 Lu Wang
Lu Wang Pan Wu
Pan Wu Yingying Su
Yingying Su Yi Wei
Yi Wei Xi Guo
Xi Guo Lan Yang
Lan Yang Min Wang
Min Wang Bin Liu
Bin Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 June 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.885543
The genus Cronobacter includes seven species; however, the strains of Cronobacter sakazakii, Cronobacter malonaticus, and Cronobacter turicensis were highly correlated with clinical infections. Rapid and reliable identification of these three species of Cronobacter is important in monitoring and controlling diseases caused by these bacteria. Here, we identified four pairs of novel marker genes for the Cronobacter genus, C. sakazakii, C. malonaticus, and C. turicensis based on large-scale comparative genomic analysis from 799 Cronobacter and 136,146 non-Cronobacter genomes, including 10 Franconibacter and eight Siccibacter, which are close relatives of Cronobacter. Duplex and multiplex PCR methods were established based on these newly identified marker genes. The reliability of duplex and multiplex PCR methods was validated with 74 Cronobacter and 90 non-Cronobacter strains. Strains of C. sakazakii, C. malonaticus, and C. turicensis could be detected accurately at both the genus and species level. Moreover, the newly developed methods enable us to detect 2.5 × 103 CFU/ml in pure culture. These data indicate that the accurate and sensitive established methods for Cronobacter can serve as valuable tools for the identification of these strains recovered from food, environmental, and clinical samples.
Cronobacter, belonging to the family Enterobacteriaceae, is a genus of Gram-negative, motile, facultative anaerobic, opportunistic, foodborne pathogens that can cause bacteremia, meningitis, and necrotizing enterocolitis in neonates (Forsythe, 2018). Cronobacter has been isolated from various environments (Killer et al., 2015; Singh et al., 2015; Ling et al., 2018; Li et al., 2020), and several disease cases have been associated with the ingestion of Cronobacter-contaminated dry food products, such as powdered milk formula (Drudy et al., 2006; Forsythe, 2018). Despite the low incidence of infection, the mortality of Cronobacter infection in neonates can be as high as 27%–80% (Drudy et al., 2006; Masood et al., 2015). The Cronobacter species of serious clinical significance are Cronobacter sakazakii, Cronobacter malonaticus, and Cronobacter turicencis, and other four species of the genus (Cronobacter universalis, Cronobacter dublinensis, Cronobacter muytjensii, and Cronobacter condimenti) are primarily environmental commensals with low clinical significance (Sonbol et al., 2013; Feeney et al., 2014; Forsythe, 2018; Li et al., 2020). Thus, reliable methods to identify C. sakazakii, C. malonaticus, and C. turicensis are critical to reduce mortality and transmission of diseases caused by Cronobacter spp.
Molecular detection methods are more useful tools than traditional methods to increase our understanding of the epidemiology of a bacterium important to public health. These protocols are usually designed to amplify DNA fragments of certain genes contained in genomes of the pathogen of interest. Over the past decade, a range of molecular methods based on genes, such as 16S rDNA (Malorny and Wagner, 2005), 23S rDNA (Derzelle et al., 2007), MMS (Seo and Brackett, 2005), rpoB (Stoop et al., 2009), ompA (Zimmermann et al., 2014), fusA (Li et al., 2017), cgcA (Carter et al., 2013), ygrB (Huang et al., 2013), etc. have been developed to identify Cronobacter spp. These approaches can be used as alternatives to traditional culture-based detection methods or can be used to confirm results generated by traditional approaches. However, only a few of these methods are able to simultaneously detect and differentiate species within the Cronobacter genus. rpoB had been used to detect six species of Cronobacter (Stoop et al., 2009); however, a two-step PCR procedure was needed to differentiate between C. sakazakii and C. malonaticus. Even though gyrB (Huang et al., 2013) was utilized for direct species identification of C. sakazakii and C. dubliniensis, the method cannot distinguish between other species. Primers were designed based on cgcA (Carter et al., 2013) to identify Cronobacter at the species level; however, non-specific amplicons occurred. Thus, it is necessary to identify novel specific markers for Cronobacter spp. and develop efficient identification methods according to these markers.
As high-throughput genome sequencing technologies continue to improve, the number of sequenced microbial genomes has continued to increase dramatically over the past decade. This makes it possible to employ an in silico large scale comparative genomic approach coupled with in vitro PCR validation to facilitate the translation of genomic data into diagnostic marker gene discoveries. In this study, a low-cost and simple attempt was made to identify novel diagnostic marker genes specific for Cronobacter spp.
A total of 164 bacteria isolates, including 74 Cronobacter strains (62 C. sakazakii, five C. dubliniensis, three C. malonaticus, two C. turicensis, and two C. universalis) and 90 non-Cronobacter strains (18 Enterobacter cloacae, 36 Enterobacter aerogenes, and 36 Escherichia coli), were used in this study for in vitro validation (Supplementary Table S1). Moreover, 799 Cronobacter (578 C. sakazakii, 100 C. malonaticus, 60 C. dubliniensis, 35 C. turicensis, 15 C. muytjensii, nine C. universalis, and two C. condimenti) and 136,146 non-Cronobacter genomes belonging to 31 genera were used for large-scale in silico comparative genomic analysis (Supplementary Tables S2, S3). These non-Cronobacter genomes include 10 Franconibacter, eight Siccibacter, and 810 E. cloacae, which are close relatives of genus Cronobacter.
Single-copy core genes found using OrthoFinder v2.3.3 (Emms and Kelly, 2015) were used as original data for construction of a phylogenetic tree. Enterobacter cloacae ATCC 13047™ and ECNIH2 (GenBank accession number GCA_000025565.1 and GCA_000724505.1, respectively) served outgroups, as it is the species closely related to the Cronobacter genus (Joseph et al., 2012). MAFFT v7 with “G-INS-I” alignment method (Katoh and Standley, 2013) was used for creating multiple sequence alignments for each core gene and resulting alignments were concatenated. Thereafter, RAxML v8 with GTR (General Time Reversible) evolution model (Stamatakis, 2014) was applied to construct a phylogenetic maximum likelihood tree. The tree and subtrees were plotted with the R package’s ggtree (Yu et al., 2016). To confirm the degree of genomic relatedness and clarify relationships between the species of Cronobacter, ANIb values (the average nucleotide identity values based on BLAST) for all possible pairs of genomes were calculated using the program FastANI v.1.0 (Jain et al., 2018).
Single-copy core genes in the Cronobacter genus were identified using OrthoFinder. Large-scale blast score ratio (LS-BSR) software was used to identify highly-conserved genes in the Cronobacter genus compared with other non-Cronobacter bacteria (Sahl et al., 2014). This was run against the assembled genomes of 799 Cronobacter isolates and 136,146 non-Cronobacter isolates belonging to 31 genera. Thereafter, the matrix generated by LS-BSR was processed using a script developed in house to evaluate and visualize the highly conserved genes across the data set. Genes with an average blast score ratio (BSR) value >0.9 in all Cronobacter genomes and <0.1 in all non-Cronobacter genomes were considered highly conserved genes in Cronobacter. These genes were further screened manually (genes with the smallest BSR value <0.8 in any Cronobacter genome or the largest BSR value >0.4 in any non-Cronobacter genome were excluded) and searched against the full National Center for Biotechnology Information (NCBI) nucleotide database to confirm their specificity. Two promising conserved genes were selected for identification of the Cronobacter genus.
As shown above, highly conserved single-copy core genes in each species of C. sakazakii, C. malonaticus, and C. turicensis compared with other non-Cronobacter species were identified using OrthoFinder and LS-BSR. To explore whether these highly conserved genes were specific for target species and absent in the other six species of Cronobacter, these genes were analyzed using LS-BSR once more. Moreover, these genes, with an average BSR value > 0.9 in target species genomes and <0.4 in the other six Cronobacter species genomes, were considered as candidate marker genes. These candidate marker genes were screened manually once more (genes with the smallest BSR value < 0.8 in all Cronobacter genomes or largest BSR value > 0.4 in all non-Cronobacter genomes were excluded) and searched against the NCBI nucleotide database to confirm their specificity. The two most promising conserved genes of each species were selected for identification of C. sakazakii, C. malonaticus, and C. turicensis.
Multiple sequence alignment of genus- and species-specific gene alleles was performed to obtain conserved regions, which were used for primer design by Primer Premier 6.0 (Supplementary Figure S1). Thereafter, the specificity of theory sequences of amplicons was verified using BLAST and primer sequences were evaluated for their ability to form homo- and heterodimers as well as hairpins using the oligo-analyzer. Desalted primers were synthesized from Invitrogen. Primer sequences and corresponding theory amplicon sizes are shown in Table 1.
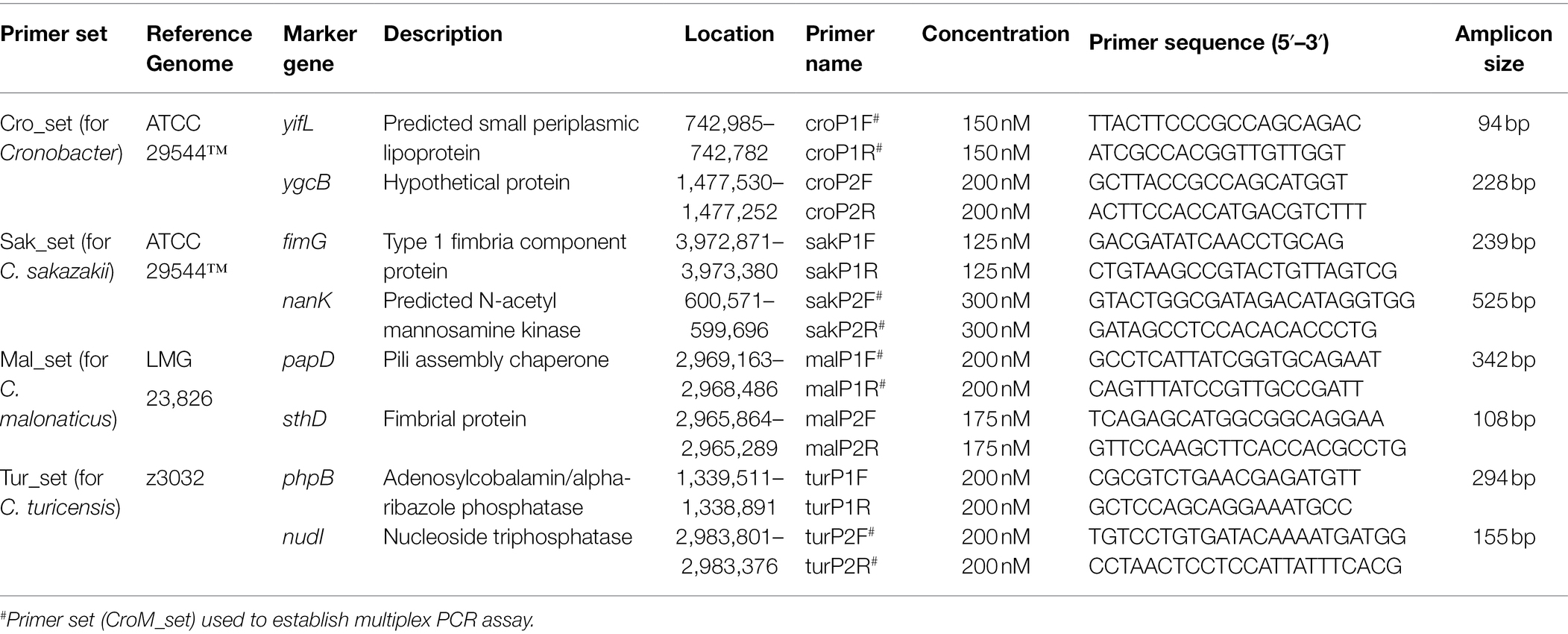
Table 1. Primer sets utilized in PCR assay and their target genes.
Specificity of each primer set was assessed by running a PCR assay on a panel of bacterial strains consisting of 74 Cronobacter and 90 non-Cronobacter isolates. PCR mixtures contained 10 μl Ex Taq Master Mix (Takara, the premixed solution contains Ex Taq DNA Polymerase, 2 × PCR Buffer, and 200 μM dNTPs), 0.5 μl of each primer (optimal concentration shown in Table 1), 1 μl bacterial template (1 × 106 CFU/ml), and RNase/DNase free water to adjust the volume to 20 μl. All PCR runs included a negative control without template. PCR reactions were run as follows: initial hot start (94°C for 10 min), amplification for 35 cycles (94°C for 30 s, 59.5°C for 30 s, and 72°C for 60 s), and final extension (72°C for 10 min). Annealing temperatures were optimized by gradually increasing the temperature from 50 to 65°C in the assay. PCR products were examined using agarose gel electrophoresis and visualized after ethidium bromide staining. For sensitivity verification, pure cultures of C. sakazakii ATCC 29544™, C. malonaticus LC03, and C. turicensis LC08 with a concentration of 2.5 × 106 CFU/ml were serially diluted 10-fold to 2.5 × 102 CFU/ml and used as PCR templates.
To confirm that 799 genomes belonged to corresponding species of Cronobacter, a maximum-likelihood phylogenetic tree was constructed, based on 223 single-copy core genes. Phylogenetic analysis showed that the genus was divided into seven clades corresponding to seven species of Cronobacter (Figure 1). To explore the genomic similarities among phylogenetic clades further, ANIb values of all genome assemblies were calculated using fastANI (Figure 1). All intraclade ANIb values exceeded the commonly used 95% species-level threshold (Kalyantanda et al., 2015) for C. sakazakii (97.29%–99.99%), C. malonaticus (96.33%–99.99%), C. turicensis (95.99%–99.99%), C. dubliniensis (96.91%–99.99%), C. muytjensii (98.72%–99.99%), C. universalis (98.20%–99.99%), and C. condimenti (99.93%–99.97%), showing that each clade represented a single species.
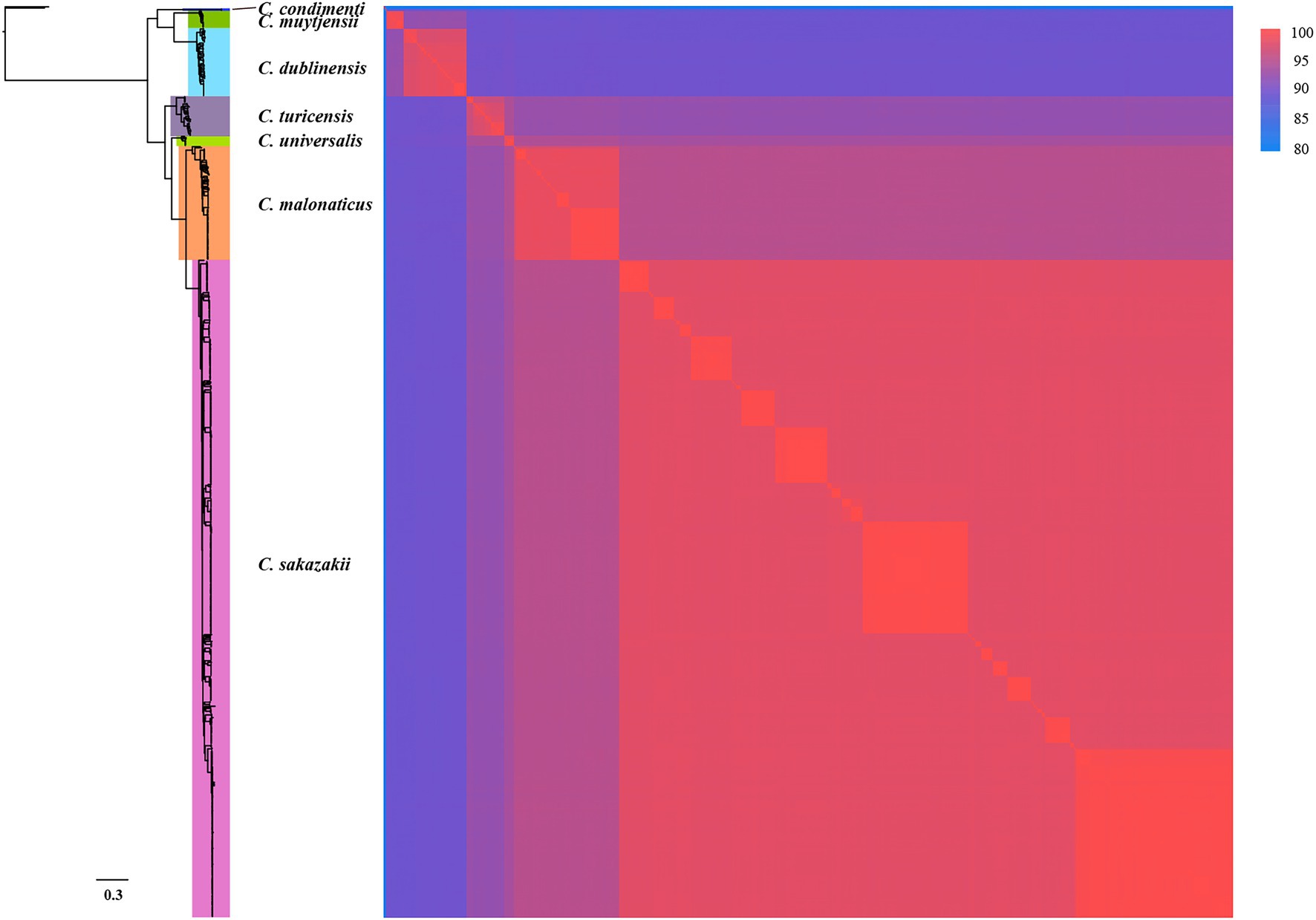
Figure 1. Whole-genome phylogeny of seven species of Cronobacter. The maximum-likelihood phylogeny tree was constructed based on single-nucleotide polymorphisms extracted from 799 genomes, excluding those resulting from recombination (two Enterobacter cloacae strains were used as the outgroup). ANIb values of the 799 Cronobacter genomes was calculated using FastANI.
Three hundred and ninety-one conserved single-copy core genes were identified from 799 accessible Cronobacter genomes in the PubMLST database using OrthoFinder. To explore whether these conserved single-copy core genes were specific for the Cronobacter genus, we used the large-scale BLAST score ratio (LS-BSR) to evaluate genes present in the 799 isolates of the Cronobacter genus yet absent in 136,146 isolates, belonging to 31 genera of non-Cronobacter common environmental microbes and pathogens. According to the BSR value, 78 genes were highly conserved in Cronobacter species (average BSR value > 0.9 in Cronobacter and <0.1 in non-Cronobacter; Figure 2A). To find genes that uniquely existed in all Cronobacter isolates and deficient in all other non-Cronobacter bacteria, genes with a BSR value <0.8 in any Cronobacter isolates and >0.4 in any non-Cronobacter isolates were excluded. We finally selected two most promising conserved genes (yifL and ygcB) as Cronobacter genus-specific marker genes after manual screening and searching against the NCBI nonredundant nucleotide database (Figures 2B,C). Characteristics of the two genus-specific genes and corresponding designed primers are shown in Table 1.
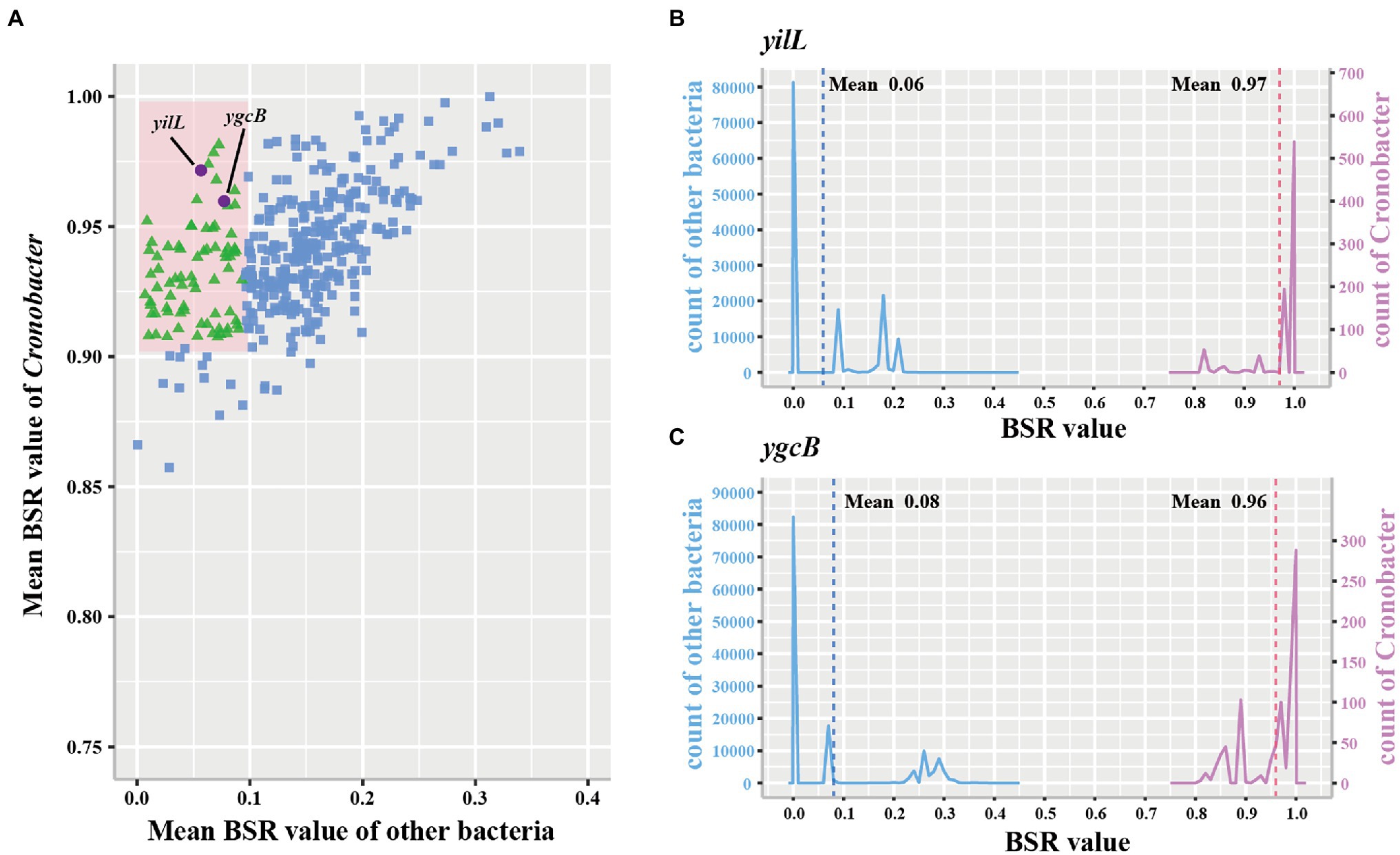
Figure 2. Conserved single-copy core genes of Cronobacter were further evaluated by large-scale blast score ratio (LS-BSR). (A) LS-BSR analysis of each conserved genes among Cronobacter and non-Cronobacter bacteria. (B,C) BSR value distribution of the Cronobacter-specific marker genes yifL and ygcB.
To examine whether marker genes exist in C. sakazakii, C. malonaticus, and C. turicensis, further analysis was performed. A total of 1,002, 2,555, and 3,238 conserved single-copy core genes were identified from 578 C. sakazakii, 100 C. malonaticus, and 35 C. turicensis genomes using OrthoFinder, respectively. Using LS-BSR, we discovered 134, 683, 1,110 genes that were conserved in C. sakazakii, C. malonaticus, and C. turicensis yet absent in other non-Cronobacter bacteria (Figures 3A,C,E; using a threshold of the average BSR value >0.9 in each target species and <0.1 in non-Cronobacter bacteria). To identify genes that were only conserved in genomes of each target species, the LS-BSR comparison of genomes between each target species and the remaining six species of Cronobacter demonstrated that 5, 14, 44 genes were highly conserved in C. sakazakii, C. malonaticus, and C. turicensis, respectively (Figures 3B,D,F). To confirm specificity, these candidate genes were screened manually and searched against the NCBI nonredundant nucleotide database and the two most promising conserved genes were selected as species-specific marker genes for each species (fimG and nanK for C. sakazakii, papD and sthD for C. malonaticus, and phpB and nudI for C. turicensis; Supplementary Figures S2–S4). Characteristics of the three pairs of species-specific genes and corresponding designed primers are shown in Table 1.
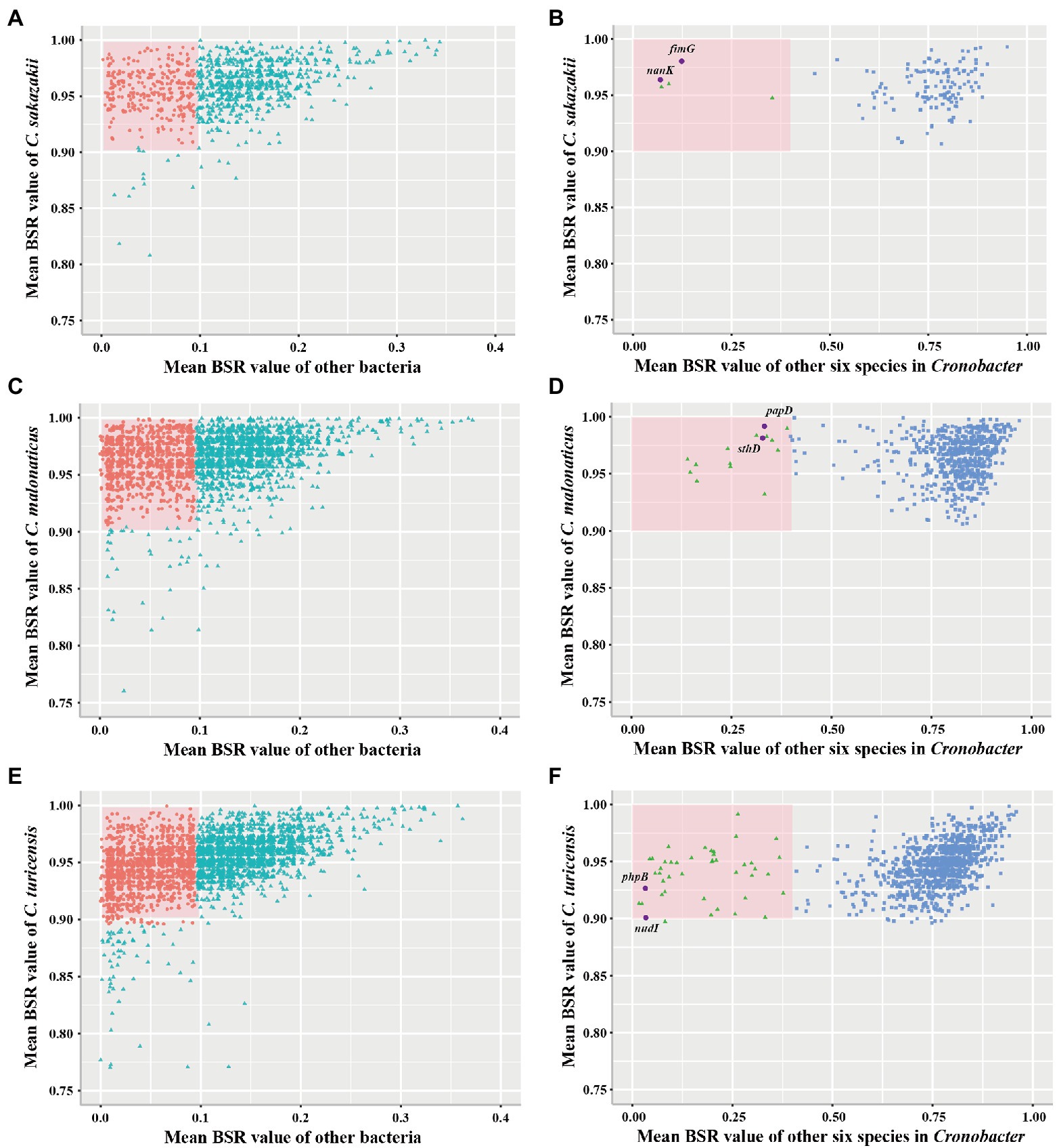
Figure 3. Conserved single-copy core genes of Cronobacter sakazakii, Cronobacter malonaticus, and C. turicensis were further evaluated by LS-BSR. (A,C,E) LS-BSR analysis of each conserved genes among target species and non-Cronobacter bacteria. (B,D,F) LS-BSR analysis of each gene analyzed again among target species and other six species of Cronobacter.
The duplex PCR assay was developed based on genus or species marker genes to evaluate the specificity of designed primers. Seventy-four Cronobacter strains and 90 non-Cronobacter strains, closely related to Cronobacter, were utilized to evaluate specificity of genus- and species-specific primer sets (each primer set contains two primer pairs, shown in Table 1). The specificity of each primer set was crosstested with isolates of target species of Cronobacter and non-target species of Cronobacter and non-Cronobacter. The results showed that each primer set successfully amplified their target genes with correct amplicon sizes and without non-specific band (Figure 4; Supplementary Table S4). Primer set Cro_set could detect all isolates of Cronobacter, and primer sets Sak_set, Mal_set, and Tur_set could only detect corresponding isolates from C. sakazakii, C. malonaticus, and C. turicensis, respectively (Figure 4; Supplementary Table S4). To confirm specificity, 90 non-Cronobacter strains were tested and all produced no PCR products (Supplementary Table S4).
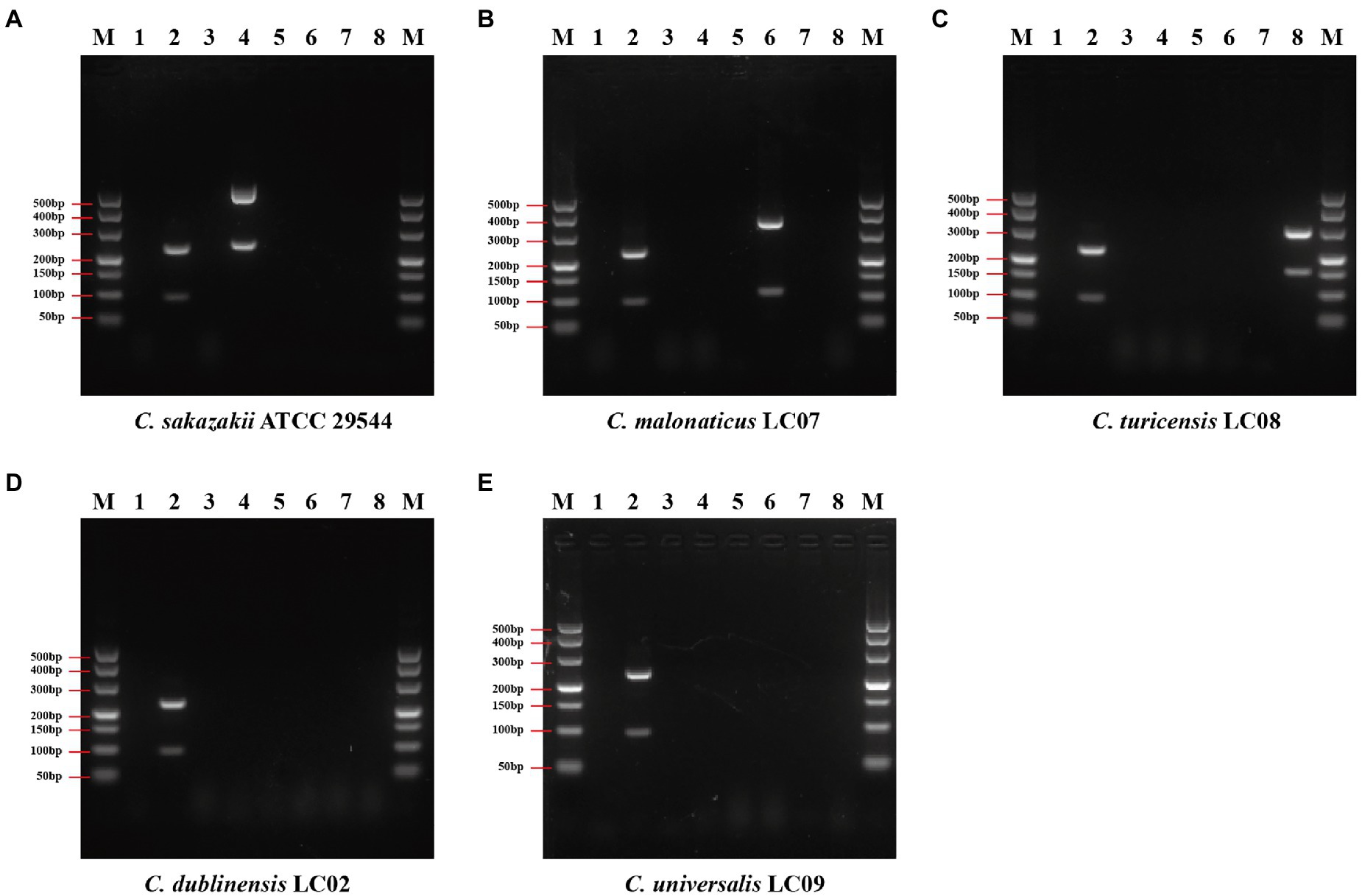
Figure 4. Results of duplex PCR based on genus- and species-specific genes. (A) Cronobacter sakazakii ATCC 29544™, (B) Cronobacter malonaticus LC07, (C) C. turicensis LC08, (D) C. dubliniensis LC02, and (E) Cronobacter universalis LC09. Lane M: DNA marker; lanes 1, 3, 5, and 7: negative control without template; lane 2: Cronobacter genus primer set Cro_set; lane 4: C. sakazakii species primer set Sak_set; lane 6: C. malonaticus species primer set Mal_set; and lane 8: C. turicensis species primer set Tur_set.
To differentiate species of Cronobacter using only one PCR reaction accurately, a multiplex primer set CroM_set (shown in Table 1), including four specific primer pairs, croP1F/croP1R, sakP2F/sakP2R, malP1F/malP1R, and turP2F/turP2R, were selected based on amplicon sizes to develop the multiplex PCR assay. Primer pairs were mixed and used to screen 2.5 × 105 CFU/ml pure culture from 74 Cronobacter isolates. The results showed that all target isolates produced the expected PCR products and non-target isolates gave no PCR products (Supplementary Table S4). Representative PCR results using 10 Cronobacter pure cultures as templates are shown in Figure 5. To confirm specificity, 90 non-Cronobacter strains were tested and all produced no PCR products (Supplementary Table S4).
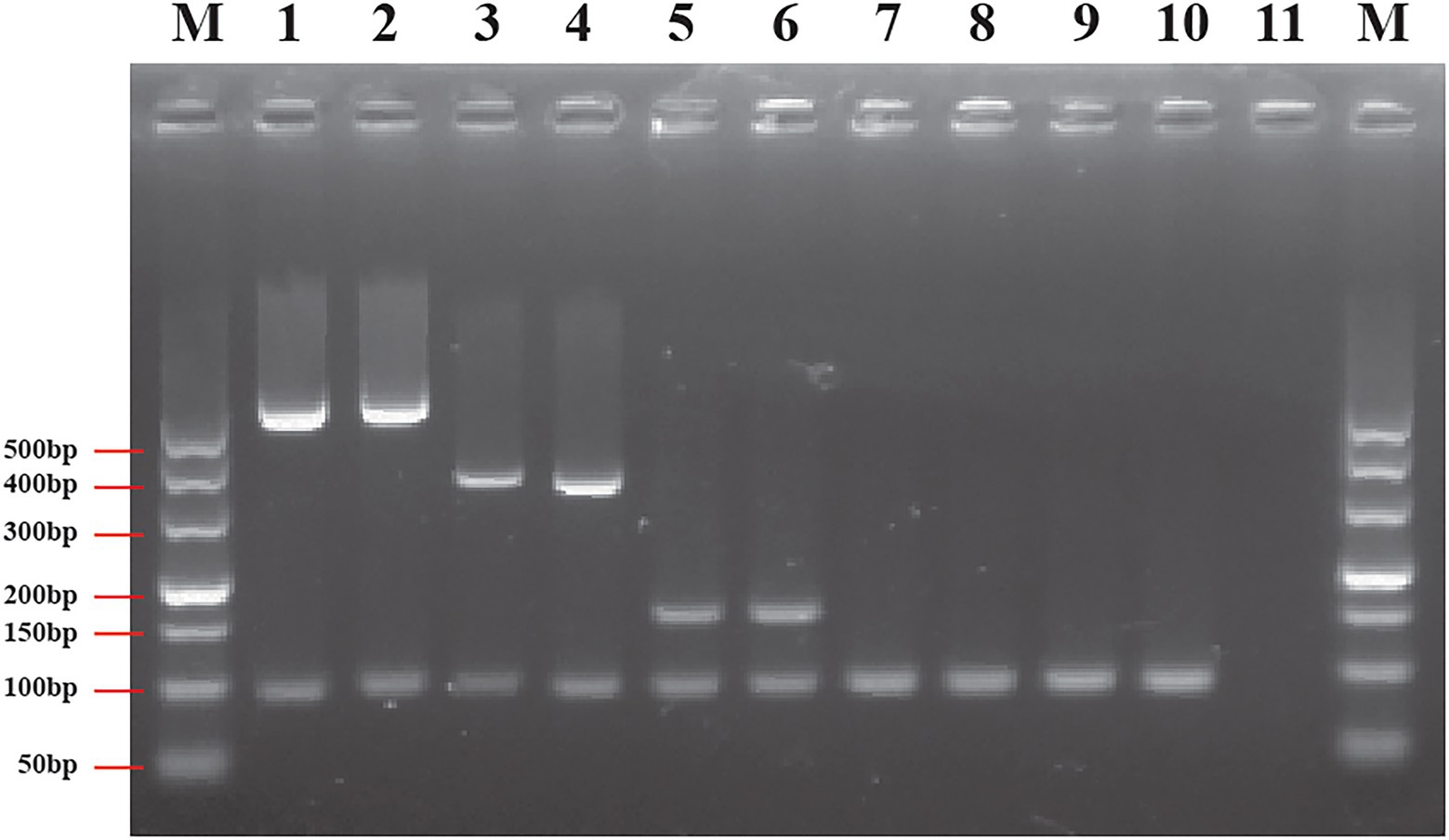
Figure 5. Results of multiplex PCR based on genus- and species-specific genes. Lane M: DNA marker; lane 1: Cronobacter sakazakii ATCC 29544™; lane 2: C. sakazakii ATCC BAA-894™; lane 3: Cronobacter malonaticus LC03; lane 4: C. malonaticus LC07; lane 5: Cronobacter turicensis LC08; lane 6: C. turicensis LC12; lane 7: Cronobacter dubliniensis LC01; lane 8: C. dubliniensis LC02; lane 9: Cronobacter universalis LC09; lane 10: C. universalis LC10; and lane 11: negative control without template.
The sensitivity (limit of detection) of duplex and multiplex PCR assay for the identification of Cronobacter spp. was evaluated using a serial 10-fold dilution in the range of 2.5 × 107–2.5 × 102 CFU/ml of pure cultures. The representative PCR assay using pure cultures of C. sakazakii ATCC 29544™, C. malonaticus LC07, and C. turicensis LC08 is shown in Figure 6. Although results were unstable when using 2.5 × 102 CFU/ml pure culture of isolates from different Cronobacter species, visible and clear amplicons (positive signals) were generated with ≥2.5 × 103 CFU/ml pure culture for all PCR reactions, indicating the limit of detection of both duplex (Figure 6A) and multiplex (Figure 6B) PCR method that had high sensitivity.
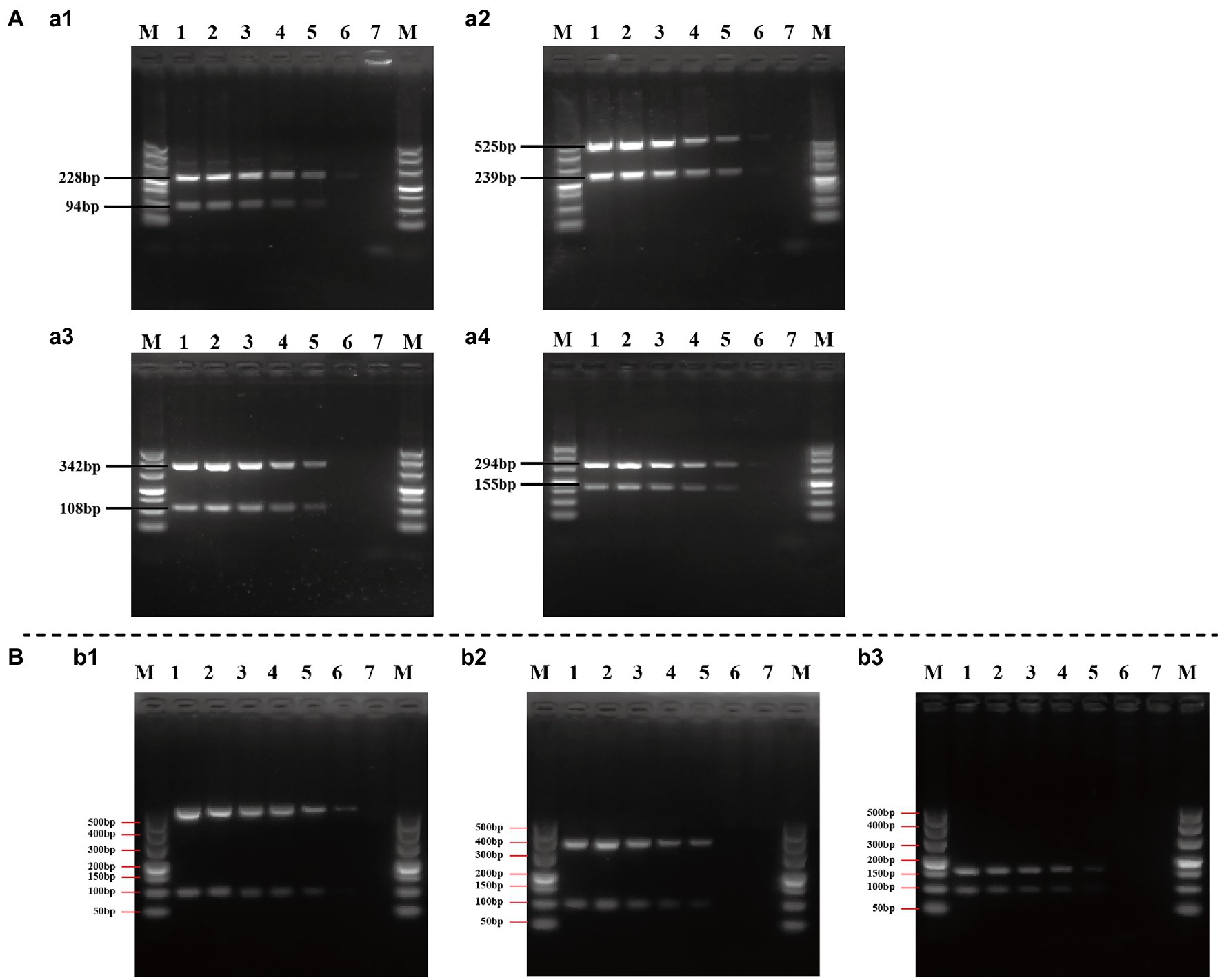
Figure 6. Evaluation of the sensitivity of duplex (A) and multiplex (B) PCR assay. (a1–a4): Cronobacter sakazakii ATCC 29544™, C. sakazakii ATCC 29544™, Cronobacter malonaticus LC07, and Cronobacter turicensis LC08 as template to evaluate the sensitivity of duplex PCR assay based on Cro_set, Sak_set, Mal_set, and Tur_set; (b1–b3): C. sakazakii ATCC 29544™, C. malonaticus LC07, and C. turicensis LC08 as template to evaluate the sensitivity of multiplex PCR assay based on Cro_set. Lane M: DNA marker; lanes 1–6: template of pure culture from 2.5 × 107 to 2.5 × 102 CFU/ml; and lane 7: negative control without template.
Cronobacter has been isolated from various environments, including foods and clinical sources, and several disease cases have been associated with the ingestion of Cronobacter-contaminated foods, such as powdered milk formula (Kalyantanda et al., 2015). Detection of Cronobacter spp., especially C. sakazakii, C. malonaticus, and C. turicensis, has become increasingly important in food safety and clinical diagnosis.
Methods based on PCR have been widely used for detection of pathogens because they are simple and rapid compared with conventional culture-based methods (Petti, 2007). In recent decades, molecular methods based on genes, such as 16S rDNA, MMS, fusA, rpoB, ompA, gyrB, cgcA, etc., have been used for the identification of Cronobacter (Malorny and Wagner, 2005; Seo and Brackett, 2005; Derzelle et al., 2007; Stoop et al., 2009; Sonbol et al., 2013; Zimmermann et al., 2014; Li et al., 2017), however, only a few are used to differentiate Cronobacter at the species level. Therefore, it is essential to identify specific novel marker genes for Cronobacter spp. Owing to advancements in high-throughput sequencing technologies; it is possible to employ large-scale genome sequences for identification of highly specific marker genes. Goay et al. (2016) have identified five novel Salmonella Typhi-specific genes as markers for diagnosis of typhoid fever based on comparative genomic analysis; Hazen et al. (2016) have found several genes specific for lethal enteropathogenic E. coli using LS-BSR. Therefore, comparative genomic analysis would be practical to screen specific marker genes for rapid and precise detection of pathogens. Although Shang et al. (2021) obtained several Cronobacter species-specific genes through pan-genome analysis; screening threshold was based on 95% (not 100%) of the target genomes and 5% (not 0%) of the non-target genomes. Lee et al. (2022) identified 16 genes specific for C. sakazakii, but these genes were screened only from 17 genomes of Cronobacter. Therefore, the present study was the first approach to screen novel marker genes for both at genus and species level of Cronobacter based on large-scale genomic analysis from 799 genomes of Cronobacter and 136,146 genomes of non-Cronobacter. Thereafter, duplex and multiplex PCR methods were established according to these marker genes. Moreover, the specificity of duplex and multiplex PCR methods was validated with 74 Cronobacter and 90 non-Cronobacter strains. The results showed that C. sakazakii, C. malonaticus, and C. turicensis could be detected accurately at both the genus and species level, and C. universalis and C. dublinensis could be detected accurately at the genus level. However, it is difficult to evaluate the detection of C. muytjensii and C. condimenti due to the lack of strains. The sensitivity of duplex and multiplex PCR assay was also determined and the detection limit was 2.5 × 103 CFU/ml for pure culture.
In summary, we successfully screened out Cronobacter genus- and species-specific marker genes using large-scale genomic analysis, and the specificity and sensitivity of these selected targets were evaluated using duplex and multiplex PCR. Thus, the established methods described here were proved to be reliable and sensitive for the identification of Cronobacter spp.
The datasets presented in this study can be found in online Cronobacter PubMLST database (https://pubmlst.org/organisms/cronobacter-spp/). The names of the repositories and accession numbers can be found in the Supplementary Material.
LW, MW, and BL designed the research. PW, YS, and YW performed the research. MW and XG provided technical support and insights. LW and LY analyzed the data. LW, XG, LY, and BL wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (NSFC) Program (32070130 and 81772148), Young Scholar of Tianjin (20JCJQJC00180), the Committee on Science and Technology of Tianjin (19YFZCSN00080), and Health Commission of Hubei Province Foundation (WJ2019H528).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.885543/full#supplementary-material
Supplementary Figure S1 | Design of PCR primers based on genus- and species-specific genes. Primer positions and directions are indicated as red arrows; genes lengths are indicated as black lines; variable and conserved (from magenta to purple) nucleotide positions are highlighted in the color stripe.
Supplementary Figure S2 | Large-scale BLAST score ratio (LS-BSR) analysis of Cronobacter sakazakii specific marker genes fimG (A) and nanK (B). (a) BSR value distribution of target marker genes in C. sakazakii, (b) BSR value distribution of target marker genes in the other six species of Cronobacter, and (c) BSR value distribution of target marker genes in other bacteria.
Supplementary Figure S3 | Large-scale BLAST score ratio (LS-BSR) analysis of C. malonatious specific marker genes papD (A) and sthD (B). (a) BSR value distribution of target marker genes in Cronobacter malonaticus, (b) BSR value distribution of target marker genes in the other six species of Cronobacter, and (c) BSR value distribution of target marker genes in other bacteria.
Supplementary Figure S4 | Large-scale BLAST score ratio (LS-BSR) analysis of C. turicensis specific marker genes phpB (A) and nudI (B). (a) BSR value distribution of target marker genes in C. turicensis, (b) BSR value distribution of target marker genes in the other six species of Cronobacter, and (c) BSR value distribution of target marker genes in other bacteria.
Carter, L., Lindsey, L. A., Grlm, C. J., Sathymoorthy, V., Jarvls, K. G., Goplnath, G., et al. (2013). Multiplex PCR assay targeting a diguanylate cyclase-encoding gene, cgcA, to differentiate species within the genus Cronobacter. Appl. Environ. Microbiol. 79, 734–737. doi: 10.1128/AEM.02898-12
Derzelle, S., Dilasser, F., Maladen, V., Soudrie, N., Leclercq, A., Lombard, B., et al. (2007). Comparison of three chromogenic media and evaluation of two molecular-based identification systems for the detection of Enterobacter sakazakii from environmental samples from infant formulae factories. J. Food Prot. 70, 1678–1684. doi: 10.4315/0362-028X-70.7.1678
Drudy, D., Mullane, N. R., Quinn, T., Wall, P. G., and Fanning, S. (2006). Enterobacter sakazakii: an emerging pathogen in powdered infant formula. Clin. Infect. Dis. 42, 996–1002. doi: 10.1086/501019
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Feeney, A., Kropp, K. A., O’connor, R., and Sleator, R. D. (2014). Cronobacter sakazakii: stress survival and virulence potential in an opportunistic foodborne pathogen. Gut Microbes 5, 711–718. doi: 10.4161/19490976.2014.983774
Forsythe, S. J. (2018). Updates on the Cronobacter genus. Annu. Rev. Food Sci. Technol. 9, 23–44. doi: 10.1146/annurev-food-030117-012246
Goay, Y. X., Chin, K. L., Tan, C. L. L., Yeoh, C. Y., Ja'afar, J. A. N., Zaidah, A. R., et al. (2016). Identification of five novel Salmonella Typhi-specific genes as markers for diagnosis of typhoid fever using single-gene target PCR assays. Biomed. Res. Int. 2016:8905675. doi: 10.1155/2016/8905675
Hazen, T. H., Donnenberg, M. S., Panchalingam, S., Antonio, M., Hossain, A., Mandomando, I., et al. (2016). Genomic diversity of EPEC associated with clinical presentations of differing severity. Nat. Microbiol. 1:15014. doi: 10.1038/nmicrobiol.2015.14
Huang, C.-H., Chang, M.-T., and Huang, L. (2013). Use of novel species-specific PCR primers targeted to DNA gyrase subunit B (gyrB) gene for species identification of the Cronobacter sakazakii and Cronobacter dublinensis. Mol. Cell. Probes 27, 15–18. doi: 10.1016/j.mcp.2012.08.004
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Joseph, S., Sonbol, H., Hariri, S. H., Desai, P. T., Mcclelland, M., and Forsythe, S. J. (2012). Diversity of the Cronobacter genus as revealed by multilocus sequence typing. J. Clin. Microbiol. 50, 3031–3039. doi: 10.1128/JCM.00905-12
Kalyantanda, G., Shumyak, L., and Archibald, L. K. (2015). Cronobacter species contamination of powdered infant formula and the implications for neonatal health. Front. Pediatr. 3:56. doi: 10.3389/fped.2015.00056
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Killer, J., Skrivanova, E., Hochel, I., and Marounek, M. (2015). Multilocus sequence typing of Cronobacter strains isolated from retail foods and environmental samples. Foodborne Pathog. Dis. 12, 514–521. doi: 10.1089/fpd.2014.1884
Lee, J.-I., Kim, S.-S., and Kang, D.-H. (2022). Development of DNA probes to detect Cronobacter sakazakii based on comparative genomics and its application in food samples. Food Control 137:108853. doi: 10.1016/j.foodcont.2022.108853
Li, X., Cui, J., Du, X., Cui, Z., Huang, Y., and Kan, B. (2017). Duplex real-time PCR method for the differentiation of Cronobacter sakazakii and Cronobacter malonaticus. J. Food Prot. 80, 50–56. doi: 10.4315/0362-028X.JFP-16-171
Li, Y., Zhang, L., Hu, Y., Hong, C., Xie, A., Wu, Y., et al. (2020). Prevalence and genetic characteristics of Cronobacter spp. from food and human clinical stool samples in Wenzhou, China 2008-2018. Food Microbiol. 89:103432. doi: 10.1016/j.fm.2020.103432
Ling, N., Li, C., Zhang, J., Wu, Q., Zeng, H., He, W., et al. (2018). Prevalence and molecular and antimicrobial characteristics of Cronobacter spp. isolated from raw vegetables in China. Front. Microbiol. 9:1149 doi: 10.3389/fmicb.2018.01149
Malorny, B., and Wagner, M. (2005). Detection of Enterobacter sakazakii strains by real-time PCR. J. Food Prot. 68, 1623–1627. doi: 10.4315/0362-028X-68.8.1623
Masood, N., Moore, K., Farbos, A., Paszkiewicz, K., Dickins, B., Mcnally, A., et al. (2015). Genomic dissection of the 1994 Cronobacter sakazakii outbreak in a French neonatal intensive care unit. BMC Genomics 16:750. doi: 10.1186/s12864-015-1961-y
Petti, C. A. (2007). Detection and identification of microorganisms by gene amplification and sequencing. Clin. Infect. Dis. 44, 1108–1114. doi: 10.1086/512818
Sahl, J. W., Caporaso, J. G., Rasko, D. A., and Keim, P. (2014). The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. PeerJ 2:e332. doi: 10.7717/peerj.332
Seo, K. H., and Brackett, R. E. (2005). Rapid, specific detection of Enterobacter sakazakii in infant formula using a real-time PCR assay. J. Food Prot. 68, 59–63. doi: 10.4315/0362-028X-68.1.59
Shang, Y., Ye, Q., Wu, Q., Pang, R., Zhou, B., Wang, C., et al. (2021). PCR and multiplex PCR assays for the detection of Cronobacter species using specific targets obtained by a bioinformatics approach. Food Control 125:107896. doi: 10.1016/j.foodcont.2021.107896
Singh, N., Goel, G., and Raghav, M. (2015). Prevalence and characterization of Cronobacter spp. from various foods, medicinal plants, and environmental samples. Curr. Microbiol. 71, 31–38. doi: 10.1007/s00284-015-0816-8
Sonbol, H., Joseph, S., Mcauley, C. M., Craven, H. M., and Forsythe, S. J. (2013). Multilocus sequence typing of Cronobacter spp. from powdered infant formula and milk powder production factories. Int. Dairy J. 30, 1–7. doi: 10.1016/j.idairyj.2012.11.004
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stoop, B., Lehner, A., Iversen, C., Fanning, S., and Stephan, R. (2009). Development and evaluation of rpoB based PCR systems to differentiate the six proposed species within the genus Cronobacter. Int. J. Food Microbiol. 136, 165–168. doi: 10.1016/j.ijfoodmicro.2009.04.023
Yu, G., Smith, D. K., Zhu, H., Guan, Y., Lam, T. T. Y., and Mcinerny, G. (2016). Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. doi: 10.1111/2041-210X.12628
Keywords: Cronobacter spp., foodborne pathogen, genomic analysis, marker gene, PCR
Citation: Wang L, Wu P, Su Y, Wei Y, Guo X, Yang L, Wang M and Liu B (2022) Detection of Genus and Three Important Species of Cronobacter Using Novel Genus- and Species-Specific Genes Identified by Large-Scale Comparative Genomic Analysis. Front. Microbiol. 13:885543. doi: 10.3389/fmicb.2022.885543
Edited by:
Alexandre Leclercq, Institut Pasteur, FranceReviewed by:
Laurent Guillier, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), FranceCopyright © 2022 Wang, Wu, Su, Wei, Guo, Yang, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Wang, d2FuZ21AbmFua2FpLmVkdS5jbg==; Lan Yang, c3VubnlsYW4wN0AxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.