 Mildred Castellanos
Mildred Castellanos Theodore B. Verhey
Theodore B. Verhey Madeleine Goldstein
Madeleine Goldstein George Chaconas
George Chaconas- 1Department of Biochemistry and Molecular Biology, Cumming School of Medicine, Snyder Institute for Chronic Diseases, University of Calgary, Calgary, AB, Canada
- 2Department of Microbiology, Immunology and Infectious Diseases, Cumming School of Medicine, Snyder Institute for Chronic Diseases, University of Calgary, Calgary, AB, Canada
The Lyme disease spirochete Borrelia burgdorferi, encodes an elaborate antigenic variation system that promotes the ongoing variation of a major surface lipoprotein, VlsE. Changes in VlsE are continual and always one step ahead of the host acquired immune system, which requires 1–2 weeks to generate specific antibodies. By the time this happens, new VlsE variants have arisen that escape immunosurveillance, providing an avenue for persistent infection. This antigenic variation system is driven by segmental gene conversion events that transfer information from a series of silent cassettes (vls2-16) to the expression locus, vlsE. The molecular details of this process remain elusive. Recombinational switching at vlsE is RecA-independent and the only required factor identified to date is the RuvAB branch migrase. In this work we have used next generation long-read sequencing to analyze the effect of several DNA replication/recombination/repair gene disruptions on the frequency of gene conversions at vlsE and report a requirement for the mismatch repair protein MutL. Site directed mutagenesis of mutL suggests that the putative MutL endonuclease activity is required for recombinational switching at vlsE. This is the first report of an unexpected essential role for MutL in a bacterial recombination system and expands the known function of this protein as well as our knowledge of the details of the novel recombinational switching mechanism for vlsE variation.
Introduction
Obligate parasites have an essential need for ongoing association with a host. In the case of pathogens this translates into the necessity for strategies to maintain a persistent infection. One such pathogenic ruse employed by several types of bacteria, protozoa, and fungi is antigenic variation whereby the pathogen continually changes a major surface protein to generate variants that escape the acquired immune system of the host (Deitsch et al., 2009; Cahoon and Seifert, 2011; Prucca et al., 2011; Vink et al., 2012; Volz et al., 2012; Glover et al., 2013; Horn, 2014; Norris, 2014; McCulloch et al., 2015; Obergfell and Seifert, 2015; Gargantini et al., 2016; Palmer et al., 2016; Devlin et al., 2017; Wahlgren et al., 2017; Sima et al., 2019; Chaconas et al., 2020; Lopez et al., 2021). Borrelia burgdorferi, the Lyme disease spirochete encodes a single diversity generating recombination system of this type for the surface-bound lipoprotein VlsE [see Norris (2014) and Chaconas et al. (2020) for recent reviews]. Continual variation of VlsE was first described in and has been characterized primarily in the type strain B. burgdorferi B31 (Zhang et al., 1997). In this strain, the vls locus is located on the linear plasmid lp28-1 and variability of VlsE is driven by changes at the DNA level of the vlsE gene. This occurs through unidirectional segmental gene conversion events (recombinational switching) that transfer information from a series of 15 silent cassettes corresponding to the variable region of vlsE (Figure 1), potentially resulting in over ∼1040 possible unique VlsE sequences (Verhey et al., 2018a). Recombinational switching does not occur at significant levels during growth in culture, or in ticks, but requires an animal infection to induce the ongoing process, which continues long-term in a mammalian infection (see Norris, 2014; Chaconas et al., 2020). The signal that induces activation of recombination at vlsE remains unknown. Antigenic variation of VlsE in B. burgdorferi is required for persistent infection of an immunocompetent murine host (Purser and Norris, 2000; Bankhead and Chaconas, 2007; Rogovskyy et al., 2015; Magunda and Bankhead, 2016) and may operate through an epitope-shielding mechanism (Lone and Bankhead, 2020). In the absence of VlsE, infection proceeds at the wild-type level, but spirochete clearance occurs between 12 and 21 days post-infection (Lawrenz et al., 2004; Bankhead and Chaconas, 2007).
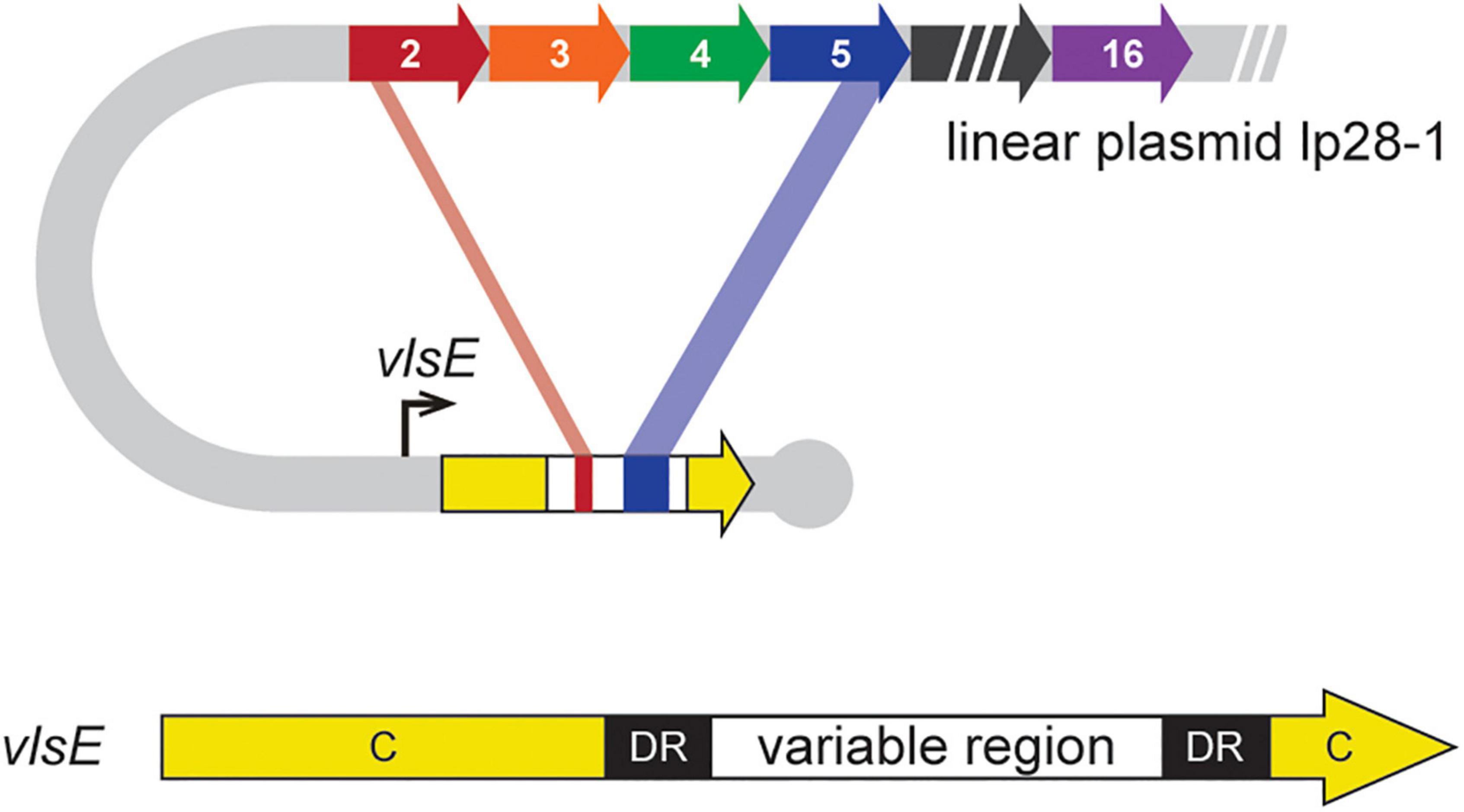
Figure 1. Schematic of DNA rearrangements underlying antigenic variation in B. burgdorferi. The vls locus is located near the right hairpin telomere on lp28-1 (Zhang et al., 1997). It is composed of an expression locus (vlsE) that contains a variable region flanked by 17-bp direct repeats (DR) and constant regions (C). Upstream of vlsE are 15 homologous, but different, silent cassettes corresponding to the variable region of vlsE, which carry information for unidirectional segmental gene conversion events to generate chimeric VlsE antigens (Norris, 2014; Chaconas et al., 2020). Figure modified from Verhey et al. (2018b).
Although discovered 25 years ago, the molecular details by which the segmental gene conversion events underlying antigenic variation in B. burgdorferi remain largely uncharacterized. Recombinational switching at vlsE is homology-driven, but surprisingly, RecA-independent (Liveris et al., 2008; Dresser et al., 2009). The only identified required factor for the reaction is the RuvAB branch migrase (Dresser et al., 2009; Lin et al., 2009), which is shared with the better characterized pilE antigenic variation system of Neisseria gonorrhoeae (see Chaconas et al., 2020). In spite of the paucity of identified protein factors that mediate recombinational switching, recent advances have been made in characterization of the reaction products through the use of next generation (NG) PacBio long-read sequencing technology. NG sequencing coupled with VAST software (variable antigen sequence tracer), a unique pipeline allowing analysis of large data sets of full-length vlsE sequences and inference of silent cassette sources of recombined sequences (Castellanos et al., 2018; Verhey et al., 2018a,b, 2019), has revealed a variety of important new information on switching at vlsE [reviewed in Chaconas et al. (2020)].
In this work we have applied our NG sequencing pipeline to the analysis of switching at vlsE in several mutant B. burgdorferi strains that previously displayed reduced persistence in wild-type C3H/HeN mouse infections. The current study demonstrates a requirement for MutL for recombinational switching, particularly for the putative MutL endonuclease. This is the first report of a role for MutL in bacterial recombination, raises the possibilities that MutL may participate in other recombination activities and expands the known protein factors needed for gene conversion at vlsE.
Results
Properties of Recombinational Switching at vlsE in Various B. burgdorferi Replication/Recombination/Repair Mutant Strains
In this work we analyzed recombinational switching at vlsE by next generation DNA sequencing in six B. burgdorferi B31 5A4 strains carrying mutations in genes encoding the DNA metabolizing proteins mutL (DNA mismatch repair), recJ (single-strand specific exonuclease), sbcC (subunit of SbcCD nuclease), sbcD (subunit of SbcCD nuclease), priA (primosomal protein N’), and bbg32 (putative replicative DNA helicase) that previously demonstrated a decrease in persistence at 21 days after infection of wild-type C3H/HeN mice (Dresser et al., 2009). This decrease in persistence is consistent with a decrease in recombinational switching that is more accurately analyzed with NG-sequencing than in our earlier study using small sample sizes and Sanger sequencing. The strains with decreased persistence were also used to infect SCID mice, which lack an acquired immune response. The SCID background removes selective pressure from these mice and allows mutants with decreased switching at vlsE to survive in the host by eliminating immune clearance of strains not carrying the most recent VlsE variants. Infection of SCID mice, therefore, provides the best avenue for analysis of recombinational switching without perturbations from acquired immunity. Frozen glycerol cultures of spirochetes recovered from our previous infection of SCID mice (Dresser et al., 2009) were grown from week 0 to week 5. For each strain, genomic DNA was isolated from spirochete cultures obtained at 0 and 5 weeks post-infection. For each gene disruption two B. burgdorferi clones were obtained, and two mice were infected per clone. Week 5 genomic DNA was pooled separately for the two replicate mice, from four tissues: bladder, ear, heart, and joint. Week 0 genomic DNA was isolated independently for both clones. A 776 bp amplicon encompassing the vlsE variable region and part of the constant region on both sides of the gene was amplified by polymerase chain reaction (PCR) from each sample as previously described (Verhey et al., 2018a), using barcoded primers (Supplementary Table 1) to identify each sample (Supplementary Table 2). The vlsE amplicons were sequenced using PacBio long read technology and the data analyzed using our previously reported pipeline and VAST software (Verhey et al., 2018a,b, 2019).
The level of recombinational switching at vlsE in the SCID mice, where there is no immune selection for any switch variants, is shown in Figure 2. At 5 weeks post-infection there were on average about three switches (distinct patches of sequence changes that match sequences in the silent cassettes) per vlsE amplicon in the wild-type strain. As previously noted and as expected for switching at vlsE (Verhey et al., 2018a,b, 2019), almost all the observed SNPs were found in the variable regions of vlsE, for the wild-type and mutant strains rather than in the constant portions of the gene, which do not contribute to antigenic variation (Supplementary Figure 1).
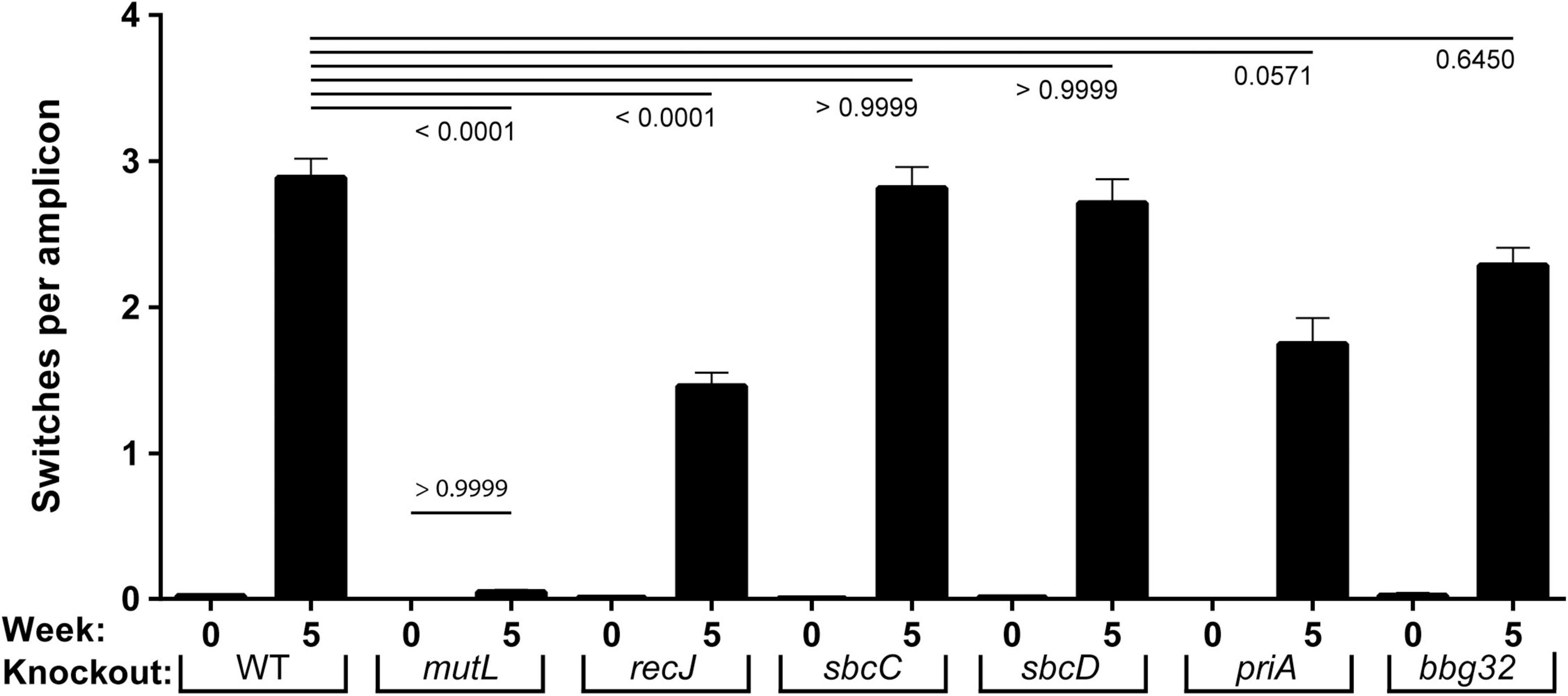
Figure 2. Frequency of vlsE switching events in strains with mutant replication/recombination/repair genes. Inferred recombination are shown as a frequency per read events (gene conversion tracts per amplicon) for WT and six mutant strains of B. burgdorferi prior to (week 0) and after infection (week 5). Mean ± SEM are shown for each population of full-length vlsE amplicon sequences. The p values for each mutant versus the wild-type strain at 5 weeks is shown at the top of the graph. The p value for comparison of mutL at 0 and 5 weeks is shown above the mutL bars. The p values were determined using Dunn’s multiple comparison test.
An unexpected result from the sequencing was the 97% reduction in switching activity to background levels in the mutL strain at week 5, which was more severe than our previously estimated 36% decrease using a smaller sample size and a classical DNA sequencing approach (Dresser et al., 2009). This decrease (Figure 2) was highly significant compared to WT (p < 0.0001) and was not significantly higher than the background switching at time zero (p > 0.9999). In the recJ strain we saw a 52% decrease in switching (p < 0.0001), similar to our previously reported 47% decrease (Dresser et al., 2009). For sbcC and sbcD, wild-type levels of switching were displayed. For priA a 41% decrease in switching, on the threshold of significance (p = 0.057), was observed and for bbg32 a smaller (28%), but not significant decrease occurred. The sbc and priA strains were previously reported to undergo switching at wild-type levels with smaller sample size (Dresser et al., 2009).
We also analyzed the change in parental vlsE sequence for each mutant between 0 and 5 weeks (Supplementary Figure 2) and found the inverse of what we observed in Figure 2: as expected, those mutants demonstrating higher levels of switching at vlsE showed lower levels of the parental vlsE sequence at 5 weeks. We also analyzed the number of distinct vlsE variants at 5 weeks post-infection as an additional measure of the efficiency of recombinational switching at vlsE (Supplementary Figure 3). Again, the mutant strains demonstrating the highest levels of switching in Figure 2, showed the greatest number of switch variants.
To further characterize the product of switching in the various mutants, the length of the recombination tract was estimated for each mutant as previously described (Verhey et al., 2018b). The uncorrected mean switch length (Verhey et al., 2018b) for the mutants and the wild-type (Figure 3) agreed well with our previously reported value of 16.4 bp in SCID mice, with the exception of the mutL strain, which displayed a somewhat longer (22.4 bp) but statistically insignificant difference in tract length (p = 0.55). This may be because only a few switch variants (11) were recovered with the mutL strain.
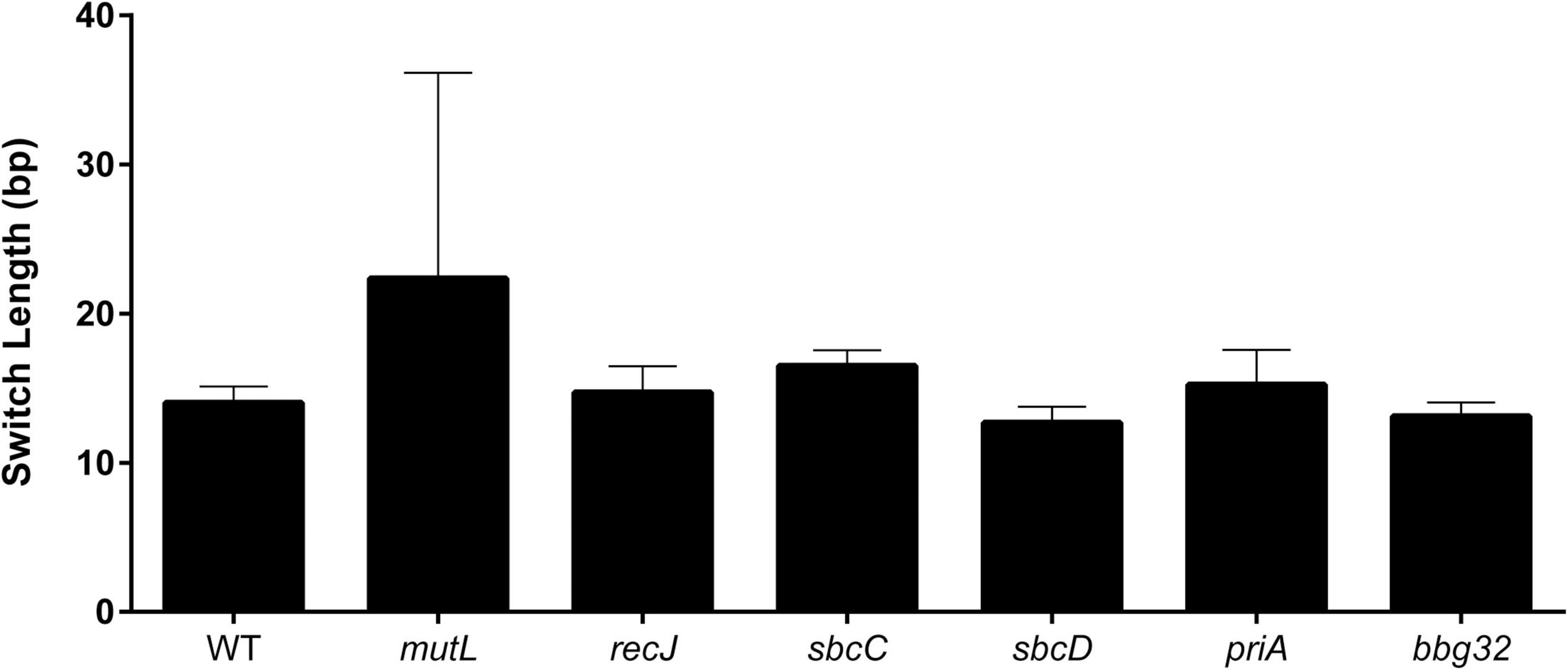
Figure 3. Length of recombination tracts in strains with mutant replication/recombination/repair genes. Recombination tracts were inferred by minimizing the number of recombination tracts required to explain the SNPs of a given read as previously described (Verhey et al., 2018b). The minimum length of each recombination tract is shown as the mean ± SEM for vlsE sequences at 5 weeks post-infection.
We then analyzed the silent cassette usage in each of the mutants except mutL (Figure 4). There was no significant difference in cassette usage for any of the mutants at any of the 15 silent cassettes. Finally, we analyzed the frequency of non-templated SNPs generated by each mutant (Figure 5). The non-templated changes are generated by error-prone repair and the number is correlated with the level of switch events [26], reminiscent of mutagenic changes associated with mitotic gene conversion in yeast [32]. This complicates a simple analysis to determine whether the various gene disruptions influence the level of non-templated SNPs, since the mutants used show variable levels of templated SNPs. We therefore plotted the frequency of non-templated SNPs against the frequency templated SNPs (Figure 5). Regardless of the recombinational switching level, if the non-templated SNP level per templated switch remains constant for each mutant, the data would be expected to lie on a diagonal line. As shown in the plot, a least squares regression line accommodates the data agree well with a Pearson correlation coefficient, r = 0.9086 indicating a similar non-templated SNP frequency per templated SNP for the WT and all the mutants tested, with the exception of bbg32. This mutant displayed a 44% decrease from the line of best fit, outside of the 95% confidence limits, suggesting that deletion of the replicative helicase encoded by lp28-2 may result in a less than twofold decrease in non-templated SNP frequency. Because of the multiplicity of helicases and possible pinch-hitting of one helicase for another, assessment of helicase activity in the generation of non-templated SNPs may obscure a possible role in these experiments.
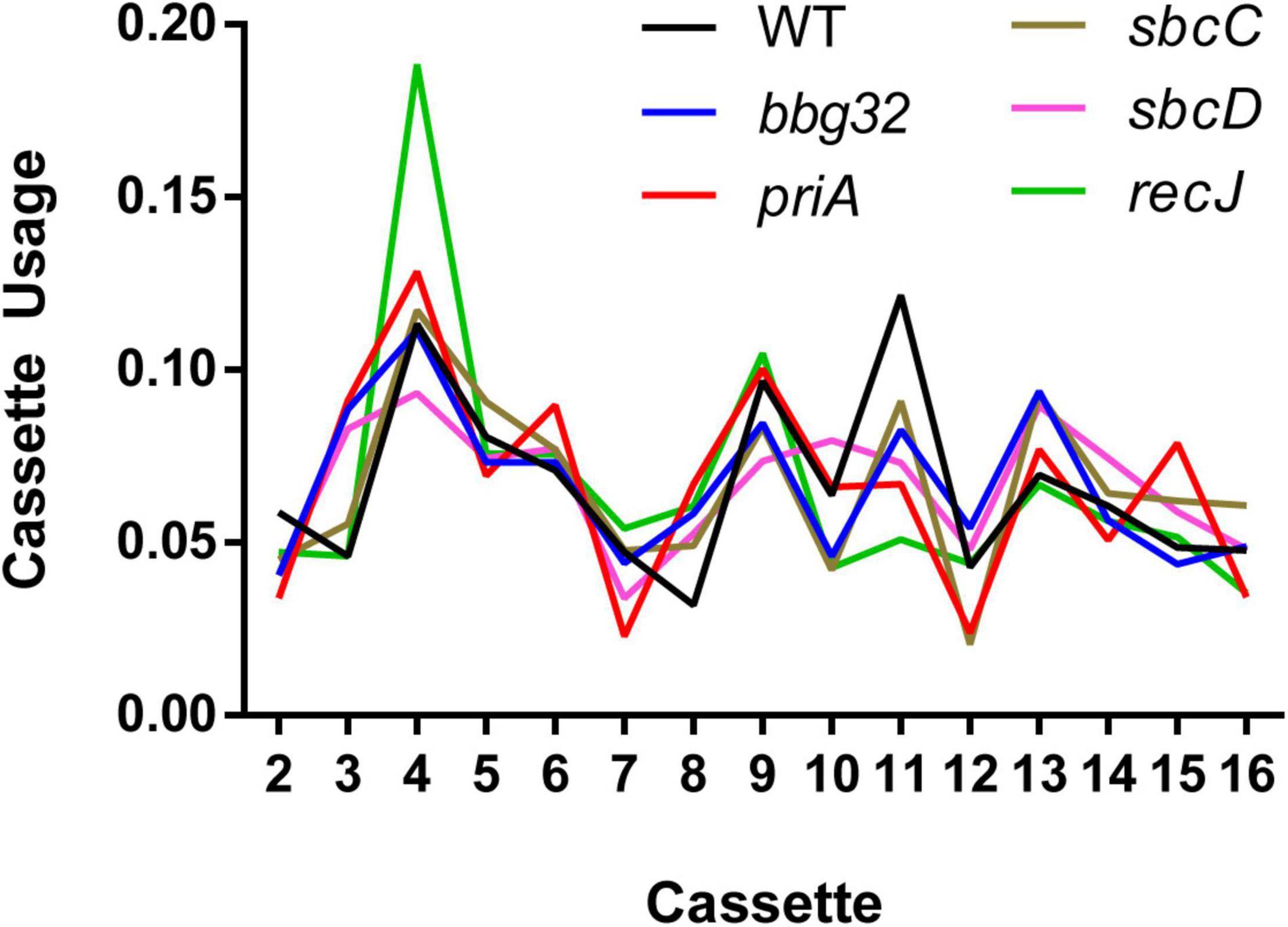
Figure 4. Cassette usage for inferred recombination tracts. For each inferred recombination event, we identified the most likely originating cassette(s). The plot shows the relative usage of each silent cassette as a proportion compared to other cassettes for each mutant strain. Recombination events having N possible origins were spread out over all possible silent cassettes with a weight of 1/N. The mutL gene disruption was omitted due to low switching activity resulting in a small sample size of only 11 recombination events.
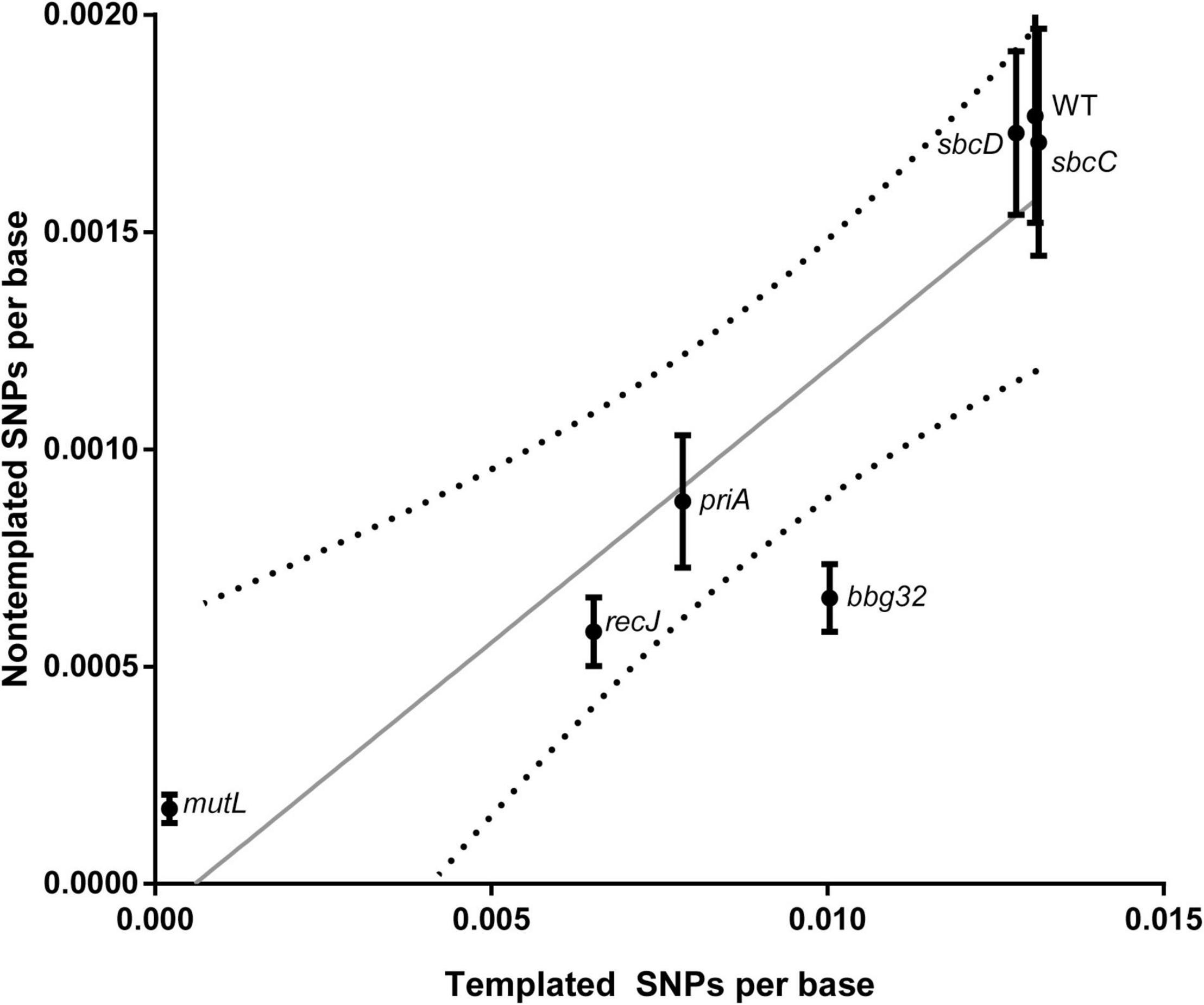
Figure 5. Frequency of non-templated SNPs versus templated SNPs at 5 weeks for WT and each gene disruption mutant. The unit of observation is each base sequenced, and the mean ± SEM is shown. A least-squares regression line and associated 95% confidence bands (dotted lines) are shown.
Generation of MutL Mutants Specifically Defective in Adenosine Triphosphate Binding, Adenosine Triphosphate Hydrolysis, DNA Nicking, or Interaction With the β-Sliding Clamp
To determine which functional region(s) of MutL (Fukui et al., 2011; Guarne, 2012; Banasik and Sachadyn, 2014; Guarne and Charbonnier, 2015) are involved in recombinational switching at vlsE, we generated chromosomal mutL mutants in the endogenous mutL locus, with specific changes in motifs of the protein predicted to be involved in adenosine triphosphate (ATP) binding, ATP hydrolysis, endonuclease activity, or in the β-clamp binding motif. The mutations introduced were based upon MutL mutations that affected mismatch repair in the MutL prototype from Bacillus subtilis (Pillon et al., 2010, 2011; Bolz et al., 2012). The B. burgdorferi mismatch repair system lacks a MutH ortholog as does B. subtilis, and the MutL proteins from these species contain an endonuclease motif (Figure 6) believed to be responsible for nicking of mismatched DNA once identified by MutS. In selecting the amino acids in MutL to mutate, we started with previously characterized mutations in B. subtilis and chose residues that were identical (or conserved in the case of L479) in B. burgdorferi and which demonstrated a phenotype in B. subtilis with single amino acid changes in the putative ATP binding, ATP hydrolysis and endonuclease motifs (Supplementary Figure 4). Several residues were changed in the β-clamp binding motif to most effectively neutralize the MutL- β-clamp interaction (Pillon et al., 2010).
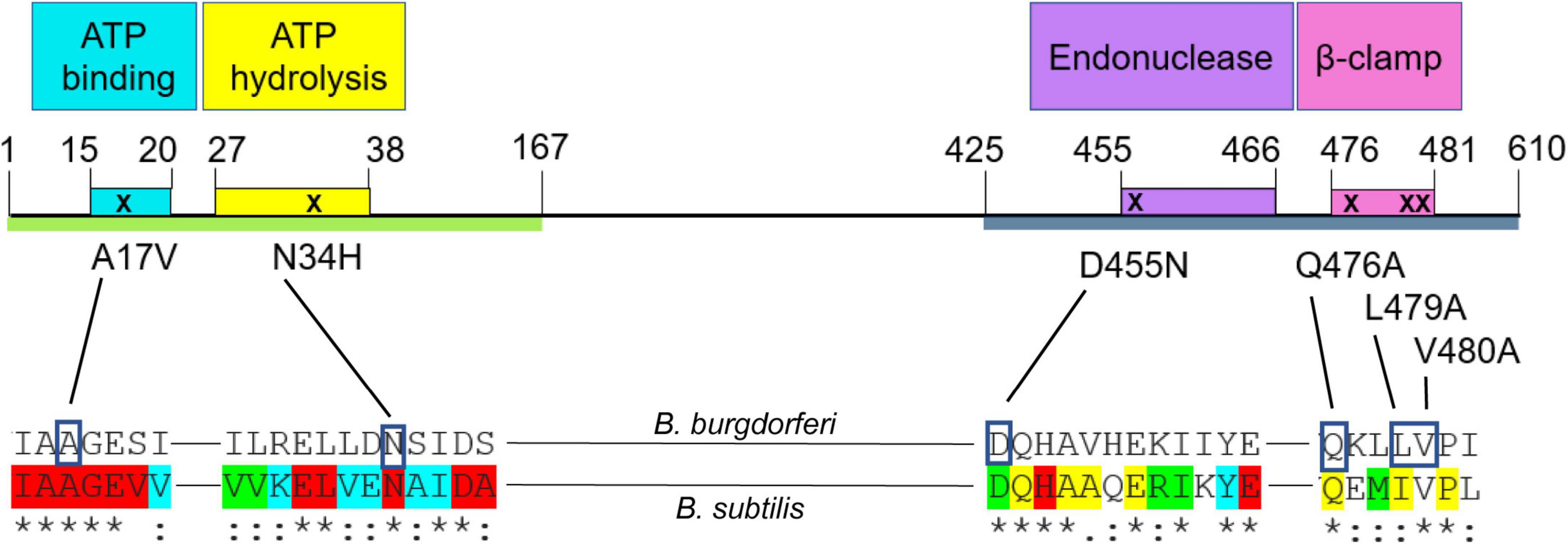
Figure 6. Schematic of Borrelia burgdorferi MutL protein and point mutations introduced in this study. The green line represents the N-terminal (ATPase) domain of MutL (Bolz et al., 2012) and the blue one, the C-terminal domain (endonuclease) domain (Pillon et al., 2010, 2011). The colored boxes represent the conserved motifs that confer different activities to the protein (Pillon et al., 2010, 2011; Bolz et al., 2012). Conserved residues in a lineup of about 200 MutL orthologs (Banasik and Sachadyn, 2014) are indicated at the bottom of the figure. The colors show different degrees of conservation, where red indicates an identical status in all the sequences included in the analysis. The blue color indicates only conservative amino acid changes in 100% of the proteins; green indicates only conservative amino acid changes in more than 90% of the sequences analyzed and yellow shows those residues with only conservative changes in 60–90% of the sequences analyzed. The asterisks indicate identical residues between the B. burgdorferi and B. subtilis alignment, the colons denote amino acids with conserved but non-identical changes.
Site-directed mutagenesis of the large 1,830 bp mutL gene was not successful in our early attempts, despite our previous experience constructing site directed mutants in the B. burgdorferi hrpA gene (Salman-Dilgimen et al., 2013). However, success was achieved using the general strategy shown in Figure 7 to generate chromosomal mutations (see section “Experimental Procedures” for details), which may also be useful for mutating other difficult B. burgdorferi genes. To facilitate site-directed mutations in the large (1,830 bp) coding region, we first introduced the point mutations at the complementary ends of double stranded fragments that when annealed would regenerate the full mutL gene. These fragments were cloned in pJET (Figure 7A). With the mutated versions in pJET, the resulting mutL genes were transferred to pOK12 through conventional cloning (Figures 7B,C). Subsequently the gentamicin resistance cassette was inserted downstream of mutL (Figure 7D) followed by the downstream gene bb0212 (to serve as a recombination target) after the gent cassette (Figure 7E). This resulted in the mutL donor plasmids pMC148, pMC149, pMC150, and pMC151 (Supplementary Table 3), which were used to transform B. burgdorferi B31 5A4 for gentamicin resistance. Clones were further analyzed by PCR for double recombination events (Figure 7F) as described in section “Experimental Procedures” to recover the point mutations in the endogenous mutL gene of the B. burgdorferi chromosome (Figure 7G).
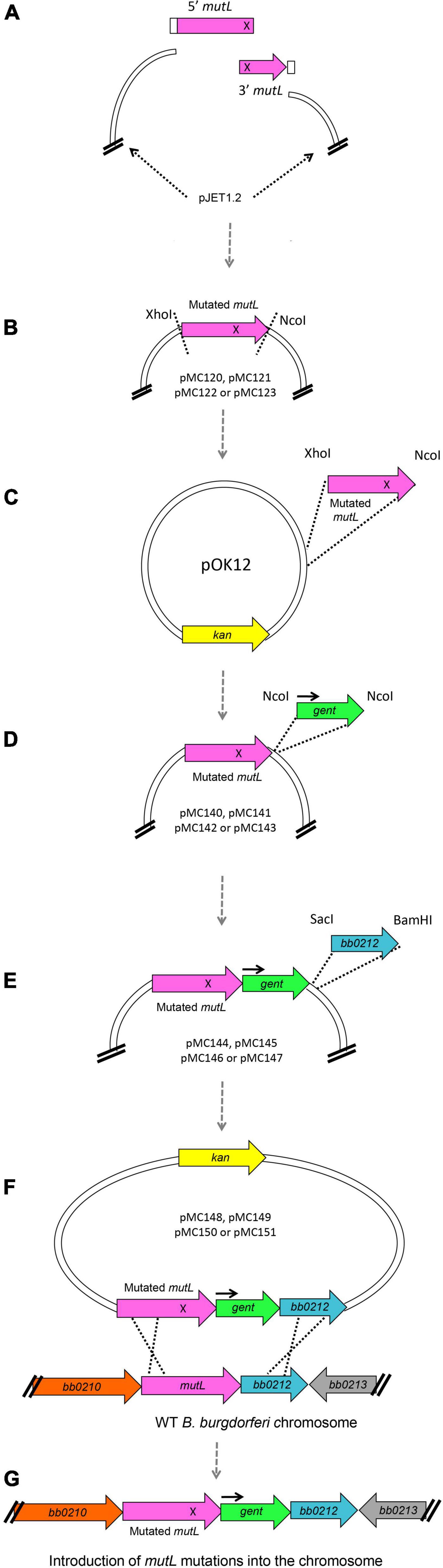
Figure 7. Schematic of plasmid construction and introduction of point mutations into B. burgdorferi chromosomal mutL. (A) To generate the E. coli plasmids carrying point mutations in mutL we designed a two-insert strategy where two mutL double stranded fragments carrying complementary base pair changes (represented as an X) were ligated with each other and to pJET1.2 using the HiFi DNA Assembly mix from NEB. This assembly mix contains a 5′-3′ exonuclease that generates single stranded 3′ overhangs. These overhangs are able to anneal for a high-fidelity polymerase to extend the overhangs and fill in the gaps; finally, a DNA ligase seals the nicks. (B) The pJET plasmids containing the mutated versions of mutL were then digested with XhoI and NcoI, and (C) the mutated mutL inserts were then cloned into pOK12, (D) generating the plasmids pMC140, pMC141, pMC142, and pMC143 (see Supplementary Table 3). We then inserted the gentamicin resistance cassette with its own promoter (black arrow) downstream of mutL at the NcoI site. (E) Finally, we cloned the bb0212 gene downstream of the gent cassette to maintain the natural gene order in the B. burgdorferi chromosome. (F) Plasmids pMC148, pMC149, pMC150, and pMC151 were used to transform B. burgdorferi B31 wild-type clone 5A4 [20] and (G) GentR-KanS transformants were further analyzed for double recombination events.
Analysis of Recombinational Switching in MutL Mutants in the Motifs for Adenosine Triphosphate Binding, Adenosine Triphosphate Hydrolysis, DNA Nicking, or Interaction With the β-Sliding Clamp
Following infection of SCID mice with two clones for each mutant (except the ATP hydrolysis mutant), spirochetes were recovered from blood, ear, or bladder. DNA was prepared and vlsE gene amplicons were sequenced and analyzed by PacBio long read sequencing and VAST software (Verhey et al., 2018a,b) (see section “Experimental Procedures”). As expected, the wild-type strain showed increasing switching at vlsE over time and the mutL deletion strain did not show switching above background levels (Figure 8). Interestingly, the putative ATP binding mutant showed no decrease in switching at vlsE from the wild-type strain, however, the ATP hydrolysis mutant displayed a significant decrease in recombinational switching. These results were perplexing, because if ATP hydrolysis is required for MutL to promote switching at vlsE, then ATP binding should also be required. The simplest proposal for the discrepancy is that the single A17V mutation in B. burgdorferi MutL (Figure 6) is not sufficient to disrupt ATP binding, even though it does so in the orthologous B. subtilis MutL. Another possibility is that the ATP-off (or ADP-on) state is active in vlsE switching. A concern with the ATP binding versus hydrolysis comparison was that only a single mutant clone in the ATP hydrolysis motif was used in the data shown in Figure 8. We therefore repeated the SCID mouse infection with two additional mutant clones and analyzed the results using classical sequencing of independent switch variants (Dresser et al., 2009). The mutant clone used for Figure 8 and the two additional ATP hydrolysis mutant clones all demonstrated a dramatic decrease in vlsE switching (Supplementary Figure 5), confirming our initial result shown in Figure 8.
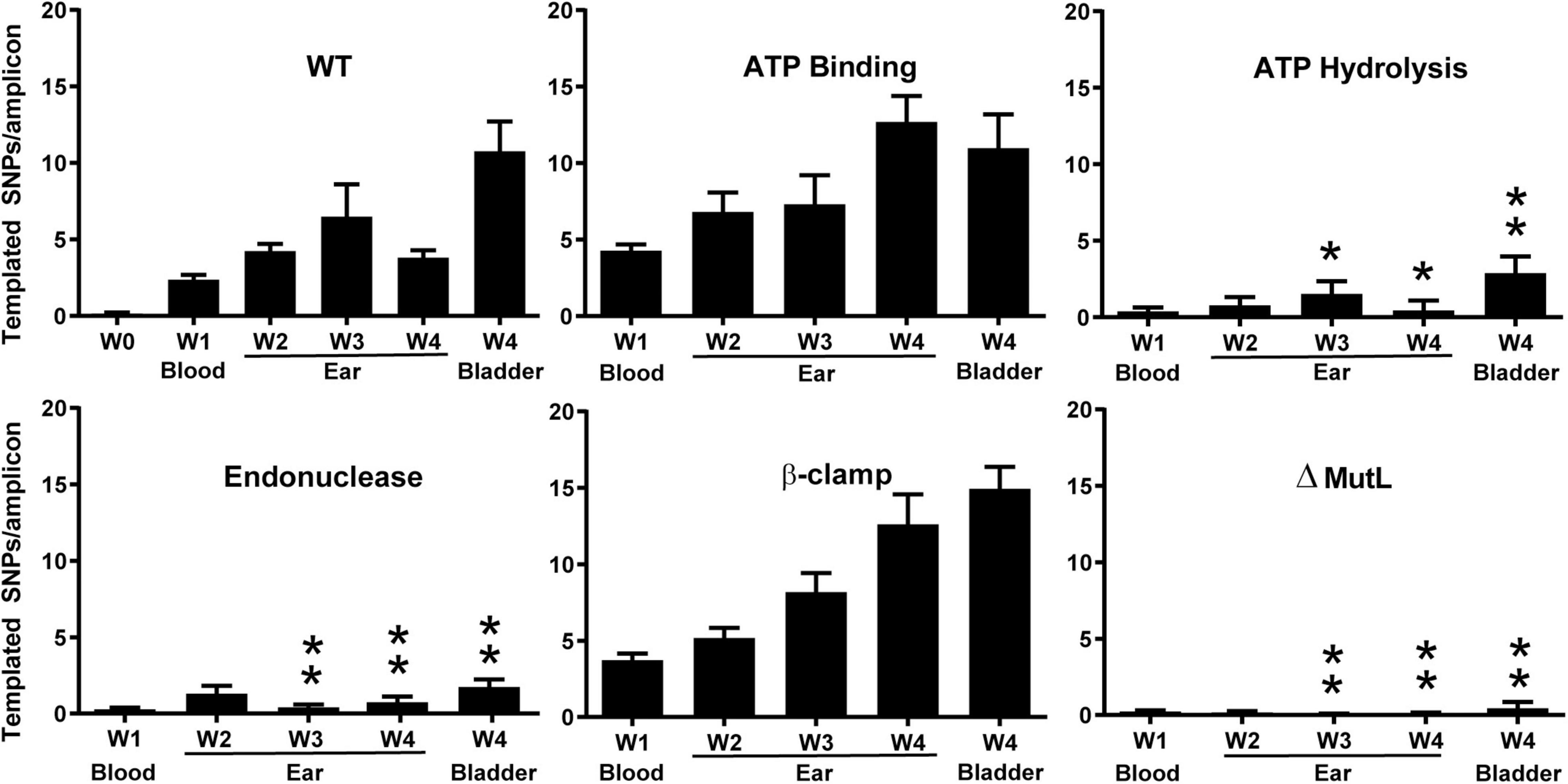
Figure 8. The effect of mutations in MutL on recombinational switching at vlsE as measured by the number of templated SNPs per amplicon from NGS sequencing at weeks 1 in mouse blood and at weeks 1–4 in mouse ear and at week 4 in bladder. The wild-type week 0 sample was from spirochetes that were not used to infect mice. B. burgdorferi carrying the mutations described in Figure 6 was used to infect SCID mice as described in section “Experimental Procedures.” Mean ± SEM are shown. P values were determined for differences between WT and mutL mutants in the corresponding tissue type and time. A single asterisk indicates p < 0.05 and two asterisks indicated p < 0.01. P values were determined using two-sample t-test with unequal variances.
The most interesting result from the analysis of the mutL mutants was the apparent requirement of the putative MutL endonuclease activity for recombinational switching at vlsE. A dramatic and significant decrease in templated SNPs was observed with the D455N endonuclease mutant. This is a conversion of the aspartic acid to the corresponding amide that is not expected to affect the three-dimensional structure of the protein but is expected to disrupt the metal ion binding required for hydrolytic activity, suggesting that the DNA endonuclease activity is required for switching at vlsE. Finally, the triple mutation in the β-clamp binding motif of MutL did not reduce the level of recombinational switching.
Discussion
MutL Is Required and RecJ Appears to Play a Role in vlsE Gene Conversion
The MutL protein (Fukui et al., 2011; Guarne, 2012; Banasik and Sachadyn, 2014; Guarne and Charbonnier, 2015) and its orthologs are involved in mismatch repair (Iyer et al., 2006; Modrich, 2006, 2016), an important process in all organisms whereby mis-incorporated bases from replication errors are removed and the original DNA sequence is restored. The bacterial MutS protein is responsible for recognizing mismatches and in organisms that encode it, MutH cleaves the DNA backbone for subsequent processing. In organisms lacking a MutH ortholog, MutL encodes the endonuclease activity to nick the DNA for entry of an exonuclease and removal of the area containing the mismatched base, preparing the area for resynthesis and correction. The phenotype of a mutL mutant in prokaryotes is a dramatic increase in mutation frequency (Schaaper, 1993). This is expanded in eukaryotes by a dramatic increase in cancers, such as in Lynch syndrome (Olave and Graham, 2021). The pronounced reduction in recombinational switching to background levels in the vlsE gene in the absence of MutL (Figure 2 and Supplementary Figures 2, 3) was surprising, especially because deletion of key interacting partner, MutS, did not affect either the switching frequency or the infectivity of mutS spirochetes in immunocompetent mice (Dresser et al., 2009). Taken together, these results point to an unexpected role for MutL that is outside its usual mismatch repair function (since MutS is not involved) and raises the possibility that MutL may be directed to vlsE, the silent cassettes, or a synapse between them, by a yet unidentified protein. This work reports for the first time a function for MutL in bacterial DNA recombination.
In addition to an important role for MutL, the absence of RecJ (Lovett, 2011; Cheng et al., 2016) resulted in a significant 50% decrease in switching in vlsE (Figure 2 and Supplementary Figures 2, 3). It is noteworthy that the RecJ 5′ to 3′ exonuclease often functions at nicks generated by MutL (Fukui et al., 2011; Guarne, 2012) suggesting that their paired activity may be important for recombinational switching. The failure of the recJ mutant to completely block gene conversion at vlsE is not surprising since RecJ shares overlapping functions with other cellular exonucleases (Lovett, 2011) that might pinch-hit in its absence.
Finally, disruption of the PriA replication restart protein (Michel et al., 2018; Windgassen et al., 2018) resulted in a 41% decrease in switching (Figure 1), on the threshold of significance (p = 0.057). However, changes in parental DNA sequences (Supplementary Figure 2) or distinct variants (Supplementary Figure 3), relative to wild-type did not vary significantly, leaving a possible role for the PriA helicase as an open question at this time.
The Putative MutL Endonuclease Is Required for vlsE Gene Conversion
To further probe the role of MutL in recombinational switching at vlsE, site directed mutations were generated in the putative ATP binding, ATP hydrolysis, endonuclease, and β-clamp binding motifs previously identified and characterized in B. subtilis (Figure 6). Interestingly, mutations in either the ATP binding or β-clamp binding motifs did not display a decrease in recombinational switching at vlsE. In contrast, mutagenesis of both the endonuclease and ATP hydrolysis motifs resulted in significant dramatic decreases in gene conversion (Figure 8). Our favored interpretation of the discrepancy in the ATP-interacting motifs is that the conservative change in the ATP binding motif of the A17V mutant is ineffectual at blocking ATP binding. Another possibility is that the ATP-off (or ADP-on) state is active in vlsE switching. However, we also cannot rule out that the single amino acid change in the hydrolysis mutant (N34H) is disruptive to the overall protein structure, thereby abrogating gene conversion by an indirect effect rather than specifically blocking ATPase activity. This possibility seems less likely to us in view of the demonstrated specificity of the mutation in B. subtilis MutL. The lack of an effect of the ATP binding mutant in contrast to the dramatic phenotype of the ATP hydrolysis mutant will require further analysis by genetic and/or biochemical approaches not currently available in vivo or in vitro for B. burgdorferi MutL.
The endonuclease activity of MutL (in bacteria lacking MutH) unlocks the double helix for exonucleolytic removal of mis-incorporated bases. B. burgdorferi lacks a MutH ortholog and its MutL N-terminal domain carries a clearly recognizable endonuclease motif (Figure 6). Substitution of aspartic acid 455 with an asparagine removed an essential catalytic residue based upon structure and function of the B. subtilis enzyme (Pillon et al., 2010). The dramatic decrease in gene conversion from the substitution of the acid with an amide (change of the hydroxyl group to an amino group) at position 455 strongly suggests that the putative endonuclease activity is required for gene conversion at vlsE (Figure 8). This type of mutagenic approach in the metal ion binding site of many hydrolytic enzymes has been used to identify active site residues. The ATPase region in the N-terminal domain of MutL (Ban et al., 1999; Fukui et al., 2011) has a role in modulating the endonuclease structure and function in the C-terminal domain of MutL (Pillon et al., 2010; Yamamoto et al., 2011; Bolz et al., 2012) and we speculate that the B. burgdorferi ATP hydrolysis mutant manifests its phenotypic effect by blocking the endonuclease activity.
Finally, mutation of three residues in the β-clamp binding motif (Figure 6) did not display any negative effects on gene conversion at vlsE (Figure 8). The β-clamp can modulate the endonuclease activity of B. subtilis MutL (Pillon et al., 2011, 2015; Almawi et al., 2019) and the results suggest (if the mutations disrupt MutL- β-clamp interactions) that the β-clamp does not play a role in recombinational switching at vlsE. The efficacy of this triple MutL mutant in abrogating β-clamp interactions will need to be confirmed by genetic or biochemical approaches.
Role of the Putative MutL Endonuclease in Recombinational Switching
Endonuclease activities are common initiators of DNA recombination reactions. The generation of double strand breaks (Haber, 2018; Wright et al., 2018; Li et al., 2019) or single strand discontinuities (Maizels and Davis, 2018) can promote recombination either by providing a DNA end for strand invasion, allowing an entry point for an exonuclease or by cleaving branched junctions to resolve recombination intermediates, as is the case in eukaryotic meiotic recombination. The MutL endonuclease could potentially be involved in either the initiation or resolution steps of recombination or both. The endonuclease activity of MutL is non-specific and has not been reported to recognize any specific sequence or DNA lesion (Fukui et al., 2011; Guarne, 2012). How then might MutL be recruited to the vlsE locus to participate in generating segmental gene conversion events? MutL has been reported to interact with several other proteins including MutS, UvrD and the DNA polymerase β-clamp. One option for targeting is through protein-protein interactions (Friedhoff et al., 2016), although this still leaves open the question of how the vls locus is recognized in the first place. A second possibility might be a direct recruitment whereby MutL directly recognizes the vls locus. We have previously reported an exceedingly high preponderance of strand-specific G-runs of 3 nucleotides or longer in vlsE and in the silent cassette coding strands (Walia and Chaconas, 2013; Chaconas et al., 2020). It is tempting to speculate that assembly of transient G-quadruplex structures from the multitude of G-runs might provide a target for recruitment of MutL, which through its endonuclease could generate a free DNA end for strand invasion to initiate a recombination reaction. A recent report of specific binding of Escherichia coli MutL to G4 DNA (Pavlova et al., 2020) suggests that this possibility is worth further investigation by biochemical characterization of the B. burgdorferi MutL protein.
Although a role for MutL in recombination has not been previously reported in bacteria, several reports of a role for MutL orthologs in recombination in higher organisms have (Argueso et al., 2004; Nishant et al., 2008; Zakharyevich et al., 2012; Ranjha et al., 2014; Rogacheva et al., 2014; Duroc et al., 2017; Manhart et al., 2017; Toledo et al., 2019; Cannavo et al., 2020; Kulkarni et al., 2020; Rahman et al., 2020). Of particular interest is the consistent implication of the endonuclease activity of MutL orthologs at a late step of homologous recombination as a double Holliday junction resolving enzyme that is activated by PCNA (Kulkarni et al., 2020), the eukaryotic equivalent of the bacterial β-clamp protein. Interestingly, our triple substitution (Figure 6) in the β-clamp interacting domain of MutL displayed recombinational switching at wild-type levels (Figure 8) suggesting the possibility that perhaps the endonuclease activity may not be involved in the junction resolution step at vlsE.
Previously we and the Norris lab (Dresser et al., 2009; Lin et al., 2009) had shown that the RuvAB complex, which can branch-migrate Holliday junctions, was a key factor in vlsE diversification. In many bacteria, RuvAB acts in conjunction with the Holliday junction resolvase, RuvC; but a RuvC ortholog is apparently absent from B. burgdorferi. It is possible that another structure-specific endonuclease could act in place of RuvC, and indeed, as noted above, in eukaryotes, the MutL homolog complex Mlh1-Mlh3, has been shown to cleave such branched structures (Argueso et al., 2004; Nishant et al., 2008; Zakharyevich et al., 2012; Ranjha et al., 2014; Rogacheva et al., 2014; Duroc et al., 2017; Manhart et al., 2017; Toledo et al., 2019; Cannavo et al., 2020; Kulkarni et al., 2020; Rahman et al., 2020).
Another possibility is that recombinogenic lesions are generated by the processing of site-specific cytidine deaminations, and depend on MutL. The vls gene arrangement in B. burgdorferi is strikingly similar to the immunoglobulin genes in chickens, where a single V(D)J segment is diversified by gene conversions from an array of adjacent pseudogenes. In the avian system, these gene conversions are initiated by chromosome breaks created by the mismatch repair-dependent processing of dU::G base pairs that are created by the activation-induced cytidine deaminase (AID; Di Noia and Neuberger, 2007; Arakawa and Buerstedde, 2009; Lanning and Knight, 2015).
The required role of MutL for recombination in a recombination-driven antigenic variation pathway is uniquely reported here for B. burgdorferi. This, plus additional unique features of this reaction such as its RecA independence (Liveris et al., 2008; Dresser et al., 2009) and the dispensability of a variety of other proteins required by the well-characterized N. gonorrhoeae system for antigenic variation (Obergfell and Seifert, 2015; Chaconas et al., 2020), make further studies on gene conversion in B. burgdorferi an exciting frontier, ripe for further study and development of biochemical and genetic analyses.
Experimental Procedures
Construction of Plasmids Carrying mutL Mutations
All strains and plasmids used in this study are noted in Supplementary Table 3 and were propagated in BSK-II prepared in-house and supplemented with 6% rabbit serum (Barbour, 1984). Strains were stored at −80°C in medium containing 20% glycerol. B. burgdorferi constructs were made in the B31 5A4 strain (Purser and Norris, 2000). Gene disruption mutants were generated by allelic exchange as previously described (Dresser et al., 2009). To generate mutations in various B. burgdorferi MutL motifs (Supplementary Table 3), we introduced point mutations in the regions for ATP binding, ATP hydrolysis activity, the endonuclease activity and the region interacting with the DNA polymerase β-clamp (Figure 6, Supplementary Table 4, and Supplementary Figure 4). As a general strategy, primers were designed to amplify the full mutL (or full mutL plus part of the upstream gene, depending on the domain to be mutated) in two parts with overlapping sequences so they could be joined without “scars” other than the point mutations. The point mutations were introduced at the overlapping sequences that the primers carry (Supplementary Table 4 and Figure 7). The primer combinations are as follows: for the ATP Binding site we used primers B3049 and B3050 (left side, 643 bp), and B3051 and B3052 (right side, 1,824 bp), for the ATP hydrolysis KO mutant we used primers B3049 and B3053 (left side, 689 bp), and B3054 and B3052 (right side, 1,778 bp). For the endonuclease point mutation we used primers B3055 and B3056 (left side, 1,117 bp), and B3057 and B3052 (right side, 512 bp). Finally, to mutate the β-clamp binding domain we used primers B3055 and B3058 (left side, 1,193 bp), and B3059 and B3052 (right side, 433 bp). The 5′ and 3′ ends of mutL also have overlapping sequences to pJET1.2 (NEB) so they could be sub-cloned first. Amplification conditions for the mutL PCRs are as follows: a denaturation step at 95°C for 2 min, followed by 30 cycles at 95°C for 15 s, 50 or 54°C for 15 s and 68°C for 60 s, finished by a final extension cycle at 68°C for 5 min. Phusion High Fidelity DNA Polymerase from NEB was used for all the PCRs described here. PCR products were gel purified when they displayed non-specific bands (Zymoclean Gel DNA recovery kit) or cleaned with a PCR column (Qiagen PCR purification kit). Ligations were performed using the NEBuilder HiFi DNA Assembly master mix by adding 0.14 pmols per insert (right and left sides of mutL for each domain to be mutated) and 0.07 pmols of pJET1.2 (NEB) in a final volume of 20 μL. Ligations were incubated at 50°C for 15 min and 3 μL were used to transform chemically competent DH5-α cells. Candidates were analyzed by digesting the plasmid DNA with BglII (NEB) for 2 h at 37°C. Those that had the expected band sizes were then sequenced to confirm the presence of the point mutations. Sanger sequencing was done by using the pJET1.2 Forward and Reverse primers using a 60°C annealing temperature.
Subsequently, the full mutL gene was transferred to the vector pOK12 (Vieira and Messing, 1991) which is unable to replicate in B. burgdorferi. To do this, we digested the pOK12 and pJET::mutL constructs with XhoI and NcoI with 10 units of each enzyme overnight. Bands were purified from 1% agarose gels (Zymoclean Gel DNA recovery kit) and ligated with T4 DNA ligase from NEB for 2 h at room temperature. We used 50 ng of pOK12 with a 1:1 vector:insert molar ratio. 5 μL of the ligation reactions were used to transform competent DH5-α cells. Candidate colonies were analyzed through digestion patterns with XhoI and NcoI. In this way we generated plasmids pMC140, pMC141, pMC142, and pMC143 (Supplementary Table 3). The next step was to clone the gentamicin cassette downstream of point mutated mutL in these constructs. The gent cassette was amplified with oligos B3061 and B3094, introducing NcoI sites on both sides (Supplementary Table 4), by 30 cycles at 95°C for 10 s, 60°C for 10 s, and 72°C for 25 s, and followed by a final extension cycle at 72°C for 5 min. The gentamicin product and plasmids pMC140, pMC141, pMC142, and pMC143 were digested overnight with NcoI and gel purified. Ligations were performed in molar ratios 1:5 vector:insert with T4 DNA ligase at 16°C overnight. 5 μL of the reactions were used to transform competent DH5-α. Since the gentamicin cassette could be cloned in either orientation, we selected those that had it in the same direction as mutL. To this end we used the restriction enzyme NdeI and incubated the reactions for 2 h at 37°C. We called the new plasmids: pMC144, pMC145, pMC146, and pMC147 (Supplementary Table 3).
The final step in the construction of the domain knock-out vectors was to clone bb0212 downstream of the gentamicin cassette; this is the gene located downstream of mutL in the wild type configuration. bb0212 was amplified with oligos B3101 and B3102 (Supplementary Table 4), which contained the SacI and BamHI restriction sites, respectively. Amplification conditions were done with a first denaturation step at 95°C for 2 min, followed by 30 cycles at 95°C for 10 s, 55°C for 10 s, and 68°C for 25 s, finished with a final extension cycle at 68°C for 5 min. Plasmids pMC144, pMC145, pMC146, and pMC147 and the bb0212 gene were digested with SacI and BamHI overnight and purified from 1% agarose gels. Ligations were performed at molar ratios of 1:3 vector:inserts and incubated 2 h at room temperature. 5 μL was used to transform competent DH5-α cells and candidate colony plasmid DNAs were digested with either BamHI or SpeI. Those clones that produced the expected digestion patterns were then sent sequenced to verify the presence of the desired mutations and the integrity of the remainder of the mutL gene. The oligos used to sequence mutL were: B3107, B3108, B3109, B3110, and B3052 (Supplementary Table 4). Final plasmids were named plasmids pMC148, pMC149, pMC150, and pMC151.
Introduction of mutL Domain Mutants Into the B. burgdorferi Chromosome
Allelic exchange was performed as previously described (Samuels et al., 2018). The construction strategy used in this work is outlined in Figure 7. The gene of interest was amplified and cloned into a vector unable to replicate in B burgdorferi. Then an antibiotic cassette was inserted in the middle of the gene to interrupt it, but conserving homologous regions flanking the antibiotic cassette that serve as recombination targets. This construct was used to transform B. burgdorferi strain B31 5A4 (Purser and Norris, 2000; see Supplementary Table 1). For allelic exchange, 50 μg of plasmids pMC148, pMC149, pMC150 and 40 μg of pMC151 were used in the transformations. To screen for transformants that had acquired the mutations, we designed primers that contain the point mutations at the 3′ end of the relevant primers such that amplification occurred only if the mutation had been introduced but with the wild-type gene. Primers used to screen the ATP binding and ATP hydrolysis domain mutants were B3113 and B3107, and B3107 and B3114, respectively. The PCR program used for them was a two-step PCR for 30 cycles at 95°C for 10 s and 68°C for 20 s of annealing/extension. To screen for the endonuclease mutation, we used primers B3115 and B3117 with an annealing temperature of 58°C, while the denaturing and extension steps were the same as for the ATP binding and hydrolysis mutants. Finally, for the β-clamp binding site mutation, we used primers B3116 and B3117 at an annealing temperature of 50°C. All PCRs were carried out with Phusion polymerase from NEB. After identifying at least five candidates per construct, we grew them to saturation and purified the genomic DNA with two extractions of phenol-chloroform-isoamyl alcohol (25:24:1) and precipitation with isopropanol. Once the DNA was purified we did PCRs to screen for the presence of gentamicin and the absence of the kanamycin cassette by using the following conditions: 2 min at 95°C, followed by 30 cycles of 10 s at 95°C, 10 s at 50°C and 30 s at 68°C, and finally one cycle of 5 min at 68°C. PCR bands including the point mutations were amplified and sent for Sanger sequencing to confirm that the mutations were present. To this end we used oligos B3107 and B3122 for the ATP binding and ATP hydrolysis mutants, and B3109 and B3124 for the endonuclease and the β-clamp binding domain mutants. PCR conditions were 2 min at 95°C, followed by 30 cycles of 10 s at 95°C, 10 s at 60°C, and 20 s at 68°C, to finalize with one cycle of 5 min at 68°C. The same oligos were used to sequence the bands at 60°C.
Further characterization was performed to ascertain that our mutants were not merodiploid where only one recombination event occurred, instead of two, producing a cointegrate rather than an allelic replacement. To analyze possible merodiploidy we used the primers B3102 and B3110 where a 2,176 bp band would indicate an allelic replacement, while a 1,472 bp band would reveal the presence of wild-type copies of mutL. The PCRs were performed by incubating at 2 min at 95°C, followed by 30 cycles of 10 s at 95°C, 10 s at 60°C, and 60 s at 68°C, and finalized with one cycle of 5 min at 68°C. For the β-clamp binding site domain mutation, another primer set was used under the same conditions described above (B3110 and B3128).
Finally, we assessed plasmid content using a multiplex PCR assay (Bunikis et al., 2011) for all our mutant clones to confirm that they had not lost essential plasmids for infectivity and persistence. PCR conditions were as follows: one cycle to denature the DNA at 95°C for 2 min, then 30 cycles at 95°C for 30 s, 60°C for 60 s, 68°C for 60 s, and a final cycle at 68°C for 5 min.
Mouse Infections and Isolation of B. burgdorferi Genomic DNA
Mouse infections for Figures 2–5 were previously described (Dresser et al., 2009). Spirochetes were cultured from the frozen glycerol stocks from the SCID mouse infections from week 0 to week 5 (Dresser et al., 2009), and genomic DNA was isolated. For each gene disruption two B. burgdorferi clones were obtained, and two mice were infected per clone. Week 5 genomic DNA was pooled from spirochete cultures from four tissues: bladder, ear, heart, and joint. For the more recent mutL mutant infections, a total of 24 Fox Chase SCID male mice (C.B-17 SCID) were purchased from Charles River Laboratories. Two independent clones of each construct were individually grown to infect mice with 104 spirochetes in exponential growth phase in BSK-II medium. Additionally, two more clones belonging to the parental isogenic 5A4 strain and two clones belonging to the full mutL knock out were also used as positive and negative controls, respectively. Two mice were infected per clone, for a total of four mice per mutant (Supplementary Table 2). The inoculation was done by subcutaneous injection into the backs of mice at 4–6 weeks of age. DietGel® Boost (ClearH2O, Westbrook, ME, United States) was added to the cages as a supplement to help maintain weight. Tissue samples were collected at week 1 (50 μl of blood from the tail vein), week 2 and week 3 (ear punches), and week 4 post-infection (ear punches and bladder). Each of these samples was grown in BSK-II with 6% rabbit serum and antibiotics (0.02 mg ml–1 phosphomycin, 0.05 mg ml–1 rifampicin, and 2.5 μg ml–1 amphotericin B) to which B. burgdorferi is naturally resistant. Genomic DNA was purified from 8 ml cultures by phenol-chloroform extraction followed by isopropanol precipitation (Verhey et al., 2018a).
vlsE Polymerase Chain Reaction Amplification and Sequencing
The barcoded primers used in the present work were previously described in Verhey et al. (2018a) and Supplementary Table 1. The primers amplify a 776 bp band from vlsE, and contain barcodes that allow sample identification. We used a paired barcode scheme, where the combination of the forward and the reverse primer uniquely identified each sample (Supplementary Table 2). PCRs were performed for 30 cycles either with a three-step (Verhey et al., 2018a) or two-step (Dresser et al., 2009) cycle as previously described. Phusion Polymerase from New England Biolabs (NEB) was used for all amplifications. PCR products were quantified by agarose gel densitometry using Image-J, with standard curves made from DNA ladders from NEB following staining with ethidium bromide. Following quantification, samples were pooled, concentrated on a Qiagen QIAquick PCR Purification kit (Qiagen), run in an agarose gel and gel-purified without staining using either the Qiagen or Monarch (NEB) gel extraction kit. The recovered material was quantified by ethidium bromide staining and gel densitometry. The amplicon mixture was sequenced by PacBio long-read technology using the RS II instrument at Génome Québec Innovation Centre and three SMRT cells (Figures 2–5) and using the Sequel II instrument and one SMRT cell for Figure 8. Sanger sequencing of vlsE was performed as previously reported (Dresser et al., 2009).
Bioinformatics
PacBio sequencing data was processed from raw BAM files obtained from Génome Québec. To generate DNA strand-specific circular consensus sequences in FASTQ format, we used CCS v6.0.0 (Pacific Biosciences)1, and specified the –by-strand option. For Figures 2–5, we additionally filtered consensus reads with a minimum read quality of 0.99999 (equivalent to Q50), but set the –min-passes filter to 0 and the –min-snr filter to 0. For Figure 8 we specified a minimum read quality of 0.99975 (equivalent to Q36) but retained the default –min-passes filter of 3 and the –min-snr filter of 2.5. Sequences were demultiplexed using fdemux2, which detects unique asymmetric barcode pairs from PacBio reads. To maintain highly accurate demultiplexing, exactly two barcodes in the expected orientation were required, and an edit distance of 2 was specified to allow for some errors. Since any one barcode is at least 6 edits away from another, this should prevent false positives in demultiplexing.
Sequences were then trimmed to include 5′ and 3′ 16-mer flanking sequences which are invariant in the dataset due to their location in the constant region and inclusion in the PCR primers. These sequences were CGACGGGGAAACCAGA for the 5′ end of the amplicon and AGGCTGCTAGTAAAGA for the 3′ end.
Sequence data was then analyzed using our previously reported Variable Antigen Sequence Tracer software (Verhey et al., 2018a). Statistics were computed using GraphPad Prism 6.0.
Accession Numbers
PacBio sequencing data is available in the NCBI Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/sra: DNA replication and repair mutant sequencing, SRR18680908; MutL mutant sequencing, SRR18680907.
Data Availability Statement
All relevant data are within the manuscript and Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
All animal experimentation was carried out in accordance with the principles outlined in the most recent policies and Guide to the Care and Use of Experimental Animals by the Canadian Council on Animal Care. The animal protocols (AC16-0068 and AC20-0031) were approved by the Animal Care Committee of the University of Calgary.
Author Contributions
GC, TV, and MC: conception or design of the work. TV, MC, and MG: data collection. GC and TV: data analysis and interpretation. GC, TV, MC, and MG: drafting the article and final approval of the version to be published. GC: critical revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the Canadian Institutes of Health Research (PJT-153336) to GC. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Xi Tan for help with the SCID mouse infections for the two additional ATP hydrolysis mutants and Genevieve Chaconas for technical assistance. We would like to acknowledge Geneviève Geneau and Joëlle Nelcy from Génome Québec for preparing and sequencing PacBio amplicon libraries. We would also like to thank Jim Haber and Yuko Nakajima for helpful discussions and comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.888494/full#supplementary-material
Footnotes
References
Almawi, A. W., Scotland, M. K., Randall, J. R., Liu, L., Martin, H. K., Sacre, L., et al. (2019). Binding of the regulatory domain of MutL to the sliding beta-clamp is species specific. Nucleic Acids Res. 47, 4831–4842. doi: 10.1093/nar/gkz115
Arakawa, H., and Buerstedde, J. M. (2009). Activation-induced cytidine deaminase-mediated hypermutation in the DT40 cell line. Philos Trans R Soc Lond B Biol. Sci. 364, 639–644. doi: 10.1098/rstb.2008.0202
Argueso, J. L., Wanat, J., Gemici, Z., and Alani, E. (2004). Competing crossover pathways act during meiosis in Saccharomyces cerevisiae. Genetics 168, 1805–1816. doi: 10.1534/genetics.104.032912
Ban, C., Junop, M., and Yang, W. (1999). Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell 97, 85–97. doi: 10.1016/s0092-8674(00)80717-5
Banasik, M., and Sachadyn, P. (2014). Conserved motifs of MutL proteins. Mutat Res. 769, 69–79. doi: 10.1016/j.mrfmmm.2014.07.006
Bankhead, T., and Chaconas, G. (2007). The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol. Microbiol. 65, 1547–1558. doi: 10.1111/j.1365-2958.2007.05895.x
Barbour, A. G. (1984). Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 57, 521–525.
Bolz, N. J., Lenhart, J. S., Weindorf, S. C., and Simmons, L. A. (2012). Residues in the N-terminal domain of MutL required for mismatch repair in Bacillus subtilis. J. Bacteriol 194, 5361–5367. doi: 10.1128/JB.01142-12
Bunikis, I., Kutschan-Bunikis, S., Bonde, M., and Bergström, S. (2011). Multiplex PCR as a tool for validating plasmid content of Borrelia burgdorferi. J. Microbiol. Methods 86, 243–247. doi: 10.1016/j.mimet.2011.05.004
Cahoon, L. A., and Seifert, H. S. (2011). Focusing homologous recombination: pilin antigenic variation in the pathogenic Neisseria. Mol. Microbiol. 81, 1136–1143. doi: 10.1111/j.1365-2958.2011.07773.x
Cannavo, E., Sanchez, A., Anand, R., Ranjha, L., Hugener, J., Adam, C., et al. (2020). Regulation of the MLH1-MLH3 endonuclease in meiosis. Nature 586, 618–622. doi: 10.1038/s41586-020-2592-2
Castellanos, M., Verhey, T. B., and Chaconas, G. (2018). A Borrelia burgdorferi mini-vls system that undergoes antigenic switching in mice: investigation of the role of plasmid topology and the long inverted repeat. Mol. Microbiol. 109, 710–721. doi: 10.1111/mmi.14071
Chaconas, G., Castellanos, M., and Verhey, T. B. (2020). Changing of the guard: How the lyme disease spirochete subverts the host immune response. J. Biol. Chem. 295, 301–313. doi: 10.1074/jbc.REV119.008583
Cheng, K., Xu, H., Chen, X., Wang, L., Tian, B., Zhao, Y., et al. (2016). Structural basis for DNA 5 -end resection by Recj. elife 5:e14294. doi: 10.7554/eLife.14294
Deitsch, K. W., Lukehart, S. A., and Stringer, J. R. (2009). Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 7, 493–503. doi: 10.1038/nrmicro2145
Devlin, R., Marques, C. A., and McCulloch, R. (2017). Does DNA replication direct locus-specific recombination during host immune evasion by antigenic variation in the African trypanosome? Curr. Genet. 63, 441–449. doi: 10.1007/s00294-016-0662-7
Di Noia, J. M., and Neuberger, M. S. (2007). Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76, 1–22. doi: 10.1146/annurev.biochem.76.061705.090740
Dresser, A. R., Hardy, P.-O., and Chaconas, G. (2009). Investigation of the role of DNA replication, recombination and repair genes in antigenic switching at the vlsE locus in Borrelia burgdorferi: an essential role for the RuvAB branch migrase. PLoS Pathogens 5:e1000680. doi: 10.1371/journal.ppat.1000680
Duroc, Y., Kumar, R., Ranjha, L., Adam, C., Guerois, R., Md Muntaz, K., et al. (2017). Concerted action of the MutLbeta heterodimer and Mer3 helicase regulates the global extent of meiotic gene conversion. elife 6:e21900. doi: 10.7554/eLife.21900
Friedhoff, P., Li, P., and Gotthardt, J. (2016). Protein-protein interactions in DNA mismatch repair. DNA Repair (Amst) 38, 50–57. doi: 10.1016/j.dnarep.2015.11.013
Fukui, K., Shimada, A., Iino, H., Masui, R., and Kuramitsu, S. (2011). “Biochemical Properties of MutL, a DNA Mismatch Repair Endonuclease,” in DNA Repair: On the Pathways to Fixing DNA Damage and Errors, ed. F. Storici (Rijeka: Intech).
Gargantini, P. R., Serradell, M. D. C., Rios, D. N., Tenaglia, A. H., and Lujan, H. D. (2016). Antigenic variation in the intestinal parasite Giardia lamblia. Curr. Opin. Microbiol. 32, 52–58. doi: 10.1016/j.mib.2016.04.017
Glover, L., Hutchinson, S., Alsford, S., McCulloch, R., Field, M. C., and Horn, D. (2013). Antigenic variation in African trypanosomes: the importance of chromosomal and nuclear context in VSG expression control. Cell Microbiol. 15, 1984–1993. doi: 10.1111/cmi.12215
Guarne, A. (2012). The functions of MutL in mismatch repair: the power of multitasking. Prog Mol. Biol. Transl. Sci. 110, 41–70. doi: 10.1016/B978-0-12-387665-2.00003-1
Guarne, A., and Charbonnier, J. B. (2015). Insights from a decade of biophysical studies on MutL: Roles in strand discrimination and mismatch removal. Prog Biophys Mol. Biol. 117, 149–156. doi: 10.1016/j.pbiomolbio.2015.02.002
Haber, J. E. (2018). DNA Repair: The search for homology. Bioessays 40:e1700229. doi: 10.1002/bies.201700229
Horn, D. (2014). Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 195, 123–129. doi: 10.1016/j.molbiopara.2014.05.001
Iyer, R. R., Pluciennik, A., Burdett, V., and Modrich, P. L. (2006). DNA mismatch repair: functions and mechanisms. Chem. Rev. 106, 302–323. doi: 10.1021/cr0404794
Kulkarni, D. S., Owens, S. N., Honda, M., Ito, M., Yang, Y., Corrigan, M. W., et al. (2020). PCNA activates the MutLgamma endonuclease to promote meiotic crossing over. Nature 586, 623–627. doi: 10.1038/s41586-020-2645-6
Lanning, D. K., and Knight, K. L. (2015). Diversification of the primary antibody repertoire by AID-mediated gene conversion. Results Probl. Cell Differ. 57, 279–293. doi: 10.1007/978-3-319-20819-0_12
Lawrenz, M. B., Wooten, R. M., and Norris, S. J. (2004). Effects of vlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Infect Immun. 72, 6577–6585. doi: 10.1128/IAI.72.11.6577-6585.2004
Li, J., Sun, H., Huang, Y., Wang, Y., Liu, Y., and Chen, X. (2019). Pathways and assays for DNA double-strand break repair by homologous recombination. Acta Biochim Biophys Sin (Shanghai) 51, 879–889. doi: 10.1093/abbs/gmz076
Lin, T., Gao, L., Edmondson, D. G., Jacobs, M. B., Philipp, M. T., and Norris, S. J. (2009). Central role of the Holliday junction helicase RuvAB in vlsE recombination and infectivity of Borrelia burgdorferi. PLoS Pathogens 12:e1000679. doi: 10.1371/journal.ppat.1000679
Liveris, D., Mulay, V., Sandigursky, S., and Schwartz, I. (2008). Borrelia burgdorferi vlsE antigenic variation is not mediated by RecA. Infect. Immun. 76, 4009–4018. doi: 10.1128/IAI.00027-08
Lone, A. G., and Bankhead, T. (2020). The Borrelia burgdorferi VlsE Lipoprotein Prevents antibody binding to an arthritis-related surface antigen. Cell Rep. 30, 3663–3670. doi: 10.1016/j.celrep.2020.02.081
Lopez, J., Hovius, J. W., and Bergstrom, S. (2021). Pathogenesis of relapsing fever. Curr. Issues Mol. Biol. 42, 519–550. doi: 10.21775/cimb.042.519
Lovett, S. T. (2011). The DNA Exonucleases of Escherichia coli. EcoSal Plus 4:10.1128/ecosallus.4.4.7. doi: 10.1128/ecosalplus.4.4.7
Magunda, P. R. H., and Bankhead, T. (2016). Investigating the potential role of non-vls genes on linear plasmid 28–1 in virulence and persistence by Borrelia burgdorferi. BMC microbiol. 16:180. doi: 10.1186/s12866-016-0806-4
Maizels, N., and Davis, L. (2018). Initiation of homologous recombination at DNA nicks. Nucleic Acids Res. 46, 6962–6973. doi: 10.1093/nar/gky588
Manhart, C. M., Ni, X., White, M. A., Ortega, J., Surtees, J. A., and Alani, E. (2017). The mismatch repair and meiotic recombination endonuclease Mlh1-Mlh3 is activated by polymer formation and can cleave DNA substrates in trans. PLoS Biol. 15:e2001164. doi: 10.1371/journal.pbio.2001164
McCulloch, R., Morrison, L. J., and Hall, J. P. (2015). DNA Recombination strategies during antigenic variation in the African trypanosome. Microbiol. Spectr. 3:Mdna3–Mdna0016. doi: 10.1128/microbiolspec.MDNA3-0016-2014
Michel, B., Sinha, A. K., and Leach, D. R. F. (2018). Replication fork breakage and restart in Escherichia coli. Microbiol. Mol. Biol. Rev. 82:e00013–e18. doi: 10.1128/MMBR.00013-18
Modrich, P. (2006). Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 281, 30305–30309. doi: 10.1074/jbc.R600022200
Modrich, P. (2016). Mechanisms in E. coli and human mismatch repair (nobel lecture). Angew Chem. Int. Ed Engl. 55, 8490–8501. doi: 10.1002/anie.201601412
Nishant, K. T., Plys, A. J., and Alani, E. (2008). A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae. Genetics 179, 747–755. doi: 10.1534/genetics.108.086645
Norris, S. J. (2014). vls Antigenic variation systems of lyme disease Borrelia: eluding host immunity through both random, segmental gene conversion and framework heterogeneity. Microbiol. Spectr. 2:jbc.REV119.008583. doi: 10.1128/microbiolspec.MDNA1123-0038-2014
Obergfell, K. P., and Seifert, H. S. (2015). Mobile DNA in the Pathogenic Neisseria. Microbiol Spectr 3:MDNA3–MDNA0015.
Olave, M. C., and Graham, R. P. (2021). Mismatch repair deficiency: The what, how and why it is important. Genes Chromosomes Cancer 1–8. doi: 10.1002/gcc.23015 [Epub ahead of print].
Palmer, G. H., Bankhead, T., and Seifert, H. S. (2016). Antigenic variation in bacterial pathogens. Microbiol. Spectr. 4:10.1128/microbiolsec.VMBF–0005–2015. doi: 10.1128/microbiolspec.VMBF-0005-2015
Pavlova, A. V., Monakhova, M. V., Ogloblina, A. M., Andreeva, N. A., Laptev, G. Y., Polshakov, V. I., et al. (2020). Responses of DNA mismatch repair proteins to a stable g-quadruplex embedded into a DNA Duplex Structure. Int. J. Mol. Sci. 21:8773. doi: 10.3390/ijms21228773
Pillon, M. C., Babu, V. M., Randall, J. R., Cai, J., Simmons, L. A., Sutton, M. D., et al. (2015). The sliding clamp tethers the endonuclease domain of MutL to DNA. Nucleic Acids Res. 43, 10746–10759. doi: 10.1093/nar/gkv918
Pillon, M. C., Lorenowicz, J. J., Uckelmann, M., Klocko, A. D., Mitchell, R. R., Chung, Y. S., et al. (2010). Structure of the endonuclease domain of MutL: unlicensed to cut. Mol. Cell 39, 145–151. doi: 10.1016/j.molcel.2010.06.027
Pillon, M. C., Miller, J. H., and Guarne, A. (2011). The endonuclease domain of MutL interacts with the beta sliding clamp. DNA Repair (Amst) 10, 87–93. doi: 10.1016/j.dnarep.2010.10.003
Prucca, C. G., Rivero, F. D., and Lujan, H. D. (2011). Regulation of antigenic variation in Giardia lamblia. Annu Rev Microbiol 65, 611–630. doi: 10.1146/annurev-micro-090110-102940
Purser, J. E., and Norris, S. J. (2000). Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad Sci. U.S.A. 97, 13865–13870. doi: 10.1073/pnas.97.25.13865
Rahman, M. M., Mohiuddin, M., Shamima Keka, I., Yamada, K., Tsuda, M., Sasanuma, H., et al. (2020). Genetic evidence for the involvement of mismatch repair proteins, PMS2 and MLH3, in a late step of homologous recombination. J. Biol. Chem. 295, 17460–17475. doi: 10.1074/jbc.RA120.013521
Ranjha, L., Anand, R., and Cejka, P. (2014). The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J. Biol. Chem. 289, 5674–5686. doi: 10.1074/jbc.M113.533810
Rogacheva, M. V., Manhart, C. M., Chen, C., Guarne, A., Surtees, J., and Alani, E. (2014). Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease. J. Biol. Chem. 289, 5664–5673. doi: 10.1074/jbc.M113.534644
Rogovskyy, A. S., Casselli, T., Tourand, Y., Jones, C. R., Owen, J. P., Mason, K. L., et al. (2015). Evaluation of the Importance of VlsE antigenic variation for the enzootic cycle of Borrelia burgdorferi. PLoS One 10:e0124268. doi: 10.1371/journal.pone.0124268
Salman-Dilgimen, A., Hardy, P. O., Radolf, J. D., Caimano, M. J., and Chaconas, G. (2013). HrpA, an RNA helicase involved in RNA processing, is required for mouse infectivity and tick transmission of the lyme disease spirochete. PLoS Pathog 9:e1003841. doi: 10.1371/journal.ppat.1003841
Samuels, D. S., Drecktrah, D., and Hall, L. S. (2018). Genetic transformation and complementation. Methods Mol. Biol. 1690, 183–200. doi: 10.1007/978-1-4939-7383-5_15
Schaaper, R. M. (1993). Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J. Biol. Chem. 268, 23762–23765. doi: 10.1016/s0021-9258(20)80446-3
Sima, N., McLaughlin, E. J., Hutchinson, S., and Glover, L. (2019). Escaping the immune system by DNA repair and recombination in African trypanosomes. Open Biol. 9:190182. doi: 10.1098/rsob.190182
Toledo, M., Sun, X., Brieno-Enriquez, M. A., Raghavan, V., Gray, S., Pea, J., et al. (2019). A mutation in the endonuclease domain of mouse MLH3 reveals novel roles for MutLgamma during crossover formation in meiotic prophase I. PLoS Genet 15:e1008177. doi: 10.1371/journal.pgen.1008177
Verhey, T. B., Castellanos, M., and Chaconas, G. (2018a). Analysis of recombinational switching at the antigenic variation locus of the Lyme spirochete using a novel PacBio sequencing pipeline. Mol. microbiol. 107, 104–115. doi: 10.1111/mmi.13873
Verhey, T. B., Castellanos, M., and Chaconas, G. (2018b). Antigenic variation in the lyme disease spirochete: new insights into the mechanism of recombinational switching with a suggested role for error-prone repair. Cell Reports 23, 2595–2605. doi: 10.1016/j.celrep.2018.04.117
Verhey, T. B., Castellanos, M., and Chaconas, G. (2019). Antigenic variation in the lyme spirochete: detailed functional assessment of recombinational switching at vlsE in the JD1 strain of Borrelia burgdorferi. Mol. Microbiol. 111, 750–763. doi: 10.1111/mmi.14189
Vieira, J., and Messing, J. (1991). New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene 100, 189–194. doi: 10.1016/0378-1119(91)90365-i
Vink, C., Rudenko, G., and Seifert, H. S. (2012). Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol. Rev. 36, 917–948. doi: 10.1111/j.1574-6976.2011.00321.x
Volz, J. C., Bartfai, R., Petter, M., Langer, C., Josling, G. A., Tsuboi, T., et al. (2012). PfSET10, a Plasmodium falciparum methyltransferase, maintains the active var gene in a poised state during parasite division. Cell Host Microbe 11, 7–18. doi: 10.1016/j.chom.2011.11.011
Wahlgren, M., Goel, S., and Akhouri, R. R. (2017). Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat. Rev. Microbiol. 15, 479–491. doi: 10.1038/nrmicro.2017.47
Walia, R., and Chaconas, G. (2013). Suggested role for G4 DNA in recombinational switching at the antigenic variation locus of the lyme disease spirochete. PLoS One 8:e57792. doi: 10.1371/journal.pone.0057792
Windgassen, T. A., Wessel, S. R., Bhattacharyya, B., and Keck, J. L. (2018). Mechanisms of bacterial DNA replication restart. Nucleic Acids Res. 46, 504–519. doi: 10.1093/nar/gkx1203
Wright, W. D., Shah, S. S., and Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 293, 10524–10535. doi: 10.1074/jbc.TM118.000372
Yamamoto, T., Iino, H., Kim, K., Kuramitsu, S., and Fukui, K. (2011). Evidence for ATP-dependent structural rearrangement of nuclease catalytic site in DNA mismatch repair endonuclease MutL. J. Biol. Chem. 286, 42337–42348. doi: 10.1074/jbc.M111.277335
Zakharyevich, K., Tang, S., Ma, Y., and Hunter, N. (2012). Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell 149, 334–347. doi: 10.1016/j.cell.2012.03.023
Keywords: Lyme disease, borreliosis, Borrelia, antigenic variation, vlsE, gene conversion, mutL
Citation: Castellanos M, Verhey TB, Goldstein M and Chaconas G (2022) The Putative Endonuclease Activity of MutL Is Required for the Segmental Gene Conversion Events That Drive Antigenic Variation of the Lyme Disease Spirochete. Front. Microbiol. 13:888494. doi: 10.3389/fmicb.2022.888494
Received: 02 March 2022; Accepted: 18 April 2022;
Published: 19 May 2022.
Edited by:
Axel Cloeckaert, Institut National de Recherche pour l’Agriculture, l’Alimentation et l’Environnement (INRAE), FranceReviewed by:
Peter Kraiczy, Goethe University Frankfurt, GermanyBrian Stevenson, University of Kentucky, United States
D. Scott Samuels, University of Montana, United States
Steven J. Norris, University of Texas Health Science Center at Houston, United States
Eva Sapi, University of New Haven, United States
Janakiram Seshu, University of Texas at San Antonio, United States
Copyright © 2022 Castellanos, Verhey, Goldstein and Chaconas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George Chaconas, Y2hhY29uYXNAdWNhbGdhcnkuY2E=
†These authors have contributed equally to this work