 Jia Yang
Jia Yang Abolfazl Masoudi
Abolfazl Masoudi Hao Li1
Hao Li1 Yifan Gu
Yifan Gu Can Wang
Can Wang Zhijun Yu
Zhijun Yu Jingze Liu
Jingze Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 02 September 2022
Sec. Terrestrial Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.913349
Pinus bungeana is a native but endangered plant species in China, with high ornamental value and adaptability to drought and cold. The relationship between the soil community structure and endophytic microbes in the tissues of P. bungeana under different health statuses is poorly understood. In this study, the endophytic bacterial and fungal communities of P. bungeana under different health statuses were compared and analyzed in the Xiong'an New Area. Using high-throughput deep sequencing [16S and internal transcribed spacer (ITS) rRNA] techniques, the effect of the health status of P. bungeana on the microbial communities in bulk soil, rhizospheric soil, roots, stems, and leaves was determined in this study. We observed that the diversity of the bacterial and fungal communities of the aboveground parts (stems and leaves) of healthy P. bungeana plants was much higher than that of the unhealthy plants. However, the diversity of bacterial and fungal communities in the belowground parts (bulk soil, rhizospheric soil, and roots) showed almost no difference in microbial community richness, indicating that the possible cause of illness was transmitted in a “top-down” manner. Furthermore, there were significant differences in the microbial diversity and community structure in different ecological niches of P. bungeana (P < 0.01). Proteobacteria and Actinobacteria were the dominant bacterial phyla, while Ascomycota, Basidiomycota, and Mortierellomycota were the predominant fungal phyla. Redundancy analysis (RDA) revealed that soil organic matter (SOM), total phosphorous (TP), total potassium (TK), total nitrogen (TN), water content (WC), power of hydrogen (pH), total carbon (TC), and the ratio of carbon to nitrogen (C/N) were significantly correlated with the composition of the microbial communities. Altogether, these results provide a scientific basis for further studies on the mechanism underlying the “aboveground–underground” microbial interactions in plantation forests, which can aid in promoting the healthy and sustainable development of the Millennium Xiulin forest in the Xiong'an New Area.
Plants and their endophytic microflora usually form a “holobiont” with complicated interactions and evolution (Vandenkoornhuyse et al., 2015; Hassani et al., 2018). Interactions between host plants and microbiota can be divided into three categories, namely, negative (pathogenic), positive, and neutral interactions (Zheng and Gong, 2019). Of these interactions, symbiosis is considered synergistic and aids both interacting partners (Lopez et al., 2012). For instance, it has been reported that microbial communities play a crucial role in promoting the growth and development of Arabidopsis thaliana (Durán et al., 2018), grapevine (Rolli et al., 2015), and citrus root (Zhang et al., 2017); improving plant resistance; and promoting sustainable and healthy production (Weyens et al., 2009; Brader et al., 2014; Naveed et al., 2014). Furthermore, microbial populations can improve nutrient transport and utilization by host plants and promote host resistance against biotic and abiotic stresses. The host plants, in turn, provide a stable niche habitat, energy, and carbon source for the microbial population (Raaijmakers et al., 2009; Peiffer et al., 2013; Philippot et al., 2013).
Each plant compartment, including roots, stems, leaves, flowers, and seeds, represents a unique niche of microbial entities and possesses unique microbial communities (Lindow and Brandl, 2003). Moreover, the endophytic microbiota shows significant variations in different tissues and organs (Ottesen et al., 2013). Soil is rich in microflora (Zarraonaindia et al., 2015; Mangeot-Peter et al., 2020); for the successful colonization of plant tissues, soil microbiota needs to overcome the immune system of the host plant and abiotic stresses, following which some populations are eliminated from the internal microenvironment of the plant due to the lack of adaptation (Bulgarelli et al., 2013). In addition, the plant microbial community is required to adapt to the varying climatic and soil conditions. The changes in the soil microenvironment depend on specific microbiota, soil properties (pH and nutrients), soil type, light, temperature, humidity, and other environmental factors. This alteration can significantly affect plant roots and endophytic flora (Delgado-Baquerizo et al., 2018; Semenov et al., 2018; O'brien et al., 2019; Wang et al., 2021). In most cases, the microorganisms in underground tissues differ from those in the aboveground parts of plants. It has been reported that the microbiota in the aboveground parts of mature poplar is markedly different from that in the rhizospheric soil (Gottel et al., 2011). Another study demonstrated that the microbial species composition and diversity of the underground parts of Cannabis sativa are significantly different from those in the aboveground tissues (Wei et al., 2021).
In plant–soil–microorganism systems, the interaction between microbiota can affect or directly change the health status of host plants. For instance, the resistance of A. thaliana and apple fruits against Botrytis cinerea was found to be highly associated with the plant symbiont, Pseudomonas sp. (Unyarat et al., 2016). Another study reported that the application of Actinomyces sp. to nursery plants effectively reduces infection with pathogenic bacteria in the roots (Álvarez-Pérez et al., 2017). It has been reported that soil biota might protect against infections with fungal pathogens inoculated in plant roots (Rodrigo et al., 2011).
Pinus bungeana Zucc. Ex Endl. (P. bungeana) is an endangered tree species and the only three-leaf pine in East Asia. With its beautiful posture and unique bark, the timber of P. bungeana can be used to construct houses, furniture, stationery, and other materials, which have great ornamental and economic value. P. bungeana is an important species for ecological construction, with robust adaptability to dry, cold, calcareous, and mild saline–alkali soils. The plant has exceptional resistance to sulfur dioxide, hydrogen fluoride, and smoke dust pollution and plays a crucial role in forest regeneration, thereby serving as a unique tree species for the creation of environmental protection forests in industrial and mining areas (Syring et al., 2007; Yang et al., 2015; Guo et al., 2020). In a previous study, we observed highly diversified microflora in the roots of P. tabuliformis (Wang et al., 2020). However, the microbial community structure and niche differentiation in different tissues of P. bungeana in the man-made Xiong'an New Area forest remain to be elucidated.
Accumulating studies have demonstrated that plants can significantly influence the structure of the plant microbiome (Esmaeili et al., 2017; Yu and Hochholdinger, 2018) by producing different types of root exudates (Jacoby et al., 2017). However, there is a paucity of studies on the impact of the varying plant health status on the microbial communities in plantation forests. We, therefore, hypothesized that endophytic microbial communities are unique in different compartments of P. bungeana, and the health status of the plants shapes the microbial community structure and niche differentiation in different tissues. Therefore, in this study, the bacterial and fungal communities in the rhizospheric soil, bulk soil, and the inner layers of roots, stems, and leaves of P. bungeana plants of different health statuses were explored using 16S rRNA and internal transcribed spacer (ITS) gene sequencing techniques, for elucidating the complex interactions between microorganisms and the host plant. These findings will provide a scientific basis for further studies on the mechanism of “aboveground–underground” microbial interactions in plantation forests, which can aid in promoting the healthy and sustainable development of the Millennium Xiulin forest in the Xiong'an New Area.
The sampling sites were located in the Xiong'an New Area (38° 43′ to 39° 10′ N and 115° 38′ to 116° 20′ E) announced by the government of the People's Republic of China on April 1, 2017. As part of the development of the new national region, the Xiong'an New Area is another key area after the Shenzhen Special Economic Zone and the Shanghai Pudong New Area (Li et al., 2020). The Xiong'an New Area has a warm temperate continental monsoon-type climate with four seasons and significant differences between cold, warm, and humid periods and has an annual average temperature of 12.1°C, frost-free period of 173 days, and an average yearly precipitation of 560 mm (Xu et al., 2018; Xie et al., 2019). The Millennium Xiulin area was launched in November 2017 with the aim of creating an appealing ecological environment and constructing a new and impressive ecological city. P. bungeana is one of the main afforestation tree species in the Millennium Xiulin area.
In total, three blocks were selected in the Millennium Xiulin area, and healthy P. bungeana plants (H, with dark green leaves and no sign of pests or diseases) and unhealthy P. bungeana plants (U, with withered needles and yellow caducous, leaves with tan spots) were randomly selected from each block. In addition, the unhealthy plants in the study area were characterized by the mixed occurrence of brown spots, needle cast, and needle blight, which could have been caused by pathogens, a mild climate, high humidity, or poor management of the barren soil. A total of 30 samples, including healthy bulk soil (HFRS), healthy rhizospheric soil (HRS), healthy roots (HR), healthy stems (HS), healthy leaves (HL), unhealthy bulk soil (UFRS), unhealthy rhizospheric soil (URS), unhealthy roots (UR), unhealthy stems (US), and unhealthy leaves (UL), were collected from the experimental area in the September of 2021, and each sample was collected in triplicate. After removing the surface impurity, the bulk and rhizospheric parts of coniferous P. bungeana plants were collected. Underground root samples with a diameter of ~0.3 cm were extracted from depths of 0–30 cm, and stems with diameters of ~0.5 cm were selected as stem samples. The rhizospheric soil was strictly defined as the soil particles that adhered to the roots. The bulk soil was collected from the topsoil region (0–15 cm) and from regions more than 20 cm away from the roots. The leaf chamber represented all the leaves at the end and sub-end of the stem branch, which were collected using sterile scissors. Disposable gloves were worn and replaced every time different tissues were sampled. The samples were placed in a portable refrigerator, transported to the laboratory at low temperature (~−20°C), and stored at −80°C for further analyses.
The samples were processed in our laboratory according to the method described by Beckers et al. (2016). In brief, the bulk soil samples were processed by removing the plant debris and stones by sieving (<2 mm pore size) and divided into two groups: one group was air-dried and used for determining the physicochemical properties, while the other group was placed in a sterile bag and stored at −80°C. The whole root system was depleted of soil particles by shaking on a platform for 20 min at 120 rpm, and the soil particles that were directly removed from the roots represented the rhizospheric samples. The root, stem, and leaf compartments were first rinsed with sterile ddH2O for 30 s and subsequently rinsed with 70% (v/v) ethanol and sodium hypochlorite (2.5% sodium hypochlorite with 0.1% Tween 80) for 2 and 5 min, respectively. The tissues were then washed with 70% (v/v) ethanol for an additional 30 s to remove the epiphytic bacteria and fungi from the surface, following which the samples were rinsed five times with sterile ddH2O (Beckers et al., 2017). The plant samples were cut into small pieces with a sterile scalpel and then sterilized with sterile PBS (130 mM NaCl, 7 mM Na2HPO4, 3 mM NaH2PO4, at pH 7.4). After sterilization, the plant samples were homogenized and subjected to laminar airflow under sterile conditions. Finally, quadruple aliquots (1.5 mL) of homogeneous plant samples (roots, stems, and leaves) from each sample were stored at −80°C until DNA extraction (Wagner et al., 2016).
The total DNA was extracted from the bulk soil (0.4 g) and rhizospheric soil (0.4 g) of P. bungeana using a FastDNAⓇ Spin Kit (MP Biomedicals, USA), according to the standard operating instructions. All the plant tissues, including roots, stems, and leaves, were cut into fine pieces (<5 mm), and 50 mg of the tissues were ground for 3 min in liquid nitrogen using 5-mm beads. The DNA was extracted from the pulverized tissue using the same kit as previously described. The extraction was repeated two times to obtain sufficient DNA yield. The concentration and purity of the DNA were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA), and the integrity of the DNA was subsequently examined by agarose gel electrophoresis with 1% agarose gels (Cregger et al., 2018; Wang et al., 2019).
PCR amplification was performed in our laboratory, and Illumina MiSeq sequencing was performed by Majorbio company (Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China). The bacterial 16S rRNA amplicon library was generated by two-step PCR, and the first round of PCR amplification was performed using 799F (5'-AACMGGATTAGATACCCKG-3') and 1392R (5'-ACGGGCGGTGTGTRC-3') primers (Bodenhausen et al., 2013; Hartman et al., 2018). PCR amplification of the 16S rRNA gene was performed under the following conditions: initial denaturation at 95°C for 3 min, followed by 27 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 45 s, and final extension at 72°C for 10 min with a hold step at 4°C. The PCR mixtures contained 5 × 4 μL of TransStart FastPfu buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL forward primer (5 μM), 0.8 μL reverse primer (5 μM), 0.4 μL TransStart FastPfu DNA polymerase, 10 ng template DNA, and up to 20 μL ddH2O. The PCR was performed in triplicate. The PCR product was separated on 2% agarose gels and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA), according to the manufacturer's instructions. The purified amplicons were next subjected to the second round of PCR amplification using 799F (5'-AACMGGATTAGATACCCKG-3') and 1193R (5'-ACGTCATCCCCACCTTCC-3') primers to reduce the length of the amplicon (394 bp) for sequencing. The PCR conditions were the same as those used in the first round, with the exception that the number of cycles was reduced to 13. The products were purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using a Quantus™ Fluorometer (Promega, Madison, WI, USA).
The universal primers, ITS1F (5'-CTTGGTCATTTAGAGGAAGTAA-3') and ITS2R (5'-GCTGCGTTCTTCATCGATGC-3'), were selected for generating the fungal ITS amplicon library. The reaction system, purification, and quantification procedures were the same as those used for bacterial PCR. Purified amplicons were pooled in equimolar concentrations and subjected to paired-end sequencing using the Illumina MiSeq PE300 platform (Illumina, San Diego, CA, USA) by Majorbio company (Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China). Bacterial and fungal raw data were deposited at the National Center for Biotechnology Information Sequence Read Archive database under the BioProject numbers PRJNA821431 (accession number: SUB11251518) and PRJNA821418 (accession numbers: SUB11250928), respectively.
Bioinformatics analyses were performed by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The raw reads were demultiplexed, quality-filtered, and controlled using fastp version 0.20.0 (Chen et al., 2018) and merged using FLASH software, version 1.2.7 (Magoč and Salzberg, 2011), with the following procedure: (i) 300-bp reads were truncated at any site with an average quality score of <20 over a 50-bp sliding window. Truncated reads shorter than 50 bp and reads containing ambiguous characters were discarded. (ii) Overlapping sequences longer than 10 bp were assembled according to their overlapped sequence. The maximum mismatch ratio of the overlapping region was set to 0.2. The reads that could not be assembled were discarded. (iii) The samples were distinguished according to the barcode and primer sequences, and the sequence direction was adjusted for exact barcode matching and two nucleotide mismatches in primer matching.
The operational taxonomic units (OTUs) were clustered using UPARSE software, version 7.1. In order to eliminate the redundant computational steps in the intermediate processes of analysis, the non-repetitive sequences were extracted from the optimized sequences, and single non-repetitive sequences were removed. The non-repetitive sequences (excluding single sequences) were clustered based on the OTUs using a similarity threshold of 97%, and chimeras were removed during clustering to obtain the representative OTU sequences (Dickie, 2010). Chimeric sequences were identified and removed using UCHIME software, version 3 (Edgar et al., 2011), against the SILVA database (version 138), the 16S rRNA database for bacteria, and the UNITE ITS database (version 7.0) for fungi, using a confidence threshold of 70% (Wang et al., 2019). Non-bacterial and non-fungal sequences were removed from the data set (Wang et al., 2022).
All the statistical analyses were performed using a cloud platform (Majorbio Bio-Pharm Technology Co., Ltd.). Analysis of variance (ANOVA), Student's t-test, and post-hoc analyses were conducted using R version 3.5.1 (R Core Team, 2018). Statistical analyses of the rhizospheric and bulk soil data of healthy and unhealthy P. bungeana plants, and correlation analysis of soil and microbial communities were performed using SPSS version 22.0 for Windows (IBM Corp, NY, Armonk, USA). The post-hoc comparison was performed to measure significant differences in microbial richness and composition among different compartments in healthy and unhealthy plants by Tukey's honestly significant difference (HSD) test. QIIME version 1.9.1 was used to calculate the beta diversity distance, and R version 3.5.1 was used for Bray–Curtis hierarchical clustering and PCoA statistical analysis to identify the similarity or difference of the community structure among different samples. The similarities and differences in the relationships of microbial community structures in different compartments of P. bungeana were investigated with heatmaps generated using the VEGAN package. Linear discriminant analysis (LDA) coupled with effect size measurements (LEfSe) analysis was conducted to identify the statistically different biomarkers between groups (Wei et al., 2021). The alpha diversity indices (Chao, Shannon, Simpsoneven) were calculated using mothur version 1.30.2 (Schloss et al., 2011). Calculation of beta diversity and generation of Venn diagrams were performed using R version 3.5.1. The Circos table viewer was used for visualizing the Circos diagram, which depicted the distribution proportion of core microbiota in each of the plant compartments and provided a measure of the distribution proportion of each dominant species in the different samples (Krzywinski et al., 2009). Network visualization was performed to obtain information regarding the species abundance in the different compartments using Cytoscape version 3.4.0 (Shannon et al., 2003). The co-existing relationships among the species in the different niches were simultaneously determined for elucidating the similarities and differences between samples.
The pH of the soil samples was measured from a soil–water suspension (1:5 m/v) using a Benchtop FiveEasy Plus™ pH Meter (Mettler Toledo, Shanghai, China). The total carbon (TC), total nitrogen (TN), and the ratio of carbon to nitrogen (C/N) were determined using an elemental analyzer (Vario EL-3, Elementar, Germany). The soil organic matter (SOM) was measured using the dichromate oxidation method (Certini et al., 2007). The water content (WC) was determined using the drying method, while total soil phosphorus (TP) and total potassium (TK) were measured using the acid solution molybdenum-antimony colorimetric method and a flame photometer, respectively, according to China's Soil Standard protocols for forest regions (LY/T 1232–2015, LY/T 1234–2015, and LY/T 1228–2015, in Chinese).
A total of 593,512 high-quality reads were obtained for the bacterial species, with an average length of 377 bp, while 2,247,150 reads were obtained for the fungal species, with an average length of 236 bp. A total of 1,777 bacterial and 1,978 fungal OTUs were identified across all the samples. The rarefaction curves for evaluating the richness of the bacterial OTUs were generally close to saturation, indicating that the volume of the sequenced reads was reasonable (Supplementary Figure S1). The rarefaction curves showed that the alpha diversity of the fungal lineages for the HL and HS samples was higher than that of the other eight samples.
There was no noticeable difference in the bacterial richness among the HFRS, UFRS, HRS, URS, HS, and HL samples, and the bacterial richness of these samples was significantly higher than that of the other samples (P < 0.01). There was no significant difference in bacterial richness between the HR and UR samples and between the US and UL samples, which showed the lowest bacterial richness (Figure 1A). There was no significant difference in the bacterial evenness among the samples in the same niche, with the exception of the UL sample, in which the bacterial evenness was significantly lower than that of the HL samples. This indicated that bacterial evenness was affected by differences in health conditions (P < 0.01). Bacterial evenness was lowest in the root and UL samples (Figure 1B). The bacterial diversity in the rhizospheric soil, bulk soil, and stems and leaves of healthy P. bungeana plants was significantly higher than that of the other samples (P < 0.01). The bacterial diversity was lowest in the US, UL, and all root samples (Figure 1C).
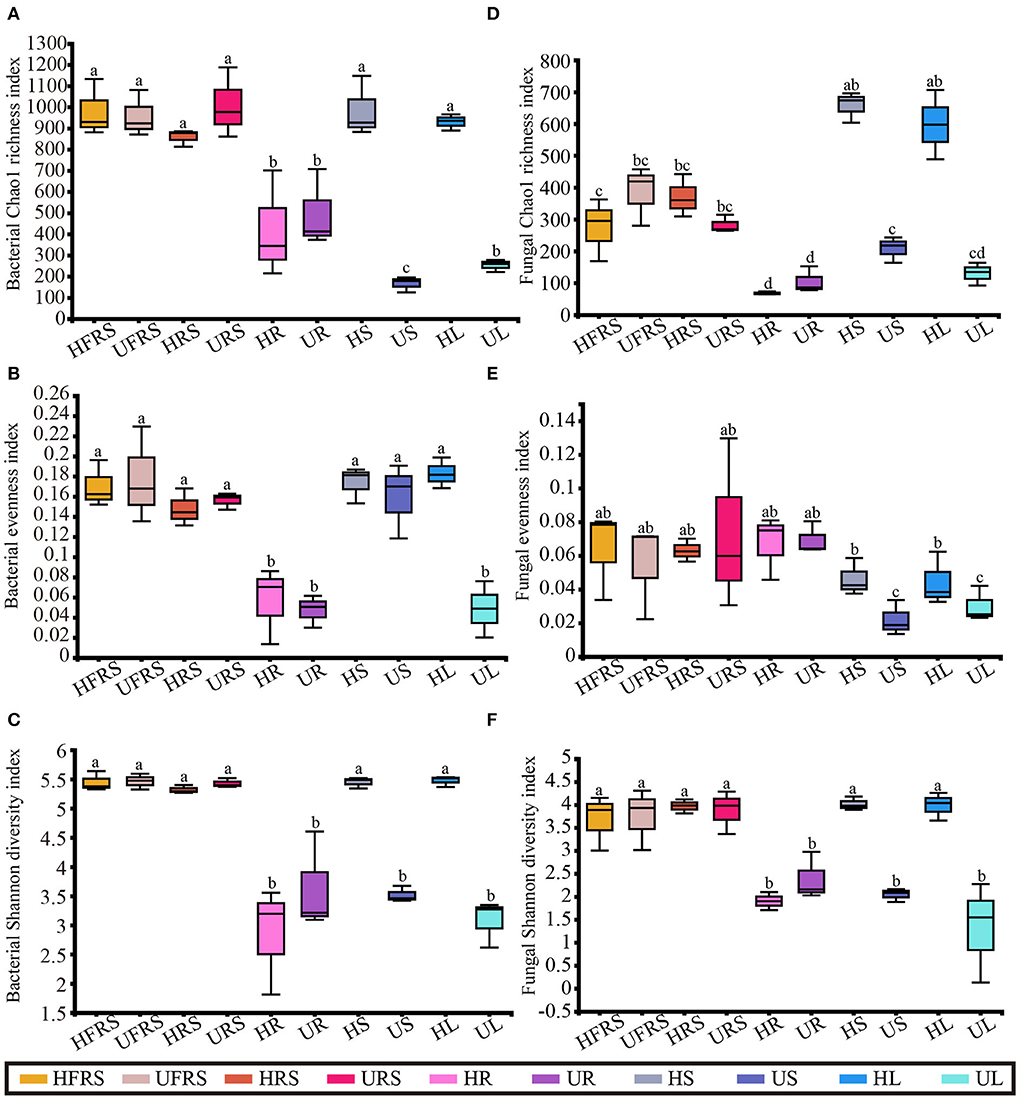
Figure 1. Microbial alpha diversity in healthy and unhealthy P. bungeana plants. (A) Bacterial Chao1 richness index. (B) Bacterial evenness index. (C) Bacterial Shannon diversity index. (D) Fungal Chao1 richness index. (E) Fungal evenness index. (F) Fungal Shannon diversity index. All the diversity indices were calculated at the OTU level. The box plots depict the first (25%) and third (75%) quartiles, the median, mean, and the maximum and minimum observed values within each data set. The alpha diversity estimates represent the results of three biological replicates for each group. The data were analyzed by one-way ANOVA and Tukey–Kramer post-hoc comparisons. Significant differences (P < 0.05) across plant compartments are indicated with lowercase letters. HFRS, healthy bulk soil; HRS, healthy rhizosphere soil; HR, healthy roots; HS, healthy stems; HL, healthy leaves; UFRS, unhealthy bulk soil; URS, unhealthy rhizosphere soil; UR, unhealthy roots; US, unhealthy stems; UL, unhealthy leaves.
There were no significant differences in fungal richness between the bulk and rhizospheric soil samples of P. bungeana at different health conditions (P > 0.05). In addition, there were no significant differences in the endophytic fungal communities in the roots of P. bungeana at two different health conditions, in different tissue niches (P > 0.05). The richness of the OTUs of the HS and HL samples was significantly higher than that of other samples (P < 0.01), whereas that of the HFRS, UFRS, HRS, and URS samples was substantially higher than that of the root samples (P < 0.01). There was no noticeable difference in the richness of the OTUs among all the root samples and among the UL, US, and UL samples (Figure 1D). There was no apparent difference in the fungal evenness in the same niches between the healthy and unhealthy plants, while the evenness in the US and UL samples was significantly lower than that of the HFRS, UFRS, HRS, URS, HR, and UR samples (P < 0.01) (Figure 1E). The fungal diversity of the HFRS, UFRS, HRS, URS, HS, and HL samples was significantly higher than that of the other samples (P < 0.01), and there were no significant differences in the fungal diversity among all the root samples and the US and UL samples (Figure 1F).
The bacterial beta diversity was evaluated at the phylum and OTU levels, while fungal beta diversity was evaluated at the class and OTU levels. Principal coordinate analysis (PCoA) revealed general similarities in the structures of bacterial and fungal communities among the samples (Figure 2). The microbial communities in the roots of P. bungeana were distinguishable, regardless of the health status of the plants. In addition, the US and UL microbial communities were separated from those of the other samples at the OTU and class levels, while the microbial communities in the HFRS, UFRS, HRS, URS, HS, and HL samples were indistinguishable. PC1 and PC2 explained 41.56 and 19.62%, respectively, of the total bacterial variation at the OTU level (Figure 2A). PC1 and PC2 explained 21.64 and 11.86%, respectively, of the total variation in fungi (Figure 2B). Hierarchical clustering of bacteria and fungi (at OTU, phylum, or class levels) revealed that the microbial communities from the root, stem, and leaf tissue samples were well clustered according to the respective plant compartments. By contrast, the other samples were not well clustered according to the respective compartments (Figures 2C,D; Supplementary Figure S2).
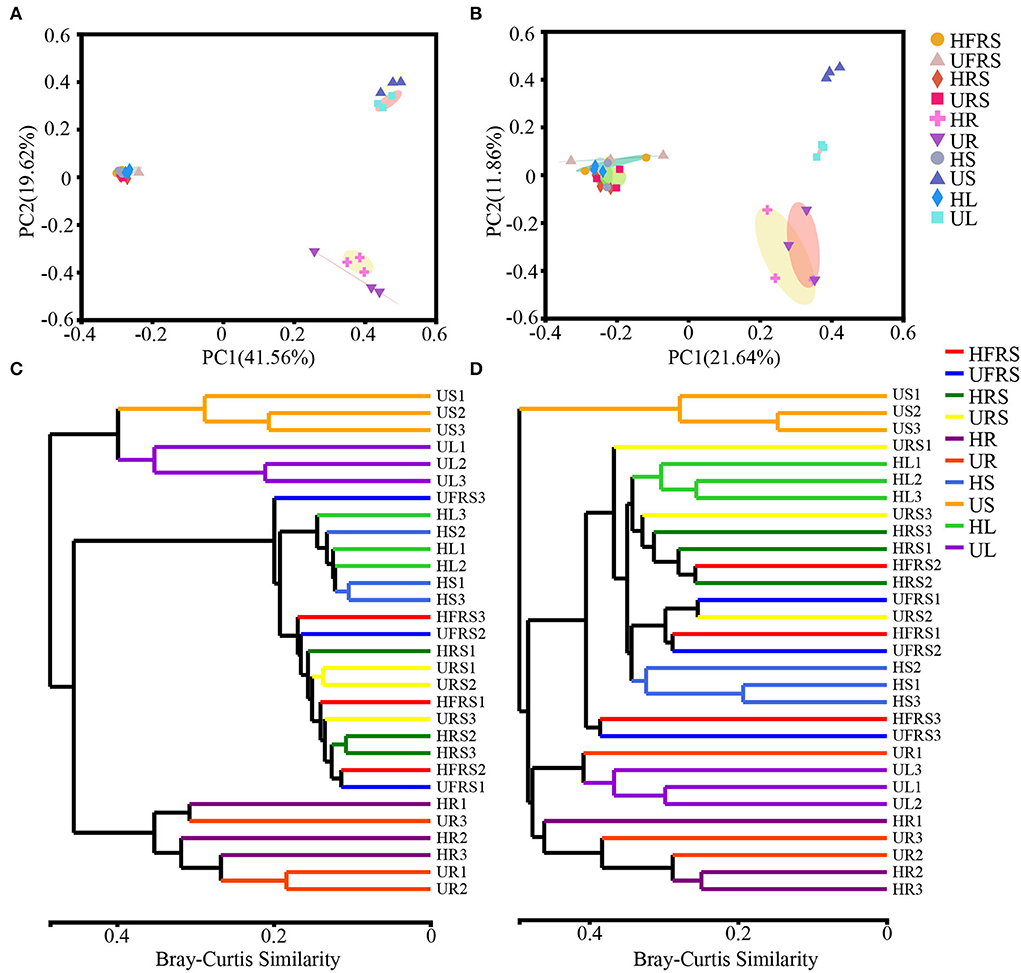
Figure 2. Composition of the microbial communities in the rhizospheric and bulk soil samples and in different plant compartments (roots, stems, and leaves) of healthy and unhealthy P. bungeana plants was analyzed by principal coordinates analysis (PCoA) and dendrograms constructed using the hierarchical clustering algorithm at the OTU level. PCoA of bacterial (A) and fungal (B) community compositions plotted based on Bray–Curtis distance. The percent of variation explained by PC1 and 2 is given as percent values at respective axes. (C) Bacterial and (D) fungal dendrograms constructed by hierarchical clustering of microbial communities. PCoA was based on the rarefaction curve of the minimum sample sequence number. Hierarchical clustering was based on the Bray–Curtis distance and superimposed on PCoA plots. The OTUs were constructed using a sequence similarity threshold of 97%. Abbreviations of variables are given in Figure 1.
A total of 31 bacterial phyla were detected in the soil samples (bulk and rhizospheric soil samples) and the niches in the different compartments (roots, stems, and leaves) of P. bungeana under different health statuses (Figure 3A). Proteobacteria (41.16%) and Actinobacteria (38.33%) were the most abundant phyla in each of the plant niches. The major phyla in bulk and rhizospheric soils were similar, with Proteobacteria (35.12%), Actinobacteria (34.18%), and Firmicutes (12.76%) being the most abundant phyla. The frequency of endophytic bacteria in the roots, stems, and leaves of P. bungeana was different. The abundance of Actinobacteria was the highest in HR samples (55.95%) and lowest in UR samples (4.27%), while Bacteroidota was the most abundant phylum in all the samples. The abundance of Chloroflexi was higher in the HS (7.00%) and HL (10.26%) samples than in the other samples, while Proteobacteria was the most abundant phyla in the US (55.49%) and UL (79.41%) samples. Of the 80 bacterial classes, Alphaproteobacteria, Actinobacteria, Gammaproteobacteria, and Thermoleophilia had a higher abundance, accounting for more than 60% of the total bacteria. Alphaproteobacteria (HR = 36.57%, UR = 37.91%) and Actinobacteria (HR = 54.46%, UR = 43.31%) were the dominant bacterial classes in the roots. Thermoleophilia was the most dominant in the stems (HS = 22.79%) and leaves (HL = 25.07%) of healthy P. bungeana plants (Figure 3C).
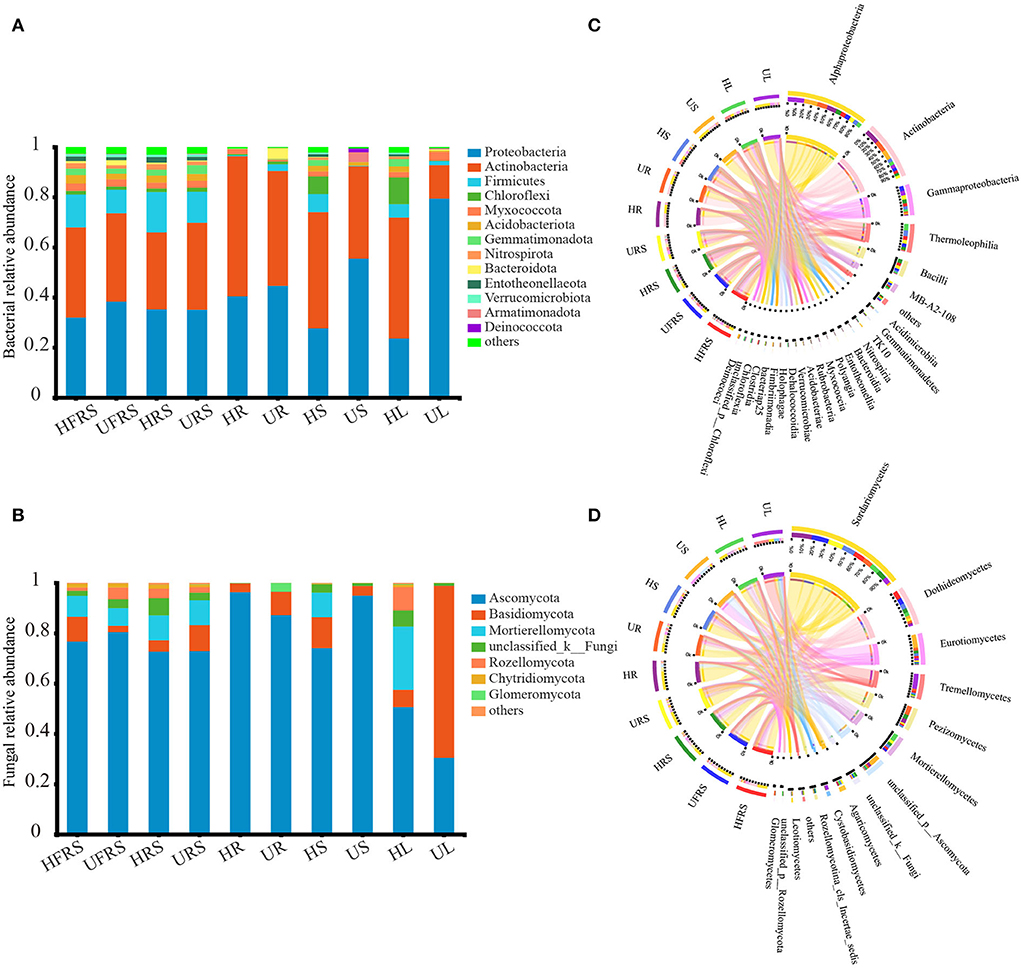
Figure 3. Phylum and class distributions of the OTUs. Relative sequence abundance of (A) bacterial and (B) fungal phyla associated with the soil (rhizospheric and bulk soil), and different plant compartments (roots, stems, and leaves) of healthy and unhealthy P. bungeana plants. The Circos plot depicts the distribution proportion of core (C) bacterial and (D) fungal classes in soil samples and different compartments of healthy and unhealthy P. bungeana plants. The core represents the microbes with major contributions. The Circos plot depicts the proportion of core microbiota in each compartment and reflects the proportion of each dominant species in different samples. The thickness of the ribbons in the Circos plots thickness represents the abundance of the taxon. The absolute tick above the inner segment and relative tick above the outer segment represent the read abundances and relative abundances of the taxa, respectively. Abbreviations of variables are given in Figure 1.
There were 14 fungal phyla in the bulk soil, rhizospheric soil, root, stem, and leaf samples of P. bungeana (Figure 3B). Ascomycota (75.64%), Basidiomycota (8.76%), and Mortierellomycota (7.82%) were the most abundant phyla in the bulk and rhizospheric soil samples. The abundance of endophytic fungi was different among the roots, stems, and leaves of P. bungeana, whereas the abundance of Ascomycota was highest in the HR sample, being 96.28%. The abundance of Basidiomycota (9.30%) and Glomeromycota (3.41%) was higher in the UR samples than in the HR samples, and the abundance of Mortierellomycota was higher in the HS and HL samples than in the other tissues. The abundance of Mortierellomycota (25.12%) was highest in the HL samples. Basidiomycota was the most dominant phylum in the UL samples (68.39%). The differences among the bulk soil samples were insignificant at the class level (P > 0.05). Sordariomycetes, Dothideomycetes, and Eurotiomycetes were the main fungal communities (Figure 3D), representing nearly 70% of all fungal profiles in the samples. The abundance of Pezizomycetes was higher in the HRS samples (15.94%), while the abundance of Agaricomycetes was higher in the URS samples (7.15%) than in the other soil samples. The relative abundance of Sordariomycetes in the endophytic fungal communities in the HR, HS, and HL samples was low, but its abundance was higher than in the UR, US, and UL samples. Pezizomycetes accounted for 29.88% of the fungal classes in the UR samples, which was higher than that observed in the other tissues. The dominant fungal classes in the US samples were Eurotiomycetes (43.25%) and Unclassified_p_ascomycota (36.11%). Mortierellomycetes accounted for 25.08% of the fungal classes in the HL samples, which was higher than that in the other tissues. In comparison, Tremellomycetes (51.10%) and Sordariomycetes (18.60%) were the dominant classes in the UL samples. The total relative abundance of all the orders, families, and genera in relation to their distribution in the different plant compartments is depicted in Supplementary Figure S3.
A total of 180 bacterial OTUs (4.44%) were shared among the five samples of healthy P. bungeana tissues (Figure 4A). Unique OTUs were most abundant in the HFRS samples (957, 23.60%) and least abundant in the HR samples (344, 8.48%). Unique OTUs were least abundant in the HRS samples (896, 22.10%) compared to that in the HS (937, 23.11%) and HL (921, 22.71%) samples. The total number of OTUs shared among unhealthy P. bungeana samples was 31 (10.38%) (Figure 4B). Unique OTUs were most abundant in the URS samples (947, 31.71%) and least abundant in the US samples (187, 6.26%) compared to that of the UFRS (936, 31.35%), UR (554, 18.55%), and UL (362, 12.12%) samples. Analyses of the total OTUs in the same niche under different health conditions revealed that the aboveground tissues, including leaves (12.84%) and stems (6.64%) had less OTUs. By contrast, the underground niches, including rhizospheric soil (65.89%), bulk soil (64.61%), and roots (33.63%), had more OTUs (Supplementary Figures S4A–E).
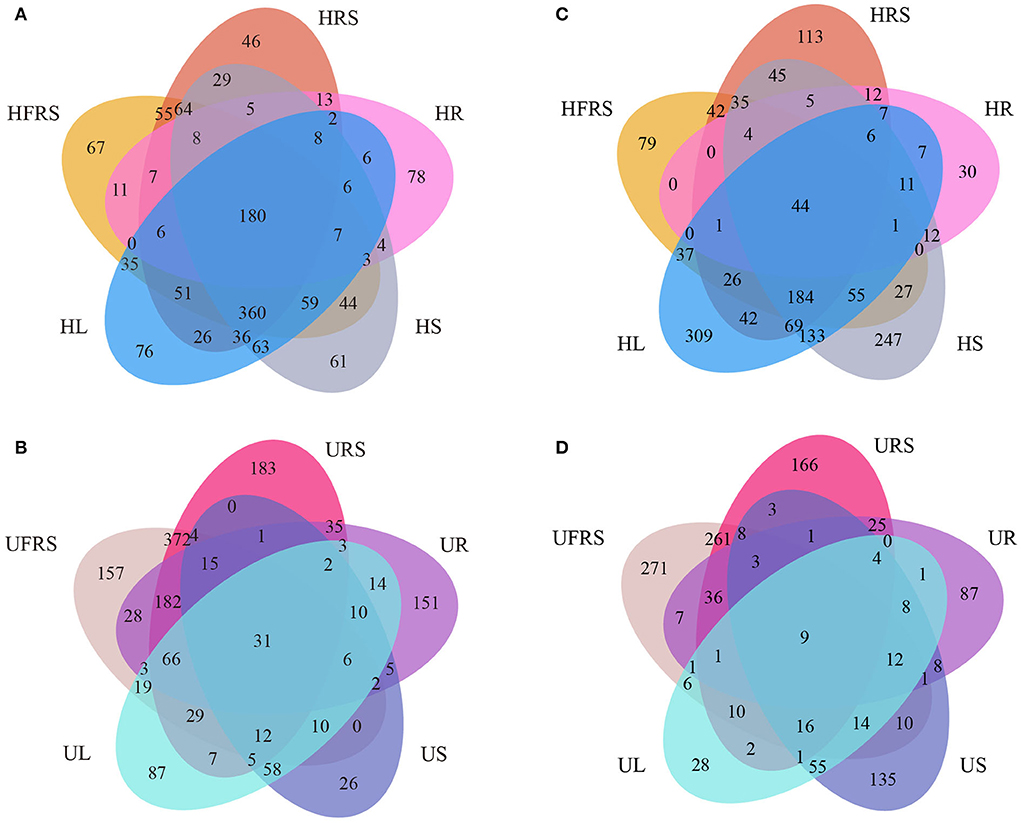
Figure 4. Venn diagrams depicting the unique and shared (A,B) bacterial and (C,D) fungal OTUs in different compartments of P. bungeana. Venn diagram depicting the bacterial OTUs in different niches of (A) healthy and (B) unhealthy P. bungeana plants. Venn diagram depicting the fungal OTUs in different niches of (C) healthy and (D) unhealthy P. bungeana plants. Abbreviations of variables are given in Figure 1.
A total of 44 fungal OTUs (1.41%) were shared among the five samples of healthy P. bungeana tissues (Figure 4C). Unique OTUs (933, 9.58%) were most abundant in HL samples and least abundant in HR samples (142, 1.06%). A total of 529 (2.60%), 636 (4.04%), and 881 (7.92%) OTUs were present in the HFRS, HRS, and HS samples, respectively. However, nine OTUs (0.48%) were shared across the different compartments of unhealthy P. bungeana plants (Figure 4D). Specifically, a total of 545 (35.37%), 178 (9.45%), 545 (28.94%), 203 (10.78%), and 291 (15.45%) unique OTUs were identified in the UFRS, UL, URS, UR, and US samples, respectively. The number of shared OTUs was abundant in the bulk soil (38.52%) compared to that in the rhizospheric soil (37.33%) and roots (27.88%) but least abundant in the stems (9.18%) and leaves (5.87%) of healthy P. bungeana plants (Supplementary Figures S4F–J).
The heatmap (Figure 5) and results of sample clustering analysis revealed that the fungal community structures were similar in the bulk and rhizospheric soil samples. However, the composition of the bacterial communities was similar in the HR and UR samples. The dominant classes of bacteria and fungi were different among the HS, US, HL, and UL samples. In addition, the abundance of bacteria and fungi in the rhizospheric soil, bulk soil, and HS and HL samples of P. bungeana at different health conditions was significantly higher than that in the other samples (P < 0.01).
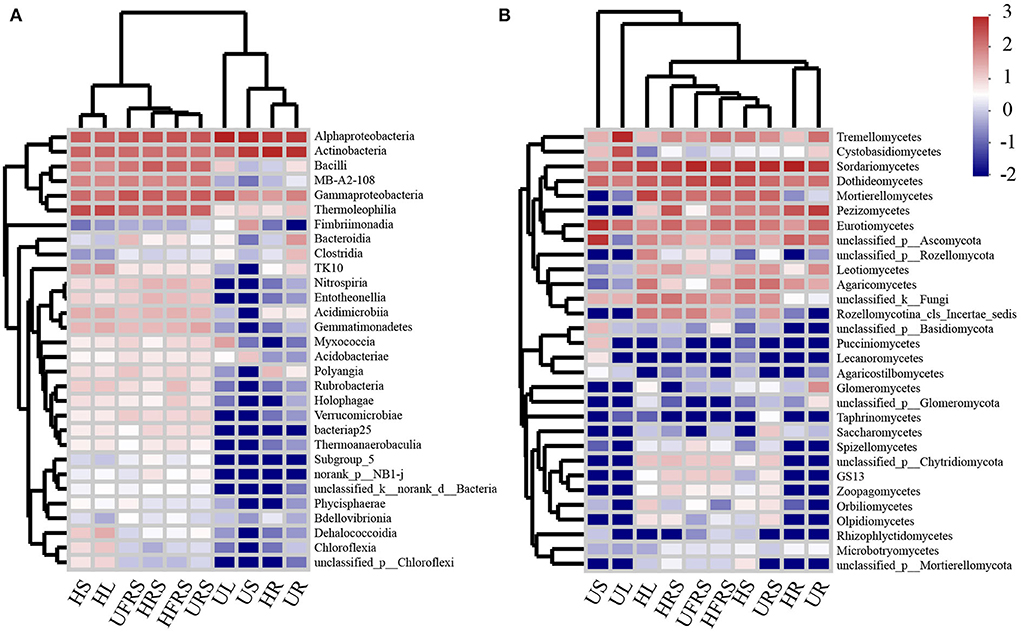
Figure 5. Heatmap analysis of the top 30 classes of (A) bacterial and (B) fungal communities in the compartments of P. bungeana in healthy and unhealthy plants. Abbreviations of variables are given in Figure 1.
LEfSe analyses revealed differences in the composition of the microbial communities in the different plant compartments among healthy and unhealthy plants. Analyses of the bacterial composition revealed that Acidobacteriota was significantly enriched in the HFRS sample (Figure 6A; Supplementary Figure S5A). Nocardioidaceae (from family to genus) and Gammaproteobacteria were significantly enriched in the UFRS sample. Firmicutes (from phylum to genus), Micrococcaceae, and Entotheonellaeota (from phylum to class) were significantly enriched in the HRS sample. The significantly enriched bacteria in the URS sample included MB-A2-108 (from class to genus), Nitrosomonadaceae (from order to family), and Gemmatimonadota (from phylum to genus). Actinobacteria (from class to genus), Proteobacteria (from phylum to genus), and Myxococcia (from class to family) were significantly enriched in the HR sample. Bacteroidota (from phylum to class), Micromonosporales (from order to genus), Pseudorhodoplanes, Bradyrhizobium sp., and Hyphomicrobiaceae were enriched in the UR sample. Gaiellales (from order to genus) was enriched in the HS sample. The enriched bacteria in the US samples were Frankiales, Micrococcales, Acetobacterales (all three from order to family), Propionibacteriales, Friedmanniella, Sphingomonas, Oxalobacteraceae, and Armatimonadota (all five from phylum to genus). Acidimicrobiia, Thermoleophilia (from class to genus), Tistrellales (from order to genus), and Chloroflexi (from phylum to genus) were enriched in the HL samples. Proteobacteria (from phylum to genus), Corynebacteriales (from order to family), and Myxococcaceae (from class to family) were enriched in the UR samples. Pleosporaceae, Gibberella, Stachybotryaceae, and Aspergillaceae were significantly enriched in the HFRS samples (Figure 6B; Supplementary Figure S5B), while eight groups of fungi were considerably enriched in the UFRS sample, including Capnodiales (from order to genus), Glomerellales (from order to genus), Sordariales, and Sarocladium sp. Cucurbitariaceae, Hypocreales_fam_incertae_sedis, and unclassfied_k_Fungi (from phylum to genus) were significantly enriched in the HRS samples. Hypocreales (from order to family) and Agaricomycetes (from class to order) were significantly enriched in the URS samples, while Ascomycota (from phylum to genus) was significantly enriched in the HR samples. A total of four groups of fungi, namely, Setophoma, Leotiomycetes (from class to order), Helotiales, and Exophiala were significantly enriched in the UR samples. Eurotiales (from order to family) was significantly enriched in the HS samples. Trichomeriaceae and unclassified_p_Ascomycota (from class to genus) were significantly enriched in the US samples. Chaetomiaceae and Mortierellomycota (from phylum to genus) were significantly enriched in the HL samples, while Tremellomycetes (from class to order) and Cystobasidiomycetes (from class to genus) were significantly enriched in the UL samples.
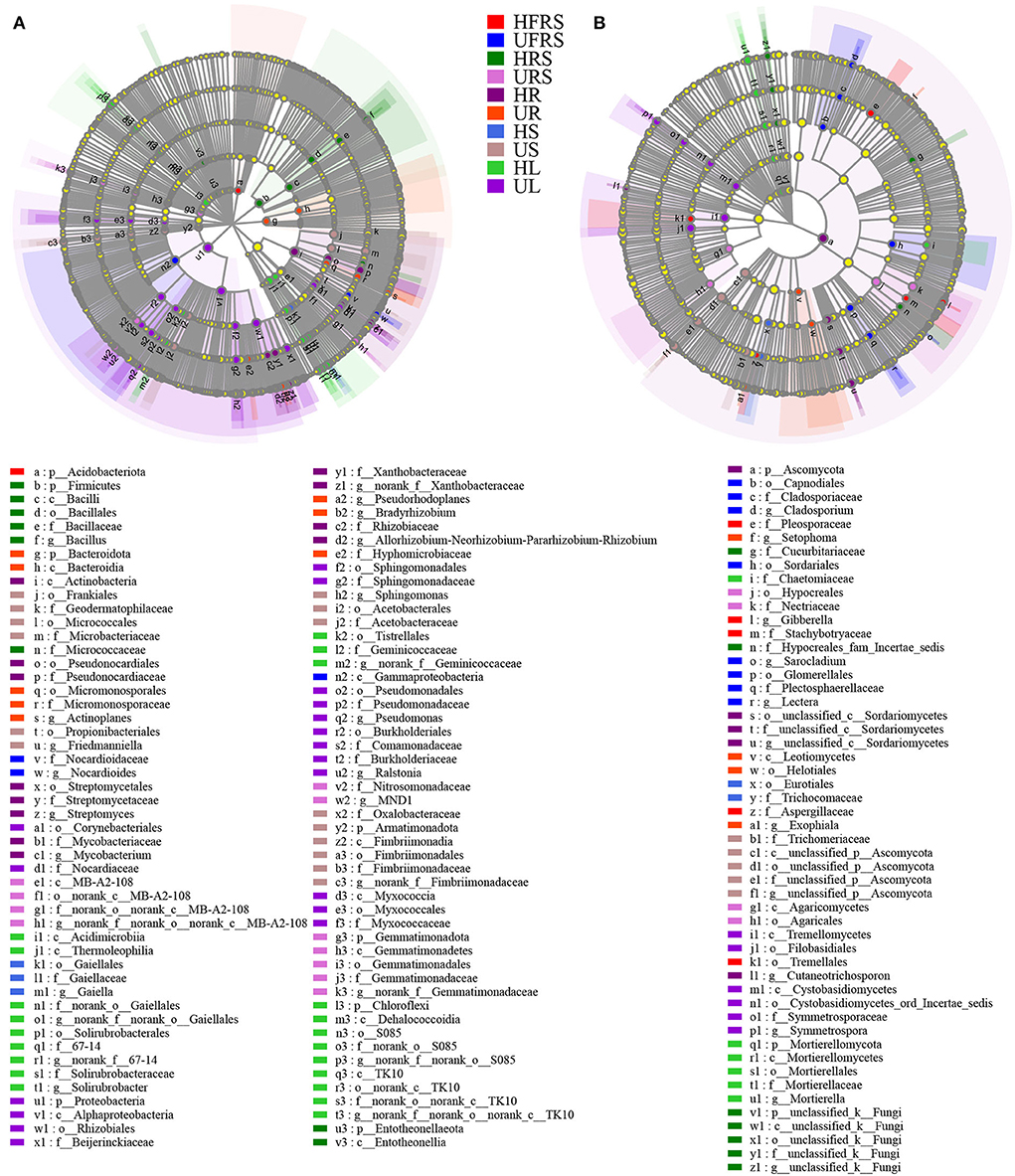
Figure 6. LEfSe (LDA > 4) showing the phylogenetic distribution of (A) bacterial and (B) fungal microbial taxa (from phylum to genus) in rhizospheric and bulk soils, and different compartments (roots, stems, and leaves) of healthy and unhealthy P. bungeana plants. LEfSe analysis was used to search for microbial taxa that were statistically different between groups. The identified microbial taxa are represented as nodes marked with letters in the LEfSe diagram. The circles radiating from the inside to the outside represent the classification levels from phylum to genus. The differently colored regions represent different samples (red, HFRS; dark blue, UFRS; dark green, HRS; pink, URS; dark purple, HR; orange, UR; light blue, HS; brown, US; light green, HL; and light purple, UL). The species with no significant differences (P > 0.05) are uniformly colored in yellow, and the different species are colored according to the group. The colors of the nodes correspond to microbial groups that play an important role in the correspondingly colored groups. Abbreviations of variables are given in Figure 1.
The pH of the rhizospheric and bulk soils of healthy and unhealthy P. bungeana plants ranged from 8.08 to 8.19, indicating that all the tested soils were alkaline. The SOM, TN, TK, and TP contents in the soil of P. bungeana were at medium levels, according to China Soil Standard protocols for forest regions (LY/T 1232–2015, LY/T 1234–2015, and LY/T 1228–2015, in Chinese). The rhizospheric soil of healthy P. bungeana plants had the highest TC content (15.37 g/kg), while the rhizospheric soil of unhealthy P. bungeana had the highest C/N (27.33), and the WC levels were approximately 10 g/kg in the rhizospheric and bulk soils of both healthy and unhealthy P. bungeana plants (Table 1). RDA demonstrated that the bacterial and fungal communities in the rhizospheric and bulk soils of P. bungeana at different health conditions were separated according to the primary environmental characteristics, including SOM, pH, TN, TP, TN, TC, WC, and C/N (Figure 7). The results of the Mantel test revealed that WC and pH were correlated with the microbial communities (P < 0.05), and that the bacterial community structure was significantly affected by the SOM content (P < 0.01). However, the TN, TP, TK, and TC had no significant effect on microbial community structures in the rhizospheric and bulk soils of healthy and unhealthy P. bungeana plants (Table 2).
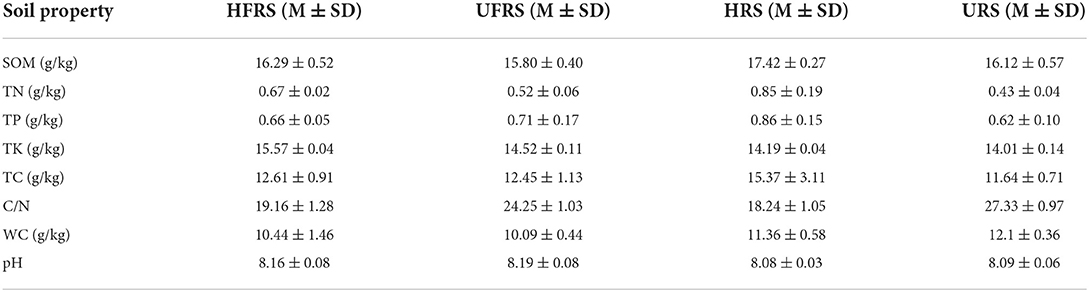
Table 1. Rhizospheric and bulk soil properties of P. bungeana.
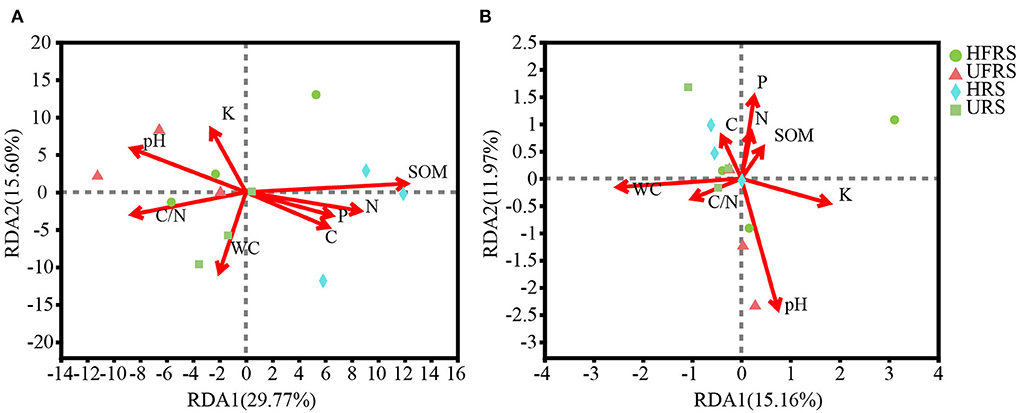
Figure 7. RDA showing the association between edaphic parameters and the composition of (A) bacterial and (B) fungal communities at the OTU level in the rhizospheric and bulk soils of healthy and unhealthy P. bungeana plants. The length of the arrows correspond to the variance that can be explained by the edaphic variable, while the direction indicates an increase in the magnitude of the edaphic variable. The OTUs were clustered using a sequence similarity threshold of 97%. Abbreviations of variables are given in Figure 1.
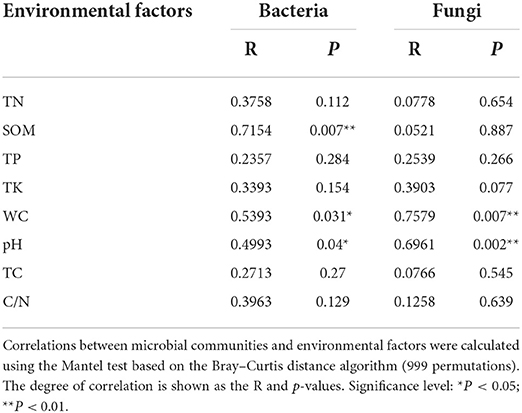
Table 2. Correlation between microbial communities and environmental factors.
In this study, the endangered conifer species, P. bungeana, in the Millennium Xiulin forest of the Xiong'an New Area was selected to study the community structure and variations in the bacterial and fungal communities in soils (bulk and rhizospheric soils) and niches in different plant compartments (roots, stems, and leaves), with the aim of elucidating the changes in the related microbial communities of trees at different health conditions. The results can provide important insights for preserving the health and promoting the sustainable development of this tree species in plantation forests.
Plant niches are significantly correlated with bacterial communities, whereas fungal communities exhibit a different trend (Devin et al., 2016; Zheng and Gong, 2019). The endospheric microbiome is affected by pathogenic infections and potentially affects microbial composition and function (Bulgari et al., 2011; Douanla-Meli et al., 2013). We observed that the bacterial diversity in the rhizospheric and bulk soils and in the stems and leaves of healthy P. bungeana plants was significantly higher than that in the roots of both healthy and unhealthy P. bungeana plants and in the stems and leaves of unhealthy P. bungeana plants. By contrast, there were no significant differences in the bacterial diversity among the rhizospheric soil (HRS and URS), bulk soil (HFRS and UFRS), HS, and HL samples. Fungal abundance in the HS and HL samples was higher than that in the soil (bulk and rhizospheric soil), root (UR and HR), US, and UL samples. The microbial communities in the US and UL samples differed significantly from those of the other samples. The majority of endophytic microorganisms in the HS and HL samples could have been derived from rainwater, air, insects, or the spread of hyphae or spores, in comparison to the microbial communities in the bulk and rhizospheric soil samples and other plant compartments (UR, HR, US, and UL). In addition, the majority of root endophytic communities are derived from the rhizospheric soil and may have experienced strict biological tissue selection prior to colonization in the roots (Compant et al., 2016; Wei and Ashman, 2018). Furthermore, interactions with the biochemical products of P. bungeana may promote the reciprocal relationship between the microorganisms in the stems and leaves of healthy P. bungeana plants. By contrast, the presence of pathogenic microorganisms in the stems and leaves of unhealthy P. bungeana may greatly prevent supplementation by other microbial communities (Cregger et al., 2018; Liu et al., 2018).
The results of the co-occurrence network (CoNet) analysis revealed that the top 20% of fungal genera characterized in healthy plants showed no significant negative interactions (Supplementary Figure S6B). However, the microbes Actinophytocola sp. (Fawzia et al., 2018), MND1, Microvirga sp., Nitrospira sp., Paenibacillus sp., Sp. Sp., Pseudorhodoplanes sp., Gaiella sp., Blastococcus sp., Solirubrobacter sp., Nocardioides sp., Rhodomicrobium sp., and Agromyces sp. in the roots showed a strong negative correlation (Supplementary Figure S6A). The endemic bacteria in the US, UL, and UR samples of unhealthy plants, including Methylobacterium-Methylorubrum sp., Br. Sp., Pedomicrobium sp., Modestobacter sp., Ps. Sp., N. sp., Amnibacterium sp. (Jiang et al., 2018), G. sp., Steroidobacterium sp., Mycobacterium sp., Actinoplanes sp., Friedmanniella sp., and Jatrophihabitans sp. (Supplementary Figure S6C), along with the endemic fungi in the US and UL samples, including Bradymyces sp., Pyrenochaetopsis sp., Fusarium sp., Symmetrospora sp., Aspergillus sp., Plectosphaerella sp., Gibellulopsis sp., Gibberella sp., Pecter asp., Schizothecium sp., Clonostachys sp., Strelitziana sp., Talaromyces sp., Schizothecium sp., Volutella sp., Filobasidium sp., Fusicolla sp., and Paracylindrocarpon sp. (Supplementary Figure S6D), and other dominant bacterial genera in soils, plant tissues, and environmental samples showed a negative correlation, resulting in a strong competitive relationship.
The age of the P. bungeana plants selected in this study ranged from 2 to 3 years. Owing to the short period of forest establishment, the plant tissues and the microbial community of the local forest soil are immature, and most of the microorganisms in the tissues may have originated from the caulosphere, phyllosphere, or anthosphere (Zheng and Gong, 2019). In addition, the microbial community of P. bungeana may be shaped by its tissue structure, physicochemical properties, specific metabolic pathways, and unique gene products of the host plant, making it possible for the plant niches to recruit certain specific microbes (Horton et al., 2014). Finally, potent abiotic factors, including nutritional availability and exposure to light, can result in marked differences in the microbial communities within the stems and leaves of healthy P. bungeana plants (Cregger et al., 2018). The rarefaction curves delineated by the OTUs in soils and tissues were highly uneven, which could be attributed to the uneven microbial colonization of plant roots and inner rings (Bais et al., 2006; Lugtenberg and Kamilova, 2009).
The bacterial and fungal communities in the soil (bulk and rhizospheric soils), and niches in the different compartments (roots, stems, and leaves) of P. bungeana differed at the OTU level. A total of 180 bacterial OTUs and 44 fungal OTUs were shared by the samples obtained from the soil and different ecological niches of healthy P. bungeana plants. The number of OTUs was higher in the stems and leaves, indicating that most of the microbial communities in stems and leaves could have originated from aerospores. A total of 31 bacterial and nine fungal OTUs were shared among the samples obtained from soils and different ecological niches of unhealthy P. bungeana plants, suggesting that the differences in the health conditions of plants altered the diversity and composition of microbial communities. Similar results have also been reported in Pisum sativum, and the study demonstrated that the composition and diversity of endophytic microbes were adversely affected in the majority of unhealthy plants (Yu et al., 2012).
A number of fungal OTUs shared by all samples from healthy plants were significantly higher than those of unhealthy plants, indicating that the health conditions of the plants had a pronounced effect on the fungal community. Furthermore, few fungal OTUs were shared among the samples collected from the aboveground sites (stems and leaves), while the fungal OTUs in the samples obtained from underground parts (bulk soil, rhizospheric soil, and roots) were similar. It is, therefore, speculated that the illness of P. bungeana could be attributed to a “top-down” disease, such as needle cast, brown blotch, or Hypoderma desmazieri Duby, suggesting that quarantine and management strategies should be strengthened, combined with biochemical control measures for eliminating pathogens.
Plant-specific traits, including internal tissue structure, plant health status, and genetic factors (Lebeis et al., 2015), can affect the abundance and differentiation of microbial communities (Zheng and Gong, 2019). It is widely recognized that soil microbes can successfully colonize host plants, and the host plant successfully attracts microbes from the soil to colonize the root plane through root exudates. Some rhizospheric microbes that are able to successfully colonize the roots pass through the plant endodermis and middle column sheath, reach the xylem vessel, and finally form the endophytic community (Compant et al., 2009). A previous study reported that there is a certain association between endophytes and soil microbes (Cocking, 2003). The results of this study indicated that there are differences and inevitable connections between the microbial composition and diversity in different niches. For instance, Proteobacteria and Actinobacteria were the most abundant bacterial phyla in the soil (bulk and rhizospheric soil) samples and niches in different compartments (roots, stems, and leaves) of healthy and unhealthy P. bungeana plants, suggesting that the soils and plant niches are linked in terms of microbial transmission. Microbes also have a certain amount of tolerance and are able to grow and reproduce in different environments, including the phyllosphere, stems, and other compartments. Owing to their high tolerance potential or the need for niche overlap, soil microflora are capable of co-existing in aboveground tissues to a greater extent (Whipps et al., 2008).
Proteobacteria and Actinobacteria are the most abundant bacterial phyla in P. bungeana Beckers et al., 2017; Wallace et al., 2018), and it has been reported that Actinobacteria can colonize any plant tissue (Dinesh et al., 2017). Different tissues of the plant can be colonized by different Actinobacteria, which might be determined by the interactions with the microbes of the host plant (Nimnoi et al., 2010). We observed that the abundance of Actinobacteria was higher in the roots than in other tissues. The rhizospheric soil was abundant in Actinomycetes, which is actively recruited by the roots. Actinomycetes such as Actinomadura glauciflava might promote root development by producing indole-3-acetic acid (IAA), or the bacteria may produce siderophores for binding environmental Fe3+ for improving nutrient absorption, thereby promoting healthy growth (Madhurama et al., 2014). However, we observed that Firmicutes was more abundant in leaves and stems. This finding is in agreement with the results of a study on tree peony (Paeonia Sect. Moutan) and could be related to air transport and root “block” (Yang et al., 2017). At the genus level, Bacillus sp., Actinop. sp., and norank_f_norank_o_Gaiellales were abundant in the soils and tissues of healthy plants (Supplementary Figure S3C). These genera have been detected in several plants and might be beneficial for plant health and growth (Rodrigo et al., 2011; Wei et al., 2021). The dominant bacterial genera in the UL and US samples were Sp. sp. and Me. sp., and their abundance might protect unhealthy plants against pathogens or suppress bacterial pathogens by secreting certain substances, including phenylalanine ammonia-lyase, β-1, 3-glucanase, and peroxidase (Tomoyuki et al., 2016; Madhaiyan et al., 2017).
Ascomycota, Basidiomycota, and Mortierellomycota were the dominant fungal phyla in the soil (bulk and rhizospheric soil), and niches in different plant compartments (roots, stems, and leaves) of healthy and unhealthy P. bungeana plants (Hardoim et al., 2015). The findings were similar to those reported in Populus trichocarpa (Allison et al., 2019) and oak (Toju et al., 2013). Ascomycota and Basidiomycota are widely distributed in different habitats and play an important role in maintaining soil balance and improving plant productivity (Wang et al., 2010; Yang et al., 2019). Exserohilum sp., Le. sp., Py. sp., and Fusi. sp., were the dominant genera in healthy plants. These core microbial taxa were shared by soil (bulk and rhizospheric soil) samples and niches in the different compartments (roots, stems, and leaves) of healthy and unhealthy P. bungeana plants and were probably vertically transmitted, indicating species conservatism to a certain degree (Wei et al., 2021). The dominant microbial genus in the stems and leaves of unhealthy P. bungeana plants was Fi. sp., which produces a killer toxin (FC-1) that is highly effective against the opportunistic fungal pathogen Cryptococcus neoformans and probably protects the health of the plant (Keszthelyi et al., 2006). However, it is well known that some pathogenic genera, including Dothistroma septosporum, Do. pini, and Lecanosticta acicola belong to the phylum Ascomycota, and can cause plant diseases (Raitelaityte et al., 2017; Van der Nest et al., 2019). The unhealthy P. bungeana plants in the study area were highly likely to be infected by these pathogenic fungi, which, in turn, altered the composition of the endophytic microbial community. Microbial interactions play an important role in plant health, and endophytic plant microbes can contribute to the growth and health of organisms through their structural and functional diversity (Santhanam et al., 2015; Agler et al., 2016). Following infections with pathogenic bacteria, specific microorganisms can be enriched in the plant tissue for the continual inhibition of pathogenic bacterial disease via the secretion of certain compounds or the expression of synthetic gene clusters (Carrión et al., 2019). As aforementioned, the microbial communities in the tissues of P. bungeana showed differences at different health conditions (e.g., Sp. sp., Me. sp., and Fi. sp.), which together protected the plant from internal and external stressors via several unknown functional traits.
The microbial diversity and abundance were lowest in the roots, which could be related to the source of endophytic microbes and their growth and expansion. A large number of microorganisms in the soil and rhizosphere were challenged during entry into roots owing to the barrier effect. By contrast, there were abundant microorganisms in the stems and leaves due to the invasion of aerial spores (Timon et al., 2013). These phenomena highlighted the differences in microbial diversity among plant-related habitats (Pinto et al., 2014; Peay et al., 2016; Hassani et al., 2018), and the lower microbial diversity in stems and leaves of unhealthy P. bungeana plants could allow the invasion of pathogenic fungi, including St. sp. and Pestalotiopsis sp., resulting in the development of resistance to other groups of microorganisms.
It is generally believed that plants have specific core microflora that dynamically responds to the environment, including soil properties (Podolich et al., 2015). Soil microbiome is a key player in the maintenance of the health of soil ecosystems, and soil properties are the main factors that affect the existence and diversity of soil microorganisms (Thiem et al., 2018; Whitaker et al., 2018). It has been proposed that soil type is one of the main driving factors for rhizospheric microbial communities (Hinsinger et al., 2009; Yaish et al., 2016). Soil microorganisms can promote material flow and energy conversion by decomposing nutrient substances to gradually improve soil quality (Bouchez et al., 2016). In this study, soil nutrients were at medium or medium to low levels. The distribution of the soil bacterial and fungal communities of P. bungeana was significantly affected by the WC (water content) and pH (P < 0.05). The soil WC was closely related to the expression of certain functional genes of the microbes (Bahram et al., 2018) and was the main factor affecting the structure and function of soil microbial communities. Water can affect soil aeration conditions, and good soil aeration conditions are the basis for the survival of most aeromicrobes (Peralta et al., 2012). Soil pH has a substantial effect on the composition of soil microbial communities, and the acid–base property of soils has a highly significant effect on the soil microbial composition (Lauber et al., 2009; Rousk et al., 2010). In addition, the SOM significantly affected the soil bacteria of P. bungeana (P < 0.01). SOM is an important soil fertility index, providing crops with required substances and improving the physical and chemical conditions of soils. It is one of the most important soil nutrients that can directly affect the soil structure, air permeability, and other properties. The SOM content was significantly positively correlated with the improvement in microbial activity and developmental potential (Bastida et al., 2015). However, no significant correlation was found between soil TN, TP, TK, TC, and C/N and the distribution of microbial communities in this study, which could be attributed to the low content of these elements in our samples.
This study highlighted the fundamental effects of the health status of P. bungeana on the microbial communities in different niches of P. bungeana in the plantation forests in the Xiong'an New Area in China. The most significant finding of this study is that the health of the plants had significant effects on the endophytic bacterial and fungal communities in the stems and leaves of P. bungeana. The results of this study demonstrated that there were no differences in the bacterial and fungal alpha diversities of the samples of bulk soil, rhizospheric soil, and roots between healthy and unhealthy P. bungeana plants. The microbial diversity was found to be higher in the stems and leaves of healthy P. bungeana plants. In addition, this study confirmed that the richness of the microbial communities in the aboveground parts (stems and leaves) of healthy P. bungeana plants was much higher than that of the unhealthy plants. By contrast, there were almost no differences in the richness of the microbial communities in the underground parts (bulk soil, rhizospheric soil, and roots), suggesting that the possible cause of illness was transmitted in a “top- down” manner in unhealthy plants. Altogether, the results provided a scientific basis for further studies on the mechanism of the “aboveground–underground” microbial interactions, which can aid in promoting the healthy and sustainable development of the Millennium Xiulin forest in the Xiong'an New Area.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA821431; https://www.ncbi.nlm.nih.gov/, PRJNA821418.
JY: writing—original draft. JY, HL, YG, and AM: methodology, software, data curation, and validation. CW and MW: soil sampling and experiment. ZY and JL: conceptualization, supervision, funding acquisition, and review and editing. All authors contributed to the article and approved the submitted version.
This study was supported by the Hebei Science and Technology Innovation Base Project (216Z2903G), the National Key Research and Development Project of China (2018YFC0506900), the National Natural Science Foundation of China (31672365), the National Natural Science Foundation of China (42071257), and the Postgraduate Innovation Foundation of Hebei (CXZZBS2022071).
We would like to thank the staff of Millennium Xiulin in the Xiong'an New Area for their enthusiastic help in sampling. We sincerely thank the editors and reviewers for their careful review and critical comments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.913349/full#supplementary-material
Agler, M. T., Ruhe, J., Kroll, S., Morhenn, C., Kim, S. T., Weigel, D., et al. (2016). Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 14, e1002352. doi: 10.1371/journal.pbio.1002352
Allison, M. V., Reese, M., Daniel, Z. Y., Zamin, K. Y., Nancy, L. E., Melissa, A. C., et al. (2019). Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 7, 1–15. doi: 10.1186/s40168-019-0668-8
Álvarez-Pérez, J. M., González-García, S., Cobos, R., Olego, M. A., Ibañez, A., Díez-Galán, A., et al. (2017). Use of endophytic and rhizosphere Actinobacteria from grapevine plants to reduce nursery fungal graft infections that lead to young grapevine decline. Appl. Environ. Microb. 83, e01564–e01517. doi: 10.1128/AEM.01564-17
Bahram, M., Hildebrand, F., Forslund, S. K., Anderson, J. L., Soudzilovskaia, N. A., Bodegom, P., et al. (2018). Structure and function of the global topsoil microbiome. Nature 560, 233–237. doi: 10.1038/s41586-018-0386-6
Bais, H. P., Weir, T. L., Perry, L. G., Gilroy, S., and Vivanco, J. M. (2006). The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 57, 233–266. doi: 10.1146/annurev.arplant.57.032905.105159
Bastida, F., Selevsek, N., Torres, I. F., Hernández, T., and García, C. (2015). Soil restorationwith organic amendments: linking cellular functionality and ecosystem processes. Sci. Rep UK 5, 249–260. doi: 10.1038/srep15550
Beckers, B., De Beek, M. O., Weyens, N., Boerjan, W., and Vangronsveld, J. (2017). Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 5, 25. doi: 10.1186/s40168-017-0241-2
Beckers, B., De Beek, M. O., Weyens, N., Van Acker, R., Van Montagu, M., Boerjan, W., et al. (2016). Lignin engineering in field-grown poplar trees affects the endosphere bacterial microbiome. P. Natl. Acad. Sci. U. S. A. 113, 2312–2317. doi: 10.1073/pnas.1523264113
Bodenhausen, N., Horton, M. W., and Bergelson, J. (2013). Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE 8, e56329. doi: 10.1371/journal.pone.0056329
Bouchez, T., Blieux, A. L., Dequiedt, S., Domaizon, I., Dufresne, A., Ferreira, S., et al. (2016). Molecular microbiology methods for environmental diagnosis. Environ. Chem. Lett. 14, 423–441. doi: 10.1007/s10311-016-0581-3
Brader, G., Compant, S., Mitter, B., Trognitz, F., and Sessitsch, A. (2014). Metabolic potential of endophytic bacteria. Curr. Opin. Biotech. 27, 30–37. doi: 10.1016/j.copbio.2013.09.012
Bulgarelli, D., Schlaeppi, K., Spaepen, S., Van Themaat, E. V. L., and Schulze-Lefert, P. (2013). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant. Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
Bulgari, D., Casati, P., Crepaldi, P., Daffonchio, D., Quaglino, F., Brusetti, L., et al. (2011). Restructuring of endophytic bacterial communities in grapevine yellows-diseased and recovered Vitis vinifera L. plants. Appl. Environ. Microb. 77, 5018–5022. doi: 10.1128/AEM.00051-11
Carrión, V. J., PerezJaramillo, J., Cordovez, V., Tracanna, V., de Hollander, M., RuizBuck, D., et al. (2019). Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 366, 606–612. doi: 10.1126/science.aaw9285
Certini, G., Forte, C., D'acqui, L. P., and Santi, C. A. (2007). Spectroscopic properties of bulk and dichromate oxidation resistant soil organic matter from an anthroposequence in a Mediterranean environment. Plant Soil. 291, 55–65. doi: 10.1007/s11104-006-9173-5
Chen, S. F., Zhou, Y. Q., Chen, Y. R., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890. doi: 10.1093/bioinformatics/bty560
Cocking, E. C. (2003). Endophytic colonization of plant roots by nitrogen-fixing bacteria. Plant Soil 252, 169–175. doi: 10.1023/A:1024106605806
Compant, S., Clément, C., and Sessitsch, A. (2009). Plant growth-promoting bacteria in the rhizo- and endosphere of plants: their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 42, 669–678. doi: 10.1016/j.soilbio.2009.11.024
Compant, S., Saikkonen, K., Mitter, B., Campisano, A., and Mercado-Blanco, J. (2016). Editorial special issue: soil, plants and endophytes. Plant Soil. 405, 1–11. doi: 10.1007/s11104-016-2927-9
Cregger, M. A., Veach, A. M., Yang, Z. K., Crouch, M. J., Vilgalys, R., Tuskan, G. A., et al. (2018). The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6, 1–14. doi: 10.1186/s40168-018-0413-8
Delgado-Baquerizo, M., Oliverio, A. M., Brewer, T. E., Benavent-González, A., Eldridge, D. J., Bardgett, R. D., et al. (2018). A global atlas of the dominant bacteria found in soil. Science 359, 320–325. doi: 10.1126/science.aap9516
Devin, C. D., Damaris, D., Citlali, F. G., Stephen, G., Scott, C., Tanja, W., et al. (2016). Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 209, 798–811. doi: 10.1111/nph.13697
Dickie, I. (2010). Insidious effects of sequencing errors on perceived diversity in molecular surveys. New Phytol. 188, 916–918. doi: 10.1111/j.1469-8137.2010.03473.x
Dinesh, R., Srinivasan, V., Te, S., Anandaraj, M., and Srambikkal, H. (2017). Endophytic actinobacteria: diversity, secondary metabolism and mechanisms to unsilence biosynthetic gene clusters. Crit. Rev. Microbiol. 43, 546–566. doi: 10.1080/1040841X.2016.1270895
Douanla-Meli, C., Langer, E., and Mouafo, F. T. (2013). Fungal endophyte diversity and community patterns in healthy and yellowing leaves of Citrus limon. Fungal Ecol. 6, 212–222. doi: 10.1016/j.funeco.2013.01.004
Durán, P., Thiergart, T., Garrido-Oter, R., Agler, M., Kemen, E., Schulze-Lefert, P., et al. (2018). Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175, 973–983. doi: 10.1016/j.cell.2018.10.020
Edgar, R., Haas, B., Clemente, J., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Esmaeili, T. A., Chatterton, S., Gossen, B. D., and McLaren, D. L. (2017). Metagenomic analysis of oomycete communities from the rhizosphere of field pea on the Canadian prairies. Can. J. Microbiol. 63, 758–768. doi: 10.1139/cjm-2017-0099
Fawzia, C. C., Bouznada, K., Noureddine, B., and Meklat, A. (2018). Planomonospora, Saccharothrix and Actinophytocola genera in Saharan soils of Algeria: isolation, taxonomic identification and antagonistic properties. J. Microb. Biotechnol. Food 7, 460–465. doi: 10.15414/jmbfs.2018.7.5.460-465
Gottel, N. R., Castro, H. F., Kerley, M., Yang, Z., Pelletier, D. A., Podar, M., et al. (2011). Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types distinct. Appl. Environ. Microb. 77, 5934–5944. doi: 10.1128/AEM.05255-11
Guo, C. C., Shen, Y. B., and Shi, F. H. (2020). Effect of temperature, light, and storage time on the seed germination of Pinus bungeana Zucc. ex Endl.: the role of seed-covering layers and abscisic acid changes. Forests 11, 1–16. doi: 10.3390/f11030300
Hardoim, P. R., van Overbeek, L. S., Berg, G., Pirttila, A. M., Compant, S., Campisano, A., et al. (2015). The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. R. 79, 293–320. doi: 10.1128/MMBR.00050-14
Hartman, K., van der Heijden, M. G. A., Wittwer, R. A., Banerjee, S., Walser, J. C., and Schlaeppi, K. (2018). Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 6, 74. doi: 10.1186/s40168-018-0456-x
Hassani, M. A., Duran, P., and Hacquard, S. (2018). Microbial interactions within the plant holobiont. Microbiome 6, 58. doi: 10.1186/s40168-018-0445-0
Hinsinger, P., Bengough, A. G., Vetterlein, D., and Young, I. M. (2009). Rhizosphere: biophysics, biogeochemistry and ecological relevance. Plant Soil 321, 117–152. doi: 10.1007/s11104-008-9885-9
Horton, M. W., Bodenhausen, N., Beilsmith, K., Meng, D., Muegge, B. D., Subramanian, S., et al. (2014). Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 5, 5320. doi: 10.1038/ncomms6320
Jacoby, R., Peukert, M., Succurro, A., Koprivova, A., and Kopriva, S. (2017). The role of soil microorganisms in plant mineral nutrition-current knowledge and future directions. Front. Plant Sci. 8, 1617. doi: 10.3389/fpls.2017.01617
Jiang, Z. K., Tuo, I., Huang, D. L., Osterman, I. A., Tyurin, A. P., Liu, S. W., et al. (2018). Diversity, novelty, and antimicrobial activity of endophytic Actinobacteria from mangrove plants in Beilun Estuary National Nature Reserve of Guangxi, China. Front. Microbiol. 9. 868. doi: 10.3389/fmicb.2018.00868
Keszthelyi, A., Ohkusu, M., Takeo, K., Pfeiffer, I., Litter, J., and Kucsera, J. (2006). Characterisation of the anticryptococcal effect of the FC-1 toxin produced by Filobasidium capsuligenum. Mycoses 49, 176–183. doi: 10.1111/j.1439-0507.2006.01227.x
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos – an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microb. 75, 5111–5120. doi: 10.1128/AEM.00335-09
Lebeis, S. L., Paredes, S. H., Lundberg, D. S., Breakfield, N., Gehring, J., McDonald, M., et al. (2015). Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349, 860–864. doi: 10.1126/science.aaa8764
Li, H. P., Wickham, J. D., Bushley, K., Wang, Z. G., Zhang, B., and Sun, J. H. (2020). New approaches in urban forestry to minimize invasive species impacts: the case of Xiong'an New Area in China. Insects 11, 1–17. doi: 10.3390/insects11050300
Lindow, S. E., and Brandl, M. T. (2003). Microbiology of the phyllosphere. Appl. Environ. Microb. 69, 1875–1883. doi: 10.1128/AEM.69.4.1875-1883.2003
Liu, Y. Q., Liu, L. N., Chen, Y., Li, C. Y., and Yang, J. (2018). Dynamic analysis of interaction between plant pathogenic fungus effector protein and plants. Mol. Plant Breed. 16, 6678–6687. doi: 10.13271/j.mpb.016.006678
Lopez, B. R., Tinoco-Ojanguren, C., Bacilio, M., Mendoza, A., and Bashan, Y. (2012). Endophytic bacteria of the rock-dwelling cactus Mammillaria fraileana affect plant growth and mobilization of elements from rocks. Environ. Exp. Bot. 81, 26–36. doi: 10.1016/j.envexpbot.2012.02.014
Lugtenberg, B., and Kamilova, F. (2009). Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 63, 541–556. doi: 10.1146/annurev.micro.62.081307.162918
Madhaiyan, M., Alex, T. H. H., Cho, H., Kwon, S. W., Whitman, W. B., and Ji, L. H. (2017). Sphingomonas jatrophae sp nov and Sphingomonas carotinifaciens sp nov, two yellow- pigmented endophytes isolated from stem tissues of Jatropha curcas L. Int. J. Syst. Evol. Micr. 67, 5150–5158. doi: 10.1099/ijsem.0.002434
Madhurama, G., Sonam, D., Urmil, P. G., and Ravindra, N. K. (2014). Diversity and biopotential of endophytic actinomycetes from three medicinal plants in India. Afr. J. Microbiol. Res. 8, 184–191. doi: 10.5897/AJMR2012.2452
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mangeot-Peter, L., Tschaplinski, T. J., Engle, N. L., Veneault-Fourrey, C., Martin, F., and Deveau, A. (2020). Impacts of soil microbiome variations on root colonization by fungi and bacteria and on the metabolome of Populus tremula 9 alba. Phytobiomes J. 4, 142–155. doi: 10.1094/PBIOMES-08-19-0042-R
Naveed, M., Mitter, B., Reichenauer, T. G., Wieczorek, K., and Sessitsch, A. (2014). Increased drought stress resilience of maize through endophytic colonization by Burkholderia phytofirmans PsJN and Enterobacter sp. FD17. Environ. Exp. Bot. 97, 30–39. doi: 10.1016/j.envexpbot.2013.09.014
Nimnoi, P., Pongsilp, N., and Lumyong, S. (2010). Endophytic actinomycetes isolated from Aquilaria crassna Pierre ex Lec and screening of plant growth promoters production. World J. Microb. Biot. 26, 193–203. doi: 10.1007/s11274-009-0159-3
O'brien, F. J. M., Almaraz, M., Foster, M. A., Hill, A. F., Huber, D. P., King, E. K., et al. (2019). Soil salinity and pH drive soil bacterial community composition and diversity along a lateritic slope in the Avon River critical zone observatory, western Australia. Front. Microbiol. 10, 1486. doi: 10.3389/fmicb.2019.01486
Ottesen, A. R., González, P. A., White, J. R., Pettengill, J. B., Li, C., Allard, S., et al. (2013). Baseline survey of the anatomical microbial ecology of an important food plant: Solanum lycopersicum (tomato). BMC Microbiol. 13, 1–11. doi: 10.1186/1471-2180-13-114
Peay, K. G., Kennedy, P. G., and Talbot, J. M. (2016). Dimensions of biodiversity in the Earth mycobiome. Nat. Rev. Microbiol. 14, 434–447. doi: 10.1038/nrmicro.2016.59
Peiffer, J. A., Spor, A., and Koren, O. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. PNAS 110, 6548–6553. doi: 10.1073/pnas.1302837110
Peralta, A. L., Matthews, J. W., Flanagan, D. N., and Kent, A. D. (2012). Environmental factors at dissimilar spatial scales influence plant and microbial communities in restored wetlands. Wetlands 32, 1125–1134. doi: 10.1007/s13157-012-0343-3
Philippot, L., Raaijmakers, J. M., Lemanceau, P., and van der Putten, W. H. (2013). Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. doi: 10.1038/nrmicro3109
Pinto, C., Pinho, D., Sousa, S., Pinheiro, M., Egas, C., and Gomes, A. C. (2014). Unravelling the diversity of grapevine microbiome. PLoS ONE 9, e85622. doi: 10.1371/journal.pone.0085622
Podolich, O., Ardanov, P., Zaets, I., Pirttilä, A. M., and Kozyrovska, N. (2015). Reviving of the endophytic bacterial community as a putative mechanism of plantresistance. Plant Soil. 388, 367–377. doi: 10.1007/s11104-014-2235-1
R Core Team (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Raaijmakers, J. M., Paulitz, T. C., Steinberg, C., Alabouvette, C., and Moënne-Loccoz, Y. (2009). The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil. 321, 341–361. doi: 10.1007/s11104-008-9568-6
Raitelaityte, K., Rutkauskas, A., Radzijevskaja, J., Žukauskiene, J., Markovskaja, S., and Paulauskas, A. (2017). The fungal pathogens causing diseases in pines. Biologija 62. 276–283. doi: 10.6001/biologija.v62i4.3414
Rodrigo, M., Marco, K., de Irene, B., Ester, D., van der Menno, V., Johannes, H. M. S., et al. (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. 332, 1097–1100. doi: 10.1126/science.1203980
Rolli, E., Marasco, R., Vigani, G., Ettoumi, B., Mapelli, F., Deangelis, M. L., et al. (2015). Improved plant resistance to drought is promoted by the root-associated microbiome as a water stress dependent trait. Environ. Microbiol. 17, 316–331. doi: 10.1111/1462-2920.12439
Rousk, J., Bth, E., Brookes, P. C., Lauber, C. L., Lozupone, C., Caporaso, J. G., et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340–1351. doi: 10.1038/ismej.2010.58
Santhanam, R., Luu, V. T., Weinhold, A., Goldberg, J., Oh, Y., and Baldwin, I. T. (2015). Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping. PNAS 112, e5013–e5020. doi: 10.1073/pnas.1505765112
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6, e27310. doi: 10.1371/journal.pone.0027310
Semenov, M. V., Chernov, T. I., Tkhakakhova, A. K., Zhelezova, A. D., Ivanova, E. A., Kolganova, T. V., et al. (2018). Distribution of prokaryotic communities throughout the chernozem profiles under different land uses for over a century. Appl. Soil Ecol. 127, 8–18. doi: 10.1016/j.apsoil.2018.03.002
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Syring, J., Del Castillo, R. F., Cronn, R., and Liston, A. (2007). Multiple nuclear loci reveal the distinctiveness of the threatened, neotropical Pinus chiapensis. Syst. Bot. 32, 703–717. doi: 10.1600/036364407783390836
Thiem, D., Gołebiewski, M., Hulisz, P., Piernik, A., and Hrynkiewicz, K. (2018). How does salinity shape bacterial and fungal microbiomes of Alnus glutinosa roots? Front. Microbiol. 9, 651. doi: 10.3389/fmicb.2018.00651
Timon, T. W., Han, A. B. W., and Jan, D. (2013). Fungal spores for dispersion in space and time. Adv. Appl. Microbiol. 85, 43–91. doi: 10.1016/B978-0-12-407672-3.00002-2
Toju, H., Yamamoto, S., Sato, H., and Tanabe, A. S. (2013). Sharing of diverse mycorrhizal and root-endophytic fungi among plant species in an oak-dominated cooltemperate forest. PLoS ONE 8, e78248. doi: 10.1371/journal.pone.0078248
Tomoyuki, M., Misue, A., Hisayuki, M., Masayuki, S., Takakazu, K., Shusei, S., et al. (2016). Metagenomic analysis revealed methylamine and ureide utilization of soybean-associated Methylobacterium. Microbes Environ. 31, 268–278. doi: 10.1264/jsme2.ME16035
Unyarat, R., Laurent, F., Artit, V., Antoine, B., Jean-Pierre, M., and Floriane, L. (2016). The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. New Phytol. 210, 1033–1043. doi: 10.1111/nph.13808
Van der Nest, A., Wingfield, M. J., Janoušek, J., and Barnes, I. (2019). Lecanosticta acicola: a growing threat to expanding global pine forests and plantations. Mol. Plant Pathol. 20, 1327–1364. doi: 10.1111/mpp.12853
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
Wagner, M. R., Lundberg, D. S., Del Rio, T. G., Tringe, S. G., Dangl, J. L., and Mitchell-Olds, T. (2016). Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7, 12151. doi: 10.1038/ncomms12151
Wallace, J., Laforest-Lapointe, I., and Kembel, S. W. (2018). Variation in the leaf and root microbiome of sugar maple (Acer saccharum) at an elevational range limit. PeerJ 6, e5293. doi: 10.7717/peerj.5293
Wang, C., Masoudi, A., Wang, M., Yang, J., Shen, R. W., Man, M., et al. (2020). Community structure and diversity of the microbiomes of two microhabitats at the root–soil interface: implications of meta-analysis of the root-zone soil and root endosphere microbial communities in Xiong'an New Area. Can. J. Microbiol. 66, 605–622. doi: 10.1139/cjm-2020-0061
Wang, C., Masoudi, A., Wang, M., Yang, J., Yu, Z. J., and Liu, J. Z. (2021). Land-use types shape soil microbial compositions under rapid urbanization in the Xiong'an New Area, China. Sci. Total Environ. 777, 145976. doi: 10.1016/j.scitotenv.2021.145976
Wang, H. Y., Guo, S. Y., Huang, M. R., Thorsten Lumbsch, H., and Wei, J. C. (2010). Ascomycota has a faster evolutionary rate and higher species diversity than Basidiomycota. Sci. China Life Sci. 53, 1163–1169. doi: 10.1007/s11427-010-4063-8
Wang, M., Masoudi, A., Wang, C., Yang, J., Zhai, Y. P., Wu, C. H., et al. (2022). Plantation type and afforestation age disclose variable influences on soil microbial compositions in man-made forests in the Xiong'an New Area, China. Land Degrad Dev. 1–16. doi: 10.1002/ldr.4372
Wang, Y. B., Zhang, W. X., Ding, C. J., Zhang, B. Y., Huang, Q. J., Huang, R. F., et al. (2019). Endophytic communities of transgenic Poplar were determined by the environment and niche rather than by transgenic events. Front. Microbiol. 10, 588. doi: 10.3389/fmicb.2019.00588
Wei, G. F., Ning, K., Zhang, G. Z., Yu, H. B., Yang, S. M., Dai, F., et al. (2021). Compartment niche shapes the assembly and network of Cannabis sativa-associated microbiome. Front. Microbiol. 12, 714993.
Wei, N., and Ashman, T. L. (2018). The effects of host species and sexual dimorphism differ among root, leaf and flower microbiomes of wild strawberries in situ. Sci. Rep UK 8, 5195. doi: 10.1038/s41598-018-23518-9
Weyens, N., van der Lelie, D., Taghavi, S., Newman, L., and Vangronsveld, J. (2009). Exploiting plant–microbe partnerships to improve biomass production and remediation. Trends Biotechnol. 27, 591–598. doi: 10.1016/j.tibtech.2009.07.006
Whipps, J. M., Hand, P., Pink, D., and Bending, G. D. (2008). Phyllosphere microbiology with special reference to diversity and plant genotype. J. Appl. Microbiol. 105, 1744–1755. doi: 10.1111/j.1365-2672.2008.03906.x
Whitaker, B. K., Reynolds, H. L., and Clay, K. (2018). Foliar fungal endophyte communities are structured by environment but not host ecotype in Panicum virgatum (switchgrass). Ecology 99, 2703–2711. doi: 10.1002/ecy.2543
Xie, S., Su, Y., Xu, W., Cai, W., Wang, X., Lu, F., et al. (2019). The effect of habitat changes along the urbanization gradient for breeding birds: an example from the Xiong'an New Area. PeerJ 7, e7961. doi: 10.7717/peerj.7961
Xu, H., Wang, M., Shi, T., Guan, H., Fang, C., and Lin, Z. (2018). Prediction of ecological effects of potential population and impervious surface increases using a remote sensing based ecological index (RSEI). Ecol. Indic. 93, 730–740. doi: 10.1016/j.ecolind.2018.05.055
Yaish, M. W., Al-Lawati, A., Jana, G. A., Vishwas, P. H., and Glick, B. R. (2016). Impact of soil salinity on the structure of the bacterial endophytic community identified from the roots of Caliph Medic (Medicago truncatula). PLoS ONE 11, e0159007. doi: 10.1371/journal.pone.0159007
Yang, C., Li, J., Liu, N., and Zhang, Y. (2019). Effects of fairy ring fungi on plants and soil in the alpine and temperate grasslands of China. Plant Soil. 441, 499–510. doi: 10.1007/s11104-019-04141-7
Yang, R., Liu, P., and Ye, W. (2017). Illumina-based analysis of endophytic bacterial diversity of tree peony (Paeonia Sect. Moutan) roots and leaves. Braz. J. Microbiol. 48, 695–705. doi: 10.1016/j.bjm.2017.02.009
Yang, Y. X., Wang, M. L., Liu, Z. L., Zhu, J., Yan, M. Y., and Li, Z. H. (2015). Nucleotide polymorphism and phylogeographic history of an endangered conifer species Pinus bungeana. Biochem. Syst. Ecol. 64, 89–96. doi: 10.1016/j.bse.2015.11.016
Yu, L., Nicolaisen, J., Larsen, J., and Ravnskov, S. (2012). Molecular characterization of root-associated fungal communities in relation to health status of Pisum sativum using barcoded pyrosequencing. Plant Soil 357, 395–405. doi: 10.1007/s11104-012-1180-0
Yu, P., and Hochholdinger, F. (2018). The role of host genetic signatures on root–microbe interactions in the rhizosphere and endosphere. Front Plant Sci. 9, 1896. doi: 10.3389/fpls.2018.01896
Zarraonaindia, I., Owens, S. M., Weisenhorn, P., West, K., Hampton-Marcell, J., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. MBIO 6, 1–10. doi: 10.1128/mBio.02527-14
Zhang, Y., Xu, J., Riera, N., Jin, T., Li, J., and Wang, N. (2017). Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 5, 97. doi: 10.1186/s40168-017-0304-4
Keywords: Pinus bungeana, microbiota, rhizosphere, niche differentiation, plant–microbe interactions
Citation: Yang J, Masoudi A, Li H, Gu Y, Wang C, Wang M, Yu Z and Liu J (2022) Microbial community structure and niche differentiation under different health statuses of Pinus bungeana in the Xiong'an New Area in China. Front. Microbiol. 13:913349. doi: 10.3389/fmicb.2022.913349
Received: 05 April 2022; Accepted: 11 August 2022;
Published: 02 September 2022.
Edited by:
Kim Yrjälä, Zhejiang Agriculture and Forestry University, ChinaReviewed by:
Shaikhul Islam, Ehime University, JapanCopyright © 2022 Yang, Masoudi, Li, Gu, Wang, Wang, Yu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhijun Yu, eXV6aGlqdW5AaGVidHUuZWR1LmNu; Jingze Liu, bGl1amluZ3plQGhlYnR1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.