 Yuan-feng Dai1,2,3
Yuan-feng Dai1,2,3 Xiao-mao Wu1*
Xiao-mao Wu1* Han-cheng Wang2*Wen-hong Li4Liu-ti Cai2Ji-xin Li5Feng Wang2*
Han-cheng Wang2*Wen-hong Li4Liu-ti Cai2Ji-xin Li5Feng Wang2* Shafaque Sehar6
Shafaque Sehar6 Imran Haider Shamsi6*
Imran Haider Shamsi6*- 1Department of Plant Protection, Institute of Crop Protection, College of Agriculture, Guizhou University, Guiyang, China
- 2Guizhou Provincial Academician Workstation of Microbiology and Health, Guizhou Academy of Tobacco Science, Guiyang, China
- 3Bijie Tobacco Company, Bijie, China
- 4Guizhou Institute of Plant Protection, Guizhou Academy of Agricultural Sciences, Guiyang, China
- 5Guizhou Tobacco Company of CNTC, China National Tobacco Corporation, Guiyang, China
- 6Zhejiang Key Laboratory of Crop Germplasm Resource, Department of Agronomy, College of Agriculture and Biotechnology, Zhejiang University, Hangzhou, China
Phyllospheric microbial composition of tobacco (Nicotiana tabacum L.) is contingent upon certain factors, such as the growth stage of the plant, leaf position, and cultivar and its geographical location, which influence, either directly or indirectly, the growth, overall health, and production of the tobacco plant. To better understand the spatiotemporal variation of the community and the divergence of phyllospheric microflora, procured from healthy and diseased tobacco leaves infected by Alternaria alternata, the current study employed microbe culturing, high-throughput technique, and BIOLOG ECO. Microbe culturing resulted in the isolation of 153 culturable fungal isolates belonging to 33 genera and 99 bacterial isolates belonging to 15 genera. High-throughput sequencing revealed that the phyllosphere of tobacco was dominantly colonized by Ascomycota and Proteobacteria, whereas, the most abundant fungal and bacterial genera were Alternaria and Pseudomonas. The relative abundance of Alternaria increased in the upper and middle healthy groups from the first collection time to the third, whereas, the relative abundance of Pseudomonas, Sphingomonas, and Methylobacterium from the same positions increased during gradual leaf aging. Non-metric multi-dimensional scaling (NMDs) showed clustering of fungal communities in healthy samples, while bacterial communities of all diseased and healthy groups were found scattered. FUNGuild analysis, from the first collection stage to the third one in both groups, indicated an increase in the relative abundance of Pathotroph-Saprotroph, Pathotroph-Saprotroph-Symbiotroph, and Pathotroph-Symbiotroph. Inclusive of all samples, as per the PICRUSt analysis, the predominant pathway was metabolism function accounting for 50.03%. The average values of omnilog units (OUs) showed relatively higher utilization rates of carbon sources by the microbial flora of healthy leaves. According to the analysis of genus abundances, leaf growth and leaf position were the important drivers of change in structuring the microbial communities. The current findings revealed the complex ecological dynamics that occur in the phyllospheric microbial communities over the course of a spatiotemporal varying environment with the development of tobacco brown spots, highlighting the importance of community succession.
Introduction
Tobacco is an important economic crop cultivated extensively for its leaves, which are dried and processed primarily for smoking in pipes, cigarettes, and cigars (Wang et al., 2013). According to FAOSTAT (FAO, 2019), about 100 countries produce tobacco, but China undoubtedly remains the major producer accounting for 39.06% (2,392,090 metric tons) of the global tobacco production (Wang et al., 2014; Xu et al., 2014). Many fungal and bacterial diseases occur on tobacco leaves during the course of tobacco production (Chen et al., 2020), even during the curing process (Rotem, 1994). One of the most important foliar fungal diseases is tobacco brown spot, which is caused by Alternaria alternata that is prone to infect senescent leaves (Ishida and Kumashiro, 1988). Initially, tobacco brown spots appear at the leaf maturity stage and are mostly observed in the lower leaves during June in China (Xu et al., 1993; He et al., 1997), then spread from the lower leaves to the top (LaMondia, 2001; Hou et al., 2016). Under high humidity conditions and appropriate conditions for infection, the spores germinate on the leaf surface in 24 h, then germ tubes were produced and penetrate the leaf cells directly through walls or stomata (Slavov et al., 2004). At last, germinating spores release toxins which could cause brown spot disease of tobacco and some small necrotic spots could be noticed 48 h after inoculation (Kodama et al., 1990). The saprophytic nature of A. alternata allows it to remain in the soil on a very large amount of lignified plant debris, over winter as mycelium and survive for several months by invading dead plant tissues. Due to the short incubation period (5–8 days) under the right conditions, A. alternata spreads swiftly and infects the stems, transmitting the disease through seeds by colonizing pedicels and capsules in extreme cases, when long periods of rain occur during harvesting (Shew and Lucas, 1991).
A number of microbial communities are established in the phyllosphere including fungi, bacteria, viruses, protists, and archaea (Berlec, 2012; Vorholt, 2012), the structure of which is strongly shaped by a myriad of abiotic and biotic factors, including space (Rastogi et al., 2012; Rumakanta et al., 2017), growth season (Copeland et al., 2015; Ding and Melchner, 2016), geographical location (Whipps et al., 2008; Chen et al., 2020), plant species (Whipps et al., 2008; Knief et al., 2010; Wellner et al., 2011), disease severities (Luo et al., 2017, 2019), pesticide treatments (Qin et al., 2019; Chen X. et al., 2021), and agricultural management (Karlsson et al., 2017). Similarly, phyllospheric microbial communities of tobacco were strongly correlated with the plant's spatiotemporally varying agro-ecosystem. Alterations in the microbial community are always accompanied by changes in the metagenomic environment of the host, exhibiting the methods through which phytopathogens invade and redirect the host's resources and functions and the strategies adopted by hosts to respond against pathogen invasion (Whipps et al., 2008). Therefore, the comprehension of these complex interactions among microbial species at multifarious levels is required to predict how variations in microbial diversity will impact the functioning of an ecosystem (Langenheder et al., 2010). Fungal communities have been reported to differ with phases of curing, i.e., the yellowing phase, the color-fixing phase, and the stem-drying phase, and with their respective collection height/position on the plant, even exhibiting some differences in tobacco petioles and lamina during the tobacco curing process (Rotem, 1994). Previous studies show contradictory results of either increment or reduction in the abundance of fungal community and diversity of tobacco leaves infected by Golovinomyces cichoracearum, Didymella segeticola, and A. alternata (Guo et al., 2020; Huang et al., 2020; Xiang et al., 2020). Moreover, pesticide treatments have been documented to influence bacterial communities colonizing the tobacco phyllosphere (Chen X. et al., 2021; Liu et al., 2021).
To date, many methods have been described to analyze the phyllospheric microbial structure and biodiversity. Normally, culturing methods are economic but also time-consuming and can only detect <1% of microorganisms (Chen et al., 2018). Previously, pathogen identification could only be performed individually or in small groups. However, with the development of molecular sequencing, the next-generation sequencing of high-throughput technique has become the main research approach to microbial classification, proving to be ultrasensitive and precise for the indepth identification of culturable and non-culturable microbes and the simultaneous quantification of microbial community structure (Jayawardena et al., 2018; Mboowa et al., 2018). This technological advancement not only puts emphasis on the micro-ecology of distinct microbial taxa but also on the collective genetic composition of microbial communities belonging to different niches or “microbiomes” (Henry et al., 2021). Several host-associated microbiomics studies affirm that microbial bio-divergence is a feature that constitutes a portion of the host organism's extended phenology (Bruijning et al., 2020) with immense implications on the health, fitness, and evolution of the host. In view of its significance on the host's function and fitness, crop scientists aspire to model and navigate host–microbiome interactions (Bruijning et al., 2020; Henry et al., 2021). Reportedly, the community-level physiological profile technique (BIOLOG) has effectively been utilized to ascertain the bacterial community's functional diversity (Zhang et al., 2010). In earlier investigations, the profiling of the tobacco phyllosphere microbiome concentrated mainly on the biodiversity analysis of field-grown tobacco leaves sampled at a single stage, which was infected by G. cichoracearum, D. segeticola, and A. alternata (Guo et al., 2020; Huang et al., 2020). Although there is a huge void in our current knowledge regarding the epidemiology of brown leaf spot disease, whether it is antagonized or synergized by other phyllospheric microbial communities during different growth stages of tobacco as the dynamics of microbial diversity is known to partake significantly in the robustness and probability of getting disease among crops, either via competitive response toward pathogens or by instigating the innate immunity of plants against them (Douglas et al., 2020).
Consequently, in the present study, our goal was to distinguish the tobacco microbial communities and the dynamic variation in microbial communities and their biodiversity in three leaf growth stages and three stalk positions. Moreover, we were interested in the microbial functional diversity during tobacco growth phases and the development of tobacco brown spots, particularly in the symbiotic relationship between microbial community and environmental factors. For this, culture-independent isolation and high-throughput techniques were employed. Furthermore, the substrate utilization profiling (BIOLOG) technique was used to determine the utilization patterns of carbon sources. This study gives a new insight into phyllospheric microbial alteration in the spatiotemporally varying ecosystems during different growing stages and provides a theoretical basis for the precise prediction of diseases and precise pesticide application.
Materials and Methods
Meteorological Observations
To record information on the relative humidity, air temperature, wind speed, and rainfall, an automatic weather station (Beijing Xinhong Tec. IT, Co., Ltd., Beijing, China) was constructed consisting of a hygroscope (MC-KWS), a thermometer (MC-KWS), an anemoscope (MC-FX), a rain gauge (MC-YL), etc. The meteorological data storage system (MC-YL) was employed to acquire and process all measurements. The following are the collection dates: the first stage (August 8, 2020), the second stage (August 30, 2020), and the third stage (September 19, 2020).
Site Description and Sample Planting
An experimental field site was selected in Bijie city (27°22'5”N,105°19'57”E), located in the northwest of Guizhou Province of China. The fields were planted with tobacco cv. Yunyan87 for almost 2 years. Seedlings were planted during April and the growth management was kept the same as other tobacco plants but devoid of pesticide treatments. The chosen experimental area measured 60 m2, which was divided into three plots (20 m2 each), and then subdivided into three rows (1.1 m space between the rows). Ten Yunyan87 seedlings were planted with a 0.55 m gap between two plants on the same row.
Sampling and Processing of N. tabacum L. Leaves
Leaf positions were determined as upper (1~6th top leaf), middle (7~12th top leaf), and lower (13~18th top leaf), measuring from the top of the flue-cured tobacco plant (Supplementary Figure 1). When tobacco brown spot was first observed (about 100 days after planting), healthy and diseased leaves infected by A. alternata from the above-mentioned three leaf positions were collected randomly from three plots at the time of 100, 122, and 142 days after planting, respectively. Two kinds of leaves were clipped carefully from the plants using sterile hand shears and 20 g of the leaves were put into 50 ml plastic centrifuge tubes, separately. The scissors were sterilized with 70% ethanol for 30 s after cutting each piece of leaf. Upon arrival in the laboratory, the sample containing tubes were placed straightaway at 4°C prior to culturing and BIOLOG, or in the case of high-throughput sequencing at −80°C.
For designating codes to samples, the letter BJY87 was used for healthy leaves and BJB87 for diseased tobacco leaves, followed by the number 1 for the first time to collect leaves, 2 for the second time, 3 for the third time, and then followed by 1 for the first replicate, 2 for the second replicate, 3 for the third replicate, and finally followed by “U” for the upper, “M” for the middle, and “D” for the lower positions, respectively. For instance, BJY8711U were healthy leaves for the first replicate collected the first time from the upper position in the field, and BJB8722D were the diseased leaves for the second replicate collected the second time from the lower position (Supplementary Table 1).
Isolation and Molecular Identification of Phyllospheric-Culturable Microorganisms
Isolation and Identification of Bacteria
Culturable bacteria were isolated by using plate coating. Bacteria were isolated on Luria-Bertani (LB) media or nutrient agar (NA). Afterward, 10 g of leaves were placed in conical flasks with 300 ml of sterilized water and shaken for 1 h at 200 rpm, then 30, 40, and 50 ml supernatants were spread on both LB and NA plates. Once the plates underwent dark conditions for 2–5 days at 28°C, colonies with the same morphology, color, and growth rates were isolated. Axenic cultures were stored in 40% glycerin with LB bacterial solution at −20°C (LB: 40% glycerin = 1:1). The bacterial DNA was extracted using TIANamp Bacteria DNA Kit (Tiangen Biotech Co. Ltd., Beijing, China) according to the operating steps. The amplification of the 16S rRNA gene of isolates was carried out based on the procedures described by Li et al. (2021). Resultant PCR products were electrophoresed on a 1% agarose gel and the execution of targeted fragment size (16S 1600 bp) purification was done using Gene JET (Thermo Fisher Scientific, Waltham, MA, United States), then sequenced and used as a query in a BLASTN search of the NCBI nr database1. Moreover, for the identification of the isolates, matches with identity values >98% were used.
Isolation and Identification of Fungi
The tissue separation method was adopted for culturable fungal isolation (Attitalla et al., 2010). Isolation was done either on potato dextrose agar (PDA) or alkyl ester agar (AEA) according to the method of Chen X. et al. (2021). Diseased and healthy tissues (5 × 5 mm pieces) were directly inoculated on both PDA and AEA plates. The remaining tissues were placed in the dark at 28°C for 5 days, and the hyphae were then ready to be transferred to a fresh plate. Pigment, growth rate, and morphological traits were the distinct criteria for fungal isolation. Purified cultures were stored at 4°C on PDA slants. Molecular identification was conducted according to the method of Huang et al. (2021). The PCR products were electrophoresed on a 1% agarose gel and the purification of the ITS 500 bp fragment was exercised using Gene JET (Thermo Fisher Scientific, Waltham, MA, United States). Subsequently, the fragments were then sequenced and used as a query in a BLASTN search of the NCBI nr database1. For the purpose of identification of each isolate, matches with identity values >98% were used.
Extraction of DNA, Amplification, and Sequencing Process
DNA was extracted from the tobacco phyllosphere using an EasyPure Plant Genomic DNA Kit (TransGen Biotech, China) with the CTAB method. Qualitative detection of the DNA solution was carried out by 2% agarose gel electrophoresis, while the quantitative detection was determined through NanoDrop ND-2000 (Thermo Fisher Scientific, Waltham, MA, United States). Amplification of the hypervariable V3–V4 region of bacterial 16S rRNA genes was performed with the primers, 341F (5'-CCTACGGGRBGCASCAG-3') and 806R (5'-GGACTACNNGGGTATCTAAT-3') (Jeanette et al., 2012). The fungal ITS1-5F region was amplified using ITS5-1737F (5'-GGAAGTAAAAGTCGTAACAAGG-3') and ITS2-2043R (5'-GCTGCGTTCTTCATCGATGC-3') (Zhao et al., 2014). All reaction tubes contained 15 μl of Phusion®High-Fidelity PCR Master Mix (New England Biolabs) with 2 μM of forward and reverse primers and 10 ng of template DNA. The thermal cycler was set at 98°C for 1 min for initial denaturation, thenceforth at 98°C for 10 s (30 cycles of denaturation), at 50°C (30 s) for annealing, at 72°C (30 s) for elongation, and finally at 72°C (5 min). Thereafter, the resultant products were purified using Qiagen Gel Extraction Kit (Qiagen, Germany). With amplified products, the library was constructed and sequenced on an Illumina NovaSeq platform, and then 250 bp paired-end reads were generated.
The script from the Novogene platform was used for clipping barcode and primer sequences by Cutadapt (V1.9.12). From each sample, the operational taxonomic units (OTUs) were determined by UPARSE (Edgar, 2013), and additional biological data were also obtained. Furthermore, the representative OTU sequences were assigned species annotations. Unit (V.7.24) assisted in counting the community composition of each sample, then calculations were made for the OTUs matrix. For full-sample similarity comparison and to analyze the alpha-beta-diversity and calculate the observed species, Chao1, Shannon, Simpson, ACE, as well as Good's-coverage indices, QIIME (V.1.9.1) was employed. R software (V.2.15.3) was utilized to illustrate the rank abundance curve, dilution curve, and species accumulation box. Using Muscle (V.3.8.31), sequence alignments for ITS locus were carried out (Edgar, 2004). Moreover, FastTree 2 (V.1.9.1) software and the maximum likelihood (ML) method was chosen for phylogenetic analysis (Price et al., 2010). PCA was displayed by WGCNA packages and ggplot2 package in R software (V.1.9.1). Analysis of fungal trophic mode was conducted using the FUNGuild database (Nguyen et al., 2016), and bacterial OTUs were put into PICRUSt to predict functional analysis (Langille et al., 2013).
Biolog Inoculation and Analysis
The BIOLOG method was employed to inspect the functional metabolic fingerprints of the community and the patterns of carbon source utilization (Classen et al., 2003). Fifty ml of 0.85% NaCl (27 mL, w/v) was mixed with 3 g of tobacco leaves (cut into pieces) and shaken for 1 h at 28°C. The mixture was then diluted at a ratio of 1:1000. Into each of the ECO micro-plate well, 150 μL of dilution was added using an electronic pipette and incubated at 28°C in darkness for 7 days. The Biolog OmniLog® system (BIOLOG, Hayward, CA, USA) used in this study was equipped with a charge-coupled device camera system and an incubator. The color level of each well was measured in omnilog units (OUs) every 30 min by OmniLog digital camera. The value of OU is the reflection of the dynamic development of microbe carbon metabolism, represented by the utilization of carbon metabolism. OU and optical density (OD620nm) are comparable and could be calculated as 500 times the OD (Cruz et al., 2021). Heatmap illustrator (HemI 1.0.3.3) was used to display the heatmap of the maximum OU values of 16 samples.
Data and Statistical Analysis
IBM SPSS Statistics 23 (IBM Corp., New York, USA) was used to analyze the data and compare the differences in alpha-diversity indices of fungal and bacterial communities (Mao et al., 2020). A multiple comparison was performed by the LSD test (P < 0.05) to compare the diversity index of different treatments. The mean values were compared and the P ≤ 0.05 was considered to be statistically significant.
Results
Diversity and Abundance of Phyllospheric-Culturable Microorganism
By tissue isolation method, 153 fungal isolates belonging to 33 genera were obtained, while plate coating yielded 99 bacterial isolates belonging to 15 genera (Supplementary Table 2). For fungi, more genera were isolated on PDA (25) than AEA (24). The most predominant genus was Cercophora comprising 9 of 75 isolates on PDA and 14 of 78 on AEA, followed by Coprinopsis with 8 isolates on PDA and 13 isolates on AEA. The third abundant genus was Alternaria with 16 isolates (8 isolates each on PDA and AEA, respectively). The rarest genera were Brunnipila, Coniochaeta, Gibellulopsis, Hypoxylon, Irpex, Peniophora, Preussia, Myrmaecium, Rhizopus, Zopfiella, Schizophyllum, and Verticillium with 1 isolate on either PDA or AEA. The number of genera at three collection stages were 13, 17, and 25, respectively, inferring that the fungal diversity gets richer as the tobacco leaves become mature. For bacteria, more genera were isolated on LB (15) than on NA (13). The most abundant genus was Pantoea accounting for up to 33.33% of the bacterial isolates (17 of 53 isolates on LB and 16 of 46 isolates on NA), followed by Pseudomonas with 26 isolates (13 isolates on LB and on NA, respectively). The rarest genus was Agrobacterium with only 1 isolate on LB.
When analyzed the collection period of samples combining healthy and diseased leaves, diversity was the highest at the third sampling stage with 26 fungal and 13 bacterial genera, followed by the first and second time (13 fungal and 10 bacterial genera, 17 fungal and 4 bacterial genera, respectively). The relative abundance (percent of isolates of a genus out of the total number of isolates) at the first time showed that Cercophora and Pseudomonas were the most abundant (37.93% of fungal isolates and 29.17% of bacterial isolates), at the second time showed that Alternaria (19.64% of fungal isolates) and Pantoea (47.83% of bacterial isolates) were the most abundant, and at the third time showed that Coprinopsis (19.12% of fungal isolates) and Pantoea (42.31% of bacterial isolates) were the most abundant.
When analyzed for leaf type, by combining samples from all collection dates as well as different leaf positions, fungal diversity was higher for diseased leaves (25 genera) than healthy leaves (23 genera). In contrast, bacterial diversity for diseased leaves (10 genera) was lower than for healthy leaves (13 genera). The relative abundance in healthy groups suggested that Coprinopsis (18.57% of fungal isolates) and Pseudomonas (44.90% of bacterial isolates) were the most abundant genera. For diseased groups, the most abundant genera were Cercophora (15.66% of fungal isolates) and Pantoea (58.00% of bacterial isolates).
A comparison of microbial abundance among positions exhibited that the numbers of fungal genera from three positions (up, middle, and lower) were 16, 18, and 22, respectively. In the case of bacteria, the numbers of genera from three positions (up, middle, and lower) were 14, 5, and 9, respectively. Combining leaf position and leaf type, the number of fungal genera from the upper and lower diseased leaves (13 and 17 genera) was much higher than healthy leaves (8 and 14 genera), whereas the number of bacterial genera from the upper diseased leaves (7 genera) was less than the healthy leaves (10 genera).
Diversity of Microbial Communities From High-Throughput Sequencing
Quality of Fungal and Bacterial Sequence Data
There was a total of 1,150,015 effective tags and 305,236,114 bases of fungi across 9 healthy samples and 7 diseased samples and the average length was 266 nt. A total of 1,321 OTUs of fungi at ≥ 97% nt identity were obtained after removing low-quality, chimeric, and rare sequences from 16 samples, resulting in an average of 71,876 tags per sample (Table 1). The fungal sequence of each sample was deposited in the SRA database under the accession # PRJNA751254. With regard to bacteria, after quality filtering and de-noising, the removal of mitochondrial, chloroplast DNA, and other non-bacterial sequences, a sum of 1,317,757 effective tags were classified into 358 OTUs at ≥ 97% nt identity, and 537,085,996 bases were retained from 16 samples (Table 1). The sequences of 46 samples were deposited in the SRA database under project accession # PRJNA751261. Once the fungal and bacterial sample numbers reached ~45, the species accumulation boxplot indicated that the plateau phase has been approached (Supplementary Figure 2), pertaining to the sufficiency of sampling efforts for the estimation of species richness and describing the fungal and bacterial diversity. With the increment of the sequencing, the rarefaction curves for all samples tended to level off (Supplementary Figure 3), representing that the fungal and bacterial composition had been accurately described by the sequence coverage.
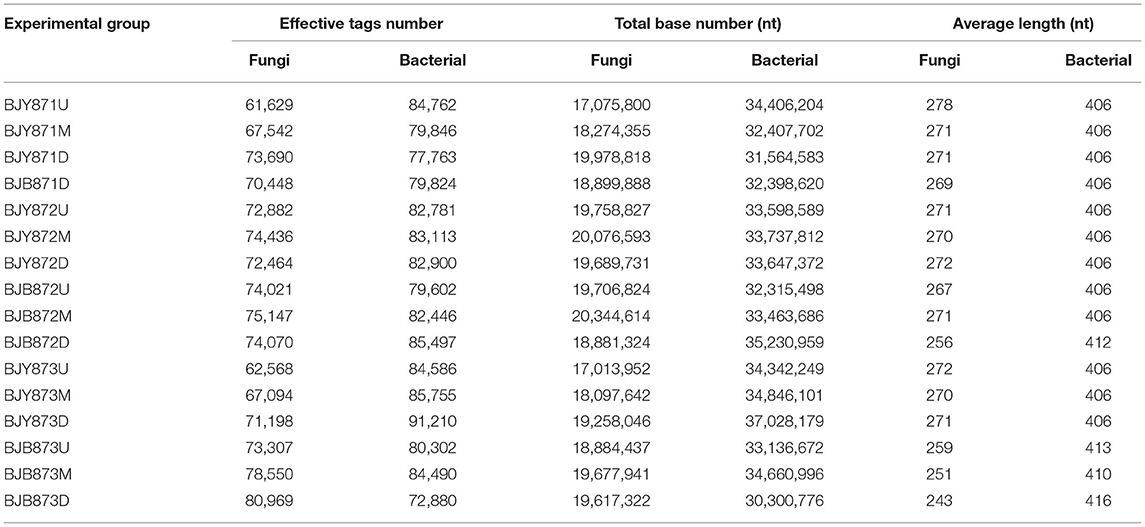
Table 1. Data quality control of sequencing samples.
Fungal and Bacterial Diversity
A comparison among sample collection periods unveiled a progressive increase in the count of fungal OTUs with time (Table 2). For fungi, the number of OTUs of diseased leaves increased significantly from the first collection time to the third time (48, 202, and 337, respectively). For bacteria, the number of OTUs of healthy leaves decreased, with a significant difference between the second and third time, while it increased from the first to the second sampling stage. For fungi, irrespective of position, healthy leaves displayed the highest diversity of OTUs contingent on the mean values of Shannon, Simpson, and Chao1 at three collection stages (Table 2). However, the diversity on diseased leaves was lower, established on the mean Shannon, Simpson, and Chao1 values in the second and third periods. The trend remained the same for the bacterial diversity of OTUs, where the lowermost diversity index values were obtained for OTUs of bacteria for diseased leaves at three collection periods.
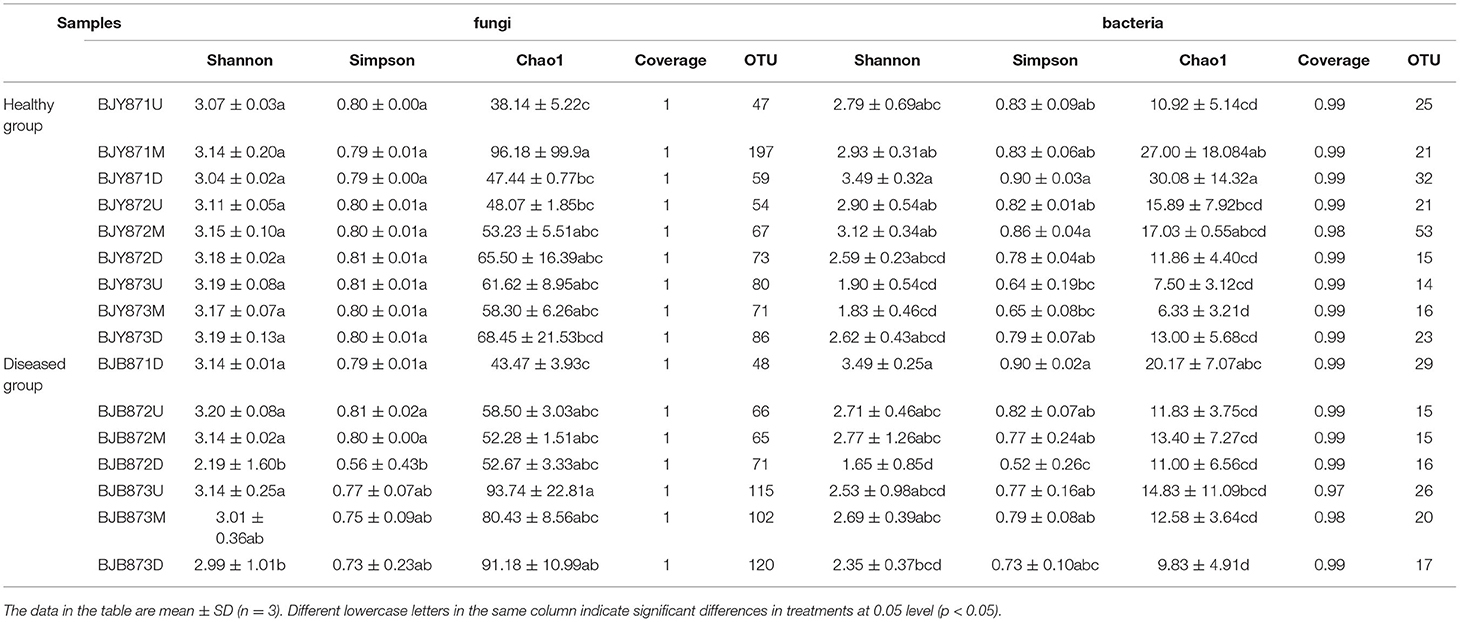
Table 2. Alpha diversity index of the ITS and 16S rRNA libraries obtained for clustering at 97% identity (OTU level).
Microbial Taxa in the Tobacco Phyllosphere
Fungal Taxa Composition of the OTUs
The distribution of different fungal phyla for the OTUs showed that 99.9% of the effective tags could be categorized under Ascomycota, Basidiomycota, Mortierellomycota, and Chytridiomycota. The ones unassigned were the members of other phyla and were highly unlikely to be true fungi (Figure 1A). In all healthy and diseased tobacco leaves, Ascomycota (78.02%) predominated the fungal communities and, to a lesser extent, Basidiomycota (21.97%), while Mortierellomycota and Chytridiomycota comprised <0.01% of the tags. Combining position, collection time, and different leaves, the average number of tags per time for the Ascomycota was 68.68, 76.54, and 85.74% in the three collection periods, respectively, indicating an increase as the tobacco leaves get older.
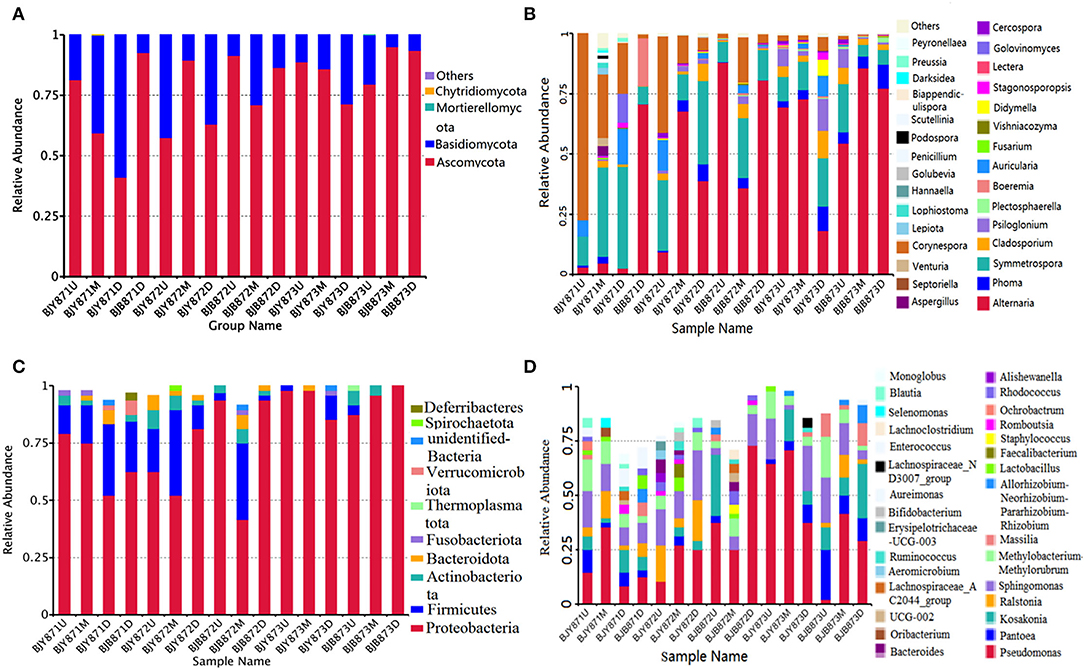
Figure 1. The relative abundance of 16 samples at phylum (top 10) and genus (top 30) levels. Different fungal phyla (A) and genus (B); different bacterial phyla (C) and genus (D). Phyla and genus making up <0.01 of total composition were classified as “Others.”
At the phylum level, the abundance of Ascomycota and Basidiomycota from healthy samples at the same position was increased during three collection stages, regardless of leaf health status, which suggests that the fungal abundance increased with the aging of tobacco leaves (Table 3). Statistical analysis revealed that there was no significant difference in the relative abundance of Ascomycota among the healthy samples with regard to different collecting stages and stalk positions; so did Basidiomycota, indicating that time-points and leaf positions could not affect the fungal community composition in the healthy leaves. For the diseased group, the relative abundance of Ascomycota from the same position was increased, and there was a significant difference between the abundance of Ascomycota in aspects of BJB871D (3.94%) and BJB873D (41.53), BJB872M (0.37%) and BJB873M (39.77%), portending that the collecting time had a significant effect on the abundance of Ascomycota in the diseased group. The abundance of Basidiomycota from 6 diseased samples had no difference except for BJB873D. A comparison of different positions at each sampling stage portended a higher relative abundance of Ascomycota from middle healthy leaves than healthy leaves from the upper and lower positions in three stages, whereas the abundance of Basidiomycota from lower healthy leaves was higher than leaves from the upper and middle healthy leaves. For diseased leaves, the abundance of Ascomycota was higher in the lower leaves than in the upper and middle leaves. While comparing two kinds of leaves from the same position, the relative abundance of Ascomycota and Basidiomycota from diseased leaves were higher than healthy leaves in three stages at three positions and the difference got more significant during three collecting stages, indicating that the time-point was a factor which could affect the fungal community composition.
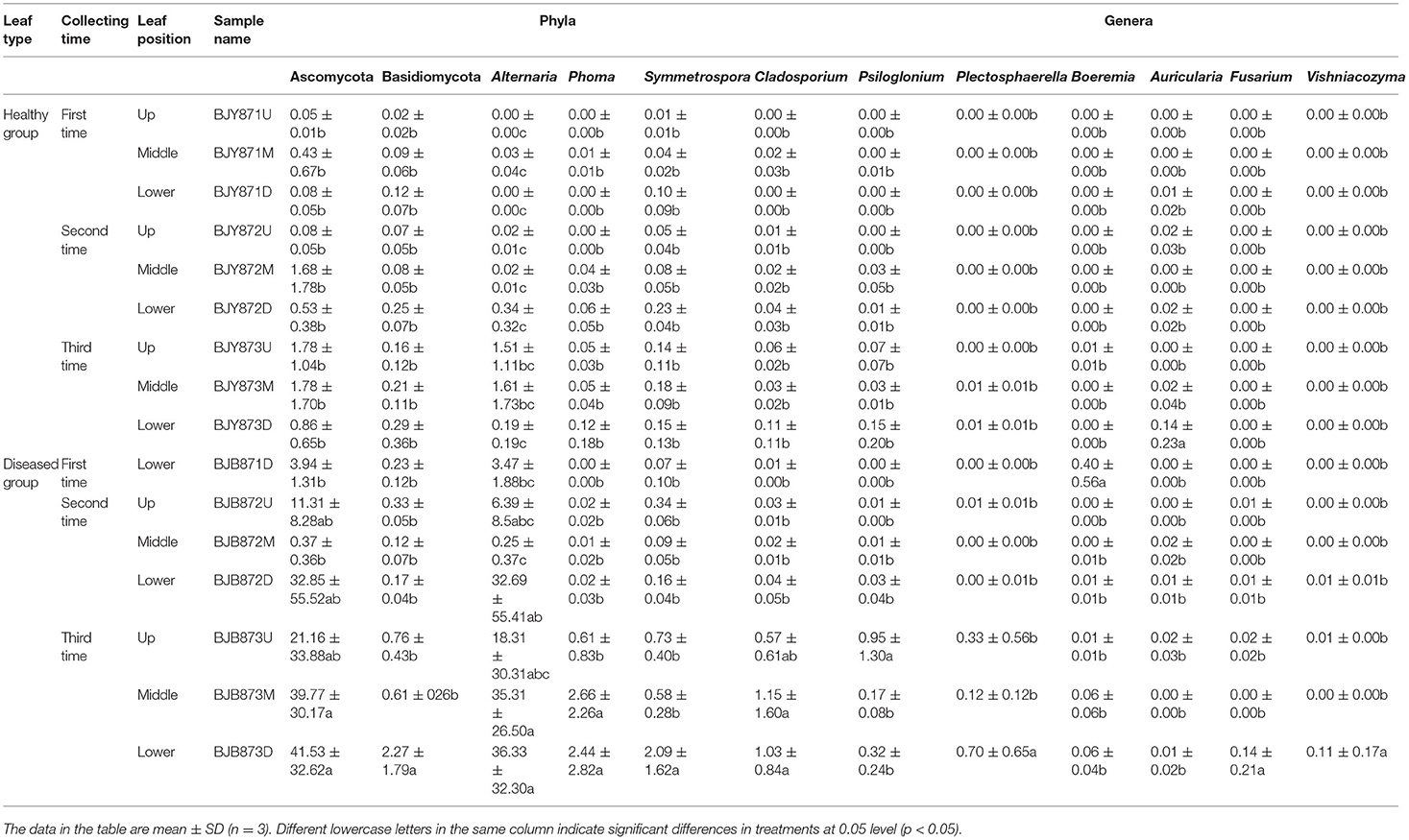
Table 3. Community compositions and relative abundance of phyllosphere microorganism from tobacco leaves infected by Alternaria alternata at fungal phyla and genera levels (%).
The differences among the communities at the OTU level from healthy and diseased samples at three collection periods are depicted in Venn diagrams (Figures 2A–C). Overall, 1,321 OTUs were acquired, where 734 belonged to the healthy and 587 to the diseased group (Table 2). There were 351 OTUs discovered for the first time, and 10.54% of them were shared OTUs. Healthy leaves from the middle position contained more fungal varieties (197 OTUs). A total of 396 OTUs were obtained for the second time, and 10.61% of them were shared OTUs. In the third stage, a total of 574 OTUs were discovered, and 9.58% of them were shared OTUs. However, the number of eukaryote OTUs in the healthy group was more in contrast to the diseased group, indicating that there was a significant difference among their fungal varieties. Of note, the number of OTUs in the healthy and diseased groups increased markedly at the second and third collection time. Apropos the second collection time, 194 OTUs were obtained from the healthy leaves, and 23.2% of them were shared OTUs; 202 OTUs were discovered from the diseased leaves, and 25.74% of them were shared OTUs. Diseased leaves from the upper position contained more fungal varieties than the healthy leaves (66 and 54 OTU, respectively). Concerning the third period, 237 OTUs were discovered from healthy leaves, and 23.63% of them were shared OTUs, while there were 337 OTUs in the diseased group, and 24.93% of them were shared. Samples from the diseased leaves (upper, middle, and lower positions) featured more fungal varieties (115, 102, and 120 OTUs, respectively) than the samples from the healthy leaves (80, 71, and 86 OTUs, respectively), as presented in Supplementary Figure 4.
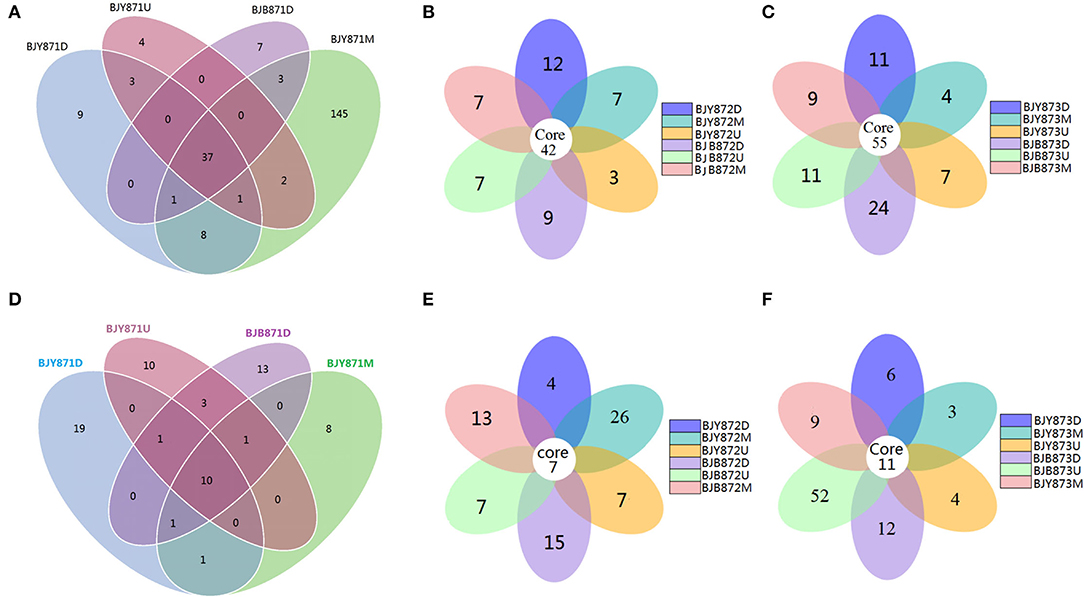
Figure 2. Venn diagram showing the number of operational taxonomic units (OTUs) detected in healthy and diseased tobacco leaves. Fungal Venn diagram in healthy and diseased groups from three positions (A–C). Bacterial Venn diagram in healthy and diseased groups from three positions (D–F).
From 16 samples, 87 genera of fungi were obtained, and the most frequent ones are shown in Figure 1B, and the abundance of the top 10 genera is shown in Table 3. Out of those 30 genera, the 10 highest numbers of tags were for Alternaria, Phoma, Symmetrospora, Cladosporium, Psiloglonium, Plectosphaerella, Boeremia, Auricularia, Fusarium, and Vishrniacozyma. Alternaria, which would include the tobacco leaf brown spot pathogen “A. alternata,” had 48.71% of the tags. According to the ML tree of the 100 most abundant fungal genera, the dominant fungi were associated with Ascomycota, followed by Basidiomycota (Figure 3A). On the part of Ascomycota, Alternaria remained the dominant genus, including A. alternata, followed by Phoma, Cladosporium, Psiloglonium, and Boeremia while Symmetrospora was the dominant genus of Basidiomycota. The abundance of the top five genera varied substantially among the samples (Figure 4) and statistical analysis for the top ten genera was shown in Table 3. Apparently, Alternaria was the dominant phyllospheric fungus in 7 diseased samples and the abundance of diseased samples from the same position was increased during three collection stages. On comparing the diseased leaves from the same collecting time, the abundance of the top ten genera did not show any differences, while when regarding to time-points, there was a significant difference between the abundance of Alternaria in aspects of BJB871D (3.47%), BJB873D (36.33%), BJB872M (0.25%), and BJB873M (35.31%), and the results were the same regarding the abundance of Phoma, Symmetrospora, Cladosporium, Fusarium, and Vishniacozyma in the lower diseased samples. The abundance of Phoma from the same position in the same leaf type increased during the course of three sampling stages; moreover, Symmetrospora was found to be the dominant genus in healthy leaves at the first stage. Furthermore, the abundance of Phoma, Symmetrospora, and Cladosporium from the same position at the third collecting stage in two kinds of leaf types had significant differences; they were higher in diseased leaves, while they had no difference at the first and second stages, indicating that that time-point was a factor which could affect the fungal genus composition. However, the abundance of the top ten genera of the healthy group had no difference during the three stages.
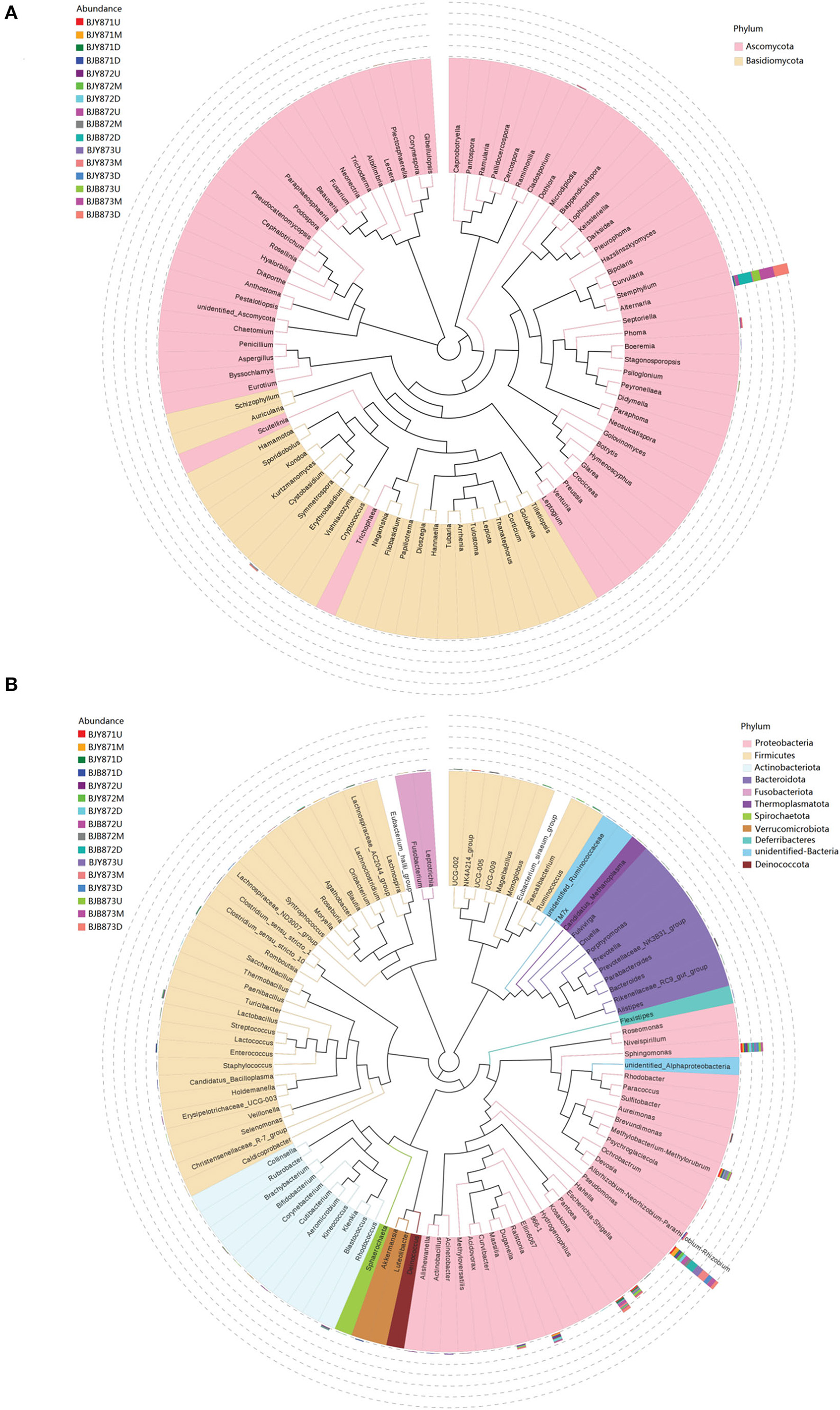
Figure 3. Maximum likelihood (ML) tree of 100 most abundant fungal (A) and bacterial (B) genera from the tobacco leaves infected with Alternaria alternata obtained by the analysis of ITS rDNA and 16S rRNA pyrosequencing data.

Figure 4. Pie charts showing the relative abundance of top 5 fungal genus of Alternaria, Phoma, Symmetrospora, Cladosporium, Psiloglonium, and “others” in the three analyzed leaf positions and at three collecting time.
Bacterial Taxa Composition of OTUs
The 16S data-set showed that a total of 97.72% effective tags could be classified into the Proteobacteria, Firmicutes, Actinobacteriota, Bacteroidota, Fusobacteriota, Thermoplasmatota, Verrucomicrobiota, Spirochaetota, and Deferribacteres. Members of the other phyla were unassigned or unidentified (Figure 1C). In all healthy and diseased leaves, the bacterial communities were dominated by the Proteobacteria (78.65%), followed by the Firmicutes (12.77%), Actinobacteriota (2.99%), and Bacteroidota (1.82%), while Fusobacteriota, Thermoplasmatota, Verrucomicrobiota, Spirochaetota, and Deferribacteres comprised <1.00% of the tags.
The abundance of main phyla and genera for 16 samples are shown in Table 4. It was found that the abundance of Proteobacteria in healthy leaves from the upper and middle positions decreased firstly, then increased at the third stage while the abundance of Firmicutes increased and then decreased. However, in lower healthy leaves, the abundance of Proteobacteria increased from 52.08% at the first collecting stage to 85.42% at the third stage, while the abundance of Firmicutes and Bacteroidota decreased continuously. In the diseased group, the abundance of Proteobacteria increased and Firmicutes decreased. Moreover, there was a significant difference between the abundance of Proteobacteria in aspects of BJB871D (69.92%) and BJB873D (100%), and the abundance of Firmicutes regarding BJB872M (33.33%) and BJB873M (0.00%), and Actinobacteriota had no differences among 16 samples. It was noted that the rhizobia of Neorhizobium, Mesorhizobium, Pararhizobium, Methylorubrum, and Methylobacterium were found by metagenomics (Figure 1D).
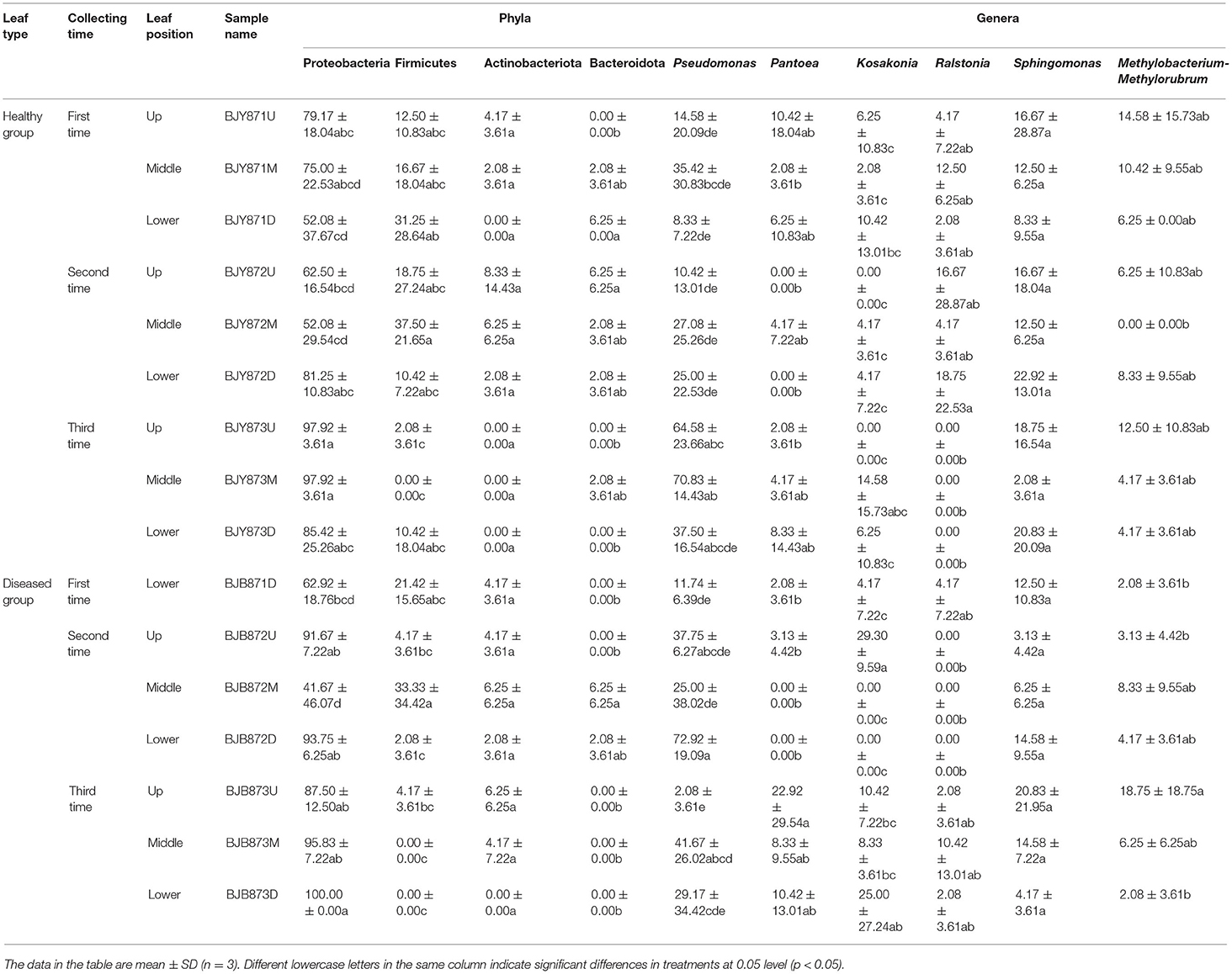
Table 4. Community compositions and relative abundance of phyllosphere microorganism from tobacco leaves infected by Alternaria alternata at bacterial phyla and genera levels (%).
Combining position, sampling time, and different leaves, the relative abundance of OTUs per time for the Proteobacteria was 67.19, 70.83, and 94.10% in the first, second, and third times, respectively, indicating an increase as the tobacco leaf get mature. While the relative abundance of OTUs per time for the Firmicutes was 20.57, 17.53, and 2.78% in the three collection stages, respectively, indicating a significant decline as tobacco leaves get mature. For healthy and diseased leaves and in all the samples, the relative abundance of Proteobacteria and Firmicutes had the same upward/down trend. The differences between the communities at the OTU level from healthy and diseased samples at three collection periods are portrayed in Venn diagrams (Figures 2D–F). A sum of 107 OTUs was detected for the first time, and 9.35% of them were shared OTUs. Healthy leaves from the lower position exhibited more bacterial varieties (32 OTUs). In the second stage, a total of 135 OTUs were discovered from the healthy and diseased leaves with 7 shared OTUs. About 89 OTUs were obtained from the healthy leaves, and 10.36% of them were shared OTUs. Healthy leaves contained more bacterial varieties than the diseased leaves (53 and 15 OTUs, respectively), from the same position (middle position). For the third stage, a total of 53 OTUs were discovered from the healthy leaves with 26.42% of them being shared, while 63 OTUs were discovered from the diseased leaves, and 36.51% of them were shared. Samples from the healthy leaves contained more bacterial varieties (78, 89, and 53 OTUs, respectively), than the samples from the diseased leaves (29, 46, and 63 OTUs, respectively), as shown in Supplementary Figure 5.
Figure 1D represents the 30 most common bacterial genera; the top ten among those were Pseudomonas, Pantoea, Kosakonia, Ralstonia, Sphingomonas, Methylobacterium-Methylorubrum, Faecalibacterium, Lactobacillus, Massilia, and Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium. Pseudomonas was the highly recurring genus in most of the healthy and diseased groups (Figure 5). There was no difference between the abundance of main genera among 16 samples based on statistical analysis (Table 4), indicating that the bacterial genera were not affected by time-point, leaf type, and stalk position. The 100 most abundant bacterial genera, according to the ML tree method, fell under the phylum of Proteobacteria, then Firmicutes, followed by Actinobacteria and Bacteroidota (Figure 3B). Pseudomonas, Sphingomonas, Kosakonia, Methylorubrum, Pantoea, and Ralstonia were the dominant genera of Proteobacteria. For Firmicutes, the dominant bacterial genera were Lactobacillus, Enterococcus, Ruminococcus, and Blautia. For the Actinobacteria and Bacteroidota, the dominant genera were Rhodococcus and Bacteroides, respectively. It is worth mentioning that Ralstonia was among the top ten bacterial genera and was detected in 10 samples.
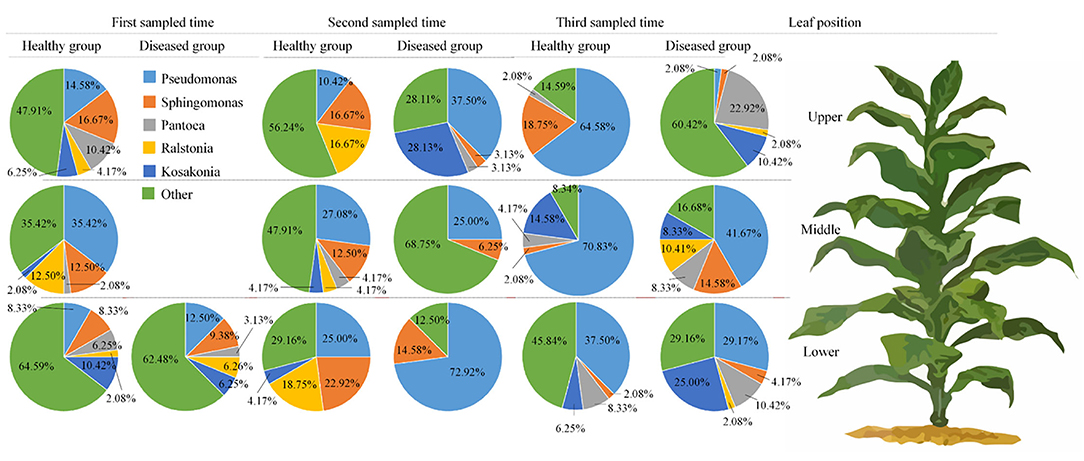
Figure 5. Pie charts showing the relative abundance of top 5 bacterial genus of Pseudomonas, sphingomonas, Pantoea, Ralstonia, Kosakonia, and “others” in the three analyzed leaf positions and at three collecting time.
Spatial Distribution of Microbial Communities
Non-metric multi-dimensional scaling (NMDS) served to evaluate the difference between samples in species complexity (Figure 6). In relation to fungal communities, all healthy samples tended to cluster together, except for three samples, which were at the middle position in the first collection stage and at the upper and lower positions in the third stage. The communities of diseased groups from the first and second stages overlapped with each other, while the communities of diseased samples collected at the third stage were evidently separated from one another (Figure 6A). Regarding bacterial communities, although all diseased and healthy groups were scattered, the samples from the same group were closer to each other, respectively (Figure 6B). The results corroborate significant differences between healthy and diseased tobacco leaves pertaining to the fungal and bacterial communities, as affected by A. alternata.
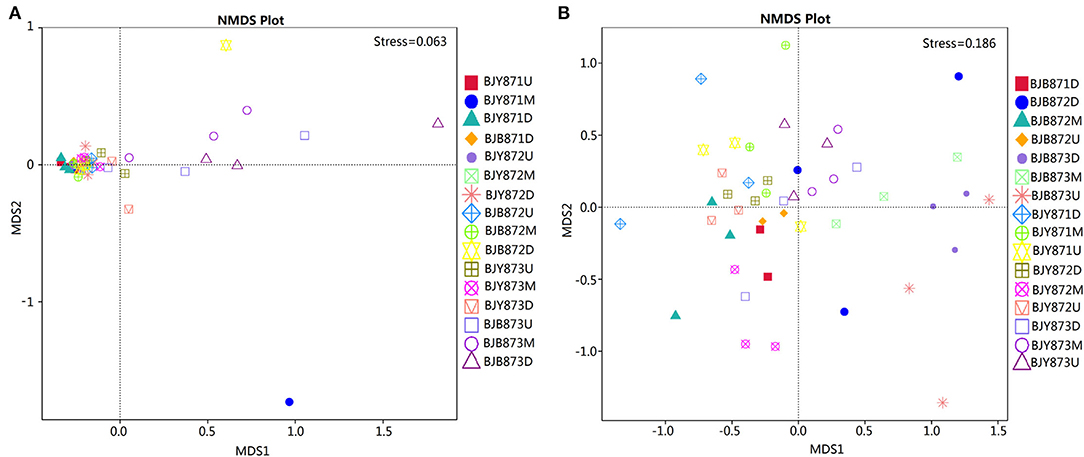
Figure 6. Non-metric multi-dimensional scaling (NMDS) analysis of the fungal (A) and bacterial (B) communities. The spot represents one sample, the distance between two spots represents the degree of deviation, and the same color represents samples in one group. Stress value is <0.2, indicating that NMDS could accurately reflect the difference of microbial communities among samples.
Microbial Functional Prediction
FUNGuild database assisted in analyzing the fungal ecological functions. According to their trophic mode, the fungal OTUs were divided into 8 groups. The gene sequences in the 16 samples were mostly involved in the pathotroph-saprotroph-symbiotroph (8.88%), followed by pathotroph-saprotroph, and pathotroph-symbiotroph at 0.42 and 0.20%, respectively. However, the unassigned sequences scored 90.16% and constituted the largest group (Figure 7). Strangely, saprotroph-symbiotroph and symbiotroph only appeared in the BJY871M, in the healthy leaves collected from the middle position at the first sampling stage. In both healthy and diseased groups, the relative abundance of pathotroph-saprotroph, pathotroph-saprotroph-symbiotroph, and pathotroph-symbiotroph increased from the first collection stage to the last.
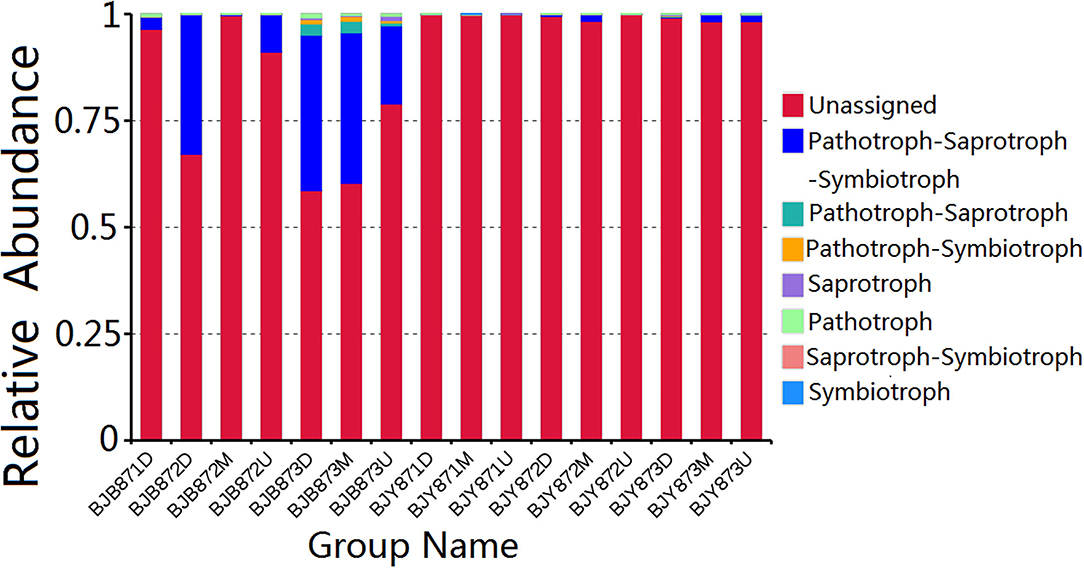
Figure 7. Relative abundance of fungal functional groups (guilds) contingent on OTU annotation table with disturbance frequency level.
Variation of bacterial functional categories was carried out by PICRUSt. There were six biological metabolism pathways based on the KEGG PATHWAY database except for the unassigned sequences (Figure 8A). The predominant pathway was metabolism function (50.03%), followed by genetic information processing (16.93%), environmental information processing (13.62%), cellular processes (2.61%), and human diseases (1.24%). At KEGG level2 function, the top thirty-five sub-functions were presented by cluster heatmap (Figure 8B). The relative abundances of the top ten sub-functions were as follows: membrane transport (11.21%), energy metabolism (10.00%), carbohydrate metabolism (8.39%), amino acid metabolism (8.34%), replication and repair (7.55%), metabolism of cofactors and vitamins (5.76%), poorly characterized (5.14%), translation (4.70%), cellular processes and signaling (3.57%), and nucleotide metabolism (3.28%). When analyzed for the position, the relative abundance of the top thirty-five sub-functions remained the same at three collection stages both in healthy and diseased groups. Similarly, when analyzed for the collection time, the relative abundance of the top thirty-five sub-functions from three positions was very similar, nullifying the likelihood of any difference among the 16 samples.
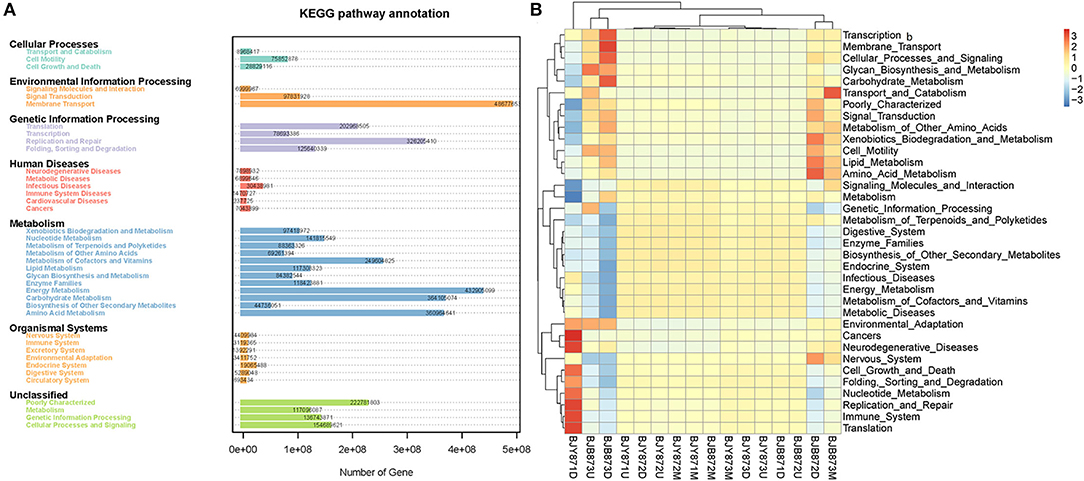
Figure 8. Variation of bacterial functional categories based on qualified sequences analyzed by PICRUSt. The KEGG pathway annotation of genes predictions from 16 samples (A). The cluster heatmap at the KEGG level 2 function (B).
Microbial Community Metabolic Profiles
This study made use of BIOLOG ECO, which is a plate of 96 wells having 3 parallels and one blank control with 31 different carbon sources, including carboxylic acids, carbohydrates, amino acids, amines, polymers, and phenolic compounds. The maximum color changes of carbon substrates observed in the fingerprint pattern provide useful carbon source utilization profiles of microbial communities (Figure 9). Among the six carbon substrate classes, the average OU values of carbohydrates (containing D-cellobiose, D-xylose, I-erythritol, α-D-lactose, D-mannitol, N-acetyl-D-glucosamine, β-methyl-D-glucoside, Glucose-1-phosphate, D-galactonic acid γ-lactone, and D,L-α-Glycerol Phosphate) were the highest in 16 samples, implying that microbial communities had strong utilization ability of carbohydrates, followed by the carboxylic acids (including D-glucosaminic acid, D-galacturonic acid, γ-hydroxy butyric acid, pyruvic acid methyl ester, α-ketobutyric acid, D-malic acid, and itaconic acid). The polymers (Tween-40, Tween-80, α-cyclodextrin, and glycogen), amines (phenylethyl-amine and putrescine), and amino acids (including L-serine, glycyl-L-glutamic acid, L-threonine, L-arginine, L-asparagine, and L-phenylalanine) were also effectively degraded by the microbial communities. The microbial communities had the lowest OU value of phenolic compounds (2-hydroxy benzoic acid and 4-hydroxy benzoic acid), suggesting that they had the lowest utilization of phenolic compounds.

Figure 9. Omnilog units (OUs) of carbon sources in BIOLOG ECO-Plate of microbial communities from different samples. The maximum OU values of 16 samples are shown.
The utilization characteristics of substrates in the microbial communities of the two leaf groups remained the same. In the healthy group, the average OU values of 6 carbon sources increased from the first to the third collection stage, as in the diseased group, probably due to the changes in microbial community composition with the environmental factors. However, the average OU values of 6 carbon sources of the healthy group were higher than the diseased group except for the phenolic compounds. It signified that the microbial community of the healthy group had higher utilization rates of carbohydrates, polymers, amino acids, carboxylic acids, and amines, but lower utilization rates of phenolic compounds. Comparisons based on the leaf position indicated that the OU average values of 6 carbon sources of middle leaves were the highest, followed by the upper and lower leaf, referring to the highest utilization of 6 carbon sources by microorganisms associated with the middle leaf. The utilization of 6 carbon sources varied among 16 samples; the sample of BJY8732M had the highest OU average values. It indicated that microbes had the highest metabolism in the maturation period among the three collection stages. The microbial community on the third sampling time showed the highest carbohydrates and carboxylic acids utilization rates, with the average OU values of 289.77 and 289.40, respectively. The sample of BJB8732D had the lowest OU average values, perhaps due to fewer available nutrients for the microorganisms in a leaf getting too old.
Discussion
In this study, three methods (culturomics, metagenomics, and BIOLOG) were employed to explore the dynamics of community diversity and functional response of phyllospheric microbes from healthy and diseased tobacco leaves infected by A. alternata in the spatio-temporal varying ecosystem (Rotem, 1994; Lv et al., 2013; Liu et al., 2020). More fungal genera were isolated from diseased leaves, while the bacterial genera from diseased leaves were less than healthy leaves by a traditional plating technique. Besides, microbial genera were the highest at the third sampling stage. In high-throughput sequencing technology, the dominant fungal and bacterial phyla were Ascomycota and Proteobacteria, respectively, which is consistent with the results of a previous study (Huang et al., 2021). The top five fungal genera were Alternaria, Phoma, Symmetrospora, Cladosporium, and Psiloglonium, whereas the top five bacterial genera were Pseudomonas, Pantoea, Kosakonia, Ralstonia, and Sphingomonas; the dominant fungal and bacterial genus were Alternaria and Pseudomonas which were consistent with the result of previous studies (Xu et al., 2014; Liu et al., 2019, 2020). More OTUs were obtained from the healthy group than the diseased group (Huang et al., 2020; Xiang et al., 2020). Moreover, the number of fungal OTUs was subject to change with an increase in disease severity. The α-diversity indices of fungi showed that there were no differences in the healthy group over time at the three leaf positions (Huang et al., 2021). A significant difference based on leaf types was observed where the relative abundance of predominant phyla, Ascomycota, and Basidiomycota from leaves infected by A. alternata, were found higher than healthy leaves from the same position at the three stages. Alternaria was the dominant genus in eleven samples except for the healthy leaves in the first stage, and at the upper and middle healthy leaves in the second stage. The relative abundance of Phoma from the same position in the same leaf type increased in three stages and had no significant difference among 9 healthy samples while it indicated a significant difference at 0.05 level in diseased samples at the lower and middle positions in the first and last stages. In terms of bacterial phyla and genera, Proteobacteria and Pseudomonas were the dominant ones in 16 samples, respectively, coinciding with Zhang's report (Zhang et al., 2020). The gene sequences in sixteen samples were mostly involved in the Pathotroph-saprotroph-symbiotroph and it was the main fungal trophic mode. The predominant pathway and sub-functions were metabolism function and membrane transport. Microbial communities had a strong utilization ability of carbohydrates with the lowest utilization of phenolic compounds.
A comparative analysis of the cultivable microbial genera counts isolated from four different media (AEA and PDA, LB, and NA) did not demonstrate any statistical difference, signifying that the media composition had no differential impact whatsoever on the detection of microorganisms. Due to complex or restricted nutritional or environmental requirements, only a minute percentage of microorganisms could be cultured in the lab (Ma et al., 2017; Li et al., 2018); concurrently, it is reported that about 0.1–3% of bacteria are readily cultivable (Wagner et al., 1993; Amann et al., 1995). In this study, 33 fungal and 15 bacterial genera were obtained by culturomics, while 87 fungal and 159 bacterial genera were found by metagenomics, showing the limitation of the culture-dependent method. The fungal genera frequently yielded by this method were Coprinopsis, Cercophora, and Alternaria, while Coprinopsis and Cercophora were non-pathogens. When comparing fungal genus counts from two kinds of leaf types in three stages, there were significant differences at the second stage; from the15 genera isolated from diseased leaves, only 8 genera isolated from healthy leaves, whereas the number of bacterial genera showed no difference. While comparing the genera between healthy and diseased groups, it was detected that Botrytis was isolated only from healthy leaves with a relative abundance of 14.29%, and it was also found in BJY872D, BJY873U, BJB873U, and BJB873D by a high-throughput technique. The total is well known that Botrytis is a causal agent of tobacco gray mold (Zhu et al., 2002). Mucor had a relative abundance of 12.05% in diseased leaves and only 1.43% in healthy ones. Likewise, Pseudomonas and Pantoea were the dominant ones in both healthy and diseased groups, with the relative abundance of Pseudomonas being 44.90 and 58.00%, and Pantoea being 6.00 and 10.20% in diseased and healthy groups, respectively. These observations suggest that Mucor and Botrytis could be indicators for the appearance of tobacco brown spot, while Pseudomonas and Pantoea could be indicators for the disease index of tobacco brown spot.
Referring to the high-throughput sequencing results, Ascomycota persisted to be the dominant fungal phylum followed by Basidiomycota and Chytridiomycota. This result is in line with Liu's findings (Liu et al., 2019). The taxa responded differently to the leaf senescence process; the number of OTUs in Ascomycota and Basidiomycota increased greatly during leaf senescence in the same stalk position from one leaf type, and were higher for the diseased leaves than the healthy leaves from the same position at one stage, implying that the abundance of phyla is related to disease index and is consistent with the above-mentioned conclusion that fungal OTUs was subject to change with an increase in disease severity. When healthy leaves at one collection stage were compared, the Ascomycota OTUs had a higher prevalence in the middle leaves, whereas for the lower healthy leaves, Basidiomycota had the highest OTUs scores. The data obtained with respect to bacterial phyla coincided with an earlier report that documents Proteobacteria, Firmicutes, and Actinobacteriota as the dominant bacterial phyla (Liu et al., 2020). The relative abundance of Proteobacteria remained the highest in samples from the final collection stage among 16 samples; it was higher for diseased leaves than healthy ones from the lower position in all stages. For Firmicutes, it was lower in the diseased leaves than healthy leaves from the same position at the second and third sampling periods, while they had no significant difference at 0.05 level (p < 0.05) in 16 samples.
At the genus level, Alternaria, Phoma, Symmetrospora, Cladosporium, and Psiloglonium were found most consistently across 16 samples, implying that they constitute the fungal “core” phyllospheric genera on tobacco. Alternaria was the most abundant fungal genus in thirteen samples except for three healthy samples collected at the first stage, followed by Symmetrospora and Phoma. It is well-known that tobacco is cultivated for its foliage which naturally gets mature from the bottom to the top, while this event of maturing or yellowing characterizes leaf senescence. During leaf senescence, certain macromolecules, i.e., proteins, lipids, nucleic acids, etc. are degraded and nutrients are released along with a change in the phyllospheric microbiome (Woo et al., 2013). High-throughput sequencing revealed that the abundance of Phoma in 16 samples from the same position increased from the first collection period to the last in both kinds of leaf types. For Alternaria, it increased in the upper and middle healthy leaves, while it increased from the first stage to the second and decreased at the third stage in the lower healthy and diseased leaves. Definitely, leaves from the upper and middle positions become older with the passage of time (i.e., from the first collection time to the third), whereas leaves in the lower position senesce quickly and could not provide sufficient nutrients for microbial sustenance, which very much explains the fluctuations in the growth patterns of A. alternata. Comparing the abundance of the same genus in the healthy group from the one in the collecting stage, the abundance of Alternaria from the middle position and Symmetrospora from the lower position was the highest among the three stalk positions. The abundance of Alternaria was the highest in the lower position in the diseased group, indicating that the abundance of Alternaria is related to the tobacco growing stage, leaf type, and leaf stalk position. There was a significant difference between the abundance of Ascomycota in aspects of BJB871D and BJB873D, BJB872M, and BJB873M; the same difference was found in Phoma, implying that the collecting time had a significant effect on the abundance of Ascomycota and Phoma in the diseased group from the lower and middle positions at 0.05 level. It should be noted that Alternaria and Symmetrospora were detected in all samples; their abundance was between 2.57 and 88.07% and 4.9 and 42.16%, respectively. The relative abundance of Symmetrospora was closely correlated with the abundance of Alternaria; when the abundance of Alternaria increased, the abundance of Symmetrospora would decrease. It is well-known that Symmetrospora species are red ballistosporous yeasts in the subphylum Pucciniomycotina of phylum Basidiomycota. They are ubiquitous in nature, grow as pinkish-red colonies, form nearly symmetrical ballistoconidia, and produce xanthophyll 2-hydroxytorularhodin and carotenoids (Weber et al., 2005; Almasoud et al., 2019). Understandably, Symmetrospora had higher relative abundances (p <0.05) in the fresh flavor style flue-cured tobacco leaves (FTLs) during the process of fermentation (Zhang et al., 2020). It is not hard to find that the abundance of “Other” from the sample at the same leaf position decreased with the age of tobacco leaves which might be related to the fact that older leaves already had lower nutrients and reduce microbial diversity.
The comparison of genera isolated by culture-dependent method and high-throughput sequencing resulted in the shared detection of eight fungal genera, and most of them were phyto-pathogens, including Cladosporium (Xu et al., 2014), Didymella (Guo et al., 2020), Alternaria (Wang et al., 2016), Fusarium (Chen et al., 2022), Cercospora (Kashiwa and Suzuki, 2021), Aspergillus (Welty and Nelson, 1971), and Penicillium (Pei et al., 2019). Independently, the culture-dependent approach showed that the top five frequently found fungal genera were Cercophora, Coprinopsis, Alternaria, Mucor, and Didymella. However, Alternaria, Phoma, Symmetrospora, Cladosporium, and Psiloglonium were particularly noticeable in the high-throughput technique. With respect to bacteria, Pseudomonas, Pantoea, and Sphingomonas were mutually detected by both techniques. In high-throughput sequencing, Pseudomonas and Sphingomonas were the commonly found bacterial genera that were noticed in all samples, with Pseudomonas being the most dominant one. Through the culture-dependent method, the frequently isolated bacterial genera were Pantoea, Pseudomonas, and Variovorax, whereas the Sphingomonas was the fifth commonly found genus. This result is in line with the findings of Liu et al. (2020), where Pantoea and Pseudomonas were declared, after high-throughput sequencing, as the dominant bacterial genera isolated from A. alternata infected leaves. Yet it is worth noting that rhizobia including Neorhizobium, Mesorhizobium, Pararhizobium, Methylorubrum, and Methylobacterium were found by metagenomics. This result is in line with the previous report that their presence has also been detected using cultivation-independent methods (Jackson et al., 2006). These genera belong to the class of α-Proteobacteria which is a group of gram-negative and rod-shaped bacteria. Members of rhizobia were commonly isolated from soil, and also from a wide variety of natural habitats, such as fresh water, sediments, plant roots, and plant stems. It had been reported that Methylobacterium could be isolated from leaf (Madhaiyan and Poonguzhali, 2014) and was the dominant microbes of FTL in the fermentation process (Zhang et al., 2020), and it imparts a range of beneficial effects on plant growth and development through various mechanisms (Madhaiyan et al., 2007). The molecular taxonomy has classified Methylobacterium as representing a line of descent in the α2 subgroup of the Proteobacteria (Madhaiyan et al., 2012). Studies showed a horizontal transfer of symbiotic genes from a Bradyrhizobium to a Mesorhizobium (Barcellos et al., 2007; Rogel et al., 2011). Most notably, Ralstonia was also identified by high-throughput sequencing in 10 samples with a relative abundance of <20%. Being a soil-borne pathogen with long incubation periods Ralstonia presumably contributes to its incidence.
Microbial functions were predicted by using the FUNGuild database and PICRUSt, where most of the fungal OTUs were recognized as pathotroph-saprotroph-symbiotroph, accompanied by pathotroph-saprotroph and pathotroph-symbiotroph. Crops may pose a risk of opportunistic fungal infection development in patients who are immunocompromised as the potential pathogenicity of those fungal crop contaminants cannot be disregarded (Ashley et al., 2018). The data signifies the presence of a large number of fungi; having phyto-pathogenic potential, the fungi are associated with tobacco phyllosphere during leaf senescence, for instance, the facultative necrotrophs, including A. alternata (the causal agent of brown spot) (Wang et al., 2016), Cercospora nicotianae that causes frogeye both on flue-cured and cigar tobacco (Fajola and Alasoadura, 1973; Zhao et al., 2020), Phoma omnivirens (the causal agent of black spot stalk) (Jiang et al., 2018), Stagonosporopsis cucurbitacearum that causes spot blight on tobacco (Wang et al., 2018), and D. segeticola that causes tobacco leaf spot (Xiang et al., 2020), along with the obligate biotroph, G. cichoracearum (the causal agent of powdery mildew) (Chen et al., 2019). Probably, those pathogens had arrived on tobacco leaves from diseased tissues in the field, whereas some could be growing either pathogenically inside the leaves or saprophytically on leaf surfaces during plant growth (Laforest-Lapointe et al., 2016). PICRUSt analysis revealed that the predominant functional pathway was metabolism function, followed by genetic and environmental information processing; moreover, the top three sub-functions were membrane transport, energy metabolism, and carbohydrate metabolism. Bacterial gene function prediction showed no significant difference among the 16 samples.
The result of substrate utilization profiling (BIOLOG) displayed that microbial communities had a strong utilization ability of carbohydrates, followed by carboxylic acids, amino acids, polymers, and amines; however, the utilization of phenolic compounds was the minimum. The substrate utilization characteristics of microbial communities in two kinds of leaf groups remained the same, which increased from the first to the third collection periods, while the healthy group had relatively higher utilization rates of carbon sources than the diseased group except for the phenolic compounds. The middle leaves had a higher utilization ability of 6 carbon sources in both leaf types, followed by the upper and lower leaves. Furthermore, a higher microbial diversity on tobacco leaves infected with tobacco brown spots during different growth stages was yielded from high-throughput sequencing than the culture-dependent method.
Conclusion
In this study, spatiotemporal variation of the community and the divergence of phyllospheric microflora were explored to be associated with three different leaf positions across three leaf growth stages (advancing gradually toward leaf senescence) in healthy or diseased leaves. More fungal and bacterial genera were obtained by metagenomics than by the culture-dependent method. Apparently, there were some differences between healthy and diseased leaf types. The abundance of Ascomycota and Basidiomycota from diseased leaves was higher than in healthy leaves in three periods in all positions by high-throughput sequencing. Moreover, the abundance of main fungal community composition was affected by time-points, and the time-point was the main factor that could affect the fungal community composition in the diseased group. The abundance of two main fungal phyla (Ascomycota and Basidiomycota) and genera (Alternaria and Phoma) from the middle and lower positions at the last stage was much higher than samples at the first and second stages in the diseased group. Function prediction analysis visualized the microbial community structure in tobacco leaves and better mining the microbe succession in varying spatiotemporal environments. Pathotroph-saprotroph-symbiotroph was the dominant trophic mode of the fungal community and metabolism function was the predominant pathway in the three stages. Carbohydrates were the main carbon source for microbes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA751254 and PRJNA751261.
Author Contributions
Y-fD, X-mW, H-cW, and IS conceived and designed the experiment. Y-fD, L-tC, and H-cW conducted the experiment and collected the samples. H-cW and J-xL performed the analysis of samples. Y-fD and H-cW analyzed the data. Y-fD wrote the first draft of the manuscript. H-cW, X-mW, FW, SS, and IS revised the manuscript. All authors read and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (32160522, 31960550, and 32160656), the China National Tobacco Corporation [110202001035(LS-04) and 110202101048(LS-08)], the Guizhou Science Technology Foundation [ZK(2021)Key036], the International Science and Technology Cooperation Base [(2020)4102], Guizhou Provincial Academician Workstation of Microbiology and Health [(2020)4004], the Guizhou Tobacco Company (2020XM22 and 2020XM03), the Cultivation Program of Guizhou University [(2019)09], the Hundred Level Innovative Talent Foundation of Guizhou Province [GCC(2022)023-1 and GCC(2022)028-1], the Sino-Pakistan Project NSFC (31961143008), the National Natural Science Foundation of China, the International (Regional) Cooperation and Exchange Program, the Research fund for International young scientists (31750110462), and the Jiangsu Collaborative Innovation Center for Modern Crop Production (JCIC-MCP) China.
Conflict of Interest
Y-fD was employed by Bijie Tobacco Company. J-xL was employed by Guizhou Tobacco Company of CNTC, China National Tobacco Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We appreciate all the colleagues for their technical support in the process of data analysis. We also thank the Upland Flue-Cured Tobacco Quality and Ecology Key Laboratory of China Tobacco, Guizhou Academy of Tobacco Science, Guiyang, China, for providing access to the experimental equipment.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.920109/full#supplementary-material
References
Almasoud, J., McArdle, L., Henne, R., Mathews, K., O'Connor, Í., McLoughlin, L. E., et al. (2019). Draft genome sequence of a red basidiomycete yeast, symmetrospora coprosmae strain UCD350, isolated from soil in Ireland. Microbiol. Resourc. Announc. 8, e01256–e01219. doi: 10.1128/MRA.01256-19
Amann, R. L., Ludwig, W., and Sciileifer, K. (1995). Phylogenetic identification and in situ detection of indivisual microbial cells without cultivation. Microbiol. Rev. 59, 143–169. doi: 10.1128/mr.59.1.143-169.1995
Ashley, B., Ponder, M. M., and Garcia-Diaz, J. (2018). Phoma infections: Classification, potential food sources, and their clinical impact. Microorganisms 6, 58. doi: 10.3390/microorganisms6030058
Attitalla, I. H., Al-Ani, L. K. T., Nasib, M. A., Balal, I. A. A., Zakaria, M., El-maraghy, S. S. M., and Rezaul, K. S. M. (2010). Screening of fungi associated with commercial grains and animal feeds in Al-Bayda Governorate, Libya. World Appl. Sci. J. 9, 746–756. Available online at: https://www.researchgate.net/publication/280253933_Screening_of_Fungi_Associated_With_Commercial_Grains_and_Animal_Feeds_in_Al-Bayda_Governorate_Libya
Barcellos, F. G., Menna, P., da Silva Batista, J. S., and Hungria, M. (2007). Evidence of Horizontal Transfer of Symbiotic Genes from a Bradyrhizobium japonicum Inoculant Strain to Indigenous Diazotrophs Sinorhizobium (Ensifer) fredii and Bradyrhizobium elkanii in a Brazilian Savannah Soil. Applied and Environ. Microbiol. 73, 2635–2643. doi: 10.1128/aem.01823-06
Berlec, A.. (2012). Novel techniques and findings in the study of plant microbiota: search for plant probiotics. Plant Sci. 193–194, 96–102. doi: 10.1016/j.plantsci.2012.05.010
Bruijning, M., Henry, L. P., Forsberg, S. K. G., Metcalf, C. J. E., and Ayroles, J. F. (2020). When the microbiome defines the host phenotype: selection on vertical transmission in varying environments. bioRxiv 9, 280040. doi: 10.1101/2020.09.02.280040
Chen, K., Sui, L. N., Zhao, Z. J., Li, L., Hu, J. D., and Li, J. H. (2022). Co-cultured fermentation of two trichoderma strains to control cucumber Fusarium wilt. Chin. J. Biol. Control 38, 108–114. doi: 10.16409/j.cnki.2095-039x.2021.06.013
Chen, Q. L., Cai, L., Wang, H. C., Cai, L. T., Goodwin, P., Ma, J., et al. (2020). Fungal composition and diversity of the tobacco leaf phyllosphere during curing of leaves. Front. Microbiol. 11, 554051. doi: 10.3389/fmicb.2020.554051
Chen, S. Y., Li, J. J., Lin, J., Bao, K. X., Fan, J. Q., Zhang, R. Q., et al. (2018). High-throughput sequencing fungal community structures in aging tobacco strips from different growing areas and stalk positions. Tobacco Science Technology 51, 12–19.
Chen, X., Wicaksono, W. D., Berg, G., and Cernava, T. (2021). Bacterial communities in the plant phyllosphere harbour distinct responders to a broad-spectrum pesticide. Sci. Total Environ. 751, 141799. doi: 10.1016/j.scitotenv.2020.141799
Chen, X. J., Wang, H. C., Xiang, L. G., Cui, M. J., Zhou, H., Dai, Y. F., and Chen, X. (2019). Investigation and host identification of plant powdery mildew pathogen in Guizhou tobacco-growing field. Chin. Tob. Sci. 40, 55–65. doi: 10.13496/j.issn.1007-5119.2019.03.008
Classen, A. T., Boyle, S. I., Haskins, K. E., Overby, S. T., and Hart, S. C. (2003). Community level physiological profiles of bacteria and fungi: plate type and incubation temperature influences on contrasting soils. FEMS Microbiol. Ecol. 44, 319–328. doi: 10.1016/S0168-6496(03)00068-0
Copeland, J. K., Yuan, L., Layeghifard, M., Wang, P. W., and Guttman, D. S. (2015). Seasonal community succession of the phyllosphere microbiome. Mol. Plant Microbe Interact. 28, 274–285. doi: 10.1094/MPMI-10-14-0331-FI
Cruz, C. D., Esteve, P., and Tammela, P. (2021). Evaluation and validation of biolog OmniLog system for antibacterial activity assays. Lett. Appl. Microbiol. 17, 589–595. doi: 10.1111/lam.13450
Ding, T., and Melchner, U. (2016). Influences of plant species, season, and location on leaf endophytic bacterial communities of non-cultivated plants. PLoS ONE 11, e0150895. doi: 10.1371/journal.pone.0150895
Douglas, G. M., Bielawski, J. P., and Langille, M. G. I. (2020). Re-evaluating the relationship between missing heritability and the microbiome. Microbiome 8, 87. doi: 10.1186/s40168-020-00839-4
Edgar, R. C.. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh,340
Edgar, R. C.. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Fajola, A. O., and Alasoadura, S. O. (1973). Chemical control of the frog-eye disease (Cercospora nicotianae) of tobacco (Nicotiana tabacum) in Nigeria. Ann. Appl. Biol. 74, 219–224. doi: 10.1111/j.1744-7348.1973.tb07741.x
FAO (2019). Food Agriculture Data on Tobacco Crops. FAOSTAT. Retrieved from http://www.fao.org/faostat/en/#data/QC
Guo, Z. N., Xie, H. L., Wang, H. C., Yu, Z. H., and Yang, X. H. (2020). Leaf spot caused by Didymella segeticola on tobacco in China. Plant Dis. 104, 1559–1560. doi: 10.1094/PDIS-11-19-2398-PDN
He, K. J., Tang, G. Z., Huang, S. Y., Pi, P., and Tang, N. R. Y. (1997). Studies on the epidemics and control of tobacco brown spot (Alternaria alternata). J. Hunan Agric. Univ. 23, 445–449.
Henry, L. P., Bruijning, M., Forsberg, S. K. G., and Ayroles, J. F. (2021). The microbiome extends host evolutionary potential. Nat. Commun. 12, 5141. doi: 10.1038/s41467-021-25315-x
Hou, Y. J., Ma, X., Wan, W. T., Long, N., Zhang, J., Tan, Y. T., et al. (2016). Comparative genomics of pathogens causing brown spot disease of tobacco: Alternaria longipes and Alternaria alternata. PLoS ONE 11, e0155258. doi: 10.1371/journal.pone.0155258
Huang, Y., Wang, H. C., Cai, L. T., Li, W., Pan, D., Xiang, L., et al. (2021). Phyllospheric microbial composition and diversity of the tobacco leaves infected by Didymella segeticola. Front. Microbiol. 12, 699699. doi: 10.3389/fmicb.2021.699699
Huang, Y., Wang, H. C., Chen, Q. L., Xiang, L. G., Guo, Z. N., Li, Z., et al. (2020). Community structure and diversity of fungi in leaves of tobacco plant infected with powdery mildew. Guizhou Agric. Sci. 48, 54–58. Available online at: http://www.gznykx.org.cn/Jweb_gznykx/CN/abstract/abstract2973.shtml
Ishida, Y., and Kumashiro, T. (1988). Expression of tolerance to the host-specific toxin of Alternaria alternata (AT toxin) in cultured cells and isolated protoplasts of tobacco. Plant Dis. 72, 892–895. doi: 10.1094/PD-72-0892
Jackson, E. F., Echlin, H. L., and Jackson, C. R. (2006). Changes in the phyllosphere community of the resurrection fern, polypodium polypodioides, associated with rainfall and wetting. FEMS Microbiol. Ecol. 58, 236–246. doi: 10.1111/j.1574-6941.2006.00152.x
Jayawardena, R. S., Purahong, W., Zhang, W., Wubet, T., Li, X. H., Liu, M., et al. (2018). Biodiversity of fungi on Vitis vinifera L. revealed by traditional and high-resolution culture-independent approaches. Fungal Diver. 90, 1–84. doi: 10.1007/s13225-018-0398-4
Jeanette, B., Kristian, K. B., Waleed, A. A. S., Peter, H., Hansen, L. H., Soren, J. S., et al. (2012). Selection for Cu-tolerant bacterial communities with altered composition, but unaltered richness, via long-term Cu exposure. Appl. Environ. Microbiol. 78, 7438–7446. doi: 10.1128/AEM.01071-12
Jiang, Y., Sang, W. J., Zeng, E. L., Wang, Y., Wang, D. F., Qin, K., et al. (2018). Identification of pathogenic fungus causing tobacco black spot stalk in Guizhou Province. Jiangsu Agric. Sci. 46, 92–95. doi: 10.15889/j.issn.1002-1302.2018.10.024
Karlsson, I., Friberg, H., Kolseth, A. K., Steinberg, C., and Persson, P. (2017). Organic farming increases richness of fungal taxa in the wheat phyllosphere. Mol. Ecol. 26, 3424–3436. doi: 10.1111/mec.14132
Kashiwa, T., and Suzuki, T. (2021). High-quality genome assembly of the soybean fungal pathogen Cercospora kikuchii. Genes Genomes Genet. 11, jkab277. doi: 10.1093/g3journal/jkab277
Knief, C., Ramette, A., Frances, L., Alonso-Blanco, C., and Vorholt, J. A. (2010). Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J. 4, 719–728. doi: 10.1038/ismej.2010.9
Kodama, M., Suzuki, T., Otani, H., Kohmoto, K., and Nishimura, S. (1990). Purification and bioassay of host-selective AT-toxin from alternaria alternata causing brown spot of tobacco. Jpn. J. Phytopathol. 56, 628–636. doi: 10.3186/jjphytopath.56.628
Laforest-Lapointe, I., Messier, C., and Kembel, S. W. (2016). Host species identity, site and time drive temperate tree phyllosphere bacterial community structure. Microbiome 4, 27. doi: 10.1186/s40168-016-0174-1
LaMondia, J. A.. (2001). Outbreak of brown spot of tobacco caused by Alternaria alternata in Connecticut and Massachusetts. Plant Dis. 85, 230. doi: 10.1094/PDIS.2001.85.2.230B
Langenheder, S., Bulling, M. T., Solan, M., and Prosser, J. I. (2010). Bacterial biodiversity-ecosystem functioning relations are modified by environmental complexity. PLoS ONE 5, e10834. doi: 10.1371/journal.pone.0010834
Langille, M. G., Zaneveld, J., Caporaso, J. G., Mcdonald, D., Knights, D., Reyes, J., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, L., Gao, L., Liu, Y., Fang, B., Huang, Y., Mohamad, O. A. A., et al. (2021). Diversity of cultivable endophytic bacteria associated with halophytes in Xinjiang of China and their plant beneficial traits. J. Arid Land 13, 790–800. doi: 10.1007/s40333-021-0016-2
Li, W. H., Jin, D. C., and Shi, C. H. (2018). Midgut bacteria in deltamethrinresistant, deltamethrin-susceptible, and field-caught populations of Plutella xylostella, and phenomics of the predominant midgut bacterium Enterococcus mundtii. Sci. Rep. 8, 4991. doi: 10.1038/s41598-018-23329-y
Liu, C., Wang, H. C., Xie, H. L., Chen, Q. L., Yu, Z. H., Sun, G. J., et al. (2019). Fungal community structure analysis of tobacco leaf infected with brown spot disease. Guizhou Agric. Sci. 47, 54–59. Available online at: http://www.gznykx.org.cn/Jweb_gznykx/CN/article/searchArticle.do
Liu, C., Wang, H. C., Xie, H. L., Xiang, L. G., Huang, Y., Chen, Q. L., et al. (2020). Biodiversity analysis of phyllosphere bacterial genus from tobacco leaves infected by brown spot disease. Tobacco Sci. Technol. 53, 8–14. doi: 10.16135/j.issn1002-0861.2019.0114
Liu, T. B., Gu, Y. B., Zhou, Z. C., Liu, Z. H., Yin, H. Q., Qin, C., et al. (2021). Ecological strategies of biological and chemical control agents on wildfire disease of tobacco (Nicotiana tabacum L.). BMC Microbiol. 21, 184–194. doi: 10.1186/s12866-021-02237-8
Luo, L. Y., Zhang, Z., Jin, D. C., Tan, X. Q., Zhang, D. Y., Tian, Y., Lu, X. Y., and Liu, Y. (2017). Phyllosphere bacterial diversities and community structures on pumpkin with different severities of powdery mildew. Acta Phytopathol. Sin. 47, 688–695. doi: 10.13926/j.cnki.apps.000066
Luo, L. Y., Zhang, Z., Wang, P., Han, Y. Q., Jin, D. C., Su, P., et al. (2019). Variations in phyllosphere microbial community along with the development of angular leaf-spot of cucumber. AMB Express 9, 1–13. doi: 10.1186/s13568-019-0800-y
Lv, D., Ma, A. Z., Tang, X. M., Bai, Z. H., Qi, H. Y., Zhuang, G. Q., et al. (2013). Profile of the culturable microbiome capable of producing acyl-homoserine lactone in the tobacco phyllosphere. J. Environ. Sci. 25, 357–366. doi: 10.1016/S1001-0742(12)60027-8
Ma, X., Jiang, M., and Liu, J. (2017). Preliminary analysis of amplicon high throughput sequencing as a method for the assessment of fungal diversity in discolored wood. Holzforschung 71, 793–800. doi: 10.1515/hf-2017-0015
Madhaiyan, M., and Poonguzhali, S. (2014). Methylobacterium pseudosasicola sp. nov. and Methylobacterium phyllostachyos sp. nov., isolated from bamboo leaf surfaces. Int. J. Syst. Evolut. Microbiol. 64, 2376–2384. doi: 10.1099/ijs.0.057232-0
Madhaiyan, M., Poonguzhali, S., and Sa, T. M. (2007). Metal tolerating methylotrophic bacteria reduces nickel and cadmium toxicity and promotes plant growth of tomato (Lycopersicon esculentum L.). Chemosphere 69, 220–228. doi: 10.1016/j.chemosphere.2007.04.017
Madhaiyan, M., Poonguzhali, S., Senthilkumar, M., Lee, J. S., and Lee, K. C. (2012). Methylobacterium gossipiicola sp. nov., a pink-pigmented, facultatively methylotrophic bacterium isolated from the cotton phyllosphere. Int. J. Syst. Evolut. Microbiol. 62, 162–167. doi: 10.1099/ijs.0.030148-0
Mao, H. L., Wang, K., Wang, Z., Peng, J., and Ren, N. Q. (2020). Metabolic function, trophic mode, organics degradation ability and influence factor of bacterial and fungal communities in chicken manure composting. Bioresour.Technol. 302, 122883. doi: 10.1016/j.biortech.2020.122883
Mboowa, G., Sserwadda, I., Amujal, M., and Namatovu, N. (2018). Human genomic loci important in common infectious diseases: role of high-throughput sequencing and genome-wide association studies. Can. J. Infect. Dis. Med. Microbiol. 60, 1875217. doi: 10.1155/2018/1875217
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). Funguild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Pei, D. L., Zhu, X. Q., Ye, F., Xu, Y. Y., Song, Y., and Li, C. W. (2019). Identification of the pathogen causing the blue mold disease of fragaria × ananassa in Shangqiu, Henan Province. J. Plant Protect. 46, 1375–1376. doi: 10.13802/j.cnki.zwbhxb.2019.2018192
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. doi: 10.1371/journal.pone.0009490
Qin, C., Tao, J., Liu, T., Liu, Y., and Meng, D. (2019). Responses of phyllosphere microbiota and plant health to application of two different biocontrol agents. AMB Express 9, 42–54. doi: 10.1186/s13568-019-0765-x
Rastogi, G., Sbodio, A., Tech, J. J., Suslow, T. V., Coaker, G. L., and Leveau, J. H. J. (2012). Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. ISME J. 6, 1812–1822. doi: 10.1038/ismej.2012.32
Rogel, M. A., Ormeño-Orrillo, E., and Martinez Romero, E. (2011). Symbiovars in rhizobia reflect bacterial adaptation to legumes. Syst. Appl. Microbiol. 34, 96–104. doi: 10.1016/j.syapm.2010.11.015
Rotem, J.. (1994). The Genus Alternaria: Biology, Epidemiology, and Pathogenicity. St. Paul, MN: APS press.
Rumakanta, S., Jørgensen, L. N., and Mogens, N. (2017). Spatiotemporal variation and networks in the mycobiome of the wheat canopy. Front. Plant Sci. 8, 1357. doi: 10.3389/fpls.2017.01357
Shew, H. D., and Lucas, G. B. (1991). Compendium of Tobacco Diseases. St. Paul, MN: American Phytopathological Society.
Slavov, S., Mayama, S., and Atanassov, A. (2004). Some aspects of epidemiology of Alternaria alternata tobacco pathotype. Biotechnol. Biotechnol. Equip. 18, 85–89. doi: 10.1080/13102818.2004.10817125
Vorholt, J. A.. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
Wagner, M., Amann, R., Lemmer, H., and Schleifer, K. H. (1993). Probing activated sludge with oligonucleotides specific for proteobacteria: inadequacy of culture-dependent methods for describing microbial community structure. Appl. Environ. Microbiol. 59, 1520. doi: 10.1128/aem.59.5.1520-1525.1993
Wang, H., Zhou, H., Yu, Z. H., Zhang, Z. F., Chen, X., and Cai, L. T. (2018). Spot blight on tobacco (Nicotiana tabacum) Caused by Stagonosporopiss cucurbitacearum in Guizhou Province of China. Plant Dis. 102, 2030. doi: 10.1094/PDIS-11-17-1793-PDN
Wang, H. C., Huang, Y. F., Wang, J., Chen, X. J., Wei, K. S., Wang, M. S., et al. (2016). Activities of azoxystrobin and difenoconazole against Alternaria alternata and their control efficacy. Crop Prot. 90, 54–58. doi: 10.1016/j.cropro.2016.08.022
Wang, H. C., Wang, J., Li, W. H., Huang, Y. F., Xia, H. Q., Wang, M. S., et al. (2014). Cladosporium cladosporioides identified in China on tobacco seeds. Plant Dis. 98, 1002. doi: 10.1094/PDIS-12-13-1203-PDN
Wang, H. C., Wang, M. S., Xia, H. Q., Yang, S. J., Guo, Y. S., Xu, D. Q., et al. (2013). First report of Fusarium wilt of tobacco caused by Fusarium kyushuense in China. Plant Dis. 97, 424. doi: 10.1094/PDIS-09-12-0835-PDN
Weber, R. W. S., Madhour, A., Anke, H., Mucci, A., and Davoli, P. (2005). 2-Hydroxytorularhodin, a new xanthophyll from the red yeast Sporobolomyces coprosmae. Helv. Chim. Acta 88:2960–2966. doi: 10.1002/hlca.200590239
Wellner, S., Lodders, N., and Kämpfer, P. (2011). Diversity and biogeography of selected phyllosphere bacteria with special emphasis on Methylobacterium spp. Syst. Appl. Microbiol. 34, 621–630. doi: 10.1016/j.syapm.2011.08.005
Welty, R. E., and Nelson, L. A. (1971). Growth of Aspergillus repens in flue-cured tobacco. Appl. Microbiol. 21, 854–859. doi: 10.1128/am.21.5.854-859.1971
Whipps, J. M., Hand, P., Pink, D., and Bending, G. D. (2008). Phyllosphere microbiology with special reference to diversity and plant genotype. J. Appl. Microbiol. 105, 1744–1755. doi: 10.1111/j.1365-2672.2008.03906.x
Woo, H. R., Kim, H. J., Nam, H. G., and Lim, P. O. (2013). Plant leaf senescence and death-regulation by multiple layers of control and implications for aging in general. J. Cell Sci. 126, 4823–4833. doi: 10.1242/jcs.109116
Xiang, L. G., Wang, H. C., Zheng, P., Cai, L. T., and Yu, Z. H. (2020). Analysis of endophytic fungi and phyllosphere fungi of flue cured tobacco leaves with brown spot disease. Acta Tabacaria Sin. 26, 93–100. doi: 10.16472/j.chinatobacco.2020.104
Xu, H., Yang, G. H., Zhang, M., Dang, L. Z., Wang, W. Y., Wang, M. F., et al. (2014). Analysis of diversity of endophytes and phyllospheric microorganism from Yunnan tobacco leaves. J. Yunnan Agric. Univer. 29, 149–154. doi: 10.3969/j.issn.1004-390X(n).2014.02.001
Xu, Y. Z., Xie, D. P., Li, H. T., and Wang, T. X. (1993). Study on the occurrence and epidemic law of tobacco brow spot and chemical control. China Tobacco1, 18–21.
Zhang, H. H., Tang, M., Chen, H., and Zheng, C. L. (2010). Inoculation with ectomycorrhizal fungi affects microbial biomass and bacterial functional diversity in the rhizosphere of Pinus tabulaeformis seedlings. Eur. J. Soil Biol. 46, 55–61. doi: 10.1016/j.ejsobi.2009.10.005
Zhang, Q., Geng, Z., Li, D., and Ding, Z. (2020). Characterization and discrimination of microbial community and co-occurrence patterns in fresh and strong flavor style flue-cured tobacco leaves. Microbiol. Open 9, e965. doi: 10.1002/mbo3.965
Zhao, Q., Chen, X., Liu, D. Y., Xia, C. J., Yang, J. G., Lv, H. K., et al. (2020). First report of Cercospora nicotianae causing frog eye spot in cigar tobacco in Hainan, China. Plant Dis. 104, 3257. doi: 10.1094/PDIS-03-20-0540-PDN
Zhao, S., Liu, D. Y., Ling, N., Chen, F. D., Fang, W. M., Shen, Q. R., et al. (2014). Bio-organic fertilizer application significantly reduces the Fusarium oxysporum population and alters the composition of fungi communities of watermelon fusarium wilt rhizosphere soil. Biol. Fertil. Soils 50, 765–774. doi: 10.1007/s00374-014-0898-7
Keywords: microbial community, Alternaria alternata, tobacco brown spot, high-throughput sequencing, microbial functional diversity, biolog-eco
Citation: Dai Y-f, Wu X-m, Wang H-c, Li W-h, Cai L-t, Li J-x, Wang F, Sehar S and Shamsi IH (2022) Spatio-Temporal Variation in the Phyllospheric Microbial Biodiversity of Alternaria Alternata-Infected Tobacco Foliage. Front. Microbiol. 13:920109. doi: 10.3389/fmicb.2022.920109
Received: 14 April 2022; Accepted: 22 June 2022;
Published: 28 July 2022.
Edited by:
Bernardo González, Adolfo Ibáñez University, ChileReviewed by:
Anandham Rangasamy, Tamil Nadu Agricultural University, IndiaShengming Liu, Henan University of Science and Technology, China
Chuanqing Zhang, Zhejiang Agriculture and Forestry University, China
Nikolaos Remmas, Democritus University of Thrace, Greece
Copyright © 2022 Dai, Wu, Wang, Li, Cai, Li, Wang, Sehar and Shamsi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-mao Wu, d3V4bTgyN0AxMjYuY29t; Han-cheng Wang, eGlhb2JhaXlhbmcxMjZAaG90bWFpbC5jb20=; Feng Wang, eWFuY2Fvd2FuZ2ZlbmdAMTYzLmNvbQ==; Imran Haider Shamsi, ZHJpbXJhbkB6anUuZWR1LmNu