aggregate in clusters and chains (Figures 2E,F). The growth curve analysis showed that wild-type E. faecalis and GFP–E. faecalis presented a typical “S”-shaped curve, indicating that the bacteria were suitable transgenic donors (Figure 2G). To examine whether E. faecalis was harmful to the host, we fed 5th-nstar larvae of B. mori GFP–tagged E. faecalis. The results showed no significant differences in larval biomass, pupation and adult emergence rates, whole cocoon weight, or cocoon shell weight between the control group and the GFP–E. faecalis–fed group (p > 0.05) (Figures 2H–L), suggesting that the bacteria were not toxic at the tested concentrations (103 CFU/mL–109 CFU/mL).
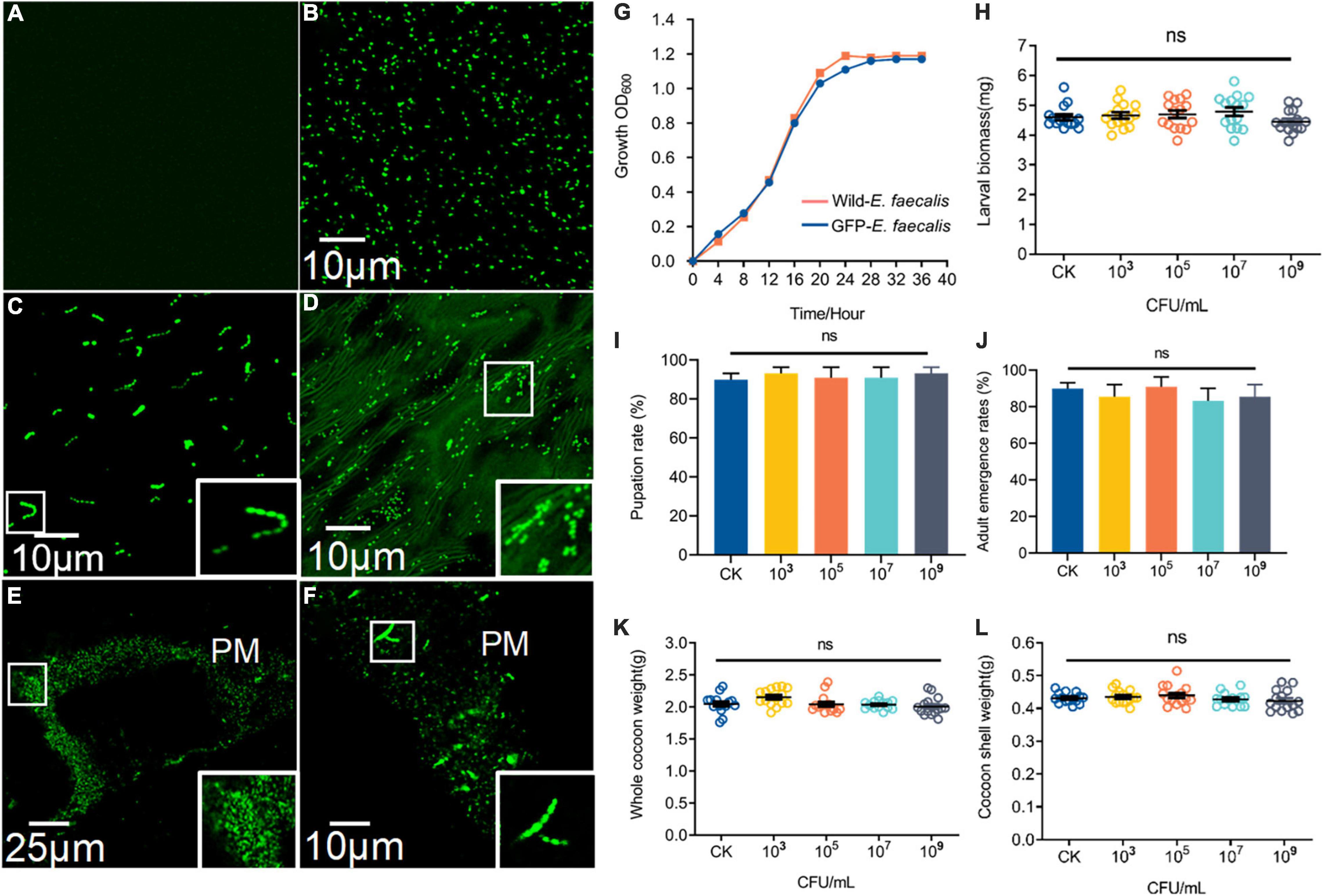
Figure 2. Characteristics and colonization of GFP–expressing E. faecalis in the B. mori intestinal tract. (A) Fluorescence micrographs of wild-type (CK) E. faecalis grown in THB. (B) Fluorescence micrographs of GFP–expressing E. faecalis under the control of pTRKH3–ermGFP. Scale bar = 10 μm. (C) GFP-E. faecalis grown for 12 h in the presence of intestinal juice and THB (1:1). Scale bar = 10 μm. (D) Fluorescent image of the gut contents of 5th-instar larvae after feeding on GFP–E. faecalis for 5 days. Scale bar = 10 μm. (E) Large clumps of GFP–E. faecalis adhEred to the peritrophic membrane of gut tissue. Scale bar = 25 μm. (F) Enterococcus with a chain–like structure was immobilized on the peritrophic membrane. Scale bar = 10 μm. (G) Shows the growth of these wild-type–E. faecalis and GFP–E. faecalis. The larval masses (H), pupation (I), whole cocoon weight (J) and adult emergence rates (K), and cocoon shell weight (L) of normal and 103, 106, and 109 CFU/mL E. faecalis–treated silkworms, respectively. Healthy silkworms treated with the same volume of sterilized water were used as the control group (CK). Different letters represent significant differences between groups according to Tukey’s honestly significant difference test (HSD, p < 0.05). Boxes show details.
GFP-E. faecalis proliferation was determined by CFU, qPCR, and FACS analysis of silkworm guts kept in the same vial for 0, 1, 3, 5, or 7 days post feeding (Figure 3A). Over time, there was a statistically significant increase in bacterial levels in the silkworm gut, except at a low concentration (103 CFU/silkworm feeding). The minimum and optimum feeding doses for colonization were found to be 105 CFU/silkworm and 107 CFU/silkworm, respectively (Figure 3B). On day 7, the total bacterial count (4.68 × 108 CFU/gut) was significantly higher than that on day 0 (5.49 × 106 CFU/gut) (t = 17.58, df = 18, p < 0.0001), showing the long-term stability of GFP–E. faecalis LX10 after 107 CFU/silkworm feeding. On the other hand, after 109 CFU/silkworm feeding, although there was a statistically significant change between day 0 and day 7, the bacterial levels only increased from 1.82 × 108 CFU/gut to 1.01 × 109 CFU/gut (t = 6.843, df = 18, p < 0.0001) (Figure 3B). Independent experimental replicates based on qPCR using universal primers for the 16S rRNA gene showed similar results for all concentrations and time points (Figure 3C).
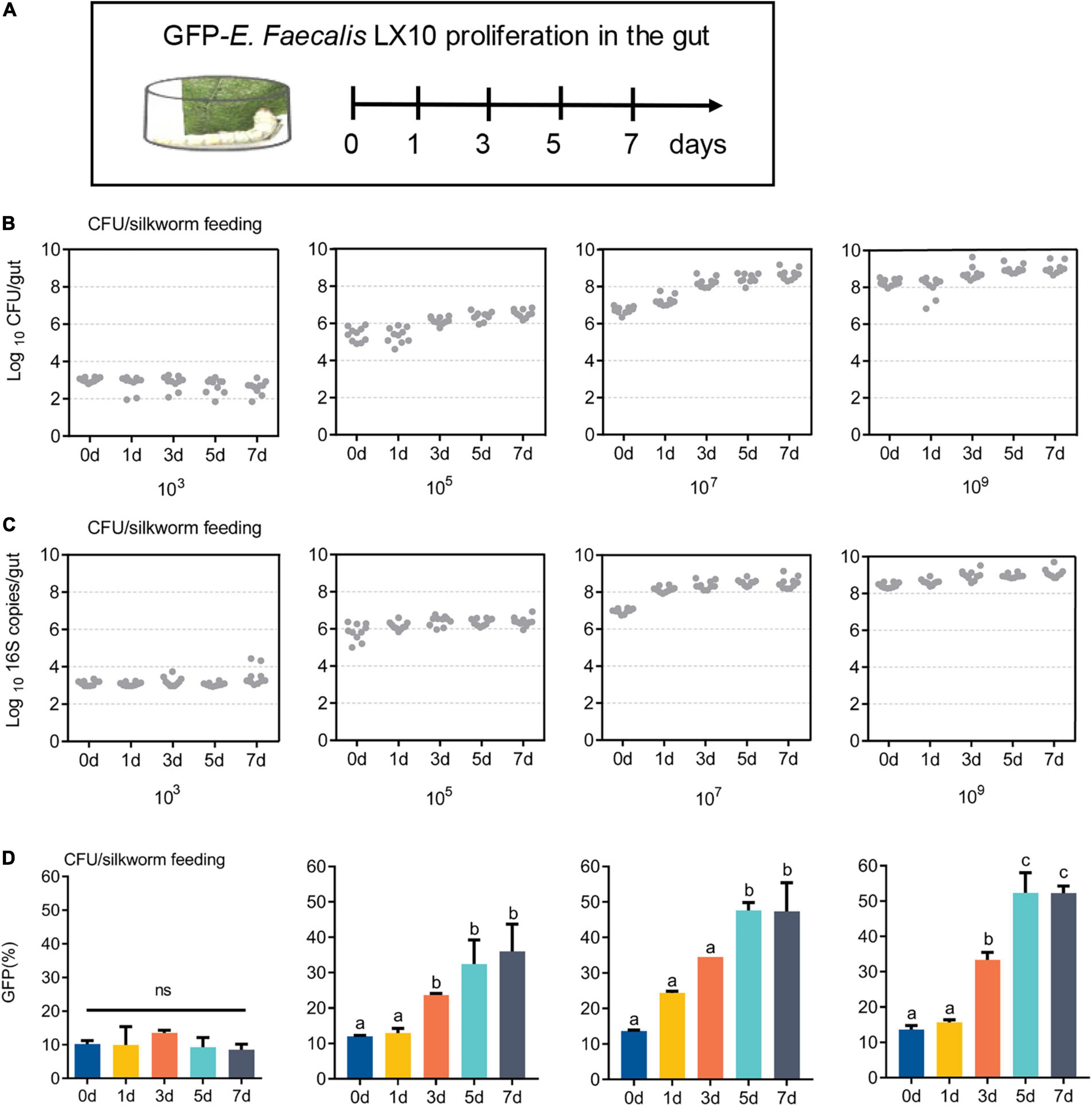
Figure 3. GFP–expressing E. faecalis stably persists and proliferates in the gut of B. mori. (A) First-instar (day 1) B. mori were inoculated with different concentrations of GFP–E. faecalis (103, 106, and 109 CFU/mL). The silkworms were raised in cages, and feces were cleaned every 4 h, as shown in the scheme. (B) The number of CFUs in individual guts was assessed by plating at d 0, 1, 3, 5, and 7. Each dot represents one individual. (C) Absolute quantification of GFP–E. faecalis 16S gene numbers in gut samples (mean with 95% confidence intervals, n = 10). (D) A flow cytometry analysis (FACS) was performed on days 0, 1, 3, 5, and 7 to determine the percentage of GFP-positive cells. CFU, colony–forming unit. Statistically significant changes were determined by ANOVA and the post hoc Tukey honestly significant difference (HSD) test.
In addition, FACS analysis showed that the GFP–E. faecalis percentage was both dose and time dependent, and this phenomenon was more pronounced when the silkworms were treated fed with 107CFU/silkworm (Figure 3D and Supplementary Figure 2). The percentage of GFP–E. faecalis increased from 13.65 to 46.5% during this period (0–7 days) (t = 197.6, df = 4, p < 0.0001). However, there was no difference in the number of GFP–E. faecalis between 0 and 7 days after 103 CFU/silkworm feeding (10.16 vs. 8.5%) (F = 1.059, p = 0.4515, Figure 3D).
General Features of the Enterococcus faecalis LX10 Genome
In total, 1.61 billion high-quality bases were generated via the whole-genome shotgun sequencing approach. The de novo assembly of the sequences successfully generated a 2,737,931 bp circular chromosome with an average gene length of 1,065 bp (Figure 4A), and one plasmid was detected (Figure 4B). A total of 2,570 predicted coding sequences (CDSs), 58 tRNA genes, 12 rRNA operons, and 58 tRNA elements were identified in the genome (including the chromosome and plasmid) of E. faecalis LX10. One chromosomal region was identified as a prophage locus (Figure 4C).
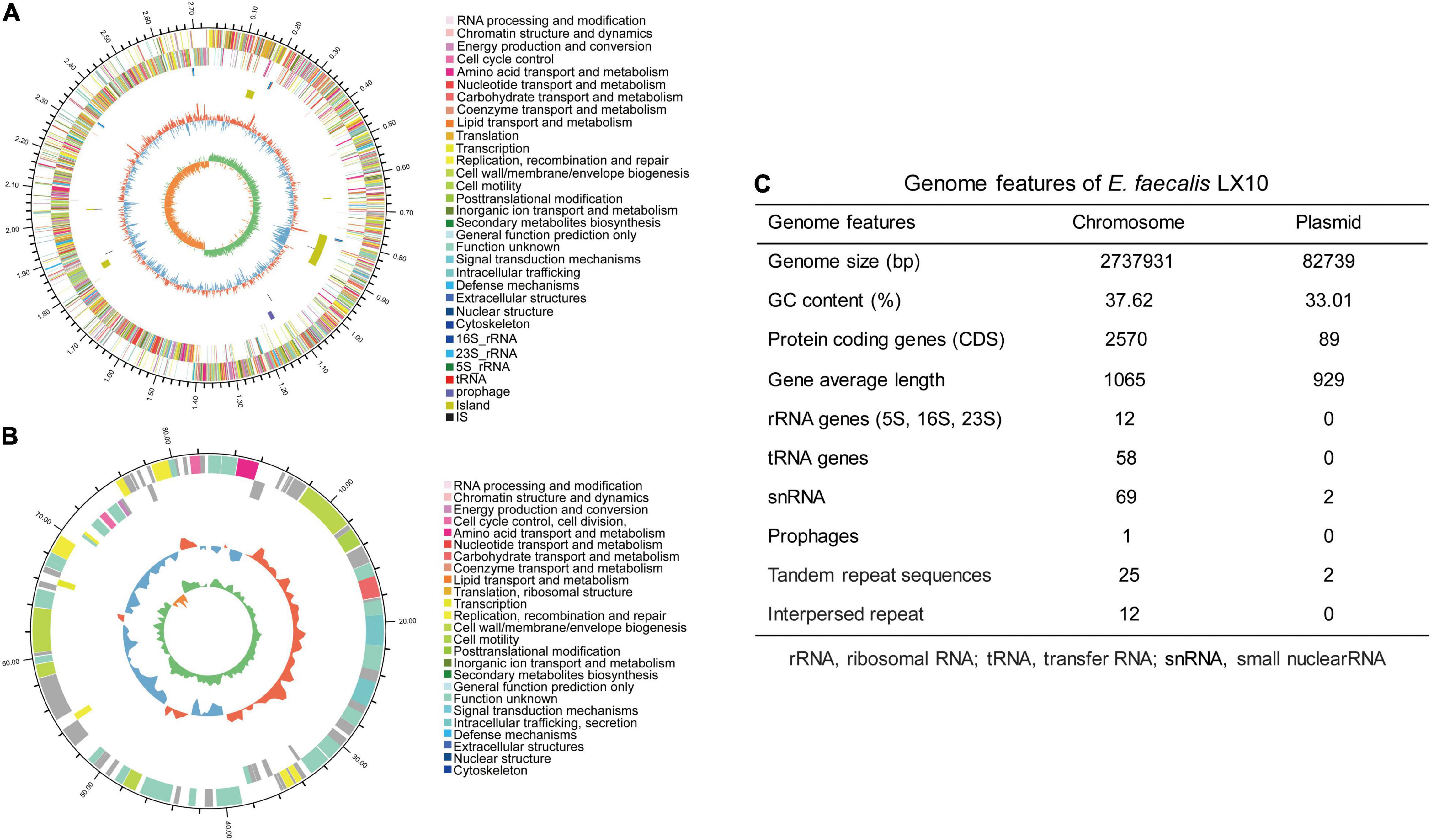
Figure 4. E. faecalis strain LX10 genome resources and characteristics. Circular representation of the chromosome (A) and plasmid (B) of E. faecalis strain LX10. The outer circle shows the size of the complete genome; second and third circles: sequences encoding the amino acids of proteins on the + and – strands, with different colors representing different COG functional classifications; fourth circle: rRNA and tRNA; fifth circle: GC content, where the outward red portion indicates that the GC content in this region is higher than the average GC content of the whole genome, and the inward blue portion indicates that the GC content in this region is lower than the whole-genome average, where the higher the peak value is, the greater the difference from the average GC content; and sixth circle: GC skew value (G–C/G + C). When the value is positive, the CDS is more likely to be transcribed from the positive chain; otherwise, the CDS is more likely to be transcribed from the negative chain. (C) Genome features of E. faecalis LX10.
The CDSs were compared with gene functions and metabolic pathways in the NR, GO, STRING, Swiss-Prot, COG, and KEGG databases. A total of 89 genes related to carbohydrate-active enzymes were annotated, including members of five CAZy families, comprising 46 glycoside hydrolases, 18 glycosyltransferases, 17 carbohydrate esterases, 7 auxiliary activities, and 1 polysaccharide lyase (Supplementary Figure 3A). A comprehensive genome-wide exploration of signal sequences, lipoprotein motifs, and other potential host cell components or binding motifs yielded an additional 217 putative surface-exposed proteins that may be associated with early colonization stages or virulence. We identified several genes (15) related to adhesin, defense, secretion, immune evasion, regulation, and iron acquisition proteins, some of which have been previously identified as colonization-related genes, whereas the other 202 genes showed no or vague functional predictions. And, the product of these genes are critical factors for host colonization (Supplementary Table 3 and Supplementary Figure 3B). Among the identified KEGG pathways, the genes involved in the global and overview maps (487 unigenes) and carbohydrate metabolism (224 unigenes) accounted for 39.22% of the predicted genes (Supplementary Figure 4A). GO analysis includes three ontologies: biological process (BP), cellular component (CC), and molecular function (MF). For example, under the CC and MF categories, the highest proportions of genes were annotated to the “integral component of membrane” (22.56%) and “ATP binding” (10.45%) terms (Supplementary Figure 4B). In the COG analysis, 2,317 COGs were classified into 19 functional categories. The analysis showed that most of the COG assigned ORFs fell under the category “Function unknown.” In the function known category, the highest and second-highest percentages of COGs were included in the carbohydrate transport and metabolism and transcription categories, which included 235 and 185 genes involved, respectively (Supplementary Figure 4C). In addition, a total of 558 genes (accounting for 20.98% of the total number of genes) were assigned to pathogenicity-related genes in the PHI database (Supplementary Figure 5A). The genes in the CARD database are genes whose presence confers resistance to antibiotics (Supplementary Figure 5B), such as lincosamide and streptogramin. The secretion system (Sec-SRP and type IV secretion system) of E. faecalis was checked based on the “Protein export (map03070)” pathway, and 10 key enzymes distributed in this pathway were identified (Supplementary Figure 6).
Effect of pH on Cell Growth and Lactic Acid Production
Cell growth was monitored under different pH-buffered (pH 6–11) conditions to monitor the effect of pH on E. faecalis LX10. The results showed that E. faecalis LX10 grew well, and the highest cell density was achieved within several hours, indicating that this strain is well adapted to the alkaline gut environment of silkworms (pH 9–11) (Figures 5A,B). In addition, E. faecalis inoculation resulted in a decrease in the gut pH [F(4, 70) = 3.594, p < 0.0001], where the 109 treatment resulted in the largest decrease relative to the CK group (9.00 vs. 10.83; t = 9.486, df = 28, p < 0.0001) (Figure 5B). Additionally, E. faecalis was able to produce lactic acid [F(5, 24) = 3.037, p = 0.029] under most tested pH conditions (pH 6–11), with the highest titer being observed at the initial pH of 8 (25.21 g/L) (Figure 5C). There was a strong, positive correlation between lactic acid production and the E. faecalis concentration (103–109 CFU/mL, pH = 11) (Figure 5D). In addition, the functional annotation of the genome revealed several genomic features associated with lactic acid production in E. faecalis LX10 as well as genes related to the uptake of related sugars and initial metabolism, which included ABC transporter ATP–binding protein, ABC transporter permease, sugar ABC transporter permease, sugar ABC transporter ATP–binding protein, sugar ABC transporter, sugar-binding protein, alcohol dehydrogenase, acetate kinase, L–lactate dehydrogenase, acetate kinase, and acetaldehyde dehydrogenase genes (Supplementary Table 4).
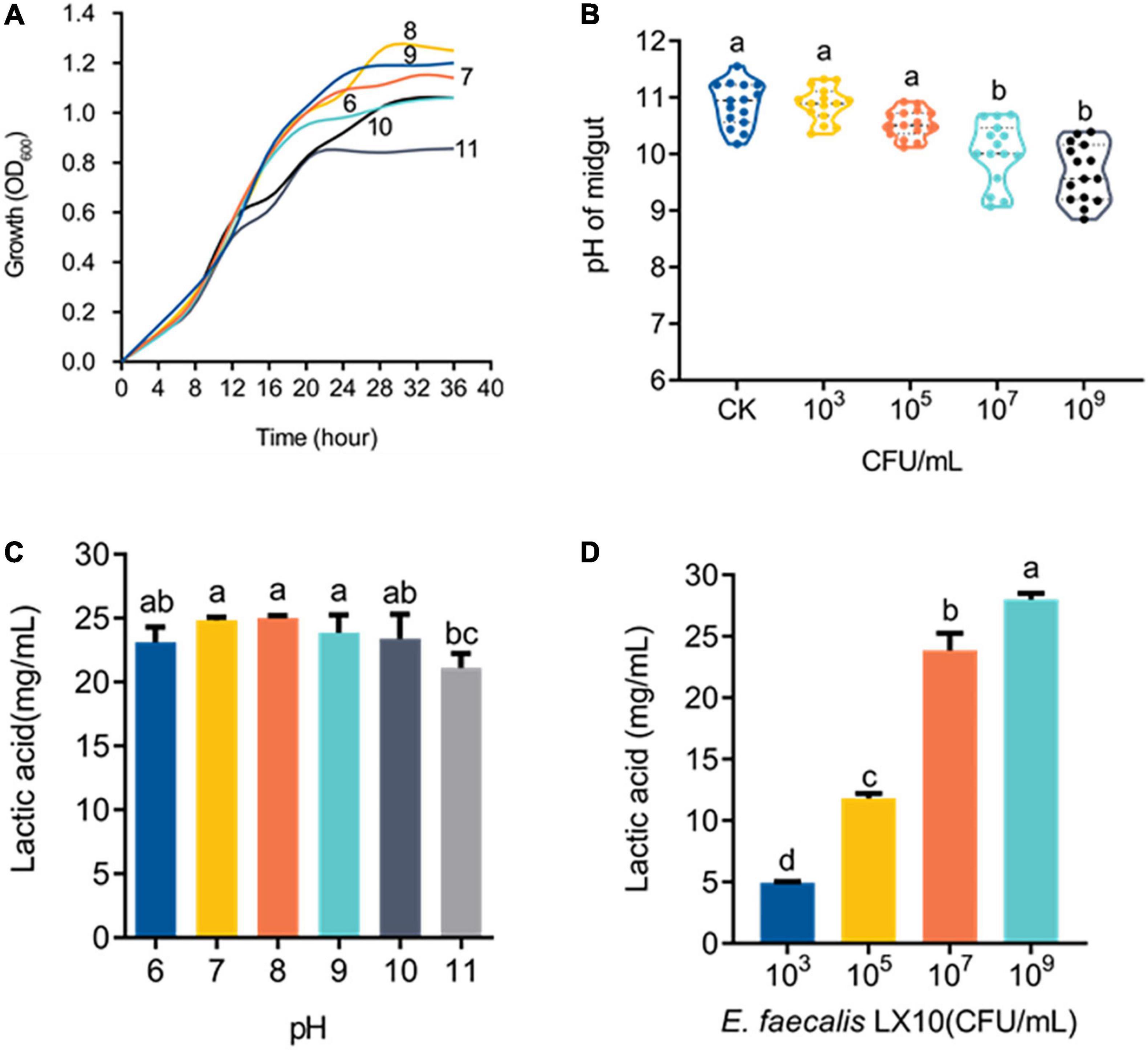
Figure 5. E. faecalis LX10 significantly reduces the intestinal pH and efficiently produces lactic acid. (A) Growth curves of the E. faecalis LX10 strain inoculated under pH-buffered conditions (pH: 6–11). (B) The pH of the germ-free silkworm gut after treatment with E. faecalis LX10 (CK, 103, 106, and 109 CFU/mL). pH values are shown as the mean ± SE (n = 10). (C) Comparison of lactic acid production by E. faecalis LX10 (107 CFU/mL) at different initial pH levels (n = 5, p > 0.05). (D) Lactic acid production by E. faecalis LX10 at different concentrations (103, 105, 107, and 109 CFU/mL; n = 5, p > 0.05). Different letters represent significant differences between groups according to Tukey’s honestly significant difference test (HSD, p < 0.05); ns not significant.
Antimicrobial Production and Bacterial Competition
The comparative analysis of genome sequences revealed a 14-gene cluster encoding a putative bacteriocin synthesis family that was enriched in E. faecalis isolates from the silkworm gut, suggesting that this bacteriocin may promote the colonization of indigenous microbiota (Figure 6A). The bacteriocin protein was purified by Ni–NATA affinity chromatography and examined by 15% SDS–PAGE. An evident band with a molecular weight of approximately 13.40 kDa was observed (lane 1 in Figure 6B and Supplementary Figure 7A), which was consistent with the predicted molecular mass of His–tagged bacteriocin.
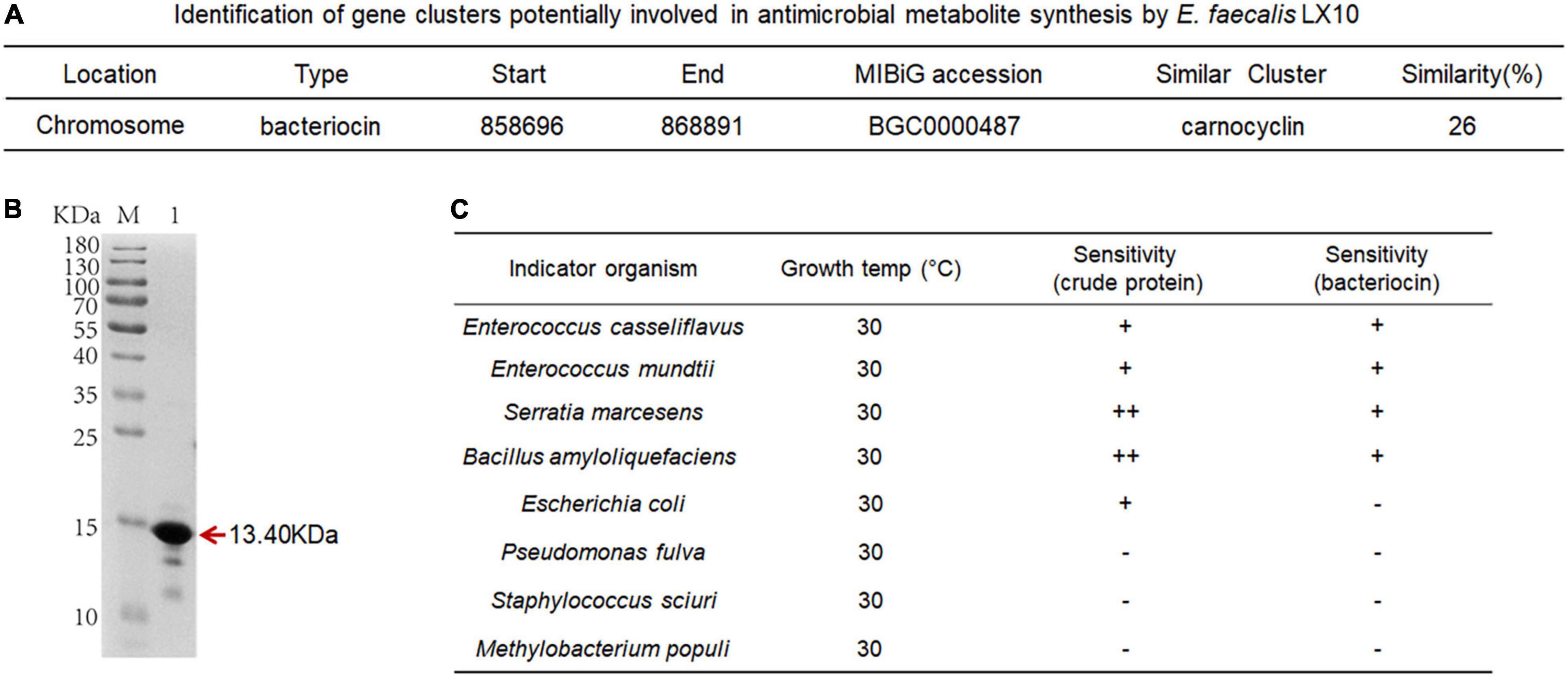
Figure 6. Characterization of bacteriocin and inhibitory activity. (A) Identification of gene clusters potentially involved in antimicrobial metabolite synthesis by E. faecalis LX10. (B) SDS–PAGE gel showing the reconstitution and purification of bacteriocin proteins. (C) Inhibition spectrum of the crude protein (2 mg/mL, 100 μL) from the cell-free culture supernatant and bacteriocin (2 mg/mL, 100 μL) produced by E. faecalis LX10. E. casseliflavus LX10, E. mundtii LX11, S. marcescens LX12, B. amyloliquefaciens, E. coli, P. fulva, S. sciuri, and M. populi as the indicator. –, no inhibition; +, inhibition; ++ good inhibition.
The inhibition assays of E. faecalis were conducted by applying the plate confrontation test. E. faecalis LX10 showed a distinct antagonistic effect against the other five representative gut bacteria (E. casseliflavus, E. mundtii, S. marcescens, B. amyloliquefaciens, and E. coli) (Figure 6C). The crude proteins and bacteriocin protein of cell-free E. faecalis LX10 inhibited the growth of indicator bacteria and resulted in a clear inhibition zone (Supplementary Figure 7B).
Genes Specifically Contributing to Gut Colonization
RT–qPCR was utilized to determine how the response of E. faecalis gene expression after colonization of the host gut. The expression of genes encoding adhesin (znuA, lepB, hssA, adhE, EbpA, and Lap), defense (cspp, tagF, and esp), immune evasion (patA and patB), regulation (BfmRS), secretion gene (prkC) and iron acquisition (ddpD and metN) proteins was measured based on their colonization-related functions (Figure 7A). After oral feeding with E. faecalis LX10, six adhesin, three defense-related, two immune evasion-related, and two regulatory genes showed substantially increased expression (Figure 7B). In particular, compared with the level observed at 0 d, the expression of BfmRS (eightfold), lepB (eightfold) and esp (17-fold) reached extremely significant levels at 5 days (t = 10.64, df = 8, p = 0.0001), 3 days (t = 25.63, df = 8, p < 0.0001) and 7 days (t = 5.120, df = 8, p = 0.0009), respectively. Two iron acquisition protein genes showed decreased or no difference in gene expression over time (p > 0.05) (Figure 7B).
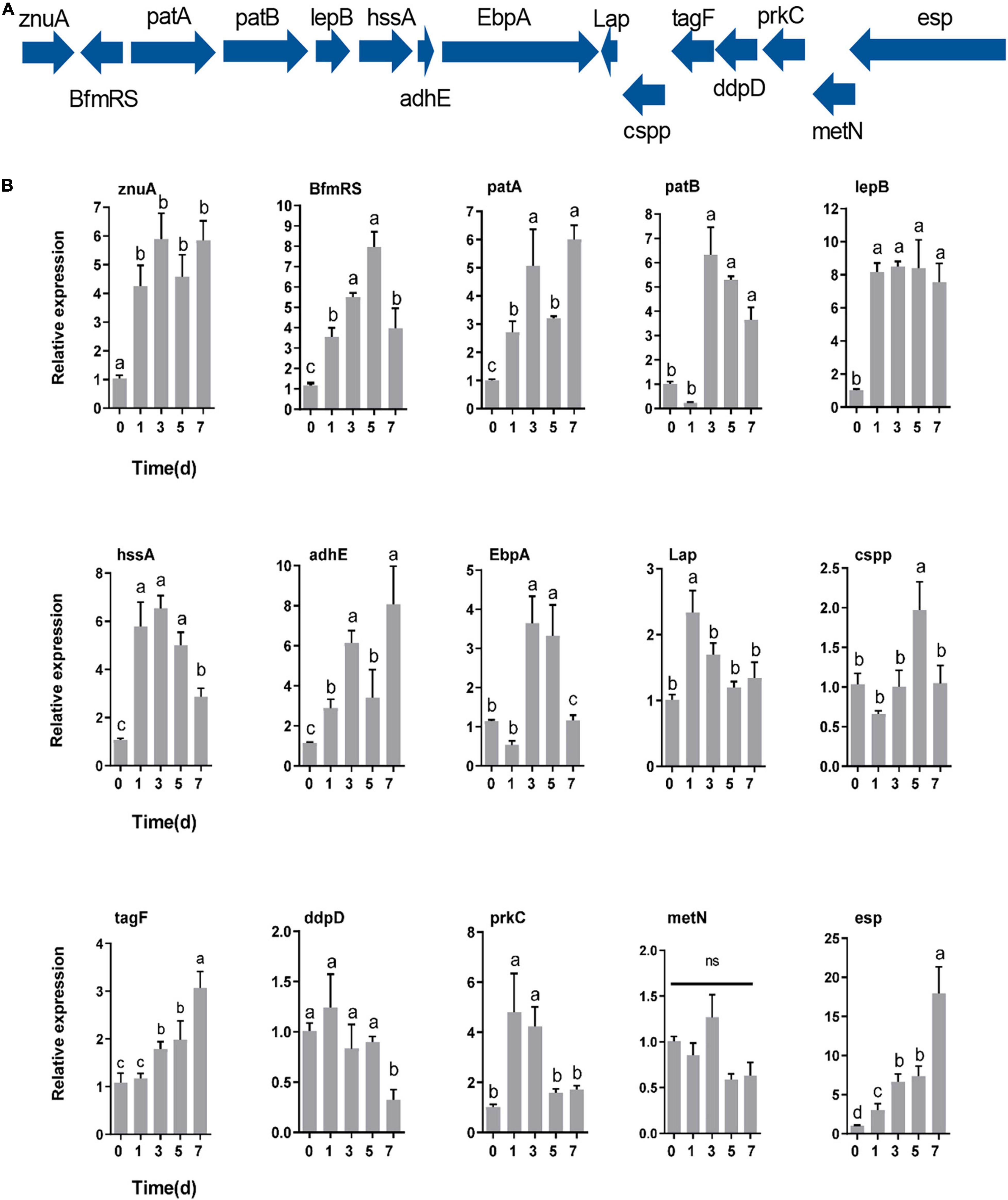
Figure 7. Genes specifically contributing to gut colonization. (A) Colonization-related gene expression at different times (0, 1, 3, 5, and 7 days) after inoculation with E. faecalis LX10 (107 CFU/mL) by RT–qPCR. (B) The mean ± standard error of the mean (SEM) of experiments performed in quintuplicate, with 10 silkworms/experiment, are shown. Variation analysis was performed by one-way ANOVA followed by Tukey’s post-hoc test. Different letters represent significant differences at p < 0.05. Adhesin genes (znuA, lepB, hssA, adhE, EbpA, and Lap), defense genes (cspp, esp, and tagF), a regulatory gene (BfmRS), a secretion system gene (prkC), immune evasion genes (patA and patB), and iron acquisition genes (ddpD and metN).
Discussion
The successful colonization of the insect gut by symbiotic bacteria is an essential step in controlling beneficial microbe systems and the health status of the host (Paniagua Voirol et al., 2018; Sauers and Sadd, 2019). In this paper, we report that the level of E. faecalis is as high as 108 copies in a normal 5th-instar larva gut, as confirmed by FISH analysis. In agreement with our results, previous studies have also found that Enterococcus is the most dominant bacterial genus in the gut microflora of silkworms (Xiang et al., 2007; Sun et al., 2016). Therefore, the dominant species, E. faecalis, is particularly likely to play a major role in facilitating the survival of its host in a changing environment during development.
Our results showed that the expression of GFP does not affect the physiology and fitness of E. faecalis, as strong fluorescence of recombinant GFP–E. faecalis was observed upon CLSM and FACS analyses. Similar results have been observed for Lactobacillus reuteri (Lizier et al., 2010) and E. mundtii (Teh et al., 2016). A recent study also showed that GFP expression did not negatively affect the bacterial function or survival of E. coli and S. marcescens (Wang S. et al., 2017; Chiapponi et al., 2020). In this study, GFP-tagged E. faecalis was introduced into the gut of B. mori, and it could stably persist and proliferate. Our recent study suggested that the minimum and optimum doses for colonization are 105 CFU/mL to 107 CFU/mL. Similarly, the minimum threshold for successful colonization for Burkholderia was shown to be 1.9 × 106 symbionts in the stinkbug Megacopta punctatissima (Hosokawa et al., 2007; Kikuchi and Yumoto, 2013). In Spodoptera littoralis, the midgut CFU count was observed to range from 1.0 × 105 during the fourth instar to 2.7 × 107 in the fifth instar after feeding on fluorescent E. mundtii (Teh et al., 2016). This suggests that different host species may influence bacterial colonization. Haoyue × Jingsong strain was used in this study, which is the most widely used and popularized multifilament bivoltine B. mori strain in China. The stable colonization of the gut by E. faecalis requires introduction within a certain threshold range. This is probably because the food intake rates of silkworms affect the speed of microbe passage through the gut and, thus, the colonization efficiency via excretion (Smith et al., 2015; Zhang et al., 2022). In addition, we found that attachment to the gut appears to be required for E. faecalis to successfully colonize the PM. The PM of insects is composed of chitinous and glycoprotein structures that form a protective barrier against intestinal bacterial infection, abrasive food particles, digestive enzymes, and oral pathogens (Nakashima et al., 2018). Additionally, the PM compartmentalizes digestive processes, allowing efficient nutrient absorption and the reutilization of hydrolytic enzymes (Hegedus et al., 2009; Kuraishi et al., 2011; Rose et al., 2020). GFP bacteria accumulated around the PM, suggesting that there may be specific, stable cell attachment sites. Interestingly, GFP–E. faecalis may switch from diplococcus to chain form under treatment with intestinal juice. Previous work sowed that certain common intestinal molecules, bile acids, and lysozyme also increase chaining and biofilm formation in E. faecalis, thereby increasing the colonized surface area (Varahan et al., 2013; McKenney et al., 2019). In brief, the increased surface area of longer chains may advantageously increase adherence to the host, as bacteria must prevent themselves from being flushed off of system surfaces to achieve successful colonization (Weiser, 2013).
In addition, E. faecalis LX10 actively secretes the antimicrobial peptide bacteriocin, which may enhance its competitive fitness by inhibiting other competing gut bacteria. Bacteriocin production is a powerful means of shaping gut communities, and the interactions and transmission between a host and its gut microbiota determine colonization success (Stephens et al., 2015). Many factors, such as the gut environment (pH and osmotic pressure) (Kikuchi and Yumoto, 2013), host-species specificity (La Rivière et al., 2015), antimicrobial agents, and host or symbiont genotypes (Wang Q. et al., 2017), may determine colonization success. We speculate that the high competitiveness of E. faecalis in the gut may also be attributed to its ability to produce bacteriocins strengthening the competitive ability of Enterococcus against other bacteria (Cebrian et al., 2012). It has been reported that the pathogen Helicobacter pylori can be eradicated by autolysins secreted from Lactobacillus acidophilus and that E. coli O157:H7 can be inhibited by Saccharomyces cerevisiae in ruminal fluid (Lorca et al., 2001; Bach et al., 2003). In addition, the gut bacterium E. mundtii of Spodoptera littoralis can secrete stable class IIa bacteriocin, which facilitates the normal development of the gut microbiota over harmful gut invading bacteria (Shao et al., 2017). Bacteriocin exerts its activity against microbes by inhibiting the formation of cell walls or causing holes to form in the cell membrane, leading to the release of cytoplasm and the death of target bacteria (Scholl, 2017; Kranjec et al., 2020). Bacteriocin protects the bacterium against its own bacteriocin activity through the action of immune-related proteins, such as ABC transporter, LanI, and LanFEG proteins, which increase the colonization of probiotic bacteria vis cell–cell interactions in the digestive tract (McAuliffe et al., 2001).
E. faecalis LX10 can grow well under alkaline conditions and can decrease the pH of the gut by producing high levels of lactic acid. Many Enterococcus species are highly competitive due to their tolerance of a wide range of temperatures and pH levels (Abdel-Rahman et al., 2015; Kridsada et al., 2022). E. faecalis LX10 produces significantly higher levels of lactic acid (18.74–28.79 g/L) at different pH levels relative to species reported in other studies. For example, 9 Enterococcus spp. were selected from various sources, and their lactic acid production at 30°C and pH 10 ranged from 11.3 to 18.3 g/L (Yokaryo and Tokiwa, 2014). One reason that bacteria produce different amounts of acid may be their different isolation sources. Lactic acid may break down membrane-bound enzymes and lead to their metabolic assimilation through the lactic acid fermentation pathway to improve food digestibility (Kridsada et al., 2022). We speculate that lactic acid plays an important role in modulating the gut environment, which is conducive to maintaining the dynamic balance of the gut microbiota under alkaline conditions (Yuksekdag et al., 2021). Furthermore, genomic analysis revealed that the sugar uptake gene responsible for lactate production was dispersed across the chromosome and plasmid of E. faecalis LX10, reflecting its metabolic flexibility in efficient lactic acid production. For instance, the pentose phosphate pathway (PP pathway) and the phosphoketolase pathway (PK pathway) were previously reported to be the two main pathways for lactic acid fermentation (Okano et al., 2009; Shinkawa et al., 2011). Key enzymes involved in the PP and PK pathways, including ABC transporter permease and transketolase, were detected in E. faecalis LX10. Therefore, we speculated that the activity of related enzymes involved in lactic acid fermentation was quite stable under high-pH conditions, which might be one of the colonization mechanisms underlying specialized adaptation to the silkworm gut niche.
The successful colonization of E. faecalis LX10 led to drastically increased expression of most adhesin genes (znuA, lepB, hssA, adhE, EbpA, and Lap), defense-related genes (cspp, tagF and esp), regulation gene (BfmRS), secretion gene (prkC) and immune evasion-related genes (patA and patB), while the expression of iron acquisition protein-encoding genes (ddpD and metN) was largely unchanged or decreased relative to the expression levels in the gut on a genome-wide scale. Enterococcus species have been found to adhere to various extracellular matrix components or host cells using adhesins such as aggregation substances, hemagglutinin, MSCRAMM, and other virulence factors that contribute to the colonization of host tissues (Nallapareddy and Murray, 2008; Somarajan et al., 2015; Deng et al., 2019; Phillips et al., 2019). For example, adhE have been reported to play a role in adherence to host tissue cells for biofilm formation (Cobo Molinos et al., 2008; Crosby et al., 2020). In vivo, it has been shown that the adhE mutant shows a significantly attenuated biofilm formation ability in a rat endocarditis model and a murine urinary tract infection model (Nallapareddy et al., 2006; Singh et al., 2007). Another study showed that cell surface proteins (cspp and esp) contribute to cell colonization by significantly enhancing biofilm formation in the presence of and by attachment to tissue-based surfaces (Tendolkar et al., 2004). The PASTA kinase protein family contains a transmembrane Ser/Thr kinase (prkC) that is crucial for cell envelope integrity, adaptation to available nutrient sources, and resistance to antimicrobial agents that target the cell wall of E. faecalis (Hall et al., 2017). The deletion of prkC in E. faecalis results in a profound gastrointestinal tract colonization defect in antibiotic–naïve mice (Banla et al., 2018). Genes involved in core enterococcal and genome plasticity are key drivers of gut colonization.
Conclusion
In conclusion, we identified a stable interaction between B. mori and E. faecalis, which will be useful for addressing important questions in the gut microbiota field that will lead to agricultural, economic, and environmental benefits. Moreover, the release of insects with stable symbionts in interventions may be effective against both pests (e.g., by improving the survival of sterile males) and pathogen vectors (e.g., by enhancing resistance to pathogens). Understanding what regulates gut colonization may be critical for the success of these approaches. Therefore, this new framework and concept for studying specific gut bacteria will contribute to the growing field of research on silkworm-microbe interactions.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA810349 and CP092784-CP092785.
Author Contributions
XL and FZ designed the project and finalized the manuscript. XZ and HF performed the experiments. XZ, JH, and AM performed the data analysis. XZ drafted the manuscript with inputs from all authors.
Funding
This work was supported by the China Agriculture Research System of MOF and MARA, and the National Natural Science Foundation of China (81572027).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.921330/full#supplementary-material
Footnotes
- ^ http://www.cbcb.umd.edu/software/glimmer/
- ^ http://www.repeatmasker.org
- ^ http://phast.wishartlab.com
- ^ http://lowelab.ucsc.edu/tRNAscan-SE/index.html
- ^ http://vicbioinformatics.com/software.barrnap.shtml
- ^ http://crispr.i2bc.paris-saclay.fr/
- ^ http://circos.ca/
References
Abdel-Rahman, M. A., Tashiro, Y., Zendo, T., Sakai, K., and Sonomoto, K. (2015). Enterococcus faecium QU 50: a novel thermophilic lactic acid bacterium for high-yield l-lactic acid production from xylose. FEMS Microbiol. Lett. 362, 1–7. doi: 10.1093/femsle/fnu030
Aksoy, S. (2018). Insect gut microbiota: accessories to the bite. Cell Host Microbe 23, 8–9. doi: 10.1016/j.chom.2017.12.016
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bach, S. J., McAllister, T. A., Veira, D. M., Gannon, V. P. J., and Holley, R. A. (2003). Effects of a Saccharomyces cerevisiae feed supplement on Escherichia coli O157:H7 in ruminal fluid in vitro. Anim. Feed Sci. Technol. 104, 179–189. doi: 10.1016/s0377-8401(02)00325-5
Banla, I. L., Kommineni, S., Hayward, M., Rodrigues, M., Palmer, K. L., Salzman, N. H., et al. (2018). Modulators of enterococcus faecalis cell envelope integrity and antimicrobial resistance influence stable colonization of the mammalian gastrointestinal tract. Infect. Immun. 86, e381–e317. doi: 10.1128/IAI.00381-17
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Bignell, D. E., Roisin, Y., and Lo, N. (2011). Biology of Termites: A Modern Synthesis. Cham: Springer.
Broderick, N. A., Raffa, K. F., Goodman, R. M., and Handelsman, J. (2004). Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl. Environ. Microbiol. 70, 293–300. doi: 10.1128/AEM.70.1.293-300.2004
Cebrian, R., Banos, A., Valdivia, E., Perez-Pulido, R., Martinez-Bueno, M., and Maqueda, M. (2012). Characterization of functional, safety, and probiotic properties of Enterococcus faecalis UGRA10, a new AS-48-producer strain. Food Microbiol. 30, 59–67. doi: 10.1016/j.fm.2011.12.002
Chen, B., Du, K., Sun, C., Vimalanathan, A., Liang, X., Li, Y., et al. (2018). Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 12, 2252–2262. doi: 10.1038/s41396-018-0174-1
Chen, B., Zhang, N., Xie, S., Zhang, X., He, J., Muhammad, A., et al. (2020). Gut bacteria of the silkworm Bombyx mori facilitate host resistance against the toxic effects of organophosphate insecticides. Environ. Int. 143:105886. doi: 10.1016/j.envint.2020.105886
Chiapponi, E., Henriot, C., Bertrand, X., Hocquet, D., and Bornette, G. (2020). Using GFP-tagged Escherichia coli to investigate the persistence of fecal bacteria in vegetated wetlands: an experimental approach. Antibiotics 9:335. doi: 10.3390/antibiotics9060335
Ciche, T. A., Kim, K. S., Kaufmann-Daszczuk, B., Nguyen, K. C., and Hall, D. H. (2008). Cell invasion and matricide during Photorhabdus luminescens transmission by Heterorhabditis bacteriophora nematodes. Appl. Environ. Microbiol. 74, 2275–2287. doi: 10.1128/AEM.02646-07
Cobo Molinos, A., Abriouel, H., Omar, N. B., Lopez, R. L., and Galvez, A. (2008). Detection of ebp (endocarditis- and biofilm-associated pilus) genes in enterococcal isolates from clinical and non-clinical origin. Int. J. Food Microbiol. 126, 123–126. doi: 10.1016/j.ijfoodmicro.2008.05.015
Crosby, H. A., Tiwari, N., Kwiecinski, J. M., Xu, Z., Dykstra, A., Jenul, C., et al. (2020). The Staphylococcus aureus ArlRS two-component system regulates virulence factor expression through MgrA. Mol. Microbiol. 113, 103–122. doi: 10.1111/mmi.14404
Deng, L., Schilcher, K., Burcham, L. R., Kwiecinski, J. M., Johnson, P. M., Head, S. R., et al. (2019). Identification of key determinants of staphylococcus aureus vaginal colonization. mBio 10:e02321-19. doi: 10.1128/mBio.02321-19
Egert, M., Wagner, B., Lemke, T., Brune, A., and Friedrich, M. W. (2003). Microbial community structure in midgut and hindgut of the humus-feeding larva of Pachnoda ephippiata (Coleoptera: Scarabaeidae). Appl. Environ. Microbiol. 69, 6659–6668. doi: 10.1128/AEM.69.11.6659-6668.2003
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects - diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Hall, C. L., Lytle, B. L., Jensen, D., Hoff, J. S., Peterson, F. C., Volkman, B. F., et al. (2017). Structure and dimerization of ireb, a negative regulator of cephalosporin resistance in Enterococcus faecalis. J. Mol. Biol. 429, 2324–2336. doi: 10.1016/j.jmb.2017.05.019
Hammer, T. J., Janzen, D. H., Hallwachs, W., Jaffe, S. P., and Fierer, N. (2017). Caterpillars lack a resident gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 114, 9641–9646. doi: 10.1073/pnas.1707186114
Hegedus, D., Erlandson, M., Gillott, C., and Toprak, U. (2009). New insights into peritrophic matrix synthesis, architecture, and function. Annu. Rev. Entomol. 54, 285–302. doi: 10.1146/annurev.ento.54.110807.090559
Hosokawa, T., Kikuchi, Y., and Fukatsu, T. (2007). How many symbionts are provided by mothers, acquired by offspring, and needed for successful vertical transmission in an obligate insect-bacterium mutualism? Mol. Ecol. 16, 5316–5325. doi: 10.1111/j.1365-294X.2007.03592.x
Kaiser, W., Huguet, E., Casas, J., Commin, C., and Giron, D. (2010). Plant green-island phenotype induced by leaf-miners is mediated by bacterial symbionts. Proc. Biol. Sci. 277, 2311–2319. doi: 10.1098/rspb.2010.0214
Kikuchi, Y., and Fukatsu, T. (2014). Live imaging of symbiosis: spatiotemporal infection dynamics of a GFP-labelled Burkholderia symbiont in the bean bug Riptortus pedestris. Mol. Ecol. 23, 1445–1456. doi: 10.1111/mec.12479
Kikuchi, Y., Hosokawa, T., and Fukatsu, T. (2007). Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl. Environ. Microbiol. 73, 4308–4316. doi: 10.1128/AEM.00067-07
Kikuchi, Y., and Yumoto, I. (2013). Efficient colonization of the bean bug Riptortus pedestris by an environmentally transmitted Burkholderia symbiont. Appl. Environ. Microbiol. 79, 2088–2091. doi: 10.1128/AEM.03299-12
Koga, R., Tsuchida, T., and Fukatsu, T. (2009). Quenching autofluorescence of insect tissues for in situ detection of endosymbionts. Appl. Entomol. Zool. 44, 281–291. doi: 10.1303/aez.2009.281
Kranjec, C., Ovchinnikov, K. V., Gronseth, T., Ebineshan, K., Srikantam, A., and Diep, D. B. (2020). A bacteriocin-based antimicrobial formulation to effectively disrupt the cell viability of methicillin-resistant Staphylococcus aureus (MRSA) biofilms. NPJ Biofilms Microbiomes 6:58. doi: 10.1038/s41522-020-00166-4
Kridsada, U., Klongklaew, A., Kodchasee, P., Pamueangmun, P., Shetty, K., and Khanongnuch, C. (2022). Enterococci as dominant xylose utilizing lactic acid bacteria in Eri silkworm midgut and the potential use of Enterococcus hirae as probiotic for Eri culture. Insect 27:136. doi: 10.3390/insects13020136
Kuraishi, T., Binggeli, O., Opota, O., Buchon, N., and Lemaitre, B. (2011). Genetic evidence for a protective role of the peritrophic matrix against intestinal bacterial infection in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 108, 15966–15971. doi: 10.1073/pnas.1105994108
La Rivière, M., Garrabou, J., and Bally, M. (2015). Evidence for host specificity among dominant bacterial symbionts in temperate gorgonian corals. Coral Reefs 34, 1087–1098. doi: 10.1007/s00338-015-1334-7
Liang, X., Sun, C., Chen, B., Du, K., Yu, T., Luang-In, V., et al. (2018). Insect symbionts as valuable grist for the biotechnological mill: an alkaliphilic silkworm gut bacterium for efficient lactic acid production. Appl. Microbiol. Biotechnol. 102, 4951–4962. doi: 10.1007/s00253-018-8953-1
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Liu, H., Chen, B., Hu, S., Liang, X., Lu, X., and Shao, Y. (2016). Quantitative proteomic analysis of germination of Nosema bombycis spores under extremely alkaline conditions. Front. Microbiol. 7:1459. doi: 10.3389/fmicb.2016.01459
Lizier, M., Sarra, P. G., Cauda, R., and Lucchini, F. (2010). Comparison of expression vectors in Lactobacillus reuteri strains. FEMS Microbiol. Lett. 308, 8–15. doi: 10.1111/j.1574-6968.2010.01978.x
Lorca, G. L., Wadstrom, T., Valdez, G. F., and Ljungh, A. (2001). Lactobacillus acidophilus autolysins inhibit Helicobacter pylori in vitro. Curr. Microbiol. 42, 39–44. doi: 10.1007/s002840010175
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–967. doi: 10.1093/nar/25.5.955
McAuliffe, O., Ross, R. P., and Hill, C. (2001). Lantibiotics: structure, biosynthesis and mode of action. FEMS Microbiol. Rev. 25, 285–308. doi: 10.1111/j.1574-6976.2001.tb00579.x
McKenney, P. T., Yan, J., Vaubourgeix, J., Becattini, S., Lampen, N., Motzer, A., et al. (2019). Intestinal bile acids induce a morphotype switch in vancomycin-resistant enterococcus that facilitates intestinal colonization. Cell Host Microbe 25, 695.e5–705.e5. doi: 10.1016/j.chom.2019.03.008
Nakashima, K., Kimura, S., Ogawa, Y., Watanabe, S., Soma, S., Kaneko, T., et al. (2018). Chitin-based barrier immunity and its loss predated mucus-colonization by indigenous gut microbiota. Nat. Commun. 9:3402. doi: 10.1038/s41467-018-05884-0
Nallapareddy, S. R., and Murray, B. E. (2008). Role played by serum, a biological cue, in the adherence of Enterococcus faecalis to extracellular matrix proteins, collagen, fibrinogen, and fibronectin. J. Infect. Dis. 197, 1728–1736. doi: 10.1086/588143
Nallapareddy, S. R., Singh, K. V., Sillanpaa, J., Garsin, D. A., Hook, M., Erlandsen, S. L., et al. (2006). Endocarditis and biofilm-associated pili of Enterococcus faecalis. J. Clin. Invest. 116, 2799–2807. doi: 10.1172/JCI29021
Nawrocki, E. P., Burge, S. W., Bateman, A., Daub, J., Eberhardt, R. Y., Eddy, S. R., et al. (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137. doi: 10.1093/nar/gku1063
Okano, K., Yoshida, S., Tanaka, T., Ogino, C., Fukuda, H., and Kondo, A. (2009). Homo-D-lactic acid fermentation from arabinose by redirection of the phosphoketolase pathway to the pentose phosphate pathway in L-lactate dehydrogenase gene-deficient Lactobacillus plantarum. Appl. Environ. Microbiol. 75, 5175–5178. doi: 10.1128/AEM.00573-09
Paniagua Voirol, L. R., Frago, E., Kaltenpoth, M., Hilker, M., and Fatouros, N. E. (2018). Bacterial symbionts in lepidoptera: their diversity, transmission, and impact on the host. Front. Microbiol. 9:556. doi: 10.3389/fmicb.2018.00556
Phillips, Z. N., Tram, G., Seib, K. L., and Atack, J. M. (2019). Phase-variable bacterial loci: how bacteria gamble to maximise fitness in changing environments. Biochem. Soc. Trans. 47, 1131–1141. doi: 10.1042/BST20180633
Rawls, J. F., Mahowald, M. A., Goodman, A. L., Trent, C. M., and Gordon, J. I. (2007). In vivo imaging and genetic analysis link bacterial motility and symbiosis in the zebrafish gut. Proc. Natl. Acad. Sci. U.S.A. 104, 7622–7627. doi: 10.1073/pnas.0702386104
Rizzo, M. A., Davidson, M. W., and Piston, D. W. (2009). Fluorescent protein tracking and detection: fluorescent protein structure and color variants. Cold Spring Harb. Protoc. 2009:pdb.top63. doi: 10.1101/pdb.top63
Robinson, C. D., Klein, H. S., Murphy, K. D., Parthasarathy, R., Guillemin, K., and Bohannan, B. J. M. (2018). Experimental bacterial adaptation to the zebrafish gut reveals a primary role for immigration. PLoS Biol. 16:e2006893. doi: 10.1371/journal.pbio.2006893
Rose, C., Casas-Sanchez, A., Dyer, N. A., Solorzano, C., Beckett, A. J., Middlehurst, B., et al. (2020). Trypanosoma brucei colonizes the tsetse gut via an immature peritrophic matrix in the proventriculus. Nat. Microbiol. 5, 909–916. doi: 10.1038/s41564-020-0707-z
Saha, S., Bridges, S., Magbanua, Z. V., and Peterson, D. G. (2008). Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294. doi: 10.1093/nar/gkn064
Sauers, L. A., and Sadd, B. M. (2019). An interaction between host and microbe genotypes determines colonization success of a key bumble bee gut microbiota member. Evolution 73, 2333–2342. doi: 10.1111/evo.13853
Scholl, D. (2017). Phage tail-like bacteriocins. Annu. Rev. Virol. 4, 453–467. doi: 10.1146/annurev-virology-101416-041632
Schretter, C. E. (2020). Links between the gut microbiota, metabolism, and host behavior. Gut Microbes 11, 245–248. doi: 10.1080/19490976.2019.1643674
Sellegounder, D., Liu, Y., Wibisono, P., Chen, C. H., Leap, D., and Sun, J. (2019). Neuronal GPCR NPR-8 regulates C. elegans defense against pathogen infection. Sci. Adv. 5:eaaw4717. doi: 10.1126/sciadv.aaw4717
Shao, Y., Chen, B., Sun, C., Ishida, K., Hertweck, C., and Boland, W. (2017). Symbiont-derived antimicrobials contribute to the control of the lepidopteran gut microbiota. Cell Chem. Biol. 24, 66–75. doi: 10.1016/j.chembiol.2016.11.015
Shinkawa, S., Okano, K., Yoshida, S., Tanaka, T., Ogino, C., Fukuda, H., et al. (2011). Improved homo L-lactic acid fermentation from xylose by abolishment of the phosphoketolase pathway and enhancement of the pentose phosphate pathway in genetically modified xylose-assimilating Lactococcus lactis. Appl. Microbiol. Biotechnol. 91, 1537–1544. doi: 10.1007/s00253-011-3342-z
Siegel, S. J., and Weiser, J. N. (2015). Mechanisms of Bacterial Colonization of the Respiratory Tract. Annu. Rev. Microbiol. 69, 425–444. doi: 10.1146/annurev-micro-091014-104209
Singh, K. V., Nallapareddy, S. R., and Murray, B. E. (2007). Importance of the ebp (endocarditis- and biofilm-associated pilus) locus in the pathogenesis of Enterococcus faecalis ascending urinary tract infection. J. Infect. Dis. 195, 1671–1677. doi: 10.1086/517524
Smith, C. C., Snowberg, L. K., Gregory Caporaso, J., Knight, R., and Bolnick, D. I. (2015). Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 9, 2515–2526. doi: 10.1038/ismej.2015.64
Somarajan, S. R., La Rosa, S. L., Singh, K. V., Roh, J. H., Hook, M., and Murray, B. E. (2015). The fibronectin-binding protein Fnm contributes to adherence to extracellular matrix components and virulence of Enterococcus faecium. Infect. Immun. 83, 4653–4661. doi: 10.1128/IAI.00885-15
Stephens, W. Z., Wiles, T. J., Martinez, E. S., Jemielita, M., Burns, A. R., Parthasarathy, R., et al. (2015). Identification of population bottlenecks and colonization factors during assembly of bacterial communities within the zebrafish intestine. mBio 6:e01163-15. doi: 10.1128/mBio.01163-15
Sun, Z., Lu, Y., Zhang, H., Kumar, D., Liu, B., Gong, Y., et al. (2016). Effects of BmCPV infection on silkworm Bombyx mori intestinal bacteria. PLoS One 11:e0146313. doi: 10.1371/journal.pone.0146313
Swinfield, T. J., Oultram, J. D., Thompson, D. E., Brehm, J. K., and Minton, N. P. (1990). Physical characterisation of the replication region of the Streptococcus faecalis plasmid pAM beta 1. Gene 87, 79–90.
Teh, B. S., Apel, J., Shao, Y., and Boland, W. (2016). Colonization of the intestinal tract of the polyphagous pest Spodoptera littoralis with the GFP-tagged indigenous gut bacterium Enterococcus mundtii. Front. Microbiol. 7:928. doi: 10.3389/fmicb.2016.00928
Tendolkar, P. M., Baghdayan, A. S., Gilmore, M. S., and Shankar, N. (2004). Enterococcal surface protein, Esp, enhances biofilm formation by Enterococcus faecalis. Infect. Immun. 72, 6032–6039. doi: 10.1128/IAI.72.10.6032-6039.2004
Varahan, S., Iyer, V. S., Moore, W. T., and Hancock, L. E. (2013). Eep confers lysozyme resistance to Enterococcus faecalis via the activation of the extracytoplasmic function sigma factor SigV. J. Bacteriol. 195, 3125–3134. doi: 10.1128/JB.00291-13
Viswanathan, S., Williams, M. E., Bloss, E. B., Stasevich, T. J., Speer, C. M., Nern, A., et al. (2015). High-performance probes for light and electron microscopy. Nat. Methods 12, 568–576. doi: 10.1038/nmeth.3365
Wang, Q., Yang, S., Liu, J., Terecskei, K., Abraham, E., Gombar, A., et al. (2017). Host-secreted antimicrobial peptide enforces symbiotic selectivity in Medicago truncatula. Proc. Natl. Acad. Sci. U.S.A. 114, 6854–6859. doi: 10.1073/pnas.1700715114
Wang, S., Dos-Santos, A. L., Huang, W., Liu, K. C., Oshaghi, M. A., Wei, G., et al. (2017). Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science 357, 1399–1402. doi: 10.1126/science.aan5478
Weiser, J. N. (2013). The battle with the host over microbial size. Curr. Opin. Microbiol. 16, 59–62. doi: 10.1016/j.mib.2013.01.001
Wellinghausen, N., Bartel, M., Essig, A., and Poppert, S. (2007). Rapid identification of clinically relevant Enterococcus species by fluorescence in situ hybridization. J. Clin. Microbiol. 45, 3424–3426. doi: 10.1128/JCM.00861-07
Xiang, H., Li, M., Zhao, Y., Zhao, L., Zhang, Y., and Huang, Y. (2007). Bacterial community in midguts of the silkworm larvae estimated by PCR/DGGE and 16S rDNA gene library analysis. Jiangxi Science 50, 222–233.
Xiao, X., Yang, L., Pang, X., Zhang, R., Zhu, Y., Wang, P., et al. (2017). A Mesh-Duox pathway regulates homeostasis in the insect gut. Nat. Microbiol. 2:17020. doi: 10.1038/nmicrobiol.2017.20
Xie, S., Vallet, M., Sun, C., Kunert, M., David, A., Zhang, X., et al. (2019). Biocontrol potential of a novel endophytic bacterium from mulberry (morus) tree. Front. Bioeng. Biotechnol. 7:488. doi: 10.3389/fbioe.2019.00488
Yeruva, T., Vankadara, S., Ramasamy, S., and Lingaiah, K. (2020). Identification of potential probiotics in the midgut of mulberry silkworm, Bombyx mori through metagenomic approach. Probiotics Antimicrob. Proteins 12, 635–640. doi: 10.1007/s12602-019-09580-3
Yokaryo, H., and Tokiwa, Y. (2014). Isolation of alkaliphilic bacteria for production of high optically pure L-(+)-lactic acid. J. Gen. Appl. Microbiol. 60, 270–275. doi: 10.2323/jgam.60.270
Yuksekdag, Z., Ahlatci, N. S., Hajikhani, R., Darilmaz, D. O., and Beyatli, Y. (2021). Safety and metabolic characteristics of 17 Enterococcus faecium isolates. Arch. Microbiol. 203, 5683–5694. doi: 10.1007/s00203-021-02536-8
Zhang, F., Sun, X. X., Zhang, X. C., Zhang, S., Lu, J., Xia, Y. M., et al. (2017). The interactions between gut microbiota and entomopathogenic fungi: a potential approach for biological control of Blattella germanica (L.). Pest Manag. Sci. 74, 438–447. doi: 10.1002/ps.4726
Zhang, X., Feng, H., He, J., Liang, X., Zhang, N., Shao, Y., et al. (2022). The gut commensal bacterium Enterococcus faecalis LX10 contributes to defending against Nosema bombycis infection in Bombyx mori. Pest Manag. Sci. 78, 2215–2227. doi: 10.1002/ps.6846
Keywords: Bombyx mori, insect, gut microbiota, Enterococcus, colonization strategies, whole-genome sequencing, bacteriocin, gene expression
Citation: Zhang X, Feng H, He J, Muhammad A, Zhang F and Lu X (2022) Features and Colonization Strategies of Enterococcus faecalis in the Gut of Bombyx mori. Front. Microbiol. 13:921330. doi: 10.3389/fmicb.2022.921330
Received: 15 April 2022; Accepted: 08 June 2022;
Published: 24 June 2022.
Edited by:
Terezia Horvathova, Institute of Soil Biology (ASCR), CzechiaReviewed by:
Rishi Drolia, Purdue University, United StatesHui-min Neoh, National University of Malaysia, Malaysia
Copyright © 2022 Zhang, Feng, He, Muhammad, Zhang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Zhang, emhhbmdmYW4wNTMxQDE2My5jb20=; Xingmeng Lu, eG1sdUB6anUuZWR1LmNu