 Gulshan Yadav
Gulshan Yadav Ruchi Singh
Ruchi Singh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 17 August 2022
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.909886
This article is part of the Research Topic Drug Resistance, Global Epidemiology and Virulence of Acinetobacter View all 14 articles
Introduction: Acinetobacter baumannii, an opportunistic pathogen, rapidly acquires antibiotic resistance, thus compelling researchers to develop alternative treatments at utmost priority. Phage-based therapies are of appreciable benefit; however, CRISPR-Cas systems are a major constraint in this approach. Hence for effective implementation and a promising future of phage-based therapies, a multifaceted understanding of the CRISPR-Cas systems is necessary.
Methods: This study investigated 4,977 RefSeq genomes of A. baumannii from the NCBI database to comprehend the distribution and association of CRISPR-Cas systems with genomic determinants.
Results: Approximately 13.84% (n = 689/4,977) isolates were found to carry the CRSIPR-Cas system, and a small fraction of isolates, 1.49% (n = 74/4,977), exhibited degenerated CRISPR-Cas systems. Of these CRISPR-Cas positive (+) isolates, 67.48% (465/689) isolates harbored type I-F1, 28.59% (197/689) had type I-F2, and 3.7% (26/689) had co-existence of both type I-F1 and type I-F2 systems. Co-existing type I-F1 and type I-F2 systems are located distantly (∼1.733 Mb). We found a strong association of CRISPR-Cas systems within STs for type I-F1 and type I-F2, whereas the type I-F1 + F2 was not confined to any particular ST. Isolates with type I-F1 + F2 exhibited a significantly high number of mean spacers (n = 164.58 ± 46.41) per isolate as compared to isolates with type I-F2 (n = 82.87 ± 36.14) and type I-F1 (n = 54.51 ± 26.27) with majority targeting the phages. Isolates with type I-F1 (p < 0.0001) and type I-F2 (p < 0.0115) displayed significantly larger genome sizes than type I-F1 + F2. A significantly reduced number of integrated phages in isolates with co-existence of type I-F1 + F2 compared with other counterparts was observed (p = 0.0041). In addition, the isolates carrying type I-F1 + F2 did not exhibit reduced resistance and virulence genes compared to CRISPR-Cas(–) and CRISPR-Cas (+) type I-F1 and type I-F2, except for bap, abaI, and abaR.
Conclusion: Our observation suggests that the co-existence of type I-F1 and F2 is more effective in constraining the horizontal gene transfer and phage invasion in A. baumannii than the isolates exhibiting only type I-F1 and only type I-F2 systems.
Acinetobacter baumannii, an opportunistic pathogen, is of great clinical relevance and is associated with hospital-acquired infections in immunocompromised patients (Munoz-Price and Weinstein, 2008). It rapidly develops resistance against all classes of antibiotics. Multidrug-resistant, extensively drug-resistant, and pan-drug-resistant strains of A. baumannii have now been prominently reported worldwide (Zarrilli et al., 2009; Tal-Jasper et al., 2016; Joshi et al., 2017; Bassetti et al., 2018; Leungtongkam et al., 2018) and obligated the World Health Organization to classify this pathogen as a critical priority pathogen for developing novel antibiotics (World Health Organization, 2017). In addition to novel antibiotics, alternative treatment strategies are also being explored (Baptista et al., 2018; Merker et al., 2020; Pires et al., 2020; Kumar et al., 2021; Micoli et al., 2021).
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) genes provide immunity against phages and other foreign genetic elements through the incorporation of spacers (Pourcel et al., 2005; Barrangou et al., 2007; Brouns et al., 2008). Approximately 36–42% of completely sequenced bacteria possess CRISPR-Cas systems (Makarova et al., 2020; Pourcel et al., 2020). The distribution of CRISPR-Cas systems in bacterial phyla is found to have differential representation across the taxonomic levels, where some groups and species are nearly devoid (< 1%) of CRISPR-Cas systems while some carry in almost all (> 95%) genomes (Burstein et al., 2016; Shehreen et al., 2019). The biological significance and basis of the irregular phyletic distribution of CRISPR–Cas systems are not yet proved.
CRISPR-Cas systems have been classified into two Classes (Class 1 and Class 2) and six types (Type I–VI) (Koonin et al., 2017; Makarova et al., 2020). Class 1 includes types I, III, and IV CRISPR-Cas systems, where type I systems are the most prevalent (∼60%) in the bacterial population (Burstein et al., 2017; Hidalgo-Cantabrana et al., 2019). Contrary to Class 2, the Class 1 type I CRISPR-Cas system depends on a multi-subunit CRISPR-associated complex for antiviral defense (Cascade), which further employs Cas3 to degrade the foreign DNA (Westra et al., 2012). Based on the signature gene type I, CRISPR-Cas systems were further divided into seven subtypes (I–A to I–G) (Makarova et al., 2020). Being present in the clinically important (i.e., Pseudomonas aeruginosa, A. baumannii) and model bacterial species (i.e., E. coli), type I-F CRISPR-Cas is one of the most extensively studied systems. Because type I-F CRISPR-Cas system was first discovered in Yersinia pestis (Haft et al., 2005; Cady et al., 2011), its Cascade components were also named Csy (CRISPR subtype Ypest). An updated classification of type I-F CRISPR-Cas system defines type I-F1 loci consisting of four genes: csy1 (cas8f1), csy2 (cas5f1), csy3 (cas7f1), and csy4 (cas6f) while type I-F2 derived from type I-F1 consists of only three genes: cas5fv (cas5f2), cas6f, and cas7fv (cas7f2), along with the universal adaptation modules (cas1 and cas2-3 gene) (Figure 1). In addition, type I-F3, the minimal variant of type I-F1, is associated with Tn7-like transposons, has cas8f3/cas5f3 fused, and lacks cas1 and cas2-3 genes (Makarova et al., 2020). Generally, only a single CRISPR-Cas system is present in the majority of the bacterial isolates. However, the co-existence of different types and subtypes was also reported in the single bacterial cell (Carte et al., 2014; Majumdar et al., 2015; Pinilla-Redondo et al., 2020). Type I-F was positively associated with the type IV-A1/2 CRISPR-Cas system and is proposed to compensate for the absence of adaptation modules in type IV (Pinilla-Redondo et al., 2020). Divisive evidence regarding the functional significance of co-occurrence of distinct CRISPR-Cas loci indicates that it can function either independently (Carte et al., 2014) or share components of the process (Deng et al., 2013).

Figure 1. Diagrammatic representation of locus structure of CRISPR-Cas in isolates with co-existence of type I-F1 and type I-F2 systems.
Along with phages, the CRISPR-Cas system can restrict horizontal gene transfer (HGT) occurring through mobile genetic elements (MGEs), thereby limiting the acquisition of potentially beneficial antibiotic-resistant genes (ARGs) (Bikard et al., 2012; Jiang et al., 2013; O’Meara and Nunney, 2019). However, during strong antibiotic selection pressure, the bacterial population often involves loss or inactivation of the CRISPR-Cas system (Jiang et al., 2013; Watson et al., 2018). As the CRISPR-Cas system is present on MGEs (Koonin and Makarova, 2017), it can be attained back through HGT and generalized transduction (García-Martínez et al., 2018; Watson et al., 2018). However, there are some species where the CRISPR-Cas system can co-exist with resistance determinants indicating that a simple rule of selection pressure cannot be universal (Shehreen et al., 2019). The trade-off between retention of CRISPR-Cas system and HGT of beneficial MGEs is being explored to design novel treatments (Lin et al., 2017; Pursey et al., 2018; Pires et al., 2020). Being the natural predators of bacteria, phages can bypass their immune system through anti-CRISPR (ACR) genes. ACR inactivates the CRISPR-Cas system and can facilitate the successful integration of phage into the bacterial genome (Pawluk et al., 2018; Zhu et al., 2018), thereby making them an ideal tool to be used against antibiotic-resistant bacterial communities.
Genome-wide association studies with resistance, virulence, and other genomic determinants are proved to be beneficial to understand the behavior of CRISPR-Cas systems. This study involves the in silico analysis of a large set of genome data (n = 4,977) available in the public domain to reveal the distribution of CRISPR-Cas in A. baumannii and to validate the hypothesis that the presence of co-existing CRISPR-Cas systems may confer a fitness advantage in A. baumannii by impacting the dynamics of HGT.
A total of 4,977 genome sequences of A. baumannii were downloaded from the NCBI RefSeq database as per availability on 18 January 2021. These genomes are either complete or assembled to different levels; complete genome (245) (containing 535 complete plasmid sequences), chromosome (23), scaffold (1,690), or contig (3,019). These sequences may also contain contigs or scaffolds from episomes. NCBI Annotation (Li et al., 2021) file and genome metadata (genome size, isolation source, host, host disease, submitter, geographic location of the sample) was collected for each isolate (Supplementary Table 1). Guanine-cytosine (GC) percentage was calculated using the pearl script available on git hub.1 Multi-locus sequence typing (MLST) was performed using Pasture PubMLST typing scheme2 (Jolley and Maiden, 2010).
The presence of CRISPR array/s was determined using a standalone command-line version of CRISPRCasFinder (v4.2.20), CRISPRDetect (v2.4), and CRISPR Recognition Tool (CRT) (v1.2) (Bland et al., 2007; Biswas et al., 2016; Couvin et al., 2018). CRISPRCasFinder, NCBI-BLASTn (v2.6.0), and NCBI RefSeq genome annotation GFF files were used to determine the presence of cas genes (McGinnis and Madden, 2004) and were classified according to the recent classification (Makarova et al., 2011, 2015, 2020). Final interpretations were made based on the same value given by any two out of the three software used. Manual interpretations were made wherever necessary (Supplementary Table 1). Anti-CRISPR (ACR) genes were identified by screening genomes against type I-F anti-CRISPRdb (Dong et al., 2018) using NCBI-tBLASTn with stringent cut-off to avoid false positives, that is, e-value ≤ 10–10 and bit score ≥ 200 (Shehreen et al., 2019). Integrated phages were discovered by ProphET, a phage estimation tool (Reis-Cunha et al., 2019).
The RealPhy (v1.13), a reference alignment-based phylogeny builder (Bertels et al., 2014) was used to construct a phylogenetic tree using whole-genome data of CRISPR-Cas (+) isolates. It directly maps short reads to a reference sequence. It extracts the single nucleotide polymorphisms (SNPs) to infer the phylogenetic tree using the maximum likelihood method for the aligned SNPs positions. We have used the merge option that combines alignments from mapping to multiple reference sequences to remove bias raised due to the alignment to a single reference genome. To visualize and annotate the phylogenetic tree, Interactive Tree Of Life (iTOL)3 was used. The phylogenetic tree was uploaded as a Newick file and annotated using tools available on the iTOL website. Data of CRISPR-Cas systems, number of spacers, and MLST groups were overlaid on the phylogenetic tree as multi-value bars and color strips, respectively (Letunic and Bork, 2021). Genome alignment for variability visualization was performed using Mauve v2.4.0 (Darling et al., 2004).
A unique non-redundant spacer set for each class was obtained by clustering spacer sequences identified by CRISPRCasFinder with an array-quality score ≥ 4.0, using CD-HIT-EST (v4.8.1) (Huang et al., 2010) with an identity-cutoff of 0.95. Spacers with Ns were removed. NCBI-BLASTn (v2.6.0) was used to predict spacer targets against phage genomes, integrative conjugative elements (ICEs), plasmids, resistance genes, and virulence genes. BLASTn hits with at least 95% sequence identity and coverage were accepted as valid targets (Shehreen et al., 2019). Representative sequences of 99 phages that show interaction with A. baumannii were downloaded from the Microbe Vs. Phage (MVP) database (Gao et al., 2018). The life cycle (i.e., temperate, virulent) for these phages was predicted with PHAGEAI (Tynecki et al., 2020). ICEs sequences were downloaded from ICEberg 2.0 database (Liu M. et al., 2019). Plasmid sequences were downloaded from a curated database of plasmid sequences containing 10,892 complete plasmids (Brooks et al., 2019). Acquired resistance gene sequences were downloaded from the ResFinder database of acquired antimicrobial resistance genes (Zankari et al., 2012). Virulence genes were downloaded from the Virulence Factor Database (Chen et al., 2005; Liu B. et al., 2019).
ResFinder (v3.0), with a default minimum threshold and coverage of 0.9 and 0.6, respectively, were used to identify acquired antibiotic resistance genes (ARGs) (Camacho et al., 2009; Zankari et al., 2012, 2017; Bortolaia et al., 2020). ResFinder identifies ARGs from 15 antibiotic drug classes where a complete gene confers resistance. The virulence gene database was obtained from Virulence Finder Database (VFDB) for Acinetobacter spp. (Chen et al., 2005; Liu B. et al., 2019).
The presence of the CRISPR-Cas system, virulence, and ARGs are coded as binary variables for each genome. An unpaired two-tailed t-test (using GraphPad Prism v6.0.1) was used to determine the association of CRISPR-Cas systems with genome size, number of spacers, and phages. One-tailed paired t-test was used to compare the differences between CRISPR-Cas (–) and CRISPR-Cas (+) genomes of the same ST type. Chi-square (χ2) test was used to determine the association of the virulence and ARGs with the CRISPR-Cas systems as described earlier (Shehreen et al., 2019).
A total of 4,977 A. baumannii genomes from the NCBI Refseq database were analyzed to determine the frequency and distribution of CRISPR, Cas, and anti-CRISPRs among A. baumannii. Based on their source of origin at the time of isolation, genomes were classified into three major categories: clinical (n = 4,015), environmental (n = 185), and other (in case of unavailability of data) (n = 777). Among all, only 13.84% (n = 689/4,977) isolates harbored a functional CRISPR-Cas system and were distributed across the globe (Figure 2). These results are in concordance with previous studies that showed the presence of CRISPR-Cas systems in ∼14% of A. baumannii isolates (Shehreen et al., 2019; Pursey et al., 2022). Genomes with CRISPR-Cas systems were further analyzed and classified into three categories based on their type: 67.48% (465/689) CRISPR-Cas positive (+) genomes carried only type I-F1, 28.59% (197/689) carried only type I-F2, and 3.7% (26/689) were found to have co-existence of both type I-F1 and type I-F2 (herein after referred as type I-F1 + F2) (Figure 3). Although co-localization of different types of CRISPR-Cas systems is common in bacteria (Carte et al., 2014; Majumdar et al., 2015; Pinilla-Redondo et al., 2020) and are proposed to cooperate to counteract viral escape (Silas et al., 2017), co-localization of variants of type I-F (i.e., I-F1 and I-F2) is rare and not reported to date. A single isolate (strain MRSN7153, United States) was found to harbor a type III-B CRISPR-Cas system and was excluded from downstream analysis. A very low proportion of ACR genes (0.68%; n = 34/4,977) were found in A. baumannii (Figure 3). Hence, further correlational studies with ACR genes were not performed. Isolates with environmental niches were categorized into hospital and natural environments categories. As expected, a significantly higher percentage (∼56%) of CRISPR-Cas presence was observed in isolates with natural environmental niche compared to clinical (∼15%) and hospital environment niche (∼9%) (Supplementary Table 2), indicating that CRISPR-Cas-mediated immunity provides a clear advantage during defense against phages (Barrangou et al., 2007).
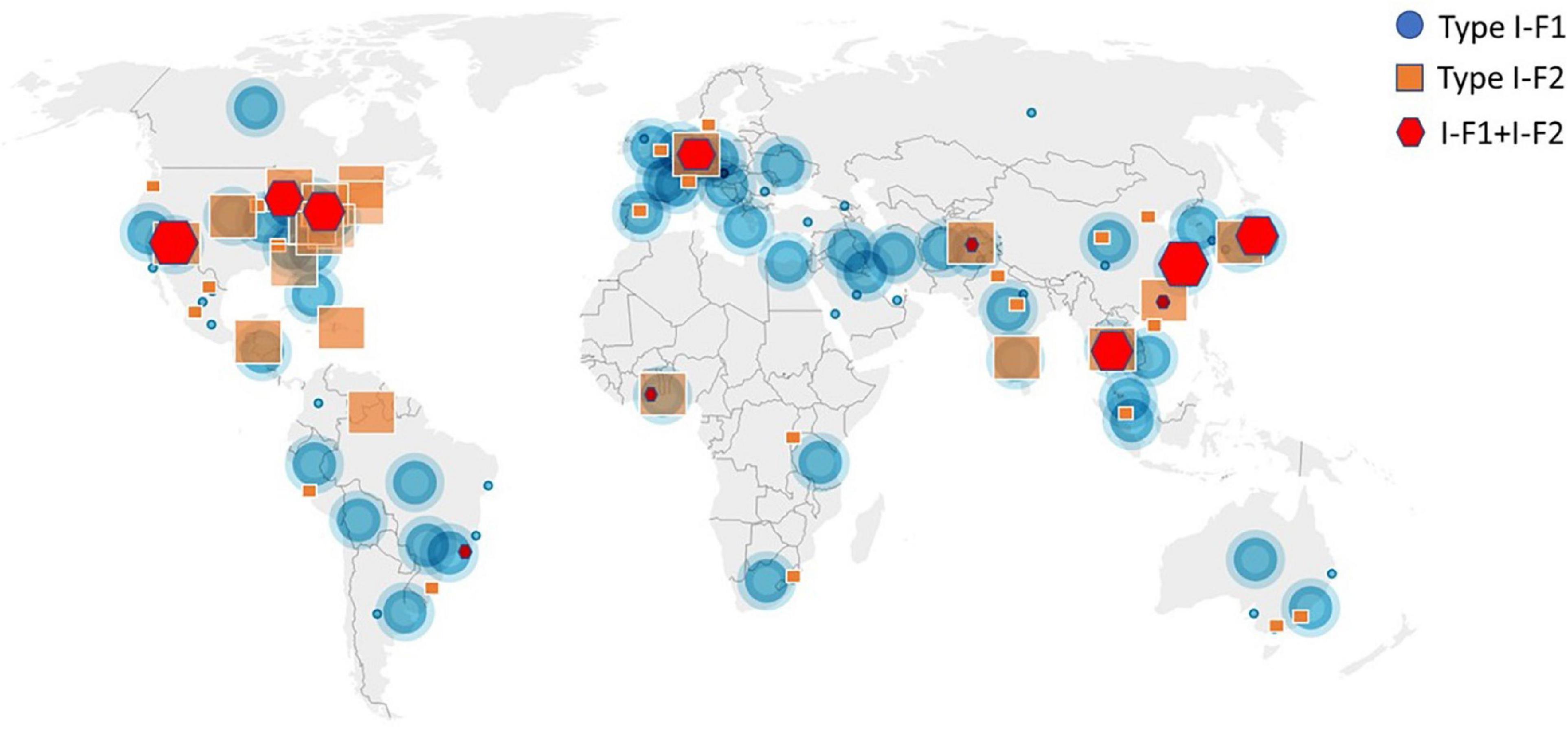
Figure 2. Geographical distribution of CRISPR-Cas (+) isolates across the world. Symbols are merged where two or more isolates coincide.
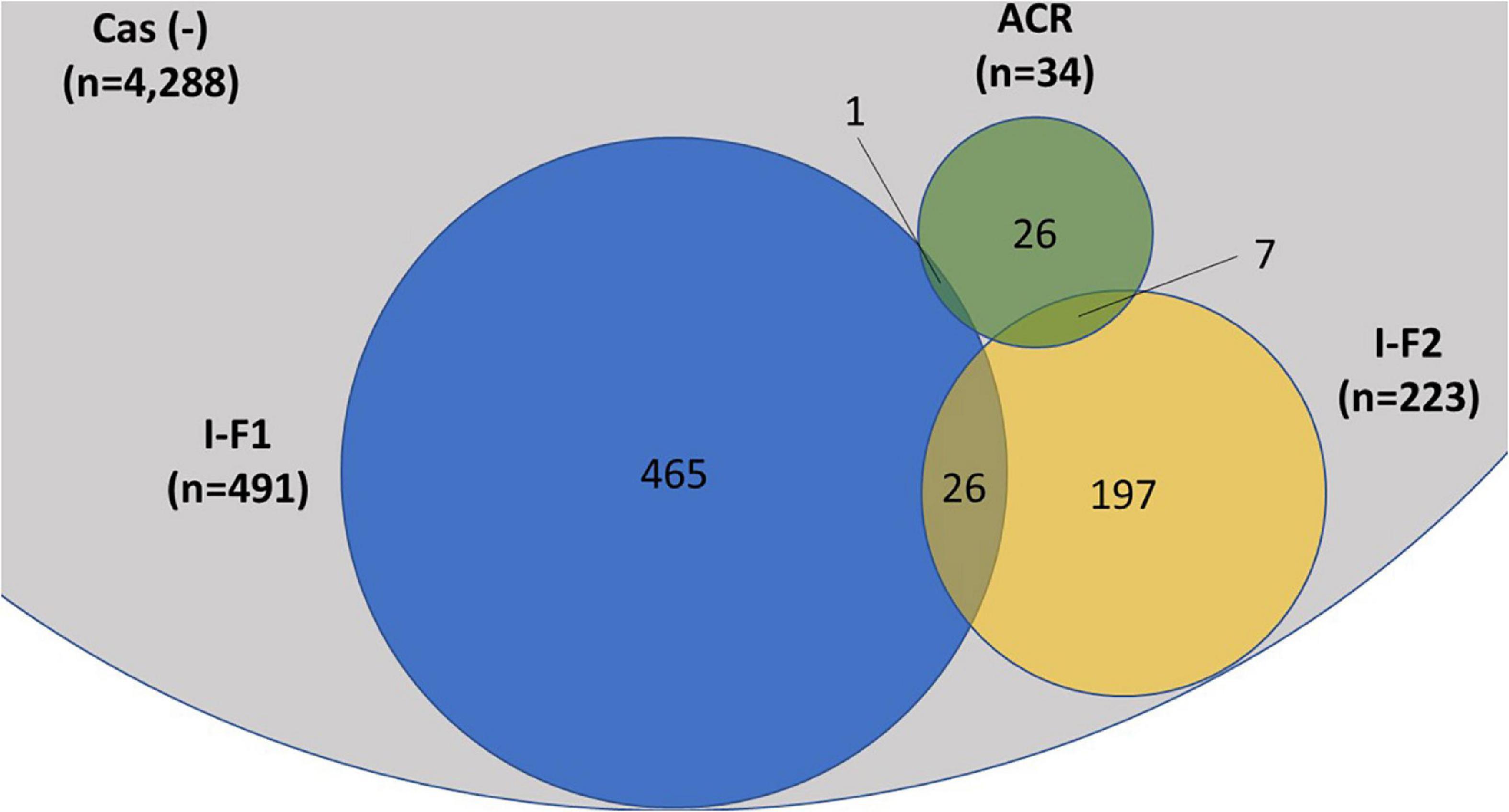
Figure 3. Distribution of CRISPR-Cas type I-F1, type I-F2, and anti-CRISPR (ACR) in the complete (n = 4,977) set of A. baumannii genomes. A single isolate was found to carry a type III-B system (data not shown). The proportions of degenerated systems (i.e., having either CRISPR or Cas cluster gene/s) are provided in Supplementary Table 2.
Recent classification based on the multiparametric analysis describes type I loci with cas3 as a signature gene and type I-F with fused cas3 and cas2 genes (Makarova et al., 2011, 2015, 2020; Koonin et al., 2017). On exploring the genomes with type I-F1 + F2 Cas gene clusters, we found that both the systems follow the same organization and features of CRISPR-Cas type I-F1 and I-F2 systems as visualized individually and are distantly (∼1.733 Mb) located in the genome (Figure 1). Interestingly, type I-F2 systems are associated with two CRISPR arrays where one is very well-adapted and spacer rich, compared to the other. In contrast, type I-F1 is only associated with a single CRISPR array. The average spacer size for arrays associated with both type I-F1 and F2 systems is ∼29 bp and belongs to the medium spacers (28–32 bp) category (Pourcel et al., 2020).
Under substantial antibiotic exposure, bacterial cells often suppress the function of the CRISPR-Cas system either by partial or complete loss of the CRISPR or Cas genes resulting in a degenerate system (Jiang et al., 2013). We found that 1.49% (74/4,977) isolates exhibited degenerated CRISPR-Cas systems, lacking either CRISPR (n = 15) or the complete set of Cas genes (n = 59) (Supplementary Table 2). We analyzed 545 plasmids from complete-level genome assemblies for the presence of the CRISPR-Cas systems and found that only 0.18% (1/545) plasmids carried a valid CRISPR array. This accounts for a very low proportion as compared to the average prevalence of 3.4% (546/15,938) across sequenced bacterial plasmids encoding the CRISPR-Cas system (Pourcel et al., 2020). None of the Cas cluster genes were found on the plasmid in A. baumannii.
A very low proportion of degenerated CRISPR-Cas systems and plasmids carrying CRISPR arrays suggest that these phenomena are rare but may occur in A. baumannii. However, a comparatively very low proportion of clinical isolates (∼15%) harboring CRISPR-Cas compared to environmental isolates (∼56%) suggests that antibiotics may exert selection pressure to lose out or selectively propagate isolates without CRISPR-Cas. Nevertheless, one can infer that the high prevalence of CRISPR-Cas among environmental isolates may be due to the abundance of phages in the environment and not the absence of antibiotics in the case of A. baumannii.
Multi-locus sequence typing relies on comparing the sequences of evolutionary conserved but polymorphism-harboring genes (Jolley and Maiden, 2010) and can be employed to compare the phylogenetic diversity among bacterial isolates (van Belkum et al., 2015). Sequence type of all A. baumannii isolates was determined using whole-genome sequence following the Pasture scheme and 4,841 isolates belonging to 314 different STs were found, 136 isolates did not belong to any defined ST. We observed that 60.63% (n = 3,018/4,977) isolates belonged to ST2 and were devoid of CRISPR-Cas except for 1 isolate which showed type I-F1. Analyzing the distribution of three classes of CRISPR-Cas system among STs, we observed that each class (type I-F1 or type I-F2) predominates within any particular ST with ST138 as an exception which showed the equivalent occurrence of both classes (Supplementary Table 3). Class I-F1 type was entirely observed in isolates with ST1 (n = 176/216), ST25 (n = 134/140), ST991 (n = 18/18), and ST20 (n = 9/10). Class I-F2 type was entirely observed in isolates with ST79 (n = 77/141), ST52 (n = 33/33), and ST16 (n = 10/25). However, type I-F1 + F2 were distributed in low frequencies across 12 different ST types, thereby not showing association toward any particular ST (Figure 4).
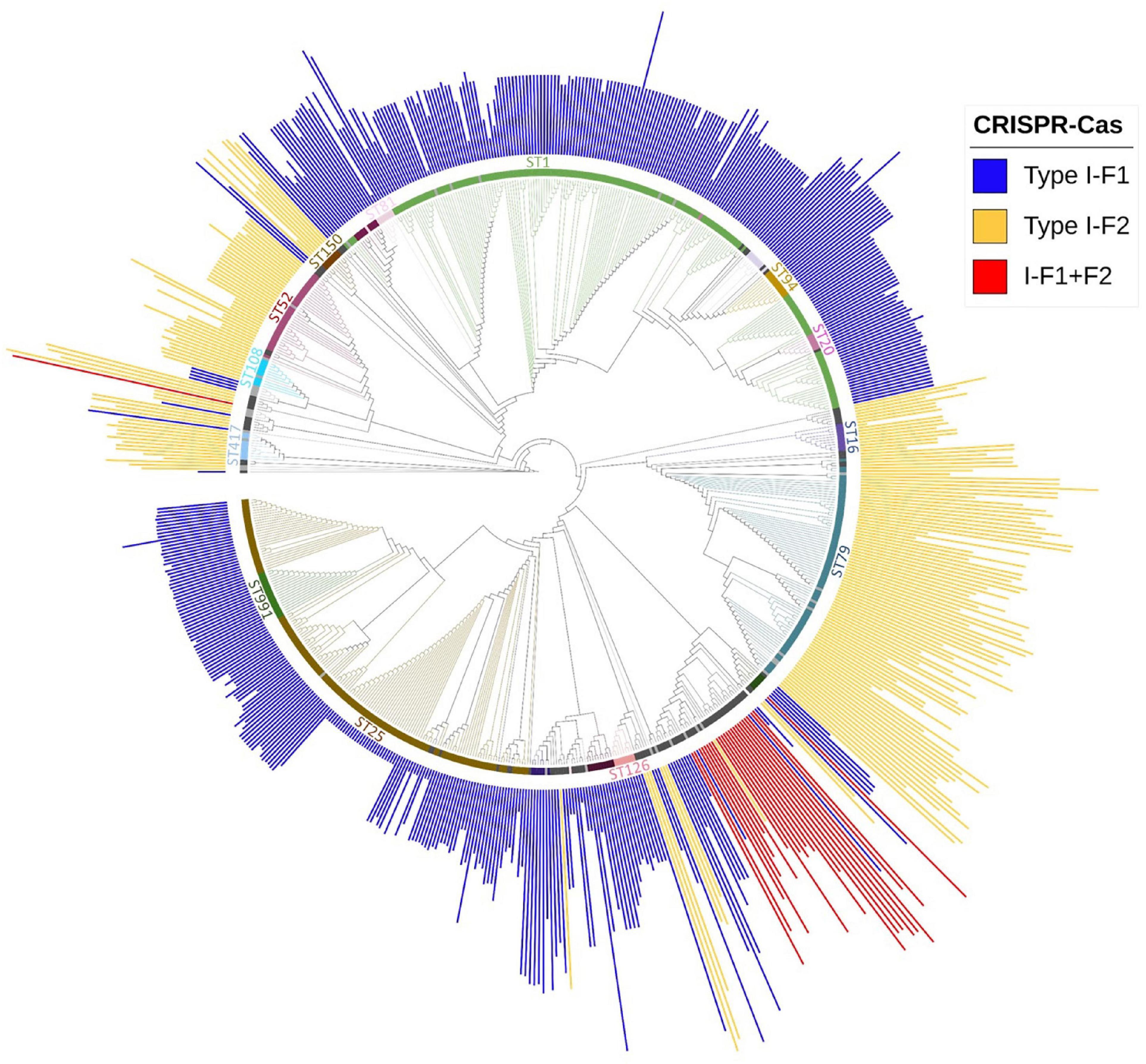
Figure 4. Phylogenetic distribution of different categories of CRISPR-Cas (+) isolates found in A. baumannii (n = 688). Major STs groups were labeled and are depicted with different colors, while lineages with ≤ 2 and ≤ 5 number of isolates with identical STs were colored with light gray and black, respectively. The height of the bars is proportionate to the total number of identified spacers in CRISPR array/s belonging to that isolate.
The presence of an active CRISPR-Cas system constrains the HGT and phage genome integration, which can limit genome expansion in bacteria and may result in comparatively smaller genome size (Ochman et al., 2000; Lerat et al., 2005; van Belkum et al., 2015; Wheatley and MacLean, 2021; Pursey et al., 2022). Nevertheless, we found that CRISPR-Cas (+) A. baumannii isolates were ∼48,982 ± 34,462 bp lengthier than CRISPR-Cas (–) isolates. These results are consistent with the previous study on A. baumannii (Pursey et al., 2022). However, categorical observations differentiate skewed data among different types of CRISPR-Cas systems. The isolates harboring only type I-F1 and only type I-F2 CRISPR-Cas systems have unusually larger genomes ∼71,461 ± 29,915 bp (p < 0.0001) and ∼21,621 ± 252,021 bp (p < 0.0115) than CRISPR-Cas (–) isolates, respectively. However, the genome size of isolates having a type I-F1 + F2 system was smallest (∼1,45,733 ± 23,750 bp smaller (p < 0.0001) in comparison with Cas (–) isolates) as compared to other classes (Figure 5A). Intra-ST analysis among prominent ST types harboring specific types of CRISPR-Cas systems confirmed the same trend (Figure 5B). The similarity in different isolates within an ST enables the identification of differential genomic determinants with relatively lower possibilities of discordant variables causing indeterminate effects of CRISPR-Cas systems (Wheatley and MacLean, 2021). Genome alignment of CRSIPR-Cas (+) and (–) isolates belonging to ST1 revealed the presence of phage, ICEs, and ARGs as the contributing factors for genome expansion, thus indicating the redundant function of CRISPR-Cas type I-F1 system (Figure 5C).
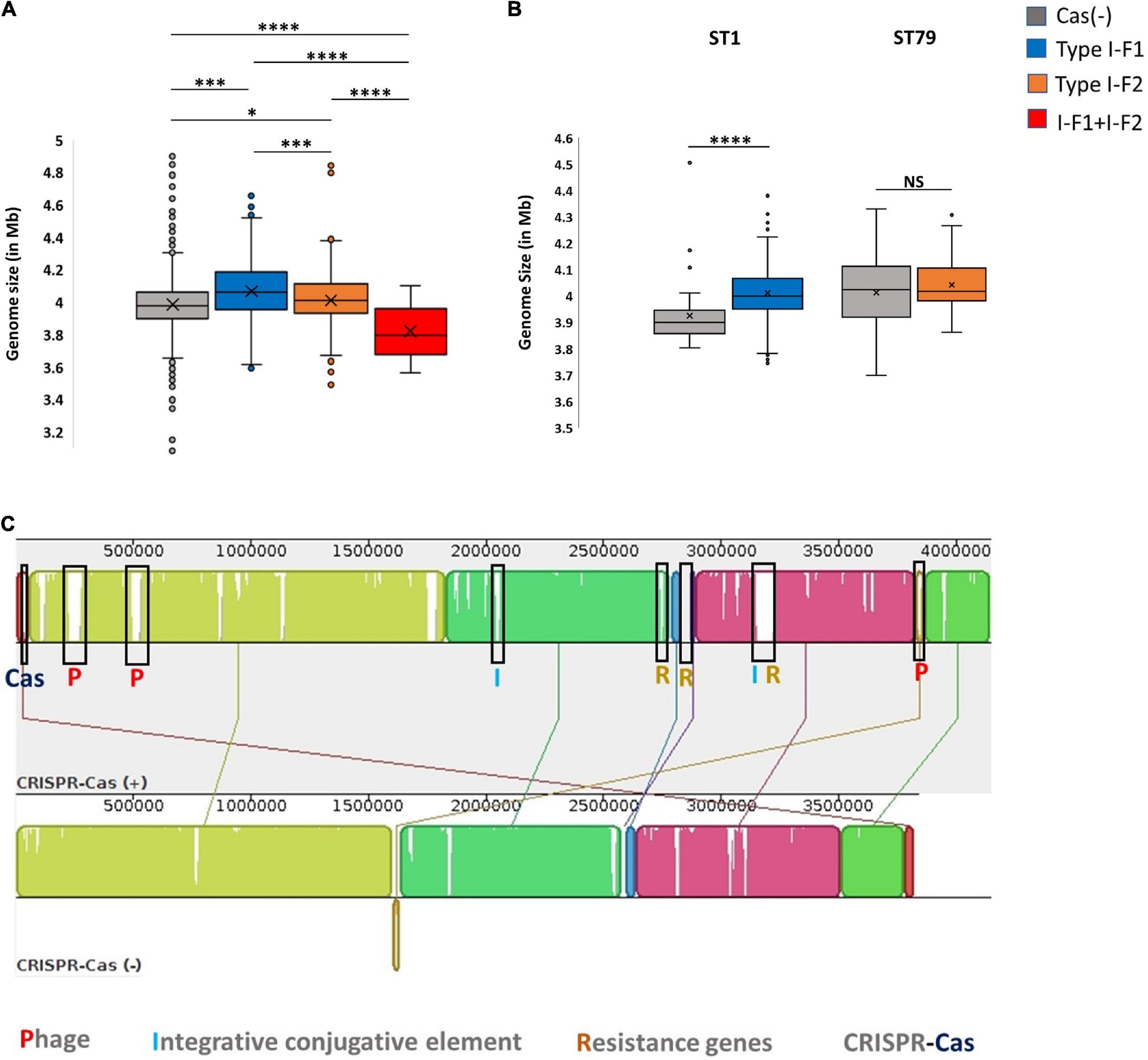
Figure 5. CRISPR-Cas and genome size. (A) Genome size comparison of CRISPR-Cas (–) and different categories of CRISPR-Cas (+) isolates. (B) Intra-ST analysis of genome size among CRISPR-Cas (–) and (+) isolates in ST1 and ST79. (C) Mauve alignment of CRISPR-Cas (+) and (–) ST1 isolates with extreme genome sizes. Where *(p < 0.05), ***(p < 0.001), ****(p < 0.0001) and NS (Not Significant).
Because the active CRISPR-Cas system restricts phages, the observed genome size data were correlated with the integrated phage genome. Average number of phages incorporated (average phage genome size) in each category, Cas (–), type I-F1, type I-F2, and type I-F1 + F2 are 3.17 ± 1.55 (41,613 ± 26,878 bp), 3.91 ± 1.88 (36,789 ± 33,141 bp), 3.91 ± 2.27 (44,580 ± 42,515 bp), and 2.80769 ± 2.1357 (27,823 ± 15,807 bp), respectively. The mean number of integrated phages in CRISPR-Cas (+) isolates of both ST1 (type I-F1) and ST79 (type I-F2) was significantly higher than CRISPR-Cas (–) isolates (type I-F1 –4.73 + 1.86 vs. 3.92 + 0.98; p = 0.005; type I-F2– 5.04 ± 1.88 and 3.87 ± 2.54; p = 0.000288). Intra-ST comparative analysis of type I-F1 + F2 in CRISPR-Cas (+) and CRISPR-Cas (–) was not performed due to the limited data set available for the associated ST types.
Although a clear decline in the average number of phages in isolates with type I-F1 + F2 was found (p = 0.0041), to confirm the activity of the CRISPR-Cas system in restricting the incorporation of phage sequences, we also substantiated the results with phage genome size incorporated in each class, which demarcated a reduction in the size of the phage genome incorporated into the A. baumannii isolates with type I-F1 + F2 (Figure 6). The unusually high genome size and integrated phage genomes in isolates with either CRISPR-Cas type I-F1 or I-F2 compared with Cas (-) isolates needs further in-depth studies. We did not find any significant difference in the number of integrated phages in type I-F1 or type I-F2; indeed, their co-existence was more efficient in limiting phage entry, as evidenced by a significantly low number of integrated phages. However, co-occurrence may be associated with the synergistic/additive activity and improving the CRISPR-Cas system’s efficacy but requires more deep, comprehensive, and experimental support.
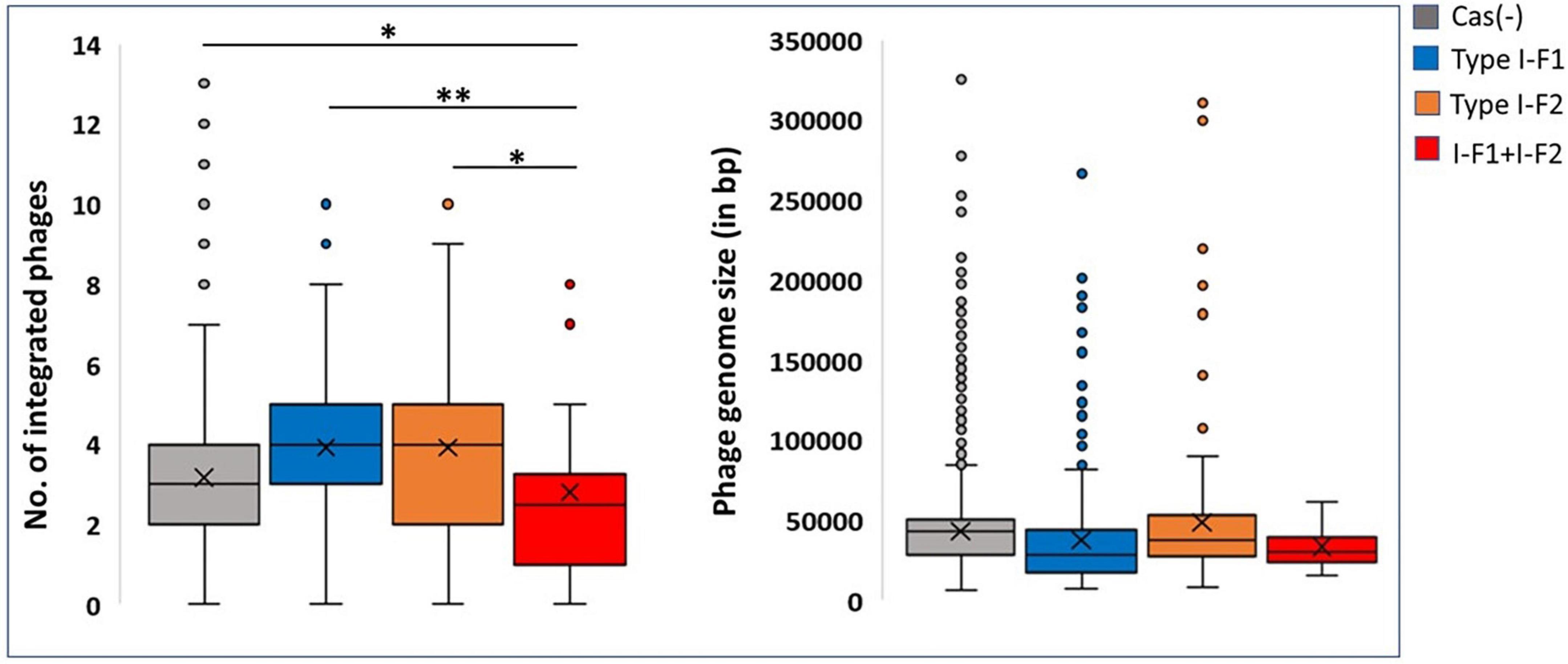
Figure 6. CRISPR and phages: Number of incorporated phages in genomes (left side) and the total genome size of the incorporated phage (right side). Where *(p < 0.05) and **(p < 0.01).
Antimicrobial resistance and virulence are important bacterial traits that help survive and infect the host. Bacteria develop antibiotic resistance either by acquiring resistance genes or through mutations in their genome. It is believed that the CRISPR-Cas system inhibits the acquisition of resistance genes but does not affect the emergence of mutations mediating antibiotic resistance. We investigated the impact of CRISPR-Cas affecting the acquisition of virulence genes and ARGs. In line with previous studies (Shehreen et al., 2019), our extensive analysis did not find an association of CRISPR-Cas (+) isolates with any particular antibiotic class (log frequency ratios ranged from –0.2 to + 0.2) (Supplementary Figure 1A). This suggests that CRISPR-Cas systems do not hinder the dissemination of resistance genes in A. baumannii. Similarly, we did not find an association of virulence genes with CRISPR-Cas (+) isolates. However, on analyzing the virulence gene frequencies among three CRISPR-Cas types, we found a strong negative association among isolates with type I-F1 + F2 for biofilm-associated protein (bap) and quorum-sensing genes, abaI and abaR, with a log frequency ratio of –0.8826, –1.0828, and –1.3767, respectively (Supplementary Figure 1B). The complete gene pool and their respective frequencies for each class of antibiotics and virulence genes found in A. baumannii are listed in Supplementary Table 4.
Overall, these results suggest that CRISPR-Cas in A. baumannii is not associated with limiting resistance and virulence gene uptake except among type I-F1 + F2 isolates for bap and quorum-sensing genes (abaI and abaR). Our results are consistent with previous studies showing a negative association among CRISPR-Cas (+) isolates for these genes with some modalities (Mangas et al., 2019; Leungtongkam et al., 2020; Tyumentseva et al., 2021).
CRISPR arrays consist of repeats and spacers where repeats were driven intrinsically, while spacers are proved to be acquired from bacteriophages and other mobile genetic MGEs that can provide memory-based immunity to the bacterium. We identified and counted incorporated spacers from each CRISPR-Cas (+) isolate within a valid (evidence level 4) CRISPR array/s. We found a significantly high number of mean spacers (n = 164.58 ± 46.41) per isolate in type I-F1 + F2 as compared to isolates with type I-F2 (n = 82.87 ± 36.14) and type I-F1 (n = 54.51 ± 26.27) (Figure 7A). Hyperactivity of type I-F2 CRISPR-Cas (+) isolates than type I-F1 in acquiring spacers may be correlated with the absence of csy1 gene (in type I-F2) involved in the formation of Csy complex that negatively regulates Cas1/2-3 complex which functions in adaptation of the CRISPR arrays (Rollins et al., 2017). While in isolates with type I-F1 + F2, a synergistic effect could explain the observed higher spacers per isolate.
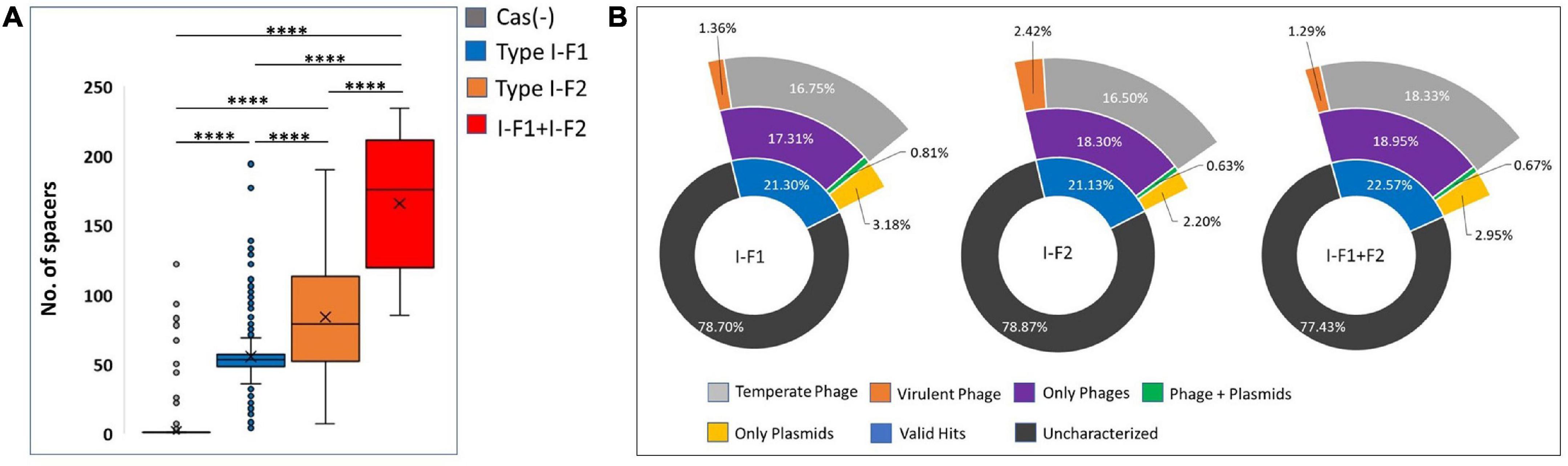
Figure 7. (A) Number of spacers incorporated in CRISPR array/s. (B) The predicted targets of unique spacers in CRISPR-Cas (+) genomes across different categories of isolates harboring CRISPR-Cas systems. Where ****(p < 0.0001).
Spacerome with unique spacers from each category, that is, type I-F1 + F2 (n = 1,338), I-F2 (n = 2,224), and I-F1 (n = 2,094), were clustered. The number of unique spacers was higher in isolates with type I-F1 + F2 (31.26%), compared to type I-F2 (13.6%) and type I-F1 (8.28%), however, the high number of unique spacers in a population of isolates with CRISPR-Cas type I-F1 + F2 could not be correlated with the higher number of unique spacers per isolate and may be due to small number of isolates belonging to different ST types.
The unique spacers from each defined category were assessed for their potential targets, namely, phages, ICEs, plasmids, virulence factors, and resistance genes. We found that only approximately 21.67% of spacers had valid target hits. Also, it was evident that a single spacer can have a target for an element category, that is, either phage or plasmids. Only a few (∼0.70%) spacers were found to target both phage and plasmids (Figure 7B). The highest proportion of spacers was predicted to target phages in each class. The targeted phage type was further classified based on temperate and virulent groups. Temperate phages are the most common targets for spacers in each category. The limited number of spacers targeting the virulent phages could be attributed to their low abundance (n = 5/99) (Figure 7B). In-depth analysis revealed that a single spacer could have multiple targets (a range of 1–37 targets for phages). Spacers that have more than one target against phages are comparatively high in isolates with type I-F1 + F2 (64%), followed by type I-F2 (60%) and type I-F1 (56%). No valid hit was found to target ICEs, resistance, or virulence gene against spacers sets. Maximum remaining spacers had no identifiable target and were designated as dark matter, representing an uncharacterized microbial element (Shmakov et al., 2020).
The presence of spacers with multiple targets reflects the effective management of spacers with remarkable plasticity in A. baumannii.
Broad-scale comparisons across the diversity of A. baumannii revealed the distribution (Figure 3) and presence of co-existing CRISPR-Cas systems associated with a higher number of spacers, smaller genome size, and reduced number of integrated phages. It is well reported that CRISPR-Cas systems provide bacteria with an edge against phages. However, it can also target MGEs and may be simply a by-product of the system. We did not find any spacer that can directly target resistance or virulence genes, whereas spacers targeting plasmids that can facilitate the horizontal transfer of resistance and virulence-related genes were observed. However, neither negative nor positive association in CRISPR-Cas (+) isolates with resistance and virulence genes were found, with few exceptions for bap, abaI, and abaR only in type I-F1 + F2 isolates. These contrasting results indicate the existence of cryptic mechanisms for regulating spacers that can target plasmids to acquire and maintain resistance and virulence genes without compromising the phage-based memory in A. baumannii. In silico data analysis suggested that the co-existence of CRISPR-Cas type I-F1 and F2 systems in A. baumannii imparts the hyperactivity against phages without affecting the presence of resistance genes that may significantly hinder the potential of phage-based therapies and the trade-off capabilities. Further research regarding novel treatment strategies should be driven considering the co-existence of CRISPR-Cas systems in A. baumannii.
The outcomes of this study correspond to the sequenced A. baumannii genomes, including scaffold-level assemblies available in the public domain assessed on 18 January 2021, and oversight newly added and un-sequenced A. baumannii population. Notably, the data are inclined toward clinical isolates due to under-represented environmental isolates. Our study shows that type I-F1 and I-F2 CRISPR-Cas systems in co-existence are distantly located; however, this distance may vary on incorporating more complete-level genome assemblies in the dataset. Understanding of complex and diverse CRISPR-Cas systems is rapidly evolving; our analysis does not account for unidentified types and subtypes of the CRISPR-Cas system and anti-CRISPR genes originating from phages. Hence the effect of anti-CRISPR genes is underestimated due to unknown anti-CRISPR proteins associated with the type I-F CRISPR-Cas system. Restricted phage entry as evidenced by a significantly low number of integrated phages in isolates with type I-F1 + F2 was determined in silico and requires experimental validation. This study identified maximum spacers with unknown targets, which depict the underrepresented or uncharacterized microbial community. Discovering such new elements may change the dynamics of targets corresponding to spacers being incorporated.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
GY and RS conceived the idea and designed the study. GY collected and analyzed the data and wrote the manuscript draft. RS reviewed and edited the final manuscript. Both authors contributed to the article and approved the submitted version.
This work received Intramural support from the ICMR-National Institute of Pathology, New Delhi.
GY was grateful to University Grants Commission (UGC), India, for providing the research fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.909886/full#supplementary-material
Baptista, P. V., Mccusker, M. P., Carvalho, A., Ferreira, D. A., Mohan, N. M., Martins, M., et al. (2018). Nano-strategies to fight multidrug resistant bacteria—“A Battle of the Titans”. Front. Microbiol. 9:1441. doi: 10.3389/fmicb.2018.01441
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Bassetti, M., Righi, E., Vena, A., Graziano, E., Russo, A., and Peghin, M. (2018). Risk stratification and treatment of ICU-acquired pneumonia caused by multidrug-resistant/extensively drug-resistant/pandrug-resistant bacteria. Curr. Opin. Crit. Care 24, 385–393. doi: 10.1097/MCC.0000000000000534
Bertels, F., Silander, O. K., Pachkov, M., Rainey, P. B., and Van Nimwegen, E. (2014). Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 31, 1077–1088. doi: 10.1093/molbev/msu088
Bikard, D., Hatoum-Aslan, A., Mucida, D., and Marraffini, L. A. (2012). CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microb. 12, 177–186. doi: 10.1016/j.chom.2012.06.003
Biswas, A., Staals, R. H., Morales, S. E., Fineran, P. C., and Brown, C. M. (2016). CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics 17:356. doi: 10.1186/s12864-016-2627-0
Bland, C., Ramsey, T. L., Sabree, F., Lowe, M., Brown, K., Kyrpides, N. C., et al. (2007). CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 8:209. doi: 10.1186/1471-2105-8-209
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Brooks, L., Kaze, M., and Sistrom, M. (2019). A curated, comprehensive database of plasmid sequences. Microbiol. Resour. Announc. 8, e1325–e1318. doi: 10.1128/MRA.01325-18
Brouns, S. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J., and Snijders, A. P. (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. doi: 10.1126/science.1159689
Burstein, D., Harrington, L. B., Strutt, S. C., Probst, A. J., Anantharaman, K., Thomas, B. C., et al. (2017). New CRISPR–Cas systems from uncultivated microbes. Nature 542, 237–241. doi: 10.1038/nature21059
Burstein, D., Sun, C. L., Brown, C. T., Sharon, I., Anantharaman, K., Probst, A. J., et al. (2016). Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun. 7:10613. doi: 10.1038/ncomms10613
Cady, K., White, A., Hammond, J., Abendroth, M., Karthikeyan, R., Lalitha, P., et al. (2011). Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 157:430. doi: 10.1099/mic.0.045732-0
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Carte, J., Christopher, R. T., Smith, J. T., Olson, S., Barrangou, R., Moineau, S., et al. (2014). The three major types of CRISPR-Cas systems function independently in CRISPR RNA biogenesis in S treptococcus thermophilus. Mol. Microbiol. 93, 98–112. doi: 10.1111/mmi.12644
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Deng, L., Garrett, R. A., Shah, S. A., Peng, X., and She, Q. (2013). A novel interference mechanism by a type IIIB CRISPR-Cmr module in S ulfolobus. Mol. Microbiol. 87, 1088–1099. doi: 10.1111/mmi.12152
Dong, C., Hao, G.-F., Hua, H.-L., Liu, S., Labena, A. A., Chai, G., et al. (2018). Anti-CRISPRdb: a comprehensive online resource for anti-CRISPR proteins. Nucleic Acids Res. 46, D393–D398.
Gao, N. L., Zhang, C., Zhang, Z., Hu, S., Lercher, M. J., Zhao, X.-M., et al. (2018). MVP: a microbe–phage interaction database. Nucleic Acids Res. 46, D700–D707. doi: 10.1093/nar/gkx1124
García-Martínez, J., Maldonado, R. D., Guzmán, N. M., and Mojica, F. J. (2018). The CRISPR conundrum: evolve and maybe die, or survive and risk stagnation. Microb. Cell 5:262. doi: 10.15698/mic2018.06.634
Haft, D. H., Selengut, J., Mongodin, E. F., and Nelson, K. E. (2005). A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 1:e60. doi: 10.1371/journal.pcbi.0010060
Hidalgo-Cantabrana, C., Goh, Y. J., and Barrangou, R. (2019). Characterization and repurposing of type I and type II CRISPR–Cas systems in bacteria. J. Mol. Biol. 431, 21–33. doi: 10.1016/j.jmb.2018.09.013
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Jiang, W., Maniv, I., Arain, F., Wang, Y., Levin, B. R., and Marraffini, L. A. (2013). Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 9:e1003844. doi: 10.1371/journal.pgen.1003844
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 11:595. doi: 10.1186/1471-2105-11-595
Joshi, P. R., Acharya, M., Kakshapati, T., Leungtongkam, U., Thummeepak, R., and Sitthisak, S. (2017). Co-existence of bla OXA-23 and bla NDM-1 genes of Acinetobacter baumannii isolated from Nepal: antimicrobial resistance and clinical significance. Antimicrob. Resist. Infect. Control 6:21. doi: 10.1186/s13756-017-0180-5
Koonin, E. V., and Makarova, K. S. (2017). Mobile genetic elements and evolution of CRISPR-Cas systems: all the way there and back. Genome Biol. Evol. 9, 2812–2825.
Koonin, E. V., Makarova, K. S., and Zhang, F. (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78. doi: 10.1093/gbe/evx192
Kumar, M., Sarma, D. K., Shubham, S., Kumawat, M., Verma, V., Nina, P. B., et al. (2021). Futuristic non-antibiotic therapies to combat antibiotic resistance: a review. Front. Microbiol. 12:609459. doi: 10.3389/fmicb.2021.609459
Lerat, E., Daubin, V., Ochman, H., and Moran, N. A. (2005). Evolutionary origins of genomic repertoires in bacteria. PLoS Biol. 3:e130. doi: 10.1371/journal.pbio.0030130
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Leungtongkam, U., Thummeepak, R., Kitti, T., Tasanapak, K., Wongwigkarn, J., and Styles, K. M. (2020). Genomic analysis reveals high virulence and antibiotic resistance amongst phage susceptible Acinetobacter baumannii. Sci. Rep. 10, 1–11. doi: 10.1038/s41598-020-73123-y
Leungtongkam, U., Thummeepak, R., Wongprachan, S., Thongsuk, P., Kitti, T., and Ketwong, K. (2018). Dissemination of bla OXA-23, bla OXA-24, bla OXA-58, and bla NDM-1 Genes of Acinetobacter baumannii Isolates from Four Tertiary Hospitals in Thailand. Microb. Drug Resist. 24, 55–62. doi: 10.1089/mdr.2016.0248
Li, W., O’neill, K. R., Haft, D. H., Dicuccio, M., Chetvernin, V., Badretdin, A., et al. (2021). RefSeq: expanding the prokaryotic genome annotation pipeline reach with protein family model curation. Nucleic Acids Res. 49, D1020–D1028. doi: 10.1093/nar/gkaa1105
Lin, D. M., Koskella, B., and Lin, H. C. (2017). Phage therapy: an alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Therapeut. 8:162. doi: 10.4292/wjgpt.v8.i3.162
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Liu, M., Li, X., Xie, Y., Bi, D., Sun, J., Li, J., et al. (2019). ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 47, D660–D665. doi: 10.1093/nar/gky1123
Majumdar, S., Zhao, P., Pfister, N. T., Compton, M., Olson, S., Glover, C. V., et al. (2015). Three CRISPR-Cas immune effector complexes co-exist in Pyrococcus furiosus. RNA 21, 1147–1158. doi: 10.1261/rna.049130.114
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Makarova, K. S., Wolf, Y. I., Iranzo, J., Shmakov, S. A., Alkhnbashi, O. S., Brouns, S. J., et al. (2020). Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. doi: 10.1038/s41579-019-0299-x
Mangas, E. L., Rubio, A., Álvarez-Marín, R., Labrador-Herrera, G., Pachón, J., and Pachón-Ibáñez, M. E. (2019). Pangenome of Acinetobacter baumannii uncovers two groups of genomes, one of them with genes involved in CRISPR/Cas defence systems associated with the absence of plasmids and exclusive genes for biofilm formation. Microb. Genomics 5:e000309. doi: 10.1099/mgen.0.000309
McGinnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25. doi: 10.1093/nar/gkh435
Merker, M., Tueffers, L., Vallier, M., Groth, E. E., Sonnenkalb, L., Unterweger, D., et al. (2020). Evolutionary approaches to combat antibiotic resistance: opportunities and challenges for precision medicine. Front. Immunol. 11:1938. doi: 10.3389/fimmu.2020.01938
Micoli, F., Bagnoli, F., Rappuoli, R., and Serruto, D. (2021). The role of vaccines in combatting antimicrobial resistance. Nat. Rev. Microbiol. 19, 287–302. doi: 10.1038/s41579-020-00506-3
Munoz-Price, L. S., and Weinstein, R. A. (2008). Acinetobacter infection. New Engl. J. Med. 358, 1271–1281. doi: 10.1056/NEJMra070741
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
O’Meara, D., and Nunney, L. (2019). A phylogenetic test of the role of CRISPR-Cas in limiting plasmid acquisition and prophage integration in bacteria. Plasmid 104:102418. doi: 10.1016/j.plasmid.2019.102418
Pawluk, A., Davidson, A. R., and Maxwell, K. L. (2018). Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 16, 12–17.
Pinilla-Redondo, R., Mayo-Muñoz, D., Russel, J., Garrett, R. A., Randau, L., Sørensen, S. J., et al. (2020). Type IV CRISPR–Cas systems are highly diverse and involved in competition between plasmids. Nucleic Acids Res. 48, 2000–2012. doi: 10.1093/nar/gkz1197
Pires, D. P., Costa, A. R., Pinto, G., Meneses, L., and Azeredo, J. (2020). Current challenges and future opportunities of phage therapy. FEMS Microbiol. Rev. 44, 684–700. doi: 10.1093/femsre/fuaa017
Pourcel, C., Salvignol, G., and Vergnaud, G. (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. doi: 10.1099/mic.0.27437-0
Pourcel, C., Touchon, M., Villeriot, N., Vernadet, J.-P., Couvin, D., Toffano-Nioche, C., et al. (2020). CRISPRCasdb a successor of CRISPRdb containing CRISPR arrays and cas genes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res. 48, D535–D544. doi: 10.1093/nar/gkz915
Pursey, E., Dimitriu, T., Paganelli, F. L., Westra, E. R., and Van Houte, S. (2022). CRISPR-Cas is associated with fewer antibiotic resistance genes in bacterial pathogens. Philos. Trans. R. Soc. B 377:20200464. doi: 10.1098/rstb.2020.0464
Pursey, E., Sünderhauf, D., Gaze, W. H., Westra, E. R., and Van Houte, S. (2018). CRISPR-Cas antimicrobials: challenges and future prospects. PLoS Pathogens 14:e1006990. doi: 10.1371/journal.ppat.1006990
Reis-Cunha, J. L., Bartholomeu, D. C., Manson, A. L., Earl, A. M., and Cerqueira, G. C. (2019). ProphET, prophage estimation tool: a standalone prophage sequence prediction tool with self-updating reference database. PLoS One 14:e0223364. doi: 10.1371/journal.pone.0223364
Rollins, M. F., Chowdhury, S., Carter, J., Golden, S. M., Wilkinson, R. A., and Bondy-Denomy, J. (2017). Cas1 and the Csy complex are opposing regulators of Cas2/3 nuclease activity. Proc. Natl Acad. Sci. U.S.A. 114, E5113–E5121. doi: 10.1073/pnas.1616395114
Shehreen, S., Chyou, T.-Y., Fineran, P. C., and Brown, C. M. (2019). Genome-wide correlation analysis suggests different roles of CRISPR-Cas systems in the acquisition of antibiotic resistance genes in diverse species. Philos. Trans. R. Soc. B 374:20180384. doi: 10.1098/rstb.2018.0384
Shmakov, S. A., Wolf, Y. I., Savitskaya, E., Severinov, K. V., and Koonin, E. V. (2020). Mapping CRISPR spaceromes reveals vast host-specific viromes of prokaryotes. Commun. Biol. 3, 1–9. doi: 10.1038/s42003-020-1014-1
Silas, S., Lucas-Elio, P., Jackson, S. A., Aroca-Crevillen, A., Hansen, L. L., Fineran, P. C., et al. (2017). Type III CRISPR-Cas systems can provide redundancy to counteract viral escape from type I systems. Elife 6:e27601.
Tal-Jasper, R., Katz, D. E., Amrami, N., Ravid, D., Avivi, D., Zaidenstein, R., et al. (2016). Clinical and epidemiological significance of carbapenem resistance in Acinetobacter baumannii infections. Antimicrob. Agents Chemother. 60, 3127–3131. doi: 10.1128/AAC.02656-15
Tynecki, P., Guziñski, A., Kazimierczak, J., Jadczuk, M., Dastych, J., and Onisko, A. (2020). PhageAI-bacteriophage life cycle recognition with machine learning and natural language processing. BioRxiv[preprint] doi: 10.1101/2020.07.11.198606
Tyumentseva, M., Mikhaylova, Y., Prelovskaya, A., Tyumentsev, A., Petrova, L., Fomina, V., et al. (2021). Genomic and phenotypic analysis of multidrug-resistant Acinetobacter baumannii clinical isolates carrying different types of CRISPR/Cas systems. Pathogens 10:205. doi: 10.3390/pathogens10020205
van Belkum, A., Soriaga, L. B., Lafave, M. C., Akella, S., Veyrieras, J.-B., Barbu, E. M., et al. (2015). Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. MBio 6, e1796–e1715. doi: 10.1128/mBio.01796-15
Watson, B. N., Staals, R. H., and Fineran, P. C. (2018). CRISPR-Cas-mediated phage resistance enhances horizontal gene transfer by transduction. MBio 9, e02406–e02417. doi: 10.1128/mBio.02406-17
Westra, E. R., Van Erp, P. B., Künne, T., Wong, S. P., Staals, R. H., Seegers, C. L., et al. (2012). CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell 46, 595–605. doi: 10.1016/j.molcel.2012.03.018
Wheatley, R. M., and MacLean, R. C. (2021). CRISPR-Cas systems restrict horizontal gene transfer in Pseudomonas aeruginosa. ISME J. 15, 1420–1433. doi: 10.1038/s41396-020-00860-3
World Health Organization (2017). Guidelines for the prevention and control of carbapenem-resistant Enterobacteriaceae, Acinetobacter baumannii and Pseudomonas aeruginosa in health care facilities. Geneva: World Health Organization.
Zankari, E., Allesøe, R., Joensen, K. G., Cavaco, L. M., Lund, O., and Aarestrup, F. M. (2017). PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 72, 2764–2768. doi: 10.1093/jac/dkx217
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644.
Zarrilli, R., Giannouli, M., Tomasone, F., Triassi, M., and Tsakris, A. (2009). Carbapenem resistance in Acinetobacter baumannii: the molecular epidemic features of an emerging problem in health care facilities. J. Infection Dev. Count. 3, 335–341. doi: 10.3855/jidc.240
Keywords: Acinetobacter baumannii, CRISPR-Cas, co-existence, type I-F1, type I-F2
Citation: Yadav G and Singh R (2022) In silico analysis reveals the co-existence of CRISPR-Cas type I-F1 and type I-F2 systems and its association with restricted phage invasion in Acinetobacter baumannii. Front. Microbiol. 13:909886. doi: 10.3389/fmicb.2022.909886
Received: 31 March 2022; Accepted: 25 July 2022;
Published: 17 August 2022.
Edited by:
Remy A. Bonnin, Université Paris-Saclay, FranceReviewed by:
Duolong Zhu, Baylor College of Medicine, United StatesCopyright © 2022 Yadav and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruchi Singh, cnVjaGlzcEBnbWFpbC5jb20=, cnVjaGlzaW5naC5uaXBAZ292Lmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.