 Weiwei Cao
Weiwei Cao Yuanfeng Cai
Yuanfeng Cai Zhihua Bao
Zhihua Bao Shuwei Wang
Shuwei Wang Xiaoyuan Yan
Xiaoyuan Yan Zhongjun Jia
Zhongjun Jia- 1State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Ministry of Education Key Laboratory of Ecology and Resource Use of the Mongolian Plateau & Inner Mongolia Key Laboratory of Grassland Ecology, School of Ecology and Environment, Inner Mongolia University, Hohhot, China
- 4Inner Mongolia Key Laboratory of Environmental Pollution Control and Waste Resource Reuse, Inner Mongolia University, Hohhot, China
The bioavailability of nitrogen constrains primary productivity, and ecosystem stoichiometry implies stimulation of N2 fixation in association with carbon sequestration in hotspots such as paddy soils. In this study, we show that N2 fixation was triggered by methane oxidation and the methanotrophs serve as microbial engines driving the turnover of carbon and nitrogen in rice roots. 15N2-stable isotope probing showed that N2-fixing activity was stimulated 160-fold by CH4 oxidation from 0.27 to 43.3 μmol N g–1 dry weight root biomass, and approximately 42.5% of the fixed N existed in the form of 15N-NH4+ through microbial mineralization. Nitrate amendment almost completely abolished N2 fixation. Ecophysiology flux measurement indicated that methane oxidation-induced N2 fixation contributed only 1.9% of total nitrogen, whereas methanotrophy-primed mineralization accounted for 21.7% of total nitrogen to facilitate root carbon turnover. DNA-based stable isotope probing further indicated that gammaproteobacterial Methylomonas-like methanotrophs dominated N2 fixation in CH4-consuming roots, whereas nitrate addition resulted in the shift of the active population to alphaproteobacterial Methylocystis-like methanotrophs. Co-occurring pattern analysis of active microbial community further suggested that a number of keystone taxa could have played a major role in nitrogen acquisition through root decomposition and N2 fixation to facilitate nutrient cycling while maintaining soil productivity. This study thus highlights the importance of root-associated methanotrophs as both biofilters of greenhouse gas methane and microbial engines of bioavailable nitrogen for rice growth.
Introduction
Rice roots serve as one of the major carbon inputs in paddy fields, also playing important roles in maintaining nutrient availability and crop yields through microbial recycling (Witt et al., 2000). It has been estimated that plant-associated matter could contribute 1,700–3,470 kg organic carbon ha–1, of which 12–14% could be of roots origin (Kimura et al., 2004). Root-derived soil organic carbon (SOC) is often considered more stable than that from aboveground plant residues (Gale and Cambardella, 2000; Rasse et al., 2005; Kätterer et al., 2011). In the past decade, numerous studies have focused on the influence of plant carbon input on the native microbial biomass carbon (MBC) and SOC sequestration (Lu et al., 2003; Guenet et al., 2010; Ge et al., 2012), particularly the dynamic changes of chemical composition (e.g., lignin, nonstructural carbohydrates) during rice root decomposition (Lu et al., 2010; Park et al., 2020). However, microbial mechanisms underlying the coupling of carbon and nitrogen are poorly understood, though most studies have been done with respect to the root organic carbon (ROC) allocation (CO2, dissolved organic carbon, SOC, MBC) or the root-derived nitrogen release (NH4+, NO3–, dissolved organic nitrogen, gas N).
Plant-microbe interactions are crucial for plant growth, productivity, phytoremediation, and the soil carbon sequestration (Bulgarelli et al., 2012; Lundberg et al., 2012; Trivedi et al., 2013). Root-associated microbes catalyze nutrient cycling (C, N, P, S, Fe) (Baya et al., 1981; Scheid et al., 2004) and play a key role on plant growth (Ding et al., 2019; Moreau et al., 2019). For instance, plant growth-promoting bacteria include Clostridium, Bacteroidetes (Rui et al., 2009), Proteobacteria, Sphingobacteria, and Actinobacteria (Li et al., 2011; Guo et al., 2020). Nevertheless, culture-dependent technique has significantly hampered taxonomic identification of microorganisms in association with root decomposition (Kimura et al., 1989; Murakami et al., 1990). The rapid advance of high-throughput sequencing in combination with stable isotope probing (SIP) thus provides powerful tools in recent years to establish direct links of carbon and nitrogen flows to phylogenetic identities of plant-associated microbial communities.
Rice paddies contribute 10–25% of global CH4 emissions, and up to 90% of the methane could be consumed by methane-oxidizing bacteria (MOB, methanotrophs) in rice roots due to radial oxygen loss (Holzapfel-Pschorn et al., 1986). The vast amount of methane oxidation in rice roots could intensify methane-driven carbon metabolism at multitrophic levels. For example, methylotrophic Hyphomicrobiaceae and phylogenetically distinct guilds of nitrogen-fixing Rhizobia have been revealed as key players driving CH4-derived carbon transfer (Murase and Frenzel, 2007; Lee et al., 2021). Moreover, plant residues are often characterized with the exceptionally high C/N ratios, leading to severe deficiency of nitrogen for microbial growth (Zechmeister-Boltenstern et al., 2015). For example, soil microbial biomass often had a value of C:N ratios about 8–9 (Cleveland and Liptzin, 2007), while rice root could be as high as 27 (Lu et al., 2010). Plant root-associated microbes could have thus likely evolved N2-fixing function to meet N demand. The ability to fix N2 in methanotrophs has long been demonstrated in pure cultures (Davis et al., 1964; Murrell and Dalton, 1983), and molecular analysis revealed distinct evolutionary trajectories of nitrogenase genes for MOB (Auman et al., 2001; Dedysh et al., 2004). A rough estimate has suggested that methanotrophy could have likely contributed 33–47% of reactive N input in water-submerged peatlands (Larmola et al., 2014), whereas our previous study showed 11.5% of plant N in rice fields was from MOB on the basis of nitrogen dilution pool extrapolation (Bao et al., 2014; Minamisawa et al., 2016). However, N2 fixation occurred most likely in association with root decomposition and respiration to relieve environmental stresses in complex environment (Fog, 1988; Ramirez et al., 2012; Finn et al., 2015), and the solid evidence for methanotrophy-induced carbon and nitrogen coupling is still missing.
SIP is a powerful mean to trace carbon and nitrogen flow, and to establish direct link of ecologically important processes to physiologically active taxa in complex environment (Dumont and Murrell, 2005; Lu et al., 2006). Here, to investigate the influence of methane oxidation on root carbon and nitrogen turnover and identify the active microorganisms, dual DNA-SIP (13CH4 and 15N2) was conducted. The rice roots were collected at the seedling stage and incubated in N-free or N-replete liquid medium with 13CH4 and 15N2. Root decomposition and microbial metabolism were thoroughly determined by the root biomass, microbial biomass, gases production, and dissolved compounds, respectively.
Materials and Methods
Rice Root Sampling and Pretreatment
Rice samples at the seedling stage (30 days after sowing in the field) were collected from the Changshu Agro-Ecological Experimental Station of the Chinese Academy of Sciences, Jiangsu Province, China (31.5497N, 120.6984E). Seeds were pre-germinated 48 h and sown into the field in waterlogged condition from May 1 to June 6, 2019. The rice seedlings were fertilized with urea, calcium superphosphate, and potassium chloride in 433.9, 535.7, and 133.9 kg ha–2, respectively, on May 15, supplemented by 112.5 kg ha–2 of urea on May 24, and 187.5 kg ha–2 of urea on June 2. The entire rice plants were brought back to laboratory in cold storage. After washing the loosely adherent soil particles with tap water by gentle shaking, the roots were cut off and put into 50 ml centrifuge tubes with 40 ml deionized sterile water, followed by vortex for 10 min and centrifugation at 5,000 rpm for 10 min. The fresh roots were then transferred to a new 50 ml centrifuge tube with 40 ml deionized sterile water, subjected to sonication at 40 Hz for 30 s, and centrifuged again at 5,000 rpm for 10 min to collect root pellet. The sonication and centrifugation procedures were repeated twice to ensure the complete removal of soils on the roots as previously described (Bao et al., 2014; Edwards et al., 2015). The soil-free roots were cut into 1–2 cm short pieces and divided into two subsamples. One subsample was used for microcosm incubation, whereas the other was frozen at –80°C for DNA extraction to investigate the root-associated microbial communities.
Microcosm Construction for 13CH4 and 15N2 Stable Isotope Probing
We set three treatments to incubate the rice roots in nitrate mineral salts (NMS) or nitrate-free mineral salts (NFMS) media (Whittenbury et al., 1970) to enhance microbial growth. In brief, 1 g of fresh roots was placed into a 120-ml serum bottle amended with 40 ml medium. Treatment NoCH4: NFMS medium+10% 15N2+10% O2; treatment 13CH4: NFMS medium+10% 13CH4+10% 15N2+10% O2; and treatment 13CH4+NO3–: NMS medium+10% 13CH4+10% 15N2+20% O2. One atmosphere pressure was achieved for all serum bottles using inert argon gas. The labeled gas was purchased from the Cambridge Isotope Laboratories company (99% 15N-labeled for 15N2 and 99% 13C-labeled for 13CH4). The headspace gases were evacuated and flushed with argon gas before microcosm construction, and the serum bottles were sealed with rubber stoppers and aluminum caps. All treatments were conducted in triplicate bottles in a shaker with 150 rpm at 30°C for 30 days in dark.
Measurement of Carbon and Nitrogen Turnover in Rice Root
Microbial-Respired CO2 From Root Organic Carbon Decomposition and CH4 Oxidation
Headspace gases (1 ml) in the serum bottles were sampled on days 0 and 30, respectively, for the determination of the CO2 emission during microcosm incubation. It includes root-derived CO2 and 13CH4-derived 13CO2 in treatments with CH4 amended. Concentrations of CO2 were measured using a gas chromatography (Agilent 7890, United States) equipped with a Porapak Q column and a thermal conductivity detector with N2 as the carrier gas. The column temperature was set at 60°C, and the detector and injector were set at 150°C. The isotope excess of 13C-CO2 was measured using a MAT253 stable isographic quality spectrometer (Thermo Fisher Scientific, Dreieich, Germany).
Inorganic N Species (NH4+, NO3– and NO2–)
After microcosm incubation, all of the roots and medium in the serum bottles were transferred to 50 ml centrifuge tubes and were centrifuged at 5,000 rpm for 10 min to collect roots, as well as microbial biomass (undissolved). The supernatants (dissolved compounds) were collected for measurement of inorganic nitrogen, including NH4+, NO3–, and NO2–. The concentrations were determined by a continuous flow analytical system (San++System, Skalar, Holland), and the 15N isotope excess was determined using IsoPrime 100 stable isotope ratio mass spectrometer (Elementar, Germany). Nitrite was always below the detection limit for all treatments, except for the 13CH4+NO treatment, whereas ammonium was detected in all the treatments.
Total Organic Carbon and Total Organic Nitrogen
The root system pellets after centrifugation as mentioned above were thoroughly rinsed to remove the dissolved carbon and nitrogen (DOC, DON, and inorganic N species). The rinsed pellets were then ground into powders in liquid nitrogen. Approximately 0.2 g powders were set apart for DNA extraction, whereas the rest of the pellet powders were completely freeze-dried by Freeze Dryers (Alpha 1-2 LD plus, CHIRIST, Germany). The dry powders were passed through a sieve with 0.15 mm pore size for the determination of Total Organic Carbon (TOC) and Total Organic Nitrogen (TON) by FlashSmart Elemental Analyzer (Thermo Fisher Scientific, Dreieich, Germany). It should be noted that DOC was calculated as the difference between the undissolved TOC of root system at day 0 and CO2 plus undissolved TOC at day 30, and DON as the difference between undissolved TON of root system at day 0 and inorganic nitrogen plus undissolved TON at day 30. The isotope excess of 13C-TOC and 15N-TON was analyzed by Flash 2000 elemental analyzer coupled to a Delta V Advantage isotope ratio mass spectrometer (Thermo Fisher Scientific, Dreieich, Germany).
Ecophysiology Flux Calculation of Methanotrophy-Directed Carbon and Nitrogen Metabolism in CH4-Consuming Rice Root
Flux of 13CH4 Consumption (TCH4)
Total CH4 consumption includes 13CH4-derived biomass in root system TOC, 13CO2 production in the headspace, and the 13C-dissolved organic carbon in liquid medium, including cell lysis, extracellular polymers (EPS), and intermediates. It was calculated as follows:
where TCH4 is total CH4 consumption, expressed as μmol C g–1 dry weight biomass; ATOC, atom excess of 13C in TOC, %; CTOC, TOC content, %; ACO2, atom excess of 13C in CO2, %; CCO2, the detected concentration of the total CO2 emission, μmol l–1; Vgas, the volume of the headspace gas of the incubation bottles (80 ml, 0.08 l); M, molar mass of 13C, g mol–1; and m, the dry weight biomass of root pellets, g. It should be noted that 13C-dissolved organic carbon was not taken into account in our results, whereas its proportion was relatively low (<5%) as previously reported (He et al., 2020).
Microbial Carbon Utilization Efficiency of CH4-Derived C
Carbon utilization efficiency (CUE) during methanotrophy is defined as the production of 13C-organic carbon per mole of CH4 oxidized (Trimmer et al., 2015), which is calculated as follows:
where the meaning of the designations is the same as that in Equation 1.
15N2 Fixation (TN2)
It includes 15N-microbial biomass nitrogen (15N-MBN) in root system, and 15N-inorganic nitrogen in liquid medium released from microbial mineralization of the fixed organic 15N. It is calculated as follows:
where TN2 is expressed as μmol N g–1 d.w.b.; the designations ANH4, ANO3, and ANO2 are the atom excess of 15N in the inorganic nitrogen, %; the designations CNH4, CNO3, and CNO2 are the concentrations of the inorganic nitrogen, mg l–1; Vl is the volume of liquid medium (40 ml, 0.04 l); M, molar mass of 15N, g mol–1; and m, the dry weight biomass of root pellets, g. 15N atom abundance was not determined if the bulk concentration of inorganic N was below detection limit (Supplementary Table 1).
Methanotrophic N2-Fixing Efficiency
Methanotrophic N2-fixing efficiency was defined as the amount of 15N2 fixed when one mole of methane was oxidized, calculated as the ratio of TCH4 to TN2. This value was employed to make a rough estimate of methane oxidation-induced N2 fixation budget in paddy field globally.
DNA-Stable Isotope Probing and High-Throughput Sequencing
DNA Extraction
For day 0 and incubated root samples, approximately 0.2 g of the ground powders were used for bacterial genome DNA extraction using FastDNA Spin Kit for Soil (MP Bi) according to the instructions of the kit. The extracted DNA was dissolved in 50 μl deionized sterile water, and its concentration and quality were determined by a UV spectrophotometer (NanoDrop ND-1000). The obtained DNA extracts were 10–50 ng μl–1 with A260/A280 in the range of 1.70–1.85.
DNA-SIP
The DNA obtained above were applied to ultra-high-speed density gradient centrifugation according to Jia and Conrad (2009), Xia et al. (2011). Brief steps are 2.0 μg of DNA was mixed with CsCl to form a centrifuge solution with an initial buoyancy density of 1.725 g ml–1, which was then transferred to a 5.1 ml centrifuge tube, and placed in a Beckman ultra-high-speed centrifuge for centrifugation at 45,000 rpm at 20°C for 44 h. Centrifuged solutions were divided into 15 fractions with different buoyancy density and collected into 1.5 ml sterile Eppendorf tubes. Each fraction was washed with PEG-6000 to remove CsCl as well as further washed with 70% ethanol to get clean DNA. Finally, the DNA was dissolved in 30 μl sterile water, stored at –20°C for subsequent 13C-DNA identification through quantitative polymerase chain reaction (PCR) of pmoA and nifH genes.
PCR and Illumina Sequencing
Bacterial primer sets of 515F/907R (Stubner, 2002) for 16S rRNA gene, A189f/mb661r (Costello and Lidstrom, 1999) for pmoA gene, and polF/polR (Poly et al., 2001) for nifH gene were used to investigate the total microbial communities, methane-oxidizing, and nitrogen-fixing microbes, respectively. The changes in gene abundances among different treatments as well as the different fractions of isotope labeled DNA were analyzed by quantitative PCR. Quantitative PCR reaction system includes 10 μl of SYBR Premix Ex Taq (Takara), 0.5 μl of primers (10 μM), 1 μl of DNA templates, and 8 μl of sterile water. The amplification curve and melting curve analysis were also performed after PCR cycling procedures for each reaction. All amplifications were conducted in triplicate, with amplification efficiencies in the range of 80–100%, R2 in 0.990–0.999, using CFX96 Optical Real-Time Detection System (Bio-Rad, Laboratories Inc., Hercules, CA, United States). High-throughput sequencing was employed to investigate the microbial community compositions of total DNA and 13C-DNA. The PCR products were gel purified by the Agarose Gel DNA Purification Kit (TaKaRa) and then mixed in equimolar ratios for library preparation. Paired-end sequencing (2 × 300 bp) was conducted using the Illumina MiSeq system. The details for primers and PCR conditions are given in Supplementary Table 2.
Bioinformatics Analyses
16S rRNA and nifH Genes
All raw sequences were processed using the Quantitative Insights Into Microbial Ecology pipeline (Caporaso et al., 2010). The quality control procedure removed reads with average quality score < 20 containing mismatched primers and ambiguous bases. Chimeras were eliminated using VSEARCH program (Rognes et al., 2016). The high-quality sequences were then clustered into operational taxonomic units (OTUs) at 97% similarities for 16S rRNA gene (Edgar, 2010) or 95% for nifH gene. For 16S rRNA gene, 1,282,215 clean reads were obtained for all of the 18 DNA samples. SILVA database (version 1.9.5) was used for taxonomic affiliation (Quast et al., 2013). Principal component analysis (PCA) of 16S rRNA gene community was conducted in Canoco5.1 The alpha diversity indices were calculated using the function ‘‘diversity’’ in R package ‘‘vegan.’’ For nifH gene, online Functional Gene Pipeline/Repository (FunGene2) was used for taxonomic analysis. The sequences of representative OTUs were then selected for phylogenetic tree construction with the MEGA5.2 software with bootstrapping of 1,000 replicates (Tamura et al., 2011).
pmoA Gene
Raw sequence files were processed using the Mothur software (version 1.33.3). The commands “trim.seqs” (minlength = 400 and qaverage = 30) and “split groups” were used for quality control and sample splitting (Schloss et al., 2009). These reads were then processed using the online version of FunGene Pipeline (Fish et al., 2013) to check the chimera using VSEARCH (Rognes et al., 2016) and to correct frameshifts using FramBot (Wang et al., 2013). OTU clustering based on amino acid sequences was performed using the UCLUST algorithm (Edgar, 2010) at a 7% cutoff value (Degelmann et al., 2010). Taxonomic affiliation was performed using the Bayesian method based on a database containing 6,628 pmoA and pmoA-related sequences of pure culture and uncultured methanotrophic ecotypes (Dumont et al., 2014).
Co-occurrence Network Analysis
Network structures of 16S rRNA gene sequences for all of the total DNA, including day 0 and incubated samples, were calculated in SpiecEasi R package. SpiecEasi (Sparse InversE Covariance estimation for Ecological Association and Statistical Inference) is a novel statistical method developed specifically for compositional data and network inference; it is done under the assumption of sparsity using sparse neighborhood and inverse covariance selection algorithms (Kurtz et al., 2015). The resulting adjacency matrices were converted into network objects using the R igraph package. Cytoscape (v 3.8.2) was used for visualizing the networks and identifying network modules (Shannon et al., 2003). Two topological parameters were used to estimate the role of the nodes, namely, within-module connectivity Zi and the among-module connectivity Pi (Guimera and Nunes Amaral, 2005). Module hubs were critical to their own module coherence (Zi > 2.5, Pi ≤ 0.62), connectors connected modules together and were important to network coherence (Zi ≤ 2.5, Pi > 0.62), and network hubs were vital to both the network and their own module coherence (Zi > 2.5, Pi > 0.62), whereas others are defined as peripherals (Zi ≤ 2.5, Pi ≤ 0.62) (Olesen et al., 2007). Module abundance was calculated as the sum of relative abundance of the microbial taxa (OTU) that belonged to it under different treatments. The relationships between relative abundance of each ecological cluster and C, N flow were analyzed using Pearson’s correlation test.
Statistical Analysis
Different forms of carbon and nitrogen contents and gene copy numbers were presented as mean ± standard deviation of triplicate values for each treatment. Statistical analyses were performed using IBM SPSS Statistics 19.0 (SPSS Inc., Cary, NC, United States). We tested significant differences by a one-way ANOVA with the least significant difference test and a p-value of less than 0.05 was considered statistically significant.
Results
Microbial Carbon Metabolism and N2 Fixation in CH4-Consuming Rice Root
The atom percent of 13CO2 increased from the background level of 1.08% in the absence of CH4 (NoCH4) to 15.05 and 23.78% in the presence of 13CH4 and 13CH4+NO3–, respectively (Figure 1A). Biomass synthesis of 13CH4-C resulted in significant enrichment of 13C-TOC from 1.08% under the NoCH4 treatment to 4.65 and 5.36% under the 13CH4 and 13CH4+NO3– treatments, respectively (Figure 1B). It indicated that MOB-assimilated 13C-CH4 contributed 3.57% of the ROC. The net CH4 consumption was assessed as the sum of 13CO2 flux and root 13C-TOC content with 1,238 and 1,734 μmol g–1 d.w.b. under the 13CH4 and 13CH4+NO3– treatments, respectively (Figure 1C). The carbon utilization efficiencies were 53 and 38% under the 13CH4 and 13CH4+NO3– treatments, respectively (Figure 1C).
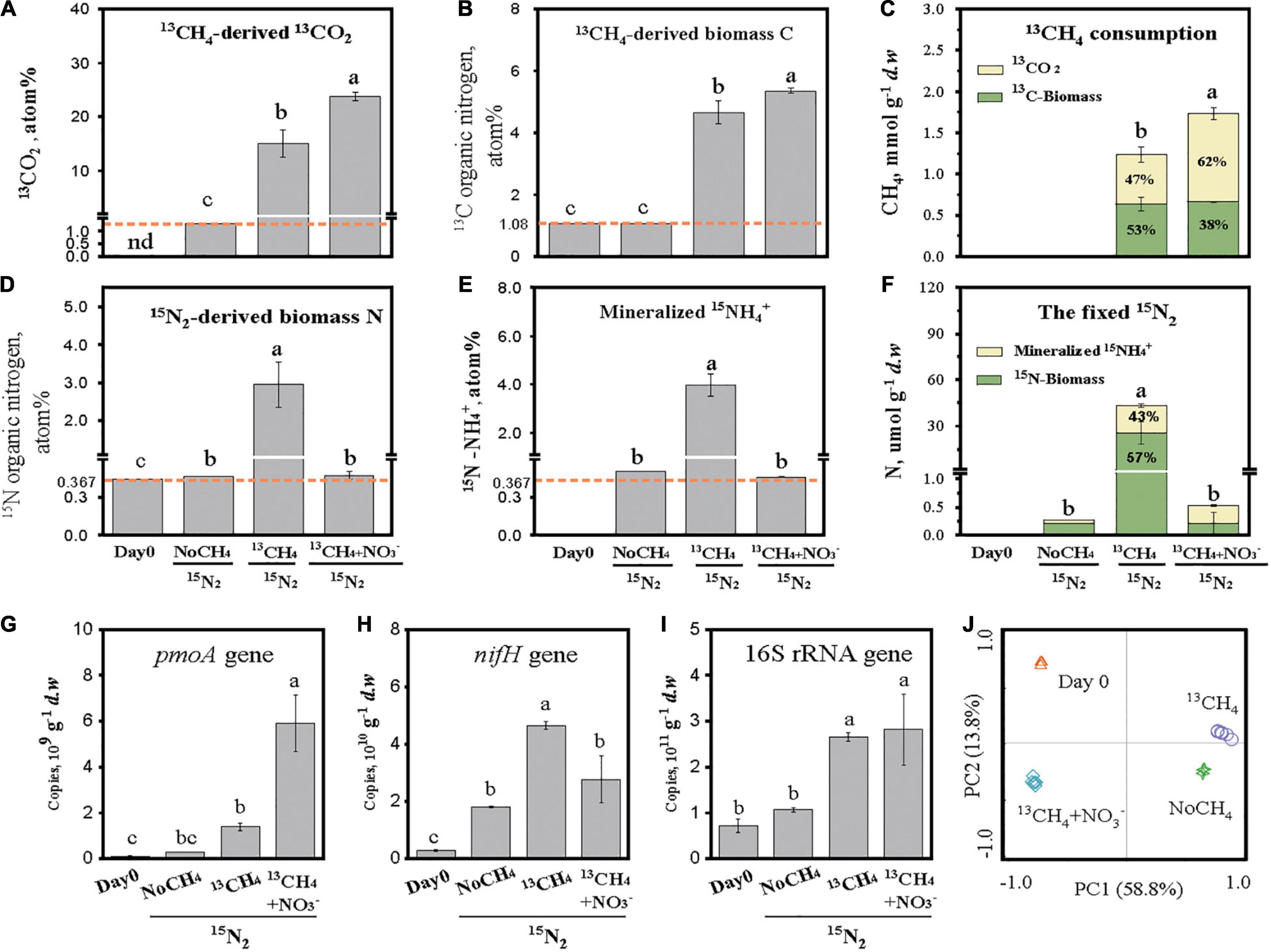
Figure 1. CH4-derived carbon flow and N2 fixation in CH4-consuming rice roots. The designations “NoCH4,” “13CH4,” and “13CH4+ NO3–” refer to the microcosms incubated without CH4, with 10% (v/v) 13CH4, and with 10% (v/v) 13CH4 and KNO3 fertilization, respectively. The atom percent of stable isotope C and N is shown as 13CH4-derived 13CO2 (A), 13CH4-derived biomass 13C (B), 13CH4 consumption as the sum of 13CH4-derived 13CO2 and 13CH4-derived biomass 13C (C),15N2-derived biomass 15N (D), mineralized 15N-NH4+ from the fixed organic 15N (E), and the fixed 15N2 as the sum of 15N2-derived biomass 15N and mineralized 15N-NH4+ (F). 13CO2 was calculated on the basis of the headspace CO2 concentration and its 13C atom percent. 13C-MBC was calculated on the basis of the total undissolved organic carbon content in root and its 13C atom percent. 15N-microbial biomass nitrogen (MBN) was calculated on the basis of the total undissolved organic nitrogen content in root and its 15N atom percent. The 15NH4+ represents the ammonium released from mineralization of 15N-cellular materials from 15N2-fixers and was calculated on the basis of the NH4+ concentration and its 15N atom percent. The abundance of methane-oxidizing bacteria, N2 fixers, and total root-associated microorganisms was determined by real-time quantitative PCR of the biomarker genes pmoA (G), nifH (H), and 16S rRNA (I), respectively. Principal component analysis (PCA) plots of 16S rRNA gene sequences were used to assess community shifts of total root-associated microorganisms in rice roots (J). The error bars represent the standard deviations of three microcosms. Different letters above the columns indicate a significant difference among different treatments (p < 0.05). n.d., not detected.
N2 fixation was significantly stimulated by methane oxidation as the atom percent of 15N-TON in rice roots increased drastically from the background level of 0.369% to the 2.951% in 13CH4 treatment (Figure 1D). In the absence of methane (NoCH4), the atom percent of 15N-TON remained unchanged, whereas nitrate amendment (13CH4+NO3–) almost abolished completely 15N enrichment in TON at 0.385% (Figure 1D). Intriguingly, 15N-NH4+ increased drastically from the background level of 0.369% to the 3.98% in 13CH4 treatment (Figure 1E), indicating rapid mineralization of 15N-TON. After 30 days incubation, the 15N-TON contents were 0.204, 24.9, and 0.210 μmol g–1 d.w.b. under the NoCH4, 13CH4, and 13CH4+NO3– treatments, respectively, and the 15N-NH4 contents were 0.06, 18.4, and 0.32 μmol g–1 d.w.b., respectively (Figure 1F). The fixed N was thus 164-fold higher under 13CH4 than under NoCH4 treatment. Moreover, 42.5% of the fixed N was microbially mineralized as bioavailable 15NH4+ in CH4-consuming rice roots (Figure 1F).
The population sizes of MOB and N2 fixers were assessed by real-time quantitative PCR analysis of functional pmoA and nifH gene biomarkers. Being consistent with CH4 consumption (Figure 1C) and N2 fixation (Figure 1F), a significant increase was observed for the pmoA gene (16-fold) and nifH gene (20-fold) under 13CH4 incubation (Figures 1G,H). Nonetheless, 13CH4+NO3– had the highest pmoA gene copies, whereas the nifH gene abundance was lower than that under the 13CH4 treatment, suggesting selective growth of MOB carrying no nifH genes under 13CH4+NO3–. The 16S rRNA gene abundance of the total microbial communities was also stimulated during methane oxidation regardless of nitrate amendment (Figure 1I), and obvious shifts in community structure were observed among the different treatments by PCA (Figure 1J).
Methane Oxidation as an Engine Driving Carbon and Nitrogen Flows in Rice Root
On day 0, ROC and MBC were considered undissolved and accounted for 98.6 and 1.4% of TOC, respectively (Figure 2A). Remarkable decomposition of ROC occurred during 30 days incubation, and the majority was recovered at day 30 as the dissolved ROC with 56.8, 62.4, and 66.8% of TOC under NoCH4, CH4, and CH4+NO3– treatments, respectively (Figures 2B–D). Root-associated methane oxidation triggered 2.5–6.5% more mineralization of ROC than the NoCH4 control (Figures 2B–D). Meanwhile, ROC-respired CO2 accounted for 6.6–10.1% of TOC, whereas 13CO2 from methane oxidation was below 1.9%. Intriguingly, root-associated MBC remained largely unchanged from 1.0 to 1.4% of TOC, whereas 13CH4-derived MBC accounted for 1.1 and 1.2% under CH4 and CH4+NO3– treatments, respectively (Figures 2C,D). It thus indicated strong stimulations of methanotrophy-directed ROC decomposition.
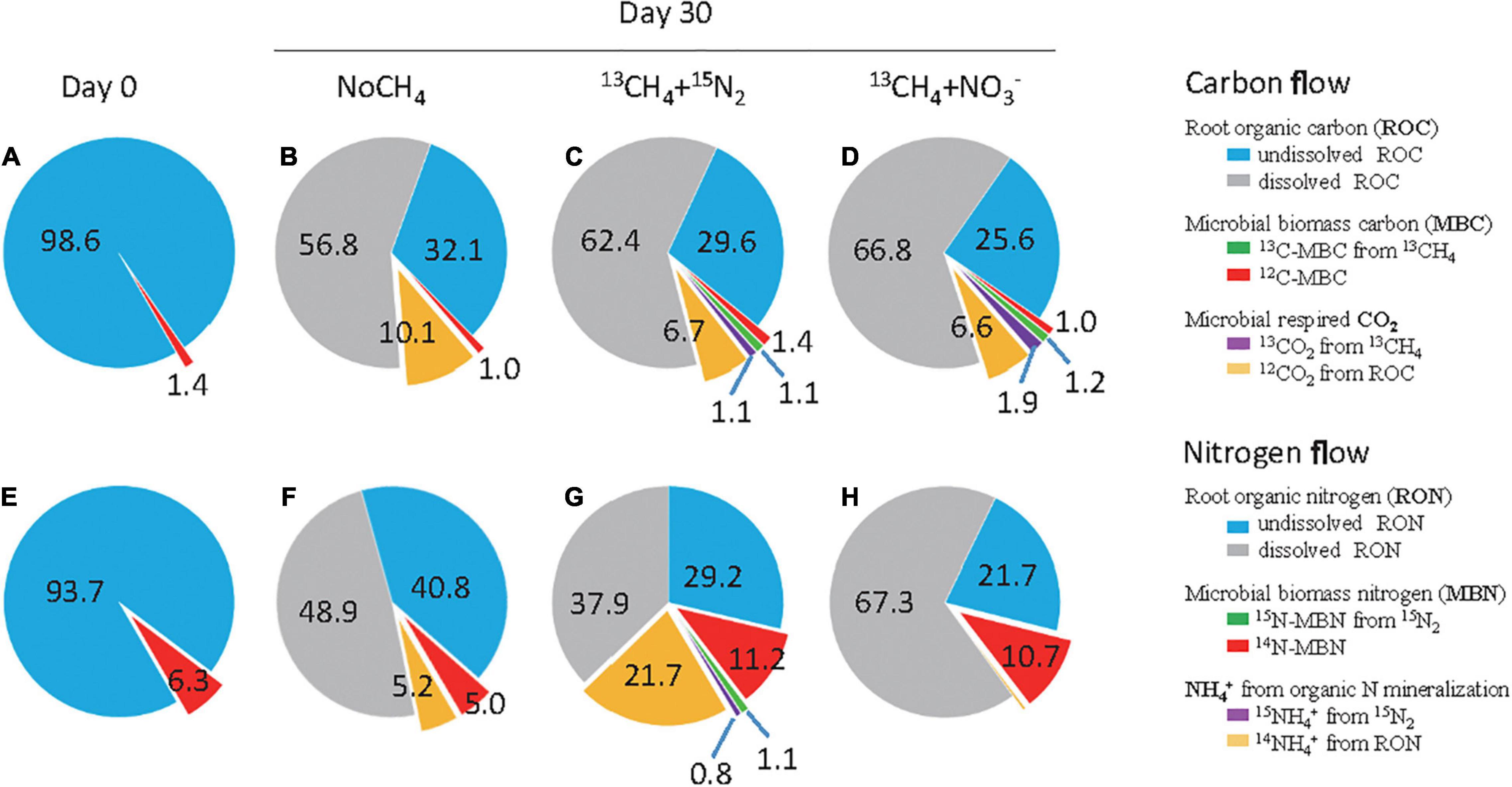
Figure 2. Methanotrophy-directed flow of carbon and nitrogen by rice root-associated microbes. The carbon and nitrogen percentage in rice roots at day 0 (A,E) and under three treatments of NoCH4 (B,F), 13CH4 (C,G), and 13CH4+NO3– (D,H) at day 30. Briefly, the undissolved root organic carbon (ROC) and nitrogen (RON) were determined on the basis of dry weight. The 12C-MBC was estimated by real-time quantification of microbial cells assuming that each cell has the dry weight of 6.2 × 10– 13 g (Knief and Dunfield, 2005) and contains 3.82 copies of 16S rRNA genes (Sun et al., 2013). 12CO2 from ROC was calculated by the difference between the total headspace CO2 and the 13CO2. The 14N-MBN was estimated by real-time quantification of microbial cells in a way similar to 12C-MBC. The 14NH4+ from RON mineralization was determined by the difference between the total NH4+ content and 15NH4+ in aqueous phase. The descriptions for other designations are the same as that in Figure 1.
Similarly, rapid mineralization of the undissolved root organic nitrogen (RON) was catalyzed by methanotrophy-mediated microbial communities, leading to a significant decline in RON from 93.7% of TON on day 0 (Figure 2E) to 40.8% (NoCH4), 29.2% (13CH4), and 21.7% (13CH4+NO3–) on day 30 (Figures 2F–H). The resulting NH4+-N content was 3.63, 15.5, and 0.26 μmol N bottle–1 under the NoCH4, 13CH4, and 13CH4+NO3– treatments, (Supplementary Table 1), and accounted for 5.2, 21.7, and 0.36% of TON, respectively (Figures 2F–H). The inorganic N was predominated by RON-mineralized 14NH4+-N with approximately 15.0 μmol N bottle–1, and methanotrophy-induced fixation of 15N2 was only 0.55 μmol N bottle–1 (Supplementary Table 1). Notably, MBN was also stimulated significantly from 6.3% of TON on day 0 to 11.2 and 10.7% on day 30 under 13CH4 and 13CH4+NO3– treatments, respectively, whereas CH4-induced 15N-MBN remained to be as low as 1.1% of TON. In contrast, nitrate amendment completely abolished mineralization of RON to inorganic 14NH4+-N (Figure 2H).
Active Methanotrophs in CH4-Consuming Rice Root
High-throughput sequencing of 16S rRNA gene indicated that more diverse species were induced during methane oxidation and nitrogen fixation. The Shannon index was higher after incubation than that in the fresh roots while the Simpson index displayed an opposite result (Supplementary Table 3). Intriguingly, the relative abundance of methanotrophs remained at a relatively low level, although a significant increase occurred from 0.05 to 0.87% after incubation (Supplementary Table 3).
High-throughput sequencing of pmoA gene amplicons revealed significant changes in MOB community structure in rice roots (Figure 3A). Over the incubation period, type I Methylomonas-like MOB increased drastically from 1.2 to 80% under 13CH4, whereas type II Methylocystis-like MOB increased from 25.3 to 88.6% under 13CH4+NO3– treatment (Figure 3A). Similar results were obtained for 16S rRNA gene analysis (Figure 3A). Notably, in NoCH4 treatment, MOB members within the uncultured rice paddy cluster (RPC-1 group) showed a significant increase in pmoA genes from 2.8 to 28.6%, whereas 16S rRNA genes indicated apparent stimulation of Methylomonas-like MOB.
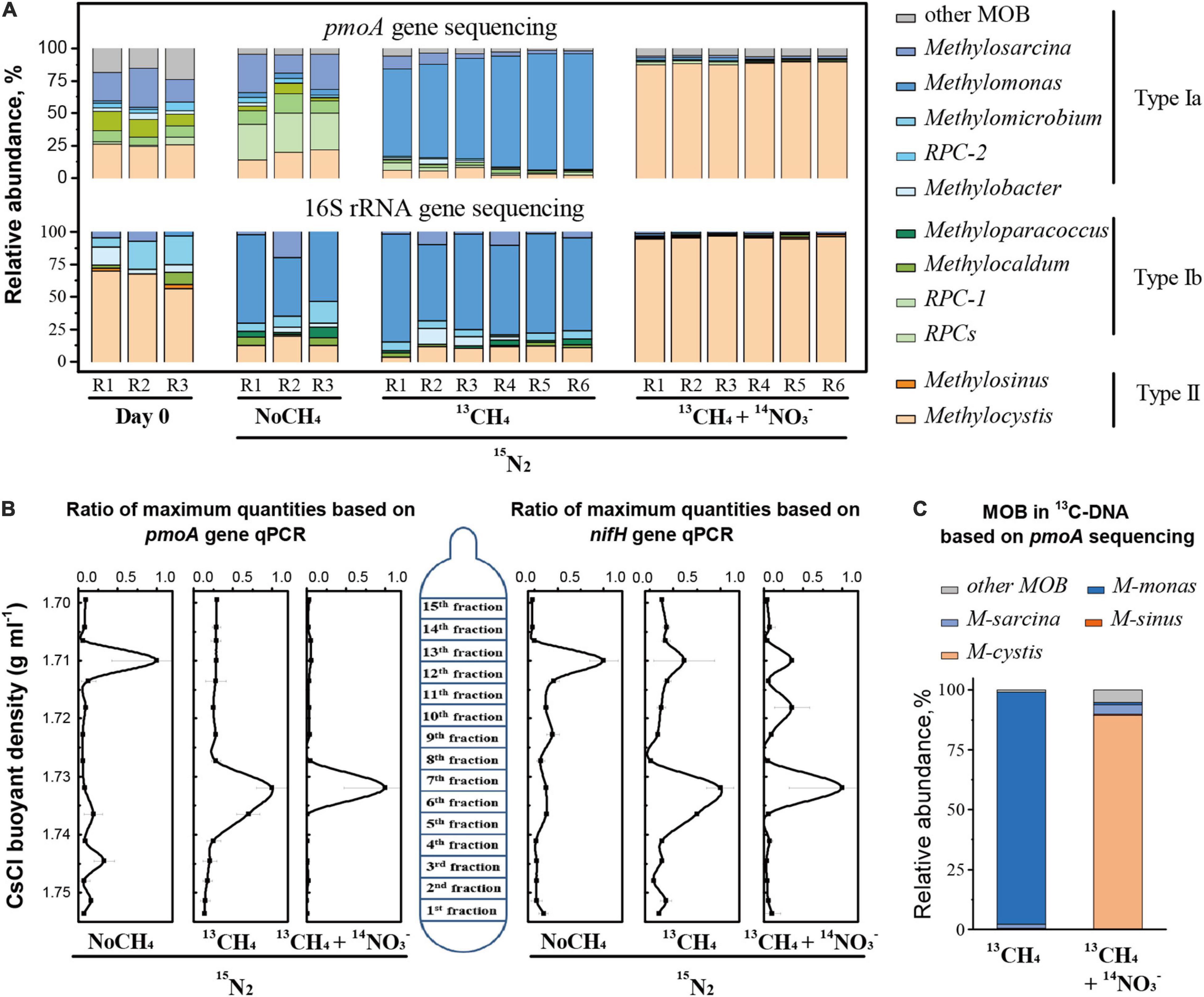
Figure 3. Population dynamics of active methanotrophs in CH4-consuming rice roots. Methanotrophic communities in the roots over a 30-day incubation period under different treatments on the basis of pmoA and 16S rRNA gene sequencing (A). Abundance of the pmoA and nifH genes as a function of buoyancy density of the fractionated DNA following ultracentrifugation of the total DNA (B). Taxonomic identities and relative abundance of 13C-labeled active methane-oxidizing bacteria (MOB) in 13C-DNA (C). Designations “R1–R6” represent biological duplication. Error bars represent the standard deviations of three technical duplicates. The designations “NoCH4,” “13CH4,” and “13CH4+ NO3–” are the same as those in Figure 1.
DNA-SIP relying on cell propagation provided further support. The 13C-labeled MOB were resolved by real-time quantitative PCR analysis of the pmoA and nifH genes as a function of the buoyant density of the DNA gradient following ultracentrifugation of the total DNA extracted from rice roots (Figure 3B). A single peak of pmoA genes occurred in the 13C-labeled “heavy” DNA (fractions 6–7) under 13CH4, whereas the unlabeled peak of pmoA genes remained in the “light” DNA under NoCH4. The peak was visualized clearly in the 13C-labeled “heavy” fraction DNA (seventh fraction) under 13CH4+NO3– (Figure 3B). Similar patterns were observed for nifH gene abundance as a function of DNA buoyant density. High-throughput sequencing of 13C-pmoA genes further indicated that active MOB were phylogenetically affiliated with Methylomonas and Methylocystis under the 13CH4 and 13CH4+NO3– treatments, respectively (Figure 3C).
Keystone Species Associated With Carbon and Nitrogen Turnover in Rice Root
After incubation for 30 days, statistical analysis showed that 314 root-associated OTUs significantly increased under NoCH4 treatment compared to that of day 0 fresh roots (Figure 4A). Similarly, a total of 65 under 13CH4 (Figure 4B) and 104 OTUs under 13CH4+NO3– (Figure 4C) showed statistically significant increases relative to NoCH4 treatment. As for NoCH4 treatment, Anaerolineaceae-like OTU emerged as the most abundant phylotype, accounting for 8.6% of the total microbial abundance, while Bacteroidete-like OTU exhibited the highest 461-fold increase from 0.004 to 1.7% (Figure 4D, Supplementary Table 4). As for 13CH4 treatment, Bacteroidete-like OTU predominated root-associated microbial communities with 14.3% of total microbial abundance and showed the most significant increase of 8.4-fold (Figure 4E and Supplementary Table 4). As for 13CH4+NO3– treatment, Xanthobacteraceae dominated root-associated microbial communities (20.9% of total microbial abundance) and exhibited the 91-fold increase (Figure 4F and Supplementary Table 4).
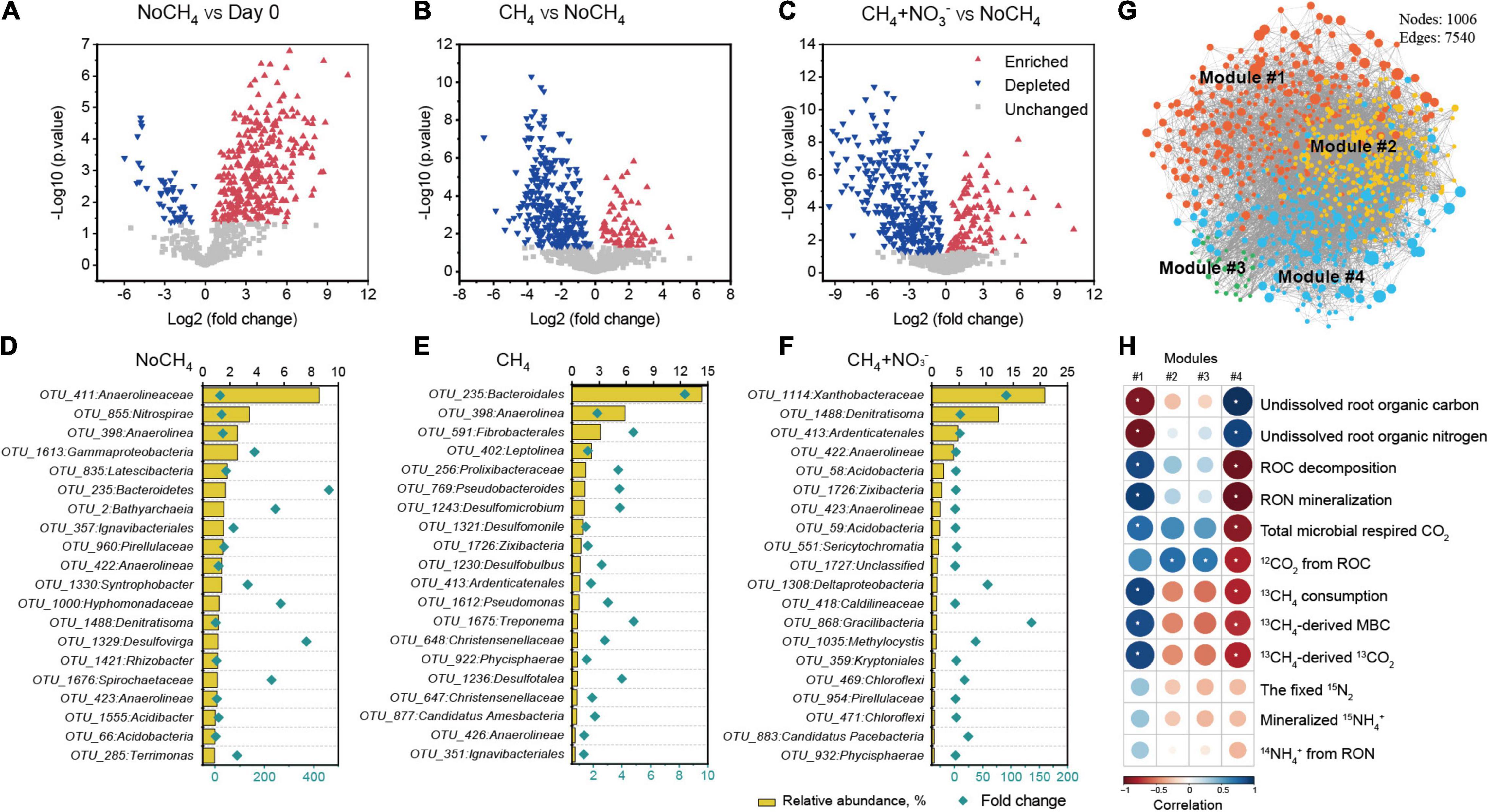
Figure 4. Population dynamics of microbial communities during methanotrophy-directed carbon and nitrogen turnover in rice root. Volcano plot shows significantly changed microbial taxa (operational taxonomic unit, OTU) over a 30-day incubation period for pairwise comparison of root microcosms including NoCH4 on day 30 vs. day 0 fresh roots (A), CH4 vs. NoCH4 on day 30 (B), and CH4+NO3– vs. NoCH4 on day 30 (C). The significantly changed OTUs satisfying the criteria of log 2 (fold-change) value > 0 or < 0 and p < 0.05 are considered enriched (red triangle) or depleted (blue triangle), respectively. The unchanged OTUs are represented by gray squares. The top 20 most abundant OTUs are shown, indicating the significantly enriched taxa under treatments of NoCH4 (D), CH4 (E), and CH4+ NO3– (F). In each figure, the relative abundances of the OTUs are shown as column while the fold changes are displayed as diamonds. The detailed taxonomy information of the OTUs is listed in Supplementary Table 4. Network diagram is constructed with the colored nodes representing four ecological modules (G), and the size of the node corresponds to the number of connections (degree). Heatmap showing the Person’s correlation between carbon or nitrogen flows and relative abundance of the main ecological clusters (H). Correlations (r values) are indicated with gradient colors ranging from red (-1) to blue (1), p < 0.05 is marked on the map with stars. The designations in the heatmap were the same as that in Figure 1 or Figure 2, while “ROC decomposition” is the sum of dissolved ROC and 12CO2 from ROC. “RON mineralization” is the sum of dissolved RON and 14NH4+ from RON.
Co-occurring pattern analysis of total microbial community further suggested these significantly increased taxa could have played a major role in root organic matter decomposition with rapid growth rates and reproduction ability (Figure 4G). Four ecological clusters (modules) strongly co-occurring with each other (modules #1, #2, #3, and #4) during root methane oxidation. These four modules were characterized by phylogenetically distinct keystone taxa (Supplementary Figure 1). For instance, module #1 contained Anaerovorax and the unclassified Gammaproteobacteria, whereas module #2 was defined by Lokiarchaeia, Galbitalea, and Chloroflexi (Supplementary Figure 1). Methane oxidation resulted in significant changes in modules. The abundance of module #1 was significantly stimulated for all NoCH4, CH4, and CH4+NO3– treatments, whereas module #4 showed decreasing trends (Supplementary Figure 2). Modules #2 and #3 were more abundant in NoCH4 treatment.
Significant correlations between C and N flows and modules suggested a methanotrophy-primed assembly of distinct communities. The abundance of module #1 was negatively correlated with the undissolved ROC and RON, but positively associated with root decomposition, including ROC decomposition, RON mineralization, total CO2 respiration, and RON-derived 14NH4+ production. Moreover, there was a positive relationship between module #1 and methanotrophy, including methane consumption, 13C-MBC production, and 13CO2 evolution. Intriguingly, the opposite trend was observed for module #4. Meanwhile, modules #2 and 3 were positively correlated with ROC-derived CO2 (Figure 4H).
Discussion
Our results provide compelling evidence that methanotrophy-directed carbon and nitrogen turnover in rice roots could have played a pivotal role in the self-sustaining productivity of paddy soil. MOB-mediated 13C-CH4 flow contributed 3.57% of the rice ROC (Figure 1B), being reasonably similar to 10% of Sphagnum moss carbon (Raghoebarsing et al., 2005). It also induced strong N2-fixing activity and contributed 2.58% of the RON under nitrogen-deplete condition (Figure 1D). Ecophysiology flux and DNA-SIP analysis indicated that methanotrophy-primed MOB and other strongly related taxa could have played a key role in root decomposition and N2 fixation to facilitate nutrients cycling while maintaining soil productivity.
It was shown in this study that methane oxidation stimulated biological nitrogen fixation while promoting microbial mineralization of rice RON under nitrogen constraint. A 50-year assessment of the global nitrogen budget for rice production systems indicated that 24% of rice N resulted from nonsymbiotic nitrogen fixation (Ladha et al., 2016). It has been estimated that annual net methane emission during the rice-growing season is 150 kg ha–1 (Sun et al., 2020), and up to 90% of methane has been recycled through methane oxidization before being released into the atmosphere (Conrad, 2009). Methanotrophy-induced N2 fixation was thus roughly estimated to be 40.7 kg N ha–1 in this study, which was close to 45 kg N ha–1 under near in situ field conditions (Bei et al., 2013). Compared to a C/N ratio of approximately 5 in a previous study in peatland (Larmola et al., 2014), the higher C/N ratio of 29 in the rice root triggered N2 fixation of 43.3 μmol N g–1 dry weight root biomass by gammaproteobacterial Methylomonas-like MOB during methane oxidation. MOB were widely regarded as the biofilters of greenhouse gas CH4 in flooded paddies or wetlands, and we provide an experimental evidence for great nitrogen-fixing potential of MOB in this study. The contribution of methanotrophy to nitrogen input deserves more attention in paddy fertilization management. Moreover, up to 42.5% of the newly fixed N existed in the form of 15N-NH4+, suggesting rapid microbial biomass N mineralization under methane oxidation. Therefore, methanotrophy could improve the rice self-sustaining system by providing more inorganic nitrogen that is directly used by plants, especially in the root or rhizosphere with high methane-producing and -oxidizing activities. These results linked the methane oxidation to crop productivity more closely in paddy fields.
A large amount of NH4+ was released though root decomposition under methane oxidation, which promoted the ratio of NH4+ mineralization from 5.2 (NoCH4) to 21.7% under nitrogen-deplete conditions (Figure 2B). This might be explained by nitrogen mining hypothesis between plant and microbial co-evolution. For example, plants have to be adapted to environment through interaction with soil microorganisms with defined ecosystem C:N:P stoichiometry (Chen and Chen, 2021). Microorganisms are supposed to have much stronger functional flexibility to acquire nitrogen from external environment to alleviate stoichiometric N constraints under excess C condition (Zechmeister-Boltenstern et al., 2015). It was confirmed by our results that microbial N2-fixing activity was stimulated under methane oxidation with low nitrogen condition. Besides, excessive carbon input would have intensified nitrogen recycling of microbial necromass (Kaiser et al., 2014; Cui et al., 2020). MOB, as biological engine of rice root decomposition, were likely to stimulate the flow of microbial carbon and nitrogen at different trophic levels, promoting the nutrient recycle and release. In fact, up to 42.5% of the newly fixed organic 15N was re-mineralized into NH4+ (Figure 1F). Moreover, decreasing microbial carbon use efficiency of community metabolism could also be a speculative feedback of nitrogen deficiency (Manzoni et al., 2012). Similarly in this study, 10.1% of ROC was converted into CO2 through microbial respiration under NoCH4 condition, whereas it was significantly reduced due to methanotrophy-induced N2 fixation or nitrate addition (Figure 2A). According to the law of ecological stoichiometry during organic matter degradation, the decomposition rate of organic carbon was higher than that of organic nitrogen, resulting in progressively reduced C:N ratio, and net N loss occurred when C:N ratio was 33–68 with variations across different litter types (Moore et al., 2011). Meanwhile, nitrogen release was found dominantly driven by initial tissue N content, where 1.02–1.98% of nitrogen content was supposed to exhibit nitrogen release (Parton et al., 2007). In this study, rice root with a relatively low C:N ratio of 20 and high N content of 1.73% was used, which may also contribute the nitrogen release during root mineralization.
DNA-SIP revealed a direct link between methane oxidation and taxonomic identities of active microorganisms. It has long been assumed that alphaproteobacterial MOB could be favored under nutrient-poor conditions, whereas gammaproteobacterial MOB rapidly propagates under rich substrate conditions (Murrell and Dalton, 1983). In stark contrast, we observed strong stimulation of Methylomonas-like MOB under nitrogen-fixing condition, and Methylocystis-like MOB showed preferential growth in nitrate-rich condition. The results might be explained by the rapid degradation or turnover of newly produced methanotrophic cells in the root decomposition system as well as the oxygen condition or other unknown factors in this study. It must be stated that the community catalyzing N2 fixation was not exclusively methane oxidizers, although the coupling of methanotrophy-primed carbon flow with N2 fixation was clearly demonstrated in this study. nifH gene sequencing and networking analysis showed that Burkholderia and Anaeromyxobacter could also have played a key role in N2 fixation, in addition to methanotrophic diazotrophs (Supplementary Figure 3). The contribution of MOB for nitrogen fixation should be delineated by approaches with higher resolutions such as fluorescent in situ hybridization (FISH) and Nanoscale Secondary Ion Beam Masspectrometry (NanoSIMS) at single-cell level (Kapili et al., 2020). Multiple samplings at different times would also be of help to elucidate the assembly patterns of active microbial communities in rice paddy field.
Co-occurring pattern analysis indicated that non-methanotrophs played important roles in the C and N turnover. Anaerolineaceae and Bacteroidetes preferred to grow under low nitrogen conditions while an uncultured Xanthobacteraceae taxon was more abundant in nitrate amended condition (Figures 4A–F). Anaerolineaceae belongs to the phylum Chloroflexi, which were predicted to have the ability to degrade diverse plant compounds, such as cellulose, starch, long-chain sugar, and pyrogallol (Hug et al., 2013; Angel et al., 2018). Bacteroidetes were detected in the soil with rice root decomposition (Rui et al., 2009) and was thought to be copiotrophs of r-strategy with high growth rate under nutrient-rich conditions (Fierer et al., 2007). These functional guilds could also secrete diverse arrays of carbohydrate-active enzymes for rapid growth (Larsbrink and McKee, 2020). Xanthobacteraceae was one of the very few taxa grown on chemically recalcitrant substrate lignin (Goldfarb et al., 2011). Other specialist taxa might be responsible for the turnover of recalcitrant carbon of root residues, despite their low abundance as slowly growing K-strategist (Koch, 2001). Ecological modules provide compelling evidences to elucidate the interactions and organizations of the microbial communities that might be associated with distinct functions (Faust and Raes, 2012). Module #1 showed strong association with the C and N turnover, with its keystone species including Anaerovorax and uncultured Gammaproteobacteria. These microorganisms are well known for degradation of nitrogen-rich chitin (Dai et al., 2016), implying their strong capability of nitrogen mining. In addition, Anaerovorax was known to ferment putrescine to acetate, butyrate, molecular hydrogen, and ammonia (Matthies et al., 2000), and these metabolic products might have been used as substrate to facilitate the growth of more diverse heterotrophs. It seems plausible that Anaerovorax served as the keystone taxa driving carbon and nitrogen turnover in this study. It should be noted that a number of uncultured or unclassified phylotypes was revealed in co-occurring pattern, and cultivation-dependent approach would be a key to better understand the couplings of carbon and nitrogen in rice root and soil ecosystems.
Conclusion
Our findings, based on dual SIP with 13CH4 and 15N2, confirmed the methanotrophy-mediated nitrogen fixation in rice root. A large proportion of fixed organic nitrogen was mineralized into the inorganic forms during this process, which could alleviate nitrogen constraint of root carbon turnover. High-throughput sequencing of the microorganisms revealed that Methylomonas-like methanotrophs dominated the nitrogen fixation and participated in the root decomposition together with other co-occur groups. Our study indicates that methanotrophy-induced turnover of carbon and nitrogen played an important role in the self-sustaining productivity of paddy soil. It thus provides a starting point for a more sophisticated assay to optimize field management for climate-smart agriculture of rice paddies.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SRR14456817–SRR14456854 and https://www.ncbi.nlm.nih.gov/, SRR18151151–SRR18151142.
Author Contributions
ZJ and WC designed the research and analyzed data. WC performed research. YC and ZB helped data mining. SW and XY offered the access to the long-term field experimental site and helped experimental design. WC wrote the first draft. ZJ finalized the manuscript. All authors have seen and approved the final version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (91751204, 41877062, 41530857, and 41807104).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Rong Huang, Xi Wang, Deling Sun, and Xiaoli Sun at the Institute of Soil Science for helpful discussion and sample analysis. We also thank the staff from the Changshu Agro-Ecological Experimental Station of the Chinese Academy of Sciences for the access to the rice field for root sampling.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.885087/full#supplementary-material
Footnotes
References
Angel, R., Panhölzl, C., Gabriel, R., Herbold, C., Wanek, W., Richter, A., et al. (2018). Application of stable-isotope labelling techniques for the detection of active diazotrophs. Environ. Microbiol. 20, 44–61. doi: 10.1111/1462-2920.13954
Auman, A. J., Speake, C. C., and Lidstrom, M. E. (2001). nifH sequences and nitrogen fixation in type I and type II methanotrophs. Appl. Environ. Microbiol. 67, 4009–4016. doi: 10.1128/aem.67.9.4009-4016.2001
Bao, Z., Watanabe, A., Sasaki, K., Okubo, T., Tokida, T., Liu, D., et al. (2014). A rice gene for microbial symbiosis, Oryza sativa CCaMK, reduces CH4 flux in a paddy field with low nitrogen input. Appl. Environ. Microbiol. 80, 1995–2003. doi: 10.1128/aem.03646-13
Baya, A. M., Boethlin, R. S., and Ramos-Cormenzana, A. (1981). Vitamin production in relation to phosphate solubilization by soil bacteria. Soil Biol. Biochem. 13, 527–531. doi: 10.1016/0038-0717(81)90044-4
Bei, Q. C., Liu, G., Tang, H. Y., Cadisch, G., Rasche, F., and Xie, Z. B. (2013). Heterotrophic and phototrophic 15N2 fixation and distribution of fixed 15N in a flooded rice-soil system. Soil Biol. Biochem. 59, 25–31. doi: 10.1016/j.soilbio.2013.01.008
Bulgarelli, D., Rott, M., Schlaeppi, K., Ver Loren Van Themaat, E., Ahmadinejad, N., Assenza, F., et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, X., and Chen, H. Y. H. (2021). Plant mixture balances terrestrial ecosystem C:N:P stoichiometry. Nat. Commun. 12:4562. doi: 10.1038/s41467-021-24889-w
Cleveland, C. C., and Liptzin, D. (2007). C:N:P stoichiometry in soil: is there a “Redfield ratio” for the microbial biomass? Biogeochemistry 85, 235–252. doi: 10.1007/s10533-007-9132-0
Conrad, R. (2009). The global methane cycle: recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 1, 285–292. doi: 10.1111/j.1758-2229.2009.00038.x
Costello, A. M., and Lidstrom, M. E. (1999). Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl. Environ. Microbiol. 65, 5066–5074. doi: 10.1128/aem.65.11.5066-5074.1999
Cui, J., Zhu, Z., Xu, X., Liu, S., Jones, D. L., Kuzyakov, Y., et al. (2020). Carbon and nitrogen recycling from microbial necromass to cope with C:N stoichiometric imbalance by priming. Soil Biol. Biochem. 142, 107720. doi: 10.1016/j.soilbio.2020.107720
Dai, Y., Yan, Z., Jia, L., Zhang, S., Gao, L., Wei, X., et al. (2016). The composition, localization and function of low-temperature-adapted microbial communities involved in methanogenic degradations of cellulose and chitin from Qinghai-Tibetan Plateau wetland soils. J. Appl. Microbiol. 121, 163–176. doi: 10.1111/jam.13164
Davis, J. B., Coty, V. F., and Stanley, J. P. (1964). Atmospheric nitrogen fixation by methane-oxidizing bacteria. J. Bacteriol. 88, 468–472. doi: 10.1128/jb.88.2.468-472.1964
Dedysh, S. N., Ricke, P., and Liesack, W. (2004). NifH and NifD phylogenies: an evolutionary basis for understanding nitrogen fixation capabilities of methanotrophic bacteria. Microbiology 150, 1301–1313. doi: 10.1099/mic.0.26585-0
Degelmann, D. M., Borken, W., Drake, H. L., and Kolb, S. (2010). Different atmospheric methane-oxidizing communities in European Beech and Norway Spruce soils. Appl. Environ. Microbiol. 76, 3228–3235. doi: 10.1128/aem.02730-09
Ding, L. J., Cui, H. L., Nie, S. A., Long, X. E., Duan, G. L., and Zhu, Y. G. (2019). Microbiomes inhabiting rice roots and rhizosphere. FEMS Microbiol Ecol 95:fiz040. doi: 10.1093/femsec/fiz040
Dumont, M. G., and Murrell, J. C. (2005). Stable isotope probing - linking microbial identity to function. Nat. Rev. Microbiol. 3, 499–504. doi: 10.1038/nrmicro1162
Dumont, M. G., Luke, C., Deng, Y. C., and Frenzel, P. (2014). Classification of pmoA amplicon pyrosequences using BLAST and the lowest common ancestor method in MEGAN. Front. Microbiol. 5:34. doi: 10.3389/fmicb.2014.00034
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Finn, D., Page, K., Catton, K., Strounina, E., Kienzle, M., Robertson, F., et al. (2015). Effect of added nitrogen on plant litter decomposition depends on initial soil carbon and nitrogen stoichiometry. Soil Biol. Biochem. 91, 160–168. doi: 10.1016/j.soilbio.2015.09.001
Fish, J. A., Chai, B. L., Wang, Q., Sun, Y. N., Brown, C. T., Tiedje, J. M., et al. (2013). FunGene: the functional gene pipeline and repository. Front Microbiol. 4:291. doi: 10.3389/fmicb.2013.00291
Fog, K. (1988). The effect of added nitrogen on the rate of decomposition of organic matter. Biol. Rev. 63, 433–462. doi: 10.1111/j.1469-185X.1988.tb00725.x
Gale, W. J., and Cambardella, C. A. (2000). Carbon dynamics of surface residue- and root-derived organic matter under simulated no-till. Soil Sci. Soc. Am. J. 64, 190–195. doi: 10.2136/sssaj2000.641190x
Ge, T., Yuan, H., Zhu, H., Wu, X., Nie, S. A., Liu, C., et al. (2012). Biological carbon assimilation and dynamics in a flooded rice-Soil system. Soil Biol. Biochem. 48, 39–46. doi: 10.1016/j.soilbio.2012.01.009
Goldfarb, K. C., Karaoz, U., Hanson, C. A., Santee, C. A., Bradford, M. A., Treseder, K. K., et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol. 2:94. doi: 10.3389/fmicb.2011.00094
Guenet, B., Neill, C., Bardoux, G., and Abbadie, L. (2010). Is there a linear relationship between priming effect intensity and the amount of organic matter input? Appl. Soil Ecol. 46, 436–442. doi: 10.1016/j.apsoil.2010.09.006
Guimera, R., and Nunes Amaral, L. A. (2005). Functional cartography of complex metabolic networks. Nature 433, 895–900. doi: 10.1038/nature03286
Guo, T., Zhang, Q., Ai, C., He, P., and Zhou, W. (2020). Microbial utilization of rice root residue-derived carbon explored by DNA stable-isotope probing. Eur. J. Soil Sci. 72, 460–473. doi: 10.1111/ejss.12970
He, R., Su, Y., Leewis, M. C., Chu, Y. X., Wang, J., Ma, R. C., et al. (2020). Low O2 level enhances CH4-derived carbon flow into microbial communities in landfill cover soils. Environ. Pollut. 258:12. doi: 10.1016/j.envpol.2019.113676
Holzapfel-Pschorn, A., Conrad, R., and Seiler, W. (1986). Effects of vegetation on the emission of methane from submerged paddy soil. Plant Soil 92, 223–233. doi: 10.1007/BF02372636
Hug, L. A., Castelle, C. J., Wrighton, K. C., Thomas, B. C., Sharon, I., Frischkorn, K. R., et al. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1:22. doi: 10.1186/2049-2618-1-22
Jia, Z., and Conrad, R. (2009). Bacteria rather than Archaea dominate microbial ammonia oxidation in an agricultural soil. Environ. Microbiol. 11, 1658–1671. doi: 10.1111/j.1462-2920.2009.01891.x
Kaiser, C., Franklin, O., Dieckmann, U., and Richter, A. (2014). Microbial community dynamics alleviate stoichiometric constraints during litter decay. Ecol. Lett. 17, 680–690. doi: 10.1111/ele.12269
Kapili, B. J., Barnett, S. E., Buckley, D. H., and Dekas, A. E. (2020). Evidence for phylogenetically and catabolically diverse active diazotrophs in deep-sea sediment. ISME J. 14, 971–983. doi: 10.1038/s41396-019-0584-8
Kätterer, T., Bolinder, M. A., Andrén, O., Kirchmann, H., and Menichetti, L. (2011). Roots contribute more to refractory soil organic matter than above-ground crop residues, as revealed by a long-term field experiment. Agric. Ecosyst. Environ. 141, 184–192. doi: 10.1016/j.agee.2011.02.029
Kimura, M., Murakami, H., and Wada, H. (1989). Microbial colonization and decomposition processes in rice rhizoplane. I. microbial colonization associated with lateral root emergence. Soil Sci. Plant Nutr. 35, 63–70. doi: 10.1080/00380768.1989.10434737
Kimura, M., Murase, J., and Lu, Y. (2004). Carbon cycling in rice field ecosystems in the context of input, decomposition and translocation of organic materials and the fates of their end products (CO2 and CH4). Soil Biol. Biochem. 36, 1399–1416. doi: 10.1016/j.soilbio.2004.03.006
Knief, C., and Dunfield, P. F. (2005). Response and adaptation of different methanotrophic bacteria to low methane mixing ratios. Environ. Microbiol. 7, 1307–1317. doi: 10.1111/j.1462-2920.2005.00814.x
Kurtz, Z. D., Muller, C. L., Miraldi, E. R., Littman, D. R., Blaser, M. J., and Bonneau, R. A. (2015). Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 11:e1004226. doi: 10.1371/journal.pcbi.1004226
Ladha, J. K., Tirol-Padre, A., Reddy, C. K., Cassman, K. G., Verma, S., Powlson, D. S., et al. (2016). Global nitrogen budgets in cereals: a 50-year assessment for maize, rice, and wheat production systems. Sci. Rep. 6:19355. doi: 10.1038/srep19355
Larmola, T., Leppanen, S. M., Tuittila, E.-S., Aarva, M., Merila, P., Fritze, H., et al. (2014). Methanotrophy induces nitrogen fixation during peatland development. Proc. Natl. Acad. Sci. U.S.A. 111, 734–739. doi: 10.1073/pnas.1314284111
Larsbrink, J., and McKee, L. S. (2020). Bacteroidetes bacteria in the soil: glycan acquisition, enzyme secretion, and gliding motility. Adv. Appl. Microbiol. 110, 63–98. doi: 10.1016/bs.aambs.2019.11.001
Lee, S., Sieradzki, E. T., Nicolas, A. M., Walker, R. L., Firestone, M. K., Hazard, C., et al. (2021). Methane-derived carbon flows into host-virus networks at different trophic levels in soil. Proc. Natl. Acad. Sci. U.S.A. 118:e2105124118. doi: 10.1073/pnas.2105124118
Li, Y., Lee, C. G., Watanabe, T., Murase, J., Asakawa, S., and Kimura, M. (2011). Identification of microbial communities that assimilate substrate from root cap cells in an aerobic soil using a DNA-SIP approach. Soil Biol. Biochem. 43, 1928–1935. doi: 10.1016/j.soilbio.2011.05.016
Lu, H., Wu, W., Chen, Y., Zhang, X., Devare, M., and Thies, J. E. (2010). Decomposition of Bt transgenic rice residues and response of soil microbial community in rapeseed-rice cropping system. Plant Soil 336, 279–290. doi: 10.1007/s11104-010-0476-1
Lu, Y., Rosencrantz, D., Liesack, W., and Conrad, R. (2006). Structure and activity of bacterial community inhabiting rice roots and the rhizosphere. Environ. Microbiol. 8, 1351–1360. doi: 10.1111/j.1462-2920.2006.01028.x
Lu, Y., Watanabe, A., and Kimura, M. (2003). Carbon dynamics of rhizodeposits, root- and shoot-residues in a rice soil. Soil Biol. Biochem. 35, 1223–1230. doi: 10.1016/s0038-0717(03)00184-6
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Manzoni, S., Taylor, P., Richter, A., Porporato, A., and Agren, G. I. (2012). Environmental and stoichiometric controls on microbial carbon-use efficiency in soils. New Phytol. 196, 79–91. doi: 10.1111/j.1469-8137.2012.04225.x
Matthies, C., Evers, S., Ludwig, W., and Schink, B. (2000). Anaerovorax odorimutans gen. nov., sp. nov., a putrescine-fermenting, strictly anaerobic bacterium. Int. J. Syst. Evol. Microbiol. 50, 1591–1594. doi: 10.1099/00207713-50-4-1591
Minamisawa, K., Imaizumi-Anraku, H., Bao, Z., Shinoda, R., Okubo, T., and Ikeda, S. (2016). Are symbiotic methanotrophs key microbes for N acquisition in paddy rice root? Microbes Environ. 31, 4–10. doi: 10.1264/jsme2.ME15180
Moore, T. R., Trofymow, J. A., Prescott, C. E., Titus, B. D., and Group, C. W. (2011). Nature and nurture in the dynamics of C, N and P during litter decomposition in Canadian forests. Plant Soil 339, 163–175. doi: 10.1007/s11104-010-0563-3)
Moreau, D., Bardgett, R. D., Finlay, R. D., Jones, D. L., and Philippot, L. (2019). A plant perspective on nitrogen cycling in the rhizosphere. Funct Ecol 33, 540–552. doi: 10.1111/1365-2435.13303
Murakami, H., Kimura, M., and Wada, H. (1990). Microbial colonization and decomposition processes in rice rhizoplane. Soil Sci. Plant Nutr. 36, 441–450. doi: 10.1080/00380768.1990.10416912
Murase, J., and Frenzel, P. (2007). A methane-driven microbial food web in a wetland rice soil. Environ. Microbiol. 9, 3025–3034. doi: 10.1111/j.1462-2920.2007.01414.x
Murrell, J. C., and Dalton, H. (1983). Nitrogen-fixation in obligate methanotrophs. J. Gen. Microbiol. 129, 3481–3486.
Olesen, J. M., Bascompte, J., Dupont, Y. L., and Jordano, P. (2007). The modularity of pollination networks. Proc. Natl. Acad. Sci. U.S.A. 104, 19891–19896. doi: 10.1073/pnas.0706375104
Park, H.-J., Lim, S.-S., Kwak, J.-H., Lee, K.-S., In Yang, H., Kim, H.-Y., et al. (2020). Biomass, chemical composition, and microbial decomposability of rice root and straw produced under co-elevated CO2 and temperature. Biol. Fertil. Soils 56, 991–1005. doi: 10.1007/s00374-020-01471-y
Parton, W., Silver, W. L., Burke, I. C., Grassens, L., Harmon, M. E., Currie, W. S., et al. (2007). Global-scale similarities in nitrogen release patterns during long-term decomposition. Science 315, 361–364. doi: 10.1126/science.1134853
Poly, F., Monrozier, L. J., and Bally, R. (2001). Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res. Microbiol. 152, 95–103. doi: 10.1016/s0923-2508(00)01172-4
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Raghoebarsing, A. A., Smolders, A. J. P., Schmid, M. C., Rijpstra, W. I. C., Wolters-Arts, M., Derksen, J., et al. (2005). Methanotrophic symbionts provide carbon for photosynthesis in peat bogs. Nature 436, 1153–1156. doi: 10.1038/nature03802
Ramirez, K. S., Craine, J. M., and Fierer, N. (2012). Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob. Change Biol. 18, 1918–1927. doi: 10.1111/j.1365-2486.2012.02639.x
Rasse, D. P., Rumpel, C., and Dignac, M.-F. (2005). Is soil carbon mostly root carbon? Mechanisms for a specific stabilisation. Plant Soil 269, 341–356. doi: 10.1007/s11104-004-0907-y
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahe, F. (2016). VSEARCH: a versatile open source tool for metagenomics. Peerj 4:e2584. doi: 10.7717/peerj.2584
Rui, J., Peng, J., and Lu, Y. (2009). Succession of bacterial populations during plant residue decomposition in rice field soil. Appl. Environ. Microbiol. 75, 4879–4886. doi: 10.1128/AEM.00702-09
Scheid, D., Stubner, S., and Conrad, R. (2004). Identification of rice root associated nitrate, sulfate and ferric iron reducing bacteria during root decomposition. FEMS Microbiol. Ecol. 50, 101–110. doi: 10.1016/j.femsec.2004.06.001
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-Source, Platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Stubner, S. (2002). Enumeration of 16S rDNA of Desulfotomaculum lineage 1 in rice field soil by real-time PCR with SybrGreen™ detection. J. Microbiol. Methods 50, 155–164. doi: 10.1016/S0167-7012(02)00024-6
Sun, D.-L., Jiang, X., Wu, Q. L., and Zhou, N.-Y. (2013). Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 79, 5962–5969. doi: 10.1128/AEM.01282-13
Sun, J., Wang, M., Xu, X., Cheng, K., Yue, Q., and Pan, G. (2020). Re-estimating methane emissions from Chinese paddy fields based on a regional empirical model and high-spatial-resolution data. Environ. Pollut. 265:115017. doi: 10.1016/j.envpol.2020.115017
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Trimmer, M., Shelley, F. C., Purdy, K. J., Maanoja, S. T., Chronopoulou, P. M., and Grey, J. (2015). Riverbed methanotrophy sustained by high carbon conversion efficiency. ISME J. 9, 2304–2314. doi: 10.1038/ismej.2015.98
Trivedi, P., Anderson, I. C., and Singh, B. K. (2013). Microbial modulators of soil carbon storage: integrating genomic and metabolic knowledge for global prediction. Trends Microbiol. 21, 641–651. doi: 10.1016/j.tim.2013.09.005
Wang, Q., Quensen, J. F., Fish, J. A., Lee, T. K., Sun, Y. N., Tiedje, J. M., et al. (2013). Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. Mbio 4:e00592-13. doi: 10.1128/mBio.00592-13
Whittenbury, R., Phillips, K. C., and Wilkinson, J. F. (1970). Enrichment, isolation and some properties of methane-utilizing bacteria. J. Gen. Microbiol. 61, 205–218. doi: 10.1099/00221287-61-2-205
Witt, C., Biker, U., Galicia, C. C., and Ottow, J. C. G. (2000). Dynamics of soil microbial biomass and nitrogen availability in a flooded rice soil amended with different C and N sources. Biol. Fertil. Soils 30, 520–527. doi: 10.1007/s003740050031
Xia, W., Zhang, C., Zeng, X., Feng, Y., Weng, J., Lin, X., et al. (2011). Autotrophic growth of nitrifying community in an agricultural soil. ISME J. 5, 1226–1236. doi: 10.1038/ismej.2011.5
Keywords: methanotrophs, carbon and nitrogen flow, methane, nitrogen fixation, stable isotope probing
Citation: Cao W, Cai Y, Bao Z, Wang S, Yan X and Jia Z (2022) Methanotrophy Alleviates Nitrogen Constraint of Carbon Turnover by Rice Root-Associated Microbiomes. Front. Microbiol. 13:885087. doi: 10.3389/fmicb.2022.885087
Received: 27 February 2022; Accepted: 21 April 2022;
Published: 18 May 2022.
Edited by:
Yurong Liu, Huazhong Agricultural University, ChinaReviewed by:
Peng Bao, Institute of Urban Environment (CAS), ChinaRahul Bahulikar, BAIF Development Research Foundation, India
Copyright © 2022 Cao, Cai, Bao, Wang, Yan and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongjun Jia, amlhQGlzc2FzLmFjLmNu