 Sabin Khanal
Sabin Khanal Sanjay Antony-Babu2
Sanjay Antony-Babu2 Xin-Gen Zhou
Xin-Gen Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 04 May 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.874120
This article is part of the Research Topic Basidiomycete Fungi: From Biosystematics and Biodiversity to Biotechnology View all 21 articles
Rice (Oryza sativa) is the second leading cereal crop in the world and is one of the most important field crops in the US, valued at approximately $2.5 billion. Kernel smut (Tilletia horrida Tak.), once considered as a minor disease, is now an emerging economically important disease in the US. In this study, we used multi-locus sequence analysis to investigate the genetic diversity of 63 isolates of T. horrida collected from various rice-growing areas across in the US. Three different phylogeny analyses (maximum likelihood, neighbor-joining, and minimum evolution) were conducted based on the gene sequence sets, consisting of all four genes concatenated together, two rRNA regions concatenated together, and only ITS region sequences. The results of multi-gene analyses revealed the presence of four clades in the US populations, with 59% of the isolates clustering together. The populations collected from Mississippi and Louisiana were found to be the most diverse, whereas the populations from Arkansas and California were the least diverse. Similarly, ITS region-based analysis revealed that there were three clades in the T. horrida populations, with a majority (76%) of the isolates clustering together along with the 22 Tilletia spp. from eight different countries (Australia, China, India, Korea, Pakistan, Taiwan, The US, and Vietnam) that were grouped together. Two of the three clades in the ITS region-based phylogeny consisted of the isolates reported from multiple countries, suggesting potential multiple entries of T. horrida into the US. This is the first multi-locus analysis of T. horrida populations. The results will help develop effective management strategies, especially breeding for resistant cultivars, for the control of kernel smut in rice.
Rice (Oryza sativa L.) is one of the most important crops with a worldwide production of 509 million metric tons annually (FAOSTAT, 2019). Rice provides a major source of energy for more than half of the world population (FAOSTAT, 2019). In 2019, the US rice production was estimated to be 10 million metric tons. Arkansas, California, Louisiana, Mississippi, Missouri, and Texas are the major rice producers in the US (USDA, 2020). Rice kernel smut, caused by Tilletia horrida Tak., causes partial or full bunt in rice grains, resulting in a direct reduction in grain yield and quality (Whitney and Cartwright, 2018). Rice kernel smut was first reported in 1896 in Japan (Takahashi, 1896); currently, rice kernel smut is widespread across rice-growing countries and its distribution is expected to be wider than recently reported (Carris et al., 2006).
Average losses from rice kernel smut have been reported around 15%; however, losses as high as 87 and 100% in Pakistan and China have been reported (Biswas, 2003). Major losses from kernel smut are due to the depletion in grain quality with countries restricting the maximum permissible limit for the smutted grains. Milled rice in the US has a restriction of 3% smutted rice (USDA, 2020). Similarly, certified rice seeds in India have a restriction of 0.5% smutted rice grains (Chahal, 2001). Historically kernel smut was considered a minor disease; however, persistent occurrence and frequent outbreaks of the disease in recent years have made kernel smut as one of the most economically important diseases in rice in many countries (Elshafey, 2018; Wang et al., 2019b; Allen et al., 2020; Zhou et al., 2020). In the US, kernel smut occurrence and severity have been on the rise for the past decade and pose a serious threat to the US rice production (Espino, 2019, 2020; Allen et al., 2020; Way and Zhou, 2020). In 2021, severe outbreaks of kernel smut occurred widely across the Texas rice areas and southwest Louisiana, with the percentage of affected panicles ranged up to 50% and the infected kernels ranged up to 20 percent (Zhou et al., 2021a). In states such as Arkansas and California where rice industry is valued as billion-dollar industry, potential economic losses are even higher (USDA, 2020). With continual increase in acreage compounded with the use of susceptible cultivars, kernel smut has also threatened organic rice production in California and Texas, the two leading states in the US organic rice production (Zhou et al., 2021b).
Rice kernel smut is caused by a basidiomycota fungus, belonging to Tilletia genus and Tilletiaceae family. More than 80 genera and 4,200 species of smut fungi have been reported as the pathogens to many plant species (Vánky, 1987). Phylogenetically Tilletia species have been considered to separate their lines from those of other smut fungi, Ustilago and Sporisorium (Roux et al., 1998). Tilletia horrida forms thick walled dark teliospores which can be present widely on the soil, plant debris, and rice seeds (Carris et al., 2006; Whitney and Cartwright, 2018). Kernel smut taxonomy has been turbulent through the years of many studies. Tilletia horrida Tak., was first described by Takahashi in 1896; however, over the years, various authors reclassified the fungus to different genus and species: T. barclayana (Bref.) Sacc. & Syd., Neovossia barclayana (Bref.), and Neovossia horrida (Tak.; Tullis and Johnson, 1952). Through the years, T. barclayana and T. horrida have been interchangeably used to describe kernel smut of rice. However, a distinction between T. horrida and T. barclayana has been demonstrated by various molecular and phylogenetic studies (Levy et al., 2001; Carris et al., 2006). Currently, T. horrida has been more commonly used to describe kernel smut of rice in the literature (Wang et al., 2015, 2019a,b; Allen et al., 2020).
Molecular phylogeny through multi-locus sequence typing (MLST) offers an excellent means to parse bacterial population structure with the use of housekeeping gene sequences and hence found a rightful reliable place in disease epidemiology (Maiden et al., 1998). MLST characterizes bacterial strain by their unique allelic profiles by measuring the variations in housekeeping genes. MLST provides a discriminatory power to differentiate different bacterial strains. Although use of MLST is less prevalent in mycology, it has also become a useful tool for studying to understand the fungal populations (Taylor and Fisher, 2003). The method represents an important tool to determine the population of fungi that are pathogenic to humans (Bougnoux et al., 2003; Bain et al., 2007) and plants (Kellner et al., 2011; Choi et al., 2013; Sun et al., 2013; Gurjar et al., 2021). MLSA has also been used to study the genetic diversity of various smut fungi (Kellner et al., 2011; Sun et al., 2013; Gurjar et al., 2021; Sedaghatjoo, 2021). Previous phylogenetic studies of T. horrida populations have been rare. One phylogenetic study conducted with T. horrida isolates collected from seven different provinces in China did not find any genetic variation (Wang et al., 2018). In the current study, we used the MLSA approach to understand the genetic diversity of the T. horrida populations in the US. Understanding the genetic diversity will help in designing and improving rice breeding programs to develop new cultivars with improved kernel smut resistance and in developing effective chemical management strategies for control of kernel smut. The results of our multi-gene phylogeny analyses showed, for the first time, the presence of genetic diversity in the rice kernel smut populations in the US, with all the T. horrida isolates clustering into four different genetic groups.
Rice grain samples were collected in the 2018 and 2019 growing seasons from six major different rice-growing states in the US (Figure 1; Table 1). Rice grain samples showing the symptoms of kernel smut were brought to the Plant Pathology Lab at the Texas A&M AgriLife Research Center, Beaumont, Texas. Sixty-three fungal isolates were isolated from the infected rice grain samples. Putative T. horrida were isolated from teliospores in 2% water-agar, based on the procedure described previously (Chahal et al., 1993). Germination of teliospores was visually confirmed under microscope after 3 days of incubation. Primary sporidia that germinated from the single teliospores were transferred to potato dextrose agar (PDA) plates and incubated for growth at 28o C for 14 days. Mycelium was stored in a solution comprising of 2% of sucrose and 20% of glycerol solution in −80°C for long-term storage.
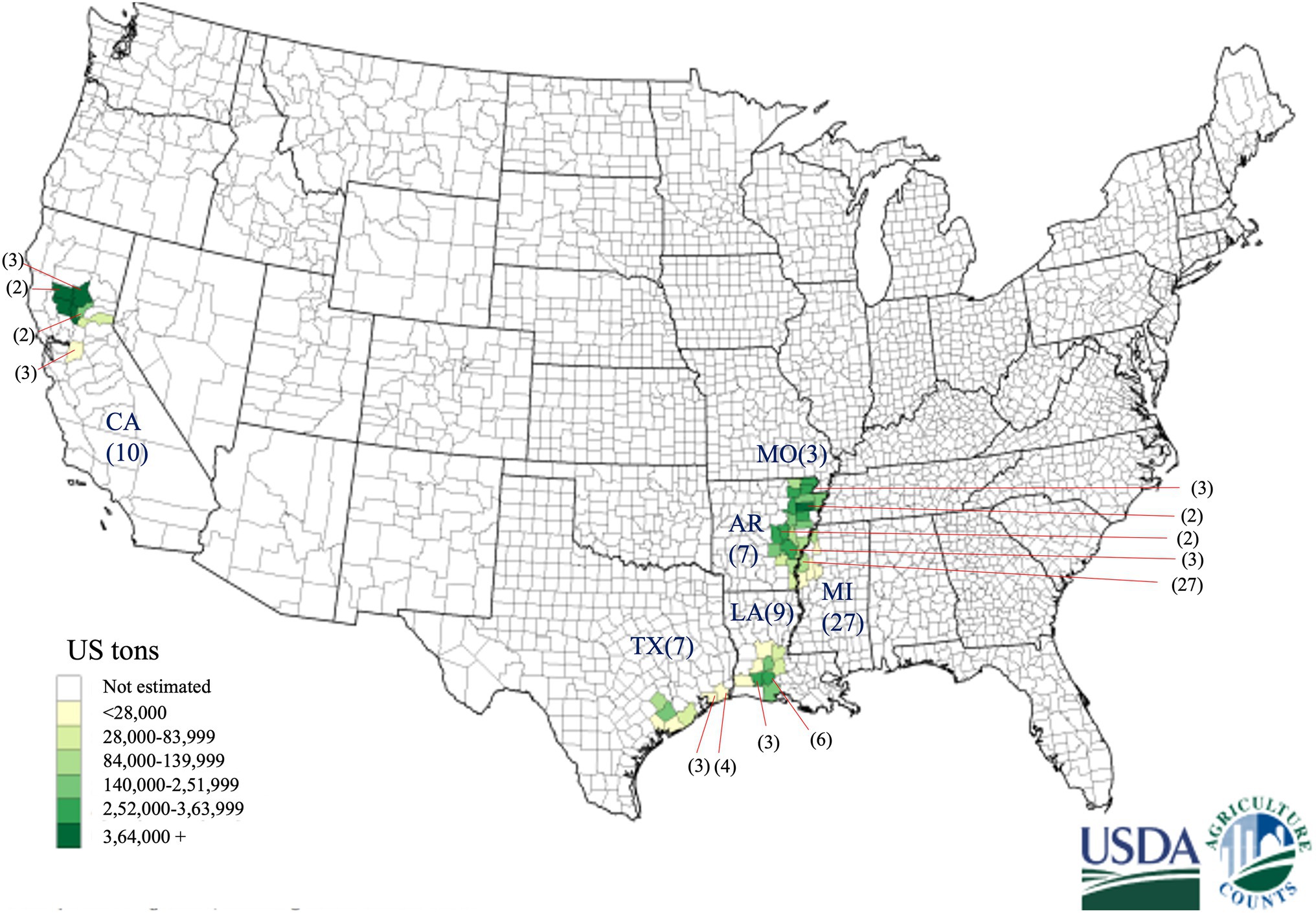
Figure 1. Geographical distribution of 63 Tilletia horrida isolates in Arkansas (AR), California (CA), Louisiana (LA), Mississippi (MI), Missouri (MO), and Texas (TX), covering almost all rice-growing areas in the US. Gradient shading areas inside each state represent the rice production in the US in 2019 provided by the USDA National Agricultural statistics services. The number in the parentheses represents the number of isolates from each county pointed by the red arrow.
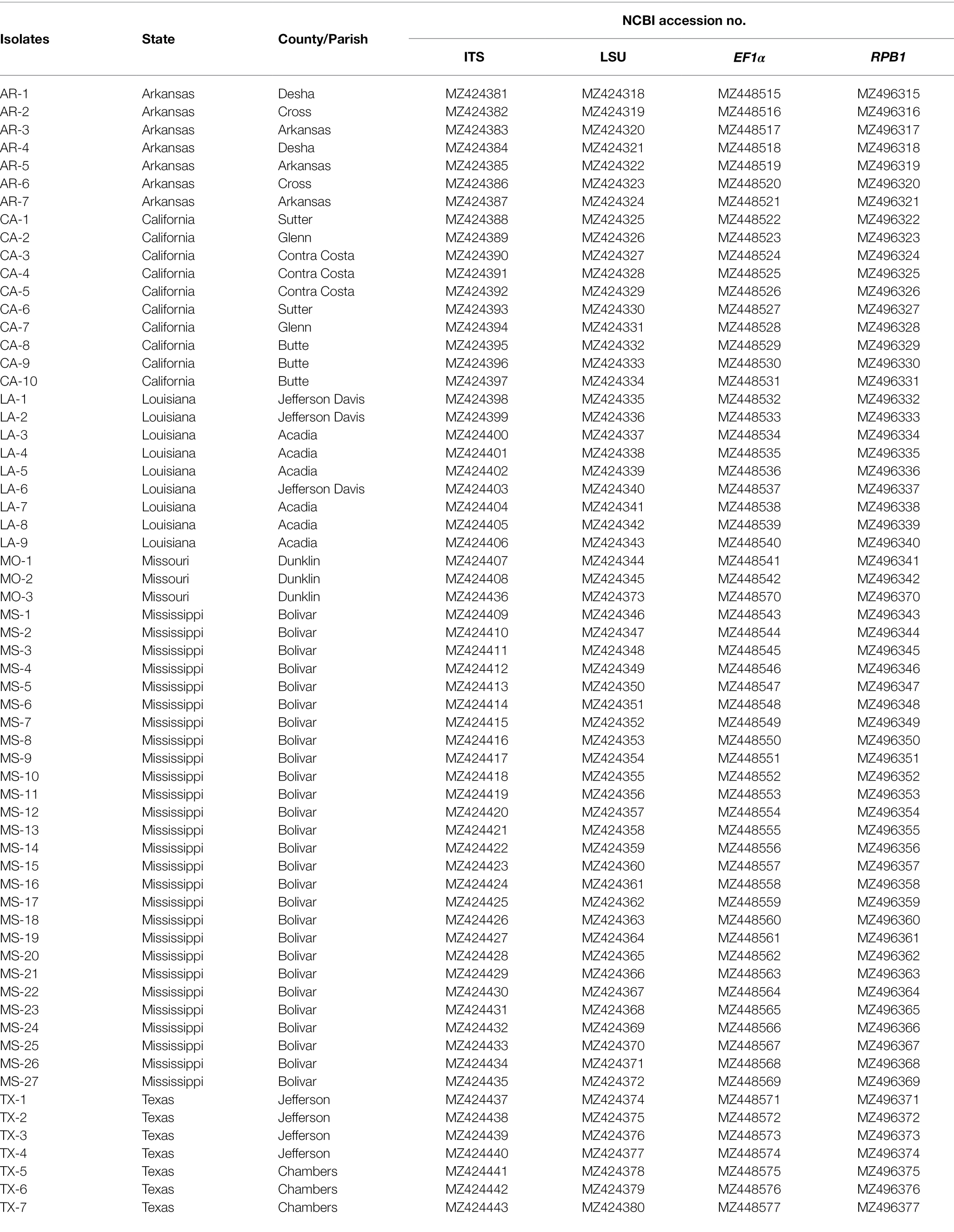
Table 1. Geographic origin and NCBI accession number of 63 isolates of Tilletia horrida sequenced in this study.
Tilletia horrida isolates growing in PDA plates for 14 days were used for DNA extraction. Mycelium was collected by washing the culture plates with 1% NaCl solution and 100 mg (fresh weight) of the mycelium mass were used for the DNA extraction. DNA was extracted using the fungi/yeast genomic isolation kit (Norgen Biotek Corp., ON, Canada) following the manufacturer’s protocol. The quality of the DNA was checked using the Spectramax quickdrop spectrophotometer (Molecular Devices LLC, San Jose, CA).
Genomic DNA of the T. horrida isolates was amplified by PCR using four different genomic regions, consisting of two protein-coding genes: translation elongation factor 1-α (EF-1α) and the largest subunit of RNA polymerase II (RPB1), and two rRNA regions: ITS1 through 2 regions and D1/D2 domains of the large subunit (LSU) rRNA. The primers used in this study were obtained from previous studies (Fell et al., 2000; Wang et al., 2014) and the conserved primer sequence website of the Vilgalmys Mycology lab-Duke University.1 Each PCR reaction mixture was composed of 3 μl of DNA adjusted between 10–50 ng/μl, 12.5 μl of 2x KAPA 2G master mix (KAPA Biosystems, Roche Sequencing, Wilmington, MA, United States), 1.25 μl of forward, 1.25 μl of reverse primers, and 8 μl of water to bring the total reaction volume to 25 μl. PCR parameters for amplifying EF-1α and RPB1 were used in this study were the same as described previously (Wang et al., 2014). Amplification for ITS and LSU were performed as follows: initial denaturation at 94°C for 5 min followed by 40 cycles of denaturation at 30s at 94°C, annealing 15 s at 53.5°C, and elongation at 30s at 72°C; and final elongation at 72°C at 5 min. All PCR amplification was conducted in Biometra TOne Thermocycler (Analytikjena, Jena, Germany). All PCR products were run in 1% agarose gel and visualized in blue light. All PCR products were purified from the electrophoresis gel with Zymoclean Gel DNA recovery kits (Zymoresearch, Irvine, CA, United States) according to manufacturer’s recommendations. The purified PCR products were sequenced using capillary Sanger’s sequencing protocol by external sequencing service provider, Eton Biosciences Inc. (San Diego, CA, United States).
Sequences were manually curated and trimmed for noises at the 5′ and 3′ ends. Consensus sequences from forward and reverse reads were generated by Benchling online.2 Sequences were aligned with MAFFT v7.475 (Katoh and Standley, 2013) with accurate alignment method, L-INS-I, built-in MAFFT function of “—adjustdirection” was used to orient the nucleotide sequences in same direction. All sequence alignments were edited and adjusted manually in MEGAX (Kumar et al., 2018). Three different phylogeny analyses were conducted as: Maximum Likelihood (ML; Felsenstein, 1981), Neighbor-Joining (NJ; Saitou and Nei, 1987), and Minimum Evolution (ME; Rzhetsky and Nei, 1993). ML analysis was performed with RaxML version 8.2.12 (Stamatakis, 2014). RaxML analysis was conducted for 1,000 bootstrap replicated with rapid bootstrap analysis with GTRCAT substitution approximation. NJ and ME analyses were performed in R 4.0.3 (R Core Team, 2020) with APE package version 5.4–1(Paradis et al., 2004) using Rstudio (R Studio Team, 2020). NJ analysis was performed in default mode, whereas ME was performed with balanced function (Desper and Gascuel, 2004). Tree topologies were visualized and edited using FigTree v1.4.4 (Rambaut, 2018). Overall, three different sequences sets were used to construct the phylogeny trees.
Aligned individual sequences were concatenated in different combinations to form three datasets: (1) Ribosomal RNA datasets of 1,075 bp formed by combination of LSU and ITS (including 5.8 rRNA) in that order; (2) Multi-gene datasets of 1,615 bp formed by combination of two protein-coding genes EF1-α and RPB1 in that order; and (3) Multi-gene datasets of 2,690 bp were formed by combining all four sequences, order of genes EF1-α, RPB1, LSU, and ITS (including 5.8S rRNA). Nucleotide sequence length was approximately 895, 720, 545, and 530 bp for EF1-α, RPB1, LSU, and ITS (including 5.8S rRNA), respectively. All datasets were subjected to all three phylogeny constructions such as ML, NJ, and ME. Tilletia horrida strain QB1 (Bio project no: PRJNA280382; Wang et al., 2015) was used as the reference. Tilletia controversa strain DAOMC 236426 (Bio project no: PRJNA393324; Nguyen et al., 2019) was used as an outgroup in the final tree.
In order to take advantage of the multiple T. horrida ITS sequences in the database (with no corresponding protein-coding gene sequences), we performed an ITS region-along sequence analyses. ITS sequences of all 63 T. horrida isolates from this study were subjected to the National Center for Biotechnology Institute (NCBI) BLAST (Altschul et al., 1990). All the hits in the NCBI results were downloaded for the analysis. Multiple entries in the result were cross-referenced based on the accession numbers and the duplicates were removed. A total of 172 unique accession numbers of various Tilletia spp. were downloaded from NCBI using BioPython 1.78 (Cock et al., 2009) in Python 3.8.5 (Van Rossum and Drake, 2009). Based on the preliminary tree branching pattern, a final ITS region-only phylogeny tree was constructed using 26 T. horrida sequences, six T. barclayana sequences, and one T. australiensis sequence (Table 2). Preliminary ITS region-only phylogeny tree with all 172 Tilletia spp. isolates, NCBI accession numbers, and other information are available in supplementary (Supplementary Table S1; Figure S2).
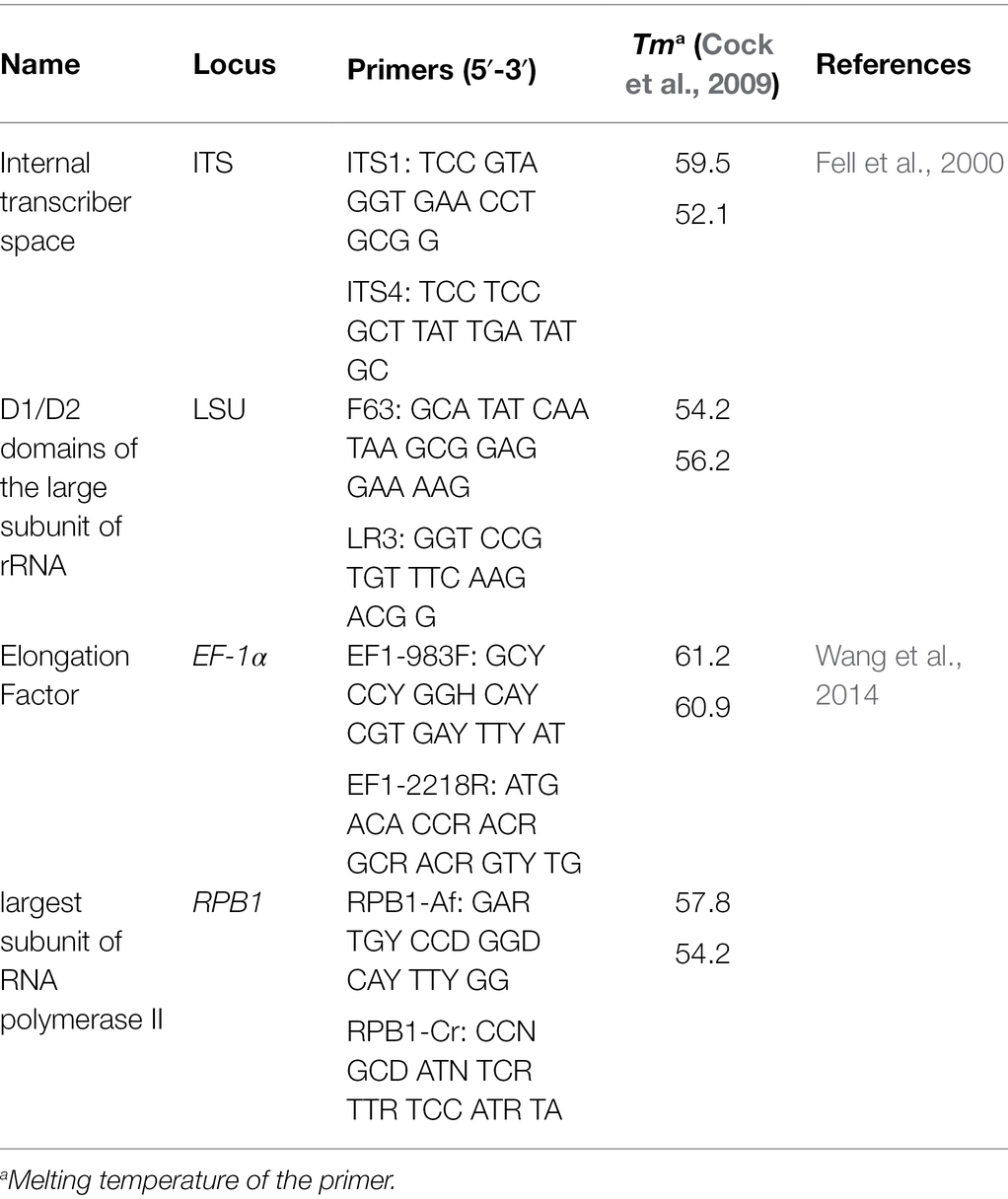
Table 2. Primers of ITS, LSU, EF-1α, and RPB1 used in this study.
DnaSP v6.0 (Rozas et al., 2017) was used to determine nucleotide diversity and the minimum number of recombination events. Similarly, DnaSP v 6.0 (Rozas et al., 2017) was also used for the calculation of class I neutrality tests: Tajima’s D and Fu and Li’s D* and F*, for detecting departure from the mutation/drift equilibrium (Tajima, 1989; Fu and Li, 1993). For the above-mentioned calculation, only T. horrida isolates were considered in multi-gene sequence sets and T. controversa was used as an outgroup as needed. However, for ITS region-only sequence sets, T. barclayana strain 104 was used as an outgroup as needed.
All the sequenced genes have been deposited into the National Center for Biotechnology Institute (NCBI) database under the following accession numbers: LSU, MZ424318–MZ424380, ITS, MZ424381–MZ424443, EF1, MZ448515–MZ448577, and RPB1, MZ496315–MZ496377.
We took advantage of the ITS region sequences available on multiple isolates in the databases and conducted phylogenetic analysis first based on the ITS region-only. For the analysis, we downloaded 172 various Tilletia spp. from NCBI and constructed a phylogenetic tree. Most of the isolates formed species-specific clades (Supplementary Figure S2). Hence, the final tree was constructed with 63 T. horrida isolates collected from this study, along with a subset of the sequences from the NCBI database, including 26 T. horrida isolates, six T. barclayana isolates, and one T. australiensis isolate downloaded from the NCBI database. All T. barclayana isolates were included in the final tree due to taxonomy controversy of the kernel smut fungus. Phylogeny based on ITS region (Figure 2) shows three different groups of the isolates. Most isolates (76%) were clustered together along with 22 isolates from eight different countries (Australia, China, India, Korea, Pakistan, Taiwan, the US, and Vietnam) in Clade III (Figure 2; Table 3). Out of the remaining 15 isolates, 11 T. horrida isolates collected in this study were clustered together in clade II. All the isolates clustered in clade II were from the current study. Clade I was grouped by clustering of seven isolates from China, one from Japan, and four T. horrida isolates from the current study (Figure 2; Table 3).
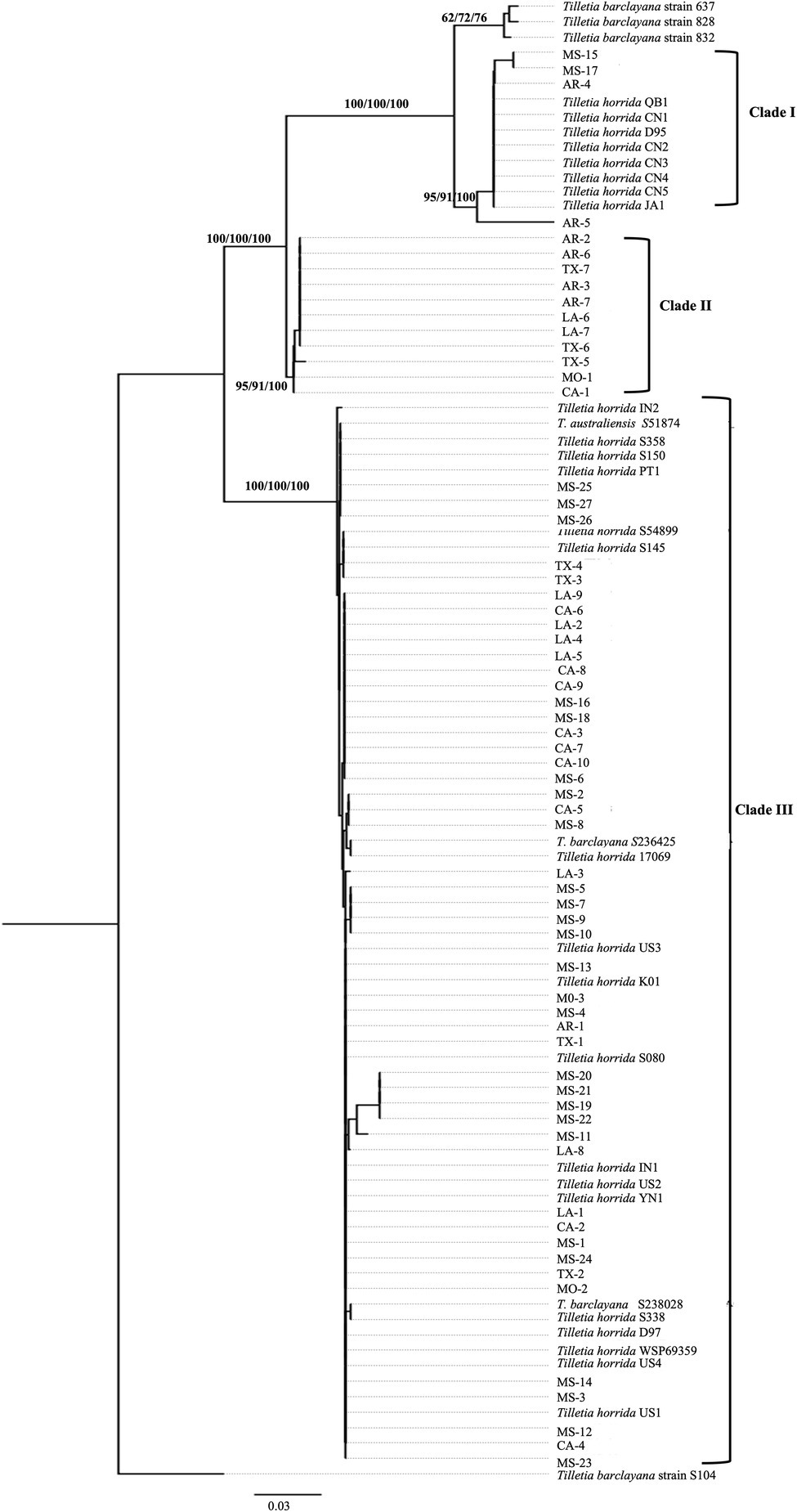
Figure 2. ITS region-based neighbor-joining phylogenetic tree with 63 T. horrida isolates collected Arkansas (AR), California (CA), Louisiana (LA), Mississippi (MS), Missouri (MO), and Texas (TX) in the US in the current study, along with 33 ITS sequences obtained from the NCBI database. T. horrida strain QB1 was used as a reference isolate and T. barclayana S104 was used as an outgroup. The scale bar represents the number of substitutions per site. The values on the branches indicate the percentage of trees based on 1,000 bootstrap replicates on ML/NJ/ME, respectively. Only branches values with >50% replicates are shown.
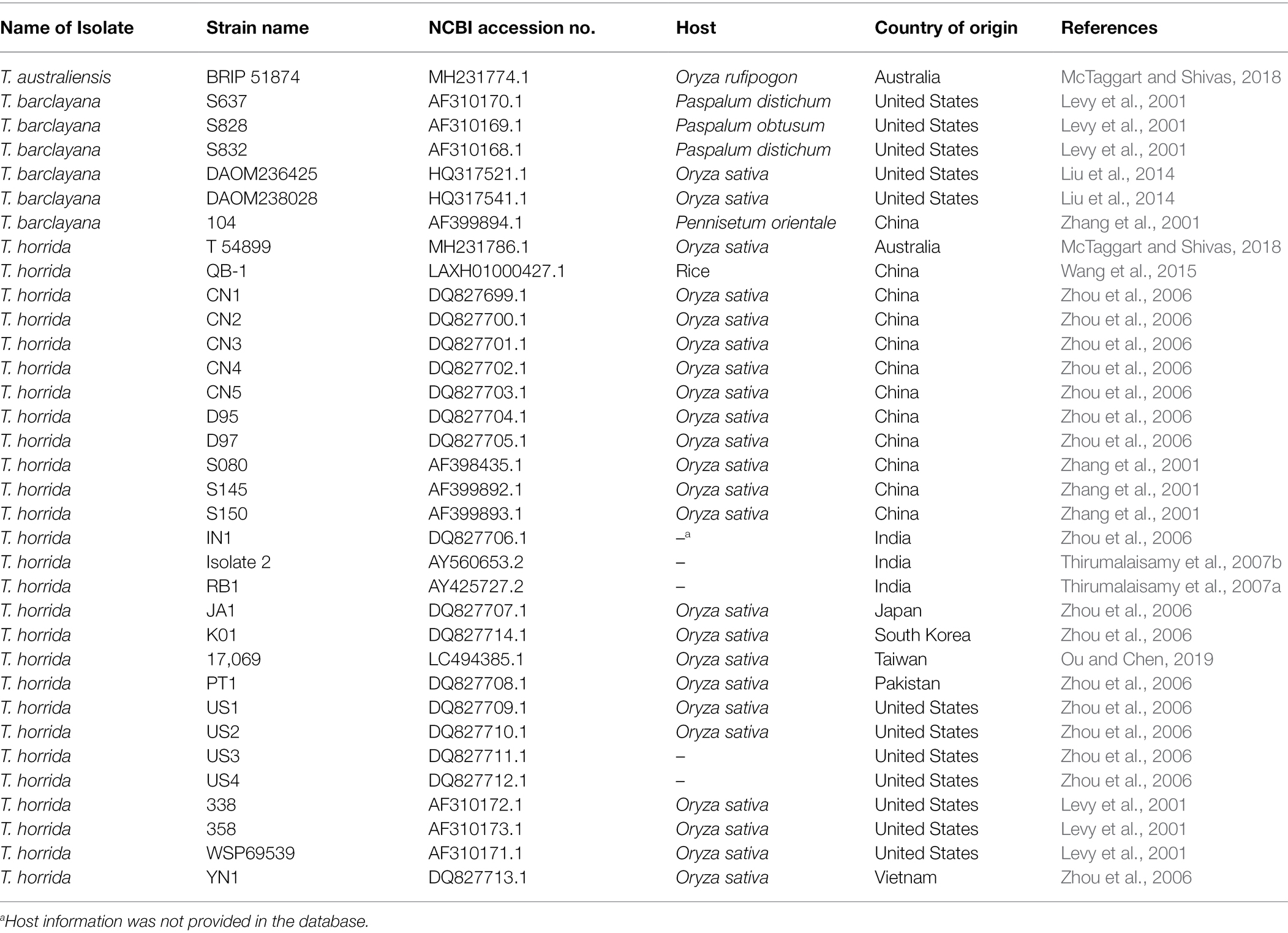
Table 3. NCBI accession number, host, and country of origin of the strains of Tilletia australiensis, T. barclayana, and T. horrida used in the final ITS-only region phylogenetic analysis.
The concatenated sequence EF1α-RPB1-LSU-ITS of 63 T. horrida isolates with 2,960 bp nucleotide was aligned and a phylogenetic tree was calculated with three different phylogenetic analyses. Phylogenetic analyses clustered the 63 isolates into five different groups (Figure 3). Most isolates (59%) were grouped together in clade V, which consists of the isolates collected from five different states (California, Louisiana, Mississippi, Missouri, and Texas). Most isolates collected from Mississippi (19/27), California (9/10), and Texas (4/7) were clustered in clade V. The isolates collected from Louisiana (4/9) and Missouri (1/3) were also grouped in clade V (Figure 3; Table 1).
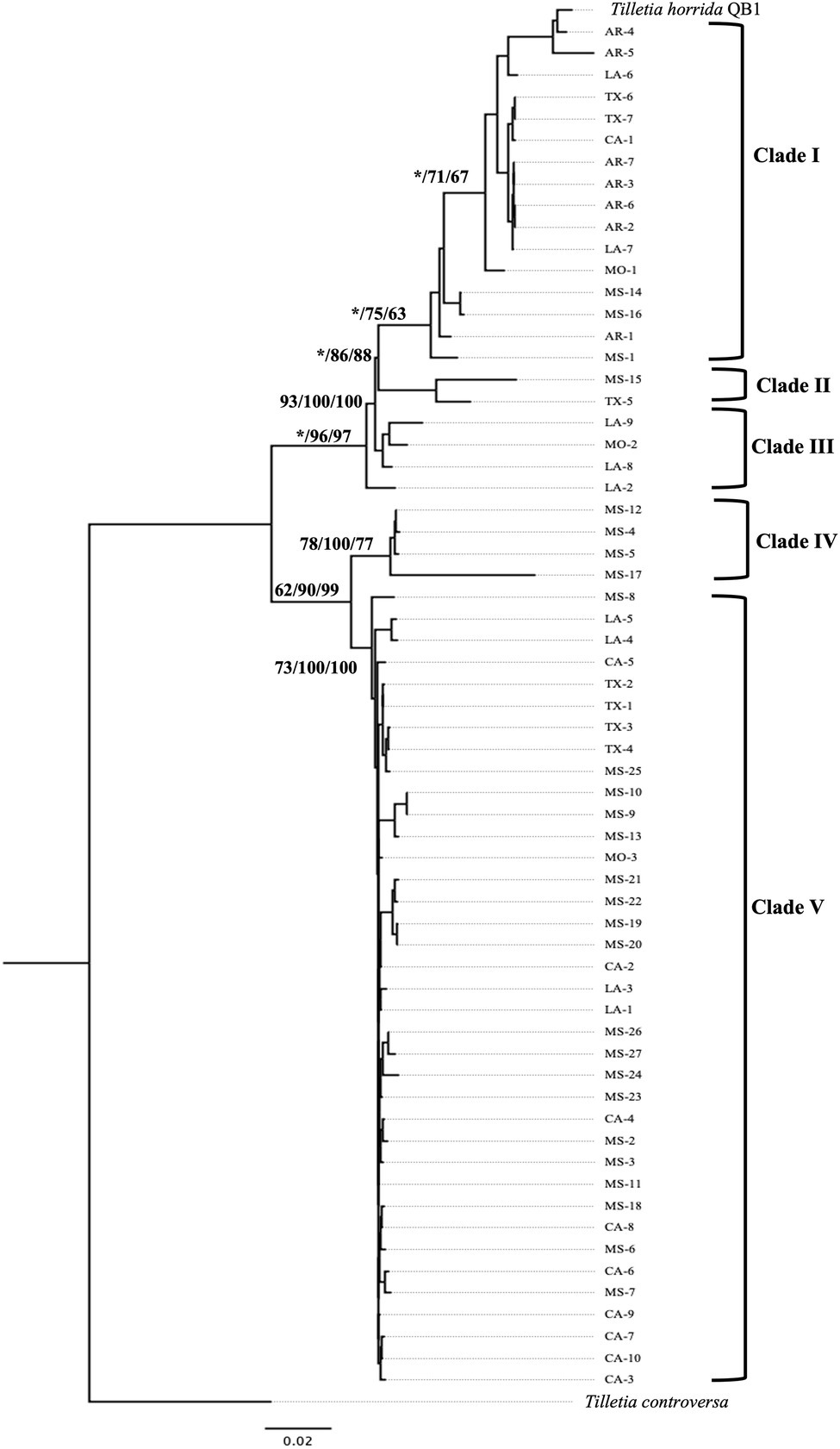
Figure 3. Neighbor-joining phylogenetic tree of concatenated sequences with EF-1α, RPB1, LSU, and ITS of 63 isolates collected from Arkansas (AR), California (CA), Louisiana (LA), Mississippi (MS), Missouri (MO), and Texas (TX) in the US. T. horrida strain QB1 was used as a reference isolate and T. controversa strain DAOMC 236426 was used as an outgroup. The scale bar represents the number of substitutions per site. The value on the branches indicates the percentage of trees based on 1,000 bootstrap replicates on ML/NJ/ME, respectively. Only branches values with >50% replicates are show; “*” indicates that the branch value is less than 50%.
Similarly, 27% of the 63 T. horrida isolates were clustered in clade I along with the reference isolate T. horrida strain QB1. All T. horrida isolates collected from Arkansas (7/7) were grouped in clade I, along with a few isolates collected from California (1/9), Louisiana (2/9), Missouri (1/3), Mississippi (3/27), and Texas (2/7). Additionally, clade III and clade IV clustered with four isolates each from Mississippi and Louisiana. Clade III consisted of the isolates collected from Louisiana (3/9) and Missouri (1/3), whereas all the isolates grouped in clade IV were collected from Mississippi (4/27). Two isolates, MS-15 and TX-5, were the only isolates to be grouped in clade II (Figure 3; Table 1).
Multi-locus phylogeny of the concatenated of LSU-ITS (including 5.8S) sequences of 63 isolates of T. horrida with 1,065 bp nucleotide was aligned and phylogenetic analyses were conducted. Our rRNA regions-based phylogenetic analysis clustered the 63 T. horrida isolates in four clades (Figure 4). Most of the isolates (76%) were grouped together in clade IV. Most isolates collected from Mississippi (20/27), California (9/10), Louisiana (7/9), and Texas (4/7), along with the isolates collected from Arkansas (1/7) and Missouri (2/3) were clustered together in clade IV (Figure 4; Table 1).
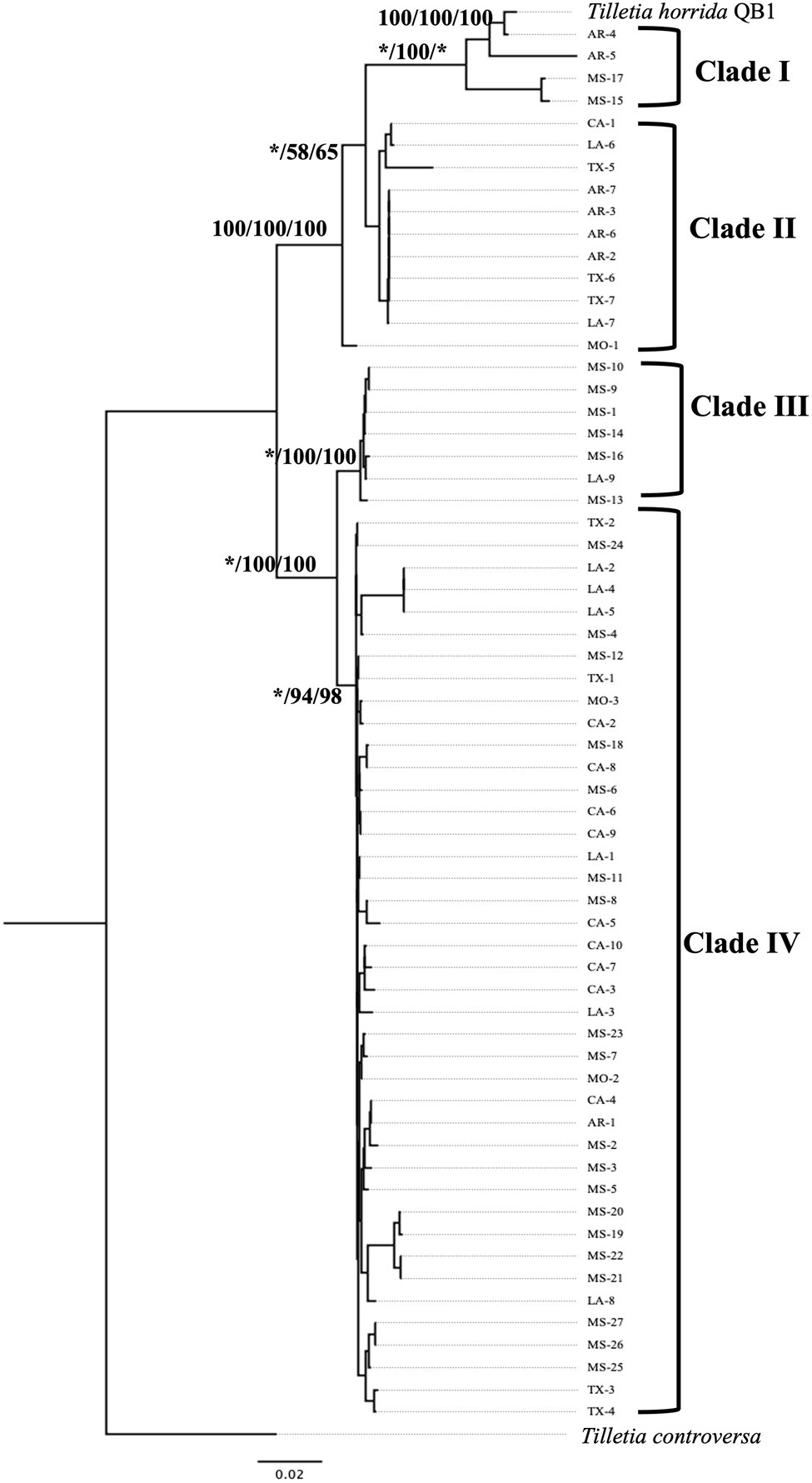
Figure 4. Neighbor-joining phylogenetic tree of concatenated sequences with rRNA regions LSU, and ITS of 63 isolates collected from Arkansas (AR), California (CA), Louisiana (LA), Mississippi (MS), Missouri (MO), and Texas (TX) in the US. T. horrida strain QB1 was used as a reference isolate and T. controversa strain DAOMC 236426 was used as an outgroup. The scale bar represents the number of substitutions per site. The value on the branches indicates the percentage of trees based on 1,000 bootstrap replicates on ML/NJ/ME, respectively. Only branches values with >50% replicates are shown; “*” indicates that the branch value is less than 50%.
Similarly, two isolates from Arkansas and Mississippi each were grouped together in clade I, along with the reference isolates T. horrida strain QB1. Clade II consisted of the isolates collected from Arkansas (4/7), California (1/10), Louisiana (2/9), and Texas (3/7). Similarly, clade III consisted of five isolates from Mississippi and one isolate from Louisiana. Isolate MO-1 did not group with clades and monophyletically branched with clade I and clade II (Figure 4; Table 1).
Multi-locus phylogeny of the concatenated of EF1-α and RPB1 sequences of 63 isolates of T. horrida with 1,615 bp nucleotide was aligned and phylogenetic analyses were conducted. Our protein-coding gene-based phylogenetic analysis clustered the 63 T. horrida isolates in six clades (Figure 5). Most of the isolates (60%) were grouped together in clade VI. Most isolates collected from Mississippi (19/27), California (9/10), and Texas (4/7), along with isolates collected from Louisiana (4/9) and Missouri (1/3) were clustered together in clade VI (Figure 5; Table 1).
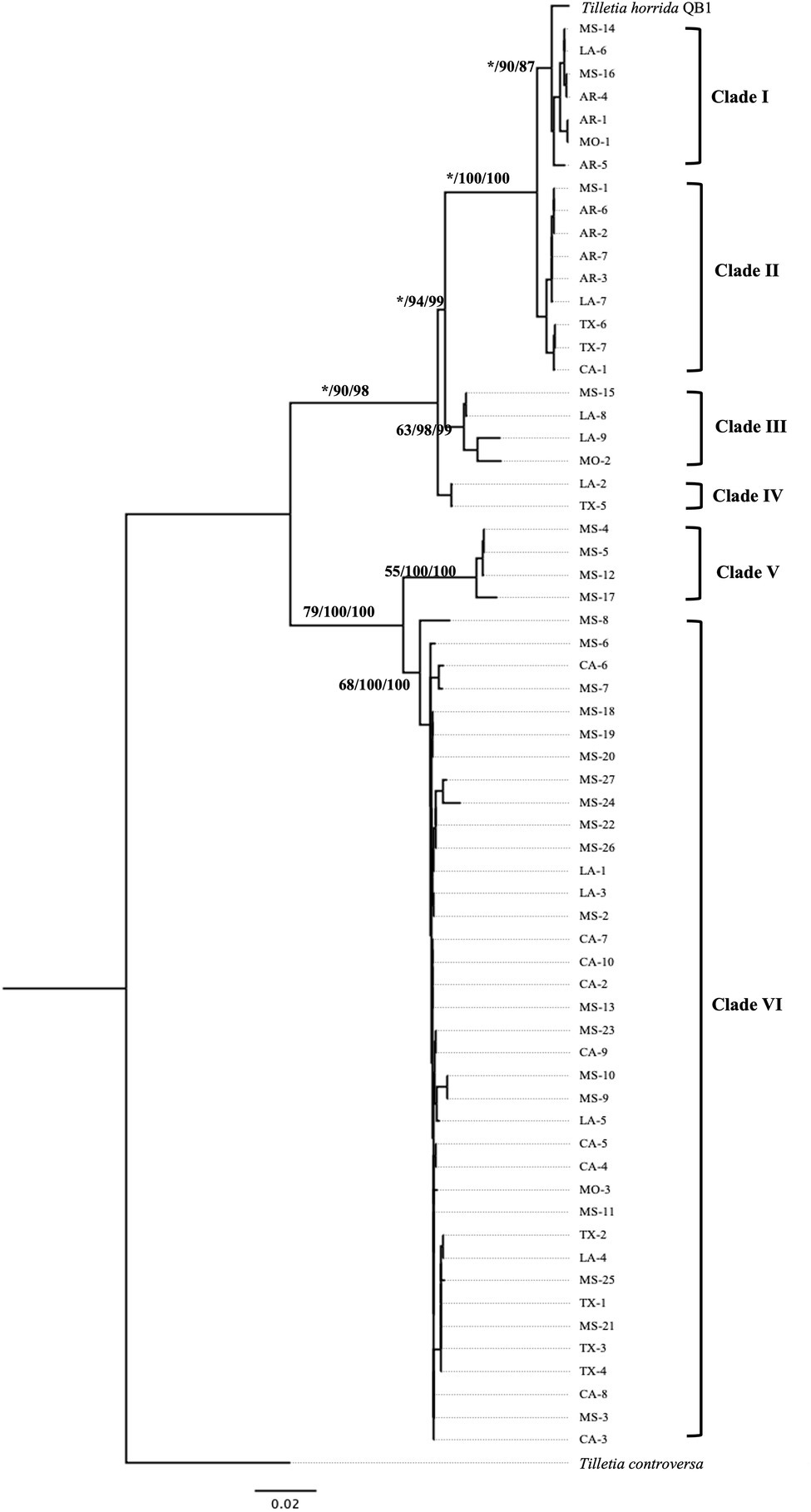
Figure 5. Neighbor-joining phylogenetic tree of concatenated sequences with protein coding genes EF-1α and RPB1of 63 isolates collected from Arkansas (AR), California (CA), Louisiana (LA), Mississippi (MS), Missouri (MO), and Texas (TX) in the US. T. horrida strain QB1 was used as a reference isolate and T. controversa strain DAOMC 236426 was used as an outgroup. The scale bar represents the number of substitutions per site. The value on the branches indicates the percentage of trees based on 1,000 bootstrap replicates on ML/NJ/ME, respectively. Only branches values with >50% replicates are show; “*” indicates that the branch value is less than 50%.
Similarly, clade I consisted of three isolates collected from Arkansas (3/7) and one isolate each from Mississippi, Missouri, and Louisiana. Similarly, clade II consisted of four isolates collected from Arkansas, and one isolate each from California, Mississippi, and Louisiana, along with two isolates from Texas. Similarly, clade III consisted of two isolates collected from Louisiana and isolate each from Missouri and Mississippi. Clade IV consisted of only two isolates, one from Louisiana and the other from Texas, whereas clade V were clustered with four isolates all collected from Mississippi.
Diversity parameters and neutrality test were calculated with concatenated sequence of all four regions and individually by DnaSP 6.0 (Rozas et al., 2017). Test of neutrality showed non-significant drift from mutation equilibrium in both Tajima’s D and Fu and Li’s D* and F* statistics for both ITS-only region and multi-locus concatenated sequence. Number of recombination event was predicted to be 4 and 50 in ITS-only sequence and multi-locus sequence sets, respectively (Table 4.)
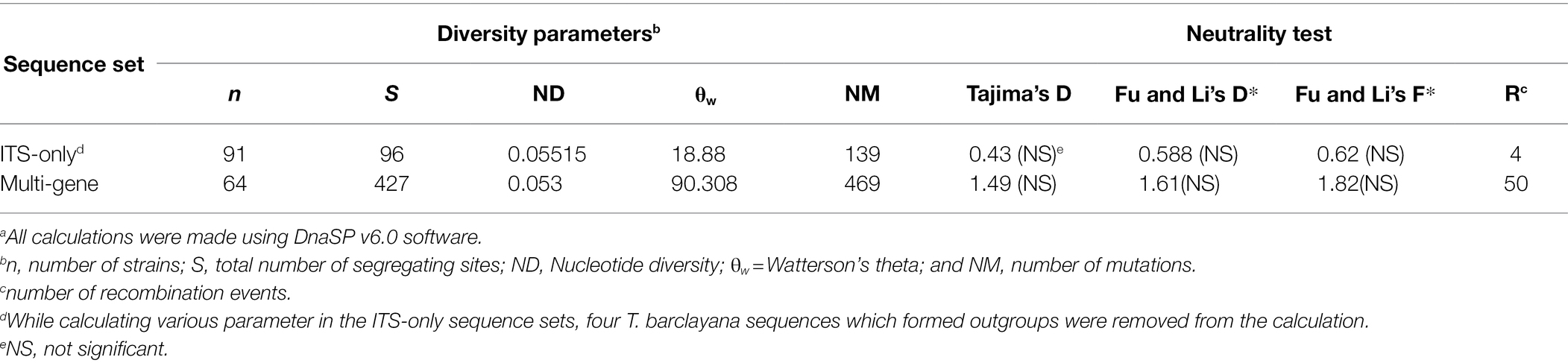
Table 4. Sequence variation statistics of the kernel smut fungal populations in the USa.
Among the individual regions nucleotide diversity (ND), Watterson’s theta (θw) and segregating sites were found to be highest in RPB1 region as compared to the other three regions. Test of neutrality showed significant drift from mutation equilibrium in both Tajima’s D and Fu and Li’s D* and F* statistics in RPB1 region, whereas only Fu and Li D* and F* statistics were significant in rRNA regions and EF1α region (Supplementary Table S2).
Kernel smut of rice has emerged as one of the most important diseases, threatening the US rice production. Economic impact of kernel smut is more significant from the loss of quality than from the loss of yield. In recent years, an increase in the severity and incidence of kernel smut and in cases of rejection of rice at selling point has been reported across the US, especially in Texas. In this study, we investigated the genetic diversity of the T. horrida populations in the US. Genomic DNA was extracted from the 63 isolates of T. horrida collected from Arkansas, California, Louisiana, Missouri, Mississippi, and Texas and subjected to multi-locus sequence analysis (MLSA). The results of our study showed the presence of genetically diverse T. horrida populations in the US. To our knowledge, this is the first study to analyze multi-locus region of the T. horrida populations in rice.
Our research reveals that there were five groups of the T. horrida populations in the US. Tilletia horrida clusters did not correspond with the geographical origin of the isolates collected. Only the isolates collected from Arkansas were clustered together in only one clade (clade I) with the reference isolate T. horrida strain QB1. Along with the Arkansas population, the isolates from California were the least diverse, whereas the population in Louisiana and Mississippi were most diverse as they grouped together in 3 out of 5 clades. Low diversity within the California population found in the current study can be attributed to quarantine practice that has been enforced in the state to prevent the introduction of disease, insect, and weed pests in rice seed into California from the southern region of the US and from foreign countries for many years (Oscar, 2006). The results of our research here are in contrast with those of Ustilaginoidea virens, the causal agent of false smut of rice, where a higher level of genetic differentiation among the U. virens populations found in the study with the isolates collected from geographically distant rice-growing areas of China (Sun et al., 2013). Unlike another bunt pathogen T. indica (Singh and Gogoi, 2011), the lack of correlation between genetic diversity and geographical specificity among the T. horrida populations in the current study may be attributed to unrestricted trading of rice seeds and lack of quarantine restriction in the southern US. In addition, the current study indicates that there are some levels of genetic differentiation in the T. horrida populations. These results have a direct implementation on the development of new rice cultivars with improved resistance to kernel smut. Future efforts toward breeding for resistant cultivars against kernel smut should consider selecting representative isolates from each of genetically diverse groups in the process of kernel smut resistance screening.
Neutrality tests suggest there was no significant departure from the mutation drift equilibrium, indicating the T. horrida population does not deviate from natural expectation in Tajima’s D and Fu and Li’s D* and F* tests (Tajima, 1989; Fu and Li, 1993). Individual phylogenetic analyses of all four regions showed higher nucleotide substitution in RPB1 and ITS as compared to the EF1⋅ and LSU regions. Along with higher nucleotide substitution per site, RPB1 also had highest nucleotide diversity (ND), Watterson’s theta (θw), and segregating sites. Similarly, based on Tajima’s D test, only RPB1 deviates from the mutation equilibrium with significant and positive Tajima’s D. Variation statistics showed 50 recombination events among the concatenated sequence. This might be due to the possible sexual recombination between different isolates. Tilleita horrida is a hemi-biotrophic fungus, which probably facilitates such recombination through mating of compatible types. However, no clear evidence of compatible sexual mating is still unknown in Tilletia horrida (Carris et al., 2006) or in similar non-systematic bunt Tilletia indica (Goates, 1988; Gupta et al., 2015).
Along with multi-locus analysis of the T. horrida populations in the US, we also analyzed 33 other Tilletia spp., including T. horrida and T. barclayana reported from eight different countries. Due to the lack of global information on multi-locus regions on the T. horrida isolates, we analyzed ITS regions-only reported in the NCBI database. Our analysis revealed that there are three groups of T. horrida isolates distributed in the world. The majority (76%) of the T. horrida isolates from this study, along with six other isolates reported previously from the US (Levy et al., 2001), was clustered together along with 15 other smut isolates from eight different countries. Similarly, four T. horrida isolates from this study were clustered together with seven T. horrida isolates reported from China (Zhou et al., 2006; Wang et al., 2015). Kernel smut is seedborne and rice is one of the most traded crops around the world (USDA, 2020). Our ITS regions-only analysis suggests potential multiple entries of the kernel smut pathogen from foreign countries into the US.
In the current study, clade III also clustered with T. australiensis isolated from wild rice (O. rufipogon) in Australia (McTaggart and Shivas, 2018) in addition to T. horrida isolates isolated from rice. ITS region sequence showed 99.75% similarity between T. australiensis and T. horrida strain 54,899 isolated from rice in Australia. Such high percentage of similarity between the kernel smut isolates from wild and cultivated rice indicates that the kernel smut fungus can infect multiple hosts and that wild rice potentially serves as an alternative host to the pathogen. The results of previously unverified reports indicate that T. horrida may infect Digitaria Haller, Leeria Sw., and Panicum L. (Tracy and Earle, 1896) and Pennisetum L. C. (Tullis and Johnson, 1952). However, until this date, there is no clear evidence that the T. horrida fungus can infect multiple hosts other than rice under natural conditions (Carris et al., 2006). Out of six T. barclayana isolates only two strains, DAOM 236425 and DAOM 238028, clustered together in clade III. Two T. barclayana strains clustered in clade III were the only T. barclayana isolated from rice (Liu, 2014), whereas other T. barclayana strains were isolated either from Paspalum spp. or Pennisetum orientale. Other strains formed a separate group from T. horrida isolates. Kernel smut isolated from other grasses clearly form different branches in the phylogenetic tree. Difference in the lineage of T. horrida and T. barclayana isolates have also been demonstrated in previous studies (Levy et al., 2001; Castlebury et al., 2005).
In conclusion, we can parse finger genetic diversity in the kernel smut fungus, T. horrida, using multi-locus sequence analysis. This is the first study analyzing the genetic diversity among the T. horrida populations in the US. Our study demonstrates the presence of genetically diverse of T. horrida isolates in different rice-growing states. Higher than 99% similarity in ITS region sequences between T. horrida isolates and between different countries may be attributed to the wide distribution of smutted rice along with global trade. The understanding of the genetic diversity of the T. horrida populations from the current study will help researchers develop effective host resistance and chemical management strategies, especially cultivar resistance, for the control of kernel smut of rice.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
SK, SA-B, SG, and X-GZ conceived and designed the experiments. SK and SG performed the isolations of the fungus and genomic DNA extractions. SK performed all sequencing experiments and wrote the manuscript. SK and SA-B analyzed the data. All authors have read and approved the manuscript.
This work was supported, in part, by USDA NIFA OREI (2015-51300-24286) and Texas Rice Research Foundation (TRRF 2018–2021).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank S. S. Uppala, J. Shi, and K. Wang and rice farmers, county extension agents, and crop consultants for assistance in collecting rice kernel smut samples in Arkansas (AR), California (CA), Louisiana (LA), Mississippi (MI), Missouri (MO), and Texas (TX). We also thank L. Espino (CA), A. Famoso (LA), J. Jiang (CA), D. L. Harrell (LA), A. A. McClung (AR), and P. L. Sanchez (CA) for providing rice kernel smut samples from their states. We thank Elek Nagy for proofreading the manuscript. Portions of this research were conducted with the advanced computing resources provided by Texas A&M High Performance Research Computing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.874120/full#supplementary-material
Allen, T. W., Growth, D. E., Wamishe, Y. A., Espino, L., Jones, G., and Zhou, X. G. (2020). Disease loss estimates from the rice producing states in the United States: 2018 and 2019. Proc. Rice Tech. Wrkg. Grp. 36:41.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bain, J. M., Tavanti, A., Davidson, A. D., Jacobsen, M., Shaw, D., Gow, N., et al. (2007). Multilocus sequence typing of the pathogenic fungus Aspergillus fumigatus. J. Clin. Microbiol. 45, 1469–1477. doi: 10.1128/JCM.00064-07
Biswas, A. (2003). Kernel smut disease of rice: current status and future challenges. Environ. Ecol. 21, 336–351.
Bougnoux, M.-E., Tavanti, A., Bouchier, C., Gow, N. A. R., Magnier, A., Davidson, A. D., et al. (2003). Collaborative consensus for optimized multilocus sequence typing of Candida albicans. J. Clin. Microbiol. 41, 5265–5266. doi: 10.1128/JCM.41.11.5265-5266.2003
Carris, L. M., Castlebury, L. A., and Goates, B. J. (2006). Nonsystemic bunt fungi-Tilletia indica and T. horrida: A review of history, systematics, and biology. Annu. Rev. Phytopathol. 44, 113–133. doi: 10.1146/annurev.phyto.44.070505.143402
Castlebury, L. A., Carris, L. M., and Vánky, K. (2005). Phylogenetic analysis of Tilletia and allied genera in order Tilletiales (Ustilaginomycetes; Exobasidiomycetidae) based on large subunitnuclear rDNA sequences. Mycologia 97, 888–900. doi: 10.1080/15572536.2006.11832780
Chahal, S., Aulakh, K. S., and Mathur, S. B. (1993). Germination of teliospores of Tilletia barclayana, the causal agent of kernel smut of rice, in relation to some physical factors. J. Phytopathol. 137, 301–308. doi: 10.1111/j.1439-0434.1993.tb01351.x
Choi, J., Park, S.-Y., Kim, B.-R., Roh, J.-H., Oh, I.-S., Han, S.-S., et al. (2013). Comparative analysis of pathogenicity and phylogenetic relationship in Magnaporthe grisea species complex. PLoS One 8:e57196. doi: 10.1371/journal.pone.0057196
Cock, P. J., Antao, T., Chang, J. T., Champman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Desper, R., and Gascuel, O. (2004). Theoretical foundation of the balanced minimum evolution method of phylogenetic inference and its relationship to weighted least-squares tree fitting. Mol. Biol. Evol. 21, 587–598. doi: 10.1093/molbev/msh049
Elshafey, R. A. S. (2018). Biology of rice kernel smut disease causal organism Tilletia barclayana and its molecular identification. J. phytopathol. Pest Manag. 5, 108–128.
Espino, L. (2019). Field sampling of kernel smut in rice and its effect on yield and quality. Phytopathology 109:1
Espino, L. (2020). Field sampling of kernel smut in rice and its effect on yield and quality. Proc. Rice Tech. Wrkg. Grp. 36:1
FAOSTAT (2019). FAOSTAT Database [Online]. Rome, Italy: FAO. Available: http://www.fao.org/faostat/en/#data/QC (Accessed August 25, 2020).
Fell, J. W., Boekhout, T., Fonseca, A., Scorzetti, G., and Statzell-Tallman, A. (2000). Biodiversity and systematics of basidiomycetous yesasts as determined by large subunit rDNA D1-D2 domain sequence analysis. Int. J. Syst. Evol. Microbiol. 50, 1351–1371. doi: 10.1099/00207713-50-3-1351
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Fu, Y. X., and Li, W. H. (1993). Statistical tests of neutrality of mutations. Genetics 133, 693–709. doi: 10.1093/genetics/133.3.693
Goates, B. J. (1988). Histology of infection of wheat by Tilletia indica, the Karnal bunt pathogen. Phytopathology 78, 1434–1441. doi: 10.1094/Phyto-78-1434
Gupta, A. K., Seneviratne, J. M., Bala, R., Jaiswal, J. P., and Kumar, A. (2015). Alteration of genetic make-up in Karnal bunt pathogen (Tilletia indica) of wheat in presence of host determinanats. Plant Pathol. J. 31, 97–107. doi: 10.5423/PPJ.OA.10.2014.0106
Gurjar, M. S., Aggarwal, R., Jain, S., Sharma, S., Singh, J., Gupta, S., et al. (2021). Multilocus sequence typing and single nucleotide polymorphism analysis in Tilletia indica isolates inciting Karnal bunt of wheat. J. Fungi 7:103. doi: 10.3390/jof702013
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kellner, R., Vollmeister, E., Feldbrügge, F., and Begerow, D. (2011). Interspecific sex in grass smuts and the genetic diversity of their pheromone-receptor systerm. PLoS Genet. 7:e1002436. doi: 10.1371/journal.pgen.1002436
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Levy, L., Castlebury, L. A., Carris, L. M., Meyer, R. J., and Pimentel, G. (2001). Internal transcribed spacer sequence-based phylogeny and polymerase chain reaction-restriction gragment length polymorphism differentiation of Tilletia walkeri and T. indica. Phytopathology 91, 935–940. doi: 10.1094/PHYTO.2001.91.10.935
Liu, M., Mccabe, E., Chapados, J. T., Carey, J., Wilson, S. K., Tropiano, R., et al. (2014). Detection and identification of selected cereal rust pathogens by TaqMan® real-time PCR. Can. J. Plant Pathol. 37, 92–105. doi: 10.1080/07060661.2014.999123
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Nat. Acad. Sci. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
McTaggart, A. R., and Shivas, R. G. (2018). Direct submission. Queensland Alliance for Agriculture and Food Innovation, University of Queensland. Bethesda (MD): National Library of medicine (US), National Center for biotechnology information.
Nguyen, H. D. T., Sultana, T., Kesanakurti, P., and Hambleton, S. (2019). Genome sequencing and comparison of five Tilletia species to identify candidate genes for the detection of regulated species infecting wheat. IMA Fungus 10:11. doi: 10.1186/s43008-019-0011-9
Oscar, J. (2006). “Rice pathology,” in Rice Breeding Progress Report and 2007 Research Proposal. ed. C. A. Biggs (United States: California Rice Research Station).
Ou, J. H., and Chen, C. Y. (2019). Survey of rice seed-borne fungi, with reference to their pathogenicity. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. Available at: https://www.ncbi.nlm.nih.gov/nuccore/LC494385.1 (Accessed January 25, 2022).
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
R Core Team (2020). R: A Language and Environment for Statitical Computing. Vienna, Austria: R Foundation for Statistical Computing.
Roux, C., Almaraz, T., and Durrieu, G. (1998). Phylogeny of Some Smuts Fungi Based on ITS [international transcribed spacer] Sequence Analysis. France: Comptes Rendus de l’Academie des Sciences Serie 3 Sciences de la Vie.
Rozas, J., Ferrer-Mata, A., Sánchez-Delbarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Rzhetsky, A., and Nei, M. (1993). Theoretical foundation of the minimum-evolution method of phylogenetic inference. Mol. Biol. Evol. 10, 1073–1095.
Saitou, N., and Nei, M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Sedaghatjoo, S. (2021). Genome Sequencing and Phylogenetic Analyses of Common and Dwarf Bunt of Wheat Provide Insights into their Genomic Diversity and Species Boundaries, and Enable the Development of Detection Assay for Tilletia Controversa. Germany: Georg-Augus-Universaität Göttingen.
Singh, D. V., and Gogoi, R. (2011). Karnal bunt of wheat (Triticum sp.): A global scenario. I. J. Agric. Sci. 81, 3–14.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, X., Kang, S., Zhang, Y., Tan, X., Yu, Y., He, H., et al. (2013). Genetic diversity and population structure of rice pathogen Ustilaginoidea virens in China. PLoS One 8:e76879. doi: 10.1371/journal.pone.0076879
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595. doi: 10.1093/genetics/123.3.585
Takahashi, Y. (1896). On Ustilago virensi Cooke and a new species of Tilletia parasitic on rice plant. Bot. Magaz. Tokyo 10:en16. doi: 10.15281/jplantres1887.10.109_16
Taylor, J. W., and Fisher, M. C. (2003). Fungal multilocus sequence typing—it’s not just for bacteria. Curr. Opin. Microbiol. 6, 351–356. doi: 10.1016/S1369-5274(03)00088-2
Thirumalaisamy, P. P., Renu Singh, D. V., and Aggarwal, R. (2007a). ITS nucleotide sequence of Tilletia horrida isolate RB1 from India. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. Available at: https://www.ncbi.nlm.nih.gov/nuccore/AY425727.2 (Accessed January 25, 2022).
Thirumalaisamy, P. P., Renu Singh, D. V., and Aggarwal, R. (2007b). Direct Submission. National Library of Medicine (US), National Center for Biotechnology Information. Available at: https://www.ncbi.nlm.nih.gov/nuccore/AY560653.2 (Accessed January 25, 2022).
Tracy, S. M., and Earle, F. S. (1896). New Species of fungi from Mississippi. Bull. Torrey Bot. Club 205, 23, doi: 10.2307/2478175
Tullis, E. C., and Johnson, A. G. (1952). Synonymy of Tilletia horrida and Neovossia barclayana. Mycologia 44, 773–788. doi: 10.1080/00275514.1952.12024236
USDA (2020). Rice Yearbook [Online]. Available at: https://www.ers.usda.gov/data-products/rice-yearbook/ (Accessed October 27, 2020).
Wang, N., Ai, P., Tang, Y., Zhang, J., Dai, X., Li, P., et al. (2015). Draft genome sequence of the rice kernel smut Tilletia horrida strain QB-1. Genome Announc. 3:15. doi: 10.1128/genomeA.00621-15
Wang, A., Pan, L., Niu, X., Shu, X., Yi, X., Yamamoto, N., et al. (2019a). Comparative secretome analysis of different smut fungi and identification of plant cell death-inducing secreted proteins from Tilletia horrida. BMC Plant Biol. 19:360. doi: 10.1186/s12870-019-1924-6
Wang, A., Pang, L., Wang, N., Ai, P., Yin, D., Li, S., et al. (2018). The pathogenic mechanisms of Tilletia horrida as revealed by comparative and functional genomics. Sci. Rep. 8:15413. doi: 10.1038/s41598-018-33752-w
Wang, A., Shu, X., Niu, X., Yi, X., and Zheng, A. (2019b). Transcriptome analysis and whole genome re-sequencing provide insights on rice kernel smut (Tilletia horrida) pathogenicity. J. Plant Pathol. 102, 155–167. doi: 10.1007/s42161-019-00401-8
Wang, Q. M., Theelen, B., Groenewald, M., Bai, F. Y., and Boekhout, T. (2014). Moniliellomycetes and malasseziomycetes, two new classes in Ustilaginomycotina. Persoonia 33, 41–47. doi: 10.3767/003158514X682313
Way, M. O., and Zhou, X. G. (2020). Rice water weevils, kernel smut and rice plant hoppers in rice farming. Available at: https://www.ricefarming.com/departments/specialists-speaking/if-you-had-smut-in-2019-plan-to-protect-crops-this-season/ (Accessed April 15, 2020).
Whitney, N. G., and Cartwright, R. D. (2018). Kernel Smut in Compendium of Rice Diseases and Pests. 2nd Edn. St. Paul, Minnesota, USA: The American Phytopathological Society.
Zhang, G. M., Yao, Y. J., Qi, P. K., and Zhang, Z. (2001). Phylogenetic analysis of nucleotide sequences from ITS region of Tilletia indica and its related species. National Library of Medicine (US), National Center for Biotechnology Information. Available at: https://www.ncbi.nlm.nih.gov/popset?DbFrom=nuccore&Cmd=Link&LinkName=nuccore_popset&IdsFromResult=15282033 (Accessed January 25, 2022).
Zhou, X. G., Khanal, S., and Imran, M. (2021a). Texas rice: severe outbreaks of kernel smut in 2021. Agfax.com-Online Ag News Source. Available at: https://agfax.com/2021/09/10/texas-rice-severe-outbreaks-of-kernel-smut-in-2021/ (Accessed September 10, 2021).
Zhou, X. G., Uppala, L. S., Liu, B., Guo, Z., Gaire, S., Lei, S., et al. (2020). In-Vitro and Field Evaluation of Fungicides for Control of Rice Kernel Smut. Proc. Rice Tech Wrkg Grp. 36:121.
Zhou, X. G., Way, M. O., McClung, A., and Duo, F. (2021b). Texas organic rice production guidelines. Texas AgriLife Research EPLP-052. Available at: https://beaumont.tamu.edu/eLibrary/Bulletins/2021_OrganicRice_Production_Guidelines.pdf (Accessed March 30, 2022).
Zhou, Y., Yang, L., Yi, J., Zhou, G., Yin, L., and Dong, Y. (2006). ITS Sequence Analysis of Tilletia horrida. Available at: https://www.ncbi.nlm.nih.gov/popset?DbFrom=nuccore&Cmd=Link&LinkName=nuccore_popset&IdsFromResult=111034999 (Accessed March 30, 2022).
Keywords: Rice, kernel smut, Tilletia, Tilletia horrida, Tilletia barclayana, genetic diversity
Citation: Khanal S, Antony-Babu S, Gaire SP and Zhou X-G (2022) Multi-Locus Sequence Analysis Reveals Diversity of the Rice Kernel Smut Populations in the United States. Front. Microbiol. 13:874120. doi: 10.3389/fmicb.2022.874120
Edited by:
Baokai Cui, Beijing Forestry University, ChinaReviewed by:
Alexander N. Ignatov, Peoples’ Friendship University of Russia, RussiaCopyright © 2022 Khanal, Antony-Babu, Gaire and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin-Gen Zhou, eHpob3VAYWVzcmcudGFtdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.