 Yang Qu
Yang Qu Yingjie Sun
Yingjie Sun Zengqi Yang
Zengqi Yang Chan Ding
Chan Ding
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 04 July 2022
Sec. Virology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.889374
This article is part of the Research Topic Cell Organelle Exploitation by Viruses During Infection, Volume II View all 5 articles
Calcium, as a second intracellular messenger, participate in various physiological and biochemical processes, including cell growth and proliferation, energy metabolism, information transfer, cell death, and immune response. Ca2+ channels or pumps in plasma and organelle membranes and Ca2+-related proteins maintain Ca2+ homeostasis by regulating Ca2+ inflow, outflow and buffering to avoid any adverse effects caused by Ca2+ overload or depletion. Thus, Ca2+ signaling also provides a target for virus invasion, replication, proliferation and release. After hijacking the host cell, viruses exploit Ca2+ signaling to regulate apoptosis and resist host immunity to establish persistent infection. In this review, we discuss cellular Ca2+ signaling and channels, interaction of calcium-associated proteins with viruses, and host cell fate, as well as the role of Ca2+ in cell death and antiviral response during viral infection.
Calcium is an important trace element in animals and is an initiator and regulator of a variety of intra- and extracellular pathways involved in several physiological activities, including heartbeat, muscle contraction, brain memory storage, and neurosecretory system signaling. Ca2+ are involved in various intracellular physiological and biochemical processes as an intracellular second messenger, maintaining cell growth and proliferation, energy metabolism and information transfer (Berridge et al., 1998; Contreras et al., 2010). Under normal physiological conditions, intracellular Ca2+ homeostasis is vital for cells. Different concentrations of Ca2+ exist between organelles to form a large Ca2+ gradient. To maintain normal cellular activities, Ca2+ present in each cellular compartment use special channels and pumps to maintain dynamic equilibrium through influx, efflux, buffering and storage. The endoplasmic reticulum (ER) acts as a cellular Ca2+ store to regulate cellular Ca2+ homeostasis. The mitochondria are Ca2+-buffering organelles that maintain Ca2+ homeostasis by absorbing and releasing Ca2+ to establish contacts with the cytoplasm and other organelles in response to various intracellular signals (Tang et al., 2015; Panda et al., 2021).
When cells are stimulated by exogenous factors or endogenous disruption of structural function, an imbalance in Ca2+ homeostasis is inevitably induced and accompanied by an elevated intracellular Ca2+ concentration (Pinton et al., 2008). This pathological increase in Ca2+ arises from elevated inward flow from the extracellular environment and massive release of intracellular Ca2+ stores. Normal transient stimuli cause an increase in Ca2+ concentration that enhances cellular metabolism and promotes ATP production, whereas large and persistent calcium overload causes ER and mitochondrial stress, activating intracellular enzyme cascades, and sequentially triggering the death process (Pinton et al., 2008; Di Benedetto et al., 2013).
Viruses are noncellular organisms that parasitize living cells and replicate using the material and energy of host cells. After hijacking the host cell, viruses rapidly exploit various host cell systems. Viruses disrupt intracellular Ca2+ homeostasis by adapting or inhibiting Ca2+ signaling pathways and other Ca2+-dependent processes, creating favorable conditions for maximum utilization of host cell resources and replication of progeny viruses (Zhou et al., 2009; Panda et al., 2021). Ca2+ plays a role in almost every step of the viral replication cycle, includes virus entry, protein expression and modification, and virion maturation and release. The interaction between viruses and Ca2+ falls into three main categories: (1) viral proteins directly or indirectly disrupt intracellular Ca2+ homeostasis by modulating calcium channels and pumps or host membrane permeability; (2) Ca2+-regulated proteins or Ca2+-dependent pathways are involved in regulation of the virus life cycle; and (3) viral proteins bind directly to Ca2+ to hijack the host and destroy the integrity of cellular structure and function (Zhou et al., 2009).
What is the consequence of Ca2+ alterations in the life cycle of host cells during viral infection? First, the moderate increase in Ca2+ concentration caused by viral infection induces Ca2+-dependent enzymatic processes or activation of Ca2+-sensitive transcription factors to promote viral replication and persistent infection (Bergqvist and Rice, 2001; Bergqvist et al., 2003; Zhou et al., 2009). Second, Ca2+ plays a crucial role in the initiation and effectuation of cell death. Various stress injuries caused by viral infections induce Ca2+ overload in mitochondria, leading to mitochondrial membrane collapse, energy metabolism disorders, and ultimately cell death, such as necrosis, apoptosis, and autophagic cell death (Raffaello et al., 2016; Bravo-Sagua et al., 2017). At the same time, the host also mobilizes Ca2+ signaling to initiate an antiviral response to resist virus invasion (Crabtree and Clipstone, 1994; Tano and Vazquez, 2011; Mathavarajah et al., 2019). The interaction between Ca2+ signals and different pattern recognition receptor (PRR) signals resists exogenous pathogenic challenges and endogenous danger signals (Kong et al., 2021). This review highlights viral disruption of cellular Ca2+ signaling networks by interacting with host calcium pathways to promote self-replication and persistent infection, and the role of Ca2+ signaling networks in regulating cell death and antiviral responses.
In the normal state, the extracellular concentration of Ca2+ is up to the millimolar level. The cytoplasmic Ca2+ concentration is maintained at >100 nM, while the concentration in the ER or sarcoplasmic reticulum (SR), as the largest intracellular Ca2+ storage organelle, is several hundred micromolar (Berridge et al., 2003; Rizzuto et al., 2012; Zampese and Pizzo, 2012; Raffaello et al., 2016). Viruses take advantage of the large cellular Ca2+ concentration in different organelles to regulate cellular calcium signaling through promoting ATP synthesis, accelerating some Ca2+-dependent enzymatic processes and upregulating Ca2+-sensitive transcriptional factors. Viruses hijack and rely on the Ca2+ signaling networks to facilitate their penetration, replication, assembly and export to establish sustained infection.
As the first barrier against viruses, the plasma membrane has a variety of ion channels involved in the exchange of substances inside and outside the cytoplasm. Cellular maintenance of Ca2+ homeostasis requires regulation of calcium channels and pumps on the plasma membrane. During their life cycle, viruses utilize various calcium channels and pumps of the host to resist the membrane barriers to create favorable conditions for themselves. Host Ca2+ channels [voltage-gated calcium channels (VGCCs), receptor-operated calcium channels (ROCs), and store-operated calcium channels (SOCs)] mediate movement of Ca2+ across the plasma membrane and entry of Ca2+ from the extracellular medium, while the plasma membrane Ca2+-ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX) extrude Ca2+ from the cell (Zhou et al., 2009; Figure 1). In this section, we discuss the significance of cellular Ca2+ signaling, channels and pumps in the viral life cycle during virus-host conflict.
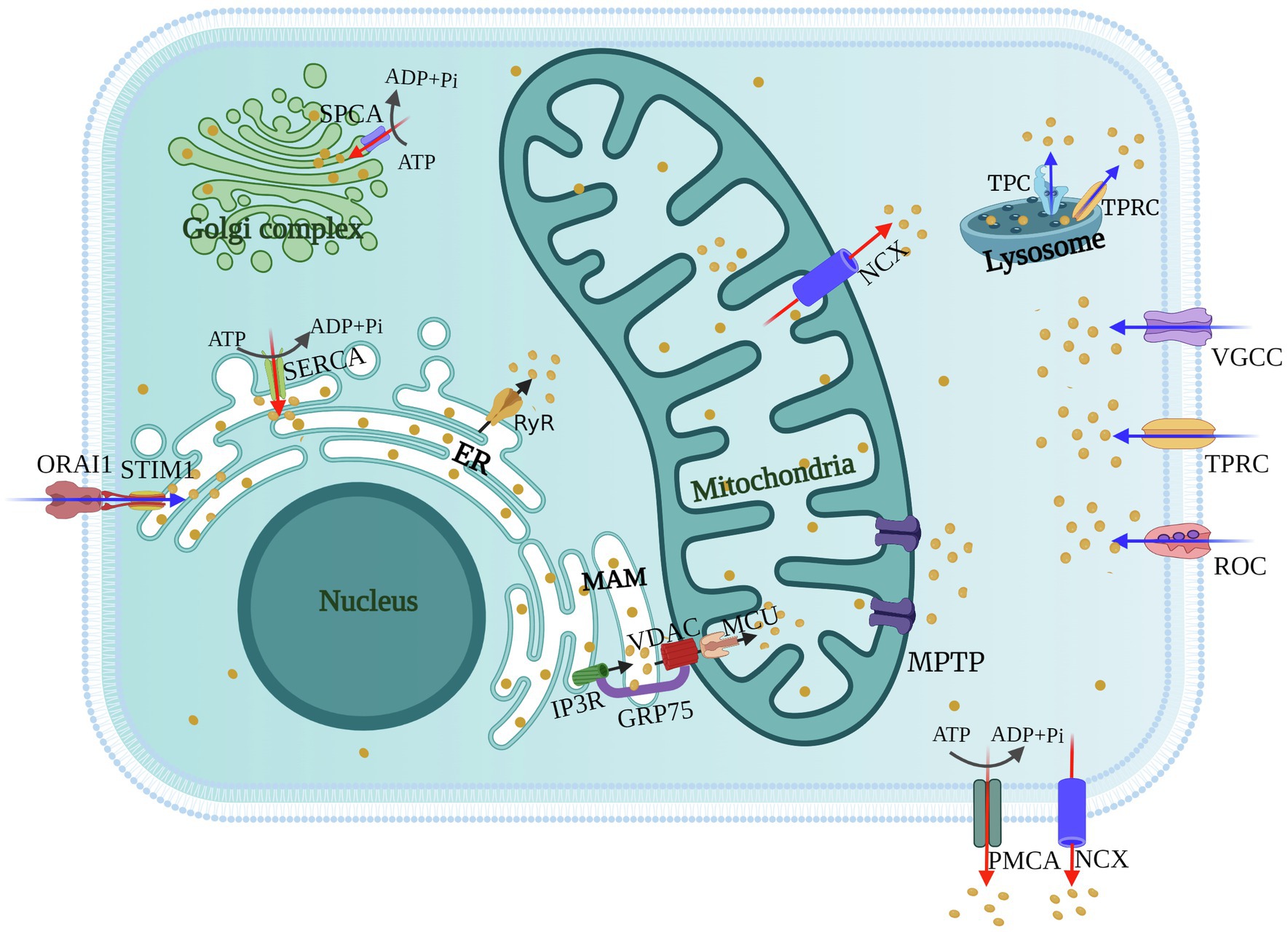
Figure 1. Schematics of cellular calcium channels and pumps. Calcium channels [voltage-gated calcium channels (VGCCs), receptor-operated calcium channels (ROCs), store-operated Ca2+ channels (SOCs), and transient receptor potential (TRP) channels] mediate the entry of Ca2+ from the extracellular environment or the release of Ca2+ from the Lysosome (blue arrows). The inositol trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) on the ER or Golgi regulate the release of Ca2+ from intracellular stores (black arrows). Ca2+ is transported between the ER and mitochondria via mitochondrial-associated membranes (MAMs), and mitochondrial uptake of Ca2+ is through voltage-dependent anion channels (VDACs) and mitochondrial calcium uniporters (MCUs). Calcium pumps [SR Ca2+-ATPase (SERCA), secretory pathway Ca2+-ATPase (SPCA), and plasma membrane Ca2+-ATPase (PMCA)] and the Na+/Ca2+ exchanger (NCX) transport Ca2+ from the cytosol to extracellular environment or intracellular stores (red arrows).
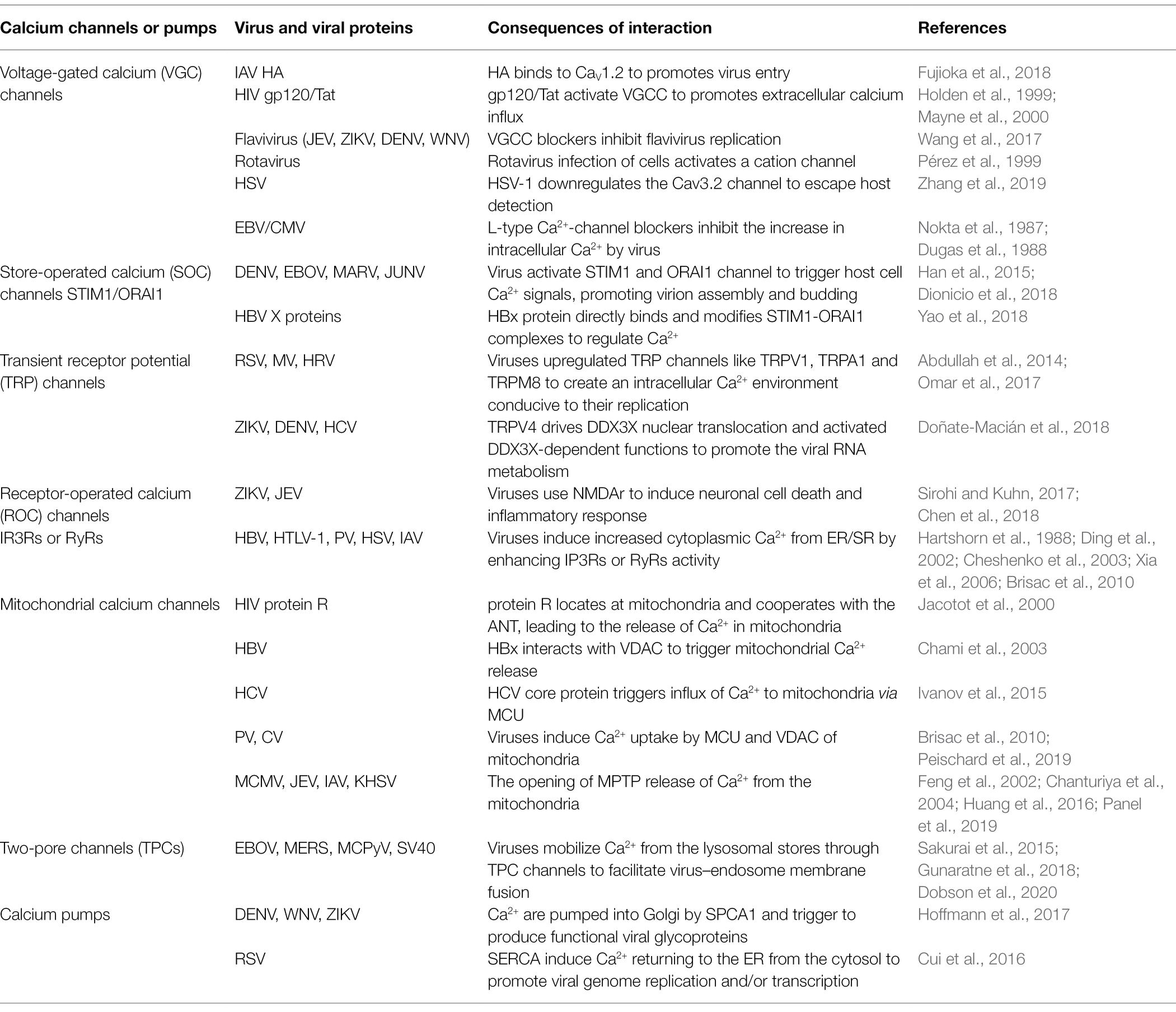
Table 1. Ca2+ channels and pumps used as targets by viruses.
Adsorption, penetration and uncoating are the first stage of viral replication. Viruses encode multiple proteins to manipulate the plasma membrane Ca2+ channels [VGCCs and two-pore channels (TPCs)] involved in regulating cellular Ca2+ uptake to increase intracellular Ca2+, thereby facilitating viral entry and replication. For example, human immunodeficiency virus type 1 (HIV-1) gp120 and Tat protein, and porcine rotavirus (RV), activate VGCCs to increase the levels of intracellular Ca2+ (Holden et al., 1999; Pérez et al., 1999; Mayne et al., 2000). The glycoprotein hemagglutinin (HA) of Alphainfluenzavirus influenzae (IAV) binds to voltage-dependent Ca2+ channel Cav1.2, triggering intracellular Ca2+ increase, and activates endocytosis to gain entry into cells (Fujioka et al., 2013, 2018; Figure 2A). Human alphaherpesvirus 1 (HSV)-1 infection results in a significant decrease in protein expression of Cav3.2 T-type Ca2+ channel subunit to escape detection by host cells (Zhang et al., 2019). Some VGCC blockers inhibit viral replication by inhibiting intracellular Ca2+ increase, such as Human gammaherpesvirus 4 (EBV), Human betaherpesvirus 5 (CMV), and flaviviruses [such as Japanese encephalitis virus (JEV), Zika virus (ZIKV), dengue virus (DENV) and West Nile virus (WNV)] (Nokta et al., 1987; Dugas et al., 1988; Wang et al., 2017). Intracellular Ca2+ oscillations are the trigger for viral penetration or uncoating (Baravalle et al., 2004; Danta, 2020). For porcine rotavirus, the critical step for uncoating and membrane permeabilization is a decrease in Ca2+ concentration of cytosol accompanying dissociation of viral Ca2+-stabilized proteins (Chemello et al., 2002; Salgado et al., 2018). The structure of some viruses contains a flexible surface loop that binds divalent Ca2+, and have been found to be essential for virus infectivity (Simpson et al., 2000).
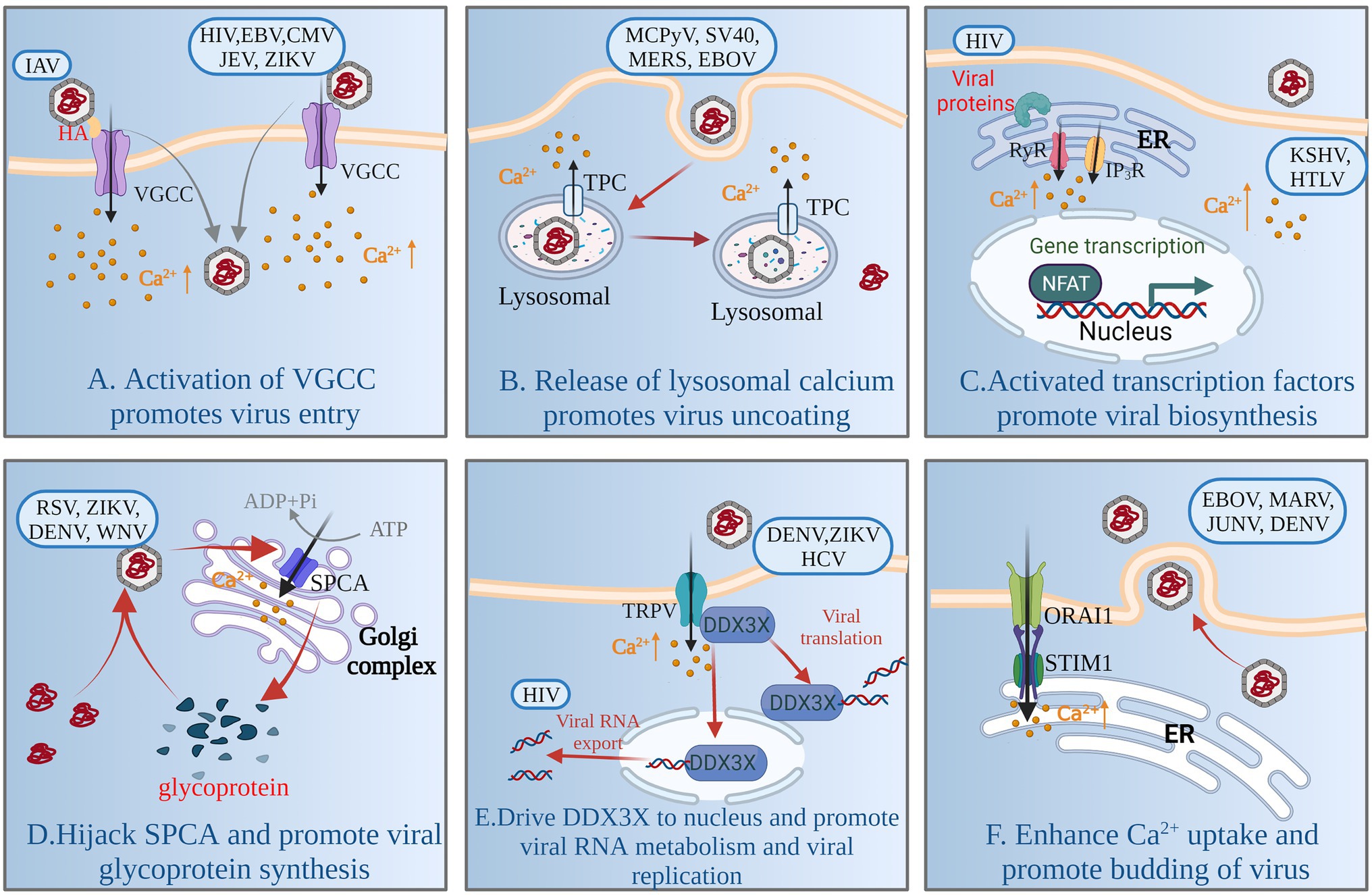
Figure 2. Model of Ca2+ signaling network involved in virus replication. By activating VGCC, the virus induces influx of extracellular Ca2+ and promotes adsorption and entry of virions (A). Viruses rely on TPC activity to mobilize Ca2+ from lysosomes and to degrade viral capsids through the lysosomal network (B). Viral infection increases cytoplasmic Ca2+, promotes activation of calcium-sensitive transcription factors (NFAT) and coactivators (P300), and induces viral RNA transcription and establishment of persistent infection (C). Viruses enhance SPCA and pump Ca2+ into the Golgi complex to promote synthesis of functional viral glycoproteins (D). Viruses activate TRPV-mediated Ca2+ influx and drive DDX3X nuclear translocation, promoting viral RNA metabolism and viral replication (E). Viruses activate the SOCE channel or promote interaction between STIM1 and ORAI1, enhancing cellular Ca2+ uptake and promoting budding of mature virus particles (F). For a complete list of definitions, see Table 1.
In the 1990s, the role of the endolysosomal system for calcium storage was discovered (Genazzani and Galione, 1996). The calcium exchange and Ca2+-mediated functional coupling also exists at the lysosome-ER interface (Raffaello et al., 2016). The mucolipin family of transient receptor potential (TRPML) channels and TPCs are involved in the release of Ca2+ from lysosomes (Patel et al., 2011; Figure 1). Many viruses mobilize Ca2+ from the endolysosomal stores through TPCs, and some viruses are dependent on the activity of TPCs to enable capsid entry into cells using endolysosomal networks, such as Alphapolyomavirus quintihominis (MCPyV), Betapolyomavirus macacae (SV40), Middle East respiratory syndrome coronavirus (MERS-CoV) and Zaire Ebola virus (EBOV; Sakurai et al., 2015; Gunaratne et al., 2018; Dobson et al., 2020; Figure 2B).
Viral biosynthesis includes mRNA transcription, and protein and DNA or RNA synthesis and metabolism, in which Ca2+-signaling networks play an important role. Some viruses or viral proteins are involved in increasing cytoplasmic Ca2+, leading to activation of the Ca2+-sensitive nuclear factor of activated T cells (NFAT) to support viral RNA transcription and establish persistent infection (Figure 2C). For example, HIV accessory protein Nef cooperates with Vpr to trigger release of ER Ca2+ that leads to activation of NFAT (Kinoshita et al., 1997; Lahti et al., 2003). Primate T-lymphotropic virus 1 (HTLV)-1 regulatory and accessory genes p12I activates NAFT (Ding et al., 2002), and upregulates expression of another Ca2+-sensitive transcription factor, P300 (Nair et al., 2006). Human gammaherpesvirus 8 (KHSV) K1 protein, is a transmembrane glycoprotein, increased cellular tyrosine phosphorylation and intracellular Ca2+ mobilization, activating NFAT and AP-1 transcription factor and producing inflammatory cytokines to promote viral dissemination (Lee et al., 2005). Furthermore, intracellular calcium triggers calcineurin-dependent signal transduction resulting in reactivation of latent KHSV infection (Zoeteweij et al., 2001).
Viruses directly activate calcium channels and pumps on some organelles to promote viral protein synthesis and modification. There are reports that glycoproteins of Paramyxoviridae, Flaviviridae and Togaviridae families fail to mature in SPCA1-deficient cells, Ca2+ is pumped into the Golgi by SPCA1, which triggers synthesis of functional viral glycoproteins that are essential for viral spread (Hoffmann et al., 2017; Figure 2D). Some respiratory viruses, such as Human orthopneumovirus (RSV), Measles morbillivirus (MV) and Human rhinovirus A, promote upregulation of TRP channels such as TRPV1, TRPA1 and TRPM8, and use TRP channels to create an intracellular Ca2+ environment conducive to their replication (Abdullah et al., 2014; Omar et al., 2017). DENV, hepacivirus C (HCV) and ZIKV or the purified viral envelope protein activate TRPV4-mediated Ca2+ influx that drives DDX3X nuclear translocation to promote viral replication; what’s more, TRPV4-DDX3X interaction regulates the nuclear export and translation of unspliced HIV-1genomic RNA (gRNA) and viral RNA metabolism (Doñate-Macián et al., 2018; Figure 2E).
Viral proteins can also bind directly to Ca2+ to promote viral biosynthesis. For example, Bluetongue virus (BTV) nonstructural phosphoprotein NS2 is a dedicated Ca2+-binding protein that significantly enhances NS2 phosphorylation, triggering viral inclusion body formation and facilitating viral replication and assembly (Rahman et al., 2020). High levels of cytosolic Ca2+ facilitates hepatitis B virus (HBV) core assembly, which is regulated by the viral multifunctional regulatory protein HBx (Choi et al., 2005). High Ca2+ levels promote Ca2+-dependent activation of proline-rich tyrosine kinase 2 and focal adhesion kinase; and induce NFAT expression, which supports HBV reverse transcription and DNA replication (Lara-Pezzi et al., 1998).
Most nonenveloped viruses release virus particles when the host cell is completely lysed, while enveloped viruses release virus particles by budding, which is accompanied by activation of Ca2+ channels and mobilization. SOCs mediated entry of Ca2+ is activated mainly by the membrane Ca2+ release-activated Ca2+ modulator 1(ORAI1) on the plasma membrane and stromal interaction molecule (STIM1) on the ER, which are stimulated by the depletion of internal Ca2+ stores. The budding process of enveloped viruses depends on the host Ca2+ signal mediated by SOCs (STIM1/ORAI1). Some viruses activate SOCs or depend on STIM1 and ORAI1 interaction to enhance cellular Ca2+ uptake for the budding of mature virus particles. Such viruses include DENV, EBOV, Marburgvirus (MARV) and Argentinian mammarenavirus (JUNV; Han et al., 2015; Dionicio et al., 2018; Figure 2F).
As previously mentioned, viral infection of cells almost inevitably leads to the production of pathological Ca2+ signals; the main source of which is increased extracellular entry and intracellular storage release. Mitochondria, as the center of cell survival and metabolism and buffer of Ca2+ signaling, participate in regulating energy synthesis, cell death, and other processes that determine cell life and death. Pathological Ca2+ accumulation and mitochondrial Ca2+ overload trigger the cell death process, including apoptosis or programmed cell death, necrosis, autophagic cell death and anoikis (Orrenius et al., 2003, 2015). Different states of abnormal Ca2+ homeostasis have different effects on cellular functions; low and transient stimulation enhances cellular metabolism and promotes ATP production, while large and continuous Ca2+ accumulation causes ER and mitochondrial stress, resulting in persistent mitochondrial damage and cell death (Pinton et al., 2008; Figure 3). Here, we focus on virus-induced mitochondrial Ca2+−overload-mediated apoptosis.
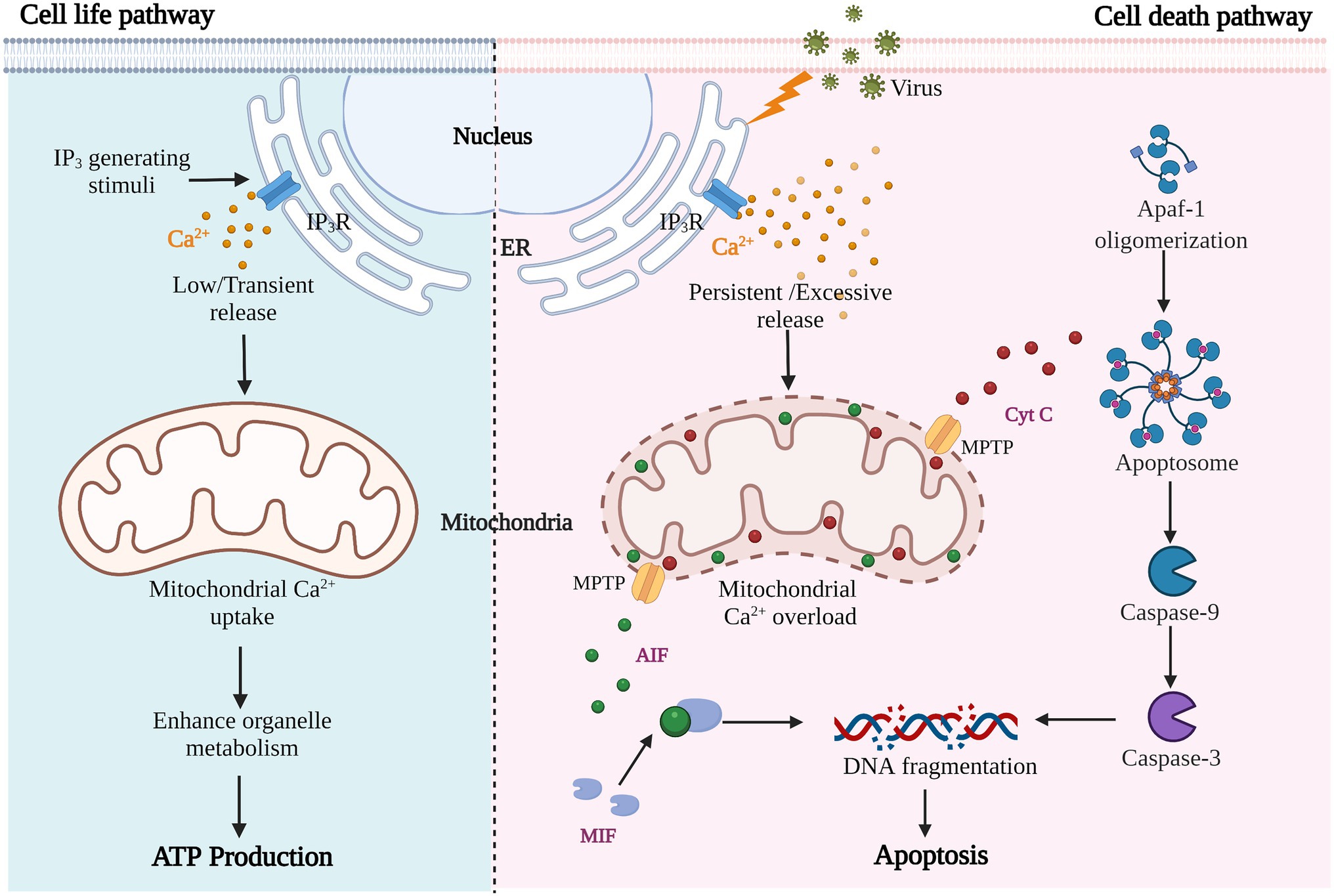
Figure 3. Results of cell metabolism or apoptosis induced by different Ca2+-related stimuli.
Apoptosis is considered to be a form of cell suicide, regulated by its own process under physiological or pathological conditions. The morphological manifestations of apoptosis are cell shrinkage, nuclear fragmentation, chromatin condensation, and formation of apoptotic bodies. Ca2+ is an important signaling molecule that regulates apoptosis during viral infection. On the one hand, viral infection induces increased extracellular Ca2+ influx and Ca2+ release from intracellular storage, and the continuously increased Ca2+ initiates apoptosis. On the other hand, the release of Ca2+ from storage disrupts the stability of intracellular structure, and many key components of the apoptotic system are activated. These two forms often coexist in virus-induced apoptosis.
Mitochondria, as the core organelles of apoptosis, are hijacked and utilized by viruses. Viruses trigger apoptosis by increasing mitochondrial Ca2+ uptake, enhancing mitochondrial membrane permeability and promoting release of apoptotic factors. Cytochrome (Cyt) C, as an activator of the caspase family, is required for caspase-dependent apoptosis. It is released into the cytoplasm and binds to apoptotic protease activating factor (Apaf)-1 to form apoptosomes, triggering the caspase cascade via caspase-9 activation (Figure 3). For example, both HBV and HCV are hepatotropic viruses that cause chronic liver disease and hepatocellular carcinoma (Scrima et al., 2018), and they promote Ca2+ uptake in mitochondria and lead to reactive oxygen species (ROS) production and apoptosis (Li et al., 2007). During HBV infection, HBx protein interacts with VDACs to trigger the release of Cyt C from the mitochondria, which triggers apoptosis. Some inhibitors of Ca2+ channels can down-regulate the proliferation of virus and avoid the occurrence of apoptosis of host cells (Chami et al., 2003; Xia et al., 2006). In the same way, enteroviral infections cause high mitochondrial Ca2+ overload, mitochondrial dysfunction, and apoptosis. When treated with the BAPTA-AM, a Ca2+ chelating agent, viral replication was also inhibited along with the alleviation of apoptosis (Brisac et al., 2010; Peischard et al., 2019). Besides, some viral proteins, such as IAV PB1-F2, induce permeabilization and destabilization of mitochondrial membranes via changes in Ca2+ homeostasis, leading to macromolecular leakage and apoptosis (Chanturiya et al., 2004). Whereas HCV NS5A protein promotes IP3R degradation, inhibiting virus-induced apoptosis and establishing chronic infection (Kuchay et al., 2018). In this process, apoptosis appears to be a host suicide defense mechanism to prevent spreading of virus.
Another promoter of cell death is apoptosis-inducing factor (AIF), which mediates the regulation of caspase-independent apoptosis (Figure 3). AIF is a mitochondrial oxidoreductase with a molecular weight of about 62 kDa anchored to the inner mitochondrial membrane (IMM) in the vicinity of Complex I. when mitochondria are damaged and the mitochondrial permeability transition pore (MPTP) is open, activated AIF is released (Susin et al., 1996). Calpain is a calcium-dependent intracellular cysteine protease that cleaves mitochondrial AIF to promote AIF activation (Cao et al., 2007; Figure 4B). The released AIF recruits macrophage migration inhibitory factor (MIF) to the nucleus, fragmentating the DNA (Yu et al., 2002; David et al., 2009; Wang et al., 2016).
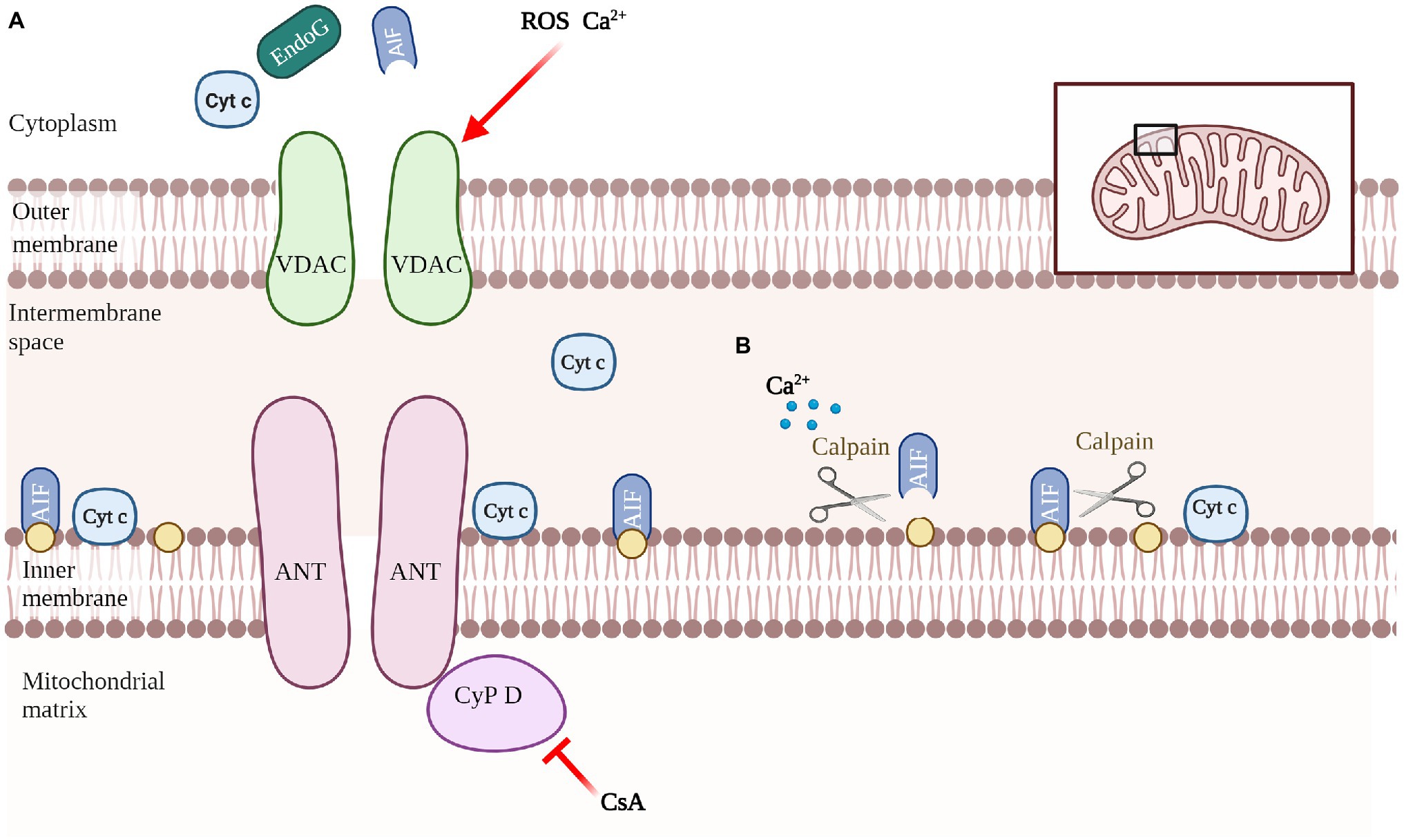
Figure 4. Schematic of mediating the release of apoptotic molecules. Composition of the nonspecific pore complex of MPTP, which triggers mitochondrial swelling and apoptotic molecules release. (A). Involvement of calcium/calpain in AIF-activated processing (B).
In conclusion, Ca2+ homeostasis imbalance, mitochondrial damage, and release of various apoptotic macromolecules are often associated with virus-induced apoptosis. From the perspective of virus, it exploits host resources by destroying intracellular Ca2+ homeostasis and regulates apoptosis to achieve the purpose of promoting its own replication. Similarly, apoptosis is also regarded by the host as an antiviral method, and the virus has achieved the establishment of persistent infection by regulating the activity of Ca2+ channels to prevent apoptosis.
Under physiological conditions, mitochondrial Ca2+ uptake is thought to serve as a safety buffer, maintaining cellular Ca2+ homeostasis in the event of a temporary intracellular Ca2+ overload, and promoting mitochondrial oxidative phosphorylation and ATP synthesis (Vasington and Murphy, 1962; Rizzuto et al., 1992). However, viral infection can lead to continuous Ca2+ accumulation and induce mitochondrial Ca2+ overload. Most of the mitochondrial Ca2+ overload comes from release from the ER, which is mainly because viral infection induces membrane contact between the ER and mitochondria (Vance, 2014). Mitochondria tightly associated with elements of ER can be isolated, and these membranes structures are frequently called mitochondria-associated membranes (MAMs) (Figure 1). IP3Rs are highly concentrated Ca2+ channels in the MAMs and play a central role that turn on/off Ca2+ release from ER stores (Mikoshiba and Hattori, 2000). Ca2+ released by ER is transported to the mitochondrial intermembrane space through the Ca2+ channel VDACs on the outer mitochondrial membrane (OMM) (Gincel et al., 2001), and the complicated MCU complex involved in Ca2+ transport through the inner mitochondrial membrane (IMM), and collaborative molecules such as cytosolic chaperone Grp75 are involved in regulation (Baughman et al., 2011; Drago et al., 2011; Poston et al., 2013). The molecular components of the uniporter comprise the pore-forming subunits MCU and dominant-negative regulator MCUb, together with calcium sensors mitochondrial calcium uptake (MICU)1, MICU2, and attachment essential MCU regulator (EMRE) (Raffaello et al., 2013; Sancak et al., 2013; Kamer and Mootha, 2015; Kamer, 2018). Apoptosis is closely related to the Ca2+ status of the mitochondria, these spatial contacts between the ER and mitochondria, are often used to function as viral targets, which triggers apoptosis by regulating the movement of Ca2+ between the ER and mitochondria (Csordás et al., 2006; Raffaello et al., 2016; Panda et al., 2021). For example, HCV, Enterovirus C (PV) and Enterovirus B trigger apoptosis through enhancing IP3R activity and promoting mitochondrial Ca2+ uptake by MCUs and VDACs (Gong et al., 2001; Brisac et al., 2010; Peischard et al., 2019). Mitochondrial Ca2+ efflux channels also play an important role in maintaining mitochondrial Ca2+ balance. Ca2+ efflux occurs through NCX and Ca2+/H+ antiporter (Khananshvili, 2013; Belosludtsev et al., 2019) (Figure 1).
Like IP3Rs, RyRs are responsible for regulating release of ER Ca2+ (Figure 1). Both IP3Rs and RyRs have similar tetramer structures and activation mechanisms, and are both activated by low Ca2+ levels and inhibited by high Ca2+ levels, so the low amount of Ca2+ released by ER further stimulates IP3Rs and RyRs activity (Fan et al., 2015). For example, HSV, HBV, HTLV-1 and IAV infections all lead to increased intracellular Ca2+, mainly due to abnormal release of ER Ca2+ caused by activation of IP3Rs (Hartshorn et al., 1988; Ding et al., 2002; Cheshenko et al., 2003; Xia et al., 2006). When ER Ca2+ storage release is increased, the ER calcium ATPase (SERCA pump) is activated and allows rapid reuptake of cytosolic Ca2+ by the ER (Courjaret and Machaca, 2014) (Figure 1). HCV core protein overexpressed in Huh7 cells induces ER Ca2+ depletion by impairing SERCA pump function (Benali-Furet et al., 2005). Depletion of intracellular Ca2+ storage also stimulates interaction between ORAI1 on the plasma membrane and STIM1 on the ER to mediate extracellular Ca2+ entry (Figure 1). In addition, some viroporins form channels on the ER that directly or indirectly increase ER permeability, leading to uncontrolled outflow of Ca2+ into the cytoplasm (Feng et al., 2002; Griffin et al., 2003; Peischard et al., 2019; Strtak et al., 2019).
In general, viral infection induces the formation of contact sites between MAMs and mitochondria, and the abnormal release of ER Ca2+ leads to mitochondrial Ca2+ overload and initiation of apoptosis.
The effect phase of apoptosis involves decreased mitochondrial membrane potential, respiratory chain uncoupling, mitochondrial permeability enhancement, and mitochondrial swelling and rupture (Hunter and Haworth, 1979). MPTP is the release channel of apoptotic molecules (such as Cyt C and AIF) and the transport channel of mitochondrial Ca2+, and its increased permeability is the decisive change in the early stage of apoptosis (Szalai et al., 1999). When viruses induce mitochondrial Ca2+ overload, accumulation of ROS and mitochondrial membrane potential dissipation, the opening of MPTP also releases Ca2+ from the mitochondria. For example, Murid betaherpesvirus 1 (Panel et al., 2019), JEV (Huang et al., 2016), IAV protein PB1-F2 (Chanturiya et al., 2004) and KHSV protein K7 (Feng et al., 2002) disrupt cytoplasmic Ca2+ levels and destroy mitochondria.
MPTP is a protein complex that connects the cytoplasm, OMM and IMM, intermembrane space of mitochondria and mitochondrial matrix, and is composed of component and regulatory molecules. The components include VDAC, adenine nucleotide translocase (ANT), and mitochondrial matrix protein cyclophilin D (Cyp D). Cyp D is also considered to be the key to MPTP opening, and is blocked by cyclosporine A (Kokoszka et al., 2004; Li et al., 2004; Baines et al., 2007) (Figure 4A). In recent years, some new molecules that may be involved in the formation of MPTP have been discovered, such as phosphate carrier, aspartate–glutamate carrier, ornithine–citrulline carrier and mitochondrial complex I on the electron transport chain (Fontaine et al., 1998; Chauvin et al., 2001). Some viral proteins locate in the mitochondrial membrane and interact directly with the MPTP to induce its opening (Jacotot et al., 2000; Chami et al., 2003).
ROS are effective activators of MPTP opening, which is induced by increased oxidative stress, and regulate Ca2+ channels in the ER and mitochondria. At the initiation stage of oxidative stress apoptosis, the transient reversible opening of MPTP increases and ROS accumulation causes damage to cells (Ma et al., 2011). MPTP continues to open irreversibly, causing mitochondrial swelling and OMM rupture, and proapoptotic proteins in the mitochondrial intermembrane are released into the cytoplasm and activate apoptotic responses. At the late stage of apoptosis, ROS are not cleared due to disorder of the antioxidant system in the mitochondria and cytoplasm. High concentrations of ROS trigger oxidative stress, resulting in mitochondrial membrane potential dissipation, mitochondrial oxidative damage, and finally induction of apoptosis (Chen et al., 2003; Lin and Beal, 2006; Stowe and Camara, 2009). In conclusion, viral infection causes abnormal release of ER Ca2+, accumulation of ROS, and excess opening of MPTP, which play a synergistic role in apoptosis.
Innate immunity is the first barrier of host defense against pathogenic microorganisms and endogenous stress responses, and it acts via different pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), retinoic-acid-inducible gene I (RIG-I)-like receptors (RLRs), cytosolic DNA sensors, and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), which cause the production of interferons (IFNs) and stimulate inflammatory cytokines after activation stimulated by microbial or cellular damage (Bowie and Unterholzner, 2008; Takeuchi and Akira, 2010; Liu et al., 2016). In addition, because Ca2+ signaling is involves in a variety of diseases, including viral infections, autoimmune diseases, and cancer, it represents an ideal target for PRRs. Conversely, intracellular Ca2+ signaling also modulates the activation of the PRR subfamily, facilitating activation of IFN regulatory factor (IRF) 3/7, initiating IFN-associated innate immune responses, and enhancing NF-κB-related inflammatory responses (Kong et al., 2021). In this section, we discuss the role of Ca2+ signaling in innate immune responses.
Among the recognized PRRs, TLRs are the most extensive and oldest form of pathogen recognition, and can recruit multiple adaptor proteins to activate transcription factors, including NF-κB, activating protein (AP)-1, and interferon (IFN) regulatory factor (IRF) family members, to cause a further inflammatory reaction and IFN-dependent antiviral immune response (Nie et al., 2018). TLRs and Ca2+ signaling interact with and regulate each other. TLRs (including TLR2, TLR3, TLR4, TLR7, TLR8 and TLR9) have been demonstrated to participate in cytokine production, activation of immunocytes, inflammation, and antiviral innate responses by regulating Ca2+ signaling (Tauseef et al., 2012; De Dios et al., 2020; Zhao et al., 2020). In particular, TLR4, which recognizes bacterial lipopolysaccharides (LPSs), is widely reported to alter cytoplasmic Ca2+ levels and activates Ca2+ signaling by regulating various Ca2+ channels. For example, endotoxin activates transient receptor potential canonical (TRPC) channels to induce Ca2+ entry in endothelial cells, secondary to TLR4-induced diacylglycerol generation (Feske et al., 2012). TLR3, TLR7, and TLR8 are RNA sensors that recognize immune-stimulated RNA and initiate downstream signals that increase the production of inflammatory cytokines and type I IFN (IFN-I). These processes are still accompanied by extracellular Ca2+ influx or release of Ca2+ from the ER and activation of associated Ca2+ channels. Especially when HIV infects CD4+ T cells, TLR7 induces increased cytoplasmic Ca2+, sensitizing activated T cell 2 nuclear factor (NFATc2) through calcination, thereby promoting HIV replication (Dominguez-Villar et al., 2015). By contrast, Ca2+ signaling also modulates TLR signaling by affecting expression of different TLR molecules or controlling TLR activation in different ways. High Ca2+ upregulates mRNA expression of TLR3 and other dsRNA sensors to augment antiviral activity in epidermal keratinocytes (Yamamura et al., 2018). Extracellular Ca2+ influx by STIM1-operated Ca2+ channels transmit the information of TLR stimuli to initiate innate immune responses (Tang et al., 2017). In general, TLRs interact with intracellular Ca2+ signals through different molecular mechanisms. It is important to note that our understanding of TLR-mediated Ca2+-signal-dependent cellular functions in different pathological processes is still insufficient. In particular, the effect of TLRs on Ca2+ signal regulation in viral infection needs more investigation.
RLRs are RNA sensor molecules that recognize the double-stranded RNAs (dsRNA), three of which are encoded in the human genome [RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2)], and enhance IFN production and play an important role in RNA virus infections (Yoneyama et al., 2005; Onomoto et al., 2021). RIG-I and MDA5 contain a tandem caspase recruitment domain, and interact with the mitochondrial antiviral signaling protein (MAVS) to activate IRF-3/7 and NF-κB, enhance IFN production, and promote transcriptional activation of proinflammatory cytokines (Yoneyama et al., 2015; Rehwinkel and Gack, 2020). Similarly, some data suggest that Ca2+ is a key regulator of the RLR pathway by regulating expression of many molecules involved in RLR signaling, or via regulation of Ca2+ channels. Studies have found that high levels of Ca2+ induce dsRNA sensors like MDA5 and RIG-I (Yamamura et al., 2018). In addition, the effects of some Ca2+ channels on RIG-I and MDA5 expression are related to Ca2+ signaling; for example, the Cav1.2 channel (a type of L-type Ca2+ channel) increases expression of RIG-I and MDA5 (Tammineni et al., 2018). Many reports have suggested that Ca2+ signaling plays an important role in the activation of RLR pathways during RNA virus infections. For example, IAV induces production of ROS to facilitate interaction of viral M2 protein with MAVS by increasing Ca2+ levels (Wang R. et al., 2019). Murine respirovirus can activate IRF3/7 by stimulating the ER to release Ca2+ through the RyR channel (Sermersheim et al., 2020). However, some studies have found that viruses recruited calcineurin, which inhibited TANK-binding kinase 1 (TBK1) phosphorylation and leading to reduced IFN-I production. Vesicular Stomatitis Virus (VSV) and HSV can restrict RLR-pathway-related antiviral innate immune response by targeting this mechanism (Huang et al., 2018). Pestivirus C infection can increase intracellular-Ca2+-level-induced autophagy through calcium/calmodulin dependent protein kinase 2 to suppress MAVS and decrease IFN-I production (Xie et al., 2021). Notably, the Ca2+ signaling pathway contributes to the regulation of RLR-mediated innate immunity, while there is no clear evidence that RLRs can control Ca2+ signaling. In addition, Ca2+ signaling can promote RLR-mediated innate immune response and reverse it in different viral infections. Therefore, it is important to illustrate the exact role of Ca2+ signaling in RNA virus regulation of RLRs in the future.
Mammalian cells recognize double-stranded DNA (dsDNA) to produce type I interferons (IFNs), suggesting that cytosolic DNA sensing is the important mechanism by which the innate immune system detects pathogens (Stetson and Medzhitov, 2006; West et al., 2015; Erdal et al., 2017). Several cytosolic DNA sensors, including cyclic GMP–AMP synthase (cGAS), melanoma 2, and DNA-dependent IFN regulatory activator, have been identified as involved in immune responses. An increasing number of studies on the activation of type I IFNs response by viral infection have focused on the cGAS–STING axis, and recent studies have shown that Ca2+ and related signaling proteins regulate cGAS–STING (Mathavarajah et al., 2019). cGAS detects reverse transcription of retroviral RNA, aberrant release of viral DNA during infection, and damage to host genomic DNA or mitochondrial DNA (Gao et al., 2013; Sun et al., 2013; Kanneganti et al., 2015). Recognition of cytosolic DNA by cGAS results in the production of second messenger cGAMP that binds to stimulator of interferon genes (STING) (Ishikawa and Barber, 2008). Activation of STING interacts with TBK1, and then STING functions as a scaffold protein for TBK1 and IRF3 assembly to stimulate phosphorylation of IRF3 and nuclear translocation and induces the transcription of IFN genes (Fitzgerald et al., 2003; Tanaka and Chen, 2012).
The current study has demonstrated that STING has a Ca2+-binding site; when STING forms a homodimer, two ions are shared between the two monomers of the protein, so Ca2+ directly participates in the regulation of STING activation (Shu et al., 2012). STING interacts with some Ca2+ channels to regulate Ca2+ flux during its activation; for example, STING interacts with Ca2+ transporters VDAC to facilitate Ca2+ uptake by mitochondria. In the resting state, STING is located in the ER and binds Ca2+-sensing transmembrane protein STIM1 and interacts with SERCA (Lee et al., 2013; Srikanth et al., 2019). In addition, changes in cytosolic Ca2+ caused by viral infection facilitate STING activation. It has been found that BAPTA-AM (an extracellular Ca2+ chelator)-mediated Ca2+ depletion and ionomycin-mediated Ca2+ elevation suppress STING-mediated IFN-β production. The function of STING is also restricted when virus-infected cells are treated with the IP3R inhibitor 2-APB or SERCA inhibitors (Hare et al., 2015; Kwon et al., 2018; Mathavarajah et al., 2019). There is growing evidence that the cGAS–STING axis is one of the cellular pathways controlled by Ca2+ signaling, and the mechanism of this pathway regulating IFN-I response in microbial infection and viral diseases will be carefully studied in the future.
The NLR family includes a variety of specific cytoplasmic sensors that detect invasive pathogens and endogenous danger signals. NOD-like receptor protein (NLRP)3 is one of the best-identified DNA sensors associated with inflammasomes in the NLR family, and is activated by invading pathogens or endogenous danger signals, leading to the formation of NLRP3 inflammasomes. The NLRP3 inflammasome is a multiprotein platform that includes NLRP3, apoptosis-associated speck-like protein (ASC) and pro-caspase-1, which leads to caspase-1-dependent secretion of proinflammatory cytokines IL-1β and IL-18 (Wen et al., 2013; Liu et al., 2018). The role of Ca2+ signaling in NLRP3 inflammasome activation has been widely reported. Lots of evidence show that Ca2+ from the extracellular environment, ER, and lysosome promote the formation and activation of NLRP3 inflammasome (Swanson et al., 2019; Li et al., 2021). For example, the cytosolic Ca2+ mediated by ion channels TRPA1 and TRPV1 facilitate the activation of the NLRP3 inflammasome (Wang M. et al., 2019); The release of the Ca2+ from ER through RyRs and IP3Rs are also observed to sensitize the NLRP3 inflammasome (Triantafilou et al., 2013a); Emanate from lysosomal Ca2+ stores regulate the production of pro-IL-1β by calcineurin, contributes to the activation of the NLRP3 inflammasome (Weber and Schilling, 2014). Until now, some data prove that viroporins enhance cytosolic Ca2+ by altering organelle membrane permeability, which promotes activation of NLRP3 inflammasomes. For example, Human rhinovirus B protein 2B targets ER and Golgi complex trigger Ca2+ release to stimulate NLRP3 inflammasome activation (Triantafilou et al., 2013b). Similarly, foot-and-mouth disease virus (FMDV) 2B protein as viroporins also promotes the flux of Ca2+, thereby stimulating NLRP3 inflammasome activation (Zhi et al., 2020). What’s more, severe acute respiratory syndrome coronavirus (SARS-CoV) envelope protein also activates the NLRP3 inflammasome by forming protein-lipid channels in ER/Golgi membranes to osmose Ca2+ (Nieto-Torres et al., 2015). So far, how intracellular Ca2+ stimulates NLRP3 activation is not fully understood. In the future, more investigations are needed to uncover the mechanisms by which Ca2+ mobilization induces NLRP3 inflammasome activation.
Viruses make full use of Ca2+ signaling networks to facilitate entry, replication, assembly and export to establish persistent infection by altering Ca2+ homeostasis. Accumulation of pathological Ca2+ signals caused by viral infection can trigger cellular antiviral response and cell death. Therefore, Ca2+-signaling networks have become an important and effective target for the treatment of viral infections. Inhibitors or activators that act on different Ca2+ channels have potential as antiviral agents, which may help in the development of antiviral drugs. Ca2+, as a signaling messenger, is transferred between organelles and participates in the regulation of various cellular activities. Mitochondria are involved in many life activities including energy synthesis, cell death and immune response, understanding that Ca2+ regulates mitochondrial involvement in the immune response and cell death during viral infection is crucial. However, there are limited research data available on the molecular mechanisms of Ca2+ interaction with the Golgi complex, peroxisomes and lysosomes, these organelles can also be targeted and hijacked by viruses. Therefore, more work is warranted to illustrate the exact effect of Ca2+ on organelle signaling during viral infection. With improving understanding of Ca2+ signaling pathways and the development of Ca2+ signal monitoring technology, the effect of Ca2+ signal changes on cell fate in the process of viral infection will become an important topic.
CD and ZY conceived the review concept and drafted the article. YQ wrote the original draft and prepared figures. YS edited and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Shanghai Agriculture Applied Technology Development Program, China (G20180207), the National Natural Science Foundation of China (grant no. 32030108), and the Foundation of Key Laboratory of Veterinary Biotechnology (no. shklab202001).
The authors declare this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript. Figures were drafted using BioRender.com. Retrieved from https://app.biorender.com/biorender-templates.
Abdullah, H., Heaney, L. G., Cosby, S. L., and McGarvey, L. P. (2014). Rhinovirus upregulates transient receptor potential channels in a human neuronal cell line: implications for respiratory virus-induced cough reflex sensitivity. Thorax 69, 46–54. doi: 10.1136/thoraxjnl-2013-203894
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J., and Molkentin, J. D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555. doi: 10.1038/ncb1575
Baravalle, G., Brabec, M., Snyers, L., Blaas, D., and Fuchs, R. (2004). Human rhinovirus type 2-antibody complexes enter and infect cells via Fc-γ receptor IIB1. J. Virol. 78, 2729–2737. doi: 10.1128/JVI.78.6.2729-2737.2004
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Belosludtsev, K. N., Dubinin, M. V., Belosludtseva, N. V., and Mironova, G. D. (2019). Mitochondrial Ca2+ transport: mechanisms, molecular structures, and role in cells. Biochemistry 84, 593–607. doi: 10.1134/S0006297919060026
Benali-Furet, N. L., Chami, M., Houel, L., De Giorgi, F., Vernejoul, F., Lagorce, D., et al. (2005). Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24, 4921–4933. doi: 10.1038/sj.onc.1208673
Bergqvist, A., and Rice, C. M. (2001). Transcriptional activation of the interleukin-2 promoter by hepatitis C virus core protein. J. Virol. 75, 772–781. doi: 10.1128/JVI.75.2.772-781.2001
Bergqvist, A., Sundström, S., Dimberg, L. Y., Gylfe, E., and Masucci, M. G. (2003). The hepatitis C virus core protein modulates T cell responses by inducing spontaneous and altering T-cell receptor-triggered Ca2+ oscillations. J. Biol. Chem. 278, 18877–18883. doi: 10.1074/jbc.M300185200
Berridge, M. J., Bootman, M. D., and Lipp, P. (1998). Calcium-a life and death signal. Nature 395, 645–648. doi: 10.1038/27094
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Bowie, A. G., and Unterholzner, L. (2008). Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922. doi: 10.1038/nri2436
Bravo-Sagua, R., Parra, V., López-Crisosto, C., Díaz, P., Quest, A. F., and Lavandero, S. (2017). Calcium transport and signaling in mitochondria. Compr. Physiol. 7, 623–634. doi: 10.1002/cphy.c160013
Brisac, C., Téoulé, F., Autret, A., Pelletier, I., Colbère-Garapin, F., Brenner, C., et al. (2010). Calcium flux between the endoplasmic reticulum and mitochondrion contributes to poliovirus-induced apoptosis. J. Virol. 84, 12226–12235. doi: 10.1128/JVI.00994-10
Cao, G., Xing, J., Xiao, X., Liou, A. K., Gao, Y., Yin, X. M., et al. (2007). Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J. Neurosci. 27, 9278–9293. doi: 10.1523/jneurosci.2826-07.2007
Chami, M., Ferrari, D., Nicotera, P., Paterlini-Bréchot, P., and Rizzuto, R. (2003). Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J. Biol. Chem. 278, 31745–31755. doi: 10.1074/jbc.M304202200
Chanturiya, A., Basanez, G., Schubert, U., Henklein, P., Yewdell, J., and Zimmerberg, J. (2004). PB1-F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J. Virol. 78, 6304–6312. doi: 10.1128/JVI.78.12.6304-6312.2004
Chauvin, C., De Oliveira, F., Ronot, X., Mousseau, M., Leverve, X., and Fontaine, E. (2001). Rotenone inhibits the mitochondrial permeability transition-induced cell death in U937 and KB cells. J. Biol. Chem. 276, 41394–41398. doi: 10.1074/jbc.M106417200
Chemello, M. E., Aristimuño, O. C., Michelangeli, F., and Ruiz, M.-C. (2002). Requirement for vacuolar H+-ATPase activity and Ca2+ gradient during entry of rotavirus into MA104 cells. J. Virol. 76, 13083–13087. doi: 10.1128/JVI.76.24.13083-13087.2002
Chen, Q., Chai, Y., Mazumder, S., Jiang, C., Macklis, R., Chisolm, G., et al. (2003). The late increase in intracellular free radical oxygen species during apoptosis is associated with cytochrome c release, caspase activation, and mitochondrial dysfunction. Cell Death Differ. 10, 323–334. doi: 10.1038/sj.cdd.4401148
Chen, Z., Wang, X., Ashraf, U., Zheng, B., Ye, J., Zhou, D., et al. (2018). Activation of neuronal N-methyl-D-aspartate receptor plays a pivotal role in Japanese encephalitis virus-induced neuronal cell damage. J. Neuroinflammation 15:238. doi: 10.1186/s12974-018-1280-8
Cheshenko, N., Del Rosario, B., Woda, C., Marcellino, D., Satlin, L. M., and Herold, B. C. (2003). Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 163, 283–293. doi: 10.1083/jcb.200301084
Choi, Y., Park, S. G., Yoo, J.-H., and Jung, G. (2005). Calcium ions affect the hepatitis B virus core assembly. Virology 332, 454–463. doi: 10.1016/j.virol.2004.11.019
Contreras, L., Drago, I., Zampese, E., and Pozzan, T. (2010). Mitochondria: the calcium connection. Biochim. Biophys. Acta Bioenerg. 1797, 607–618. doi: 10.1016/j.bbabio.2010.05.005
Courjaret, R., and Machaca, K. (2014). Mid-range Ca2+ signalling mediated by functional coupling between store-operated Ca2+ entry and IP3-dependent Ca2+ release. Nat. Commun. 5:3916. doi: 10.1038/ncomms4916
Crabtree, G. R., and Clipstone, N. A. (1994). Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu. Rev. Biochem. 63, 1045–1083. doi: 10.1146/annurev.bi.63.070194.005145
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Cui, R., Wang, Y., Wang, L., Li, G., Lan, K., Altmeyer, R., et al. (2016). Cyclopiazonic acid, an inhibitor of calcium-dependent ATPases with antiviral activity against human respiratory syncytial virus. Antiviral Res. 132, 38–45. doi: 10.1016/j.antiviral.2016.05.010
Danta, C. C. (2020). Calcium channel blockers: a possible potential therapeutic strategy for the treatment of alzheimer’s dementia patients with SARS-CoV-2 infection. ACS Chem. Nerosci. 11, 2145–2148. doi: 10.1021/acschemneuro.0c00391
David, K. K., Andrabi, S. A., Dawson, T. M., and Dawson, V. L. (2009). Parthanatos, a messenger of death. Front. Biosci. 14, 1116–1128. doi: 10.2741/3297
De Dios, R., Nguyen, L., Ghosh, S., McKenna, S., and Wright, C. J. (2020). CpG-ODN-mediated TLR9 innate immune signalling and calcium dyshomeostasis converge on the NF-κB inhibitory protein IκBβ to drive IL1α and IL1β expression. Immunology 160, 64–77. doi: 10.1111/imm.13182
Di Benedetto, G., Scalzotto, E., Mongillo, M., and Pozzan, T. (2013). Mitochondrial Ca2+ uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 17, 965–975. doi: 10.1016/j.cmet.2013.05.003
Ding, W., Albrecht, B., Kelley, R. E., Muthusamy, N., Kim, S. J., Altschuld, R. A., et al. (2002). Human T-cell lymphotropic virus type 1 p12(I) expression increases cytoplasmic calcium to enhance the activation of nuclear factor of activated T cells. J. Virol. 76, 10374–10382. doi: 10.1128/JVI.76.20.10374-10382.2002
Dionicio, C. L., Peña, F., Constantino-Jonapa, L. A., Vazquez, C., Yocupicio-Monroy, M., Rosales, R., et al. (2018). Dengue virus induced changes in Ca(2+) homeostasis in human hepatic cells that favor the viral replicative cycle. Virus Res. 245, 17–28. doi: 10.1016/j.virusres.2017.11.029
Dobson, S. J., Mankouri, J., and Whitehouse, A. (2020). Identification of potassium and calcium channel inhibitors as modulators of polyomavirus endosomal trafficking. Antiviral Res. 179:104819. doi: 10.1016/j.antiviral.2020.104819
Dominguez-Villar, M., Gautron, A. S., de Marcken, M., Keller, M. J., and Hafler, D. A. (2015). TLR7 induces anergy in human CD4(+) T cells. Nat. Immunol. 16, 118–128. doi: 10.1038/ni.3036
Doñate-Macián, P., Jungfleisch, J., Pérez-Vilaró, G., Rubio-Moscardo, F., Perálvarez-Marín, A., Diez, J., et al. (2018). The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat. Commun. 9:2307. doi: 10.1038/s41467-018-04776-7
Drago, I., Pizzo, P., and Pozzan, T. (2011). After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 30, 4119–4125. doi: 10.1038/emboj.2011.337
Dugas, B., Delfraissy, J. F., Calenda, A., Peuchmaur, M., Wallon, C., Rannou, M. T., et al. (1988). Activation and infection of B cells by Epstein-bBarr virus. Role of calcium mobilization and of protein kinase C translocation. J. Immunol. 141, 4344–4351.
Erdal, E., Haider, S., Rehwinkel, J., Harris, A. L., and McHugh, P. J. (2017). A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 31, 353–369. doi: 10.1101/gad.289769.116
Fan, G., Baker, M. L., Wang, Z., Baker, M. R., Sinyagovskiy, P. A., Chiu, W., et al. (2015). Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527, 336–341. doi: 10.1038/nature15249
Feng, P., Park, J., Lee, B. S., Lee, S. H., Bram, R. J., and Jung, J. U. (2002). Kaposi’s sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol. 76, 11491–11504. doi: 10.1128/JVI.76.22.11491-11504.2002
Feske, S., Skolnik, E. Y., and Prakriya, M. (2012). Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 12, 532–547. doi: 10.1038/nri3233
Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E., Golenbock, D. T., et al. (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496. doi: 10.1038/ni921
Fontaine, E., Eriksson, O., Ichas, F., and Bernardi, P. (1998). Regulation of the permeability transition pore in skeletal muscle mitochondria: modulation by electron flow through the respiratory chain complex I. J. Biol. Chem. 273, 12662–12668. doi: 10.1074/jbc.273.20.12662
Fujioka, Y., Nishide, S., Ose, T., Suzuki, T., Kato, I., Fukuhara, H., et al. (2018). A sialylated voltage-dependent Ca(2+) channel binds hemagglutinin and mediates influenza a virus entry into mammalian cells. Cell Host Microbe 23, 809–818.e5. doi: 10.1016/j.chom.2018.04.015
Fujioka, Y., Tsuda, M., Nanbo, A., Hattori, T., Sasaki, J., Sasaki, T., et al. (2013). A Ca2+-dependent signalling circuit regulates influenza a virus internalization and infection. Nat. Commun. 4:2763. doi: 10.1038/ncomms3763
Gao, D., Wu, J., Wu, Y. T., Du, F., Aroh, C., Yan, N., et al. (2013). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906.
Genazzani, A. A., and Galione, A. (1996). Nicotinic acid-adenine dinucleotide phosphate mobilizes Ca2+ from a thapsigargin-insensitive pool. Biochem. J. 315, 721–725. doi: 10.1042/bj3150721
Gincel, D., Zaid, H., and Shoshan-Barmatz, V. (2001). Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358, 147–155. doi: 10.1042/bj3580147
Gong, G., Waris, G., Tanveer, R., and Siddiqui, A. (2001). Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-κB. Proc. Natl. Acad. Sci. U. S. A. 98, 9599–9604. doi: 10.1073/pnas.171311298
Griffin, S. D., Beales, L. P., Clarke, D. S., Worsfold, O., Evans, S. D., Jaeger, J., et al. (2003). The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 535, 34–38. doi: 10.1016/s0014-5793(02)03851-6
Gunaratne, G. S., Yang, Y., Li, F., Walseth, T. F., and Marchant, J. S. (2018). NAADP-dependent Ca2+ signaling regulates middle east respiratory syndrome-coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium 75, 30–41. doi: 10.1016/j.ceca.2018.08.003
Han, Z., Madara, J. J., Herbert, A., Prugar, L. I., Ruthel, G., Lu, J., et al. (2015). Calcium regulation of hemorrhagic fever virus budding: mechanistic implications for host-oriented therapeutic intervention. PLoS Pathog. 11:e1005220. doi: 10.1371/journal.ppat.1005220
Hare, D. N., Collins, S. E., Mukherjee, S., Loo, Y.-M., Gale, M. Jr., Janssen, L. J., et al. (2015). Membrane perturbation-associated Ca2+ signaling and incoming genome sensing are required for the host response to low-level enveloped virus particle entry. J. Virol. 90, 3018–3027. doi: 10.1128/JVI.02642-15
Hartshorn, K. L., Collamer, M., Auerbach, M., Myers, J. B., Pavlotsky, N., and Tauber, A. I. (1988). Effects of influenza A virus on human neutrophil calcium metabolism. J. Immunol. 141, 1295–1301.
Hoffmann, H. H., Schneider, W. M., Blomen, V. A., Scull, M. A., Hovnanian, A., Brummelkamp, T. R., et al. (2017). Diverse viruses require the calcium transporter SPCA1 for maturation and spread. Cell Host Microbe 22, 460–470.e5. doi: 10.1016/j.chom.2017.09.002
Holden, C. P., Haughey, N. J., Nath, A., and Geiger, J. D. (1999). Role of Na+/H+ exchangers, excitatory amino acid receptors and voltage-operated Ca2+ channels in human immunodeficiency virus type 1 gp120-mediated increases in intracellular Ca2+ in human neurons and astrocytes. Neuroscience 91, 1369–1378. doi: 10.1016/S0306-4522(98)00714-3
Huang, S.-H., Lien, J.-C., Chen, C.-J., Liu, Y.-C., Wang, C.-Y., Ping, C.-F., et al. (2016). Antiviral activity of a novel compound CW-33 against Japanese encephalitis virus through inhibiting intracellular calcium overload. Int. J. Mol. Sci. 17:1386. doi: 10.3390/ijms17091386
Huang, H., Xiong, Q., Wang, N., Chen, R., Ren, H., Siwko, S., et al. (2018). Kisspeptin/GPR54 signaling restricts antiviral innate immune response through regulating calcineurin phosphatase activity. Sci. Adv. 4:eaas9784. doi: 10.1126/sciadv.aas9784
Hunter, D. R., and Haworth, R. A. (1979). The Ca2+-induced membrane transition in mitochondria: III. Transitional Ca2+ release. Arch. Biochem. Biophys. 195, 468–477. doi: 10.1016/0003-9861(79)90373-4
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. doi: 10.1038/nature07317
Ivanov, A. V., Smirnova, O. A., Petrushanko, I. Y., Ivanova, O. N., Karpenko, I. L., Alekseeva, E., et al. (2015). HCV core protein uses multiple mechanisms to induce oxidative stress in human hepatoma Huh7 cells. Viruses 7, 2745–2770. doi: 10.3390/v7062745
Jacotot, E., Ravagnan, L., Loeffler, M., Ferri, K. F., Vieira, H. L., Zamzami, N., et al. (2000). The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 191, 33–46. doi: 10.1084/jem.191.1.33
Kamer, K. J. (2018). Regulation of the Mitochondrial Calcium Uniporter by the MICU1/MICU2 Complex: From Biochemistry to Human Disease. Harvard University.
Kamer, K. J., and Mootha, V. K. (2015). The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 16, 545–553. doi: 10.1038/nrm4039
Kanneganti, T. D., Kundu, M., and Green, D. R. (2015). Innate immune recognition of mtDNA: an undercover signal? Cell Metab. 21, 793–794. doi: 10.1016/j.cmet.2015.05.019
Khananshvili, D. (2013). The SLC8 gene family of sodium–calcium exchangers (NCX)—structure, function, and regulation in health and disease. Mol. Aspects Med. 34, 220–235. doi: 10.1016/j.mam.2012.07.003
Kinoshita, S., Su, L., Amano, M., Timmerman, L. A., Kaneshima, H., and Nolan, G. P. (1997). The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 6, 235–244. doi: 10.1016/S1074-7613(00)80326-X
Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., et al. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. doi: 10.1038/nature02229
Kong, F., You, H., Zheng, K., Tang, R., and Zheng, C. (2021). The crosstalk between pattern-recognition receptor signaling and calcium signaling. Int. J. Biol. Macromol. 192, 745–756. doi: 10.1016/j.ijbiomac.2021.10.014
Kuchay, S., Saeed, M., Giorgi, C., Li, J., Hoffmann, H.-H., Pinton, P., et al. (2018). NS5A promotes constitutive degradation of IP3R3 to counteract apoptosis induced by hepatitis C virus. Cell Rep. 25, 833–840.e3. doi: 10.1016/j.celrep.2018.09.088
Kwon, D., Sesaki, H., and Kang, S.-J. (2018). Intracellular calcium is a rheostat for the STING signaling pathway. Biochem. Biophys. Res. Commun. 500, 497–503. doi: 10.1016/j.bbrc.2018.04.117
Lahti, A. L., Manninen, A., and Saksela, K. (2003). Regulation of T cell activation by HIV-1 accessory proteins: Vpr acts via distinct mechanisms to cooperate with Nef in NFAT-directed gene expression and to promote transactivation by CREB. Virology 310, 190–196. doi: 10.1016/S0042-6822(03)00164-8
Lara-Pezzi, E., Armesilla, A. L., Majano, P. L., Redondo, J. M., and López-Cabrera, M. (1998). The hepatitis B virus X protein activates nuclear factor of activated T cells (NF-AT) by a cyclosporin a-sensitive pathway. EMBO J. 17, 7066–7077. doi: 10.1093/emboj/17.23.7066
Lee, B.-S., Lee, S.-H., Feng, P., Chang, H., Cho, N.-H., and Jung, J. U. (2005). Characterization of the Kaposi’s sarcoma-associated herpesvirus K1 signalosome. J. Virol. 79, 12173–12184. doi: 10.1128/JVI.79.19.12173-12184.2005
Lee, M. N., Roy, M., Ong, S. E., Mertins, P., Villani, A. C., Li, W., et al. (2013). Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach. Nat. Immunol. 14, 179–185. doi: 10.1038/ni.2509
Li, Y., Boehning, D. F., Qian, T., Popov, V. L., and Weinman, S. A. (2007). Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 21, 2474–2485. doi: 10.1096/fj.06-7345com
Li, C., Chen, M., He, X., and Ouyang, D. (2021). A mini-review on ion fluxes that regulate NLRP3 inflammasome activation. Acta Biochim. Biophys. Sin. Shanghai 53, 131–139. doi: 10.1093/abbs/gmaa155
Li, Y., Johnson, N., Capano, M., Edwards, M., and Crompton, M. (2004). Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem. J. 383, 101–109. doi: 10.1042/BJ20040669
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Liu, J., Qian, C., and Cao, X. (2016). Post-translational modification control of innate immunity. Immunity 45, 15–30. doi: 10.1016/j.immuni.2016.06.020
Liu, Q., Zhang, D., Hu, D., Zhou, X., and Zhou, Y. (2018). The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 103, 115–124. doi: 10.1016/j.molimm.2018.09.010
Ma, Q., Fang, H., Shang, W., Liu, L., Xu, Z., Ye, T., et al. (2011). Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J. Biol. Chem. 286, 27573–27581. doi: 10.1074/jbc.M111.241794
Mathavarajah, S., Salsman, J., and Dellaire, G. (2019). An emerging role for calcium signalling in innate and autoimmunity via the cGAS-STING axis. Cytokine Growth Factor Rev. 50, 43–51. doi: 10.1016/j.cytogfr.2019.04.003
Mayne, M., Holden, C. P., Nath, A., and Geiger, J. D. (2000). Release of calcium from inositol 1,4,5-trisphosphate receptor-regulated stores by HIV-1 tat regulates TNF-alpha production in human macrophages. J. Immunol. 164, 6538–6542. doi: 10.4049/jimmunol.164.12.6538
Mikoshiba, K., and Hattori, M. (2000). IP3 receptor-operated calcium entry. Sci. STKE 2000:pe1. doi: 10.1126/stke.2000.51.pe1
Nair, A. M., Michael, B., Datta, A., Fernandez, S., and Lairmore, M. D. (2006). Calcium-dependent enhancement of transcription of p300 by human T-lymphotropic type 1 p12I. Virology 353, 247–257. doi: 10.1016/j.virol.2006.06.005
Nie, L., Cai, S. Y., Shao, J. Z., and Chen, J. (2018). Toll-like receptors, associated biological roles, and signaling networks in non-mammals. Front. Immunol. 9:1523. doi: 10.3389/fimmu.2018.01523
Nieto-Torres, J. L., Verdiá-Báguena, C., Jimenez-Guardeño, J. M., Regla-Nava, J. A., Castaño-Rodriguez, C., Fernandez-Delgado, R., et al. (2015). Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485, 330–339. doi: 10.1016/j.virol.2015.08.010
Nokta, M., Eaton, D., Steinsland, O. S., and Albrecht, T. (1987). Ca2+ responses in cytomegalovirus-infected fibroblasts of human origin. Virology 157, 259–267. doi: 10.1016/0042-6822(87)90268-6
Omar, S., Clarke, R., Abdullah, H., Brady, C., Corry, J., Winter, H., et al. (2017). Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS One 12:e0171681. doi: 10.1371/journal.pone.0171681
Onomoto, K., Onoguchi, K., and Yoneyama, M. (2021). Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell. Mol. Immunol. 18, 539–555. doi: 10.1038/s41423-020-00602-7
Orrenius, S., Gogvadze, V., and Zhivotovsky, B. (2015). Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 460, 72–81. doi: 10.1016/j.bbrc.2015.01.137
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Regulation of cell death: the calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565. doi: 10.1038/nrm1150
Panda, S., Behera, S., Alam, M. F., and Syed, G. H. (2021). Endoplasmic reticulum & mitochondrial calcium homeostasis: the interplay with viruses. Mitochondrion 58, 227–242.
Panel, M., Ruiz, I., Brillet, R., Lafdil, F., Teixeira-Clerc, F., Nguyen, C. T., et al. (2019). Small-molecule inhibitors of cyclophilins block opening of the mitochondrial permeability transition pore and protect mice from hepatic ischemia/reperfusion injury. Gastroenterology 157, 1368–1382. doi: 10.1053/j.gastro.2019.07.026
Patel, S., Ramakrishnan, L., Rahman, T., Hamdoun, A., Marchant, J. S., Taylor, C. W., et al. (2011). The endo-lysosomal system as an NAADP-sensitive acidic Ca2+ store: role for the two-pore channels. Cell Calcium 50, 157–167. doi: 10.1016/j.ceca.2011.03.011
Peischard, S., Ho, H. T., Theiss, C., Strutz-Seebohm, N., and Seebohm, G. (2019). A kidnapping story: how Coxsackievirus B3 and its host cell interact. Cell. Physiol. Biochem. 53, 121–140. doi: 10.33594/000000125
Pérez, J. F., Ruiz, M. C., Chemello, M. E., and Michelangeli, F. (1999). Characterization of a membrane calcium pathway induced by rotavirus infection in cultured cells. J. Virol. 73, 2481–2490. doi: 10.1128/JVI.73.3.2481-2490.1999
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., and Rizzuto, R. (2008). Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418. doi: 10.1038/onc.2008.308
Poston, C. N., Krishnan, S. C., and Bazemore-Walker, C. R. (2013). In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 79, 219–230. doi: 10.1016/j.jprot.2012.12.018
Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., et al. (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376. doi: 10.1038/emboj.2013.157
Raffaello, A., Mammucari, C., Gherardi, G., and Rizzuto, R. (2016). Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 41, 1035–1049. doi: 10.1016/j.tibs.2016.09.001
Rahman, S. K., Kerviel, A., Mohl, B.-P., He, Y., Zhou, Z. H., and Roy, P. (2020). A calcium sensor discovered in bluetongue virus nonstructural protein 2 is critical for virus replication. J. Virol. 94, e01099–e01020. doi: 10.1128/JVI.01099-20
Rehwinkel, J., and Gack, M. U. (2020). RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551. doi: 10.1038/s41577-020-0288-3
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Rizzuto, R., Simpson, A. W., Brini, M., and Pozzan, T. (1992). Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327. doi: 10.1038/358325a0
Sakurai, Y., Kolokoltsov, A. A., Chen, C.-C., Tidwell, M. W., Bauta, W. E., Klugbauer, N., et al. (2015). Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995–998. doi: 10.1126/science.1258758
Salgado, E. N., Rodriguez, G. B., Narayanaswamy, N., Krishnan, Y., and Harrison, S. C. (2018). Visualization of calcium ion loss from rotavirus during cell entry. J. Virol. 92, e01327–e01418. doi: 10.1128/JVI.01327-18
Sancak, Y., Markhard, A. L., Kitami, T., Kovács-Bogdán, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. doi: 10.1126/science.1242993
Scrima, R., Piccoli, C., Moradpour, D., and Capitanio, N. (2018). Targeting endoplasmic reticulum and/or mitochondrial Ca2+ fluxes as therapeutic strategy for HCV infection. Front. Chem. 6:73. doi: 10.3389/fchem.2018.00073
Sermersheim, M., Kenney, A. D., Lin, P. H., McMichael, T. M., Cai, C., Gumpper, K., et al. (2020). MG53 suppresses interferon-β and inflammation via regulation of ryanodine receptor-mediated intracellular calcium signaling. Nat. Commun. 11:3624. doi: 10.1038/s41467-020-17177-6
Shu, C., Yi, G., Watts, T., Kao, C. C., and Li, P. (2012). Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 19, 722–724. doi: 10.1038/nsmb.2331
Simpson, A. A., Chandrasekar, V., Hébert, B., Sullivan, G. M., Rossmann, M. G., and Parrish, C. R. (2000). Host range and variability of calcium binding by surface loops in the capsids of canine and feline parvoviruses. J. Mol. Biol. 300, 597–610. doi: 10.1006/jmbi.2000.3868
Sirohi, D., and Kuhn, R. J. (2017). Can an FDA-approved Alzheimer's drug be repurposed for alleviating neuronal symptoms of Zika virus? MBio 8:e00916-17. doi: 10.1128/mBio.00916-17
Srikanth, S., Woo, J. S., Wu, B., El-Sherbiny, Y. M., Leung, J., Chupradit, K., et al. (2019). The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 20, 152–162. doi: 10.1038/s41590-018-0287-8
Stetson, D. B., and Medzhitov, R. (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103. doi: 10.1016/j.immuni.2005.12.003
Stowe, D. F., and Camara, A. K. (2009). Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid. Redox Signal. 11, 1373–1414. doi: 10.1089/ars.2008.2331
Strtak, A. C., Perry, J. L., Sharp, M. N., Chang-Graham, A. L., Farkas, T., and Hyser, J. M. (2019). Recovirus NS1-2 has viroporin activity that induces aberrant cellular calcium signaling to facilitate virus replication. mSphere 4:e00506. doi: 10.1128/mSphere.00506-19
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. doi: 10.1126/science.1232458
Susin, S. A., Zamzami, N., Castedo, M., Hirsch, T., Marchetti, P., Macho, A., et al. (1996). Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 184, 1331–1341. doi: 10.1084/jem.184.4.1331
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. doi: 10.1038/s41577-019-0165-0
Szalai, G., Krishnamurthy, R., and Hajnóczky, G. (1999). Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J. 18, 6349–6361. doi: 10.1093/emboj/18.22.6349
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tammineni, E. R., Carrillo, E. D., Soto-Acosta, R., Angel-Ambrocio, A. H., García, M. C., Bautista-Carbajal, P., et al. (2018). The β4 subunit of Cav1.2 channels is required for an optimal interferon response in cardiac muscle cells. Sci. Signal. 11:eaaj1676. doi: 10.1126/scisignal.aaj1676
Tanaka, Y., and Chen, Z. J. (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5:ra20. doi: 10.1126/scisignal.2002521
Tang, S., Chen, T., Yang, M., Wang, L., Yu, Z., Xie, B., et al. (2017). Extracellular calcium elicits feedforward regulation of the toll-like receptor-triggered innate immune response. Cell. Mol. Immunol. 14, 180–191. doi: 10.1038/cmi.2015.59
Tang, S., Wang, X., Shen, Q., Yang, X., Yu, C., Cai, C., et al. (2015). Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 458, 186–193. doi: 10.1016/j.bbrc.2015.01.092
Tano, J. Y., and Vazquez, G. (2011). Requirement for non-regulated, constitutive calcium influx in macrophage survival signaling. Biochem. Biophys. Res. Commun. 407, 432–437. doi: 10.1016/j.bbrc.2011.03.048
Tauseef, M., Knezevic, N., Chava, K. R., Smith, M., Sukriti, S., Gianaris, N., et al. (2012). TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 209, 1953–1968. doi: 10.1084/jem.20111355
Triantafilou, K., Hughes, T. R., Triantafilou, M., and Morgan, B. P. (2013a). The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 126, 2903–2913. doi: 10.1242/jcs.124388
Triantafilou, K., Kar, S., van Kuppeveld, F. J., and Triantafilou, M. (2013b). Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 49, 923–934. doi: 10.1165/rcmb.2013-0032OC
Vance, J. E. (2014). MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. doi: 10.1016/j.bbalip.2013.11.014
Vasington, F. D., and Murphy, J. V. (1962). Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 237, 2670–2677. doi: 10.1016/S0021-9258(19)73805-8
Wang, Y., An, R., Umanah, G. K., Park, H., Nambiar, K., Eacker, S. M., et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly (ADP-ribose) polymerase-1. Science 354:aad6872. doi: 10.1126/science.aad6872
Wang, S., Liu, Y., Guo, J., Wang, P., Zhang, L., Xiao, G., et al. (2017). Screening of FDA-approved drugs for inhibitors of Japanese encephalitis virus infection. J. Virol. 91, e01055–e01017. doi: 10.1128/jvi.01055-17
Wang, M., Zhang, Y., Xu, M., Zhang, H., Chen, Y., Chung, K. F., et al. (2019). Roles of TRPA1 and TRPV1 in cigarette smoke-induced airway epithelial cell injury model. Free Radic. Biol. Med. 134, 229–238. doi: 10.1016/j.freeradbiomed.2019.01.004
Wang, R., Zhu, Y., Lin, X., Ren, C., Zhao, J., Wang, F., et al. (2019). Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 15, 1163–1181. doi: 10.1080/15548627.2019.1580089
Weber, K., and Schilling, J. D. (2014). Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 289, 9158–9171. doi: 10.1074/jbc.M113.531202
Wen, H., Miao, E. A., and Ting, J. P.-Y. (2013). Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 39, 432–441. doi: 10.1016/j.immuni.2013.08.037
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
Xia, W., Shen, Y., Xie, H., and Zheng, S. (2006). Involvement of endoplasmic reticulum in hepatitis B virus replication. Virus Res. 121, 116–121. doi: 10.1016/j.virusres.2006.01.020
Xie, B., Zhao, M., Song, D., Wu, K., Yi, L., Li, W., et al. (2021). Induction of autophagy and suppression of type I IFN secretion by CSFV. Autophagy 17, 925–947. doi: 10.1080/15548627.2020.1739445
Yamamura, Y., Morizane, S., Yamamoto, T., Wada, J., and Iwatsuki, K. (2018). High calcium enhances the expression of double-stranded RNA sensors and antiviral activity in epidermal keratinocytes. Exp. Dermatol. 27, 129–134. doi: 10.1111/exd.13456
Yao, J. H., Liu, Z. J., Yi, J. H., Wang, J., and Liu, Y. N. (2018). Hepatitis B virus X protein upregulates intracellular calcium signaling by binding C-terminal of Orail protein. Curr. Med. Sci. 38, 26–34. doi: 10.1007/s11596-018-1843-z
Yoneyama, M., Kikuchi, M., Matsumoto, K., Imaizumi, T., Miyagishi, M., Taira, K., et al. (2005). Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858. doi: 10.4049/jimmunol.175.5.2851
Yoneyama, M., Onomoto, K., Jogi, M., Akaboshi, T., and Fujita, T. (2015). Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 32, 48–53. doi: 10.1016/j.coi.2014.12.012
Yu, S.-W., Wang, H., Poitras, M. F., Coombs, C., Bowers, W. J., Federoff, H. J., et al. (2002). Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263. doi: 10.1126/science.1072221
Zampese, E., and Pizzo, P. (2012). Intracellular organelles in the saga of Ca2+ homeostasis: different molecules for different purposes? Cell. Mol. Life Sci. 69, 1077–1104. doi: 10.1007/s00018-011-0845-9
Zhang, Q., Hsia, S. C., and Martin-Caraballo, M. (2019). Regulation of T-type Ca(2+) channel expression by interleukin-6 in sensory-like ND7/23 cells post-herpes simplex virus (HSV-1) infection. J. Neurochem. 151, 238–254. doi: 10.1111/jnc.14697
Zhao, L. X., Jiang, M., Bai, X. Q., Cao, D. L., Wu, X. B., Zhang, J., et al. (2020). TLR8 in the trigeminal ganglion contributes to the maintenance of trigeminal neuropathic pain in mice. Neurosci. Bull. 37, 550–562. doi: 10.1007/s12264-020-00621-4
Zhi, X., Zhang, Y., Sun, S., Zhang, Z., Dong, H., Luo, X., et al. (2020). NLRP3 inflammasome activation by foot-and-mouth disease virus infection mainly induced by viral RNA and non-structural protein 2B. RNA Biol. 17, 335–349. doi: 10.1080/15476286.2019.1700058
Zhou, Y., Frey, T. K., and Yang, J. J. (2009). Viral calciomics: interplays between Ca2+ and virus. Cell Calcium 46, 1–17. doi: 10.1016/j.ceca.2009.05.005
Keywords: virus, calcium homeostasis, calcium channels, calcium pumps, cell death, innate immune, antiviral responses
Citation: Qu Y, Sun Y, Yang Z and Ding C (2022) Calcium Ions Signaling: Targets for Attack and Utilization by Viruses. Front. Microbiol. 13:889374. doi: 10.3389/fmicb.2022.889374
Edited by:
Miguel A. Martín-Acebes, Instituto Nacional de Investigación y Tecnología Agroalimentaria (INIA), SpainReviewed by:
Tai-Ting Woo, University of Michigan, United StatesCopyright © 2022 Qu, Sun, Yang and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chan Ding, c2hvdmVsZGVlbkBzaHZyaS5hYy5jbg==; Zengqi Yang, eXpxODE2MkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.